
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis, biological activities and computational studies of bis-Schiff base derivatives of 4-hydroxyacetophenone: insights from an in vitro, molecular docking and dynamics simulation approach†
Gul Badshah‡
a,
Aftab Alam‡b,
Muhammad Ayaza,
Ahmed A. Elhenawy c,
Imtiaz Ahmadd,
Shujaat Ahmade,
Muhammad Usmana,
Ashwag S. Alanazif,
Abdul Latifa,
Mumtaz Ali*a and
Manzoor Ahmad*a
c,
Imtiaz Ahmadd,
Shujaat Ahmade,
Muhammad Usmana,
Ashwag S. Alanazif,
Abdul Latifa,
Mumtaz Ali*a and
Manzoor Ahmad*a
aDepartment of Chemistry, University of Malakand, P.O. Box 18800, Dir Lower, Khyber Pakhtunkhwa, Pakistan. E-mail: mumtazali@gmail.com; manzoorahmad@uom.edu.pk
bDepartment of Chemistry, Rawalpindi Women University, Rawalpindi, Pakistan
cChemistry Department, Faculty of Science, Al-Azhar University, Cairo, Egypt
dPrograma de Pós-Graduação em Bioquímica e Bioprospecção, Universidade Federal de Pelotas, Campus Universitário Capão do Leão S/N, Pelotas, RS CEP 96010-900, Brazil
eDepartment of Pharmacy, Shaheed Benazir Bhutto University, Sheringal, Dir (Upper), Khyber Pakhtunkhwa, Pakistan
fDepartment of Pharmaceutical Sciences, College of Pharmacy, Princess Nourah bint Abdulrahman University, Riyadh 11671, Saudi Arabia
First published on 25th June 2025
Abstract
This study is based on the synthesis and acetyl and butyryl cholinesterase inhibitory activities of some bis-Schiff base derivatives of 4-hydroxyacetophenone. All the synthesized products (2a–j) were structurally analysed by means of modern spectroscopic methods, including 1H- and 13C-NMR and EI-MS, and finally tested for their ability to inhibit cholinesterase enzymes. In the series, six compounds—2j (IC50 = 15.86 ± 0.38 and 29.23 ± 0.04 μM), 2b (IC50 = 18.58 ± 0.21 and 35.31 ± 0.01 μM), 2a (IC50 = 44.36 ± 0.33 and 77.93 ± 1.46 μM), 2f (IC50 = 48.37 ± 0.06 and 76.35 ± 1.17 μM), 2g (IC50 = 62.28 ± 0.42 and 98.71 ± 1.18 μM), and 2e (IC50 = 98.21 ± 0.01 and 135.7 ± 2.61 μM)—were found to be the most promising inhibitors of acetyl and butyryl cholinesterase enzymes compared with the standard drug galantamine (IC50 = 104.8 ± 1.83 and 156.8 ± 1.83 μM), while the remaining compounds were found to be good-to-less active. Compound 2j displayed the most significant inhibition against AChE and BuChE among the tested bis-Schiff base derivatives, thus emerging as a superior compound to the standard galantamine. The highest activity of this compound is because of the favourable molecular interactions such as strong electrophilicity, high softness and a small energy gap. Molecular docking indicates that the compound 2j acts as a dual inhibitor owing to the formation of hydrophobic and polar interactions. The key structural features that include bromo benzyl and 2-methoxyphenol groups play a vital role in its efficacy, making it a more powerful inhibitor than the standard galantamine.
Introduction
Alzheimer's disease (AD) is a multidimensional neurodegenerative condition characterized by social disorders, advanced mental failure, a range of neuropsychiatric indications, and loss in everyday activities.1 Scientifically, it arises from a multifaceted interaction between environmental aspects and genetic dispositions.2 Usually diagnosed after the age of 56 years, AD affects about 10% of individuals aged 65 years and older, making it a noticeable age-associated sickness and the leading cause of dementia in the elderly.3 Amongst individuals above 80 years, AD incidence increases to 30% or more. In emerging countries, AD is the main cause of death, followed by cardiovascular diseases, cancer and cerebrovascular accidents.4 Around 35 million people in the world currently suffer from AD, and this number is predicted to increase to 107 million by 2050.5,6 AD is closely related to cardiac risk factors, including hypertension and high serum cholesterol levels.7,8 This disease is categorized by the progressive degeneration of neurons primarily due to reduced cholinergic neurotransmission. Assuming the multifactorial pathogenesis of AD, pioneering drug discovery plans have been established to target the disease over multi-functional mediators.9 These mediators display dual inhibitory actions in contradiction with acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE) enzymes involved in AD development.10,11 Acetylcholine plays a fundamental role in mental and memory purposes, and a decrease in its levels significantly contributes to the cholinergic dysfunction underlying the cognitive impairments linked with AD.12–14The sister enzyme of acetylcholinesterase (AChE), which is butyrylcholinesterase (BuChE), has gathered emergent attention in the management of Alzheimer's disease (AD) owing to its developing character in the initial phases of anticholinesterase healings.15–17 Higher BuChE enzymatic action is usually detected in patients with obesity, hyperlipidaemia and hypertension although condensed activity is well-known in those on beta-blocker therapy or with acute myocardial infarction.18,19 Lessened BuChE activity can unfavourably disturb cholinergic neurotransmission, causing symptoms such as death (in severe cases), vomiting, fever, dizziness, and blurred vision.20,21 Conversely, the selective inhibition of BuChE has been established to escape unwanted adverse effects, signifying that it is a potentially beneficial target.22 Highly specific BuChE inhibitors might be needed for the development of state-of-the-art treatments for AD.12 Together with cholinesterase inhibitors, their continued growth might lead to improved clinical consequences, raising the panorama of upcoming AD care, which is further effective.10
Bis-Schiff bases, di-imines or azines are well-known organic compounds that contain two azomethine groups in their core structures.23 These compounds can be prepared by refluxing amines (primary) with carbonylic compounds in acidic or basic media.24 Bis-Schiff bases may be symmetric or unsymmetric depending on the nature of the attached groups.25 Among other organic compounds, bis-Schiff bases are important in sensing,26 material science,27 and medicinal chemistry.28 Bis-Schiff base compounds have gained attention in the field of medicinal chemistry, with a diverse range of biological activities, i.e., anti-oxidant,29,30 anti-urease,24 anti-diabetic,31 and anti-viral32 properties.
Recently, our group reported the synthesis and diverse biological activities of compounds with bis-Schiff base functionality; for example, flurbiprofen bis-Schiff bases (a) were synthesized and tested for their in vivo anti-inflammatory and analgesic activities with prominent results.33 Similarly, bis-Schiff base compounds with ethyl phenyl ketone moiety were reported as anti-oxidant (b) and anti-urease (c) agents;33,34 however, bis-Schiff bases of the marketed ibuprofen drug (d) were published as potential anticancer agents.35 Furthermore, 4-nitroacetophenone-based bis-Schiff bases (e) were reported as α-glucosidase inhibitors,36 while bis-Schiff bases of 2,4-dihydroxy acetophenone (f) were documented as potent anti-diabetic agents37 (Fig. 1). Owing to the diverse biological activities of bis-Schiff bases, in the current study, we report the synthesis and cholinesterase (acetyl and butyryl) inhibitory activity of some novel bis-Schiff bases based on the 4-hydroxyacetophenone nucleus.
 | ||
| Fig. 1 Compounds containing the bis-Schiff base functionality with their biological activities. | ||
Results and discussion
Chemistry
In this study, several novel azine derivatives based on the 4-hydroxyacetophenone moiety are successfully synthesized in outstanding yields by applying a multi-step procedure. Initially, 4-hydroxy acetophenone was refluxed in a DMF solvent containing potassium carbonate with 4-bromobenzyl bromide to obtain 1-(4-((4-bromobenzyl)oxy)phenyl)ethan-1-one (1) in high yield, which was further refluxed with an extra amount of hydrazine hydrate in ethanol solvent to obtain 1-(4-((4-bromobenzyl)oxy)phenyl)ethylidene)hydrazine (2). Finally, different aromatic substituted aldehydes were refluxed with the desired compound (2) having a catalytic quantity of acetic acid in ethanol solvent to obtain the azine derivatives (2a–j) in excellent yields (Scheme 1). The thin layer chromatographic method was utilized to determine the progress of the reactions. These derivatives were characterized using modern spectroscopic techniques, including EI-MS, 13C-NMR and 1H-NMR, and finally subjected to their cholinesterase (AChE and BuChE) activity (Table 1). | ||
| Scheme 1 Synthetic procedure of bis-Schiff base derivatives (1, 2 and 2a–j). | ||
| C. no | Ar | Acetylcholinesterase | Butyrylcholinesterase |
|---|---|---|---|
| IC50 ± SEM (μM) | IC50 ± SEM (μM) | ||
| 2a |  |
44.36 ± 0.33 | 77.93 ± 1.46 |
| 2b |  |
18.58 ± 0.21 | 35.31 ± 0.01 |
| 2c |  |
150.2 ± 2.26 | 175.4 ± 2.96 |
| 2d |  |
139.0 ± 2.33 | 161.8 ± 0.07 |
| 2e |  |
98.21 ± 0.01 | 135.7 ± 2.61 |
| 2f |  |
48.37 ± 0.06 | 76.35 ± 1.17 |
| 2g |  |
62.28 ± 0.42 | 98.71 ± 1.18 |
| 2h |  |
128.2 ± 0.98 | 166.3 ± 1.20 |
| 2i |  |
141.8 ± 0.77 | 177.0 ± 3.53 |
| 2j |  |
15.86 ± 0.38 | 29.23 ± 0.04 |
| Standard | Galantamine | 104.8 ± 1.83 | 156.8 ± 1.83 |
In vitro acetyl and butyryl cholinesterase inhibitory activities
All the synthesized product compounds (2a–j) were screened against acetyl and butyryl cholinesterase inhibitory action. In the series, six compounds, including 2j (IC50 = 15.86 ± 0.38 and 29.23 ± 0.04 μM), 2b (IC50 = 18.58 ± 0.21 and 35.31 ± 0.01 μM), 2a (IC50 = 44.36 ± 0.33 and 77.93 ± 1.46 μM), 2f (IC50 = 48.37 ± 0.06 and 76.35 ± 1.17 μM), 2g (IC50 = 62.28 ± 0.42 and 98.71 ± 1.18 μM), and 2e (IC50 = 98.21 ± 0.01 and 135.7 ± 2.61 μM), were found as the most promising inhibitors of acetyl and butyryl cholinesterase enzymes compared with the standard drug galantamine (IC50 = 104.8 ± 1.83 and 156.8 ± 1.83 μM). Similar to this, the remaining four compounds 2h, 2d, 2i and 2c attributed less to least inhibitory activity with IC50 values of 128.2 ± 0.98 and 166.3 ± 1.20, 139.0 ± 2.33 and 161.8 ± 0.07, 141.8 ± 0.77 and 177.0 ± 3.53 μM, 150.2 ± 2.26 and 175.4 ± 2.96 μM, respectively (Table 1). Utilizing the log inhibitory concentration on the X-axis and absorbance on the Y-axis, a dose response curve formed and the findings are shown in Fig. 2. This figure shows that the activities have increased in conjunction with the desired concentrations. Nevertheless, the analysis was performed effectively based on the IC50 values. Acetyl and butyryl cholinesterase inhibitory action on these derivatives was compared based on their individual IC50 values. | ||
| Fig. 2 Dose response curve of AChE and BuChE inhibition of the product derivatives (2a–j). | ||
Structure activity relationship
The structure activity relationship (SAR) studies of synthetic derivatives emphasize understanding in what way explicit structural characteristics affect their biological activities. By investigating the disparities in the attached substituents, molecular frameworks and functional groups, our goal is to identify key structural features that inhibit or improve the anticipated pharmacological properties. This SAR examination offers insights into the optimization of the selectivity, potency as well as stability of the product compounds, guiding additional alterations for enriched therapeutic effectiveness.In the synthetic library, six compounds were attributed potent dual inhibitory activities against AChE and BuChE enzymes, with compound 2j (IC50 = 15.86 ± 0.38 and 29.23 ± 0.04 μM) exhibiting the highest potency. The greater activity of compound 2j could be attributed to the existence of electron donating methoxy and hydroxyl groups located at the meta and para locations of the benzene ring, respectively, that probably contribute to improving the binding efficacy and affinity of the enzymes. A minor fall in the inhibitory activity was perceived for compound 2b (IC50 = 18.58 ± 0.21 and 35.31 ± 0.01 μM), which could be attributed to the change in the position of the electron donating group. In compound 2b, the hydroxyl substituent is sited at the ortho position, while the methoxy substituent remains at the meta site of the benzene ring. This positional alteration may influence the interaction of the compounds with the active sites of the enzymes, thereby diminishing their inhibitory power to some extent.
Similarly, a comparative investigation of compounds 2a (IC50 = 44.36 ± 0.33 and 77.93 ± 1.46 μM), with 2f (IC50 = 48.37 ± 0.06 and 76.35 ± 1.17 μM), and 2g (IC50 = 62.28 ± 0.42 and 98.71 ± 1.18 μM), demonstrated that compound 2a displays the uppermost AChE inhibition likely owing to the existence of a diethyl amino substituent at the para position of the benzene ring that improves the binding interactions. Compound 2f indicates a minor decrease in the activity potentially because of the para-substituted methoxy group, while a further noticeable reduction is detected with compound 2g, which could be due to the para methyl group that may not contribute as efficiently to the binding interactions of the enzyme (Fig. 3). By comparing compounds 2d (IC50 = 139.0 ± 2.33 and 161.8 ± 0.07 μM) and 2c (IC50 = 150.2 ± 2.26 and 175.4 ± 2.96 μM), both derivatives have electron withdrawing substituents; compound 2d shows greater inhibition than 2c. This improved activity in compound 2d might be ascribed to the existence of a chlorine group at the para position of the benzene ring, which could improve the interfaces with the enzymes. Besides, compound 2c containing a bromine atom at para and a fluorine atom at ortho position displayed lesser inhibition, and this could be probably due to the spatial and electronic effects of the attached substituents, which might interfere with the optimal interaction of the enzymes.
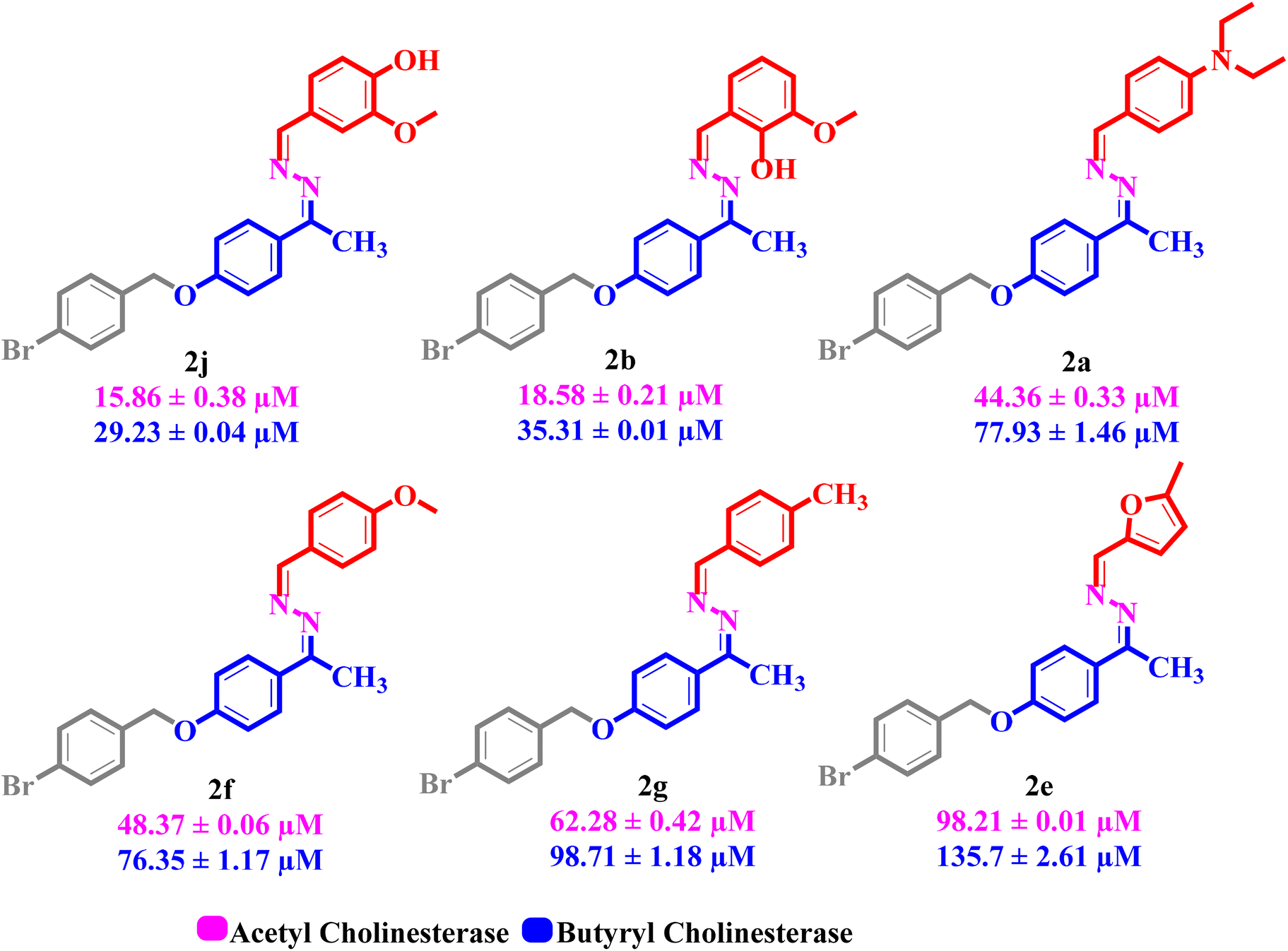 | ||
| Fig. 3 The most active dual inhibitors (AChE and BuChE) in the synthesized series. | ||
Electronic properties of bis-Schiff bases of 4-hydroxyacetophenone
The inhibition of cholinesterase enzymes, specifically acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE), is a critical therapeutic strategy for managing neurodegenerative disorders, such as Alzheimer's disease. Bis-Schiff bases derived from 4-hydroxyacetophenone have emerged as promising candidates owing to their structural diversity and potential interactions with enzyme active sites. This study investigates the relationship between the chemical reactivity indices of six bis-Schiff bases (2a, 2b, 2e, 2f, 2g and 2j) and their biological activities against AChE and BuChE. The analysis leverages time-dependent density functional theory (TD-DFT) calculations using the wB97X-D3/6-311G++ basis set to evaluate key parameters, such as the energy gap (ε = ELUMO − EHOMO), hardness (η), softness (σ), electronegativity (χ) and electrophilicity (ω). These parameters are compared with the standard drug galantamine to elucidate structure–activity relationships (Fig. 4). | ||
| Fig. 4 Heat map for electronic parameters for most active compounds compared with the standard drug galantamine. | ||
Lower energy gaps correlate with higher inhibition efficiencies, underscoring the importance of molecular flexibility and the ease of electron redistribution. Among the compounds studied, galantamine exhibits the largest energy gap (5.602 eV), which is consistent with its relatively lower reactivity and high stability. Conversely, compounds (2g and 2f) have the smallest energy gap (3.759 eV and 3.787 eV, respectively), suggesting enhanced reactivity and a greater propensity for electronic interactions with target enzymes. Compounds (2a, 2b and 2e) exhibit intermediate energy gaps (4.253–4.126 eV), indicating moderate reactivity. Notably, the methoxy-substituted derivatives (2f and 2j) display slightly lower energy gaps (3.787 and 3.789 eV, respectively), while compound (2g), which is methyl-substituted, has the smallest energy, which may enhance their ability to engage in hydrogen bonding and π–π stacking interactions at the enzyme active site.
Hardness (η) measures resistance to charge transfer, while softness (σ) quantifies susceptibility to deformation under external perturbations. Compounds with lower hardness values (higher softness) tend to be more reactive, while softer molecules exhibit greater adaptability to the enzyme environment, enhancing selectivity and potency. For instance, compound (2f) exhibits the lowest hardness (1.893 eV) and highest softness (0.264), correlating with its potent inhibitory activity against both AChE (48.37 ± 0.06%) and BuChE (76.35 ± 1.17%). In contrast, galantamine, with the highest hardness (2.801 eV) and lowest softness (0.179), demonstrates reduced reactivity but maintains excellent inhibitory efficacy owing to its optimized binding mode within the enzyme pocket. Electronegativity (χ) reflects the tendency of a molecule to attract electrons, while electrophilicity (ω) quantifies its capacity to accept electrons. Higher electrophilicity often correlates with a stronger nucleophilic attack by amino acid residues in the enzyme active site. Compound (2g) exhibits the highest electrophilicity (3.719), potentially facilitating stronger interactions with the negatively charged residues in the enzyme pocket. Similarly, compound (2f) shows elevated electrophilicity (3.606), supporting its superior inhibitory activity. Higher electrophilicity values correspond to stronger enzyme–ligand interactions, particularly for compounds (2f, 2g and 2e). Generally, compound (2j) shows the highest AChE and BuChE inhibitory activity, which demonstrates moderate values for all reactivity indices. This suggests that a balance between stability and reactivity may be crucial for optimal ChE inhibition. The presence of a 2-methoxyphenol group (2j) enhances cholinesterase inhibition and maintains moderate reactivity, supporting hydrogen bonding and π–π interactions with AChE's peripheral anionic site, complementing its low η and high ω. Compounds with higher electronegativity and electrophilicity (2g and 2f) show reduced inhibitory activity, suggesting that excessive reactivity may be detrimental. The N,N-diethyl aniline group in compound (2a) results in lower inhibitory activity possibly owing to steric hindrance or reduced interaction with the enzyme active site.
The study of frontier molecular orbitals (FMOs), specifically HOMO and LUMO, is crucial in understanding the electronic properties and reactivity of organic compounds. In this analysis, we compare the FMOs of six synthesized compounds (2a, 2b, 2e, 2f, 2g and 2j) with the reference compound galantamine, focusing on their activity against acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE).
The distribution of HOMO and LUMO can be visualized using molecular orbital diagrams (Fig. 5). In most compounds, the HOMO is localized on the electron-rich regions of the molecule, such as the aromatic rings and electron-donating groups (e.g., methoxy groups). The distribution of the HOMO can provide insights into the molecule's nucleophilicity, as regions with high electron density are more likely to interact with electrophilic sites on the enzyme. The LUMO typically represents the region of the molecule most likely to accept electrons. In these compounds, the LUMO is often localized on the conjugated system, including the Schiff base moiety and the aromatic rings. The distribution of the LUMO can provide insights into the molecule's electrophilicity and its potential to interact with nucleophilic sites on the enzyme. For instance, in compound (2a), the HOMO is primarily localized on the aromatic ring and the diethyl aniline moiety, indicating regions of high electron density. The LUMO, however, is delocalized over the hydrazono group and the bromobenzyl substituent, suggesting potential sites for electrophilic attack. Similarly, in compound (2b), the HOMO is concentrated in the methoxyphenol group, reflecting its electron-rich nature. The LUMO is spread across the hydrazono linkage and the bromobenzyl moiety, which is consistent with the observed reactivity patterns. In compound (2j), the HOMO is concentrated on the methoxyphenol group, similar to compound (2b), but the LUMO shows a different pattern owing to the altered substitution pattern.
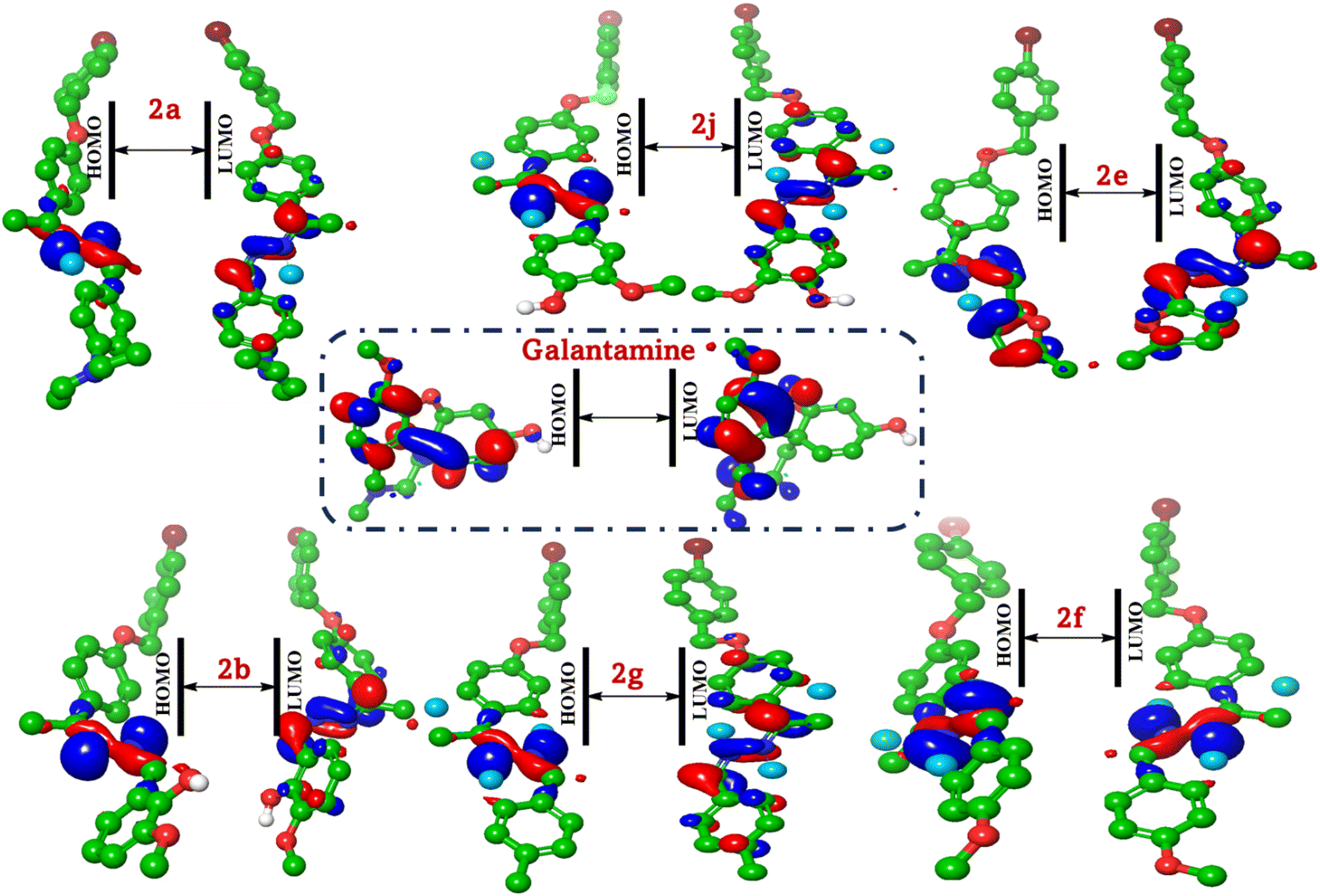 | ||
| Fig. 5 FMO based on the HOMO/LUMO analysis of the most active compounds (2a, 2b, 2e, 2f, 2g and 2j) and galantamine. | ||
For compound (2e), the presence of the furan ring alters the distribution of both HOMO and LUMO. The HOMO is more evenly distributed across the molecule, while the LUMO shows a significant contribution from the furan ring, which may influence its interaction with cholinesterase. Compounds (2f and 2g) display similar trends with the methoxy and methyl groups influencing the HOMO and LUMO distributions. The HOMO is localized on these electron-donating groups, while the LUMO is more delocalized. Galantamine, as a standard, has a distinct HOMO–LUMO distribution that correlates with its known activity against cholinesterase. The HOMO is localized on the nitrogen-containing heterocycle, while the LUMO is delocalized over the entire molecule, facilitating its binding to the enzyme active site. The provided FMO data offer a qualitative assessment. A quantitative analysis of the FMO energies and electron densities would provide more precise insights into the reactivity of the compounds. Thus, molecular docking simulations are essential for validating the predicted interactions and assessing the actual biological activity of compounds.
MEP properties of bis-Schiff bases of 4-hydroxyacetophenone
The molecular electrostatic potential (MEP) reveals the distribution of charge along the molecular skeleton, which can be correlated with the observed activities against cholinesterase.MEP is a crucial tool for understanding the reactivity and interaction capabilities of molecules, particularly in the context of drug design. This analysis compares the MEP of the most active compounds (2a, 2b, 2e, 2f, 2h and 2j) with the reference compound galantamine, focusing on their potential as cholinesterase inhibitors. The distribution of charge along the molecular skeleton and its relation to biological activity is also discussed. For compound (2a), the MEP map shows significant negative potential (red) around the diethyl aniline moiety, indicating a high electron density in this region. The positive potential (blue) is localized on the bromobenzyl substituent, suggesting potential sites for interaction with the enzyme. In compound (2b), the methoxy phenol group exhibits high negative potential, reflecting its electron-rich nature (Fig. 6).
 | ||
| Fig. 6 Insights into the charge distribution on the molecular surface. | ||
The positive potential is concentrated on the hydrazono linkage and the bromobenzyl moiety, which is consistent with the observed reactivity patterns. Compound (2e) displays different charge distributions owing to the presence of the furan ring. The negative potential is more evenly distributed across the molecule, while the positive potential is localized on the bromobenzyl substituent and the methyl groups of the furan ring. Compounds (2f and 2g) show similar trends, with the methoxy and methyl groups influencing the charge distribution. The negative potential is concentrated on these electron-donating groups, while the positive potential is more delocalized. In compound (2j), the methoxy phenol group again shows high negative potential, which is similar to compound (2b), but the positive potential is distributed differently owing to the altered substitution pattern. Galantamine, as a standard, has a distinct charge distribution that correlates with its known activity against cholinesterase. The negative potential is localized on the nitrogen-containing heterocycle, while the positive potential is spread over the entire molecule, facilitating its binding to the enzyme active site.
From the above data, the 4-bromobenzyloxy group in compound (2a) withdraws electrons via inductive effects, creating a polarized region. This may align with AChE's catalytic anionic site (CAS), which attracts cationic species. Compound (2a) with protonatable amine mimics the positive charge of galantamine, enhancing binding. Methoxy group in compounds (2b, 2f and 2j) and methyl group in compound (2g) increase electron density in aromatic rings by facilitating π–π stacking with AChE's Trp84/86 residues. The hydroxyl groups in compounds (2b and 2j) act as H-bond donors, mimicking acetylcholine's ester carbonyl interactions. The N–N bridge offers H-bonding potential. Conjugation with ethylidene delocalizes electrons, creating a planar structure that may fit AChE's narrow gorge. Bulky substituents, such as diethyl in compound (2a), could hinder binding unless accommodated by the peripheral site. The furan ring in compound (2e) is less electron rich than benzene derivatives but may engage in hydrophobic interactions. Its methyl group enhances lipophilicity, potentially improving blood–brain barrier penetration.
Docking analysis of bis-Schiff bases against AChE and BuChE
The inhibition of cholinesterase enzymes, specifically AChE (PDB: 4EY6 (ref. 38)) and BuChE, (PDB: 4BDS (ref. 39)), is a critical therapeutic strategy for managing neurodegenerative disorders, such as Alzheimer's disease. This study investigates the binding affinities and interaction mechanisms of a series of bis-Schiff bases (2a, 2b, 2e, 2f, 2g and 2j) derived from 4-hydrox-yacetophenone, with a focus on their potential as cholinesterase inhibitors. Docking studies provide insights into the binding affinities and interaction mechanisms of the investigated compounds with AChE and BuChE. The binding affinities, represented by the free binding energy (ΔG), root-mean-square deviation (RMSD), hydrogen bond energy (H.B), and receptor-H-bond interaction energy (EInt), are summarized in Fig. 7. | ||
| Fig. 7 Heat map for different docking affinity for most active bis-Schiff bases, standard galantamine and tacrine into 4EY6 and 4BDS. | ||
Acetylcholinesterase (AChE)
This analysis evaluates the 3D binding modes of six hydrazine derivatives (2a, 2b, 2e, 2f, 2g and 2j) compared to galantamine and tacrine against AChE (PDB:2PH9). The binding interactions with key residues (Trp147, Cys190) and their correlation with inhibitory activity (IC50) are explored to identify the most promising compound.Compound (2j) shows a binding affinity of −6.585 kcal mol−1 and an RMSD of 1.553 Å. The 2-methoxyphenol group in the most active compound (2j) forms H-bonds with Asp164 (2.1 Å) and Glu34 (2.5 Å). The 4-bromobenzyl moiety is oriented towards the PAS, which engages in π–π stacking with Tyr93 (distance: 3.8 Å). The hydrazine linker stabilizes the conformation via hydrophobic interactions with Ile118. Optimal H-bonding with aromatic stacking, and its orientation within the binding site, allows for optimal positioning, leading to strong binding affinity and high activity (Fig. 8).
 | ||
| Fig. 8 Binding modes of the most active compounds (2a, 2b, 2e, 2f, 2g, and 2j) and the standard galantamine and their alignment into the active site of acetylcholinesterase (AChE, PDB: 4EY6). | ||
Compound (2b) shows a slightly higher binding affinity (−6.860 kcal mol−1) compared to compound (2a), with a lower RMSD (1.836 Å). The 2-methoxy-6-methylphenol group facilitates H-bonding by donating an H-bond to Val148 (2.3 Å) and π–π stacking with Trp147. The N,N-diethyl aniline moiety forms π–π stacking with Tyr188 and stabilizes the conformation via hydrophobic interactions with Cys190. The bromobenzyl group partially overlaps with Tyr93 (4.2 Å), reducing π–π efficiency. This results in slightly lower potency compared to compound (2j) likely due to suboptimal engagement with key active site residues. Compound (2a) exhibits a binding affinity (ΔG) of −6.787 kcal mol−1, with an RMSD of 1.941 Å. The presence of the N,N-diethyl aniline moiety likely contributes to hydrophobic interactions, enhancing binding stability. The bromobenzyl group is positioned near the peripheral anionic site (PAS) to form π–π stacking with Tyr195, while the Br moiety forms a halogenated interaction with Asn197, and the bulky N,N-diethyl aniline moiety is oriented toward the catalytic anionic site (CAS). This group causes steric clashes with Trp147, limiting π–π interactions. For compound (2f) with a binding affinity of −7.071 kcal mol−1 and an RMSD of 1.693 Å, the 4-methoxybenzylidene group likely enhances hydrogen bonding and electrostatic interactions. It forms H-bonds with Val148 and π–π stacking interactions with Trp147. The 4-methoxybenzyl moiety enhances these interactions. Its orientation within the binding site allows for optimal positioning, leading to strong binding affinity and high activity. Compound (2e) displays a binding affinity of −6.702 kcal mol−1 and an RMSD of 1.295 Å. The 2,5-dimethylfuran moiety introduces unique electronic properties; it occupies the PAS, forming π–π stacking with Tyr188, while the hydrazone linker interacts with Tyr93. Compound (2g) exhibits a binding affinity of −7.047 kcal mol−1 and a higher RMSD (2.605 Å). The 4-methylbenzylidene group contributes to hydrophobic interactions, stabilizing the complex. Standard galantamine demonstrates the highest binding affinity (−7.585 kcal mol−1) with a low RMSD (1.049 Å), indicating strong interaction with AChE. Tacrine shows a binding affinity of −5.368 kcal mol−1 and an RMSD of 1.457 Å (Table S2; ESI†).
Butyrylcholinesterase (BuChE)
The analysis focuses on interactions with key residues (Trp98, Ser79) in the BuChE active site to identify the most potent inhibitor. Compound (2a) displays a binding affinity (ΔG) of −8.689 kcal mol−1 with an RMSD of 1.893 Å. The N,N-diethyl aniline moiety likely contributes to hydrophobic interactions, enhancing binding stability. Compound (2b) shows a binding affinity of −6.247 kcal mol−1 and a lower RMSD (1.245 Å). The 2-methoxy-6-methylphenol group may facilitate hydrogen bonding, thereby improving interaction with BuChE. Compound (2e) exhibits a binding affinity of −7.464 kcal mol−1 and an RMSD of 1.762 Å. The 2,5-dimethylfuran moiety introduces unique electronic properties, potentially influencing binding dynamics. Compound (2f) displays a binding affinity of −8.205 kcal mol−1 and an RMSD of 1.741 Å. The 4-methoxy benzylidene group likely enhances hydrogen bonding and electrostatic interactions. Compound (2g) shows a binding affinity of −8.211 kcal mol−1 and an RMSD of 1.472 Å. The 4-methyl benzylidene group contributes to hydrophobic interactions, stabilizing the complex. Compound (2j) exhibits a binding affinity of −8.060 kcal mol−1 and an RMSD of 1.471 Å. The 2-methoxy phenol group may enhance hydrogen bonding, improving interaction with BuChE. Standard galantamine demonstrates a binding affinity of −5.314 kcal mol−1 with an RMSD of 1.635 Å. Tacrine shows a binding affinity of −4.502 kcal mol−1 and an RMSD of 1.199 Å. Compound (2f) shows the strongest hydrogen bonding (−30.134 kcal mol−1), followed closely by 2b (−29.683 kcal mol−1) and 2j (−29.050 kcal mol−1). These values significantly exceed those of galantamine (−9.130 kcal mol−1) and tacrine (−7.249 kcal mol−1). Compound (2j) demonstrates the strongest overall interaction energy (−12.714 kcal mol−1), followed by 2b (−12.045 kcal mol−1) and 2f (−11.914 kcal mol−1). However, galantamine shows a stronger interaction energy (−23.164 kcal mol−1), suggesting that its inhibitory mechanism may involve additional factors beyond those captured in this docking analysis (Fig. 9). Galantamine binds peripherally in the active site gorge, forming π–π interactions with Trp82 and weak hydrogen bonds with Asp164. Its moderate potency (IC50 = 156.8 nM) reflects the partial engagement of the catalytic triad. The reference inhibitor tacrine occupies the catalytic triad, stacking with Trp82. The 4-bromobenzyl moiety in 2j donates H-bonds to His438 (2.7 Å). The N,N-diethyl aniline group moiety in compound (2j) engages in π–π stacking with Trp82 (3.6 Å), mimicking tacrine's interaction with Trp82. The hydrazine linker stabilizes hydrophobic interactions with Glu197. The superior potency arises from dual polar and aromatic interactions, enabling deeper penetration into the catalytic site than galantamine (Fig. 9). The 6-methoxyphenol group in 2b forms a single H-bond with Ser79 (2.4 Å), but steric clashes with Trp82 reduce π–π efficiency (4.1 Å). The bulky N,N-diethyl aniline group causes steric hindrance near Phe329, disrupting π–π interactions (distance: 5.2 Å). The 4-methoxybenzyl group in 2f forms moderate π–π stacking with Phe329 (4.0 Å), while the bromobenzyl moiety is oriented towards the PAS via interaction with Tyr332 (Table S2; ESI†). | ||
| Fig. 9 Binding mode for the most active modes of the most active compounds (2a, 2b, 2e, 2f, 2g, and 2j) and the standards galantamine and tacrine, with their alignment in the active site of 4BSD. | ||
Compound (2g) forms H–π with Tyr440 (4.8 Å) and π–π stacking interactions with Trp82. The 4-methyl benzyl moiety contributes to these interactions. However, its activity is lower compared to the reference compounds owing to less favourable interactions with the catalytic triad. The 5-methyl furan in 2e exhibits poor π–π interactions (distance: 5.5 Å from Trp231) and no H-bonds with His 438, while minimal hydrophobic stabilization is due to the furan's planar rigidity.
Compound 2j exhibited the strongest dual inhibition, driven by H-bonds with Asp164 and π–π stacking with Tyr93, while 2f and 2g showed moderate activity owing to weaker hydrophobic interactions (Table 2). Galantamine higher ΔG (−7.585 kcal mol−1) reflects its optimized binding mode but lower experimental potency (IC50 = 104.8 μM), underscoring 2j's superior efficacy.
| C. no | AChE ΔG (kcal mol−1) | BuChE ΔG (kcal mol−1) | Key AChE interactions | Key BuChE interactions |
|---|---|---|---|---|
| 2a | −6.787 | −8.689 | Hydrophobic interactions with Ile118, π–π stacking with Tyr195 | Hydrophobic interactions with Cys190, π–π stacking with Tyr332 |
| 2b | −6.860 | −6.247 | H-Bond with Val148, π–π stacking with Trp147 | H-Bond with Ser79, hydrophobic interactions with Phe329 |
| 2e | −6.702 | −7.464 | π–π stacking with Tyr188, hydrazine linker interactions with Tyr93 | Unique electronic properties with 2,5-dimethylfuran moiety |
| 2f | −7.071 | −8.205 | H-Bonds with Val148, π–π stacking with Trp147 | Strong H-bonding, π–π stacking with Phe329 |
| 2g | −7.047 | −8.211 | Hydrophobic interactions with Ile118, π–π stacking with Tyr195 | Hydrophobic interactions with Phe329, π–π stacking with Trp82 |
| 2j | −6.585 | −8.060 | H-Bonds with Asp164 and Glu34, π–π stacking with Tyr93 | Strong H-bonding with Ser79, π–π stacking with Trp82 |
| Galantamine | −7.585 | −5.314 | Peripheral binding in active site gorge, π–π interactions with Trp82 | Binds peripherally, weak H-bonds with Asp164 |
| Tacrine | −5.368 | −4.502 | Binds to catalytic triad, π–π stacking with Trp82 | Occupies catalytic triad |
Experimental
General
Analytical grade solvents, reagents and chemicals were brought from Merck, BDH, and Sigma Aldrich, respectively. Starting material 4-hydroxyacetophenone with CAS no: 99-93-4 was purchased from Sigma Aldrich, with 99% purity. Silica gel plates (Merck, aluminium plates: 60F254) were utilized to determine the progress of the reactions, and UV light at 254 nm was used for spot visualization. The melting points of these derivatives were determined using the SMP10 melting point apparatus, and the molar masses of the compounds were confirmed through high-resolution electrospray ionization mass spectrometry (Singapore made, Q-TOF 6530, Agilent LC). Avance Bruker (Zurich; Switzerland) 500 and 125 MHz spectrophotometers were employed to determine the proton- and 13C-NMR spectra of the synthetic derivatives, respectively. The parameters used in the proton-NMR discussion are as follows: coupling constant J is taken in hertz (Hz); the chemical shift value is in parts per million (ppm); triplet = t, singlet = s, doublet = d, and multiplet = m.Bis-Schiff base derivatives were synthesized by refluxing compound (2) (0.15 g, 0.00047 mole) with several benzaldehydes (0.00047 mole) in ethanol containing acetic acid as a catalyst. The reaction mixture was refluxed for 3–4 h, and the formation of the products was thoroughly checked using the TLC technique. Once the reaction was completed, the solution was decanted to ice cubes; coloured precipitates were formed, filtered, rinsed with distilled water, and left to dry overnight. The product compounds were recrystallized with absolute ethanol to obtain them in pure form. The structures of the compounds were fully verified through mass and NMR (1H and 13C) spectroscopy.
Spectra data of the compounds
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N–, 1H), 7.85 (d, J = 9.5 Hz, ArH, 2H), 7.68 (d, J = 9.0 Hz, ArH, 2H), 7.50 (d, J = 8.5 Hz, ArH, 2H), 7.30 (d, J = 9.5 Hz, ArH, 2H), 6.96 (d, J = 8.5 Hz, ArH, 2H), 6.67 (d, J = 8.0 Hz, ArH, 2H), 5.04 (s, –OCH2, 2H), 3.40 (q, J = 8.0 Hz, –CH2CH3, 4H), 2.50 (s, –CH3, 3H), 1.18 (t, J = 8.0 Hz, CH3CH2–). 13C-NMR (125 MHz, CDCl3): δ 162.89, 159.79, 158.88, 149.67, 135.81, 131.73, 130.14, 129.05, 128.30, 121.92, 114.56, 111.11, 69.24, 44.47, 14.93, 12.59. EI-MS m/z (% rel. abund.): 479 (M+, 1), 464.1 (18), 308.1 (100), 267.1 (18), 223.1 (10), 168.9 (38), 159.0 (18), 133.0 (25), 90.0 (18).N–, 1H), 7.89 (d, J = 9.5 Hz, ArH, 2H), 7.50 (d, J = 9.5 Hz, ArH, 2H), 7.30–7.23 (m, ArH, 2H), 6.98–6.86 (m, ArH, 5H), 5.05 (s, –OCH2, 2H), 3.92 (s, –OCH3, 3H), 2.46 (s, –CH3, 3H). 13C-NMR (125 MHz, CDCl3): δ 165.63, 162.60, 160.53, 149.78, 135.53, 131.76, 130.58, 129.02, 128.85, 124.04, 123.58, 115.16, 114.25, 69.90, 56.12, 25.02, 15.35. EI-MS m/z (% rel. abund.): 454 (M+), 331.0,10 303.9 (8), 283.0 (42), 223.0 (10), 170.8 (100), 150.0 (15), 89.9 (22).N–, 1H), 7.99–7.97 (m, ArH, 2H), 7.89 (d, J = 9.5 Hz, ArH, 2H) 7.50 (d, J = 8.5 Hz, ArH, 3H), 7.39–7.24 (m, ArH, 4H), 6.98–6.89 (m, ArH, 4H), 5.05 (s, –OCH2, 2H), 2.47 (s, –CH3, 3H). 13C-NMR (125 MHz, CDCl3): δ 162.81 (d, JC–F = 248 Hz), 160.43, 155.04, 155.01, 150.43, 135.59, 131.76, 130.61, 129.04, 128.73, 128.08, 127.90, 126.72, 122.01, 119.83, 114.79, 69.27, 25.02, 15.15. EI-MS m/z (% rel. abund.): 504 (M+), 333.1 (10), 170.9 (100), 106.9 (10), 89.9 (18).N–, 1H), 7.88 (d, J = 9.5 Hz, ArH, 2H), 7.76–7.74 (m, ArH, 3H), 7.51–7.24 (m, ArH, 8H), 6.98 (d, J = 9.0 Hz, ArH, 2H), 5.05 (s, –OCH2, 2H), 2.49 (s, –CH3, 3H). 13C-NMR (125 MHz, CDCl3): δ 164.83, 161.04, 156.81, 137.34, 135.64, 133.29, 131.77, 130.62, 129.74, 129.14, 129.01, 128.64, 126.72, 69.28, 26.34, 15.15. EI-MS m/z (% rel. abund.): 441.4 (M+, 1), 435.1 (20), 266.0 (8), 168.9 (100), 89.9 (18).N–, 1H), 7.87 (d, J = 8.5 Hz, ArH, 2H), 7.50 (d, J = 8.5 Hz, ArH, 2H), 7.30–7.24 (m, ArH, 2H), 6.99 (d, J = 8.0 Hz, ArH, 4H), 6.73 (d, J = 8.5 Hz, ArH, 1H), 5.04 (s, –OCH2, 2H), 2.39 (s, –CH3, 3H), 2.50 (s, –CH3, 3H). 13C-NMR (125 MHz, CDCl3): δ 164.35, 160.18, 156.21, 148.68, 147.46, 138.62, 135.67, 131.74, 129.02, 128.57, 121.97, 114.66, 108.67, 69.93, 26.34, 15.09, 14.00. EI-MS m/z (% rel. abund.): 412 (M+, 1), 266.1 (8), 168.9 (100), 149.0 (5), 90.0 (22).N–, 1H), 7.91 (d, J = 8.0 Hz, ArH, 2H), 7.85 (d, J = 8.0 Hz, ArH, 2H), 7.69 (d, J = 7.5 Hz, ArH, 2H), 7.14–6.90 (m, ArH, 6H), 5.03 (s, –OCH2, 2H), 3.54 (s, –CH3, 3H). 13C-NMR (125 MHz, CDCl3): δ 162.9, 161.3, 149.4, 135.7, 131.8, 131.1, 129.3, 128.7, 128.0, 127.6, 127.0, 122.0, 114.4, 70.9, 55.8, 15.4. EI-MS m/z (% rel. abund.): 437.1 (M+, 1), 266.1 (8), 170.9 (100), 90.0 (18).N–, 1H), 7.95 (d, J = 8.0 Hz, ArH, 2H), 7.93 (d, J = 8.5 Hz, ArH, 2H), 7.88 (d, J = 8.0 Hz, ArH, 2H), 7.23 (d, J = 8.0 Hz, ArH, 2H), 7.14–6.96 (m, ArH, 4H), 5.08 (s, –OCH2, 2H), 2.55 (s, –CH3, 3H). 13C-NMR (125 MHz, CDCl3): δ 162.4, 135.3, 132.0, 131.9, 130.8, 130.7, 129.2, 128.4, 126.8, 122.3, 114.9, 114.7, 114.6, 69.5, 69.4, 26.5, 25.2. HRMS (ESI+): 421.0713 [M + H]+ calcd for C23H21BrN2O: 421.3380.N–, 1H), 8.31 (d, J = 8.0 Hz, ArH, 1H), 8.30–7.83 (m, ArH, 4H), 7.51 (d, J = 8.0 Hz, ArH, 2H), 7.60 (t, J = 9.0 Hz, ArH, 2H), 7.32 (d, J = 9.0 Hz, ArH, 2H), 7.25 (d, J = 8.0 Hz, ArH, 4H), 5.05 (s, –OCH2, 2H), 3.54 (s, –CH3, 3H). 13C-NMR (125 MHz, CDCl3): δ 168.1, 163.0, 162.4, 161.7, 137.0, 135.6, 134.3, 132.8, 131.8, 131.2, 131.0, 129.9, 129.6, 129.2, 128.9, 128.4, 127.7, 127.1, 125.7, 125.0, 68.56, 24.15. EI-MS m/z (% rel. abund.): 457.2 (M+, 1), 435.0 (20), 266.0 (7), 168.9 (100), 89.9 (18).N–, 1H), 8.03 (d, J = 8.0 Hz, ArH, 2H), 7.99 (d, J = 8.5 Hz, ArH, 2H), 7.92 (d, J = 9.0 Hz, ArH, 2H), 7.90–7.26 (m, ArH, 6H), 5.07 (s, –OCH2, 2H), 2.55 (s, –CH3, 3H). 13C-NMR (125 MHz, CDCl3): δ 162.7, 135.6, 132.3, 132.2, 131.0, 130.5, 129.6, 129.5, 128.8, 122.6, 115.4, 115.3, 115.0, 114.9, 69.8, 26.8. EI-MS m/z (% rel. abund.): 437.0 (M+, 1), 422.0 (8), 394.0 (30), 289.0 (18), 266.0 (16), 168.9 (100), 90.0 (10).N–, 1H), 7.94 (d, J = 8.5 Hz, ArH, 2H), 7.88 (d, J = 8.5 Hz, ArH, 2H), 7.53–25 (m, ArH, 6H), 6.96 (d, J = 7.5 Hz, ArH, 1H), 5.08 (s, –OCH2, 2H), 4.00 (s, –OCH3, 3H), 2.55 (s, –CH3, 3H). 13C-NMR (125 MHz, CDCl3): δ 161.3, 151.0, 149.4, 149.3, 135.7, 131.8, 131.1, 129.3, 128.7, 128.5, 127.0, 122.9, 117.0, 114.4, 112.1, 70.8, 56.2, 15.7. HRMS (ESI+): 452.1898 [M + H]+ calcd for C23H21BrN2O3: 452.3360.
N–, 1H), 7.85 (d, J = 9.5 Hz, ArH, 2H), 7.68 (d, J = 9.0 Hz, ArH, 2H), 7.50 (d, J = 8.5 Hz, ArH, 2H), 7.30 (d, J = 9.5 Hz, ArH, 2H), 6.96 (d, J = 8.5 Hz, ArH, 2H), 6.67 (d, J = 8.0 Hz, ArH, 2H), 5.04 (s, –OCH2, 2H), 3.40 (q, J = 8.0 Hz, –CH2CH3, 4H), 2.50 (s, –CH3, 3H), 1.18 (t, J = 8.0 Hz, CH3CH2–). 13C-NMR (125 MHz, CDCl3): δ 162.89, 159.79, 158.88, 149.67, 135.81, 131.73, 130.14, 129.05, 128.30, 121.92, 114.56, 111.11, 69.24, 44.47, 14.93, 12.59. EI-MS m/z (% rel. abund.): 479 (M+, 1), 464.1 (18), 308.1 (100), 267.1 (18), 223.1 (10), 168.9 (38), 159.0 (18), 133.0 (25), 90.0 (18).N–, 1H), 7.89 (d, J = 9.5 Hz, ArH, 2H), 7.50 (d, J = 9.5 Hz, ArH, 2H), 7.30–7.23 (m, ArH, 2H), 6.98–6.86 (m, ArH, 5H), 5.05 (s, –OCH2, 2H), 3.92 (s, –OCH3, 3H), 2.46 (s, –CH3, 3H). 13C-NMR (125 MHz, CDCl3): δ 165.63, 162.60, 160.53, 149.78, 135.53, 131.76, 130.58, 129.02, 128.85, 124.04, 123.58, 115.16, 114.25, 69.90, 56.12, 25.02, 15.35. EI-MS m/z (% rel. abund.): 454 (M+), 331.0,10 303.9 (8), 283.0 (42), 223.0 (10), 170.8 (100), 150.0 (15), 89.9 (22).N–, 1H), 7.99–7.97 (m, ArH, 2H), 7.89 (d, J = 9.5 Hz, ArH, 2H) 7.50 (d, J = 8.5 Hz, ArH, 3H), 7.39–7.24 (m, ArH, 4H), 6.98–6.89 (m, ArH, 4H), 5.05 (s, –OCH2, 2H), 2.47 (s, –CH3, 3H). 13C-NMR (125 MHz, CDCl3): δ 162.81 (d, JC–F = 248 Hz), 160.43, 155.04, 155.01, 150.43, 135.59, 131.76, 130.61, 129.04, 128.73, 128.08, 127.90, 126.72, 122.01, 119.83, 114.79, 69.27, 25.02, 15.15. EI-MS m/z (% rel. abund.): 504 (M+), 333.1 (10), 170.9 (100), 106.9 (10), 89.9 (18).N–, 1H), 7.88 (d, J = 9.5 Hz, ArH, 2H), 7.76–7.74 (m, ArH, 3H), 7.51–7.24 (m, ArH, 8H), 6.98 (d, J = 9.0 Hz, ArH, 2H), 5.05 (s, –OCH2, 2H), 2.49 (s, –CH3, 3H). 13C-NMR (125 MHz, CDCl3): δ 164.83, 161.04, 156.81, 137.34, 135.64, 133.29, 131.77, 130.62, 129.74, 129.14, 129.01, 128.64, 126.72, 69.28, 26.34, 15.15. EI-MS m/z (% rel. abund.): 441.4 (M+, 1), 435.1 (20), 266.0 (8), 168.9 (100), 89.9 (18).N–, 1H), 7.87 (d, J = 8.5 Hz, ArH, 2H), 7.50 (d, J = 8.5 Hz, ArH, 2H), 7.30–7.24 (m, ArH, 2H), 6.99 (d, J = 8.0 Hz, ArH, 4H), 6.73 (d, J = 8.5 Hz, ArH, 1H), 5.04 (s, –OCH2, 2H), 2.39 (s, –CH3, 3H), 2.50 (s, –CH3, 3H). 13C-NMR (125 MHz, CDCl3): δ 164.35, 160.18, 156.21, 148.68, 147.46, 138.62, 135.67, 131.74, 129.02, 128.57, 121.97, 114.66, 108.67, 69.93, 26.34, 15.09, 14.00. EI-MS m/z (% rel. abund.): 412 (M+, 1), 266.1 (8), 168.9 (100), 149.0 (5), 90.0 (22).N–, 1H), 7.91 (d, J = 8.0 Hz, ArH, 2H), 7.85 (d, J = 8.0 Hz, ArH, 2H), 7.69 (d, J = 7.5 Hz, ArH, 2H), 7.14–6.90 (m, ArH, 6H), 5.03 (s, –OCH2, 2H), 3.54 (s, –CH3, 3H). 13C-NMR (125 MHz, CDCl3): δ 162.9, 161.3, 149.4, 135.7, 131.8, 131.1, 129.3, 128.7, 128.0, 127.6, 127.0, 122.0, 114.4, 70.9, 55.8, 15.4. EI-MS m/z (% rel. abund.): 437.1 (M+, 1), 266.1 (8), 170.9 (100), 90.0 (18).N–, 1H), 7.95 (d, J = 8.0 Hz, ArH, 2H), 7.93 (d, J = 8.5 Hz, ArH, 2H), 7.88 (d, J = 8.0 Hz, ArH, 2H), 7.23 (d, J = 8.0 Hz, ArH, 2H), 7.14–6.96 (m, ArH, 4H), 5.08 (s, –OCH2, 2H), 2.55 (s, –CH3, 3H). 13C-NMR (125 MHz, CDCl3): δ 162.4, 135.3, 132.0, 131.9, 130.8, 130.7, 129.2, 128.4, 126.8, 122.3, 114.9, 114.7, 114.6, 69.5, 69.4, 26.5, 25.2. HRMS (ESI+): 421.0713 [M + H]+ calcd for C23H21BrN2O: 421.3380.N–, 1H), 8.31 (d, J = 8.0 Hz, ArH, 1H), 8.30–7.83 (m, ArH, 4H), 7.51 (d, J = 8.0 Hz, ArH, 2H), 7.60 (t, J = 9.0 Hz, ArH, 2H), 7.32 (d, J = 9.0 Hz, ArH, 2H), 7.25 (d, J = 8.0 Hz, ArH, 4H), 5.05 (s, –OCH2, 2H), 3.54 (s, –CH3, 3H). 13C-NMR (125 MHz, CDCl3): δ 168.1, 163.0, 162.4, 161.7, 137.0, 135.6, 134.3, 132.8, 131.8, 131.2, 131.0, 129.9, 129.6, 129.2, 128.9, 128.4, 127.7, 127.1, 125.7, 125.0, 68.56, 24.15. EI-MS m/z (% rel. abund.): 457.2 (M+, 1), 435.0 (20), 266.0 (7), 168.9 (100), 89.9 (18).N–, 1H), 8.03 (d, J = 8.0 Hz, ArH, 2H), 7.99 (d, J = 8.5 Hz, ArH, 2H), 7.92 (d, J = 9.0 Hz, ArH, 2H), 7.90–7.26 (m, ArH, 6H), 5.07 (s, –OCH2, 2H), 2.55 (s, –CH3, 3H). 13C-NMR (125 MHz, CDCl3): δ 162.7, 135.6, 132.3, 132.2, 131.0, 130.5, 129.6, 129.5, 128.8, 122.6, 115.4, 115.3, 115.0, 114.9, 69.8, 26.8. EI-MS m/z (% rel. abund.): 437.0 (M+, 1), 422.0 (8), 394.0 (30), 289.0 (18), 266.0 (16), 168.9 (100), 90.0 (10).N–, 1H), 7.94 (d, J = 8.5 Hz, ArH, 2H), 7.88 (d, J = 8.5 Hz, ArH, 2H), 7.53–25 (m, ArH, 6H), 6.96 (d, J = 7.5 Hz, ArH, 1H), 5.08 (s, –OCH2, 2H), 4.00 (s, –OCH3, 3H), 2.55 (s, –CH3, 3H). 13C-NMR (125 MHz, CDCl3): δ 161.3, 151.0, 149.4, 149.3, 135.7, 131.8, 131.1, 129.3, 128.7, 128.5, 127.0, 122.9, 117.0, 114.4, 112.1, 70.8, 56.2, 15.7. HRMS (ESI+): 452.1898 [M + H]+ calcd for C23H21BrN2O3: 452.3360.Cholinesterase inhibitory activities
The acetyl and butyryl cholinesterase inhibitory activities are carried out according to the procedure in the literature.13Computational methodology
Quantum chemical calculations were performed using the Jaguar software package.40 Initial electronic structure optimizations and property analyses were conducted with the B3LYP hybrid functional in conjunction with the 6-311G++(d,p) basis set, which accounts for polarization and diffuse effects critical for accurate molecular modelling. To ensure the robustness of the simulations, validation was carried out using the dispersion-corrected wB97X-D3 functional at the same basis set level, thereby addressing long-range interactions and van der Waals forces. The optimized geometries obtained from these calculations were rigorously analysed to determine key structural parameters (bond lengths, angles, and dihedral angles) and electronic properties, including frontier molecular orbital energies (HOMO–LUMO) and molecular electrostatic potential (MEP) distributions. The convergence criteria for energy and gradient thresholds confirmed the reliability of the optimized structures, ensuring their suitability for subsequent quantum mechanical and spectroscopic investigations.Molecular docking protocol
Molecular docking simulations were conducted using the Glide module13 (Schrödinger Suite) to investigate ligand binding within the active sites of acetylcholinesterase (AChE) and butyrylcholinesterase (BuChE). The initial preparation of the crystallized PDB structure involved the removal of water molecules and co-crystallized inhibitors, followed by the addition of hydrogen atoms to optimize protonation states. The co-crystallized ligand was excised to generate an unoccupied active site, into which the target ligands were subsequently redocked. A grid box spanning 20 Å × 20 Å × 20 Å was centred on the active site to ensure adequate spatial coverage for ligand flexibility and conformational sampling. Charges were assigned using the CHARMM force field, and the ChemPLP scoring function was employed to evaluate binding affinities, prioritizing steric complementarity, hydrogen bonding, and hydrophobic interactions. To ensure docking accuracy, the ligand poses were generated using the structure with the lowest root-mean-square deviation (RMSD) relative to the crystallized ligand. This approach facilitated the identification of energetically favourable binding modes, enabling a robust analysis of ligand–receptor interactions.Conclusion
In this study, we effectively synthesized some bis-Schiff base derivatives of 4-hydroxyacetophenone through multi-step reactions. Initially, 4-hydroxyacetophenone was treated with 4-bromobenzyl bromide in the presence of potassium carbonate in DMF solvent to obtain product (1) in high yield. The second step involved the reaction between compound (1) and hydrazine hydrate to afford the product (2), which was further treated with different aromatic aldehydes in ethanol solvent using some drops of acetic acid to obtain bis-Schiff bases in excellent to good yields. All the compounds are structurally deduced utilizing state-of-the-art spectroscopic techniques, including 1H-, 13C-NMR and EI-MS, and screened for their in vitro acetyl and butyrylcholinesterase inhibitory activities. Among the series, six compounds (2j, 2b, 2a, 2f, 2g, and 2e) exhibited excellent inhibition in the range of IC50 from 15.86 ± 0.38 and 29.23 ± 0.04 μM to 98.21 ± 0.01 and 135.7 ± 2.61 μM. Compound 2j displayed a potent inhibition effect against the two enzymes, underlining its potential as a lead candidate for drug development. Compound (2j) arises as the most powerful BuChE inhibitor, which shows robust hydrogen bonding through its 2-methoxyphenol group, imitating the interactions of galantamine with Glu234 and effective π–π stacking of its bromo benzyl moiety with Trp195, similar to the interactions of tacrine with Trp84. Its hydrazine linker improves hydrophobic complementarity and steadying interfaces with Ile118. In contrast, the methoxy and methyl groups of compounds (2b) increase hydrophobicity but introduce steric hindrance, while the electron rich furan in compound (2e) endorses charge transfer but lacks hydrogen-bond donors. Para substituents in compounds (2f and 2g) develop lipophilicity but have weaker H-bonding than 2j. Reasonable docking examination confirms the superior activity and binding affinity of compound (2j), deserving further optimization for potential drug development.Data availability
Data of the compounds are available in the ESI.†Author contributions
Conceptualization: Aftab Alam and Manzoor Ahmad; methodology: Gul Badshah, Muhammad Ayaz and Muhammad Usman., biological activities: Imtiaz Ahmad; software: Ahmed A. Elhenawy and Ashwag S. Alanazi; data curation: Abdul Latif, Shujaat Ahmad and Mumtaz Ali; writing-original draft: Aftab Alam and Muhammad Ayaz, writing-review and editing: Ashwag S. Alanazi, Manzoor Ahmad and Mumtaz Ali. All the authors have read and agreed to the published version of the manuscript.Conflicts of interest
The authors declare no conflicts of interest.Acknowledgements
The authors extend their appreciation to Princess Nourah bint Abdulrahman University researcher supporting project number (PNURSP2025R342), Princess Nourah bint Abdulrahman University, Riyadh, Saudi Arabia, for supporting this work. The authors are also grateful to the Pakistan Council of Scientific and Industrial Research (PCSIR) for providing the characterization facilities.References
- C. A. Lane, J. Hardy and J. M. Schott, Alzheimer's disease, Eur. J. Neurol., 2018, 25(1), 59–70 CrossRef CAS.
- T. Storr, Multifunctional compounds for the treatment of Alzheimer's disease, Can. J. Chem., 2021, 99(1), 1–9 CrossRef CAS.
- M. Hatami, M. Mortazavi, Z. Baseri, B. Khani, M. Rahimi and S. Babaei, Antioxidant compounds in the treatment of Alzheimer's disease: natural, hybrid, and synthetic products, Evid. base Compl. Alternative Med., 2023, 2023(1), 8056462 CrossRef PubMed.
- Y. Liu, G. Uras, I. Onuwaje, W. Li, H. Yao and S. Xu, et al., Novel inhibitors of AChE and Aβ aggregation with neuroprotective properties as lead compounds for the treatment of Alzheimer's disease, Eur. J. Med. Chem., 2022, 235, 114305 CrossRef CAS PubMed.
- F. Rahim, H. Ullah, M. Taha, A. Wadood, M. T. Javed and W. Rehman, et al., Synthesis and in vitro acetylcholinesterase and butyrylcholinesterase inhibitory potential of hydrazide based Schiff bases, Bioorg. Chem., 2016, 68, 30–40 CrossRef CAS PubMed.
- M. Taha, F. Rahim, N. Uddin, I. U. Khan, N. Iqbal and M. Salahuddin, et al., Exploring indole-based-thiadiazole derivatives as potent acetylcholinesterase and butyrylcholinesterase enzyme inhibitors, Int. J. Biol. Macromol., 2021, 188, 1025–1036 CrossRef CAS PubMed.
- S. Hameed, K. M. Khan, P. Taslimi, U. Salar, T. Taskin-Tok and D. Kisa, et al., Evaluation of synthetic 2-aryl quinoxaline derivatives as α-amylase, α-glucosidase, acetylcholinesterase, and butyrylcholinesterase inhibitors, Int. J. Biol. Macromol., 2022, 211, 653–668 CrossRef CAS.
- D. Vicente-Zurdo, N. Rosales-Conrado, M. E. León-González, L. Brunetti, L. Piemontese and A. R. Pereira-Santos, et al., Novel rivastigmine derivatives as promising multi-target compounds for potential treatment of Alzheimer's disease, Biomedicines, 2022, 10(7), 1510 CrossRef CAS.
- S. Wang, X. Kong, Z. Chen, G. Wang, J. Zhang and J. Wang, Role of Natural Compounds and Target Enzymes in the Treatment of Alzheimer's Disease, Molecules, 2022, 27(13), 4175 CrossRef CAS.
- A. Nordberg, C. Ballard, R. Bullock, T. Darreh-Shori and M. Somogyi, A review of butyrylcholinesterase as a therapeutic target in the treatment of Alzheimer's disease, The primary care companion for CNS disorders, 2013, 15(2), 26731 CrossRef.
- D. G. Wilkinson, P. T. Francis, E. Schwam and J. Payne-Parrish, Cholinesterase inhibitors used in the treatment of Alzheimer's disease: the relationship between pharmacological effects and clinical efficacy, Drugs Aging, 2004, 21, 453–478 CrossRef CAS.
- G. Marucci, M. Buccioni, D. Dal Ben, C. Lambertucci, R. Volpini and F. Amenta, Efficacy of acetylcholinesterase inhibitors in Alzheimer's disease, Neuropharmacology, 2021, 190, 108352 CrossRef CAS.
- M. Khan, H. Gohar, A. Alam, A. Wadood, A. Shareef and M. Ali, et al., Para-substituted thiosemicarbazones as cholinesterase inhibitors: synthesis, in vitro biological evaluation, and in silico study, ACS Omega, 2023, 8(5), 5116–5123 CrossRef CAS PubMed.
- F. Begum, M. Yousaf, S. Iqbal, N. Ullah, A. Hussain and M. Khan, et al., Inhibition of Acetylcholinesterase with Novel 1, 3, 4, Oxadiazole Derivatives: A Kinetic, In Silico, and In Vitro Approach, ACS Omega, 2023, 8(49), 46816–46829 CrossRef CAS PubMed.
- J. G. Fernandez-Bolanos and O. Lopez, Butyrylcholinesterase inhibitors as potential anti-Alzheimer’s agents: an updated patent review (2018-present), Expert Opin. Ther. Pat., 2022, 32(8), 913–932 CrossRef CAS PubMed.
- X. H. Gao, J. J. Tang, H. R. Liu, L. B. Liu and Y. Z. Liu, Structure–activity study of fluorine or chlorine-substituted cinnamic acid derivatives with tertiary amine side chain in acetylcholinesterase and butyrylcholinesterase inhibition, Drug Dev. Res., 2019, 80(4), 438–445 CrossRef CAS PubMed.
- L. Kang, X.-H. Gao, H.-R. Liu, X. Men, H.-N. Wu and P.-W. Cui, et al., Structure–activity relationship investigation of coumarin–chalcone hybrids with diverse side-chains as acetylcholinesterase and butyrylcholinesterase inhibitors, Mol. Diversity, 2018, 22, 893–906 CrossRef CAS PubMed.
- K. Schlake, J. Teller, L. Hinken, H. Laser, R. Lichtinghagen and A. Schäfer, et al., Butyrylcholinesterase activity in patients with postoperative delirium after cardiothoracic surgery or percutaneous valve replacement-an observational interdisciplinary cohort study, BMC Neurol., 2024, 24(1), 80 CrossRef PubMed.
- B. Li, E. G. Duysen, M. Carlson and O. Lockridge, The butyrylcholinesterase knockout mouse as a model for human butyrylcholinesterase deficiency, J. Pharmacol. Exp. Ther., 2008, 324(3), 1146–1154 CrossRef CAS PubMed.
- C. Wattmo, L. Minthon and Å. K. Wallin, Mild versus moderate stages of Alzheimer's disease: three-year outcomes in a routine clinical setting of cholinesterase inhibitor therapy, Alzheimer's Res. Ther., 2016, 8, 1–15 CrossRef PubMed.
- Q. Q. Lu, Y. M. Chen, H. R. Liu, J. Y. Yan, P. W. Cui and Q. F. Zhang, et al., Nitrogen-containing flavonoid and their analogs with diverse B-ring in acetylcholinesterase and butyrylcholinesterase inhibition, Drug Dev. Res., 2020, 81(8), 1037–1047 CrossRef CAS.
- W. J. Deardorff, E. Feen and G. T. Grossberg, The use of cholinesterase inhibitors across all stages of Alzheimer's disease, Drugs Aging, 2015, 32, 537–547 CrossRef CAS.
- S. Gul, F. Jan, A. Alam, A. Shakoor, A. Khan and A. F. AlAsmari, et al., Synthesis, molecular docking and DFT analysis of novel bis-Schiff base derivatives with thiobarbituric acid for α-glucosidase inhibition assessment, Sci. Rep., 2024, 14(1), 3419 CrossRef.
- S. Gul, S. Maab, H. Rafiq, A. Alam, M. U. Rehman and M. Assad, et al., Exploring bis-Schiff Bases with Thiobarbiturate Scaffold: In Vitro Urease Inhibition, Antioxidant Properties, and In Silico Studies, Russ. J. Bioorg. Chem., 2024, 50(5), 1627–1638 CrossRef CAS.
- M. Ismail, R. Ahmad, A. Latif, A. A. Khan, A. Alam and F. A. Ozdemir, et al., Synthesis, antibacterial activities and theoretical study of polyhydroquinoline derivatives, ChemistrySelect, 2023, 8(41), e202300954 CrossRef CAS.
- A. Ghorai, J. Mondal, R. Saha, S. Bhattacharya and G. K. Patra, A highly sensitive reversible fluorescent-colorimetric azino bis-Schiff base sensor for rapid detection of Pb2+ in aqueous media, Anal. Methods, 2016, 8(9), 2032–2040 RSC.
- M. Shehata, M. Adam, K. Abdelhady and M. Makhlouf, Facile synthesis, characterizations, and impedance spectroscopic features of Zn(II)-bis Schiff base complex films towards photoelectronic applications, J. Solid State Electrochem., 2019, 23, 2519–2531 CrossRef CAS.
- X. Zhong, J. Yi, J. Sun, H.-L. Wei, W.-S. Liu and K.-B. Yu, Synthesis and crystal structure of some transition metal complexes with a novel bis-Schiff base ligand and their antitumor activities, Eur. J. Med. Chem., 2006, 41(9), 1090–1092 CrossRef CAS.
- K. M. Khan, M. Khan, N. Ambreen, F. Rahim, B. Muhammad and S. Ali, et al., Bis-Schiff bases of isatins: a new class of antioxidant, J. Pharma Res., 2011, 4(10), 3402–3404 CAS.
- M. U. Rehman, A. Alam, S. A. A. Shah, A. Ali, Q. Ali and A. F. AlAsmari, et al., Discovering the DPPH Free Radical Scavenging Activity of Azine Derivatives Bearing Ethyl Phenyl Ketone Moiety, Russ. J. Bioorg. Chem., 2024, 50(5), 1639–1645 CrossRef CAS.
- M. Khan, G. Ahad, A. Alam, S. Ullah, A. Khan and U. Salar, et al., Synthesis of new bis(dimethylamino)benzophenone hydrazone for diabetic management: in vitro and in silico approach, Heliyon, 2024, 10(1), e23323 CrossRef CAS PubMed.
- S. A. Hassan and D. M. Aziz, Synthesis of new series bis-3-chloro-β-lactam derivatives from symmetrical bis-Schiff bases as effective antimicrobial agents with molecular docking studies, Science Journal of University of Zakho, 2021, 9(3), 128–137 CrossRef.
- A. Alam, M. Ali, A. Latif, N. U. Rehman, A. J. Shah and I. A. Khan, et al., Discovery of (S)-flurbiprofen-based novel azine derivatives as prostaglandin endoperoxide synthase-II inhibitors: synthesis, in vivo analgesic, anti-inflammatory activities, and their molecular docking, Bioorg. Chem., 2023, 141, 106847 CrossRef CAS PubMed.
- R. Ahmad, M. Khan, A. Alam, A. A. Elhenawy, A. Qadeer and A. F. AlAsmari, et al., Synthesis, molecular structure and urease inhibitory activity of novel bis-Schiff bases of benzyl phenyl ketone: a combined theoretical and experimental approach, Saudi Pharm. J., 2023, 31(8), 101688 CrossRef CAS PubMed.
- M. Ayaz, A. Alam, A. M. Zainab, A. Javed and M. S. Islam, et al., Biooriented synthesis of ibuprofen-clubbed novel bis-schiff base derivatives as potential hits for malignant glioma: in vitro anticancer activity and in silico approach, ACS Omega, 2023, 8(51), 49228–49243 CrossRef CAS PubMed.
- A. Alam, M. Ali, A. Latif, N. U. Rehman, S. Saher and A. Khan, et al., Novel Bis-Schiff’s base derivatives of 4-nitroacetophenone as potent α-glucosidase agents: design, synthesis and in silico approach, Bioorg. Chem., 2022, 128, 106058 CrossRef CAS PubMed.
- S. Gul, A. Alam, M. Assad, A. A. Elhenawy, M. S. Islam and S. A. A. Shah, et al., Exploring the synthesis, molecular structure and biological activities of novel bis-Schiff base derivatives: a combined theoretical and experimental approach, J. Mol. Struct., 2024, 1306, 137828 CrossRef CAS.
- J. Cheung, M. J. Rudolph, F. Burshteyn, M. S. Cassidy, E. N. Gary and J. Love, et al., Structures of human acetylcholinesterase in complex with pharmacologically important ligands, J. Med. Chem., 2012, 55(22), 10282–10286 CrossRef CAS PubMed.
- F. Nachon, E. Carletti, C. Ronco, M. Trovaslet, Y. Nicolet and L. Jean, et al., Crystal structures of human cholinesterases in complex with huprine W and tacrine: elements of specificity for anti-Alzheimer's drugs targeting acetyl-and butyryl-cholinesterase, Biochem. J., 2013, 453(3), 393–399 CrossRef CAS PubMed.
- A. D. Bochevarov, E. Harder, T. F. Hughes, J. R. Greenwood, D. A. Braden and D. M. Philipp, et al., Jaguar: a high-performance quantum chemistry software program with strengths in life and materials sciences, Int. J. Quantum Chem., 2013, 113(18), 2110–2142 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01367d |
| ‡ These authors have equally contributed to the work. |
| This journal is © The Royal Society of Chemistry 2025 |