
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceA Spin-polarized DFT study of functionalized MXenes as effective anchor materials in lithium-sulfur batteries†
Yize Niu,
Ying Jiang,
Feihu Zou,
Weiqi Song,
Yue Zhao,
Hongye Zhang,
Qiang Li * and
Yuanyuan Pan*
* and
Yuanyuan Pan*
College of Physics, Center for Marine Observation and Communications, Qingdao University, Qingdao, China. E-mail: liqiang@qdu.edu.cn; panyy@qdu.edu.cn
First published on 28th April 2025
Abstract
Lithium-sulfur (Li-S) batteries have attracted great enthusiasm in recent years due to ultra-high theoretical energy densities, abundant sulfur electrode resources and low price. Despite the severe shuttle effect of lithium polysulfides (LiPSs), the poor conductivity of S8 and its intermediate products, and the relatively slow dynamics, pose significant challenges for the commercial application of Li-S batteries. Here, functionalized MXenes M2CT2 (M = V, Cr, Mn, and Mo; T = F and O) used as the sulfur host are studied to build multifunctional cathodes via spin-polarized first-principles calculation. Through analyzing the adsorption energy and configuration of S8/Li2Sn adsorbed M2CT2, it is found that spin polarization is indispensable to the Li-S battery calculation of MXenes with transition metals. With the spin polarization calculation, the M2CT2 exhibit moderate anchoring strengths and stable adsorption structures, which effectively mitigates the polysulfide shuttle phenomenon. The low decomposition barriers of Li2S (0.27–1.00 eV) and low diffusion barriers of Li+ (0.11–0.44 eV) of M2CT2 are observed, which effectively improve the rate performance of batteries. Among the studied MXenes, V2CO2 and Mo2CO2 are the best choices of host materials for LiPSs with metallic characteristics, outstanding electrocatalysis performance, low decomposition barriers of Li2S, and diffusion barriers of Li+. This work provides important insights into spin-polarized electrode materials for enhanced energy storage capabilities by investigating the application of intrinsic magnetic MXene compounds.
Introduction
With the continuous growth of energy demand, it is an urgent task to develop high-performance and affordable rechargeable batteries. Lithium-sulfur (Li-S) batteries have attracted substantial research interest in recent years, owing to their high theoretical capacity and specific energy.1–4 Based on the two-electron transfer redox reaction involving multiple steps, the theoretical capacity of Li-S batteries can reach up to 1675 mA h g−1. Moreover, sulfur is abundant on earth and inexpensive.5,6 As a result, the combination of high theoretical capacity and low cost endows Li-S batteries with great application potential in the field of commercial batteries.7 However, there is still a long journey ahead for Li-S batteries to achieve commercial application, which is hindered by several thorny issues. For example, the electrical conductivities of S8 and Li2Sn are poor, and the reaction kinetics is sluggish due to the high decomposition energy barrier of Li2S.8,9 During the cycling process, intermediate products of lithium polysulfides (LiPSs) are constantly generated. These intermediates can dissolve in the electrolyte, resulting in a reduction of active sulfur species. In particular, long-chain LiPSs (Li2Sn, n = 4, 6, and 8) will migrate to the lithium metal anode and deposit there by shuttling through the separator, which also results in a rapid decline in the battery's capacity.10,11 In the future, more efforts should be devoted to suppressing the shuttle effect. An ideal sulfur composite cathode is capable of suppressing the shuttle effect by moderately anchoring Li2Sn, enhancing the sulfur reduction reaction (SRR), and improving the reaction kinetics.12–14MXenes, a novel class of two-dimensional transition metal carbides and nitrides, exhibit remarkable capabilities in electrochemical energy storage systems. These materials combine metallic-grade conductivity with ultrafast charge migration kinetics and exceptional surface charge storage capacity, attributes that have positioned them at the forefront of energy research since their 2011 discovery.15 Following Gogotsi's pioneering synthesis of Ti3C2 in 2011, titanium-based MXenes have dominated Li-S battery research due to their exceptional interfacial compatibility and sulfur confinement capabilities.16 After the first synthesis of Ti3C2, M3C2 has been extensively studied and is considered to be a cathode material for Li-S batteries with excellent performance. Recent experimental studies proved that M2C is thinner than M3C2, which has also proven to be a good anchoring material for Li-S batteries with high specific capacity and long cycle life.17,18 MXenes synthesized via HF etching typically exhibit surface terminations dominated by oxygen (O) and fluorine (F) groups, which could enhance material stability, optimize interface interaction and improve electrochemical performance.19,20 According to previous reports, Ti2C(OH)2 and V2C(OH)2 exhibit excessively strong interfacial interactions with LiPSs, inducing premature decomposition of active species.21,22 Conversely, oxygen-terminated MXenes like Ti2CO2 and Ti2NO2 demonstrate optimal chemisorption energetics for LiPSs anchoring. Ti2CF2 and Ti2NF2 achieve balanced binding interactions, with systematic analyses revealing that O- and F-terminated MXenes collectively offer superior polysulfide confinement capabilities compared to OH-terminated MXenes.23,24 The interaction between the polysulfides and the hydroxide-ternibated or no functionalized MXene is very strong, which could distort the polysulfides and the MXene materials. Therefore, they cannot effectively suppress the shuttle effect.25–27 Recently, V2NO2 and V2NF2 MXenes have been widely investigated due to low decomposition barriers of Li2S and diffusion barriers of Li+, which could accelerate reaction kinetics during the discharge and charging process. The structural diversity of MXenes motivates systematic exploration of their electrochemical properties, particularly focusing on surface-engineered variants with functional groups. It is an interesting task to explore other MXene materials that possess outstanding electrochemical performance, particularly those MXene materials with a large number of functional groups and those that have undergone surface modification.28,29
In our work, a series of M2CT2 (M = V, Cr, Mn, and Mo; T = F and O) are selected as sulfur hosts to construct multifunctional cathode material via spin-polarized first-principles calculations. We note the adsorption energy of S8/Li2Sn adsorbed M2CT2 is greatly reduced from non-spin-polarized system to spin-polarized system. Spin-polarized systems provide a critical framework for comprehensively elucidating interfacial phenomena in transition metal composites at atomic scales. The adsorption energy of S8/Li2Sn adsorbed M2CT2 range from 0.63 eV to 5.15 eV, which indicates the M2CT2 could inhibit the shuttle effect and reduce the loss of active sulfur species in the polarization system. We find that S8 can be efficiently reduced to Li2S with a small rare-limiting step (0.51–1.37 eV), indicating the superior sulfur reducing ability of M2CT2. Moreover, the decomposition barriers of Li2S and migration barriers of Li+ for M2CO2 are lower than those for M2CF2 during charging process, which ultimately promotes redox kinetics during cycling. Based on the finding presented above, we identified V2CO2 and Mo2CO2 as promising candidates for host materials with moderate anchoring strength and excellent electrocatalytic performance. Systematic analysis of MXene functionalities in this work provides actionable strategies for tailoring their interfacial properties toward sulfur cathode optimization.
Computational details
Spin-polarized density functional theory (DFT) calculations are carried out using the Vienna Ab Initio Simulation Package (VASP), employing the Perdew–Burke–Ernzerhof (PBE) generalized gradient approximation (GGA) functional to describe exchange-correlation effects.30,31 The core-electron interactions are treated through the projector-augmented wave (PAW) methodology, with a plane-wave basis set truncated at 500 eV kinetic energy. For Brillouin zone integration in the 4 × 4 supercell's two-dimensional geometry, a Monkhorst–Pack grid of 5 × 5 × 1 k-points is implemented.32 To better characterize the strongly correlated 3d electron systems, we incorporate the Hubbard U correction within the GGA + U framework.33 According to the previous works, the U values of V, Cr, Mn, and Mo atoms are set to be 3, 4, 3, and 3 eV, respectively.22,34–36 Long-range van der Waals interactions are accounted for using the DFT-D3 empirical dispersion correction scheme.37 A minimum vacuum spacing of 20 Å perpendicular to the surface plane is maintained to eliminate artificial periodic interactions. Structural optimizations are performed until achieving convergence thresholds of 1.0 × 10−5 eV per atom for total energy and 0.01 eV Å−1 for residual forces. The migration mechanisms of Li+ on M2CT2 surfaces are investigated through the climbing image nudged elastic band (CI-NEB) method, which enables precise determination of minimum energy pathways and activation barriers.38 The thermodynamic stability of Li2S adsorption on V2CO2 and Mo2CO2 is simulated by ab initio molecular dynamics (AIMD) at 300 K. The NVT ensemble simulations are conducted for 5 ps with a time step of 1.0 fs, corresponding to 5000 simulation steps. Detailed calculation equation of adsorption energies (Eads), charge density difference (CDD), and the equations of Gibbs free energy change (ΔG) are comprehensively derived in the ESI† section.Results and discussions
Configuration, magnetic ground state and electronic conductivity of M2CT2
Investigating MXenes with different magnetic transition metal constituents offers a strategic pathway to leverage their compositional versatility for improving sulfur electrochemistry in Li-S systems. Therefore, the M2C(F/O)2 with the transition metals (M = V, Cr, Mn, and Mo) are chosen as sulfur hosts in our work. The monolayer M2C MXene adopts a P3m1-symmetric honeycomb lattice, featuring a stratified architecture where hexagonally arranged carbon atoms are intercalated between two metallic M layers, as structurally resolved in Fig. 1a.39 There are five possible configurations for terminal groups (T = F and O) absorbed on the M atom in Fig. S1†: (a) T atoms locate right above the M atoms (Top sites); (b) T atoms locate at the hollow sites of adjacent C atoms (Hcp sites); (c) T atoms locate at the hollow sites of contralateral M atoms (Fcc sites); (d) T atoms located at the Fcc and Hcp sites (Fcc–Hcp); (e) T atoms located at the Hcp and Top sites (Fcc–Hcp). The stable configurations of M2CT2 are Fcc structures, except for Cr2CO2 and Mo2CO2, which are Hcp structures as shown in Fig. 1c. The basic information of their lattice parameters is listed in Table S1.† | ||
| Fig. 1 (a) Top and side views of MXene monolayer with different adsorption sites (hollow site (H), carbon-top site (C) and transition metal-top (T)). (b) Geometrical structures of S8 and Li2Sn molecules. (c) Side view of the stable atomic structures of V2CT2, Cr2CT2, Mn2CT2, and Mo2CT2 with the terminal groups T = O and F. Yellow, green, dark red, brown, light red, white, blue, dark purple and light purple color balls represent the S, Li, V, C, O, F, Cr, Mn and Mo atoms, respectively. | ||
Considering that magnetic transition metal in the M2CT2, five types of magnetic ground state structures are calculated in Fig. 2: non-magnetic (NM), ferromagnetic (FM), and antiferromagnetic (AFM1, 2, and 3). After relaxation, it is found that stable structures of V2CO2 and Mo2CO2 are NM, while the stable structures of other M2CT2 are AFM, except that Mn2CF2 and Cr2CO2 are FM structures. The calculated magnetic ground states are same as the results of the previous studies.34,40–42 According to the crystal field theory, the magnetic exchange interactions in the 2D magnetic materials mainly originate from two kinds of mechanisms:43–45 (i) the super-exchange ferromagnetism between the nearest intra-layered M atoms via the outmost oxygen atom (e.g., Cr–O–Cr and Mn–F–Mn). (ii) The antiferromagnetic interactions between the nearest interlayered M atoms via the carbon atom (e.g. Cr–C–Cr and Mn–C–Mn). In this case, the ferromagnetic super-exchange processes start to compete with the antiferromagnetic super-exchange mechanism.
 | ||
| Fig. 2 Magnetic state configurations of M2CT2: (a–e) NM, FM, AFM1, AFM2, AFM3, respectively (taking a 2 × 2 V2CF2 supercells as an example). The directions of the arrow represent the spin up and down. | ||
Meanwhile, the limited conductivity of sulfur represents another key challenge impeding the advancement of Li-S batteries. To further study the electronic structural properties of M2CT2, we calculate the projected density of states (PDOS) of M2CT2. As shown in Fig. 3, V2CO2 and Mo2CO2 show significant metallic conductivity, while the others are semiconductors, except for Mn2CF2, which is a special semimetal. The DOS of the M2CT2 mainly comes from the d-orbital of the transition metal. Furthermore, it is found that the dxy and dx2−y2 orbitals are degenerated, while the dxz and dyz orbitals are degenerated at their respective energy levels as presented in Fig. S2.†
 | ||
| Fig. 3 Projected density of states of (a) V2CF2, (b) Cr2CF2, (c) Mn2CF2, (d) Mo2CF2, (e) V2CO2, (f) Cr2CO2, (g) Mn2CO2, and (h) Mo2CO2. The Fermi level is set at zero. | ||
Anchoring of M2CT2 to inhibit shuttle effect
During Li-S battery discharge, LiPSs dissolve into the electrolyte and shuttle toward the anode through the separator, leading to progressive depletion of electroactive sulfur species. Optimal cathode materials require appropriate adsorption energies to immobilize LiPSs and prevent active sulfur depletion.46,47 Before to study the anchor ability of M2CT2 for S8 and Li2Sn, the S8 and Li2Sn are optimized in 10 × 10 × 10 Å3 cell, and the relaxed structures are shown in Fig. 1b. The S–S bond length in S8 is determined to be 2.06 Å. The Li–S bond lengths in Li2S8, Li2S6, Li2S4, Li2S2 and Li2S are determined to be 2.36 Å, 2.39 Å, 2.38 Å, 2.22 Å and 2.09 Å, respectively. These structures of our work are consistent with the previous reports.40,48,49To evaluate the anchor performance of the functionalized M2CT2, we calculated the adsorption energy (Eads) of S8 and Li2Sn on these M2CT2 surface as a key parameter, which is defined in the ESI.† The positive values of the adsorption energies indicate that adsorption could proceed spontaneously, and moderate adsorption energy represents good anchoring ability. The Eads with spin-polarized and non-spin-polarized calculations are listed in Table 1 and Table S2,† respectively. The calculated Eads for S8 and Li2Sn on V2CT2 and Cr2CT2 are well consistent with those from the previous report.50 For the adsorption of the S8 and Li2Sn, a similar binding trend is observed in all the calculated M2CT2 as shown in Fig. 4. For all the calculated M2CT2, the Eads values of the S8 are comparable. In addition, we note that the Eads values increase with a decrease in the size of LiPSs.
| MXene | S8 | Li2S8 | Li2S6 | Li2S4 | Li2S2 | Li2S |
|---|---|---|---|---|---|---|
| V2CF2 | 0.80 | 1.14 | 1.05 | 1.47 | 2.08 | 2.27 |
| Cr2CF2 | 0.76 | 1.01 | 0.97 | 0.91 | 1.24 | 1.41 |
| Mn2CF2 | 0.63 | 1.17 | 1.16 | 1.99 | 2.35 | 2.58 |
| Mo2CF2 | 0.88 | 1.19 | 1.12 | 1.10 | 1.72 | 2.50 |
| V2CO2 | 1.02 | 2.07 | 2.02 | 3.30 | 3.48 | 4.37 |
| Cr2CO2 | 1.04 | 2.40 | 2.12 | 3.64 | 4.10 | 3.73 |
| Mn2CO2 | 0.89 | 2.37 | 2.30 | 3.70 | 4.27 | 4.01 |
| Mo2CO2 | 1.38 | 3.44 | 3.13 | 4.38 | 4.24 | 5.15 |
 | ||
| Fig. 4 The absorption energies of S8/Li2Sn absorbed on (a) M2CF2 and (b) M2CO2 monolayer. The dotted and solid lines represent the absorption energy with the spin-polarized and the non-spin-polarized calculations, respectively. | ||
It is also found that the Eads of S8 and Li2Sn on M2CT2 surface with the spin-polarized calculations are significantly reduced compared with those of the non-spin-polarized calculations. The Eads of Li2Sn on V2CF2 and Mn2CF2 are reduced by 0.73–1.14 eV from the non-spin-polarized system to the spin-polarized system, while the Eads values of Li2Sn absorbed on Cr2CO2 decreases by 0.62–2.44 eV. Meanwhile, the Eads of Li2Sn absorbed on Cr2CF2 and Mo2CF2 decreases by 2.10–3.55 eV, which is nearly three times that of V2CF2 and Mn2CF2.50–52 The effect of the spin-polarization also reflected from the relaxed configuration of M2CT2–S8/Li2Sn. As presented in Fig. 5 and Fig. S3.† The atomic structure of Mn2CO2 cannot even maintain at the NM state. The short-chain polysulfides break their bonds, and the substrate M2CT2 undergo severe deformation under the spin-polarized calculation. The spin polarization system has a significant effect on the absorption process for the magnetic M2CT2. Therefore, we take the accurate spin-polarized calculation in the subsequent. The adsorption strength of the M2CT2 for Li2Sn follows the sequence: Mn2CF2 > V2CF2> Mo2CF2 > Cr2CF2 and Mo2CO2 > Mn2CO2 > Cr2CO2 > V2CO2, respectively. It clearly shows that the Eads of S8/Li2Sn on M2CT2 are much smaller than that on M2C.52 The enhanced covalent bonding characteristics between Li and S atoms in low-order LiPSs facilitate accelerated electron transfer from Li to S, creating vacant d-orbitals capable of accepting electrons from transition metal (T) atoms. Computational analyses reveal that short-chain LiPSs (Li2S2/Li2S) and Li2S4 adopt specific geometric arrangements where dual lithium atoms remain proximal to the substrate surface, demonstrating strong chemisorption behavior. Conversely, long-chain species (Li2S8 and Li2S6) exhibit monodentate adsorption configurations with a single lithium atom near the interface, accompanied by horizontally oriented S8 molecular adsorption. Crucially, all adsorbed LiPSs configurations maintain structural integrity during electrochemical cycling, showing no signs of dissociation. It ensures reliable polysulfide retention on M2CT2 surfaces throughout charge/discharge processes.
 | ||
| Fig. 5 The optimized structures of S8/Li2Sn adsorption on (a) V2CT2, (b) Cr2CT2, (c) Mn2CT2, and (d) Mo2CT2 monolayer with the spin-polarized calculations. | ||
To effectively suppress polysulfide shuttling, the adsorption energetics of high-order Li2Sn (n = 4, 6, and 8) on the sulfur host matrix should surpass those with conventional ether-based electrolytes 1,3-dioxolane/1,2-dimethoxyethane (DOL/DME), thereby preventing electrolyte-mediated active material dissolution and capacity fade.53 Given the preferential solubility of high-sulfur-content LiPSs in organic electrolytes, we systematically evaluated the adsorption energetics of long-chain LiPSs (Li2S8, Li2S6, and Li2S4) on DOL and DME. Structural optimizations of both isolated electrolyte molecules and LiPS-anchored complexes are shown in Fig. S4,† which reveal comparable polysulfide-trapping capabilities between DOL and DME, with adsorption energies spanning 0.75–0.90 eV. These values are significantly lower than those observed for M2CO2 substrates and marginally reduced compared to M2CF2 interfaces, highlighting the limited chemical confinement capacity of organic electrolytes relative to engineered MXene surfaces. Based on the aforementioned analysis, M2CT2 is capable of anchoring S8/Li2Sn with a suitable Eads value, while, it can preserve the integrity of the adsorption structures. In a tug-of-war with solvent molecules, M2CO2 shows stronger chemical affinity for the LiPSs, which reveals its potential to serve as a type of host material for sulfur cathodes.
In order to explore the essence of the adsorption behavior between S8/Li2Sn and the surface of M2CT2, the CDD between M2CT2 monolayer and S8/Li2Sn are calculated as shown in Fig. 6 with the calculation details located in ESI†. The S8 and M2CF2 interface exhibits negligible charge transfer, signifying the absence of chemical bond formation between the adsorbed sulfur species and the substrate. In contrast, S8/Li2Sn adsorption on M2CO2 exhibits significantly enhanced interfacial electron redistribution. Lithiation induces pronounced electron accumulation at Li2Sn/M2CT2 interfaces, accompanied by intensified charge transfer indicative of Li–T (T = F/O) covalent bond formation. Concurrently, expanding electron-deficient domains within Li2Sn reflect destabilization of Li–S and S–S bonding interactions. It can be deduced that chemical bonds are established between Li2Sn and M2CT2 during the lithiation process, which agrees with the trend of the adsorption energy. Moreover, the decomposition pathways of LiPSs are critically governed by two concurrent mechanisms: interfacial stabilization through Li–T (T = F/O) covalent bond formation and structural destabilization via progressive cleavage of Li–S and S–S bonds within Li2Sn species.
 | ||
| Fig. 6 Side views of CDD maps of S8 and Li2Sn on (a) V2CT2, (b) Cr2CT2, (c) Mn2CT2, and (d) Mo2CT2 monolayer (isosurface = ± 0.002 au for Li2Sn, and isosurface = ± 0.0002 au for S8). The yellow and blue region indicate electron accumulation and depletion, respectively. | ||
The electronic structures of the substrate undergo changes following LiPSs adsorption, which is investigated by analyzing the PDOS of M2CT2 after Li2S adsorption. The PDOS of M2CT2 monolayer after the adsorption of Li2S are shown in Fig. 7. Compared with the PDOS of M2CT2 monolayer, the Fermi level of M2CT2-Li2S has shifted upwards with the electronic state of S atoms appearing in the band gap for the Mo2CF2−, Cr2CF2−, Mn2CO2−, Mo2CO2−, and Cr2CO2-Li2S systems. Therefore, the band gap of V2CF2 and Cr2CF2 became narrow, while Mo2CF2, Cr2CO2, and Mn2CO2 exhibit metallic characters after the adsorption of Li2S. Overall, the conductivity of M2CT2 is enhanced with the adsorption of Li2S, which is benefit for the electron transportation.
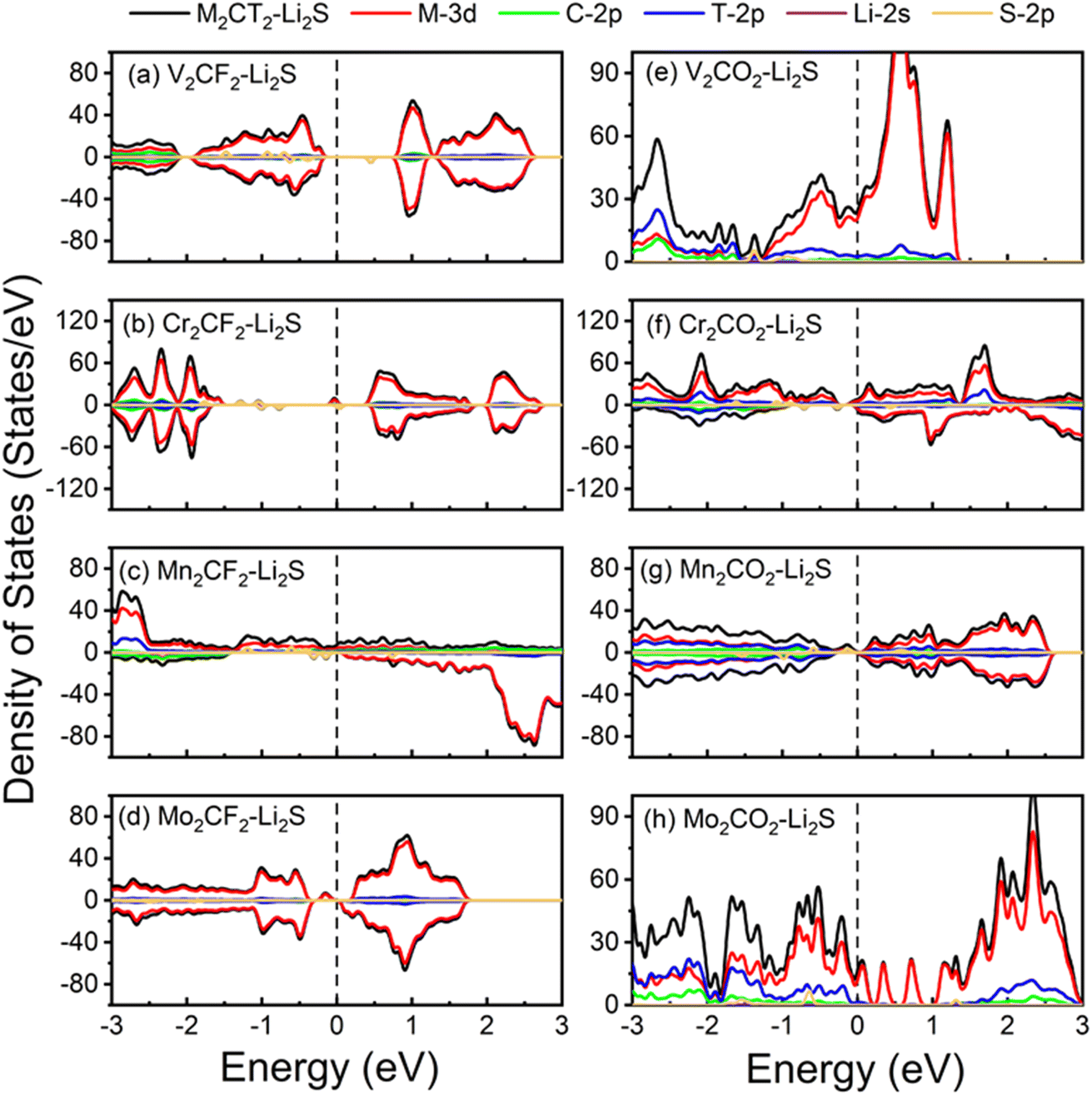 | ||
| Fig. 7 Projected density of states of (a–h) M2CT2 monolayer after the adsorption of Li2S, respectively. The Fermi level is set at zero. | ||
Electrocatalysis of M2CT2 on discharge/charging kinetics
The chemisorption intensity of Li2Sn species critically modulates electrochemical reaction kinetics. Overly robust binding interactions may restrict surface mobility of redox-active intermediates, consequently impairing charge transfer efficiency. Such kinetic constraints during sulfur redox cycling constitute a fundamental performance limitation in Li-S battery systems.54,55 The SRR pathways on M2CT2 surfaces are systematically mapped through density functional theory (DFT) calculations, with the thermodynamic Gibbs free energy change (ΔG) for each reaction intermediate from S8 to Li2S quantitatively presented in Fig. 8 and Table S3.† The calculation route of the SRR refers to the previous reports.51,52 Obviously, the steps from S8 to Li2S8 are all spontaneous exothermal on all the calculated M2CT2, which indicates that the S8 can be readily converted into Li2S8. All reduction steps from Li2S8 to Li2S are found to be endothermic, with the sole exception of the Li2S6 → Li2S4 step. The rate-limiting step is identified as Li2S4 → Li2S2 for V2CO2, Mo2CF2, and Mo2CO2, whereas for V2CF2, Cr2CF2, Cr2CO2, Mn2CF2, and Mn2CO2, the Li2S2 → Li2S step served as the kinetic bottleneck. The endergonic Li2S2 → Li2S transition stems from the inherent kinetic limitations of solid-state conversion processes, aligning with established theoretical frameworks in sulfur electrochemistry.56,57 The Gibbs free energy change barrier (ΔGbarrier) for the rate-limiting step follow the sequence: Mo2CF2 (0.51 eV) < Mn2CF2 (0.78 eV) < V2CF2 (0.84 eV) < Cr2CF2 (0.88 eV) < V2CO2 (0.89 eV) < Mn2CO2 (1.23 eV) < Mo2CO2 (1.25 eV) < Cr2CO2 (1.37 eV). It is easy to find that the M2CF2 have lower rate-limiting step reaction energy barrier than those of M2CO2, which proves that the F atom greatly enhances the catalytic activity of the SRR. | ||
| Fig. 8 The calculation of Gibbs free energy change (ΔG) of SRR on MXene monolayers. | ||
The elevated dissociation energy barrier of Li2S, the terminal discharge product, significantly impedes electrochemical cycling reversibility.58,59 This necessitates efficient delithiation kinetics (Li2S → LiS + Li+ + e−) to sustain charging efficiency. Fig. 9 quantitatively delineates the atomic-scale decomposition pathways of Li2S on M2CT2 monolayers, revealing the critical decomposition energy barriers. The minimum decomposition barriers of Li2S are 0.27, 0.32, 0.51, and 0.53 eV on Mo2CO2, V2CO2, Cr2CO2 and Mn2CO2, respectively, while they are 0.74, 0.80, 0.90, and 1.00 eV on Mn2CF2, Mo2CF2, V2CF2 and Cr2CF2, respectively. Similar to the decomposition barrier of Li2S on Ti3C2F2 (0.90 eV) and Ti3C2O2 (0.41 eV),60 all the M2CT2 greatly reduce the decomposition barriers, which indicate great catalytic effect for the decomposition reaction. Studies attribute this phenomenon to MXenes' metal-to-carbon (M/C) ratio, where elevated transition metal content (M) enhances Li2S dissociation kinetics through optimized d-band electronic interactions.27 The results indicate that the M2CT2 could promote the decomposition of Li2S, which enhances the electrochemical kinetics performance of Li-S batteries.
 | ||
| Fig. 9 Decomposition energies and decomposition paths of Li2S on (a) V2CF2, (b) Cr2CF2, (c) Mn2CF2, (d) Mo2CF2, (e) V2CO2, (f) Cr2CO2, (g) Mn2CO2, and (h) Mo2CO2 monolayer. | ||
The charging mechanism initiates with the delithiation of the terminal discharge product Li2S, where ion migration dynamics critically govern decomposition and nucleation processes. Of particular significance is the interfacial Li+ diffusion kinetics across the cathode substrate, prompting systematic analysis of Li+ migration energy barriers. Therefore, the activation energy barriers for Li+ diffusion across the cathode are quantified through NEB simulations. Three kinds of the migration paths of Li+ are considered for Fcc and Hcp structure as presented in Fig. S5 (a) and (b),† respectively, and the diffusion barriers with different migration paths are shown in Fig. 10. It is found that the path 3 (the path 6) is the most difficult migration path for Fcc (Hcp) structure, while the diffusion barriers of the path 1 (the path 4) and the path 2 (the path 5) are comparable for all the calculated M2CT2. As shown in Fig. S5,† on V2CT2, Cr2CF2, Mn2CT2 and Mo2CF2, the Li+ tend to move along the path 2 (C1 → M → C2), while on Cr2CO2 and Mo2CO2, the Li+ prefers to be along the path 5 (H1 →C→ H2). The smallest diffusion barriers of Li+ on the different M2CT2 follow the sequence: Mo2CO2 (0.11 eV) < Cr2CO2 (0.12 eV) < V2CO2 (0.13 eV) < Mn2CF2 (0.16 eV) < Mn2CO2 (0.25 eV) < V2CF2 (0.32 eV) < Cr2CF2 (0.33 eV) < Mo2CF2 (0.44 eV). These relatively low diffusion barriers of Li+ promise the rapid diffusion of Li+ on the M2CT2 surface and conversion of LiPSs during the charge process.
 | ||
| Fig. 10 Diffusion barriers with different migration paths of Li+ on (a) V2CF2, (b) Cr2CF2, (c) Mn2CF2, (d) Mo2CF2, (e) V2CO2, (f) Cr2CO2, (g) Mn2CO2, and (h) Mo2CO2 monolayer. | ||
The decomposition of Li2S, the diffusion of Li+, and the catalytic activity of the SRR of the studied M2CT2 are compared with that of the available F/O-functionalized MXenes as listed in Table S4.† (ref. 27 and 61) It is found that the decomposition barriers of Li2S for M2CT2 (0.27–1.00 eV) are apparently lower than those of V2NF2 (2.31 eV), V2NO2 (1.55 eV), and graphene (1.81 eV). Regarding the migration dynamics of Li+, the diffusion barriers of Li+ on the M2CT2 substrate (0.11 eV–0.44 eV) are close to those of V2NF2 (0.17 eV), V2NO2 (0.21 eV), and graphene (0.30 eV). Thus, M2CT2 show great kinetic performance during the discharge/charging process with relatively small decomposition barriers of Li2S and diffusion barriers of Li+. What's more, the ΔGbarrier of M2CT2 (0.51 eV–1.37 eV) are significantly lower than those of V2NF2 (2.14 eV) and V2NO2 (1.88 eV), while they are comparable or lower than that of graphene (1.07 eV). Considering the above factors, V2CO2 and Mo2CO2 have the best electrocatalytic performance among the studied M2CT2, in which the decomposition barriers of Li2S of V2CO2 and Mo2CO2 are only 0.32 eV and 0.27 eV, respectively, and the diffusion barriers of Li+ of V2CO2 and Mo2CO2 are 0.13 eV and 0.11 eV, respectively. Moreover, the V2CO2 and Mo2CO2 are metallic, which is beneficial for a fast charge/discharge rate. Therefore, V2CO2 and Mo2CO2 are the best choices for Li-S battery cathode material in the studied M2CT2.
The structural stability of electrode materials critically determines their cycling lifespan within operational voltage windows. To further investigate the stability as cathode materials for Li-S batteries, we calculate the energy of Li2S adsorption on V2CO2 and Mo2CO2 through AIMD simulations at room temperature (300 K). As illustrated in Fig. S6,† the total energy fluctuations progressively diminish during simulation, while the structural frameworks remain intact throughout the process. Therefore, it is demonstrated that both V2CO2 and Mo2CO2 maintain excellent thermodynamic stability with Li2S adsorption, and further validated their promise as highly prospective host materials for Li-S batteries.
Conclusion
In summary, we systematically explore potential of M2CT2 (M = V, Cr, Mn, and Mo; T = F and O) as anchoring materials and catalysts for Li-S batteries using DFT spin-polarized calculations. A moderate Eads for Li2Sn on M2CT2 species are observed, which would restrain the shuttling of LiPSs and prevent capacity fading and enhance the cycling capability for Li-S batteries. The decomposition barriers of Li2S (0.27 to 1.00 eV) and the Li+ diffusion barriers of M2CT2 (0.11 to 0.44 eV) are relatively low, which would accelerate the formation and decomposition of solid Li2S and hence redox reaction kinetics. It is also found that the M2CO2 exhibit lower decomposition barriers of Li2S and diffusion barriers of Li+ than M2CF2, especially for V2CO2 and Mo2CO2. Moreover, the structures of Li2S adsorbed V2CO2 and Mo2CO2 exhibit good thermodynamic stability at room temperature. All studied M2CT2 exhibit relatively low ΔGbarrier during the reaction process (0.51 to 1.37 eV), which would promote the conversion of LiPSs. Generally, the studied oxygen-terminated MXenes exhibit superior electrochemical performance compared to their fluorine-terminated MXenes. V2CO2 and Mo2CO2 are identified as the most promising candidates for Li-S battery host materials in the explored M2CT2. Our study not only provides critical insights for advancing M2CT2 implementation in Li-S battery cathodes, but also expands further theoretical investigations into MXene-based materials.Data availability
The data that supports the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work is supported by the National Natural Science Foundation of China (No. 11904409 and 51572296), Youth Innovation Technology Project of Higher School in Shandong Province (2022KJ139), Postdoctoral Research Foundation of China (No. 2018M642721), and Shandong Postdoctoral Funded Project (201901012).References
- B. Ding, J. Wang, Z. Fan, S. Chen, Q. Lin, X. Lu, H. Dou, A. Kumar Nanjundan, G. Yushin, X. Zhang and Y. Yamauchi, Mater. Today, 2020, 40, 114–131 Search PubMed
.
- W. Jin, X. Zhang, M. Liu, Y. Zhao and P. Zhang, Energy Storage Mater., 2024, 67, 103223 Search PubMed
- R. Kumar, J. Liu, J.-Y. Hwang and Y.-K. Sun, J. Mater. Chem. A, 2018, 6, 11582–11605 RSC
- A. D. Pathak, E. Cha and W. Choi, Energy Storage Mater., 2024, 72, 103711 CrossRef
- S. C. Kim, X. Gao, S. L. Liao, H. Su, Y. Chen, W. Zhang, L. C. Greenburg, J. A. Pan, X. Zheng, Y. Ye, M. S. Kim, P. Sayavong, A. Brest, J. Qin, Z. Bao and Y. Cui, Nat. Commun., 2024, 15, 1268 CrossRef CAS PubMed
- Z. Lv, P. Wang, J. Wang, S. Tian and T. Yi, J. Ind. Eng. Chem., 2023, 124, 68–88 CrossRef CAS
- J. Li, Y. Qu, C. Chen, X. Zhang and M. Shao, Nanoscale, 2021, 13, 15–35 RSC
- J. T. Kim, A. Rao, H. Y. Nie, Y. Hu, W. Li, F. Zhao, S. Deng, X. Hao, J. Fu, J. Luo, H. Duan, C. Wang, C. V. Singh and X. Sun, Nat. Commun., 2023, 14, 6404 CrossRef CAS PubMed
- Y. Chen, T. Wang, H. Tian, D. Su, Q. Zhang and G. Wang, Adv. Mater., 2021, 33, 2003666 CrossRef CAS PubMed
- R. B. Nuwayhid, J. Yeom, H. O. Ford, Z. G. Neale, M. W. Swift, N. Bernstein, R. Carter and J. W. Long, RSC Appl. Interfaces, 2025, 2, 472–483 RSC
- Z. Wu, M. Liu, W. He, T. Guo, W. Tong, E. Kan, X. Ouyang, F. Qiao, J. Wang, X. Sun, X. Wang, J. Zhu, A. Coskun and Y. Fu, Nat. Commun., 2024, 15, 9535 CrossRef CAS PubMed
- J. Chai, J. Du, Q. Li, N. Han, W. Zhang and B. Tang, Energy Fuel., 2021, 35, 15455–15471 CrossRef CAS
- S. Li, D. Leng, W. Li, L. Qie, Z. Dong, Z. Cheng and Z. Fan, Energy Storage Mater., 2020, 27, 279–296 Search PubMed
- Y. Pan, Y. Zhu, Y. Li, H. Liu, Y. Cong, Q. Li and M. Wu, Appl. Surf. Sci., 2023, 610, 155507 Search PubMed
- M. Naguib, M. Kurtoglu, V. Presser, J. Lu, J. Niu, M. Heon, L. Hultman, Y. Gogotsi and M. W. Barsoum, Adv. Mater., 2011, 23, 4248–4253 CrossRef CAS PubMed
- W. Meng, X. Liu, H. Song, Y. Xie, X. Shi, M. Dargusch, Z.-G. Chen, Z. Tang and S. Lu, Nano Today, 2021, 40, 101273 Search PubMed
- X. Liang, A. Garsuch and L. F. Nazar, Angew. Chem., Int. Ed., 2015, 54, 3907–3911 CrossRef CAS PubMed
- X. Zhao, M. Liu, Y. Chen, B. Hou, N. Zhang, B. Chen, N. Yang, K. Chen, J. Li and L. An, J. Mater. Chem. A, 2015, 3, 7870–7876 RSC
- W. Cui, Z.-Y. Hu, R. R. Unocic, G. Van Tendeloo and X. Sang, Chin. Chem. Lett., 2021, 32, 339–344 CrossRef CAS
- Q. Yang, S. J. Eder, A. Martini and P. G. Grützmacher, npj Mater. Degrad., 2023, 7, 6 CrossRef CAS
- D. Rao, L. Zhang, Y. Wang, Z. Meng, X. Qian, J. Liu, X. Shen, G. Qiao and R. Lu, J. Phys. Chem. C, 2017, 121, 11047–11054 CrossRef CAS
- J. Hu, B. Xu, C. Ouyang, S. A. Yang and Y. Yao, J. Phys. Chem. C, 2014, 118, 24274–24281 CrossRef CAS
- X. Liu, X. Shao, F. Li and M. Zhao, Appl. Surf. Sci., 2018, 455, 522–526 CrossRef CAS
- H. Lin, D.-D. Yang, N. Lou, S.-G. Zhu and H.-Z. Li, Ceram. Int., 2019, 45, 1588–1594 Search PubMed
- C. Shi, X. Zhang, Z. Li, T. T. Beyene, T. Zheng, Y. Liu and K. Zhu, Energy Fuels., 2024, 38, 14866–14890 CrossRef CAS
- Q. Zhang, X. Zhang, Y. Xiao, C. Li, H. H. Tan, J. Liu and Y. Wu, ACS Omega, 2020, 5, 29272–29283 Search PubMed
- D. Wang, F. Li, R. Lian, J. Xu, D. Kan, Y. Liu, G. Chen, Y. Gogotsi and Y. Wei, ACS Nano, 2019, 13, 11078–11086 CrossRef CAS PubMed
- C. Lamiel, I. Hussain, J. H. Warner and K. Zhang, Mater. Today, 2023, 63, 313–338 CrossRef CAS
- L. Zhang, J. Shi, K. Niu, P. Jia, Y. Gao and G. Gao, ACS Appl. Nano Mater., 2023, 6, 20812–20822 CrossRef CAS
- G. F. l. Kresse, Phys. Rev. B:Condens. Matter Mater. Phys., 1996, 54, 11169–11186 CrossRef CAS PubMed
- J. P. B. Perdew and K. Burke, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed
- G. F. l. Kresse, Comput. Mater. Sci., 1996, 6, 15–50 CrossRef CAS
- V. I. Anisimov and a. O. K. A. Jan Zaanen, Phys. Rev. B:Condens. Matter Mater. Phys., 1991, 44, 943–954 CrossRef CAS PubMed
- S. Bae, Y. G. Kang, M. Khazaei, K. Ohno, Y. H. Kim, M. J. Han, K. J. Chang and H. Raebiger, Mater. Today Adv., 2021, 9, 100118 CrossRef CAS
- Q. Sun, Z. Fu and Z. Yang, J. Magn. Magn. Mater., 2020, 514, 167141 CrossRef CAS
- X. Zhang, W. Meng, T. He, L. Jin, X. Dai and G. Liu, Appl. Surf. Sci., 2020, 503, 144091 Search PubMed
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, J. Chem. Phys., 2010, 132, 154104 CrossRef PubMed
- G. U. Henkelman, B. P. Uberuaga and H. Jónsson, J. Chem. Phys., 1991, 113, 9901–9904 CrossRef
- M. Khazaei, M. Arai, T. Sasaki, C. Y. Chung, N. S. Venkataramanan, M. Estili, Y. Sakka and Y. Kawazoe, Adv. Funct. Mater., 2012, 23, 2185–2192 CrossRef
- P. Gao, M. Song, X. Wang, Q. Liu, S. He, Y. Su and P. Qian, Nanomaterials, 2022, 12, 556 CrossRef CAS PubMed
- J. He, P. Lyu and P. Nachtigall, J. Mater. Chem. C, 2016, 4, 11143–11149 RSC
- Q. Sun, Z. Fu, Y. Li and Z. Yang, J. Alloys Compd., 2021, 850, 156769 CrossRef CAS
- Z. Tan, Z. Fang, B. Li and Y. Yang, ACS Omega, 2020, 5, 25848–25853 Search PubMed
- L. Zhang, Y. Liu, M. Wu and G. Gao, Adv. Funct. Mater., 2024, 2417857 Search PubMed
- L. Zhang, Y. Liu, Z. Xu and G. Gao, 2D Mater., 2023, 10, 045005 CrossRef CAS
- C. Zhang, W. Chu, X. Hong, Q. He, R. Lu, X. Liao and Y. Zhao, Chem. Eng. J., 2022, 439, 135679 CrossRef CAS
- G. Xu, X. Song, M. Jiang, R. Wang, S. Lian and X. Yang, Appl. Catal., B, 2025, 362, 124707 CrossRef CAS
- Z. Chen, S. Huang, X. Yuan, X. Gan and N. Zhou, Appl. Surf. Sci., 2021, 544, 148861 CrossRef CAS
- Z. Chen, S. Huang, B. Huang, M. Wan and N. Zhou, Appl. Surf. Sci., 2020, 509, 145319 CrossRef CAS
- Z. Chen, Z. Chang, Z. Liu and N. Zhou, Appl. Surf. Sci., 2022, 602, 154375 CrossRef CAS
- X. Zhu, M. Ge, T. Sun, X. Yuan and Y. Li, J. Phys. Chem. Lett., 2023, 14, 2215–2221 CrossRef CAS PubMed
- C. Wei, T. Fang, X. Tang, P. Wang and X. Liu, J. Phys. Chem. C, 2022, 126, 17066–17075 Search PubMed
- M. Cheviri and S. Lakshmipathi, Comput. Theor. Chem., 2021, 1202, 113323 Search PubMed
- Z. Du, X. Chen, W. Hu, C. Chuang, S. Xie, A. Hu, W. Yan, X. Kong, X. Wu, H. Ji and L. J. Wan, J. Am. Chem. Soc., 2019, 141, 3977–3985 CrossRef CAS PubMed
- G. Zhou, S. Zhao, T. Wang, S. Z. Yang, B. Johannessen, H. Chen, C. Liu, Y. Ye, Y. Wu, Y. Peng, C. Liu, S. P. Jiang, Q. Zhang and Y. Cui, Nano Lett., 2020, 20, 1252–1261 CrossRef CAS PubMed
- Z. Yi, F. Su, L. Dai, Z. Wang, L. Xie, Z. Zuo, X. Chen, Y. Liu and C.-m. Chen, Energy Storage Mater., 2022, 47, 327–335 CrossRef
- Z. Jin, T. Lin, H. Jia, B. Liu, Q. Zhang, L. Li, L. Zhang, Z. M. Su and C. Wang, ACS Nano, 2021, 15, 7318–7327 CrossRef CAS PubMed
- Y. Fu, H. Wang, D. Li, J. Shen, Y. Liu, M. Wei and Q. Hu, J. Alloys Compd., 2023, 965, 171306 CrossRef CAS
- X. T. Fang, L. Zhou, C. Chen, D. L. Danilov, F. Qiao, H. Li and P. H. L. Notten, Molecules, 2023, 28, 7304 CrossRef CAS PubMed
- H. Hong, N. A. R. Che Mohamad, K. Chae, F. Marques Mota and D. H. Kim, J. Mater. Chem. A, 2021, 9, 10012–10038 RSC
- K. Fan, Y. Ying, X. Luo and H. Huang, J. Mater. Chem. A, 2021, 9, 25391–25398 RSC
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01387a |
| This journal is © The Royal Society of Chemistry 2025 |