
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceImpact of trifluoromethyl Ugi adducts as anticancer agents: EGFR inhibition, apoptosis induction and miRNA up-regulation †
Mohammed Salah Ayoup *a,
Eman Mamdouhb,
Saied M. Solimanb,
Ibrahim Elghamrya,
Mohamed S. Nafiecd,
Amr Negma,
Amr Sonousief,
Asmaa E. Kassabe and
Laila F. Awad*b
*a,
Eman Mamdouhb,
Saied M. Solimanb,
Ibrahim Elghamrya,
Mohamed S. Nafiecd,
Amr Negma,
Amr Sonousief,
Asmaa E. Kassabe and
Laila F. Awad*b
aDepartment of Chemistry, College of Science, King Faisal University, Al-Ahsa 31982, Saudi Arabia. E-mail: mayoup@kfu.edu.sa; mohammedsalahayoup@gmail.com
bChemistry Department, Faculty of Science, Alexandria University, P. O. Box 426, Alexandria, 21321, Egypt. E-mail: laila.fathy@yahoo.com
cDepartment of Chemistry, College of Sciences, University of Sharjah, Sharjah 27272, United Arab Emirates
dChemistry Department, Faculty of Science, Suez Canal University, Ismailia 41522, Egypt
eDepartment of Pharmaceutical Organic Chemistry, Faculty of Pharmacy, Cairo University, Kasr El-Aini Street, P. O. Box 11562, Cairo, Egypt
fUniversity of Hertfordshire Hosted By Global Academic Foundation, New Administrative Capital, Cairo, Egypt
First published on 10th June 2025
Abstract
A series of novel bis-amide Ugi adducts with a Y-shaped configuration imitating the thematic feature of fourth-generation EGFR inhibitors was designed and synthesized via a one-step Ugi four-component reaction. All the synthesized Ugi adducts were evaluated for their anti-proliferative efficacy against MDA-MB-231 human breast cancer and A549 non-small cell lung cancer cell lines. Their selectivity was assessed using normal human lung epithelial cells (BEAS-2B). The Ugi adduct 5 stood up as the study hit concerning cytotoxic efficacy and selectivity. It was 1.42- and 1.20-fold more potent than 5-FU and cisplatin, respectively, against the MDA-MB-231 cell line, and 1.82- and 1.62-fold more potent than 5-FU and cisplatin, respectively, against the A549 cell line. Additionally, compound 5 demonstrated the most prominent selectivity against MDA-MB-231 and A549 cells (SI = 7 and 11.7, respectively); it was 14 to 20.9-fold safer than 5-FU and cisplatin. Accordingly, it was subjected to further enzymatic and mechanistic studies. The Ugi adduct 5 showed excellent sub-micromolar potency (IC50 = 0.19 μM) against human mutant EGFRT790M/C797S/L858R enzyme equipotent with that of osimertinib (IC50 = 0.1 μM). It exhibited a promising IC50 value of 2.1 nM against the EGFRWT enzyme, comparable to erlotinib (IC50 = 1.3 nM). In comparison to the untreated control, the Ugi adduct 5 caused a decrease in the expression of the cancer initiation, angiogenic, and metastatic markers (c-Myc, CD-44, CD-133, VEGF, and TGF) to 0.36, 0.5, 0.7, 0.5, and 0.4-fold, respectively, in MDA-MB-231 cells. Regarding A549 cells, the exposure to compound 5 showed a 0.41, 0.64, 0.58, 0.71, and 0.69-fold reduction in the expression of c-Myc, CD-44, CD-133, VEGF, and TGF markers compared to the untreated control. Compound 5 markedly elevated miRNA-132 and miRNA-200c expressions in the MDA-MB-231 cell line by 3.8 and 3.1-fold, while in A549 cells, compound 5 demonstrated enhancement of miRNA-132 and miRNA-200c expression by 2.4 and 1.9-fold changes compared to that of the control. It promoted apoptosis induction via caspase 3/9 activation (1.8 and 2.3-folds) in the A549 cell line. The molecular docking interpretations of the most potent Ugi adduct 5 in EGFRL858R/T790M/C797S (PDB ID: 6LUB) and the wild-type EGFRWT (PDB ID: 1M17) enzymes are aligned with and explain its potential to dually inhibit EGFRL858R/T790M/C797S and EGFRWT tyrosine kinases.
1. Introduction
Cancer continues to rank among the most serious public health concerns, with its incidence and mortality rates continuing to place a heavy strain on healthcare systems and populations worldwide. In the 21st century, cancer is a significant social, public health, and financial issue.1,2 Three out of every ten premature deaths (people 30–69 years old) worldwide are caused by cancer, and in 177 out of 183 countries, it is one of the top three causes of death in this age group.3 In 2022, about 20 million new cases of cancer, 9.7 million deaths, and 53.5 million prevalent cases are estimated by the Global Cancer Observatory of the International Agency for Research on Cancer (IARC).4Most cancers are caused by genetic defects; mutations can give normal cells new characteristics that cause them to become malignant. The capacity of cancer cells to invade and spread is one of the characteristics of the disease that are determined by genome instability and mutations.5 A cancerous cell can move inside tissues due to its ability to migrate and invade those tissues. These activities enable cancerous cells to spread across the blood and lymphatic vessels, disseminate throughout the body, and then proliferate metastatically in other organs.6 One of the main causes of death for breast and lung cancer patients and a defining feature of the disease is metastasis.5 Malignant cells leave the primary site and travel throughout the body during the metastasis process, creating secondary sites and seriously impairing organ function.7
Thus, in its most aggressive form, cancer is a disease of unchecked cell migration as well as unchecked cell growth. Metastasis requires a sequence of intricate steps known as the “metastatic cascade”. It is triggered by the initiation of tumor growth and angiogenesis, followed by cell migration and intravasation. Invasion is from the initial stages of the metastatic cascade, as cancer cells penetrate the basement membrane and move into the surrounding tissue through the extracellular matrix.8,9 The identification of therapeutics that co-target cancer cell proliferation, invasion, and migration pathways may provide novel treatment regimens for cancer patients.
The primary player in growth factor signaling is the epidermal growth factor receptor (EGFR). The receptor and the tyrosine kinase comprise the two functional domains of the EGFR. Via intracellular signal transduction, EGFR is necessary for regular cellular regulation and delivers extracellular signals into cells across the cell membrane.10,11
Overexpression and abnormal signaling of this receptor increase downstream consequences like angiogenesis, cell survival, and proliferation, eventually resulting in unchecked cell growth, differentiation, neovascularization, metastasis, and resistance of cancer cells.12,13
The majority of solid tumors, including breast, colon, and lung cancers, have been shown to exhibit abnormal overexpression (up to 20 times the expression of EGFR in normal cells) in addition to EGFR mutations, which are linked to a poor prognosis, so EGFR is a potential molecular therapeutic target for anticancer candidates.14–17 Gefitinib (Iressa TM)18 and erlotinib (Tarceva TM)19 (Fig. 1) with 4-anilinoquinazoline motifs are reversible inhibitors of the first generation of EGFR tyrosine kinase inhibitors (TKIs) that specifically block EGFR (Erb-B1) signaling as an EGFR subtype. They potentially treat cancer patients with EGFR overexpression, and they initially produce good responses. Most of these patients develop resistance to this class within a year because of the EGFR T790M mutation.20,21
 | ||
| Fig. 1 The four generations of EGFR inhibitors. | ||
Afatinib and dacomitinib (Fig. 1), quinazoline-based irreversible inhibitors, have been developed as second-generation EGFR inhibitors to counteract the EGFR T790M mutation caused by gefitinib or erlotinib.22,23 One of these compounds' structural characteristics is the γ-amino acrylamide framework, which can function as a Michael acceptor to create irreversible covalent bonds with the SH group in the Cys797 residue at the edge of the ATP binding pocket of the receptor. On the other hand, irreversible inhibitors exhibit strong efficacy against normal cell-found EGFRWT (wild type),24 and therefore, the usage of dosages that would be required to adequately suppress T790M is prohibited due to the toxicity (interstitial lung disease, rash, and diarrhea) that results from blocking wild-type EGFR.
To target mutant EGFR containing T790M, third-generation EGFR TKIs have recently been developed, including osimertinib (Tagrisso TM) (Fig. 1), olmutinib, naquotinib, and avitinib.25–28 This class spares the wild-type EGFR in favor of acting only on EGFR mutations, especially osimertinib, which was approved by the Food and Drug Administration (FDA) in 2015 to treat patients with non-small cell lung cancer (NSCLC) who have EGFR-activating mutations, such as the L858R point mutation or in-frame deletions of exon 19, and the EGFR T790M mutation.29 Resistance develops quickly once osimertinib is administered to patients with EGFR T790M mutation, usually throughout nine to thirteen months due to the activation of other resistance mechanisms, incorporating the C797S point mutation into serine that can obstruct the formation of covalent bonds with irreversible EGFR inhibitors, hence significantly lowering the therapeutic activity.30–32
Fourth-generation EGFR inhibitor design strategies have significantly evolved to target the T790M mutation and other resistant variations, like C797S, with an emphasis on Y-shaped designs and other molecular configuration changes. Y-shaped structures can accommodate a variety of mutations, including C797S and T790M, by interacting with distinct sub-pockets and inducing an inactive conformation of the receptor. Because of the adaptable design of Y-shaped EGFR inhibitors, they can effectively combat complicated resistance mutations like T790M/C797S. The arms of Y-shaped inhibitors are bifurcated and extend into different EGFR kinase domain sub-pockets, ensuring improved adaptability in targeting various sites, which qualifies them for conquering acquired resistance. Additionally, this configuration ensures effective binding by successfully addressing the steric barrier brought on by the large T790M residue. This design provides a wider binding profile, which guarantees interactions with the hinge region and solvent-exposed portions. By distinguishing between mutant and wild-type EGFR, Y-shaped inhibitors reduce systemic toxicity.33,34
EAI001, EAI045, JBJ-04-125-02, and TREA-0236 (Fig. 1) are fourth-generation bis-amide-based EGFR inhibitors with Y-shaped structures. They are potent non-ATP competitive inhibitors of mutant C797S and T790M EGFR. A closer examination of these compounds' design approaches (Y configuration strategy) and typical anticancer efficacy yielded important insights into the advancement of more potent EGFR inhibitors against T790M and C797S mutations.35,36 Although EGFR inhibitors that target the C797S mutation are being developed, small-molecule EGFR inhibitors with new motifs and enhanced drug-like qualities are still needed.
miRNAs are a subclass of short non-coding RNA molecules that range in length from 19 to 23 nucleotides.37 miRNAs control gene expression by directly binding to mRNAs' matching sites, which causes the target mRNAs to degrade quickly.38 miRNAs affect numerous important cellular processes, including cell proliferation, apoptosis, angiogenesis, invasion, and migration. They also have a role in several biological processes, including organism growth, immunological control, and carcinogenesis.39,40 miRNAs have received increased attention due to their numerous applications as regulatory molecules in numerous signaling transductions, including the EGFR signaling pathway acting as oncomirs or tumor suppressors in several cancer types.41 Recently, miRNAs have been associated with lung cancer cells' resistance to anti-EGFR drugs, suggesting that miRNAs could be helpful as new targets or promising predictive biomarkers for anti-EGFR treatment.41
Due to their simple synthesis, ease of structural diversification, chemical stability, and effectiveness as potential anticancer candidates,42–50 exerting their anticancer efficacy via different mechanisms such as tyrosine kinases inhibition, activation of caspases, and apoptosis induction, the Ugi scaffolds are privileged structures that earned a great interest in medicinal chemistry. The amide motif as a backbone scaffold is also important since it is the most widely used in bioactive compounds, including peptidomimetics.47
Inspired by all these findings, we have designed and synthesized a series of novel Ugi adducts (Fig. 2) with a Y-shaped configuration imitating the thematic feature of fourth-generation EGFR inhibitors (bis-amide-based scaffold featuring multiple aromatic rings with a fluorinated phenyl motif). The synthesized Ugi adducts were simply prepared via a one-step Ugi four-component reaction. They are based on bis-amides and incorporate multiple aromatic or heteroaromatic rings. Some of these aromatic rings feature different arrays of electron-donating and electron-withdrawing substituents that influence the electrical environment and spatial arrangement of the scaffolds. This diversity of substitutions may impact the anticancer potential of the target Ugi adducts and enable us to conduct a study of the structure–activity relationship. Within the framework of imitation design, we have incorporated the trifluoromethyl (CF3) group on one phenyl ring of the targeted Ugi adducts. The inclusion of CF3 moiety in the designed drug candidates often aimed to enhance their potencies.51–53 Some of the aromatic rings are incorporated in the Ugi scaffold via variable linkers that differ in length, and some of these linkers were selected to be shared as H-bond acceptors and/or donors in the EGFR active site.
 | ||
| Fig. 2 The design of targeted Ugi adducts as fourth-generation EGFR inhibitors. | ||
The synthesized Ugi adducts were assessed for their cytotoxic efficacy against A549 non-small cell lung cancer and MDA-MB-231 human breast carcinoma cell lines. Their selectivity was assessed using normal human lung epithelial cells (BEAS-2B) to investigate the synthesized compounds' safety for normal cells. The concentration that results in 50% inhibition of cell viability (IC50) values of all synthesized Ugi adducts was calculated. The most potent Ugi adduct was subjected to further enzymatic and mechanistic investigations, such as in vitro EGFR inhibition assay against human wild and human triple (L858R/T790M/C797S) mutant types and caspase 3/9 activation to assess its apoptotic induction potential. The impact of the most prominent Ugi adduct on regulating markers related to the proliferation, invasion, and migration of human cancer cells (c-Myc, CD44, CD133, VEGF, and TGF) was investigated. Its impact on the expression of miRNA-132 and miRNA-200c was also evaluated.
2. Results and discussion
2.1. Chemistry
Ugi reaction is a versatile and highly efficient synthetic strategy for synthesizing highly functionalized di-peptide-like products with structural diversity and high efficiency.54,55 Moreover, introducing different functionalized or substituents on these active peptidomimetic products may enhance their affinity and bioavailability as effective pharmaceutical agents and their selectivity and potency towards specific biological targets. Among these substituents is the trifluoromethyl (CF3) group, which is one of the most lipophilic functional groups.56–58 The inclusion of CF3 moiety in the molecules often aimed to improve metabolic stability and hydrophobicity in drug development.59 Moreover, combining aromatic trifluoromethyl moieties with other organic compounds will enhance their potency as biologically active derivatives.51–53,60–69 Accordingly, we focused here on constructing diverse functionalized Ugi products with trifluoromethyl moiety for their evaluation as anticancer products.The synthesis of the target Ugi products having the desired lipophilic trifluoromethyl group, which we expect to enhance the bioavailability of the synthesized products, has been attempted through two pathways of Ugi multi-component reaction (Schemes 1 and 2).
 | ||
| Scheme 1 Synthesis of fluorinated Ugi products 5–20. | ||
 | ||
| Scheme 2 Synthesis of Ugi products based fluorinated cinnamide 23–25. | ||
This can be either through the coupling of p-trifluoromethyl benzaldehyde, carboxylic acid derivatives, or benzaldehyde and p-trifluoromethyl cinnamic acid instead. Thus, the coupling reaction of p-trifluoromethyl benzaldehyde 1 and aniline derivative 2a–b or benzylamine 2c was carried out in ethanol under reflux for 1 h followed by the addition isonitrile 3 and the appropriate carboxylic acids 4a–f in 2,2,2-trifluoroethanol (TFE) afforded the Ugi products 5–20 in 55–80% yield within 48 under reflux (Scheme 1). Alternatively, compounds 23–25 were obtained in 65–84% yield under the same reaction conditions using benzaldehyde and p-trifluoromethyl cinnamic acid (Scheme 2) where the Ugi reaction conditions were optimized as reported previously.49
Structure elucidation of the synthesized compounds was confirmed from their spectral analyses. The NMR spectrum pattern of compounds 5–19, and 23–25 differed according to the carboxylate moiety introduced as well as the amine used. The purity of the most active compound 5 was confirmed by HRMS, which reported that the exact mass experimentally aligned with the theoretically calculated mass. However, characteristic functional signals were common. Thus, the trifluoromethyl group was assigned at δC 125.5–125.7 ppm in the 13C NMR spectra. Two amidic carbon (CON) signals were observed at δC 165.4–169.7 and 169.1–172.9 ppm, whereas the respective NH proton (CONH) resonated at δH 10.93–11.14 ppm as a D2O exchangeable singlet. Moreover, the methine proton NCHCO resonated at δH 6.24–6.75 ppm, which correlated with its carbon at δC 62.9–65.6 ppm for above mentioned compounds. The result and discussion of NMR and IR results of the other Ugi product were reported in the ESI file.†
2.2. Biological evaluation
| Compound no. | BEAS-2B | MDA-MB-231 | A549 | ||
|---|---|---|---|---|---|
| IC50a (μM) | IC50a (μM) | SI | IC50a (μM) | SI | |
| a All values are expressed as mean ± SEM. | |||||
| 5 | 70.4 ± 5.1 | 10.1 ± 0.6 | 7 | 6.2 ± 0.3 | 11.7 |
| 6 | 50.1 ± 3.6 | 54.2 ± 4.7 | 0.9 | 39.9 ± 2.7 | 1.3 |
| 7 | 75.6 ± 5.8 | 28.2 ± 1.4 | 2.7 | 20.6 ± 1.5 | 3.8 |
| 8 | 62.2 ± 4.2 | 27.1 ± 1.8 | 2.3 | 20.5 ± 1.1 | 3.1 |
| 9 | 71.6 ± 5.7 | 70.6 ± 6.1 | 1 | 46.4 ± 2.8 | 1.5 |
| 10 | 83.3 ± 6.8 | 84.4 ± 6.9 | 1 | 71.5 ± 6.8 | 1.2 |
| 11 | 82.1 ± 7.3 | 60.2 ± 4.9 | 1.4 | 53.9 ± 4.4 | 1.5 |
| 12 | 98.2 ± 8.1 | 65.3 ± 4.7 | 1.5 | 73.1 ± 6.6 | 1.3 |
| 13 | 98.5 ± 7.4 | 73.9 ± 5.3 | 1.3 | 64.1 ± 5.9 | 1.5 |
| 14 | 84.7 ± 6.9 | 74.8 ± 6.1 | 1.1 | 55.3 ± 4.2 | 1.5 |
| 15 | 69.4 ± 4.7 | 65.7 ± 5.4 | 1.1 | 61.1 ± 4.5 | 1.1 |
| 16 | 63.8 ± 4.8 | 27.3 ± 1.5 | 2.3 | 22.4 ± 0.8 | 2.9 |
| 17 | 50.2 ± 3.4 | 57.0 ± 4.8 | 0.9 | 40.8 ± 2.8 | 1.3 |
| 18 | 55.7 ± 3.5 | 25.1 ± 1.9 | 2.2 | 20.1 ± 1.4 | 2.8 |
| 19 | 50.1 ± 3.3 | 73.1 ± 5.7 | 0.7 | 46.2 ± 4.1 | 1.1 |
| 20 | 40.7 ± 2.8 | 40.2 ± 3.1 | 1 | 33.3 ± 2.1 | 1.2 |
| 23 | 30.9 ± 2.3 | 30.3 ± 2.4 | 1 | 26.1 ± 2.1 | 1.2 |
| 24 | 54.2 ± 4.1 | 22.1 ± 1.6 | 2.5 | 20.4 ± 1.7 | 2.7 |
| 25 | 129.1 ± 8.7 | 95.4 ± 8.1 | 1.4 | 90.2 ± 8.5 | 1.4 |
| Cisplatin | 5.7 ± 0.3 | 12.2 ± 0.8 | 0.47 | 10.1 ± 0.7 | 0.56 |
| 5-FU | 7.2 ± 0.5 | 14.4 ± 0.9 | 0.5 | 11.3 ± 1.1 | 0.64 |
2.2.1.1. Structure–activity relationship. A promising anti-proliferative potential demonstrated by the bis-amide Ugi scaffold design is reflected in the overall cytotoxic activity pattern. An interesting phenomenon is that the most prominent Ugi adducts (5, 7, 8, 16, 18, 23, and 24) demonstrated significant cytotoxicity against both non-small cell lung cancer (NSCLC, A549) and breast cancer (MDA-MB-231) cell lines at the same time demonstrated exceptionally the highest safety profiles. The activity profile demonstrated a higher potency of the synthesized Ugi adducts against non-NSCLC, A549 cell line than MDA-MB-231 cell line. The combinatorial diversity of Ugi components determined the anticancer profile (Fig. 3). Concerning modifications of Ugi aldehyde component, results showed that the derivatives derived from p-trifluoromethyl benzaldehyde (5, 7, 8, 16, and 18) endowed better anti-proliferative potential than those derived from benzaldehyde (23 and 24), reflecting the positive impact on the cytotoxicity of closer proximity of the trifluoromethylphenyl moiety from the bis-amide Ugi scaffold. Further combinatorial modifications are based on diversifying carboxylic acid derivatives. The incorporation of halogen (Cl or I) in the Ugi adducts greatly improved the antitumor activities (compounds 5, 7, and 8 derived from halogenated carboxylic acids). Further analysis of these compounds revealed that shifting of iodo to chloro group introduced the most active Ugi adduct 5 in the current study. Increasing the length of the linker connecting the aromatic motif of the carboxylic acid component of the Ugi adduct in compounds 9–15 endowed a remarkable reduction in anti-proliferative potency. Ugi bis-amides derived from aniline or p-toluidine (5, 7, 8, 18, 23, and 24) were more potent anticancer agents than adducts derived from benzylamine.
 | ||
| Fig. 3 The structure–activity relationship of the synthesized Ugi adducts. | ||
 | ||
| Fig. 4 (A) Effect of Ugi adduct 5 on the expression of c-Myc, CD-44, CD-133, VEGF, and TGF in MDA-MB 231 cells. (B) Effect of Ugi adduct 5 on the expression of c-Myc, CD-44, CD-133, VEGF, and TGF in A549 cells. | ||
 | ||
| Fig. 5 Effect of Ugi adduct 5 on the expression of miRNA-132 and miRNA-200c in MDA-MB 231 and A549 cells. | ||
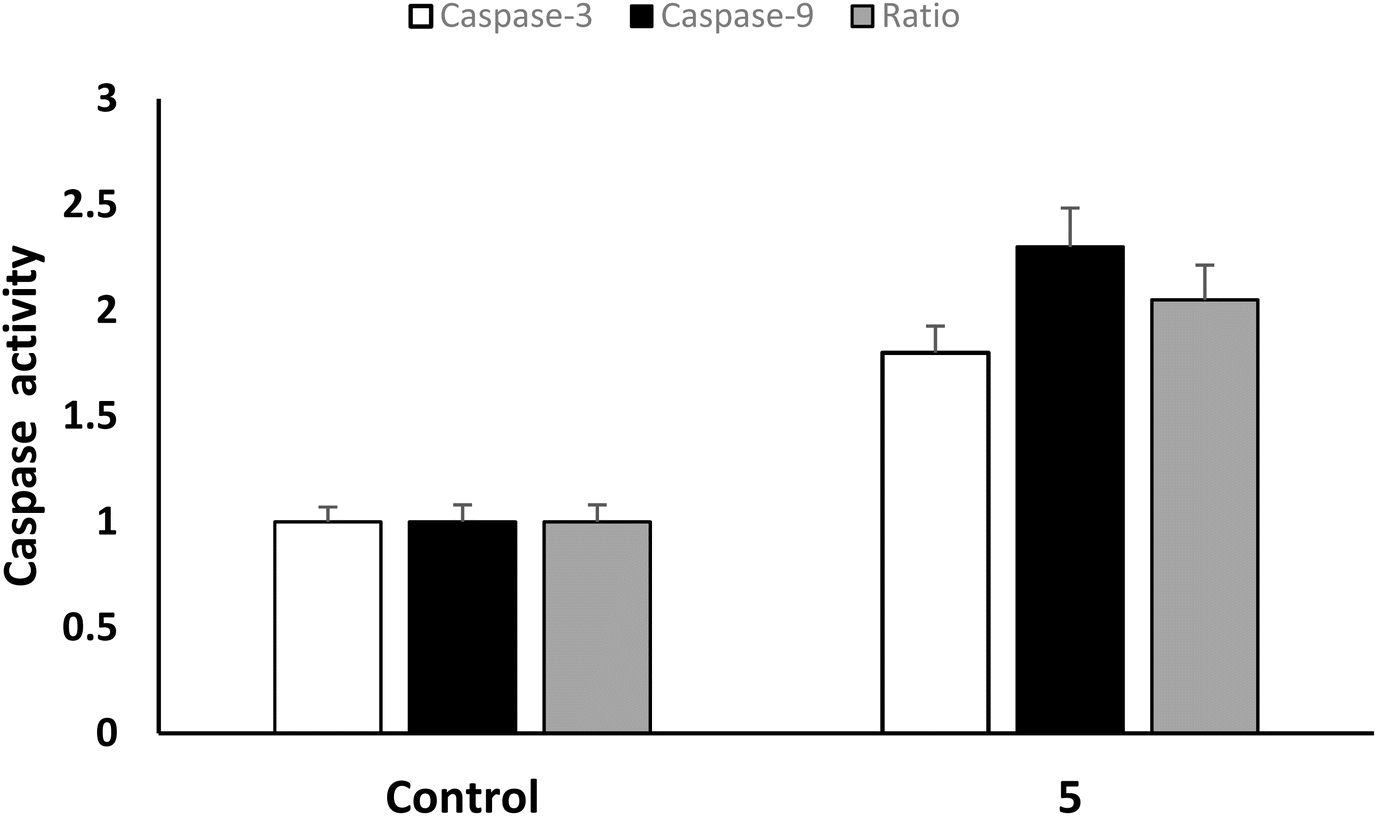 | ||
| Fig. 6 Effect of Ugi adduct 5 on the levels of caspase-3 and caspase-9 in A549 cells. | ||
 | ||
| Fig. 7 (A) 3D representation and (B) 2D representation of R-enantiomer of compound 5 binding with the binding pocket of the wild-type EGFRWT enzyme (PDB ID: 1M17). | ||
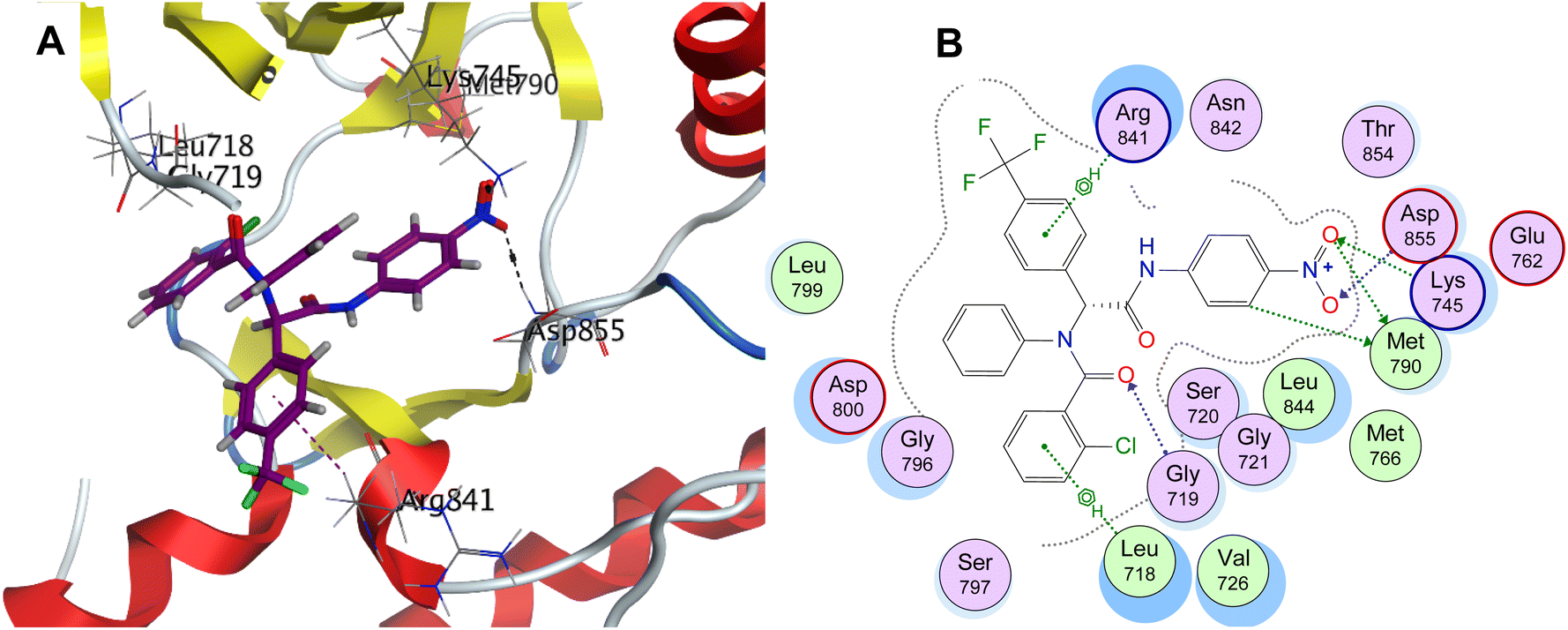 | ||
| Fig. 8 (A) 3D representation and (B) 2D representation of R-enantiomer of compound 5 binding with the binding pocket of the mutant EGFRL858R/T790M/C797S (PDB ID: 6LUB). | ||
3. Conclusion
The current study reports the synthesis of novel Ugi adducts as potential anti-proliferative agents via a one-step Ugi four-component reaction. Utilizing NMR and elemental analysis, all synthesized adducts were verified. The anti-proliferative potential of the synthesized Ugi adducts against human breast cancer MDA-MB-231 and non-small cell lung carcinoma A549 was evaluated by adopting the MTT assay. The BEAS-2B normal human lung epithelial cells were used to evaluate their selectivity. Concerning cytotoxic activity and selectivity, the Ugi adduct 5 emerged as the study hit. Against the MDA-MB-231 cell line, it was 1.42 and 1.20 times more potent than 5-FU and cisplatin, respectively. When applied to the A549 cell line, it was 1.82 and 1.62 times more potent than 5-FU and cisplatin. Additionally, compound 5 demonstrated the most prominent selectivity against MDA-MB-231 and A549 cells (SI = 7 and 11.7, respectively). Excellent sub-micromolar potency (IC50 = 0.19 μM) was demonstrated by the Ugi adduct 5 against the human mutant EGFRT790M/C797S/L858R enzyme equipotent with that of osimertinib (IC50 = 0.1 μM). It demonstrated a promising IC50 value of 2.1 nM against the EGFRWT enzyme, comparable to erlotinib (IC50 = 1.3 nM). In comparison to the untreated control, the Ugi adduct 5 caused a decrease in the expression of the cancer initiation, angiogenic, and metastatic markers (c-Myc, CD-44, CD-133, VEGF, and TGF) to 0.36, 0.5, 0.7, 0.5, and 0.4-folds, respectively in MDA-MB-231 cells. Regarding A549 cells, the exposure to compound 5 showed a 0.41, 0.64, 0.58, 0.71, and 0.69-fold reduction in the expression of c-Myc, CD-44, CD-133, VEGF, and TGF markers in comparison to untreated control. In the MDA-MB-231 cell line, compound 5 significantly increased the expression of miRNA-132 and miRNA-200c by 3.8 and 3.1-fold, respectively. In A549 cells, compound 5 showed improvements in miRNA-132 and miRNA-200c expression by 2.4 and 1.9-fold relative to the control. Through caspase 3/9 activation (1.8- and 2.3-folds), it promoted apoptosis in the A549 cell line. The molecular docking interpretations of the most potent Ugi adduct 5 in EGFRL858R/T790M/C797S (PDB ID: 6LUB) and the wild-type EGFRWT (PDB ID: 1M17) enzymes are aligned with and explain its potential to dually inhibit EGFRL858R/T790M/C797S and EGFRWT tyrosine kinases. It can be concluded that the synthesized Ugi adduct 5 represents a promising novel lead scaffold of EGFRT790M/C797S/L858R inhibitors and a possible anti-proliferative drug candidate that warrants additional investigation.4. Experimental
4.1. Chemistry
4.1.1.1. 2-Chloro-N-(2-((4-nitrophenyl)amino)-2-oxo-1-(4-(trifluoromethyl)phenyl)ethyl)-N-phenylbenzamide (5). Off-white powder (66% yield), m.p = 237–239 °C; Rf = 0.628 (EtOAc
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :n-hexane, 1:2); IR (KBr, cm−1): 3296 (N–H), 1711, 1629 (OCN); 1H NMR (500 MHz, DMSO-d6) δH: 11.10 (s, 1H, D2O exchangeable, N–H), 8.23 (d, 2H, J = 9.0 Hz, Ar–H), 7.88 (d, 2H, J = 9.0 Hz, Ar–H), 7.55 (d, 2H, J = 8.5 Hz, Ar–H), 7.42 (d, 2H, J = 7.5 Hz, Ar–H), 7.29–7.23 (m, 2H, Ar–H), 7.15–7.09 (m, 4H, Ar–H), 6.95–6.89 (m, 3H, Ar–H), 6.47 (s, 1H, NCHCO); 13C NMR (125 MHz, DMSO-d6) δC: 169.1, 168.1 (CON), 145.4, 143.0, 138.7, 138.5, 136.5, 131.7 (C–Cl), 131.1, 130.7, 129.8, 129.6, 129.5, 129.3, 129.2, 128.6, 128.5, 127.1, 125.7, 119.6 (Ar–C), 125.5 (CF3), 64.7 (NCHCO); anal. calc. for C28H19ClF3N3O4 (553.10162): C, 60.71; H, 3.46; N, 7.59. Found; C, 60.97; H, 3.61; N, 7.35. HRMS (ESI+) m/z [M + H]+ cald for C28H20ClF3N3O4+: 554.10944, found: 554.10753.
:n-hexane, 1:2); IR (KBr, cm−1): 3296 (N–H), 1711, 1629 (OCN); 1H NMR (500 MHz, DMSO-d6) δH: 11.10 (s, 1H, D2O exchangeable, N–H), 8.23 (d, 2H, J = 9.0 Hz, Ar–H), 7.88 (d, 2H, J = 9.0 Hz, Ar–H), 7.55 (d, 2H, J = 8.5 Hz, Ar–H), 7.42 (d, 2H, J = 7.5 Hz, Ar–H), 7.29–7.23 (m, 2H, Ar–H), 7.15–7.09 (m, 4H, Ar–H), 6.95–6.89 (m, 3H, Ar–H), 6.47 (s, 1H, NCHCO); 13C NMR (125 MHz, DMSO-d6) δC: 169.1, 168.1 (CON), 145.4, 143.0, 138.7, 138.5, 136.5, 131.7 (C–Cl), 131.1, 130.7, 129.8, 129.6, 129.5, 129.3, 129.2, 128.6, 128.5, 127.1, 125.7, 119.6 (Ar–C), 125.5 (CF3), 64.7 (NCHCO); anal. calc. for C28H19ClF3N3O4 (553.10162): C, 60.71; H, 3.46; N, 7.59. Found; C, 60.97; H, 3.61; N, 7.35. HRMS (ESI+) m/z [M + H]+ cald for C28H20ClF3N3O4+: 554.10944, found: 554.10753.
4.1.1.2. 2-Chloro-N-(2-((4-nitrophenyl)amino)-2-oxo-1-(4-(trifluoromethyl)phenyl)-ethyl)-N-(p-tolyl)benzamide (6). White powder (57% yield), m.p = 255–257 °C; Rf = 0.628 (EtOAc
:n-hexane, 1:2); IR (KBr, cm−1): 3369 (N–H), 1704, 1635 (OCN); 1HNMR (500 MHz, DMSO-d6) δH:11.07 (s, 1H, D2O exchangeable, N–H), 8.22 (d, 2H, J = 9.5 Hz, Ar–H), 7.87 (d, 2H, J = 9.0 Hz, Ar–H), 7.57 (d, 2H, J = 8.5 Hz, Ar–H), 7.41 (d, 2H, J = 8.5 Hz, Ar–H), 7.28–7.23 (m, 2H, Ar–H), 7.16–7.10 (m, 2H, Ar–H), 7.03 (bs, 2H, Ar–H), 6.75 (d, 2H, J = 8.0 Hz, Ar–H), 6.45 (s, 1H, NCHCO), 1.97 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δC: 169.1, 168.4 (CON), 145.3, 143.0, 138.6, 137.7, 136.6, 136.1, 131.7 (C–Cl), 130.8, 130.7, 130.7, 129.7, 129.6, 129.4, 129.3, 129.2, 129.1, 127.1, 125.7, 125.6, 119.6 (Ar–C), 125.5 (CF3), 64.7 (NCHCO), 20.8 (CH3); anal. calc. for: C29H21ClF3N3O4 (567.95): C, 61.33; H, 3.73; N, 7.40. Found; C, 61.54; H, 3.82; N, 7.23.
4.1.1.3. 2-Iodo-N-(2-((4-nitrophenyl)amino)-2-oxo-1-(4-(trifluoromethyl)phenyl)ethyl)-N-phenyl-benzamide (7). Off-white powder (77% yield), m.p = 240–243 °C; Rf = 0.628 (EtOAc
:n-hexane, 1:2); IR (KBr, cm−1): 3299 (N–H),1712, 1625 (OCN); 1H NMR (500 MHz, DMSO-d6) δH: 11.12 (bs, 1H, D2O exchangeable, N–H), 8.22 (d, 2H, J = 9.5 Hz, Ar–H), 7.87 (d, 2H, J = 9.0 Hz, Ar–H), 7.63 (d, 1H, J = 8.5 Hz, Ar–H), 7.56 (d, 3H, J = 8.5 Hz, Ar–H), 7.40 (d, 2H, J = 7.5 Hz, Ar–H), 7.21–7.13 (m, 3H, Ar–H), 6.95–6.85 (m, 4H, Ar–H), 6.47 (s, 1H, NCHCO); 13C NMR (125 MHz, DMSO-d6) δC: 170.4, 169.2 (CON), 143.0, 142.3, 139.1, 138.8, 138.5, 131.7, 131.2, 130.6, 129.5, 129.2, 129.0, 128.5, 128.4, 127.8, 125.8, 119.6 (Ar–C), 125.6 (CF3), 94.5 (C–I), 64.8 (NCHCO); anal. calc. for C28H19F3IN3O4: (645.38): C, 52.11; H, 2.97; N, 6.51. Found; C, 52.34; H, 2.74; N, 6.69.
4.1.1.4. 2-Iodo-N-(2-((4-nitrophenyl)amino)-2-oxo-1-(4-(trifluoromethyl)phenyl)ethyl)-N-(p-tolyl)-benzamide (8). Off-white powder (75% yield), m.p = 227–229 °C; Rf = 0.63 (EtOAc
:n-hexane, 1:2); IR (KBr, cm−1): 3222 (N–H), 1710, 1677 (OCN); 1H NMR (500 MHz, DMSO-d6) δH: 11.11 (bs, 1H, D2O exchangeable, N–H), 8.20 (d, 2H, J = 9.0 Hz, Ar–H), 7.85 (d, 2H, J = 8.5 Hz, Ar–H), 7.63 (d, 1H, J = 7.5 Hz, Ar–H), 7.58 (d, 2H, J = 8.5 Hz, Ar–H), 7.40 (d, 2H, J = 7.5 Hz, Ar–H), 7.16–7.08 (m, 4H, Ar–H), 6.88–6.85 (m, 1H, Ar–H), 6.75 (d, 2H, J = 7.5 Hz, Ar–H), 6.44 (s, 1H, NCHCO), 1.99 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δC: 170.5, 169.2 (CON), 143.0, 142.4, 141.0, 139.1, 138.7, 137.7, 130.9, 130.5, 129.6, 129.5, 129.0, 128.9, 127.8, 125.7, 119.6 (Ar–C), 125.6 (CF3), 94.4 (C–I), 64.7 (NCHCO), 20.9 (CH3); anal. calc. for C29H21F3IN3O4 (659.40); C, 52.82; H, 3.21; N, 6.37. Found; C, 52.95; H, 3.31; N, 6.48.
4.1.1.5. 2-(N,2-Diphenylacetamido)-N-(4-nitrophenyl)-2-(4-(trifluoromethyl)phenyl)acetamide (9). White powder (66% yield), m.p = 243–245 °C; Rf = 0.57 (EtOAc
:n-hexane, 1:2); IR (KBr, cm−1): 3331 (N–H), 1710, 1632 (OCN); 1H NMR (500 MHz, DMSO-d6) δH: 10.95 (s, 1H, D2O exchangeable, N–H), 8.19 (d, 2H, J = 9.0 Hz, Ar–H), 7.82 (d, 2H, J = 9.0 Hz, Ar–H), 7.50 (d, 2H, J = 7.5 Hz, Ar–H), 7.34 (d, 2H, J = 8.5 Hz, Ar–H), 7.21–7.14 (m, 8H, Ar–H), 7.01 (d, 2H, J = 7.0 Hz, Ar–H), 6.27 (s, 1H, NCHCO), 3.36 (d, 2H, J = 2.5 Hz, COCH2 Ph); 13C NMR (125 MHz, DMSO-d6) δC: 171.1, 169.6 (CON), 145.4, 142.9, 139.4, 138.9, 135.9, 131.7, 131.5, 129.7, 129.3, 128.7, 128.6, 126.9, 125.7, 125.6, 119.5 (Ar–C), 125.5 (CF3), 64.5 (NCHCO), 41.2 (COCH2Ph); anal. calc. for C29H22F3N3O4 (533.51): C, 65.29; H, 4.16; N, 7.88; found; C, 65.34; H, 4.23; N, 7.71.
4.1.1.6. N-(4-Nitrophenyl)-2-(2-phenyl-N-(p-tolyl)acetamido)-2-(4-(trifluoromethyl)phenyl)-acetamide (10). Off-white powder (72% yield), m.p = 233–235 °C; Rf = 0.54 (EtOAc
:n-hexane, 1:2); IR (KBr, cm−1): 3295 (N–H), 1635 (OCN); 1H NMR (500 MHz, DMSO-d6) δH: 10.94 (s, 1H, D2O exchangeable, N–H), 8.19 (d, 2H, J = 9.0 Hz, Ar–H), 7.82 (d, 2H, J = 9.0 Hz, Ar–H), 7.52 (d, 2H, J = 8.5 Hz, Ar–H), 7.34 (d, 2H, J = 8.5 Hz, Ar–H), 7.22–7.13 (m, 4H, Ar–H), 7.02–7.01 (m, 5H, Ar–H), 6.26 (s, 1H, NCHCO), 3.37 (overlap with solvent peak, 2H, CH2), 2.15 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δC: 171.3, 169.7(CON), 145.4, 142.9, 139.0, 138.0, 136.9, 135.9, 131.7, 131.1, 129.8, 129.7, 128.6, 126.9, 125.6, 119.5 (Ar–C), 125.5 (CF3), 64.8 (NCHCO), 41.0 (CH2), 21.1 (CH3); anal. calc. for C30H24F3N3O4 (547.53): C, 65.81; H, 4.42; N, 7.67; found C, 65.62; H, 4.51; N, 7.42.
4.1.1.7. N-(2-((2-((4-Nitrophenyl)amino)-2-oxo-1-(4-(trifluoromethyl)phenyl)ethyl)(phenyl)amino)-2-oxoethyl)benzamide (11). Off-white powder (60% yield), m.p = 260–262 °C; Rf = 0.48 (EtOAc
:n-hexane, 1:2); IR (KBr, cm−1): 3371 (N–H), 1702, 1675 (OCN); 1H NMR (500 MHz, DMSO-d6) δH: 10.99 (s, 1H, D2O exchangeable, CON–H), 8.64 (t, 1H, J = 5.5 Hz, D2O exchangeable, COCH2N–H), 8.20 (d, 2H, J = 9.0 Hz, Ar–H), 7.84–7.79 (m, 4H, Ar–H), 7.53–7.47 (m, 4H, Ar–H), 7.43–7.36 (m, 5H, Ar–H), 7.22–7.19 (m, 3H, Hz, Ar–H), 6.27 (s, 1H, NCHCO), 3.87, 3.84 (dd, 1H, J = 16.5 Hz, J = 6.0 Hz 1H, CHaHb), 3.58, 3.55 (dd, 1H J = 17.5 Hz, J = 5.5 Hz, CHaHb); 13C NMR (125 MHz, DMSO-d6) δC: 169.5, 169.4, 166.8 (CON), 145.4, 142.9, 138.4, 134.3, 131.9, 131.7, 131.2, 129.6, 129.0, 128.8, 127.7, 119.5 (Ar–C), 125.6 (CF3), 64.9 (NCHCO), 42.7 (NCH2); anal. calc. for C30 H23F3N4O5 (576.53); C, 62.50; H, 4.02; N, 9.72; found C, 62.73; H, 3.93; N, 9.88.
4.1.1.8. N-(2-((2-((4-Nitrophenyl)amino)-2-oxo-1-(4-(trifluoromethyl)phenyl)ethyl)(p-tolyl)amino)-2-oxoethyl)benzamide (12). Off-white powder (55% yield), m.p = 258–260 °C; Rf = 0.46 (EtOAc
:n-hexane, 1:2); IR (KBr, cm−1): 3379 (N–H), 1676, 1641 (OCN); 1HNMR (500 MHz, DMSO-d6) δH: 10.98 (s, 1H, D2O exchangeable, CON–H), 8.62 (t, 1H, J = 5.0 Hz, D2O exchangeable, COCH2N–H), 8.19 (d, 2H, J = 9.5 Hz, Ar–H), 7.83–7.79 (m, 4H, Ar–H), 7.55 (d, 3H, J = 8.5 Hz, Ar–H), 7.49 (t, 1H, J = 7.0 Hz, Ar–H), 7.42–7.36 (m, 5H, Ar–H), 7.06 (bs, 2H, Ar–H), 6.25 (s, 1H, NCHCO), 3.84, 3.81 (dd, 1H, J = 17.0 Hz, J = 6.0 Hz, CHaHb), 3.57, 3.53 (dd, 1H, J = 17.5 Hz, J = 5.0 Hz, CHaHb), 2.18 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δC: 169.6, 169.5, 166.8(CON), 145.4, 142.5, 138.5, 138.4, 135.7, 135.4, 132.0, 131.7, 130.9, 130.1, 128.8, 127.7, 119.5 (Ar–C), 125.6 (CF3), 64.9 (NCHCO), 42.6 (COCH2), 21.1(CH3); anal. calc. for C31H25F3N4O5 (590.56); C, 63.05; H, 4.27; N, 9.49. Found; C, 63.21; H, 4.31; N, 9.30.
4.1.1.9. N-(4-Nitrophenyl)-2-(N-phenyl-2-(phenylsulfonamido)acetamido)-2-(4-(trifluoromethyl)-phenyl)acetamide (13). Off-white powder (75% yield), m.p = 223–225 °C; Rf = 0.45 (EtOAc
:n-hexane, 1:2); IR (KBr, cm−1): 3253 (N–H), 1666, 1620 (OCN); 1H NMR (500 MHz, DMSO-d6) δH: 10.94 (s, 1H, D2O exchangeable, CON–H), 8.20 (d, 2H, J = 9.0 Hz, Ar–H), 7.94–7.89 (m, 1H, D2O exchangeable, CH2 N–H–SO2), 7.81 (d, 2H, J = 9.0 Hz, Ar–H), 7.63 (d, 2H, J = 7.5 Hz, Ar–H), 7.57–7.47 (m, 6H, Ar–H), 7.26–7.16 (m, 6H, Ar–H), 6.14 (s, 1H, NCHCO), 3.38 (overlap with solvent peak, 2H, NCH2); 13C NMR (125 MHz, DMSO-d6) δC: 169.2, 168.0 (CON), 145.3, 143.0, 140.7, 138.2, 137.8, 133.0, 131.6, 131.1, 129.6, 129.2, 127.1, 126.9, 119.5 (Ar–C), 125.6 (CF3), 64.7 (NCHCO), 45.1 (NCH2); anal. calc. for C29H23F3N4O6S (612.58): C, 56.86; H, 3.78; N, 9.15; found; C, 56.93; H, 3.92; N, 9.18.
4.1.1.10. N-(4-Nitrophenyl)-2-(2-(phenylsulfonamido)-N-(p-tolyl)acetamido)-2-(4-(trifluoromethyl)-phenyl)acetamide (14). Pale yellow powder (80% yield), m.p = 245–247 °C; Rf = 0.5 (EtOAc
:n-hexane, 1:2); IR (KBr, cm−1): 3254 (N–H), 1666, 1617 (OCN); 1H NMR (500 MHz, DMSO-d6) δH: 10.91 (s, 1H, D2O exchangeable, CON–H), 8.20 (d, 2H, J = 9.0 Hz, Ar–H), 7.92 (bs, 1H, D2O exchangeable, CH2 N–H–SO2), 7.80 (d, 2H, J = 9.0 Hz, Ar–H), 7.64 (d, 2H, J = 7.0 Hz, Ar–H), 7.57–7.48 (m, 8H, Ar–H) 7.25 (d, 2H, J = 8.5 Hz, Ar–H), 6.98 (bs, 1H, Ar–H) 6.12 (s, 1H, NCHCO), 3.36 (overlap with solvent peak, 2H, NCH2), 2.14 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δC: 169.2, 168.2 (CON), 145.3, 143.0, 140.8, 138.5, 138.3, 135.2, 132.9, 131.6, 130.8, 130.0, 129.6, 129.3, 129.1, 126.9, 123.3, 119.5 (Ar–C), 125.6 (CF3), 64.6 (NCHCO), 45.1 (NCH2), 21.1 (CH3); anal. calc. for C30H25F3N4O6S: (626.61); C, 57.50; H, 4.02; N, 8.94; found; C, 57.71; H, 4.33; N, 8.84.
4.1.1.11. N-Benzyl-N-(2-((4-nitrophenyl)amino)-2-oxo-1-(4-(trifluoromethyl)phenyl)-ethyl)-2-phenyl-acetamide (15). Off-white powder (70% yield), m.p = 170–172 °C; Rf = 0.57 (EtOAc
:n-hexane, 1:2); IR (KBr, cm−1): 3288 (N–H), 1708, 1630 (OCN); 1HNMR (500 MHz, DMSO-d6) δH: 10.93 (s, 1H, D2O exchangeable, CON–H), 8.17 (d, 2H, J = 8.5 Hz, Ar–H), 7.75 (d, 2H, J = 9.0 Hz, Ar–H), 7.54–7.44 (m, 4H, Ar–H), 7.26–7.07 (m, 8H, Ar–H), 6.98–6.93 (m, 2H, Ar–H), 6.24 (s, 1H, NCHCO), 4.84 (d, 1H, J = 18.5 Hz, NCHaHbPh), 4.58 (d, 1H, J = 18.5 Hz, NCHaHbPh), 3.75 (q, 2H, J = 15.5 Hz, COCH2Ph); 13C NMR (125 MHz, DMSO-d6) δC: 172.9, 169.4 (CON), 145.3, 142.9, 138.3, 135.7, 131.1, 129.9, 129.8, 128.7, 128.6, 127.2, 127.0, 126.4, 125.8, 125.7, 119.6 (Ar–C), 125.5 (CF3), 62.7 (NCHCO), 49.4 (NCH2), 40.4 (under solvent peak COCH2); anal. calc. for C30H24F3N3O4 (547.53): C, 65.81; H, 4.42; N, 7.67; found; C, 66.07; H, 4.52; N, 7.54.
4.1.1.12. N-(2-(Benzyl(2-((4-nitrophenyl)amino)-2-oxo-1-(4-(trifluoromethyl)phenyl)ethyl)amino)-2-oxoethyl)benzamide (16). Off-white powder (60% yield), m.p = 258–260 °C; Rf = 0.46 (EtOAc
:n-hexane, 1:2); IR (KBr, cm−1): 3414 (N–H), 1707, 1643 (OCN); 1HNMR (500 MHz, DMSO-d6) δH: 10.92 (s, 1H, D2O exchangeable, CON–H), 8.78 (t, 1H, J = 6.0, D2O exchangeable, CH2N–H), 8.17 (d, 2H, J = 9.0 Hz, Ar–H), 7.84 (d, 2H, J = 7.5 Hz, Ar–H), 7.75 (d, 2H, J = 9.0 Hz, Ar–H), 7.55–7.41 (m, 5H, Ar–H), 7.24–7.21 (m, 1H, Ar–H), 7.14–7.05 (m, 6H, Ar–H), 6.21 (s, 1H, NCHCO), 4.88 (d, 1H, J = 17.5 Hz, NCHaHb), 4.59 (d, 1H, J = 18.5 Hz, NCHaHb), 4.13–4.18 (2d, 1H, J = 7.5 Hz, J = 14.0 Hz, COCH2), 4.01–3.96 (m, 1H, COCH2); 13C NMR (125 MHz, DMSO-d6) δC: 171.1, 169.3, 167.3 (CON), 145.2, 142.9, 137.9, 134.3, 132.0, 131.1, 128.9, 128.7, 128.6, 127.9, 127.7, 126.5, 125.7, 119.6 (Ar–C), 125.5 (CF3), 62.9 (NCHCO), 48.7 (NCH2), 42.3 (COCH2); anal. calc. for C31H25F3N4O5 (590.56); C, 63.05; H, 4.27; N, 9.49; found; C, 63.22; H, 4.44; N, 9.31.
4.1.1.13. N-Benzyl-N-(2-((4-nitrophenyl)amino)-2-oxo-1-(4-(trifluoromethyl)phenyl)ethyl)-2-(phenylsulfonamido)acetamide (17). White powder (61% yield), m.p = 197–199 °C; Rf = 0.54 (EtOAc
:n-hexane, 1:2); IR (KBr, cm−1): 3296 (N–H), 1706, 1644 (OCN); 1H NMR (500 MHz, DMSO-d6) δH: 10.89 (s, 1H, D2O exchangeable, CON–H), 8.18 (d, 2H, J = 9.0 Hz, Ar–H), 7.98 (bs, 1H, D2O exchangeable CH2 N–H–SO2), 7.75 (d, 2H, J = 9.0 Hz, Ar–H), 7.63 (d, 2H, J = 7.5 Hz, Ar–H), 7.56 (d, 3H, J = 8.0 Hz, Ar–H), 7.49 (t, 2H, J = 7.5 Hz, Ar–H), 7.39 (d, 2H, J = 7.5 Hz, Ar–H), 7.10–7.09 (m, 2H, Ar–H), 6.94–6.74 (m, 3H, Ar–H), 6.16 (s, 1H, NCHCO), 4.69 (d, 1H, J = 18.0 Hz, CHaHb NPh), 4.48 (d, 1H, J = 18.0 Hz, CHaHb NPh), 3.80 (d, 1H, J = 17.5 Hz, CHaHb NHSO2Ph), 3.68 (d, 1H, J = 17.5 Hz CHaHb NHSO2Ph); 13C NMR (125 MHz, DMSO-d6) δC: 169.9, 169.0 (CON), 145.2, 143.0, 140.8, 139.1, 137.7, 132.9, 131.1, 129.5, 128.7, 127.3, 127.2, 127.1, 126.3, 125.8, 119.7 (Ar–C), 125.5 (CF3), 62.8 (NCHCO), 48.5 (NCH2Ph), 45.1 (CH2NH SO2Ph); anal. calc. for C30H25F3N4O6S (626.61): C, 57.50; H, 4.02; N, 8.94; found; C, 57.77; H, 4.21; N, 8.81.
4.1.1.14. N-(2-((4-Nitrophenyl)amino)-2-oxo-1-(4-(trifluoromethyl)phenyl)ethyl)-N-phenylquinoline-2-carboxamide (18). White powder (66% yield), m.p = 231–233 °C; Rf = 0.628 (EtOAc
:n-hexane, 1:2); IR (KBr, cm−1): 3265 (N–H), 1673, 1631 (OCN); 1H NMR (500 MHz, DMSO-d6) δH: 11.15 (bs, 1H, D2O exchangeable N–H), 8.23 (d, 3H, J = 9.0 Hz, Ar–H), 7.89 (d, 2H, J = 9.5 Hz, Ar–H), 7.84 (d, 1H, J = 8.5 Hz, Ar–H), 7.74 (d, 1H, J = 8.5 Hz, Ar–H), 7.66 (t, 1H, J = 7.0 Hz, Ar–H), 7.57–7.47 (m, 6H, Ar–H), 7.13 (bs, 2H, Ar–H), 6.88–6.83 (m, 3H, Ar–H), 6.53 (s, 1H, NCHCO); 13C NMR (125 MHz, DMSO-d6) δC: 169.3, 168.9 (CON), 145.4, 143.0, 140.5, 139.2, 138.5, 137.0, 136.9, 131.8, 131.6, 130.6, 129.3, 129.2, 128.4, 128.3, 128.0, 127.9, 127.5, 120.6, 119.6, 119.5 (Ar–C), 125.6 (CF3), 65.1 (NCHCO); anal. calc. for C31H21F3N4O4 (570.53): C, 65.26; H, 3.71; N, 9.82. Found; C, 65.45; H, 3.83; N, 9.59.
4.1.1.15. N-(2-((4-Nitrophenyl)amino)-2-oxo-1-(4-(trifluoromethyl)phenyl)ethyl)-N-(p-tolyl)-quinoline-2-carboxamide (19). White powder (63% yield), m.p = 213–215 °C; Rf = 0.628 (EtOAc
:n-hexane, 1:2); IR (KBr, cm−1): 3268 (N–H), 1672, 1635 (OCN); 1H NMR (500 MHz, DMSO-d6) δH: 11.13 (bs, 1H, D2O exchangeable, N–H), 8.22 (d, 3H, J = 9.0 Hz, Ar–H), 7.89 (d, 2H, J = 8.5 Hz, Ar–H), 7.84 (d, 1H, J = 7.5 Hz, Ar–H), 7.79 (d, 1H, J = 8.5 Hz, Ar–H), 7.67 (t, 1H, J = 7.5 Hz, Ar–H), 7.59–7.47 (m, 6H, Ar–H), 7.01 (bs, 2H, Ar–H), 6.69 (d, 2H, J = 7.0 Hz, Ar–H), 6.51 (s, 1H, NCHCO), 1.90 (s, 3H, CH3). 13C NMR (125 MHz, DMSO-d6) δC: 169.3, 169.1 (CON), 154.8, 146.6, 145.4, 143.0, 137.2, 137.0, 136.5, 131.8, 131.4, 130.7, 129.3, 128.9, 128.4, 128.0, 127.5, 125.6, 120.6, 119.6 (Ar–C), 125.5 (CF3), 65.7 (NCHCO), 20.9 (CH3); anal. calc. for C32H23F3N4O4 (584.56): C, 65.75; H, 3.97; N, 9.58. Found; C, 65.88; H, 4.19; N, 9.30.
4.1.1.16. {R,S}N-Benzyl-N-(2-((4-nitrophenyl)amino)-2-oxo-1-(4-(trifluoromethyl)phenyl)ethyl)-quinoline-2-carboxamide (20). White powder (80% yield), m.p = 231–233 °C; Rf = 0.628 (EtOAc
:n-hexane, 1:2); IR (KBr, cm−1): 3278 (N–H), 1618 (OCN); 1H NMR (500 MHz, DMSO-d6) δH: 11.05, 10.60(2s, 1H, D2O exchangeable N–H), 8.57, 8.39 (2d, 1H, J = 8.5 Hz, J = 8.5 Hz Ar–H), 8.21 (t, 2H, J = 8.5 Hz, Ar–H), 8.061 (d, 1H, J = 8.0 Hz, Ar–H), 7.96–7.89 (m, 1H, Ar–H), 7.84 (d, 1H, J = 8.0 Hz, Ar–H), 7.75–7.62 (m, 4H, Ar–H), 7.56–7.48 (m, 4H, Ar–H), 7.24–7.21, 7.14–7.09 (2m, 1H, Ar–H), 6.99 (s, 1H, Ar–H), 6.91 (s, 3H, Ar–H), 6.32, 6.25(2s, 1H, NCHCO) 5.21, 4.97(2d, 1H, J = 16 Hz, J = 17 Hz CHaHb – N-Ph), 4.63, 4.44 (2d, 1H, J = 17 Hz, J = 15 Hz, CHaHb N-Ph); 13C NMR (125 MHz, DMSO-d6) δC: 170.3, 169.6, 169.5, 169.0 (CON), 153.9, 153.6, 146.2, 146.1, 145.3, 145.1, 143.0, 139.3, 138.8, 138.7, 138.5, 138.2, 138.0, 131.3, 131.1, 131.0, 130.9, 129.5, 129.4, 128.8, 128.7, 128.6, 128.5, 128.4, 128.0, 127.9,127.3, 126.5, 126.3, (Ar–C), 121.7, 120.9, 119.7, 119.6 (Ar–C), 125.7, 125.6, 125.5 (CF3) , 65.5, 63.1 (NCHCO), 51.2, 48.8 (CH2); anal. calc. for C32H23F3N4O4 (584.56): C, 65.75; H, 3.97; N, 9.58, found; C, 65.82; H, 9.84; N, 9.39.
4.1.2.1. (E)-N-(2-((4-Nitrophenyl)amino)-2-oxo-1-phenylethyl)-N-phenyl-3-(4-(trifluoromethyl)-phenyl)acrylamide (23). Off-white powder (65% yield), m.p = 247–249 °C; Rf = 0.46 (EtOAc
:n-hexane, 1:2); IR (KBr, cm−1): 3445 (N–H), 1654 (OCN); 1HNMR (500 MHz, DMSO-d6) δH: 10.98 (s, 1H, D2O exchangeable, CONH), 8.20 (d, 2H, J = 9.0 Hz, Ar–H), 7.86 (d, 2H, J = 9.5 Hz, Ar–H), 7.65 (d, 2H, J = 8.5 Hz, Ar–H), 7.60 (d, 1H, J = 15.0, CH![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) CH) 7.50 (d, 2H, J = 8.0 Hz, Ar–H), 7.18–7.12 (m, 10H, Ar–H), 6.30 (s, 1H, NCHCO), 6.23 (d, 1H, J = 15.5, CHCH); 13C NMR (125 MHz, DMSO-d6) δC: 170.3, 165.4(CON), 145.7, 142.8, 140.2, 139.0, 138.9, 133.8, 131.6, 130.8, 129.2, 128.9, 128.8, 128.7, 126.4, 126.3, 122.2, 119.4 (Ar–C + CHCH), 125.6 (CF3), 65.6 (NCHCO); anal. calc. for C30H22F3N3O4 (545.52): C, 66.05; H, 4.07; N, 7.70; found; C, 65.91; H, 4.23; N, 7.61.
CH) 7.50 (d, 2H, J = 8.0 Hz, Ar–H), 7.18–7.12 (m, 10H, Ar–H), 6.30 (s, 1H, NCHCO), 6.23 (d, 1H, J = 15.5, CHCH); 13C NMR (125 MHz, DMSO-d6) δC: 170.3, 165.4(CON), 145.7, 142.8, 140.2, 139.0, 138.9, 133.8, 131.6, 130.8, 129.2, 128.9, 128.8, 128.7, 126.4, 126.3, 122.2, 119.4 (Ar–C + CHCH), 125.6 (CF3), 65.6 (NCHCO); anal. calc. for C30H22F3N3O4 (545.52): C, 66.05; H, 4.07; N, 7.70; found; C, 65.91; H, 4.23; N, 7.61.
4.1.2.2. (E)-N-(2-((4-Nitrophenyl)amino)-2-oxo-1-phenylethyl)-N-(p-tolyl)-3-(4-(trifluoromethyl)-phenyl)acrylamide (24). Off-white powder (84% yield), m.p = 227–229 °C; Rf = 0.36 (EtOAc
:n-hexane, 1:2); IR (KBr, cm−1): 3442 (N–H), 1660 (OCN); 1HNMR (500 MHz, DMSO-d6) δH: 10.96 (s, 1H, D2O exchangeable, CON–H), 8.20 (d, 2H, J = 9.0 Hz, Ar–H), 7.86 (d, 2H, J = 9.0 Hz, Ar–H), 7.66 (d, 2H, J = 8.5 Hz, Ar–H), 7.60 (d, 1H, J = 16.0 Hz CHCH) 7.51 (d, 2H, J = 8.0 Hz, Ar–H), 7.14–7.12 (m, 7H, Ar–H), 6.99 (bs, 2H, Ar–H), 6.28 (s, 1H, NCHCO) 6.27 (d, 1H, J = 16.0 Hz, CHCH), 2.17 (s, 3H, CH3); 13C NMR (125 MHz, DMSO-d6) δC: 170.3, 165.0 (CON), 145.7, 142.8, 140.2, 139.0, 137.8, 136.3, 133.9, 131.2, 130.9, 129.7, 128.9, 128.8, 126.4, 122.3, 119.3 (Ar–C + CHCH), 125.6 (CF3), 65.6 (NCHCO), 21.1 (CH3); anal. calc. for C31H24F3N3O4 (559.55): C, 66.54; H, 4.32; N, 7.51; found; C, 66.78; H, 4.29; N, 7.45.
4.1.2.3. (E)-N-Benzyl-N-(2-((4-nitrophenyl)amino)-2-oxo-1-phenylethyl)-3-(4-(trifluoromethyl)-phenyl)acrylamide (25). Off-white powder (65% yield), m.p = 245–247 °C; Rf = 0.43 (EtOAc
:n-hexane, 1:2); IR (KBr, cm−1): 3445 (N–H), 1652 (OCN); 1HNMR (500 MHz, DMSO-d6) δH: 10.93 (s, 1H, D2O exchangeable, CON–H), 8.19 (d, 2H, J = 9.0 Hz, Ar–H), 7.81 (d, 2H, J = 9.0 Hz, Ar–H), 7.75–7.66 (m, 4H, Ar–H), 7.64 (d, 1H, J = 15.0 Hz, CHCH), 7.27–7.23 (m, 5H, Ar–H), 7.11–7.09 (m, 3H, Ar–H), 7.06–7.03 (m, 1H, Ar–H), 6.96 (d, 2H, J = 7.5 Hz, Ar–H + CHCH), 6.28 (s, 1H, NCHCO), 5.04 (d, 1H, J = 17.5 Hz, NCHaHbPh), 4.6 (d, 1H, J = 17.5 Hz, NCHaHb Ph); 13C NMR (125 MHz, DMSO-d6) δC: 170.2, 167.4 (CON), 145.5, 142.8, 141.2, 139.4, 134.4, 130.4, 129.1, 128.5, 127.1, 126.5, 126.1, 122.2, 119.4 (Ar–C + CHCH), 125.6 (CF3), 63.6 (NCHCO), 48.7 (NCH2); anal. calc. for C31H24F3N3O4 (559.55); C, 66.54; H, 4.32; N, 7.51. Found; C, 66.37; H, 4.12; N, 7.68.
4.2. Biological evaluation
The biological assays were carried out according to the previously reported procedures and have been provided in the ESI;† anti-proliferative activity assay,70 EGFR inhibition assays,81,82 mRNA expression assay,83 and caspase-3/9 activation assay.844.3. Molecular docking studies
The X-ray crystallographic structures of Wild-type EGFR (PDB: 1M17)80 and mutant EGFR enzyme (PDB: 6LUB),80 with resolutions of 2.60 Å and 2.31 Å, respectively, were obtained from the RCSB Protein Data Bank (http://www.pdb.org/). Molecular modeling was performed using the Molecular Operating Environment (MOE 2022.09; Chemical Computing Group, Canada) software. Hydrogen atoms were added, and the protonation states of amino acid residues were assigned using the Protonate 3D algorithm.85 The compounds were modeled guided with the X-ray experiment of the compounds, followed by energy minimization using the MMFF94× force field. Docking studies of the synthesized compound were conducted using the MOE Dock tool, and the final ligand–enzyme complexes were selected based on interaction energy and geometric matching quality.Data availability
Data for this article, including [description of IR, NMR, experimental, elemental analysis, and methodology and docking software] are available in the attached ESI† pdf file.Conflicts of interest
The authors declare no conflict of interest.Funding
This work was supported by the Deanship of Scientific Research, Vice Presidency for Graduate Studies and Scientific Research, King Faisal University, Saudi Arabia, under the annual funding track [KFU250646].Acknowledgements
MSA acknowledges “the Deanship of Scientific Research, Vice Presidency for Graduate Studies and Scientific Research, King Faisal University, Saudi Arabia, for financial support under the annual funding track [KFU250646]”.References
- G. B. of D. C. Collaboration, JAMA Oncol., 2019, 5, 1749–1768 CrossRef PubMed.
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- F. Bray, M. Laversanne, E. Weiderpass and I. Soerjomataram, Cancer, 2021, 127, 3029–3030 CrossRef PubMed.
- F. Bray, M. Laversanne, H. Sung, J. Ferlay, R. L. Siegel, I. Soerjomataram and A. Jemal, Ca-Cancer J. Clin., 2024, 74, 229–263 CrossRef PubMed.
- D. Hanahan and R. A. Weinberg, Cell, 2011, 144, 646–674 CrossRef CAS PubMed.
- A. F. Chambers, A. C. Groom and I. C. MacDonald, Nat. Rev. Cancer, 2002, 2, 563–572 CrossRef CAS PubMed.
- A. W. Lambert, D. R. Pattabiraman and R. A. Weinberg, Cell, 2017, 168, 670–691 CrossRef CAS PubMed.
- N. V. Krakhmal, M. V. Zavyalova, E. V. Denisov, S. V. Vtorushin and V. M. Perelmuter, Cancer Invasion: Patterns and Mechanisms, 2015, vol. 7 Search PubMed.
- A. G. Clark and D. M. Vignjevic, Curr. Opin. Cell Biol., 2015, 36, 13–22 CrossRef CAS PubMed.
- B. Rude Voldborg, L. Damstrup, M. Spang-Thomsen and H. Skovgaard Poulsen, Ann. Oncol., 1997, 8, 1197–1206 CrossRef PubMed.
- J. Chen, F. Zeng, S. J. Forrester, S. Eguchi, M.-Z. Zhang and R. C. Harris, Physiol. Rev., 2016, 96, 1025–1069 CrossRef CAS PubMed.
- Md. Habban Akhter, N. Sateesh Madhav and J. Ahmad, Artif. Cells, Nanomed., Biotechnol., 2018, 46, 1188–1198 CrossRef CAS PubMed.
- S. I. Faggal, Y. El-Dash, A. Sonousi, A. M. Abdou and R. A. Hassan, RSC Med. Chem., 2024, 15(12), 4111–4125 RSC.
- S. B. Kondapaka, R. Fridman and K. B. Reddy, Int. J. Cancer, 1997, 70, 722–726 CrossRef CAS.
- R. S. Herbst, Int. J. Radiat. Oncol., Biol., Phys., 2004, 59, S21–S26 CrossRef PubMed.
- M. A. Abdelgawad, R. B. Bakr, O. A. Alkhoja and W. R. Mohamed, Bioorg. Chem., 2016, 66, 88–96 CrossRef CAS PubMed.
- T. Le and D. E. Gerber, Cancers, 2019, 11(3), 366 CrossRef CAS PubMed.
- J. Graham, M. Muhsin and P. Kirkpatrick, Nat. Rev. Drug Discovery, 2004, 3(1), 11–12 CrossRef CAS PubMed.
- J. Dowell, J. D. Minna and P. Kirkpatrick, Nat. Rev. Drug Discovery, 2005, 4, 13–14 CrossRef CAS PubMed.
- H. A. Yu, M. E. Arcila, N. Rekhtman, C. S. Sima, M. F. Zakowski, W. Pao, M. G. Kris, V. A. Miller, M. Ladanyi and G. J. Riely, Clin. Cancer Res., 2013, 19, 2240–2247 CrossRef CAS PubMed.
- D. Li, T. Shimamura, H. Ji, L. Chen, H. J. Haringsma, K. McNamara, M.-C. Liang, S. A. Perera, S. Zaghlul, C. L. Borgman, S. Kubo, M. Takahashi, Y. Sun, L. R. Chirieac, R. F. Padera, N. I. Lindeman, P. A. Jänne, R. K. Thomas, M. L. Meyerson, M. J. Eck, J. A. Engelman, G. I. Shapiro and K.-K. Wong, Cancer Cell, 2007, 12, 81–93 CrossRef CAS PubMed.
- A. Y. Helena and G. J. Riely, J. Natl. Compr. Cancer Network, 2013, 11, 161–169 CrossRef PubMed.
- S. C. M. Lau, U. Batra, T. S. K. Mok and H. H. Loong, Drugs, 2019, 79, 823–831 CrossRef CAS PubMed.
- Y. Kim, J. Ko, Z. Cui, A. Abolhoda, J. S. Ahn, S.-H. Ou, M.-J. Ahn and K. Park, Mol. Cancer Ther., 2012, 11, 784–791 CrossRef CAS PubMed.
- H.-N. Song, K. S. Jung, K. H. Yoo, J. Cho, J. Y. Lee, S. H. Lim, H. S. Kim, J.-M. Sun, S.-H. Lee, J. S. Ahn, K. Park, Y.-L. Choi, W. Park and M.-J. Ahn, J. Thorac. Oncol., 2016, 11, e45–e47 CrossRef PubMed.
- D. A. E. Cross, S. E. Ashton, S. Ghiorghiu, C. Eberlein, C. A. Nebhan, P. J. Spitzler, J. P. Orme, M. R. V Finlay, R. A. Ward, M. J. Mellor, G. Hughes, A. Rahi, V. N. Jacobs, M. R. Brewer, E. Ichihara, J. Sun, H. Jin, P. Ballard, K. Al-Kadhimi, R. Rowlinson, T. Klinowska, G. H. P. Richmond, M. Cantarini, D.-W. Kim, M. R. Ranson and W. Pao, Cancer Discovery, 2014, 4, 1046–1061 CrossRef CAS PubMed.
- T. Hirano, H. Yasuda, J. Hamamoto, S. Nukaga, K. Masuzawa, I. Kawada, K. Naoki, T. Niimi, S. Mimasu, H. Sakagami, K. Soejima and T. Betsuyaku, Mol. Cancer Ther., 2018, 17, 740–750 CrossRef CAS PubMed.
- Q. Wu, H. Jiang, S. Wang, D. Dai, F. Chen, D. Meng, P. Geng, H. Tong, Y. Zhou, D. Pan, Q. Zhou and C. Wang, Thorac. Cancer, 2020, 11, 2775–2781 CrossRef CAS PubMed.
- S. Khozin, C. Weinstock, G. M. Blumenthal, J. Cheng, K. He, L. Zhuang, H. Zhao, R. Charlab, I. Fan, P. Keegan and R. Pazdur, Clin. Cancer Res., 2017, 23, 2131–2135 CrossRef CAS PubMed.
- C. Mehlman, J. Cadranel, G. Rousseau-Bussac, R. Lacave, A. Pujals, N. Girard, C. Callens, V. Gounant, N. Théou-Anton, S. Friard, J. Trédaniel, H. Blons, C. Dujon, B. Duchemann, P. O. Schischmanoff, T. Chinet and E. Giroux Leprieur, Lung Cancer, 2019, 137, 149–156 CrossRef PubMed.
- K. S. Thress, C. P. Paweletz, E. Felip, B. C. Cho, D. Stetson, B. Dougherty, Z. Lai, A. Markovets, A. Vivancos, Y. Kuang, D. Ercan, S. E. Matthews, M. Cantarini, J. C. Barrett, P. A. Jänne and G. R. Oxnard, Nat. Med., 2015, 21, 560–562 CrossRef CAS PubMed.
- T. Grabe, J. Lategahn and D. Rauh, ACS Med. Chem. Lett., 2018, 9, 779–782 CrossRef CAS PubMed.
- M. A. Mansour, A. M. AboulMagd, S. H. Abbas, H. M. Abdel-Rahman and M. Abdel-Aziz, RSC Adv., 2023, 13(27), 18825–18853 RSC.
- Y. Jia, C. H. Yun, E. Park, D. Ercan, M. Manuia, J. Juarez, C. Xu, K. Rhee, T. Chen, H. Zhang, S. Palakurthi, J. Jang, G. Lelais, M. DiDonato, B. Bursulaya, P. Y. Michellys, R. Epple, T. H. Marsilje, M. McNeill, W. Lu, J. Harris, S. Bender, K. K. Wong, P. A. Jänne and M. J. Eck, Nature, 2016, 534, 129–132 CrossRef CAS PubMed.
- H. Patel, R. Pawara, A. Ansari and S. Surana, Eur. J. Med. Chem., 2017, 142, 32–47 CrossRef CAS PubMed.
- S. Lee, J. Kim, K. B. Duggirala, A. Go, I. Shin, B. C. Cho, G. Choi, C. H. Chae and K. Lee, Bull. Korean Chem. Soc., 2018, 39, 895–898 CrossRef CAS.
- A. Esquela-Kerscher and F. J. Slack, Nat. Rev. Cancer, 2006, 6, 259–269 CrossRef CAS PubMed.
- D. M. Pereira, P. M. Rodrigues, P. M. Borralho and C. M. P. Rodrigues, Drug Discovery Today, 2013, 18, 282–289 CrossRef CAS PubMed.
- D. Baltimore, M. P. Boldin, R. M. O'Connell, D. S. Rao and K. D. Taganov, Nat. Immunol., 2008, 9, 839–845 CrossRef CAS PubMed.
- R. H. A. Plasterk, Cell, 2006, 124, 877–881 CrossRef CAS PubMed.
- F. Han, J. He, F. Li, J. Yang, J. Wei, W. C. Cho and X. Liu, BioMed Res. Int., 2015, 2015(1), 672759 Search PubMed.
- J. Li, J. H. Huang, J. Y. Wang, Z. G. Xu, Z. Z. Chen and J. Lei, RSC Adv., 2022, 12, 33175–33179 RSC.
- F. Tahoori, R. Sheikhnejad, S. Balalaie and M. Sadjadi, Amino Acids, 2013, 45, 975–981 CrossRef CAS PubMed.
- N. Seixas, B. B. Ravanello, I. Morgan, G. N. Kaluderovic and L. A. Wessjohann, Pharmaceutics, 2019, 11(2), 59 CrossRef CAS PubMed.
- J. Wiemann, L. Fischer, J. Kessler, D. Ströhl and R. Csuk, Bioorg. Chem., 2018, 81, 567–576 CrossRef CAS PubMed.
- J. N. Gohel, K. S. Lunagariya, K. M. Kapadiya and R. C. Khunt, ChemistrySelect, 2018, 3, 11657–11662 CrossRef CAS.
- P. Brandão, A. Puerta, J. M. Padrón, M. L. Kuznetsov, A. J. Burke and M. Pineiro, Asian J. Org. Chem., 2021, 10, 3434–3455 CrossRef.
- S. De Castro, M. J. Camarasa, J. Balzarini and S. Velázquez, Eur. J. Med. Chem., 2014, 83, 174–189 Search PubMed.
- M. S. Ayoup, M. A. Fouad, H. Abdel-Hamid, E. S. Ramadan, M. M. Abu-Serie, A. Noby and M. Teleb, Eur. J. Med. Chem., 2020, 186, 111875 Search PubMed.
- M. S. Ayoup, Y. Wahby, H. Abdel-Hamid, M. M. Abu-Serie, S. Ramadan, A. Barakat, M. Teleb and M. M. F. Ismail, RSC Adv., 2023, 13, 27722–27737 RSC.
- F. R. Leroux, B. Manteau, J.-P. Vors and S. Pazenok, Beilstein J. Org. Chem., 2008, 4, 13 CrossRef PubMed.
- O. S. Artamonov, E. Y. Slobodyanyuk, D. M. Volochnyuk, I. V. Komarov, A. A. Tolmachev and P. K. Mykhailiuk, Eur. J. Org. Chem., 2014, 2014, 3592–3598 CrossRef CAS.
- F. M. Sroor, K. F. Mahrous, H. A. M. A. El-Kader, A. M. Othman and N. S. Ibrahim, Sci. Rep., 2023, 13, 17560 CrossRef CAS PubMed.
- J. Lei, J.-P. Meng, D.-Y. Tang, B. Frett, Z.-Z. Chen and Z.-G. Xu, Mol. Diversity, 2018, 22, 503–516 CrossRef CAS PubMed.
- Z. Xu, F. De Moliner, A. P. Cappelli and C. Hulme, Angew Chem. Int. Ed. Engl., 2012, 51, 8037 CrossRef CAS PubMed.
- R. J. Glyn and G. Pattison, J. Med. Chem., 2021, 64, 10246–10259 CrossRef CAS PubMed.
- H. L. Yale, J. Med. Pharm. Chem., 1959, 1, 121–133 CrossRef CAS PubMed.
- E. J. Cho, T. D. Senecal, T. Kinzel, Y. Zhang, D. A. Watson and S. L. Buchwald, Science, 2010, 328, 1679–1681 CrossRef CAS PubMed.
- N. A. Meanwell, J. Med. Chem., 2011, 54, 2529–2591 CrossRef CAS PubMed.
- Q. Wang, L. He, K. K. Li and G. C. Tsui, Org. Lett., 2017, 19, 658–661 CrossRef CAS PubMed.
- S. Schiesser, H. Chepliaka, J. Kollback, T. Quennesson, W. Czechtizky and R. J. Cox, J. Med. Chem., 2020, 63, 13076–13089 Search PubMed.
- M. Krátký, K. Svrčková, Q. A. Vu, Š. Štěpánková and J. Vinšová, Molecules, 2021, 26, 989 CrossRef PubMed.
- Y. Saito, A. Mizokami, K. Izumi, R. Naito, M. Goto and K. Nakagawa-Goto, Molecules, 2021, 26, 2812 CrossRef CAS PubMed.
- A. Abula, Z. Xu, Z. Zhu, C. Peng, Z. Chen, W. Zhu and H. A. Aisa, J. Chem. Inf. Model., 2020, 60, 6242–6250 CrossRef CAS PubMed.
- H. S. Rugo, F. Lerebours, E. Ciruelos, P. Drullinsky, M. Ruiz-Borrego, P. Neven, Y. H. Park, A. Prat, T. Bachelot, D. Juric, N. Turner, N. Sophos, J. P. Zarate, C. Arce, Y.-M. Shen, S. Turner, H. Kanakamedala, W.-C. Hsu and S. Chia, Lancet Oncol., 2021, 22, 489–498 CrossRef CAS PubMed.
- C. Möller, W. Bone, A. Cleve, U. Klar, A. Rotgeri, A. Rottmann, M.-H. Schultze-Mosgau, A. Wagenfeld and W. Schwede, ChemMedChem, 2018, 13, 2271–2280 CrossRef PubMed.
- S. R. Nasrin, T. Ishihara, A. M. R. Kabir, A. Konagaya, K. Sada and A. Kakugo, Polym. J., 2020, 52, 969–976 CrossRef CAS.
- C. Helsen, T. Van den Broeck, A. Voet, S. Prekovic, H. Van Poppel, S. Joniau and F. Claessens, Endocr.-Relat. Cancer, 2014, 21, T105–T118 CAS.
- S. M. Wilhelm, C. Carter, L. Tang, D. Wilkie, A. McNabola, H. Rong, C. Chen, X. Zhang, P. Vincent and M. McHugh, Cancer Res., 2004, 64, 7099–7109 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- X. Ma, J. Huang, Y. Tian, Y. Chen, Y. Yang, X. Zhang, F. Zhang and L. Xue, Oncogene, 2017, 36, 3159–3167 CrossRef CAS PubMed.
- G. Y. Liou, Int. J. Biochem. Cell Biol., 2019, 106, 1–7 CrossRef CAS PubMed.
- K. S. Nam, S. Oh, K. Lee, S. Yoo and I. Shin, Cell Signal, 2015, 27, 1882–1894 CrossRef CAS PubMed.
- R. Bhattacharya, F. Fan, R. Wang, X. Ye, L. Xia, D. Boulbes and L. M. Ellis, Br. J. Cancer, 2017, 117, 848–855 CrossRef CAS PubMed.
- Y. Zhao, J. Ma, Y. Fan, Z. Wang, R. Tian, W. Ji, F. Zhang and R. Niu, Mol. Oncol., 2018, 12, 305–321 CrossRef CAS PubMed.
- L. Chen, Q. Zhu, L. Lu and Y. Liu, Bioengineered, 2020, 11, 91–102 CrossRef CAS PubMed.
- S. Jurmeister, M. Baumann, A. Balwierz, I. Keklikoglou, A. Ward, S. Uhlmann, J. D. Zhang, S. Wiemann and Ö. Sahin, Mol. Cell. Biol., 2012, 32, 633–651 CrossRef CAS PubMed.
- M. Brentnall, L. Rodriguez-Menocal, R. L. De Guevara, E. Cepero and L. H. Boise, BMC Cell Biol., 2013, 14, 32 CrossRef CAS PubMed.
- K. Kashima, H. Kawauchi, H. Tanimura, Y. Tachibana, T. Chiba, T. Torizawa and H. Sakamoto, Mol. Cancer Ther., 2020, 19, 2288–2297 CrossRef CAS PubMed.
- J. Stamos, M. X. Sliwkowski and C. Eigenbrot, J. Biol. Chem., 2002, 277, 46265–46272 CrossRef CAS PubMed.
- N. I. Mansour, S. M. El-Sayed, N. S. El-Gohary, N. I. Abdel-Aziz, H. I. El-Subbagh and M. A. Ghaly, Bioorg. Chem., 2022, 127, 105966 CrossRef CAS PubMed.
- T.-H. Wang, Y.-L. Leu, C.-C. Chen, H.-J. Li, S.-C. Yang, K.-Y. Huang and C.-Y. Chen, Phytother. Res., 2022, 36, 2116–2126 CrossRef CAS PubMed.
- T. Jiang, Y.-D. Zhang, Q. Gao, J.-S. Zhou, X.-C. Zhu, H. Lu, J.-Q. Shi, L. Tan, Q. Chen and J.-T. Yu, Acta Neuropathol., 2016, 132, 667–683 CrossRef CAS PubMed.
- Y.-H. Yeh, C.-Y. Liang, M.-L. Chen, F.-M. Tsai, Y.-Y. Lin, M.-C. Lee, J.-S. Wu and C.-Y. Kuo, Food Sci. Nutr., 2019, 7, 1891–1898 CrossRef CAS PubMed.
- P. Labute, Proteins, 2009, 75, 187–205 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01428j |
| This journal is © The Royal Society of Chemistry 2025 |