
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePiperidine and valproic acid hybrid compound (F2S4-p-VPA) outperforms methotrexate as anti-proliferative and cells migration inhibition†
Martha Cecilia Rosales Hernández‡
 *a,
Raúl Horacio Camarillo López‡a,
Marycruz Olvera-Valdezb,
Leticia Guadalupe Fragoso Moralesac,
Itzia Irene Padilla Martínezd,
Mónica Adriana Torres Ramose,
Marycarmen Godínez Victoriaf,
Raúl Flores Mejíag and
José Correa Basurto*c
*a,
Raúl Horacio Camarillo López‡a,
Marycruz Olvera-Valdezb,
Leticia Guadalupe Fragoso Moralesac,
Itzia Irene Padilla Martínezd,
Mónica Adriana Torres Ramose,
Marycarmen Godínez Victoriaf,
Raúl Flores Mejíag and
José Correa Basurto*c
aLaboratorio de Biofísica y Biocatálisis, Sección de Estudios de Posgrado e Investigación, Escuela Superior de Medicina, Instituto Politécnico Nacional, Plan de San Luis y Díaz Mirón s/n, Casco de Santo Tomás, Distrito Federal, 11340, Mexico. E-mail: marcrh2002@yahoo.com.mx
bLaboratorio Nanomateriales Sustentables, Sección de Estudios de Posgrado e Investigación – Escuela Superior de Ingeniería Química e Industrias Extractivas, Instituto Politécnico Nacional, Av. Instituto Politécnico Nacional S/N, Unidad Profesional Adolfo López Mateos, CP 07708, CDMX, Mexico
cLaboratorio de Diseño y Desarrollo de Nuevos Fármacos e Innovación Biotecnológica, Sección de Estudios de Posgrado e Investigación, Escuela Superior de Medicina, Instituto Politécnico Nacional, Plan de San Luis y Díaz Mirón s/n, Casco de Santo Tomás, Distrito Federal, 11340, Mexico. E-mail: corrjose@gmail.com
dLaboratorio de Química Supramolecular y Nanociencias, Unidad Profesional Interdisciplinaria de Biotecnología del Instituto Politécnico Nacional, Av. Acueducto s/n Barrio la Laguna Ticomán, CDMX, 07340, Mexico
eDirección de Investigación, Instituto Nacional de Neurología y Neurocirugía Manuel Velasco Suárez, Ciudad de México and Centro de Investigación sobre el Envejecimiento, CINVESTAV sede sur., Av. Insurgentes Sur, 3877 Col. La Fama, 14269. Tlalpan, Mexico
fLaboratorio de Citometría de Flujo, Sección de Estudios de Posgrado e Investigación, Escuela Superior de Medicina, Instituto Politécnico Nacional, CDMX, Mexico
gLaboratorio de Inflamación y Obesidad, Escuela Superior de Medicina, Instituto Politécnico Nacional, Plan de San Luis y Díaz Mirón, Ciudad de México, 11340, Mexico
First published on 16th July 2025
Abstract
Glioblastoma and triple-negative breast cancer (TNBC) pose significant challenges in treatment due to their invasive nature and propensity for metastasis. Methotrexate (MTX) is a common chemotherapeutic agent; however, it has limited efficacy owing to its low aqueous solubility and cytostatic rather than cytotoxic effects. Valproic acid (VPA) has been used as chemotherapeutic agent, but with low potency. In this study, two novel VPA derivatives (F2S4-p-VPA and F3S4-m-VPA) were designed, chemically synthesized, and evaluated in vitro. These compounds contain tertiary amines as pharmacophore groups reminiscent of methotrexate. Cytotoxicity, migration, assays were conducted on glioblastoma (LN-18, U373) and breast cancer (MDA-MB-231) cell lines, using fibroblast (3T3-L1; non-cancer cells) as a control. Apoptosis (LN-18 and MDA-MB-231), cell cycle arrest and Bax and Bcl2 assays were carried out on LN-18 cells. Physicochemical properties of the compounds were assessed using in silico predictions. Results showed that F2S4-p-VPA exhibited better cytotoxicity than F3S4-m-VPA on both LN-18 (IC50 = 112 μM) and MDA-MB-231 (IC50 = 142 μM) cell lines, while demonstrating reduced cytotoxicity in 3T3-L1 cells. F2S4-p-VPA inhibited cell migration, outperforming MTX. Moreover, F2S4-p-VPA induced the highest rate of apoptosis in LN-18 cell, and produce the cell cycle arrest in the S and G2/M phase, showing a Bax/Bak-independent propapoptotic effect suggesting other mechanisms of cell death. Also, these novel compounds possess superior physicochemical properties to MTX and VPA. These results suggest that F2S4-p-VPA warrants further investigation in vivo and may serve as structural scaffold for the development of novel compounds for the treatment of these aggressive cancers.
1. Introduction
According to the World Health Organization, cancer is a leading cause of death globally, accounting for nearly 10 million deaths in 2020, or nearly one in six deaths. Furthermore, cancer is the leading cause of death before the age of 70 in 112 out of 183 countries.1 GLOBOCAN estimate 28.4 million new cancer cases to occur in 2040 year.2 Cancer has become a global health problem brain cancer with 308![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 102 new cases in 2020, though with fewer deaths compared to breast cancer (BC)3 which is the most common, accounting for 2.26 million new cases.1
102 new cases in 2020, though with fewer deaths compared to breast cancer (BC)3 which is the most common, accounting for 2.26 million new cases.1
Regarding brain cancer in Mexico, it ranks as the second leading cause of death in the pediatric population (below 15 years) encompassing the malignant tumors of the meninges, encephalon and other central nervous system tumors (18% in males and 15% in females).4 Specifically, glioblastoma emerges as the most common primary intracranial tumor.5 Glioblastoma multiforme (GBM) is known for its aggressiveness and lethality, characterized by rapid proliferation and resistance due invasive infiltration into surrounding brain tissue.6,7 Both pediatric and adult population experience high mortality and morbidity rates, with an incidence of 3.19 cases per 100000 individuals.8
BC has been the most common cancer among women since 2006 (ref. 9) and is one of the main reasons of morbidity and mortality in Mexico, accounting for 15.3% of new cases in 2020.3 Ductal adenocarcinoma is the most frequent type of BC in Mexican women.10 However, triple-negative breast cancer (TNBC), characterized by negative expression of estrogenic receptor (ER) and progesterone receptor (PR), and absence of amplification of the human epidermal growth factor-2 (HER2) gene, is the more prevalent in women under 40.11
These cancer types, GBM and TNBC undergo significant metastatic processes within a conducive environment characterized by factors such as hypoxia, extracellular matrix stiffness and nutrient deprivation, facilitating cell proliferation, migration, and invasion into secondary sites.12,13 Inflammation can further exacerbate chromosomal instability and promote cancer metastasis.14 Nearly 25% of the solid tumours arise from chronic inflammation, recognized as a hallmark of cancer. Elevated levels of inflammatory cells correlates with increased metastasis, often accompanied by the presence of cytokines, such as interleukin (IL)-6, tumor necrosis factor (TNF)-α, IL-1 and interferon (IFN)-γ and chemokines such as CXCL12, CXCL14 and CCL7.15 It has been reported that lipopolysaccharide (LPS) can induce migration and invasion in various cancer cells through inflammation activation, as in breast cancer.16–18 Additionally, in vitro studies have demonstrated the challenging nature of treating GBM and TNBC cells due to their high aggressiveness and propensity for invasion and migration, particularly when stimulated with TNF-α, phorbol 12-myristate 13-acetate (PMA) or LPS.7,17
Currently, there are different chemotherapy options to treat cancer as methotrexate (MTX).19 MTX is used to treat BC, including in patients whose cancer has metastasized. Formerly known as amethopterin, it is an antifolate that acts as both an anti-neoplastic agent and a suppressor of immune system.20 MTX has been combined with other anticancer drugs used to treat BC, such as mitomycin C, cyclophosphamide, and 5-fluorouracil or in combination with vitamin C.21 Additionally, MTX has been evaluated in glioma cells, decreasing cell viability in treatment for 48 h.22 However, to evaluate MTX in clinical trials of glioblastoma, high doses are necessary to cross the blood brain barrier (BBB).23
On the other hand, valproic acid (2-propylpentanoic acid; VPA) has been used either directly or synergistically as an anti-glioma agent either in vitro or in vivo studies.24,25 VPA is a short-chain fatty acid, a member of the group of histone deacetylase inhibitors (HDACIs) that has the capacity to inhibit cancer cell proliferation by modulating multiple intracellular signaling pathways and it is an agent used to limit proliferation of tumor cells.26,27 It has been demonstrated that VPA exhibits anticancer activity in a variety of human cancers, including BC and glioma principally inducing a cellular differentiation, growth arrest, and apoptosis.28 Several studies have revealed that VPA sensitizes GBM cells to chemotherapy and radiotherapy by increasing cell apoptosis and cell cycle arrest, and activating proapoptotic signaling.25,29
VPA derivatives have been explored for their anticancer properties, particularly against breast cancer. 2-Hexyl-4-pentynoic acid (HPTA), for example, proved highly effective: at just 15 μM, has the same effects of 500 μM VPA in inhibiting breast cancer cell growth and sensitizing MCF7 cells to hydroxyurea.30,31 HPTA also improved breast tumor radiosensitivity in rats by inhibiting Rad51.32 Additionally, N-(2′-hydroxyphenyl)-2-propylpentanamide (OH–VPA), a histone deacetylase inhibitor, was shown to induce nuclear HMGB1 release and alter ROS levels in HeLa cells.33 While VPA itself has shown potential in sensitizing glioblastoma to radiation therapy34,35 it's important to note that some reports indicate a limited effect of VPA on clinical outcomes for glioblastoma patients.36 Other research by M. Farroq et al. includes the synthesis and biological evaluation of various Schiff base series of valproyl derivatives for anticancer use.37
Due to high heterogeneity of GBM and TNBC characterized by their aggressiveness, developing new therapies to treat them could be a priority. The goal of this research is to evaluate new VPA derivatives (F2S4-p-VPA and F3S4-m-VPA) comparing them with MTX which is used as pharmacological therapy to treat TNBC and GBM cells (Fig. 1). While VPA has shown anti-proliferative effects on cancer cells, it is not widely used due to its low potency and cytotoxic. However, it serves as scaffold to add chemical pharmacophores present in MTX when linked to a tertiary amine yielding F2S4-p-VPA and F3S4-m-VPA; the chemical synthesis is presented in Scheme 1. Therefore, their cytotoxic activity and their ability to inhibit cell migration were tested on human malignant glioma (LN-18), human glioblastoma astrocytoma (U373), invasive ductal carcinoma (MDA-MB-231), a tumor with positive expression of epidermal growth factor (EGF) and transforming growth factor alpha (TGF-α); and fibroblast cell line (3T3-L1) non cancer cells used the later as a control. Also, the apoptosis and the expression of anti-apoptotic and apoptotic proteins and cell cycle assays with propidium iodide, were determinate by flow cytometry. The results indicated that F2S4-p-VPA exhibits better cytotoxic and cytostatic effects compared to MTX. Hence, it serves as an alternative for treating these aggressive cell types or be employed as a core for designing new compounds.
 | ||
| Fig. 1 Chemical structures of (A) methotrexate, (B) valproic acid, (C) F2S4-p-VPA, (D) F3S4-m-VPA indicating in green the chemical relationship between the structures. | ||
 | ||
| Scheme 1 Chemical synthesis of F2S4-p-VPA and F3S4-m-VPA. | ||
2. Materials and methods
2.1 Chemical synthesis
All chemical reactants and starting materials purchased from Sigma-Aldrich were directly used without further purification. All solvents were distilled prior to use. The acetone was dried by adding molecular sieves 3 Å, swirling the mixture and allowing it to sit overnight before decanting the acetone off the drying agent.Mass spectra studies were executed by staff of Escuela Superior de Medicina using a Q-TOF (model 6545) with an ESI (model G1959A) Agilent. Thin layer chromatography (TLC) was performed using commercially available pre-coated plates (TLC silica gel 60 F254). Column chromatography was carried out using Merck silica gel 60 (0.063–0.100 mm).
The synthesized compounds were characterized through 1H and 13C NMR spectroscopy. All spectra were recorded on a Varian 300 MHz spectrometer. Samples of compounds were dissolved in the indicated deuterated solvent; chemical shifts (δ) were reported in ppm downfield to tetramethylsilane. Coupling constants (J) are reported in Hertz and rounded to 0.1 Hz. Splitting patterns are abbreviated as follows: singlet (s), doublet (d), triplet (t), quintet (q), multiplet (m), broad (br) or a combination of these.
The synthesis of valproic chloride (ClVal), F2S4-p-VPA and F3S4-m-VPA is depicted in Scheme 1. The synthetic route for F2S4-p and F3S4-m compounds has been previously reported by our group.38 Briefly, the synthesis of compound F2S4-p followed a different approach to obtain the amine group in para position relative to the alkoxy group; for this purpose, the Williamson ether synthesis was performed employing 4-hydroxy-3-methoxybenzaldehyde (vanillin) and 1-(2-chloroethyl)piperidine hydrochloride (1) as the starting materials to lead the compound (2). The next step required the formation of the appropriate oxime (3) which ultimately was further reduced to the desired amine employing a catalytic hydrogenation procedure. Compound F3S4-m was obtained via the Williamson ether synthesis, in which a hydroxy-benzylamine derivative (4) reacted with 1-(2-chloroethyl) pyrrolidine hydrochloride (5) a primary alkyl halide via a nucleophilic substitution reaction. The functionalization of F2S4-p to yield F2S4-p-VPA involved an analogous acyl substitution reaction with valproic acid, facilitated by TEA, 1-ethyl-3-(3-dimethylaminopropyl)carbodiimide (EDCI), and 1-hydroxybenzotriazole (HOBt). This transformation was conducted under reflux conditions in anhydrous dichloromethane (DCM) for 72 hours, ensuring optimal coupling efficiency. The derivatization of F3S4-m to F3S4-m-VPA was accomplished via a nucleophilic acyl substitution, wherein F3S4-m reacted with ClVal in the presence of triethylamine (TEA) as a base, under anhydrous conditions in tetrahydrofuran (THF) and an inert nitrogen atmosphere, with continuous stirring at ambient temperature for 24 hours.
2.1.1.1 Synthesis of 2-propylpentanoic acid (valproic acid). Sodium valproate (1.00 g, 6.9 mmol) was dissolved in distilled water (15 mL) in a 50 mL beaker. The pH was adjusted to 4.0 by dropwise addition of 15 N HCl. The resulting VPA, appearing as a colorless liquid, separated from the aqueous layer and remained on the surface. The VPA was then extracted with ethyl acetate (2 × 15 mL) employing a 100 mL separating funnel. The organic fraction was then washed with distilled water (2 × 20 mL), dried with anhydrous Na2SO4, and filtered to remove the solid Na2SO4. The solvent was then evaporated under reduced pressure (240 mbar, 60 °C) to obtain the VPA as a colorless liquid (1.8 mL), which was used without further purification.
2.1.1.2 Synthesis of valproic chloride (ClVal). VPA (1.8 mL) was placed into a 100 mL two-neck round-bottom flask containing a magnetic stirrer (10 mm long, 7 mm wide). Thionyl chloride (0.65 mL, 9.08 mmol) was then added dropwise to the flask. The mixture was then refluxed for 2 h in dry acetone using a cooling system (ECO 30®) and a heating mantle connected to a thermal cycler (Thermolyne 45500®) to reach 80 °C. The reaction was protected from moisture using a nitrogen-filled balloon. Residual SOCl2 was removed with the aid of a Schlenk line. The resulting valproic chloride was stored at −20 °C under a nitrogen atmosphere to protect it from air until further use.
The synthesis of F2S4-p was performed according to procedures previously reported by our group38 employing peptide synthesis methodologies as these conditions are normally gentle and provide high yields.
A mixture of F2S4-p (500 mg, 1.67 mmol), EDCI (320 mg, 1.7 mmol) and HOBt (260 mg, 1.7 mmol) was dissolved with dry dichloromethane (75 mL) into a 100 mL two-neck round-bottom flask containing a magnetic stirrer (10 mm long, 7 mm wide) under an N2 atmosphere. Afterwards, VPA (235 μL, 1.8 mmol) and TEA (2.5 mL) were added to the mixture allowing the reaction to proceed 72 h under refluxing conditions. The reaction mixture was then extracted with pH-adjusted water (3 × 100 mL) to remove traces of VPA and remaining coupling reagents. The organic phase was dried with anhydrous sodium sulfate, and solvent was removed by reduced pressure. The obtained residue was purified by silica column chromatography employing a mixture of solvents, starting with hexane, and gradually transitioning to ethyl acetate as the sole solvent. Eventually, ethanol was added to achieve a 25% mixture of ethyl acetate and ethanol. The F2S4-p-VPA was obtained as pale-yellow powder (54% yield, 97% purity), mp 97 °C. IR (cm−1): 3283 (N–H stretching), 1638 (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O stretching), 1544 (aromatic CC stretches), 1515 (in plane N–H companion bend), 1237 (C–N stretch), 707 (N–H wag out of plane).1H-NMR (300 MHz, CDCl3, ppm) 6.83 (m, 3H, H-1, H-3, H-6), 5.76 (br, 1H, NH), 4.40 (d, J = 5.66, 2H, PhCH2), 4.15 (t, J = 6.44, 2H, H-7), 3.83 (s, 3H, OCH3), 2.83 (t, J = 6.43, 2H, H-8), 2.54 (t, 4H, H-9), 2.05 (q, J = 4.68, 1H, H-12), 1.62 (t, J = 6.54, 4.11, 4H, H-10), 1.47–1.23 (m, 10H, H-11, H-13, H-14),0.89 (t, J = 6.96, 6H, H-15). 13C NMR (75 MHz, CDCl3, ppm) 175.9 (CO), 149.7 (C-4),147.7 (C-3), 131.8 (C-6), 120.1 (C-1), 113.2 (C-2), 111.5 (C-5), 66.8 (C-7), 57.8 (C-8), 55.92 (OCH3), 55.12 (C-9), 47.94 (C-12), 43.24 (Ph-CH2), 35.4 (C-13), 25.9 (C-10), 24.2 (C-11), 21.0 (C-14), 14.3 (C-15). MS (ESI+): m/z calculated for ([C23H38N2O3] + H)+: 391.2955, found 391.2958, calculated for ([C23H38N2O3] + Na)+: 413.2775 found 413.2677. All proton and carbon NMR, FTIR and mass spectra for F2S4-p-VPA are available for consultation in the ESI under Sections S1 to S4.†
O stretching), 1544 (aromatic CC stretches), 1515 (in plane N–H companion bend), 1237 (C–N stretch), 707 (N–H wag out of plane).1H-NMR (300 MHz, CDCl3, ppm) 6.83 (m, 3H, H-1, H-3, H-6), 5.76 (br, 1H, NH), 4.40 (d, J = 5.66, 2H, PhCH2), 4.15 (t, J = 6.44, 2H, H-7), 3.83 (s, 3H, OCH3), 2.83 (t, J = 6.43, 2H, H-8), 2.54 (t, 4H, H-9), 2.05 (q, J = 4.68, 1H, H-12), 1.62 (t, J = 6.54, 4.11, 4H, H-10), 1.47–1.23 (m, 10H, H-11, H-13, H-14),0.89 (t, J = 6.96, 6H, H-15). 13C NMR (75 MHz, CDCl3, ppm) 175.9 (CO), 149.7 (C-4),147.7 (C-3), 131.8 (C-6), 120.1 (C-1), 113.2 (C-2), 111.5 (C-5), 66.8 (C-7), 57.8 (C-8), 55.92 (OCH3), 55.12 (C-9), 47.94 (C-12), 43.24 (Ph-CH2), 35.4 (C-13), 25.9 (C-10), 24.2 (C-11), 21.0 (C-14), 14.3 (C-15). MS (ESI+): m/z calculated for ([C23H38N2O3] + H)+: 391.2955, found 391.2958, calculated for ([C23H38N2O3] + Na)+: 413.2775 found 413.2677. All proton and carbon NMR, FTIR and mass spectra for F2S4-p-VPA are available for consultation in the ESI under Sections S1 to S4.†
2.1.2.1 Synthesis of (4-methoxy-3-(2-(pyrrolidin-1-yl)ethoxy)phenyl) methanamine (F3S4-m). The synthesis of F3S4-m has been previously reported by our group.38 The synthesis involves reacting 3-hydroxy-4-methoxybenzylamine hydrochloride with 1-(2-chloroethyl)pyrrolidine hydrochloride in the presence of K2CO3 in dry acetone under refluxing conditions for 48 h. The resulting viscous residue was purified by alumina gel column chromatography, yielding F3S4-m with a 75% yield and 96% purity.
2.1.2.2 Synthesis of F3S4-m-VPA. F3S4-m (1.31 g, 5.25 mmol) was placed into an ice-cooled 100 mL two-neck round-bottom flask containing a magnetic stirrer (10 mm long, 7 mm wide) under N2 atmosphere. Triethylamine (TEA, 0.56 mL, 12.6 mmol) was then added to the flask followed by dropwise addition of a ClVal (0.9 mL, 6.3 mmol) previously diluted in THF (3 mL) corresponding to a 1.2 mol of ClVal per each mol of 3-hydroxy-4-methoxybenzylamine hydrochloride. The mixture remained under cooled stirring for 15 min. Afterwards, the mixture was stirred for 24 h under room temperature (rt). The heterogeneous mixture was vacuumed-filtered, the solid phase was washed with THF (10 mL). All liquid fractions were collected, and solvent was removed by reduced pressure (374 mbar, 60 °C) to finally obtain a thick dark brownish liquid.
For purification of F3S4-m-VPA, 100 mg of F3S4-m-VPA was diluted in distilled hexane (20 mL) under stirring conditions over 20 min. Thereafter, the mixture was chilled over 48 h at 4 °C leading the formation to a light brown-sand colour precipitate. The solid was then removed with the aid of vacuum filtration followed by removal of traces of solvent with a Schlenk line overnight.
F3S4-m-VPA was obtained as a light brown solid (556 mg, 55.6% yield, 96% purity). m. p. 94 °C. IR (cm−1) 3290 (N–H stretching), 3082 (C–H stretch), 1635 (CO stretching), 1517 (in plane N–H companion bend), 1553 (aromatic CC stretches), 707 (N–H wag out of plane). 1H NMR (300 MHz, CDCl3, ppm); 6.83 (d, J = 1.12, 1H, H-3), 6.80 (s, 2H, H-1, H-6), 5.79 (br, NH), 4.37 (d, J = 5.64 2H, PhCH2), 4.11 (t, J = 6.01, 2H, H-7), 3.83 (s, 3H, OCH3), 2.95 (t, J = 6.15, 2H, H-8), 2.65 (t, J = 3.34, 8.66 4H, H-9), 2.07–1.96 (m, 1H, H-11), 1.80 (q, J = 14.86, 8.25, 4H, H-10), 1.69–1.32 (m, 4H, H-12), 1.32–1.14 (m, 4H, H-13), 0.87 (t, J = 7.12, 6.99, 6H, H-14). 13C NMR (75 MHz, CDCl3, ppm) δ 175.9 (CO), 148.8 (C-4),148.4 (C-5), 131.3 (C-2), 120.5 (C-1), 113.0 (C-6), 111.6 (C-3), 67.5 (C-7), 56.0 (C-8), 54.8 (C-9), 54.5 (OCH3), 47.9 (Ph-CH2), 43.2(C-11), 35.40 (C-12), 25.5 (C-10), 21.0 (C-13), 14.3 (C-14). MS (ESI+): m/z calculated for ([C22H36N2O3] + H)+: 377.2799, found: 377.2788. All proton and carbon NMR, FTIR and mass spectra for F3S4-m-VPA are available for consultation in the ESI under Sections S5 to S8.†
2.2 In vitro studies
After, 48 h of treatment, cell viability was determined by MTT assay. Firstly, the culture medium was removed, and then 20 μL of MTT solution (0.5 mg mL−1) was added to each well. The cultures were then incubated for 4 h at 37 °C. After incubation, the MTT solution was removed, and the formazan salts were dissolved with 100 μL of DMSO. The absorbance was measured at 550 nm using a Multiskan EX Thermo Scientific spectrophotometer.
 000 events were analyzed within the total cellular region in the FSC vs. SSC dot-plot.
000 events were analyzed within the total cellular region in the FSC vs. SSC dot-plot.2.3 ADMET and physicochemical properties by AdmetSar and DataWarrior
The absorption, distribution, metabolism, excretion and toxicity (ADMET) properties of the compounds (Fig. 1) were investigated using AdmetSar (version 2.0) online software and OSIRIS DataWarrior (version 5.0) PC-version software. Initially, the SMILES notations were input into AdmetSar, which utilized machine learning to predict absorption, distribution, metabolism, excretion, and toxicity properties. Simultaneously, OSIRIS DataWarrior enabled the drawing of chemical structures, facilitating the calculation of various physicochemical properties and toxicity predictions. Results from both platforms were cross-referenced to ensure robustness. This integrated approach enhances the reliability of predicted ADMET parameters, providing a comprehensive understanding of the compounds' pharmacokinetic behaviors.2.4 Statistical analysis
All results are presented as the mean ± SE. Statistical analyses were conducted using GraphPad Prism Version 5.00 software. One-way ANOVA was utilized for cell viability and apoptosis assays, with Dunnett's multiple comparison tests performed to analyse the wound-healing data, apoptosis assay. Statistical significance was defined as p < 0.05, and each experiment was repeated at least three times.3. Results
3.1 Chemical synthesis
The characterization of F2S4-p-VPA and F3S4-m-VPA was comprehensively analyzed using IR spectroscopy, NMR, and mass spectrometry, confirming their structures and high purity. F2S4-p-VPA was characterized by IR spectroscopy, showing significant peaks at 3283 cm−1 for N–H stretching and 1638 cm−1 for CO stretching, confirming the presence of amide and aromatic functionalities. The 1H NMR spectrum revealed peaks around 6.83 ppm for aromatic hydrogens, with additional signals confirming the aliphatic chain and methoxy groups. The 13C NMR spectrum featured a strong carbonyl signal at δ 175.9 ppm, along with characteristic signals for the aromatic and aliphatic regions. Mass spectrometry confirmed the molecular structure with a peak at m/z 391.2958 corresponding to the ([M] + H+), the calculated molecular weight of 391.2955. The compound was obtained with a purity of 97% as determined by NMR data analysis.
Similarly, for F3S4-m-VPA, the IR spectrum displayed key absorption bands at 3290 cm−1 (N–H stretching), 3082 cm−1 (C–H stretching), 1635 cm−1 (CO stretching), and 1517 cm−1 (aromatic CC stretching), confirming the presence of amide, alkyl, and aromatic groups. In the 1H NMR spectrum, signals were observed at 6.83 ppm for H-3 and 6.80 ppm for H-1 and H-6, consistent with aromatic protons, alongside peaks for the aliphatic chain and methoxy groups. The 13C NMR showed a carbonyl signal at δ 175.9 ppm and further signals corresponding to aromatic and aliphatic carbons. Mass spectrometry provided a molecular ion peak at m/z 377.2788 ([M] + H+), which closely matched the expected mass of 377.2799. The compound was isolated with a purity of 96% as determined by NMR data analysis. Both compounds were successfully synthesized and purified, demonstrating high yields and purity, as confirmed by the combined spectroscopic data.
3.2 Cytotoxic assays
The cytotoxic effects of VPA derivatives were compared with those of MTX by measuring the cell viability in glioblastoma cell lines (LN-18 and U373), TNBC cell line (MDA-MB-231) and a murine fibroblast cell line (3T3-L1) used this last as no cancer cells (control cells) for 48 h of treatment from 0 to 300 μM as indicated in Fig. 2.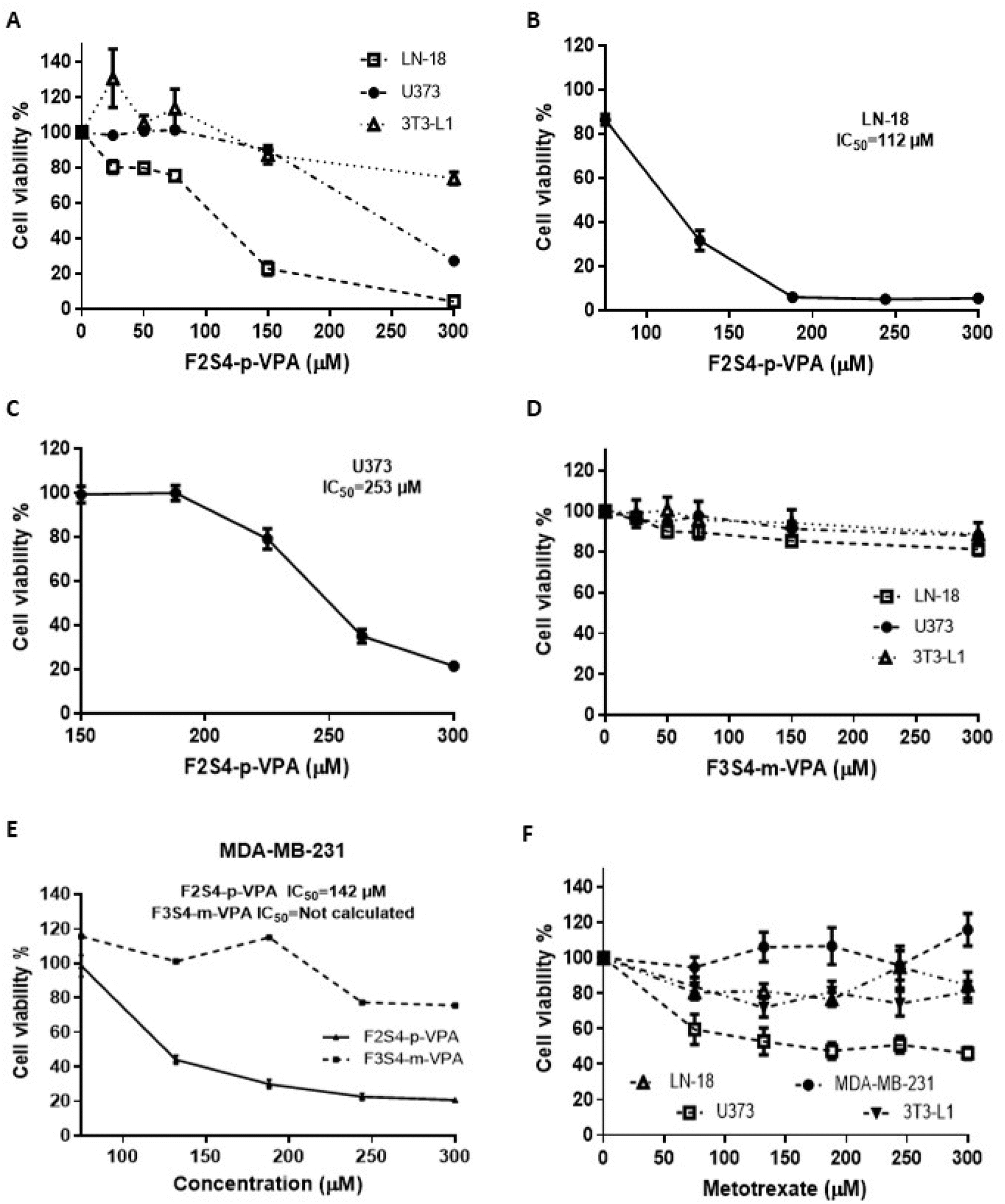 | ||
| Fig. 2 Cell viability (%) after 48 h of F2S4-p-VPA, F3S4-m-VPA, and MTX treatment at different concentrations ranging from 0 to 300 μM in glioblastoma, breast cancer, and fibroblast cells. (A) F2S4-p-VPA on U373, LN-18 and 3T3-L1 cells lines. (B) F2S4-p-VPA on LN-18 cell line. (C) F2S4-p-VPA on U373 cell line. (D) F3S4-m-VPA on U373, LN-18 and 3T3-L1 cells lines. (E) F2S4-p-VPA and F3S4-m-VPA on MDA-MB-231 cell line. (F) MTX on MDA-MB-231, LN-18, U373, and 3T3-L1 cell lines. One-way ANOVA with Dunnett's multiple comparison tests was performed and results represent the mean ± the SE from three independent experiment with n = 8, p < 0.05. | ||
The cell viability of U373 and LN-18 cells exposed to F2S4-p-VPA decreased in a concentration-dependent manner (Fig. 2A). Notably, F2S4-p-VPA exhibited a more pronounced cytotoxic effect on the LN-18 cell line than on the U373 cell line, with cell viability dropping to approximately 20% and 90% at 150 μM, respectively. A concentration of 300 μM of F2S4-p-VPA was required to induce significant cell death in the U373 cell line. The IC50 of F2S4-p-VPA against the LN-18 cell line was 112 μM (Fig. 2B) and in U373 cell line was 253 μM (Fig. 2C). Conversely, the cell viability of U373, LN-18, and 3T3-L1 cells exposed to F3S4-m-VPA from 0 to 300 μM (Fig. 2D) did not show a significant cell viability reduction.
Thus, these results showed that F2S4-p-VPA exerts a higher cytotoxic effect than F3S4-m-VPA, particularly on cancer cell lines, as evidenced by the maintenance of approximately 90% cell viability in 3T3-L1 cells at 300 μM (Fig. 2A and 2D).
The VPA derivatives were assayed on other aggressive cell line as TNBC (MDA-MB-231) with the same concentrations (0–300 μM) for 48 h. F2S4-p-VPA reduced cell viability with an IC50 of 142 μM. In contrast, F3S4-m-VPA reduced cell viability to 80% and the concentrations tested do not reach 50% (Fig. 2E).
Finally, to compare the cytotoxic effects of MTX across cell lines, it was incubated for 48 h at concentrations ranging from 0 to 300 μM (Fig. 2F). It was evident that MTX exhibited lower cytotoxicity than F2S4-p-VPA, particularly on MDA-MB-231 and 3T3-L1 cell lines. However, MTX exhibited a more potent cytotoxic effect on glioma cell lines, particularly on U373.
3.3 Cell migration assays
The effects of F2S4-p-VPA, F3S4-m-VPA, and MTX on cell migration after stimulating the cells with LPS were evaluated. Immediately after creating the wound (t = 0), the area was completely devoid of cells across all LN-18, U373, and MDA-MB-231 cell lines (Fig. 3A). Following 48 hours of LPS stimulation, LN-18 and MDA-MB-231 cells had covered the wound area, whereas U373 cells still exhibited an open wound area (Fig. 3A). Consequently, all cells were then treated with both LPS and each compound to assess whether the compounds inhibited cell migration, relative to cells treated only with LPS. | ||
| Fig. 3 LN-18, U373 and MDA-MB-231 cell lines after the treatment with LPS and F2S4-p-VPA. (A) Cells migration of LN-18, U373 and MDA-MB-231 cell lines after the treatment with LPS; the images are representative from three independent experiments, the images were taking at 4×. (B) Cells migration of LN-18, U373 and MDA-MB-231 cell lines after the treatment with F2S4-p-VPA at 185 μM + LPS and 225 μM + LPS. Percent of wound healing (C) LN-18 cells, (D) U373 cells and (E) MDA-MB-231 cells. One-way ANOVA with Dunnett's multiple comparison tests was performed and results are plotted as the mean from three independent experiments with n = 3 ± SE. ****p < 0.0001. | ||
In LN-18 cells, F2S4-p-VPA at 187 μM + LPS or 225 μM + LPS resulted in minimal wound healing. The few cells remaining in the wound area exhibited an altered morphology compared to those treated solely with LPS (ESI, S9†). This suggests that at these concentrations, F2S4-p-VPA inhibits cell migration, likely due to cell death (Fig. 3B and C). Conversely, U373 cells showed higher wound healing at 225 μM, indicating that F2S4-p-VPA was more effective at inhibiting migration at 187 μM (Fig. 3D). For MDA-MB-231 cells, F2S4-p-VPA (187 μM + LPS or 225 μM + LPS) did not effectively prevent cell migration compared to the LPS-only control. Although migration decreased at 225 μM, this reduction wasn't statistically significant (Fig. 3E). Furthermore, the morphology of MDA-MB-231 cells resembled that of the LPS-only control, unlike the observations in LN-18 cells.
Treatment of LN-18 cells with F3S4-m-VPA (187 μM + LPS or 225 μM + LPS) did not effectively inhibit cell migration, with wound healing remaining around 80% (Fig. 4A and B). For U373 cells, F3S4-m-VPA demonstrated greater efficacy in preventing cell migration at 187 μM + LPS than at 225 μM (Fig. 4A); however, this difference was not statistically significant (Fig. 4C). Notably, in MDA-MB-231 cells, F3S4-m-VPA at 225 μM significantly prevented cell migration compared to the LPS control (p < 0.020) (Fig. 4D).
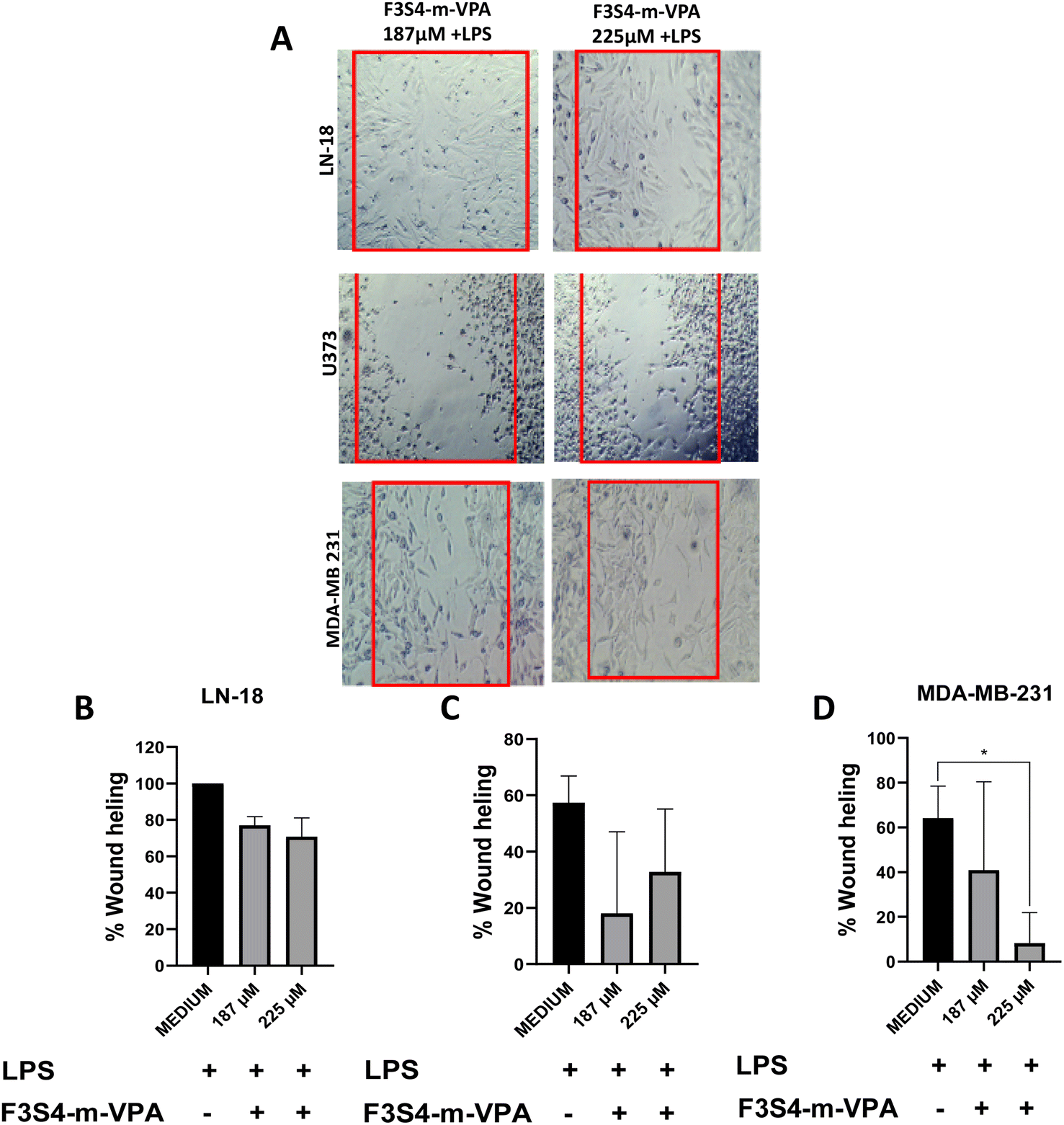 | ||
| Fig. 4 LN-18, U373 and MDA-MB-231 cell lines after the treatment with F3S4-m-VPA and LPS. (A) Cells migration; the images are representative from three independent experiments, the images are taking at 4×. Percent of wound healing (B) LN-18 cells, (C) U373 cells and (D) MDA-MB-231 cells. One-way ANOVA with Dunnett's multiple comparison tests was performed and results are plotted as the mean from three independent experiments with n = 3 ± SE. *p < 0.020. | ||
Our final evaluation focused on the effect of MTX across LN-18, U373, and MDA-MB-231 cell lines (Fig. 5A). In LN-18 cells, treatments with MTX at 187 μM + LPS or 225 μM + LPS resulted in approximately 80% wound healing. Despite this high percentage, a significant reduction in migration was observed compared to cells treated solely with LPS, suggesting a modest inhibitory effect (Fig. 5B). In contrast, U373 cells exhibited no significant differences with any MTX treatment, indicating a lack of migratory inhibition (Fig. 5C). For MDA-MB-231 cells, MTX at 225 μM was more effective in reducing wound healing than at 187 μM (Fig. 5D). Therefore, MTX demonstrated a capacity to prevent cell migration in LPS-stimulated LN-18 cells but failed to do so in MDA-MB-231 or U373 cells.
 | ||
| Fig. 5 LN-18, U373 and MDA-MB-231 cell lines after the treatment with MTX and LPS. (A) Cells migration of LN-18, U373 and MDA-MB-231 cell lines after the treatment with LPS; the images are representative from three independent experiments, the images are taking at 4×. Percent of wound healing (B) LN-18 cells, (C) U373 cells and (D) MDA-MB-231 cells. One-way ANOVA with Dunnett's multiple comparison tests was performed and results are plotted as the mean from three independent experiments with n = 3 ± SE. *p < 0.020, **p < 0.0010. | ||
Thus, F2S4-p-VPA demonstrated superior inhibition of LN-18 cell migration compared to MTX. While F2S4-p-VPA also reduced wound healing in MDA-MB-231 cells at 225 μM, its effect on cell migration was notably greater in LN-18 cells than in MDA-MB-231 cells, despite similar IC50 values in the cytotoxic assay.
3.4 Apoptosis assays
To determine whether the cytotoxic effect of VPA derivatives and MTX was associated with cell apoptosis, Annexin V-FITC/PI double staining was used with flow cytometry analysis. After 48 h of treatment on LN-18 cell line 6.0 ± 3.5% of cells were in the early apoptotic stage in the control group, while 28 ± 11.1%, 9 ± 5% and 13 ± 3.2% were observed in the F2S4-p-VPA, F3S4-m-VPA and MTX groups, respectively. The control group presented a late apoptosis percentage of 18 ± 6% whereas the F2S4-p-VPA, F3S4-m-VPA and MTX groups exhibited late apoptosis percentage of 15 ± 7%, 19 ± 4%, 25 ± 2%, respectively (Fig. 6A). The total percentage of late and early apoptosis caused by F2S4-p-VPA and F3S4-m-VPA were 43 ± 9% and 28 ± 5% respectively. This indicates that F2S4-p-VPA is capable of inducing apoptosis. | ||
| Fig. 6 Effects of F2S4-p-VPA, F3S4-m-VPA and MTX compounds on LN-18 and MDA-MB-231 cell lines. (A) LN-18, (B) MDA-MB-231 cells; both treated with compounds at 48 h and stained with Annexin V-FITC/PI. Statistical analysis of live, early, and late apoptosis and necrosis ratio of LN-18 and MDA-MB-231 cells after treatment was by one-way ANOVA with Dunnett's multiple comparison tests and all data are expressed as mean ± SE of three independent experiments with n = 3. *p < 0.0250, **p < 0.0042 compared to the control. | ||
In the MDA MB-231 cell line, only 5.6 ± 1.7% early apoptotic cells were detected in the control group, while 8.7 ± 0.7%, 21 ± 2.3% and 12 ± 8.1% apoptotic cells were identified in the presence of F2S4-p-VPA, F3S4-m-VPA and MTX groups, respectively (Fig. 6B). The total percentage of late and early apoptosis caused by F2S4-p-VPA and F3S4-m-VPA were 25 ± 0.8% and 27 ± 1.4% respectively. Thus, it was observed that F2S4-p-VPA induces a higher proportion of cells into late apoptosis, whereas F3S4-m-VPA induces more early apoptosis. This suggests that F2S4-p-VPA is more cytotoxic, leading to apoptotic cell death rather than necrosis, as the percentage of late apoptosis is lower in all cases. Then, LN-18 cells, is more sensitive for both compounds than MDA MB-231.
3.5 Cell cycle analysis by propidium iodide (PI) staining using flow cytometry
The cells cycle phases were determinate for LN-18 and 3T3-L1 as is showed in the Fig. 7. Cell cycle analysis by flow cytometry in 3T3-L1 cell line revealed slight differences between cells treated with MTX and F2S4-p-VPA (Fig. 7A) in relation with the control. The sub G0 phase shows that F2S4-p-VPA and MTX increases cell death (10.0% and 8% respectively) in relation with the control cells, however, there is no statistically significant difference were observed. In the G0–G1 phase it was higher in the control and in the cells treated with F2S4-p-VPA (59.9%) and MTX (46.8%), although it decreases around 15% with MTX. Regarding the S phase, it is observed that MTX presents a higher percentage (16.6%), which is consistent with its mechanism of action, blocking DNA synthesis. Similarly, treatment with F2S4-p-VPA also presents an increase of 6.7%, both in relation with the control cells however it was not statistically significant. Finally, in the G2–M phase, MTX and F2S4-p-VPA doesn't present a significant difference were observed compared to the control, these results indicated that MTX and F2S4-p-VPA doesn't produce a significant change in the 3T3-L1 cells.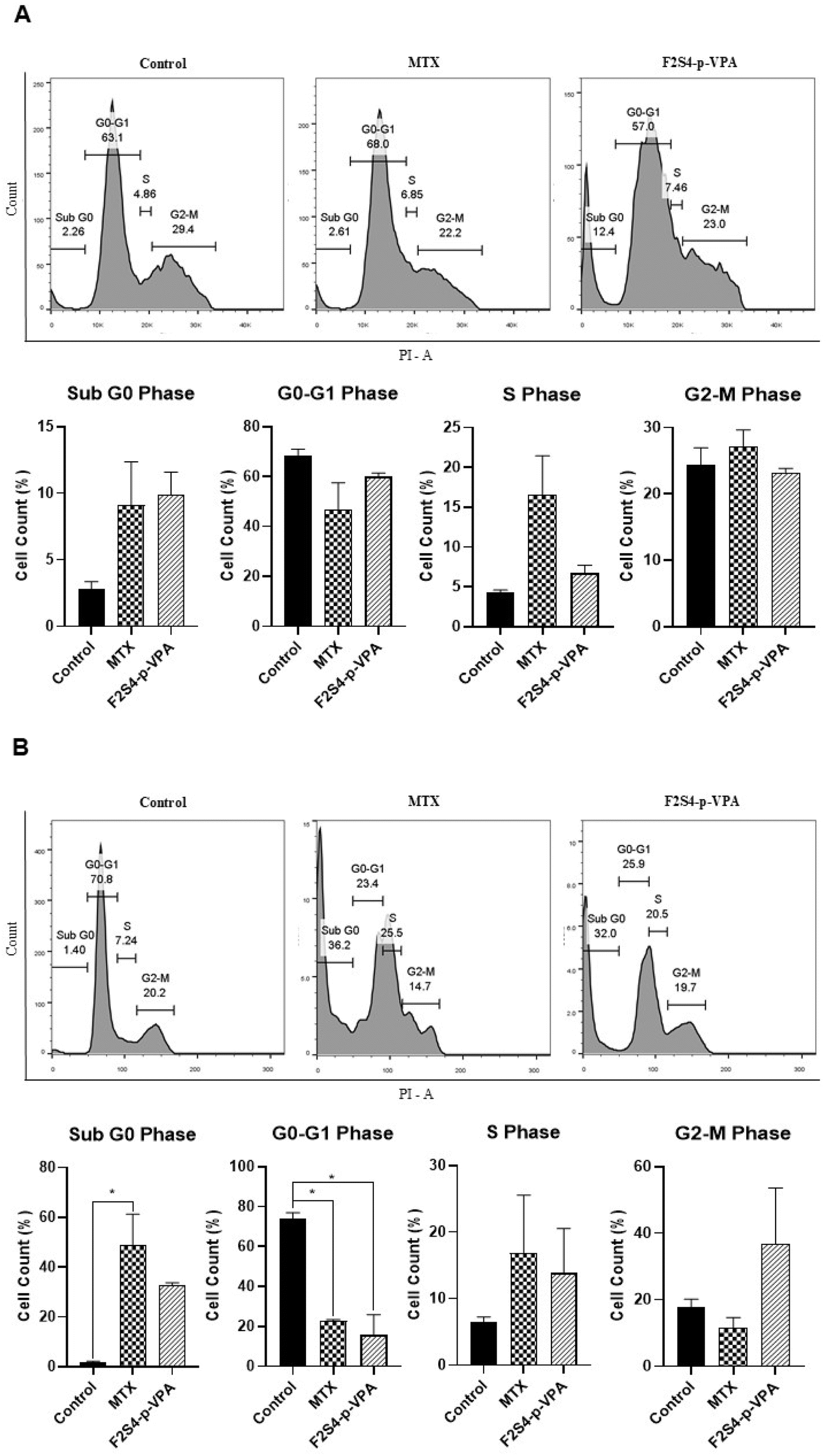 | ||
| Fig. 7 Cell Cycle assay in (A) 3T3-L1 and (B) LN-18 cells. The cells were treated with medium-DMSO (control) and with MTX or F2S4-p-VPA. The cell cycle phases were determinate 48 h after to be treated with the compounds employing propidium iodide (PI) staining using flow cytometry. The results are from three independent experiment and the statistical analysis was by one-way ANOVA with Dunnett's multiple comparison tests and all data are expressed as mean ± SEM of three independent experiments with n = 3. *p < 0.050, compared to the control. | ||
On the other hand, in the sub G0 phase of the cell cycle analysis in LN-18 cells (Fig. 7B), it is observed that MTX, compared to the control, presents a statistically significant increase in cell death (48.7%), the same, in F2S4-p-VPA cell death also increases, however, there is no statistical difference. In the G0–G1 phase, both treatments (MTX and F2S4-p-VPA) decrease drastically to 22.7% and 15.7% respectively. In the S phase, MTX and F2S4-p-VPA shows double the percentage of cells compared to the control, being this 16.8% and 15.6% respectively, suggesting that it is in this phase where cell arrest occurs. In the G2–M phase, it is observed that MTX does not accumulate cells in this phase (11.4%), because the cells are dying earlier; Unlike F2S4-p-VPA, which has a percentage of 36.7% without presenting a significant difference compared to the control.
3.6 Bax and Bcl-2 quantification protein by flow cytometry analysis
Fig. 8 shows the percentages of Bax and Bcl-2 expression obtained from the LN-18 cell line after treatment with MTX and F2S4-p-VPA. In the case of MTX, there is a slight activation of Bax (2.8%), while the expression of Bcl-2 alone and co-expression with Bax both show an average percentage of 17.8%. Most of the cells (61.6%) showed no expression of either marker, suggesting that both genes are silenced, indicating cell death by the extrinsic apoptotic pathway. The F2S4-p-VPA sample also shows low Bax activation (3.4%), and the increase in Bcl-2 alone (15.3%) which could indicate a resistance response, possibly as a defense mechanism against the induced damage. The co-expression of both genes is intermediate (20.8%), and again, the highest percentage (62.7%) showed no expression of either marker. | ||
| Fig. 8 Determination of BAX and BCL-2 protein on the LN-18 cell line treated with MTX and F2S4-p-VPA at 48 h. The results are from three independent experiment and the statistical analysis was by one-way ANOVA with Dunnett's multiple comparison tests and all data are expressed as mean ± SEM. | ||
3.7 In silico prediction of ADMET and physicochemical properties
The results of ADMET properties suggest that none of the synthesized compounds are likely to exhibit oral toxicity, as they are classified as III, while MTX is categorized as class II. Furthermore, according to ADMET predictions, the VPA derivatives can cross the BBB, unlike MTX. However, VPA derivatives may potentially induce bile salt export pump (BSEP) inhibition, which could lead to bile salt accumulation and liver toxicity, like VPA. Therefore, during their evaluation, it may be necessary to assess transaminases levels and other biomarkers as lactate dehydrogenase (LDH), to investigate whether these compounds, at the administrated doses, could induce liver damage or toxicity in other organs.43 Nevertheless, only VPA and MTX were predicted to potentially cause hepatotoxicity, whereas F2S4-p-VPA and F3S4-m-VPA were not. Additionally, only MTX was predicted to induce nephrotoxicity (Table 1A).| (A) ADMET properties obtained by AdmetSar | ||||
|---|---|---|---|---|
| F2S4-p-VPA | F3S4-m-VPA | VPA | MTX | |
| Acute oral toxicity (c) | III | III | III | II |
| Blood brain barrier | + | + | + | − |
| BSEP inhibitior | + | + | − | − |
| Hepatotoxicity | − | − | + | + |
| CYP2C9 substrate | − | − | + | − |
| CYP2D6 substrate | + | + | − | − |
| CYP3A4 substrate | + | + | − | + |
| Eye corrosion/eye irritation | −/− | −/− | +/+ | −/− |
| Glucocorticoid receptor binding | + | − | − | − |
| Nephrotoxicity | − | − | − | + |
| OATP1B1 inhibitior/OATP1B3 inhibitior | +/+ | +/+ | +/+ | +/+ |
| OATP2B1 inhibitior | − | − | + | − |
| OCT1 inhibitior/OCT2 inhibitior | −/− | −/− | −/− | −/− |
| P-glycoprotein inhibitior | − | − | − | − |
| P-glycoprotein substrate | + | + | − | + |
| Plasma protein binding | 0.692681 | 0.723911 | 0.960923 | 0.637697041 |
| Subcellular localization | Mitochondria | Mitochondria | Mitochondria | Mitochondria |
| (B) Physicochemical properties obtained by DataWarrior | ||||
|---|---|---|---|---|
| F2S4-p-VP | F3S4-m-VPA | VPA | MTX | |
| Mol weight | 390.566 | 376.539 | 144.213 | 454.446 |
| clogP |
4.2558 | 3.9138 | 2.1912 | −1.2285 |
| H-acceptors | 5 | 5 | 2 | 13 |
| H-donors | 1 | 1 | 1 | 5 |
| Total surface area | 333.67 | 319.91 | 129.66 | 333.45 |
| Relative PSA | 0.144 | 0.1502 | 0.2016 | 0.46409 |
| Polar surface area | 50.8 | 50.8 | 37.3 | 210.54 |
| Druglikeness | 2.9034 | 3.6092 | −2.6221 | −7.5894 |
| Mutagenic/tumorigenic | None/none | None/none | None/none | None/high |
| Reproductive effective/irritant | None/none | Low/none | High/none | None/none |
None of the compounds are expected to induce carcinogenesis. Only VPA is identified as a CYP2C9 substrate, as previously reported, while the VPA derivatives could potentially act as substrates for CYP2D6, as well as for CYP3A4, including MTX. However, none of the compounds are predicted to induce CYP inhibition. Nonetheless, they may potentially induce OATP1B1 and OATP1B3 inhibition, which could lead to drug accumulation and increased drug–drug interactions. However, they do not appear to inhibit OCT1 and OCT2, which are transporters for cationic substrates. The VPA derivatives and MTX were predicted to be substrates for P-glycoprotein, which could potentially reduce their efficacy by pumping the drug out of the cell and away from the target site, leading to drug resistance.44 In terms of plasma protein binding, the VPA derivatives and MTX exhibited similar results, contrary to VPA. Also, is important to consider the subcellular localization of the compounds due subcellular organelles have different functions and microenvironments. The results obtained showed that all compounds could be localized in the mitochondria (Table 1), where the major biological function is related with energy conversion and storage of calcium ions.45 However, this physiological process could be altered by the compounds and then allow that mitochondria release cyt c, allowing the pathway caspase activation producing apoptosis or well by increased the reactive oxygen species (ROS) which induce oxidative stress, destroy cellular structure and mitochondrial outer membrane permeability, and lead to apoptosis.46,47 Therefore, the applications of in silico methods play key roles in the prediction of chemical subcellular localization due to their low costs and high performance.
Regarding to the Lipinski rule, the molecular weights of all compounds fall within the allowable range of 500 g mol−1. The clogP values are lower than 5 for F2S4-p-VPA, F3S4-m-VPA and VPA aligning with Lipinski's criteria. However, MTX has a negative cLogP, indicating a preference for the aqueous phase, with a high percent (60–85%) of MTX excreted when administrated intravenously.48 About hydrogen bond donors and acceptors only MTX exceeds the Lipinski rule allowance for acceptors, with 13 acceptors compared to the allowed, while the VPA derivatives have 5 hydrogen bond acceptors. Additionally, MTX exhibits a larger polar surface area compared to VPA and its derivatives. Concerning mutagenic, tumorigenic, reproductive effects, and irritancy, only MTX and VPA have a high probability, as indicated in Table 1B. Complete results from both platforms can be found in the ESI (Table 1S and 2S†).
4. Discussion
Glioblastoma multiforme (GBM), a highly aggressive and malignant central nervous system cancer, is a leading cause of death globally, alongside other aggressive malignancies like triple-negative breast cancer (TNBC).49–51 GBM is characterized by its migratory and invasive properties and classified by the World Health Organization (WHO) into grades II–IV, with GBM representing grade IV.52,53 The urgent need for new drug options and evaluation methods for these aggressive cancers can be addressed by artificial intelligence (AI). Specifically, radiomics combined with machine learning (ML) offers a promising approach for analyzing imaging data, such as magnetic resonance imaging (MRI). While traditional image analysis is labor-intensive due to manual annotation, deep learning (DL), a subset of ML, can automate this process. DL is crucial for developing clinical evaluation models, detecting glioblastoma recurrence, and enabling tumor segmentation and feature extraction via algorithms like convolutional neural networks (CNNs). These advancements are vital for developing personalized therapeutic strategies.54In drug design, modifying ligands to enhance their efficacy is crucial for developing improved anticancer agents. Valproic acid (VPA) has shown promise for treating various cancers, including glioblastoma. However, its effectiveness often requires high millimolar concentrations to significantly reduce cell viability and proliferation.25 To overcome this limitation, our work proposes novel VPA derivatives, specifically F2S4-p-VPA (containing piperidine) and F3S4-p-VPA (containing pyrrolidine). These organic heterocyclic amines are known for their potential anticancer activity.55–57 We hypothesized that linking VPA with these amines would yield superior antiproliferative effects, given their structural relation to existing anticancer drugs like methotrexate (MTX).
The evaluation of VPA derivatives and MTX on U373 and LN-18 (GBM) and MDA-MB-231 (TNBC) cell lines revealed that F2S4-p-VPA exhibited superior cytotoxic effects compared to F3S4-m-VPA and MTX. Notably, F2S4-p-VPA selectively reduced the viability of GBM cells while showing minimal impact on healthy 3T3-L1 fibroblasts. This enhanced antiproliferative effect on TNBC cells might stem from the overexpression of HDAC8 in TNBC, an enzyme known to be inhibited by VPA.58,59 The IC50 values for F2S4-p-VPA (112 mM on LN-18; 142 mM on MDA-MB-231) indicate a stronger cytotoxic effect than both MTX and VPA (for comparison, VPA's IC50 on MDA-MB-231 is 7.29 mM).60 Furthermore, MTX's efficacy against MDA-MB-231 is limited due to cellular resistance requiring high concentrations,61,62 and its primary mode of action is cytostatic.63 Our findings for F2S4-p-VPA align with previous reports of other piperidine-derived compounds showing μM-range IC50 values against MDA-MB-231.64
The effects of VPA derivatives and MTX on cell migration were compared, using LPS as an exogenous stimulus. This is particularly relevant as TNBC cells, like MDA-MB-231, express toll-like receptor 4 (TLR4), the lipopolysaccharide (LPS) is an agonist of TLR4 then it promotes tumor migration, invasion, and metastasis. LPS has been shown to increase invasion and migration in MDA-MB-231 and MCF-7 cells.65 Furthermore, LPS upregulates the tumor-associated protein B7–H6, which is rarely found in normal tissues but is expressed in various tumor cells, including neuroblastoma and breast cancer. B7–H6 enhances glioma cell proliferation, migration, and invasion by modulating vimentin, N-cadherin, MMP-2, and MMP-9 expression66,67 and can be induced by inflammatory factors like TLR, TNF-α, and IL-1β.68 The migration assays showed that F2S4-p-VPA effectively halted the migration process even in the presence of LPS. At 187 and 225 mM of F2S4-p-VPA with LPS, the compound prevented cell migration, though its effectiveness varied depending on the cell; migration was more significantly reduced in MDA-MB-231 and LN-18 cells compared to U373 cells. These findings are important, as piperidine-containing compounds, such as piperine, are known to inhibit TNBC cell migration, suggesting that molecules with this heterocycle hold significant potential for inhibiting cancer cell migration and invasion.69
Differences in Annexin V-FITC/PI double staining assay were observed between the LN-18 and MDA-MB-231 cell lines treated with VPA derivatives and MTX as a positive control. The percentage of live cells in the control sample (untreated cells) was approximately 80%; however, upon addition of the compounds, it decreased, indicating cellular death. The concentration of the compounds used were around their IC50 values to maintain high quantities of cells. Nevertheless, the percentages obtained are consistent with those reported in other studies using MDA-MB-231 cells.70 In MDA-MB-231 cells, changes in early and late apoptosis were observed when VPA derivatives were used. Apoptosis assays showed increased levels of early apoptosis with F3S4-m-VPA and increased levels of late apoptosis with F2S4-p-VPA indicating cell death via apoptosis. However, LN-18 cells highlighted sensitivity to F2S4-p-VPA, inducing higher levels of both early and late apoptosis compared to MDA-MB 231 cells. Although the compounds exhibit differences in early and late apoptosis, there are reports suggesting that apoptotic cells are functionally equivalent throughout the cell death process, there are not fully membrane integrity, as the signals or biomarkers induced by late apoptotic cells are as potent as those induced by early apoptotic cells.71 Therefore, the treatments with these compounds resulted in noteworthy increases in the percentage of cells undergoing early apoptosis in both cell lines compared to the control groups. These findings are statistically significant (p < 0.0001) and imply that F2S4-p-VPA and F3S4-m-VPA, induce apoptosis, consistent with previous results demonstrating that piperidine and its derivatives such as 1-(2-(4-(dibenzo[b,f]thiepin-10-yl)phenoxy)ethyl)piperidine (DTPEP) increase cytochrome c and Bax and decrease bcl-2 in MDA-MB-231 and MCF-7 cells.72 In addition, pyrrolidine derivatives have also shown anticancer properties, including spiro derivatives, N-substituted pyrrolidines, metal complexes, poly substituted derivatives, and coumarin derivatives of pyrrolidine. These compounds have demonstrated IC50 values in the μM range and have been evaluated in various cancer cells such as HCT116, PC-3, HepG2, FaDu, COS-7, A-549, Huh-7, MV, MCF-7, HT-29, SW-480, SH-SY5Y, MDA-MB-231, K562, HL60, SK-OV-3, Hela, BGC-823, NCI–H1650, A2780, and MDA-MB-233.73
While piperidine derivatives are recognized as promising anticancer agents for prostate, lung, and breast cancers, their application in glioblastoma remains less explored.72,73 To address this, we further investigated cell cycle arrest and Bax/Bcl-2 protein expression in LN-18 cells. The cell cycle analysis revealed distinct effects of MTX and F2S4-p-VPA on 3T3-L1 and LN-18 cells. In 3T3-L1 fibroblasts, both treatments decreased the G0-G1 population and slightly increased S and G2-M phases, suggesting continued cell cycle progression. Conversely, in LN-18 cells, MTX significantly increased the Sub G0 population (48.7%), indicating substantial cell death. Although F2S4-p-VPA also increased cell death, this was not statistically significant. Both treatments decreased the G0–G1 population and increase the S phase in LN-18 cells, implying a loss of G0/G1 checkpoint control. However, only F2S4-p-VPA induced G2-M accumulation (36.7%). This suggests that F2S4-p-VPA induces a cell cycle arrest mechanism, consistent with previous reports for piperidine derivatives causing S or G2/M arrest. These findings indicate that F2S4-p-VPA may promoting G2/M arrest even in p53-deficient cells, which could be highly beneficial for treating resistant tumors.72,74
The determination of Bax and Bcl-2 on the LN-18 cell line with MTX and F2S4-p-VPA did not induce a marked activation of Bax, which is consistent with the previously reported p53 mutation in this cell line, as this transcription factor directly regulates Bax expression.75 Under MTX, most cells were concentrated in Q4 (−Bax/−Bcl-2), suggesting a cytotoxic response without specific activation of the intrinsic apoptotic pathway. In contrast, F2S4-p-VPA increased the proportion of cells in quadrant Q3 (−Bax/+Bcl-2), potentially reflecting the activation of cellular resistance mechanisms through Bcl-2 overexpression.76 The control group showed a co-expression of Bax and Bcl-2, possibly reflecting a balance between pro- and anti-apoptotic signals.
These findings suggest that the intrinsic apoptotic pathway is not the primary mechanism of cell death induced by our treatments, implying the involvement of other pathways, such as the extrinsic route. This contrasts with Zhang et al.'s findings, who observed that taxol treatment in LN-18 cells increased Bax and decreased Bcl-2 expression, favoring mitochondrial pathway-mediated apoptosis.77 Then, as was previously mentioned that the compound F2S4-p-VPA could induce apoptosis through of others Bax-independent pathways including the extrinsic pathway of apoptosis, (Fas/TNF-α/TRAIL)NoDISC → caspasa-8 → caspasa-3) then other assays are need to identify the mechanism by which F2S4-p-VPA induced apoptosis.78 While MTX has been reported to alter Bax/Bcl-2 levels (increasing Bax, decreasing Bcl-2) in human hepatocyte (CRL-11233) cells,79 specific information on its effects in LN-18 cells is scarce. Therefore, future studies investigating F2S4-p-VPA's mechanism of cell death in cancer cells with varying p53 statuses (mutated vs. wild-type) would significantly enhance our understanding of its cellular mechanisms.
The prediction of ADMET properties suggests that VPA derivatives may exhibit hepatotoxicity. However, further evaluation in vivo is necessary to confirm this, as different information is obtained from two software programs (AdmetSar and DataWarrior; see ESI†). Moreover, the prediction indicates that VPA derivatives might be capable of crossing the BBB, which is promising for glioblastoma treatment. Considering the Lipinski rule, the VPA derivatives demonstrate better physicochemical behaviour than MTX. This is because MTX has a negative cLogP indicating a preference for hydrophilic media and potential elimination from the body. Additionally, MTX has a higher number of H-acceptors and H-donors compared to VPA derivatives. The subcellular localization of MTX, VPA, F2S4-p-VPA and F3S4-m-VPA was predicted by in silico analyses in the mitochondria, despite that the subcellular distribution can be determinate by experimental methods employing fluorescence microscopy or histochemistry among other, these are expensive and require equipment and special methodologies.45 Then, employs in silico prediction of chemical subcellular localization would be practicable and helpful for molecular design and reduction of potential toxicity, due to in silico methods take in consideration the physicochemical properties of the molecules which are related with their chemical structures.80,81 Ultimately, after confirming the efficacy of synthesized molecules, experimental determination of subcellular localization and ADMET properties can definitively validate these predictions.
In conclusion, our findings suggest that piperidine and VPA-derived compounds, particularly F2S4-p-VPA, represent promising candidates for cancer treatment, including for p53-mutated cancers. Given that F2S4-p-VPA combines two important pharmacophores (piperidine and VPA), further investigations into its therapeutic potential are warranted.
5. Conclusion
The present study aimed to investigate the potential activity of newly designed VPA derivatives, which exhibit better physicochemical and ADMET properties than MTX and VPA. Among these compounds, F2S4-p-VPA demonstrated the most potent cytotoxic effect and effectively inhibited cell migration. Therefore, F2S4-p-VPA holds promise as a potential treatment for GBM, especially considering its predicted capability to cross the blood-brain barrier. However, further research is warranted to elucidate the mechanism of action of F2S4-p-VPA.Glossary
Glioblastoma: an aggressive type of brain cancer. Triple-negative breast cancer (TNBC): a subtype of breast cancer that lacks estrogen, progesterone, and HER2 receptors. Methotrexate (MTX): a common chemotherapeutic agent with low water solubility and cytostatic effects. Valproic acid (VPA): a chemotherapeutic agent with low potency, used to limit tumor cell proliferation. ADMET: an acronym for absorption, distribution, metabolism, excretion, and toxicity. MTT assay: a colorimetric assay for assessing cell metabolic activity, often used to measure cell proliferation and cytotoxicity. Annexin V-FITC/PI apoptosis assay: a method to detect apoptotic cells by using Annexin V conjugated to FITC (fluorescein isothiocyanate) and propidium iodide (PI) staining. One-way ANOVA (analysis of variance): a statistical method used to compare the means of three or more samples to determine if at least one sample mean is significantly different from the others. Dunnett's multiple comparison test: a statistical test used to compare multiple treatment groups to a single control group. Wound-healing assay: an in vitro test to study cell migration and the effects of treatments on cell movement. ESI (Electrospray Ionization): a technique used in mass spectrometry to produce ions. Q-TOF (Quadrupole Time-of-Flight): A type of mass spectrometer that combines a quadrupole mass analyzer with a time-of-flight mass analyzer.Abbreviations
| TNBC | Glioblastoma and triple-negative breast cancer |
| VPA | Valproic acid |
| HER2 | Human epidermal growth factor-2 |
| GBM | Glioblastoma multiforme |
| LPS | lipopolysaccharide |
| PMA | Phorbol 12-myristate 13-acetate |
| MTX | Methotrexate |
| HDACIs | Histone deacetylase inhibitors |
| U373 | Human glioblastoma astrocytoma |
| MDA-MB-231 | Invasive ductal carcinoma |
| EGF | Epidermal growth factor |
| TGF-α | Transforming growth factor alpha |
| LN-18 | Human malignant glioma |
| 3T3-L1 | Murine preadipocyte cell line |
Data availability
If additional ESI† is required, interested parties can obtain it by contacting the first author. Requests should be directed to the following email address: E-mail: marcrh2002@yahoo.com.mx. Please include specific details about the material you need and any relevant context to ensure a prompt and accurate response.Author contributions
Conceptualization, data curation, formal analysis, supervision, investigation, writing-original draft, resources, methodology, project administration, funding acquisition, writing-review, and editing. M. C. R. H. conceptualization, data curation, formal analysis, supervision, writing-original draft. R. H. C. L. investigation, data curation, writing-review, and editing. M. O. V. methodology, investigation, data curation, formal analysis. L. G. F. M. investigation, methodology, writing-original draft. I. I. P. M. formal analysis, supervision. M. T. R. supervision, investigation. M. G. V. methodology, investigation, formal analysis, supervision. R. F. M. methodology, formal analysis, supervision J. C. B. conceptualization, funding acquisition, resources, writing-review.Conflicts of interest
The authors of this research have no known competing financial interests or personal relationships that could appear to influence the work reported in this paper.Acknowledgements
This work was supported by a project grant from CONAHCYT Ciencia Básica y/o de Frontera: Paradigmas y controversias de la ciencia [2022, 319355]; SIP-IPN multidiciplinario [20240059, 2297–2024]. The authors acknowledge Oscar Aurelio Martínez Romero and Alejandra Yoselín González Muñiz for their support in this research.References
- WHO report on cancer: setting priorities, investing wisely and providing care for all, https://www.who.int/publications-detail-redirect/9789240001299, Accessed March 26, 2025.
- H. Sung, J. Ferlay, R. L. Siegel, M. Laversanne, I. Soerjomataram, A. Jemal and F. Bray, Ca-Cancer J. Clin., 2021, 71, 209–249 CrossRef PubMed.
- Cancer Today, http://gco.iarc.fr/today/home, Accessed March 26, 2025.
- Instituto Nacional de Estadística y Geografía, Estadísticas a propósito del día Mundial contra el Cáncer 2021, https://www.inegi.org.mx/app/saladeprensa/noticia.html?id=6338, Accessed March 26, 2022.
- J. Q. Beltrán, J. E. Soto-Abraham, J. Vidaurreta-Serrano, L. G. C. Macias, E. G. Apo and E. Ogando-Rivas, J. Neurosci. Rural Pract., 2018, 9, 516–521 CrossRef PubMed.
- A. D'Alessio, G. Proietti, G. Sica and B. M. Scicchitano, Cancers, 2019, 11, 469 CrossRef PubMed.
- G. Chandrika, K. Natesh, D. Ranade, A. Chugh and P. Shastry, Sci. Rep., 2016, 6, 22455 CrossRef CAS PubMed.
- L. Gómez-Caudillo, A. J. Ortega-Lozano, Á. G. Martínez-Batallar, H. Rosas-Vargas, F. Minauro-Sanmiguel and S. Encarnación-Guevara, Oncol. Rep., 2020, 44, 661–673 CrossRef PubMed.
- G. I. Uscanga-Perales, S. K. Santuario-Facio, C. N. Sanchez-Dominguez, S. Cardona-Huerta, G. E. Muñoz-Maldonado, P. Ruiz-Flores, J. R. Barcenas-Walls, L. E. Osuna-Rosales, A. Rojas-Martinez, J. F. Gonzalez-Guerrero, J. Valero-Gomez, G. S. Gomez-Macias, A. Barbosa-Quintana, O. Barboza-Quintana, R. Garza-Guajardo and R. Ortiz-Lopez, Oncol. Lett., 2019, 17, 3581–3588 CAS.
- M. T. Arceo-Martínez, J. E. López-Meza, A. Ochoa-Zarzosa and Z. Palomera-Sanchez, Gac. Mex. Oncol., 2021, 20, 101–110 Search PubMed.
- X. Li, J. Yang, L. Peng, A. A. Sahin, L. Huo, K. C. Ward, R. O'Regan, M. A. Torres and J. L. Meisel, Breast Cancer Res. Treat., 2017, 161, 279–287 CrossRef PubMed.
- S. Valastyan and R. A. Weinberg, Cell, 2011, 147, 275–292 CrossRef CAS PubMed.
- F. Arvelo, F. Sojo and C. Cotte, E Cancer Med. Sci., 2016, 10, 617 Search PubMed.
- N. M. Novikov, S. Y. Zolotaryova, A. M. Gautreau and E. V. Denisov, Br. J. Cancer, 2021, 124, 102–114 CrossRef PubMed.
- J. Liu, P. C. Lin and B. P. Zhou, Curr. Pharm. Des., 2015, 21, 3032–3040 CrossRef CAS PubMed.
- S. Jain, P. Dash, A. P. Minz, S. Satpathi, A. G. Samal, P. K. Behera, P. S. Satpathi and S. Senapati, The Prostate, 2019, 79, 168–182 CrossRef CAS PubMed.
- M. A. Seol, J. H. Park, J. H. Jeong, J. Lyu, S. Y. Han and S.-M. Oh, Oncotarget, 2017, 8, 40190–40203 CrossRef PubMed.
- X. Wu, S. Qian, J. Zhang, J. Feng, K. Luo, L. Sun, L. Zhao, Y. Ran, L. Sun, J. Wang and F. Xu, Cancer Metab., 2021, 9, 23 CrossRef PubMed.
- S. A. A. Rizvi, Y. Shahzad, A. M. Saleh and N. Muhammad, Oncology, 2020, 98, 520–527 CrossRef CAS PubMed.
- V. Yang, M. J. Gouveia, J. Santos, B. Koksch, I. Amorim, F. Gärtner and N. Vale, RSC Med. Chem., 2020, 11, 646–664 RSC.
- C. W. Wu, H. C. Liu, Y. L. Yu, Y. T. Hung, C. W. Wei and G.-T. Yiang, Oncol. Rep., 2017, 37, 2177–2184 CrossRef CAS PubMed.
- D. V. Lopes, A. de Fraga Dias, L. F. L. Silva, J. N. Scholl, J. Sévigny, A. M. O. Battastini and F. Figueiró, Purinergic Signalling, 2021, 17, 273–284 CrossRef CAS PubMed.
- F. Figueiró, C. P. de Oliveira, L. S. Bergamin, L. Rockenbach, F. B. Mendes, E. H. F. Jandrey, C. E. J. Moritz, L. F. Pettenuzzo, J. Sévigny, S. S. Guterres, A. R. Pohlmann and A. M. O. Battastini, Purinergic Signalling, 2016, 12, 303–312 CrossRef PubMed.
- S. A. Brodie and J. C. Brandes, Expert Rev. Anticancer Ther., 2014, 14, 1097–1100 CrossRef CAS PubMed.
- W. Han and W. Guan, Front. Oncol., 2021, 11, 687362 CrossRef CAS PubMed.
- Y. Sixto-López, J. A. Gómez-Vidal and J. Correa-Basurto, Appl. Biochem. Biotechnol., 2014, 173, 1907–1926 CrossRef PubMed.
- A. Wawruszak, M. Halasa, E. Okon, W. Kukula-Koch and A. Stepulak, Cancers, 2021, 13, 3409 CrossRef CAS PubMed.
- D. Valiyaveettil, M. Malik, D. M. Joseph, S. F. Ahmed, S. A. Kothwal and M. Vijayasaradhi, South Asian J. Cancer, 2018, 7, 159–162 CrossRef PubMed.
- P. Prasad, H. Vasquez, C. M. Das, V. Gopalakrishnan and J. E. A. Wolff, J. Neurooncol., 2009, 91, 279–286 CrossRef CAS PubMed.
- K. Gotfryd, M. Hansen, A. Kawa, U. Ellerbeck, H. Nau, V. Berezin, E. Bock and P. S. Walmod, Basic Clin. Pharmacol. Toxicol., 2011, 109, 164–174 CrossRef CAS PubMed.
- W. Ding, D. Lim, Z. Wang, Z. Cai, G. Liu, F. Zhang and Z. Feng, DNA Repair, 2020, 95, 102940 CrossRef CAS PubMed.
- Z. Cai, D. Lim, B. Jia, G. Liu, W. Ding, Z. Wang, Z. Tian, J. Peng, F. Zhang, C. Dong and Z. Feng, Radiat. Med. Protect., 2023, 4, 204–213 CrossRef.
- A. Contis-Montes de Oca, E. Rodarte-Valle, M. C. Rosales-Hernández, E. Abarca-Rojano, S. Rojas-Hernández, M. J. Fragoso-Vázquez, J. E. Mendieta-Wejebe, A. M. Correa-Basurto, I. Vázquez-Moctezuma and J. Correa-Basurto, Oncotarget, 2018, 9, 33368–33381 CrossRef PubMed.
- C. Li, H. Chen, Q. Tan, C. Xie, W. Zhan, A. Sharma, H. Shanker Sharma and Z. Zhang, Prog. Brain Res., 2020, 258, 369–379 Search PubMed.
- D. Thotala, R. M. Karvas, J. A. Engelbach, J. R. Garbow, A. N. Hallahan, T. A. DeWees, A. Laszlo and D. E. Hallahan, Oncotarget, 2015, 6, 35004–35022 CrossRef PubMed.
- S. Berendsen, E. Frijlink, J. Kroonen, W. G. M. Spliet, W. van Hecke, T. Seute, T. J. Snijders and P. A. Robe, Neurooncol. Adv., 2019, 1, 1–8 Search PubMed.
- M. Farooq, A. El-Faham, S. N. Khattab, A. M. Elkayal, M. F. Ibrahim, N. A. Taha, A. Baabbad, M. A. Wadaan and E. A. Hamed, Asian Pac. J. Cancer Prev., 2014, 15, 7785–7792 CrossRef PubMed.
- I. D. García Marín, R. H. Camarillo López, O. A. Martínez, I. I. Padilla-Martínez, J. Correa-Basurto and M. C. Rosales-Hernández, PLoS One, 2022, 17, e0269129 CrossRef PubMed.
- V. Patil, J. Pal and K. Somasundaram, Oncotarget, 2015, 6, 43452–43471 CrossRef PubMed.
- A. Rahimian and A. Mellati, Razavi Int. J. Med., 2017, 5, e55455 CrossRef.
- P. V. Dludla, B. Jack, A. Viraragavan, C. Pheiffer, R. Johnson, J. Louw and C. J. F. Muller, Toxicol Rep, 2018, 5, 1014–1020 CrossRef CAS PubMed.
- ImageJ, https://imagej.nih.gov/ij/index.html, accessed February 23, 2022.
- R. Rodríguez-Pérez and G. Gerebtzoff, Artif. Intell. Life Sci., 2021, 1, 100027 Search PubMed.
- A. J. Ii, A. H. Aa, A. H. Kb and C. H. At, Heliyon, 2022, 8, e09777 CrossRef PubMed.
- N. Zheng, H. N. Tsai, X. Zhang and G. R. Rosania, Mol. Pharm., 2011, 8, 1619–1628 CrossRef CAS PubMed.
- L. Yuan, C. Wang, D. Lu, X. Zhao, L. Tan and X. Chen, Aging, 2020, 18, 3662–3681 CrossRef PubMed.
- T. Chuwen, L. Yifan, L. Zhuoshu, Z. Ping and Z. Mingyi, Front. Cell Dev. Biol., 2022, 10, 832356 CrossRef PubMed.
- J. K. Aronson, Side Eff. Drugs Annu., 2014, 35, 821–861 Search PubMed.
- World Health Organization, Cancer, 2022, https://www.who.int/news-room/fact-sheets/detail/cancer, Accessed February 21, 2022.
- C. Hagemann, J. Anacker, S. Haas, D. Riesner, B. Schömig, R.-I. Ernestus and G. H. Vince, BMC Res. Notes, 2010, 3, 293 CrossRef PubMed.
- Y. Akiyama, M. Komiyama, H. Miyata, M. Yagoto, T. Ashizawa, A. Iizuka, C. Oshita, A. Kume, M. Nogami, I. Ito, R. Watanabe, T. Sugino, K. Mitsuya, N. Hayashi, Y. Nakasu and K. Yamaguchi, Oncol. Rep., 2014, 31, 1683–1690 CrossRef PubMed.
- M. Akmal, N. Hasnain, A. Rehan, U. Iqbal, S. Hashmi, K. Fatima, M. Z. Farooq, F. Khosa, J. Siddiqi and M. K. Khan, World Neurosurg., 2020, 136, 270–282 CrossRef PubMed.
- T. Komori, Neurol. Med. Chir., 2017, 57, 301–311 CrossRef PubMed.
- P. Chen, P. Wang and B. Gao, Sci. Prog., 2024, 107, 368504231223353 CrossRef PubMed.
- M. M. Abdelshaheed, I. M. Fawzy, H. I. El-Subbagh and K. M. Youssef, Future J. Pharm. Sci., 2021, 7, 188 CrossRef.
- P. Goel, O. Alam, M. J. Naim, F. Nawaz, M. Iqbal and M. I. Alam, Eur. J. Med. Chem., 2018, 157, 480–502 CrossRef CAS PubMed.
- J. Ji, F. Sajjad, Q. You, D. Xing, H. Fan, A. G. K. Reddy, W. Hu and S. Dong, Arch. Pharm., 2020, 353, 2000136 CrossRef CAS PubMed.
- G. Rahmani, S. Sameri, N. Abbasi, M. Abdi and R. Najafi, Pathol., Res. Pract., 2021, 220, 153396 CrossRef CAS PubMed.
- A. Furugen, Y. Kanno, N. Ohyama, Y. Kurosawa, N. Jinno, K. Narumi, K. Iseki and M. Kobayashi, Drug Metab. Pharmacokinet., 2021, 40, 100409 CrossRef CAS PubMed.
- A. R. Estrada-Pérez, J. B. García-Vázquez, H. L. Mendoza-Figueroa, M. C. Rosales-Hernández, C. Fernández-Pomares and J. Correa-Basurto, Int. J. Mol. Sci., 2023, 24, 14543 CrossRef PubMed.
- J. Raut, O. Sarkar, T. Das, S. M. Mandal, A. Chattopadhyay and P. Sahoo, Sci. Rep., 2023, 13, 21899 CrossRef CAS PubMed.
- M. Lindgren, K. Rosenthal-Aizman, K. Saar, E. Eiríksdóttir, Y. Jiang, M. Sassian, P. Östlund, M. Hällbrink and Ü. Langel, Biochem. Pharmacol., 2006, 71, 416–425 CrossRef CAS PubMed.
- R. Seigers, S. B. Schagen, W. Beerling, W. Boogerd, O. van Tellingen, F. S. A. M. van Dam, J. M. Koolhaas and B. Buwalda, Behav. Brain Res., 2008, 186, 168–175 CrossRef CAS PubMed.
- A. Arun, M. i. Ansari, P. Popli, S. Jaiswal, A. k. Mishra, A. Dwivedi, K. Hajela and R. Konwar, Cell Proliferation, 2018, 51, e12501 CrossRef CAS PubMed.
- H. Yang, B. Wang, T. Wang, L. Xu, C. He, H. Wen, J. Yan, H. Su and X. Zhu, PLoS One, 2014, 9, e109980 CrossRef PubMed.
- H. Wang, H. Zhi, D. Ma and T. Li, Cytokine, 2017, 92, 93–102 CrossRef CAS PubMed.
- X. Wang and R. A. Khalil, Adv. Pharmacol., 2018, 81, 241–330 CAS.
- F. Che, X. Xie, L. Wang, Q. Su, F. Jia, Y. Ye, L. Zang, J. Wang, H. Li, Y. Quan, C. You, J. Yin, Z. Wang, G. Li, Y. Du and L. Wang, Int. Immunopharmacol., 2018, 59, 318–327 CrossRef CAS PubMed.
- A. L. Greenshields, C. D. Doucette, K. M. Sutton, L. Madera, H. Annan, P. B. Yaffe, A. F. Knickle, Z. Dong and D. W. Hoskin, Cancer Lett., 2015, 357, 129–140 CrossRef CAS PubMed.
- J. I. Noh, S. K. Mun, E. H. Lim, H. Kim, D. J. Chang, J. S. Hur and S. T. Yee, J. Fungi, 2021, 7, 188 CrossRef CAS PubMed.
- V. A. Patel, A. Longacre, K. Hsiao, H. Fan, F. Meng, J. E. Mitchell, J. Rauch, D. S. Ucker and J. S. Levine, J. Biol. Chem., 2006, 281, 4663–4670 CrossRef CAS PubMed.
- S. Mitra, U. Anand, N. K. Jha, M. S. Shekhawat, S. C. Saha, P. Nongdam, K. R. R. Rengasamy, J. Proćków and A. Dey, Front. Pharmacol., 2022, 12, 772418 CrossRef PubMed.
- A. A. Bhat, I. Singh, N. Tandon and R. Tandon, Eur. J. Med. Chem., 2023, 246, 114954 CrossRef CAS PubMed.
- V. Srivastava, M. Y. Wani, A. S. Al-Bogami and A. Ahmad, J. Adv. Res., 2021, 29, 121–135 CrossRef CAS PubMed.
- I. Wybranska, A. Polus, M. Mikolajczyk, A. Knapp, A. Sliwa, B. Zapala, T. Staszel and A. Dembinska-Kiec, Hum. Cell, 2013, 26, 137–148 CrossRef CAS PubMed.
- T. J. McDonnell, A. Beham, M. Sarkiss, M. M. Andersen and P. Lo, Cell. Mol. Life Sci., 1996, 52, 1008–1017 CrossRef CAS PubMed.
- R. Zhang, N. L. Banik and S. K. Ray, Brain Res., 2008, 1239, 216–225 CrossRef CAS PubMed.
- S. Shen, Y. Shao and C. Li, Cell Death Discovery, 2023, 9, 284 CrossRef PubMed.
- A. G. Kabakci, H. Kaplan, E. Şingirik and M. G. Bozkır, J. Pharm. Res. Int., 2021, 33, 2227–2236 CrossRef.
- H. Yang, X. Li, Y. Cai, Q. Wang, W. Li, G. Liu and Y. Tang, Med. Chem. Commun., 2017, 8, 1225–1234 RSC.
- N. Zheng, H. N. Tsai, X. Zhang, K. Shedden and G. R. Rosania, Mol. Pharm., 2011, 8, 1611–1618 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Appendix A. Supplementary data. See DOI: https://doi.org/10.1039/d5ra01365h |
| ‡ These authors participated equal. |
| This journal is © The Royal Society of Chemistry 2025 |