
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation of the mechanism of methanol electrooxidation: a potential-dependent DFT study†
Wei Zhang *,
Biyi Huang,
Yang Cui,
Lifang Shen and
Shubin Yan*
*,
Biyi Huang,
Yang Cui,
Lifang Shen and
Shubin Yan*
Nanxun Innovation Institute, Zhejiang University of Water Resources and Electric Power, Hangzhou, 310018, China. E-mail: zhangw@zjweu.edu.cn; yanshb@zjweu.edu.cn
First published on 8th April 2025
Abstract
The methanol electrooxidation reaction (MER), a critical process in direct methanol fuel cells, is systematically investigated through potential-dependent density functional theory (DFT) simulations to unravel its mechanism and potential effects on Pt-based catalysts. For pure Pt, the rate-determining steps (RDSs) are identified as methanol adsorption and CO oxidation, leading to a high overpotential of 0.9 V. Alloying Pt with Cu (PtCu) significantly reduces the overpotential to 0.7 V, with CO oxidation remaining the sole RDS. Potential-dependent analysis reveals that PtCu exhibits enhanced methanol adsorption and weakened CO binding strength due to electronic structure modulation, effectively mitigating CO poisoning. Furthermore, multiple reaction pathways occur on PtCu surfaces, accelerating intermediate consumption. This work elucidates the regulatory effects of electrode potential on reaction thermodynamics, pathway selection, and adsorption behavior, providing theoretical insights for designing efficient and CO-tolerant bimetallic catalysts.
1 Introduction
The investigation of the mechanism underlying methanol electrooxidation reactions (MERs) is essential for optimizing reaction processes and designing efficient catalysts.1 Density functional theory (DFT), a powerful tool, has significantly contributed to understanding the fundamental aspects of the electrochemical MER, taking into account electrochemical environment effects and catalyst properties.2–4 Based on DFT calculations, the MER has been predicted as a slow process with multiple reaction pathways and various reaction intermediates.5 The path initiates either C–H, C–O or O–H bond cleavage (usually classified as the CO or non-CO pathway),6 which compete each other depending on specific conditions and the catalysts employed.7 Furthermore, the consumption of some key reaction intermediates is considered the rate-determining step (RDS), which controls the overall performance of the MER.8 In total, through the application of DFT calculations, the complex nature of the MER has been elucidated, highlighting the competitive pathways and the influence of key reaction intermediates, contributing to mechanism understanding and catalyst optimization.However, the existing theoretical research has neglected the crucial influence of electrode potential, leading to erroneous predictions regarding the reaction path, RDS and overpotential.9,10 This limitation can be attributed to the common adoption of a constant charge model in simulations, which neglects the significant impact of charge defects on reactivity observed in experiments conducted under the grand canonical (GC) ensemble.11,12 As the primary driving force in electrochemical reactions, the applied potential should be accurately considered in mechanism investigation.11 Consequently, all reaction simulations should be conducted within constant potential models to reflect the true potential dependence. Fortunately, several valuable potential-dependent DFT methods, such as the modified Poisson–Boltzmann method13 and homogeneous background method,14 have been proposed and used to provide more reliable and insightful results, paving the way for a deeper understanding of complex electrochemical reactions.
Alloying Pt with other metals can not only reduce the cost, but also improve tolerance towards CO poisoning. Based on DFT predictions, a bifunctional mechanism was demonstrated by Pt-based alloys, where water is activated over dopant metals in the surface to form active hydroxyl species, which then oxidize CO bound to neighboring Pt sites. Among various Pt-based alloy catalysts, PtCu alloys are increasingly attractive for the MER. The metal ratios and crystal shape are widely confirmed to have a significant influence on MER performance.15–17 An accurate and deep understanding of the catalytic mechanism is essential for PtCu catalyst design. Herein, we present a potential-dependent DFT study of the reaction mechanism of the MER on Pt-based catalysts. The potential effects on the structure, adsorption stability, overpotential, and reaction routes are compared and discussed.
2 Computational method
All simulations were performed based on the density functional theory, using the PBE-D3 functional and VASPsol implemented in the Vienna ab initio simulation package (VASP).18–20 The ion–core interactions were described using the projected augmented wave (PAW) method.21 The exchange–correlation effects were accounted for using the Perdew–Burke–Ernzerhof (PBE) functional within the generalized gradient approximation (GGA).22 In the plane wave basis set, the cutoff energy was set to 500 eV, and the 3 × 3 × 1 Monkhorst–Pack K-point mesh was applied to sample the Brillouin zone. Convergence criteria of 10−5 eV and 0.03 eV Å−2 were set for electronic iterations and force on each atom, respectively. The calculation parameters have demonstrated an accurate description of the geometry structures of bulk Pt (lattice constant: 3.968 Å), which consist well with experimental values (3.924 Å). Pt and PtCu were investigated in our work (Fig. s1†). For Pt electrodes, symmetric 5-layered Pt(100) slabs with a 2 × 2 supercell were built from the relaxed bulk crystal (containing 40 Pt atoms). PtCu electrode was constructed by replacing the surface Pt atom in Pt(100) with Cu at an atomic ratio of 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1. A vacuum region of 20 Å was placed between slabs to avoid interactions between the periodic cells. The implicit solvent of water was considered based on polarizable continuum model (PCM) as provided in VASPsol,23 since dispersion has significant effects on adsorption predictions. For the solvent parameters, the dielectric constant εr, surface tension of cavity τ and the cutoff charge density nc were set to 78.4, 0 meV Å−2 and 2.5 × 10−4 Å−3, respectively.
:1. A vacuum region of 20 Å was placed between slabs to avoid interactions between the periodic cells. The implicit solvent of water was considered based on polarizable continuum model (PCM) as provided in VASPsol,23 since dispersion has significant effects on adsorption predictions. For the solvent parameters, the dielectric constant εr, surface tension of cavity τ and the cutoff charge density nc were set to 78.4, 0 meV Å−2 and 2.5 × 10−4 Å−3, respectively.
The system work function (Ewf) is defined as the Fermi level position relative to the vacuum potential, which can be calculated using the formula Ewf = Esol − Efermi, where Esol and Efermi represent the electrostatic potential of the bulk implicit solvent and Fermi level, respectively. For an open electrochemical system with variable charges, the applied electrode potential (U) is obtained by adding positive or negative charges on the surface, and it is referenced to the standard hydrogen electrode (SHE), which can be calculated using the formula U = (Ewf − ΦSHE)/|e|, where ΦSHE is the work function of the SHE (4.44 eV).
To explore the potential dependence of MERs, we used a GC DFT method in which the GC reaction free energy is computed as a function of the electrode potential based on the homogeneous background method.14,24–26 The potential-dependent GC free energy [F(U)] can be calculated using eqn (1):
| F(U) = G(U) − Efermi(N – N0) | (1) |
| F(U) ≈ G0 − 0.5C(U − UPZC)2 | (2) |
| ΔF(Ui) = Freactant(Ui) − Fproduct(Ui) | (3) |
| G(H+ + e−) = 0.5G(H2) − |e|U | (4) |
| Fb(Ui) = Ftotal(Ui) − Fad(Ui) − Fsub(Ui) | (5) |
3 Results and analysis
3.1 Method introduction
The MER represents a crucial electrochemical process within the direct methanol fuel cell (DMFC), taking place at the anode where catalyst-coated electrodes are situated. The MER activity in heterogeneous catalysis is highly dependent on the surface composition of alloys.28 In this study, we conducted an investigation into the MER using Pt and PtCu catalysts. The onset of the MER typically involves methanol adsorption, followed by the oxidation of methanol to CO2 through a series of six-electron transfer steps. The electrons released in each step contribute to the electrical current in the electrochemical cell, while the protons generated participate in the overall reaction. The entire process exhibits a remarkably low thermodynamic potential of 0.02 V, as exemplified by the equation CH3OH + H2O → CO2 + 6H+ + 6e− (as illustrated in Fig. 1a). The complex reaction pathways can be effectively delineated using Table 1. | ||
| Fig. 1 (a) The proposed reaction route of the MER. (b) Generic representation of the relative GC free energies of reactants (blue) and products (red) as a function of the electrode potential (U). Energy curves were obtained by fitting simulation results based on formulae (2) and (4). Adding negative or positive charges to control the applied potential also leads to a decrease in the GC free energies of reactants or products relative to the zero-charge state (UPZCreactant, UPZCproduct). At the crossing point of reactants and products, η refers to the overpotential that represents the minimum potential needed in electrochemical reactions. At a certain electrode potential Ui, the corresponding GC reaction free energy can be obtained using formula (3). | ||
| Step | Reaction |
|---|---|
| (i) Methanol adsorption | CH3OH + * → CH3OH* |
| (ii) 1st electron transfer | CH3OH* → CH2OH* + H+ + e− |
| (iii) 2nd electron transfer | CH2OH* → CHOH* + H+ + e− |
| CH2OH* → CH2O* + H+ + e− | |
| (iv) 3rd electron transfer | CHOH* → COH* + H+ + e− |
| CHOH* → HCO* + H+ + e− | |
| CH2O* → HCO* + H+ + e− | |
| (v) 4th electron transfer | COH* → CO* + H+ + e− |
| HCO* → CO* + H+ + e− | |
| (vi) 5th electron transfer | CO* + H2O → COOH* + H+ + e− |
| (vii) 6th electron transfer | COOH* → CO2 + H+ + e− |
In order to study the influence of potential on the reaction mechanism, a methodology for evaluating the stabilities of reactants and products is introduced herein.14,29 As a critical performance metric in electrochemical reactions, the reversible potential of a redox process can be determined by employing calculations based on formula (3). Specifically, the GC free energies of the reactants and products are computed as a function of the electrode potential, as dictated by formula (1). Subsequently, a fitting procedure based on formula (2) is applied to obtain a continuous energy-potential curve, enabling a comprehensive analysis of the system's behavior.
Fig. 1b depicts a schematic representation illustrating the relative GC free energies of the reactants and products as a function of the electrode potential (U). The fitting curves are positioned at the apex, representing the energy state of the zero-charge system at PZC (UPZCreactant and UPZCproduct). Below the PZC, the energy curves are delineated as dashed lines, symbolizing the addition of electrons into the system. Conversely, above the PZC, the energy curves are depicted as solid lines, signifying the removal of electrons from the system. The modulation of the electron count within the system results in a corresponding adjustment in the electrode potential. At a specific potential Ui, the energy difference between the reactants and products is evaluated, providing insights into the exothermic or endothermic nature of the reaction. The intersection point of the curves signifies the equality of the GC free energy between the reactants and products, with the corresponding potential representing the thermodynamic overpotential (η). This overpotential serves as an indicator of the minimum electrode potential required for the conversion of this specific reaction step.
3.2 Potential-dependent reaction energetics analysis
We must acknowledge that obtaining experimental evidence to elucidate the identities and potential dependencies of intermediates involved in the intricate 6-electron electrooxidation steps poses a considerable challenge.30 However, employing theoretical calculations within the GC ensemble enables the determination of stable adsorption configurations for potential intermediates arising from methanol dehydrogenation via C–H and O–H bond scission. Based on Table 1, the evolutions of relative free energies for reactants and products on Pt and PtCu surfaces are depicted as functions of the electrode potential shown in Fig. 2a and b, respectively (reacting species are shown on the right side). These energy profiles provide valuable insights into the thermodynamics of MER process.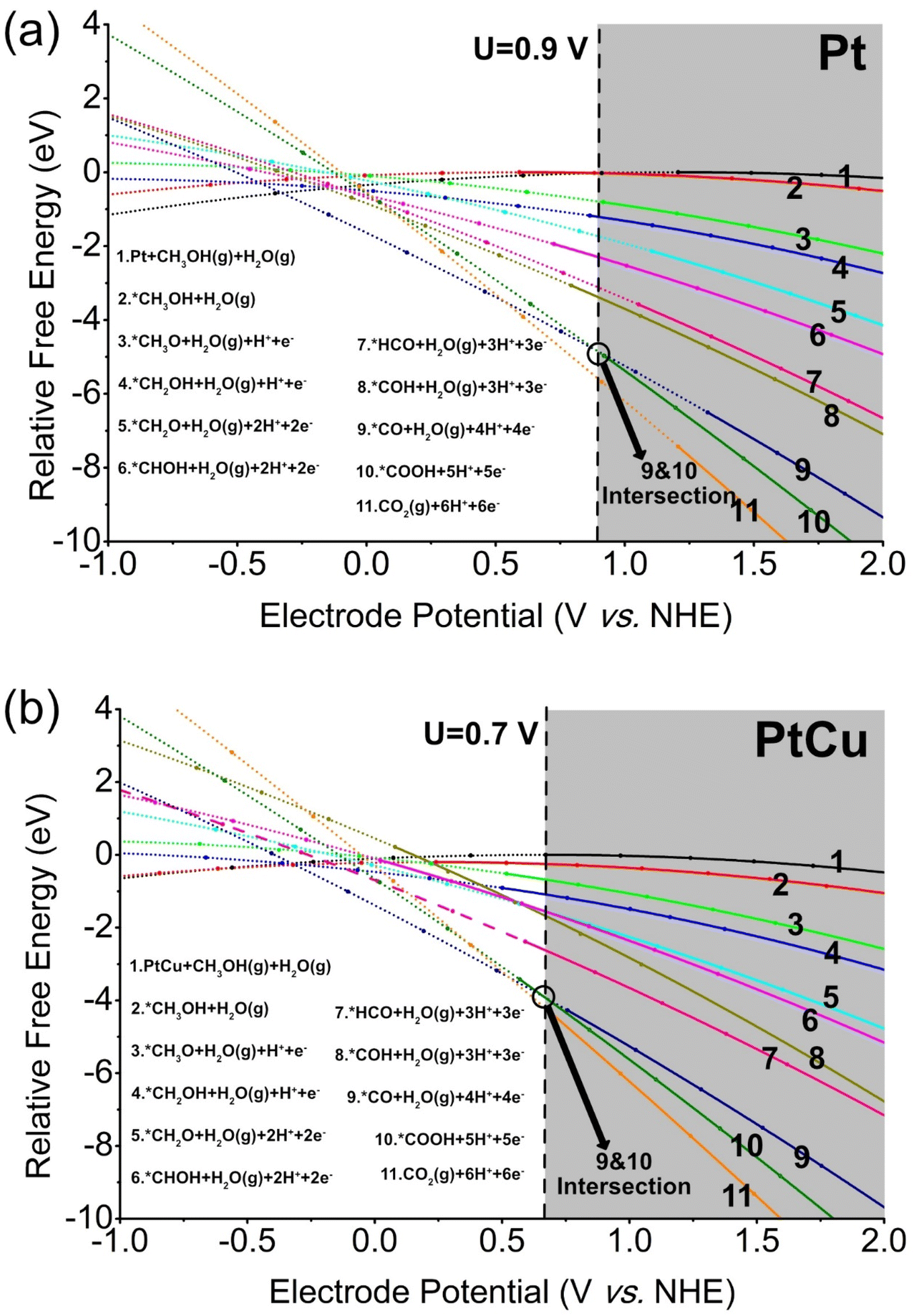 | ||
| Fig. 2 (a) The evolution of the relative free energy of the reactant or product is depicted as a function of the electrode potential, with reference to the normal hydrogen electrode (NHE), on a platinum (Pt) surface (details are shown in Table s1†). The data points obtained are fitted using a combination of formulas (2) and (4). In the plot, dashed lines represent reactants or products with negative charges, while solid lines represent those with positive charges. The grey area indicates that the MER is endothermic under the corresponding potentials. (b) The relative free energy of the reactant or product on the PtCu surface (details were shown in Table s2†). | ||
In Fig. 2a, energy curves of species 1 and species 2 intersect at U = 0.9 V, while species 9 and species 10 also intersect at U = 0.9 V. The observed intersection points indicate the occurrence of energetic balance between these species at the mentioned potential. Notably, for electrode potentials exceeding 0.9 V (grey region), the whole MER process becomes thermodynamically favorable, suggesting an exothermic nature of the overall process. Consequently, the overpotential for MER on a pure Pt catalyst is determined to be 0.9 V. It is noteworthy that both the methanol adsorption and CO oxidation on the Pt surface play pivotal roles as RDS in MER. This conclusion is consistent with experimental observations.31 Similarly, in Fig. 2b, the parabolas of species 9 and species 10 intersect at U = 0.7 V, indicating that the CO oxidation is the RDS for PtCu. This intersection point reflects the energetic equilibrium between these species at the specified potential. Importantly, it should be noted that PtCu exhibits a lower overpotential (0.7 V) compared to pure Pt (0.9 V). Additionally, the identified RDS significantly differs between Pt and PtCu, underscoring the distinct catalytic mechanisms operating in each system.
For pure Pt and PtCu, the removal of COads emerges as the RDS in the MER, thereby influencing the overall reaction rate. This phenomenon indicates that the density of COads over the catalyst surface increases at potentials below the overpotential, impeding the adsorption of fresh methanol molecules and leading to catalyst poisoning.32 It has been experimentally demonstrated that, at potentials of 0.3–0.4 V, the concentration of CO on Pt electrodes rapidly increases and reaches saturation, resulting in the rapid decay of the current.33 Notably, the RDS for PtCu exhibits a significantly lower overpotential compared with that for pure Pt, implying that COads species can be effectively consumed at lower potential. This distinctive characteristic of PtCu facilitates the efficient removal of COads, consequently weakening the adverse effects of CO poisoning, well consistent with our former experimental results.34,35
Obtained from Fig. 2, we next discuss the energetically favorable reaction paths for MER at specific electrode potentials of 0.7 V and 0.9 V shown in Fig. 3 and 4, respectively. Results highlight reaction energetics and reveal crucial intermediates and RDS involved in the MER process on Pt and PtCu surfaces.
 | ||
| Fig. 3 The free energy diagrams of methanol electrooxidation on Pt and PtCu at U = 0.7 V. | ||
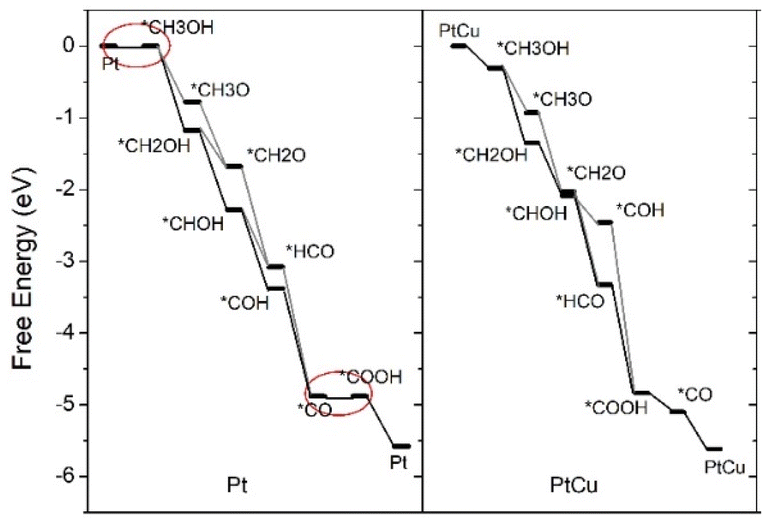 | ||
| Fig. 4 The free energy diagrams of methanol electrooxidation on Pt and PtCu at U = 0.9 V. | ||
For MER on pure Pt, the most energetically suited pathway at U = 0.7 V and U = 0.9 V involves the sequential steps: CH3OH → CH2OH → CHOH → COH → CO → COOH → CO2. The predicted pathway is consistent with other works.31,36 Notably, methanol adsorption and CO oxidation steps are endothermic (indicated by red circles) at U = 0.7 V, implying the RDS for Pt. The predicted overpotential for Pt is 0.9 V, which closely aligns with the experimental measurement of 0.91 V.37 For PtCu, the most energetically favorable pathway at U = 0.7 V and U = 0.9 V is CH3OH → CH2OH → CHOH(CH2O) → HCO → CO → COOH → CO2. As marked by red circles, the RDS in PtCu is the oxidation of COads, which achieves energy balance at a lower overpotential of 0.7 V, consistent with the experimental result of 0.76 V.37 Compared to Pt, PtCu exhibits two different dehydrogenating products of CH2OH, including CHOH and CH2O, suggesting two possible reaction paths. The presence of multiple reaction pathways implies faster consumption of reactants,38 indicating that PtCu would outperform Pt in MER.
Comparisons of the reaction paths at U = 0.7 V, obtained using the charge-variable method and the charge-neutral method, are presented in Fig. s5 and s6.† For the MER on pure Pt, the charge-neutral method accurately predicted the same reaction path; however, it failed to identify methanol adsorption as the RDS. On the PtCu surface, the charge-neutral method failed to predict the correct reaction path and underestimated the overpotential. These findings highlight the critical significance of considering charge variations in electrocatalytic processes, particularly for accurate predictions of reaction pathways and the determination of RDSs and overpotentials.39 The charge-variable method offers a more comprehensive and reliable approach to capture the complex interactions between the catalyst surface and reactant species under various electrode potentials.
In Fig. 2, it is apparent that the GC free energies associated with each step exhibit a decreasing trend as the electrode potentials increase. The position of PZC plays a crucial role in the analysis of the crossing points observed in the energy parabolas, as these points serve as key determinants for estimating the overpotential of MER. Through a meticulous examination of the crossing points vis-à-vis the PZC, it becomes possible to accurately ascertain the magnitude of the overpotential required to facilitate the progression of the MER.
In Fig. 5, we performed computational calculations to determine and compare the PZC values of clean and adsorbed Pt and PtCu. The calculated PZC for Pt was determined to be 1.21 V, which closely aligns with the reported value of 1.19 V.40 Remarkably, the PZC of PtCu surfaces was calculated to be 0.67 V. These findings underscore the influence of surface composition on the resulting PZC values. For absorbed catalysts, it has been previously demonstrated that PZC values are particularly sensitive to the presence of chemically adsorbed species, while showing limited responsiveness to physically adsorbed species.41 In line with our calculations, PZC values generally exhibit an increasing trend as the degree of surface oxidation is enhanced through the dehydrogenation of methanol. Interestingly, however, we noted that the PZC of adsorbed PtCu (red area) shifts within a much lower range than that of adsorbed Pt (grey area). The much lower PZC of pure and adsorbed PtCu leads to much earlier energetic intersection, as displayed in Fig. 2.
 | ||
| Fig. 5 The calculated zero charge potentials of pristine and adsorbed Pt and PtCu. | ||
Based on the above discussions, the MER performance in PtCu is better than that in Pt. The high catalyzing performance is owing to the low overpotential, which is displayed as the largest curve intersection in Fig. 2. Therefore, the position of energy curves is the key factor that determines the overpotential. In Fig. 6a, the potential-dependent electrochemical energy curves of clean Pt and PtCu were calculated. Interestingly, energy curve of PtCu shifts to the left. This indicates that the electrode potential of PtCu shifts within a narrower range compared with that of Pt, suggesting lower intersection and overpotential. Additionally, based on formula (2), the double-layer capacitance of Pt is calculated to be 6.03 μF cm−2, which is consistent with the experimental value of 20 μF cm−2. PtCu has a slightly lower capacitance of 5.89 μF cm−2. With the same atomic amounts, we also compared the electrode potentials of Pt and PtCu with different numbers of electrons, as shown in Fig. 6b. Upon adding the same number of electrons, PtCu demonstrated a lower potential than Pt.
 | ||
| Fig. 6 (a) The calculated potential-dependent electrochemical energy of Pt and PtCu. (b) The predicted electrode potential relative to the amount of electrons added to Pt and PtCu. | ||
3.3 Potential-dependent electronic structure analysis
Differential charge and Bader charge redistribution were conducted to reveal electronic effects that may be responsible for the predicted performance trend (Fig. s3–s5†). In Fig. s3,† the differential charge density distributions for charge-defect catalysts were calculated based on f(V) = ρ(Ne + ε) − ρ(Ne), where ρ(Ne + ε) and ρ(Ne) are the charged density grids of the system with Ne + ε and Ne electrons, respectively, and ε represents the number of electrons added in the system.26 Adding or subtracting electrons from Pt and PtCu changes the charge distributions of catalysts. Additional negative or positive charge areas accumulate at the surface of catalysts. Notably, we observed that Pt sites of PtCu alloys gain more charges than Cu sites. This indicates that the adsorption of some intermediates over Pt sites will be significantly affected in the charge-defect system.Subtracting electrons from pristine Pt decreases the electronegativity states of surface Pt atoms, and Bader charge distribution is consistent with differential charge redistribution. In the PtCu alloy, it was found that Cu donates charge to Pt and becomes positively charged (Fig. s4†). Subtracting electrons from pristine PtCu increases the chemical states of surface Pt and Cu atoms. The change in the chemical state of surface metal ultimately affects the subsequent adsorption behavior. For Pt and PtCu, subtracting electrons from catalysts increases the adsorbing strength towards methanol since electrons transferred from methanol to metal increase (Fig. s5†). Interestingly, the electropositive states of C in methanol increase when electrons are subtracted from catalysts, enhancing the electrostatic repulsion between C and H and leading to methanol dehydrogenation.
3.4 Potential-dependent adsorption energy analysis
Based on formula (5), the potential effects on adsorbing energies of methanol, H2O, and CO were calculated and illustrated in Fig. 7. Notably, adsorption energies of methanol over Pt and PtCu surfaces exhibited an increasing trend with rising electrode potentials. It is well established that methanol is characterized by its dipolar nature and electron-rich properties. Consequently, the reduction of electrons in the electrode system introduces electrostatic attraction, improving the adsorption of methanol at potentials higher than the PZC of the electrodes. At specific electrode potentials of 0.9 V and −0.6 V for Pt and PtCu surfaces, respectively, the methanol adsorption energies reach an equilibrium value of 0. Therefore, Pt and PtCu catalysts exhibit different methanol capturing abilities. However, the potential-induced enhancement of methanol adsorption energy contributes to an increased methanol loading on the catalyst surface, facilitating the dehydrogenation process. | ||
| Fig. 7 The potential-dependent adsorption free energy of (a) methanol, (b) H2O and (c) CO on Pt and PtCu. | ||
Based on our simulations, Pt surfaces exhibit relative low methanol adsorption energy, consistent with previous studies.42,43 As shown in Fig. 2a, methanol adsorption was observed at high potentials31 and was identified as the critical RDS for Pt. Earlier reports have indicated that the adsorption rate of methanol on Pt(100) surfaces is notably slower compared with that on Pt(111) and Pt(110) surfaces.31 Specifically, methanol adsorption on Pt(100) surfaces can only be detected at potentials exceeding 0.6 V, aligning with predictions derived from our computational simulations.31
In comparison to pure Pt, PtCu catalysts exhibit a considerably higher methanol adsorbing strength. As shown in Fig. s2,† upon equating the electron count between Pt and PtCu, the Pt sites on the PtCu surface demonstrate a larger charge density region relative to the pure Pt surface. Consequently, the Pt sites on PtCu catalysts become more positively charged compared with the Pt sites on Pt catalysts, thereby resulting in a stronger affinity for methanol adsorption on PtCu surfaces. This phenomenon has also been experimentally observed for PtRu electrodes, which exhibit superior methanol adsorption at high potentials than at low potentials.44
Regarding H2O adsorption, a consistent trend is observed, wherein the adsorption energy increases with the electrode potential for all three catalysts. On the Pt surface, H2O adsorption commences at 0.72 V. In agreement with experimental investigations, an increased H2O–metal interaction is observed as the potential exceeds 0.5 V, leading to the dissociation of H2O at the surface and the formation of adsorbed OH− species.45,46 Compared to Pt, PtCu exhibits significantly higher adsorption strength, even at negative potentials. This behavior can be attributed to the preferential bonding of H2O with Cu sites in PtCu alloys, where the charge density of Cu has a comparatively lesser influence than that of Pt in the charge-defect system (see Fig. s2†). Therefore, the enhanced adsorption strength exhibited by PtCu alloys facilitates H2O dissociation, particularly at lower potentials.
Similarly, the adsorption behavior of CO is highly influenced by the electrode potential and catalyst composition.47 It is widely acknowledged that the MER proceeds at a relatively slow rate, and the presence of strongly adsorbed CO on Pt, particularly at low potentials, can lead to catalyst self-poisoning. This behavior can be attributed to the strong overlap of electron density between Pt d-orbitals and CO π*-orbitals. The removal of CO typically occurs through CO electrooxidation reactions (CERs), represented by the equation CO + H2O→CO2 + 2H+ + 2e− with a slow reaction rate. As shown in Fig. 6, the adsorption strength of CO diminishes as the electrode potential becomes more positive, facilitating the electrooxidation of CO to CO2. Consistent with our simulations, extensive experimental investigations have demonstrated that Pt-based alloy catalysts exhibit a reduced adsorption energy for CO compared with pristine Pt surfaces.48,49 This is owing to the redistribution between the d-band of Pt and the d-band of the dopant metal species. Consequently, the coverage of CO on the PtCu surface is effectively reduced, thereby increasing the availability of exposed active Pt sites for the MER.
Concluding from Fig. 7, it becomes apparent that the adsorption energies of methanol are considerably lower than H2O and CO. This significant discrepancy in adsorption energies serves as the primary underlying factor contributing to the CO-poisoning phenomenon observed at low potentials and the OH-poisoning phenomenon observed at high potentials.50 In comparison to Pt, PtCu exhibits higher adsorption energies for methanol and H2O, while displaying considerably lower adsorption energy for CO.
4 Conclusions
In summary, we present a theoretical investigation on the catalytic mechanism of the MER based on DFT simulations. The potential-dependent MER, including reaction energetic thermodynamics, favorable reaction paths, electronic structures, and adsorption of some key intermediates, was thoroughly discussed for Pt and PtCu catalysts. The pure Pt catalyst exhibited a high overpotential of 0.9 V, and its MER performance was critically limited by methanol adsorption and COads removal. Compared with Pt, PtCu alloys demonstrated a significantly lower overpotential of 0.7 V. Moreover, methanol and H2O were preferentially adsorbed onto the Cu site of the PtCu alloy, resulting in much higher adsorption energies than the pure Pt catalyst, thus facilitating dehydrogenation and water activation. The introduction of Cu decreased the adsorption energy of CO, which increased the tolerance to surface CO poisoning.Data availability
All data generated or analyzed during this study are included in this published article and its ESI files.†Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The work was supported in part by the National Natural Science Foundation of China under the Grant No. 62374148, in part by the Zhejiang Provincial Natural Science Foundation of China under the Grant No. LD21F050001, Y21F040001, LGG21E060001 and LZY22E030003; the Nanxun Scholars Program for Young Scholars of ZJWEU under the Grant No. RC2024021016; and the Key Research and Development Project of Zhejiang Province under the Grant No. 2021C03019.References
- A. Hamnett, Mechanism and Electrocatalysis in the Direct Methanol Fuel Cell, Catal. Today, 1997, 38, 445–457 CrossRef CAS.
- M. Valter, B. Wickman and A. Hellman, Solvent Effects for Methanol Electrooxidation on Gold, J. Phys. Chem. C, 2021, 125, 1355–1360 Search PubMed.
- Y. Katayama, et al., Direct Observation of Surface-Bound Intermediates During Methanol Oxidation on Platinum under Alkaline Conditions, J. Phys. Chem. C, 2021, 125, 26321–26331 CAS.
- S. Sakong and A. Groß, The Importance of the Electrochemical Environment in the Electro-Oxidation of Methanol on Pt(111), ACS Catal., 2016, 6, 5575–5586 Search PubMed.
- J. Greeley and M. Mavrikakis, Competitive Paths for Methanol Decomposition on Pt(111), J. Am. Chem. Soc., 2004, 126, 3910–3919 CAS.
- W. Zhong, Y. Liu and D. Zhang, Theoretical Study of Methanol Oxidation on the Ptau(111) Bimetallic Surface: Co Pathway Vs Non-Co Pathway, J. Phys. Chem. C, 2012, 116, 2994–3000 CAS.
- A. S. Moura, J. L. C. Fajín, A. S. S. Pinto, M. D. S. Mandado and M. N. Cordeiro, Competitive Paths for Methanol Decomposition on Ruthenium: A Dft Study, J. Phys. Chem. C, 2015, 119, 27382–27391 CrossRef CAS.
- B. Beden, C. Lamy, A. Bewick and K. Kunimatsu, Electrosorption of Methanol on a Platinum Electrode. Ir Spectroscopic Evidence for Adsorbed Co Species, J. Electroanal. Chem. Interfacial Electrochem., 1981, 121, 343–347 Search PubMed.
- G. Gao and L.-W. Wang, Substantial Potential Effects on Single-Atom Catalysts for the Oxygen Evolution Reaction Simulated Via a Fixed-Potential Method, J. Catal., 2020, 391, 530–538 CAS.
- D. Kim, J. Shi and Y. Liu, Substantial Impact of Charge on Electrochemical Reactions of Two-Dimensional Materials, J. Am. Chem. Soc., 2018, 140, 9127–9131 Search PubMed.
- J. A. Gauthier, S. Ringe, C. F. Dickens, A. J. Garza, A. T. Bell, M. Head-Gordon, J. K. Nørskov and K. Chan, Challenges in Modeling Electrochemical Reaction Energetics with Polarizable Continuum Models, ACS Catal., 2019, 9, 920–931 CAS.
- Y. A. Alsunni, A. W. Alherz and C. B. Musgrave, Electrocatalytic Reduction of Co2 to Co over Ag(110) and Cu(211) Modeled by Grand-Canonical Density Functional Theory, J. Phys. Chem. C, 2021, 125, 23773–23783 CAS.
- K. Letchworth-Weaver and T. A. Arias, Joint Density Functional Theory of the Electrode-Electrolyte Interface: Application to Fixed Electrode Potentials, Interfacial Capacitances, and Potentials of Zero Charge, Phys. Rev. B, 2012, 86, 075140 Search PubMed.
- A. Hagopian, M.-L. Doublet, J.-S. Filhol and T. Binninger, Advancement of the Homogeneous Background Method for the Computational Simulation of Electrochemical Interfaces, J. Chem. Theory Comput., 2022, 18, 1883–1893 Search PubMed.
- X. Zhang, C. Wang, C. Luan, M. Liao and W. Xu, Preparation of Mof-Derived Molybdenum-Carbide-Modified Ptcu Nano-Alloy Catalysts and Their Methanol Oxidation Performance, New J. Chem., 2024, 48, 7964–7971 RSC.
- Y. Zhou and Q. Yuan, Ptcu/Pt Core/Atomic-Layer Shell Hollow Octahedra for Oxygen Reduction and Methanol Oxidation Electrocatalysis, Chem. Commun., 2024, 60, 2918–2921 RSC.
- Y. Chen, Y. Wu, Y. Chang, Z. Yang, X. Song, W. Zhou, J. Wang and H. Li, Pt3cu Alloy Anchored on Nanoporous Wo3 with High Activity and Stability in Methanol Oxidation, Int. J. Hydrogen Energy, 2024, 50, 1441–1449 CrossRef CAS.
- G. Kresse and J. Furthmüller, Efficient Iterative Schemes for Ab Initio Total-Energy Calculations Using a Plane-Wave Basis Set, Phys. Rev. B, 1996, 54, 11169–11186 CrossRef CAS PubMed.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate Ab Initio Parametrization of Density Functional Dispersion Correction (Dft-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed.
- K. Mathew, R. Sundararaman, K. Letchworth-Weaver, T. A. Arias and R. G. Hennig, Implicit Solvation Model for Density-Functional Study of Nanocrystal Surfaces and Reaction Pathways, J. Chem. Phys., 2014, 140, 084106 CrossRef PubMed.
- G. Kresse and D. Joubert, From Ultrasoft Pseudopotentials to the Projector Augmented-Wave Method, Phys. Rev. B, 1999, 59, 1758 CrossRef CAS.
- J. P. Perdew, K. Burke and M. Ernzerhof, Generalized Gradient Approximation Made Simple, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- S. Sakong, M. Naderian, K. Mathew, R. G. Hennig and A. Groß, Density Functional Theory Study of the Electrochemical Interface between a Pt Electrode and an Aqueous Electrolyte Using an Implicit Solvent Method, J. Chem. Phys., 2015, 142, 234107 CrossRef.
- J. S. Filhol and M. Neurock, Elucidation of the Electrochemical Activation of Water over Pd by First Principles, Angew. Chem., 2006, 118, 416–420 CrossRef.
- N. Lespes and J.-S. Filhol, Using Implicit Solvent in Ab Initio Electrochemical Modeling: Investigating Li+/Li Electrochemistry at a Li/Solvent Interface, J. Chem. Theory Comput., 2015, 11, 3375–3382 CrossRef CAS PubMed.
- A. Kopač Lautar, A. Hagopian and J.-S. Filhol, Modeling Interfacial Electrochemistry: Concepts and Tools, Phys. Chem. Chem. Phys., 2020, 22, 10569–10580 RSC.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jónsson, Origin of the Overpotential for Oxygen Reduction at a Fuel-Cell Cathode, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef PubMed.
- J. Suntivich, et al., Surface Composition Tuning of Au–Pt Bimetallic Nanoparticles for Enhanced Carbon Monoxide and Methanol Electro-Oxidation, J. Am. Chem. Soc., 2013, 135, 7985–7991 CrossRef CAS.
- T. Binninger and M.-L. Doublet, The Ir–Oooo–Ir Transition State and the Mechanism of the Oxygen Evolution Reaction on Iro2(110), Energy Environ. Sci., 2022, 15, 2519–2528 RSC.
- R. Rizo, J. Fernández-Vidal, L. J. Hardwick, G. A. Attard, F. J. Vidal-Iglesias, V. Climent, E. Herrero and J. M. Feliu, Investigating the Presence of Adsorbed Species on Pt Steps at Low Potentials, Nat. Commun., 2022, 13, 2550 CrossRef CAS PubMed.
- T. Iwasita, Electrocatalysis of Methanol Oxidation, Electrochim. Acta, 2002, 47, 3663–3674 Search PubMed.
- Y. Liu, Z. Duan and G. Henkelman, Computational Design of Co-Tolerant Pt3m Anode Electrocatalysts for Proton-Exchange Membrane Fuel Cells, Phys. Chem. Chem. Phys., 2019, 21, 4046–4052 RSC.
- H. Wang, T. Löffler and H. Baltruschat, Formation of Intermediates During Methanol Oxidation: A Quantitative Dems Study, J. Appl. Electrochem., 2001, 31, 759–765 CrossRef CAS.
- L. Huang, et al., Exposing Cu-Rich {110} Active Facets in Ptcu Nanostars for Boosting Electrochemical Performance toward Multiple Liquid Fuels Electrooxidation, Nano Res., 2019, 12, 1147–1153 CrossRef CAS.
- L. Huang, et al., Surface-Structure Tailoring of Ultrafine Ptcu Nanowires for Enhanced Electrooxidation of Alcohols, Sci. China Mater., 2021, 64, 601–610 CrossRef CAS.
- V. S. Bagotzky, Y. B. Vassiliev and O. A. Khazova, Generalized Scheme of Chemisorption, Electrooxidation and Electroreduction of Simple Organic Compounds on Platinum Group Metals, J. Electroanal. Chem. Interfacial Electrochem., 1977, 81, 229–238 CrossRef.
- J. Sun, J. Shi, J. Xu, X. Chen, Z. Zhang and Z. Peng, Enhanced Methanol Electro-Oxidation and Oxygen Reduction Reaction Performance of Ultrafine Nanoporous Platinum–Copper Alloy: Experiment and Density Functional Theory Calculation, J. Power Sources, 2015, 279, 334–344 CrossRef CAS.
- A. R. Poerwoprajitno, et al., A Single-Pt-Atom-on-Ru-Nanoparticle Electrocatalyst for Co-Resilient Methanol Oxidation, Nat. Catal., 2022, 5, 231–237 CrossRef CAS.
- H. Liu, F. Sun, M. Chen and H. Wang, Reconciling the Experimental and Computational Methanol Electro-Oxidation Activity Via Potential-Dependent Kinetic Mechanism Analysis, J. Mater. Chem. A, 2022, 10, 23551–23561 CAS.
- K. Shimura and H. Yoshida, Heterogeneous Photocatalytic Hydrogen Production from Water and Biomass Derivatives, Energy Environ. Sci., 2011, 4, 2467–2481 RSC.
- F. Tian, R. Jinnouchi and A. B. Anderson, How Potentials of Zero Charge and Potentials for Water Oxidation to Oh(Ads) on Pt(111) Electrodes Vary with Coverage, J. Phys. Chem. C, 2009, 113, 17484–17492 CrossRef CAS.
- X. Lu, Z. Deng, C. Guo, W. Wang, S. Wei, S.-P. Ng, X. Chen, N. Ding, W. Guo and C.-M. L. Wu, Methanol Oxidation on Pt3sn(111) for Direct Methanol Fuel Cells: Methanol Decomposition, ACS Appl. Mater. Interfaces, 2016, 8, 12194–12204 CAS.
- E. M. Karp, T. L. Silbaugh, M. C. Crowe and C. T. Campbell, Energetics of Adsorbed Methanol and Methoxy on Pt(111) by Microcalorimetry, J. Am. Chem. Soc., 2012, 134, 20388–20395 CAS.
- P. Waszczuk, A. Wieckowski, P. Zelenay, S. Gottesfeld, C. Coutanceau, J. M. Léger and C. Lamy, Adsorption of Co Poison on Fuel Cell Nanoparticle Electrodes from Methanol Solutions: A Radioactive Labeling Study, J. Electroanal. Chem., 2001, 511, 55–64 Search PubMed.
- T. Iwasita and X. Xia, Adsorption of Water at Pt(111) Electrode in Hclo4 Solutions. The Potential of Zero Charge, J. Electroanal. Chem., 1996, 411, 95–102 CrossRef.
- D. S. Mekazni, R. M. Arán-Ais, A. Ferre-Vilaplana and E. Herrero, Why Methanol Electro-Oxidation on Platinum in Water Takes Place Only in the Presence of Adsorbed Oh, ACS Catal., 2022, 12, 1965–1970 CrossRef CAS.
- B. Tran and B. R. Goldsmith, Theoretical Investigation of the Potential-Dependent Co Adsorption on Copper Electrodes, J. Phys. Chem. Lett., 2024, 15, 6538–6543 CrossRef CAS PubMed.
- J. Zheng, M. Busch, L. Artiglia, T. Skála, J. Rossmeisl and S. Agnoli, A Dft Structural Investigation of New Bimetallic Ptsnx Surface Alloys Formed on the Pt(110) Surface and Their Interaction with Carbon Monoxide, J. Phys. Chem. C, 2016, 120, 25306–25316 CrossRef CAS.
- L. Zhao, S. Wang, Q. Ding, W. Xu, P. Sang, Y. Chi, X. Lu and W. Guo, The Oxidation of Methanol on Ptru(111): A Periodic Density Functional Theory Investigation, J. Phys. Chem. C, 2015, 119, 20389–20400 CrossRef CAS.
- D. Y. Chung, K.-J. Lee and Y.-E. Sung, Methanol Electro-Oxidation on the Pt Surface: Revisiting the Cyclic Voltammetry Interpretation, J. Phys. Chem. C, 2016, 120, 9028–9035 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01511a |
| This journal is © The Royal Society of Chemistry 2025 |