
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHigh-throughput screening of FDA-approved drugs identifies colchicine as a potential therapeutic agent for atypical teratoid/rhabdoid tumors (AT/RTs)†
Phongthon Kanjanasiriratabc,
Kedchin Jearawuttanakulc,
Sawinee Seemakhanc,
Suparerk Borwornpinyocd,
Patompon Wongtrakoongateef,
Suradej Hongengcg and
Sitthivut Charoensutthivarakul *acf
*acf
aSchool of Bioinnovation and Bio-based Product Intelligence, Faculty of Science, Mahidol University, Bangkok, 10400, Thailand. E-mail: sitthivut.cha@mahidol.ac.th; Tel: +66-2-201-5899
bDepartment of Pathobiology, Faculty of Science, Mahidol University, Bangkok, 10400, Thailand
cExcellent Center for Drug Discovery (ECDD), Faculty of Science, Mahidol University, Bangkok, 10400, Thailand
dDepartment of Biotechnology, Faculty of Science, Mahidol University, Bangkok, 10400, Thailand
eDepartment of Biochemistry, Faculty of Science, Mahidol University, Bangkok, 10400, Thailand
fCenter for Neuroscience, Faculty of Science, Mahidol University, Bangkok, 10400, Thailand
gDepartment of Pediatrics, Faculty of Medicine Ramathibodi Hospital, Mahidol University, Bangkok, 10400, Thailand
First published on 17th April 2025
Abstract
Atypical teratoid/rhabdoid tumor (AT/RT) is a rare and aggressive tumor of the primary central nervous system primarily affecting children. It typically originates in the cerebellum and brain stem and is associated with a low survival rate. While standard chemotherapy has been used as a primary treatment for AT/RTs, its success rate is unsatisfactory, and patients often experience severe side effects. Therefore, there is an urgent need to develop new and effective treatment strategies. One promising approach for identifying new therapies is drug repurposing. Although many FDA-approved drugs have been repurposed for various cancers, there have been no reports of such applications for AT/RTs. In this study, a library of 2130 FDA-approved drugs was screened using a high-throughput screening system against 2D traditional cultures and 3D spheroid cultures of AT/RT cell lines (BT-12 and BT-16). From this screening, colchicine, a non-chemotherapeutic agent, was identified as a promising candidate. It exhibited IC50 values of 0.016 and 0.056 μM against 2D BT-12 and 2D BT-16 cells, respectively, and IC50 values of 0.004 and 0.023 μM against 3D BT-12 and BT-16 spheroid cultures. Additionally, the cytotoxic effects of colchicine on human brain endothelial cells and human astrocytes were evaluated, and CC50 > 20 μM was observed, which is over two orders of magnitude higher than its effective concentrations in AT/RT cells, indicating considerably lower toxicity to normal brain cells and brain endothelial cells. In conclusion, colchicine shows significant potential to be repurposed as a treatment for AT/RTs, providing a safer and more effective therapeutic option for this rare and challenging disease.
Introduction
Atypical teratoid/rhabdoid tumor (AT/RT) is a rare central nervous system (CNS) tumor that primarily affects children, particularly those under the age of 3 years. This type of tumor is classified as an atypical teratoid tumor due to its teratoid malignant properties, which arise from the abnormal development of pluripotent cells, such as germ cells and embryonal cells.1 Moreover, AT/RTs exhibit a prominent feature of rhabdoid morphology, characterized by sheets and clusters of large epithelioid cells with vesicular paranuclear inclusions and associated with a high tumor grade.2 These characteristics give the tumor its name, atypical teratoid/rhabdoid tumor (AT/RT).1 The primary cause of AT/RTs is the deletion of chromosome 22 at the Chr22q11.2 region, which contains the SMARCB1 tumor suppressor gene.3AT/RTs typically develop in two major areas of the brain: the cerebellum and brain stem.4 The cerebellum, located at the base of the brain, has a functional role in controlling movement, balance, and posture, while the brain stem controls breathing, heart rate, and muscle activities involved in seeing, hearing, walking, talking, and eating.5 Therefore, symptoms of AT/RTs often include nausea, vomiting, morning headache, drowsiness, and a loss of balance and movement such as eye or facial muscle weakness.6
Due to its rarity, the incidence of AT/RTs is often underestimated. The overall incidence rate is approximately 0.07 per 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 00000, with the highest incidence rate (0.54 per 100000) occurring in children under 1 year of age and the lowest rate (0.01 per 100000) in individuals aged 6–19 years.7 Despite its rarity, AT/RTs have a poor prognosis, with a global 4-year survival rate of just 43%.8 Currently, there is no well-defined gold standard treatment for AT/RTs. Chemotherapy is the primary treatment option, involving drugs such as vincristine, methotrexate, etoposide, cyclophosphamide, cisplatin, and doxorubicin.9–11 However, these drugs cause severe side effects including hair loss, diarrhea, nausea and vomiting, weakness, cardiac dysfunction, and abnormal neurological development.12 For this reason, there is an urgent need for new and more effective therapies.
00000, with the highest incidence rate (0.54 per 100000) occurring in children under 1 year of age and the lowest rate (0.01 per 100000) in individuals aged 6–19 years.7 Despite its rarity, AT/RTs have a poor prognosis, with a global 4-year survival rate of just 43%.8 Currently, there is no well-defined gold standard treatment for AT/RTs. Chemotherapy is the primary treatment option, involving drugs such as vincristine, methotrexate, etoposide, cyclophosphamide, cisplatin, and doxorubicin.9–11 However, these drugs cause severe side effects including hair loss, diarrhea, nausea and vomiting, weakness, cardiac dysfunction, and abnormal neurological development.12 For this reason, there is an urgent need for new and more effective therapies.
Drug repurposing has emerged as a promising and effective approach to identify new therapeutic agents. FDA-approved drugs are suitable for repurposing due to their safety profiles.13,14 Several FDA-approved drugs have already been repurposed for anticancer activity.13 For example, itraconazole, an antifungal drug, has shown strong anticancer activities in lung and prostate cancers15,16 by inhibiting mTOR activity and VEGFR2 glycosylation, which reduces angiogenesis.17,18 Nelfinavir, an anti-HIV drug, has demonstrated activity against pancreatic cancer by targeting the PI3K/AKT signaling pathway.19 However, the repurposing of FDA-approved drugs for AT/RTs has not been previously reported.
The in vitro screening of anticancer compounds has progressed beyond traditional 2D cell culture to include 3D spheroid culture. This approach has become increasingly prevalent in cancer research, including breast,20 cervical,20 colon,21 lung,22 pancreatic,23,24 and prostate cancers,25–27 among others. Compared to 2D models, 3D spheroids better mimic several features of solid tumors, such as their structural organization, biological responses, release of signaling molecules, gene expression patterns, and drug resistance. These distinctive and unique properties make 3D cell aggregates highly valuable as in vitro models for testing new anticancer therapies.28 Like solid tumors, the internal structure of spheroids consists of distinct cell layers.29,30 These include the proliferative zone in the outer layer, the quiescent zone in the middle layer, and the necrotic zone in the inner core.31 The layered structure of spheroids is a key factor in reducing the effectiveness of anticancer drugs and drug-loaded nanocarriers, as the deeper layers are less accessible to therapeutic agents. Therefore, the inclusion of 3D spheroid culture in anticancer research is a crucial step forward in developing new treatments.
The objective of this study is to identify an FDA-approved, non-chemotherapeutic drug that can effectively treat AT/RT tumor cells. To achieve this, an in-house library of 2130 FDA-approved drugs was screened using a high-throughput screening system against traditional 2D and 3D spheroid cultures of AT/RT cell lines. The cell lines used in this study, BT-12 and BT-16, are SMARCB1-deficient epithelial AT/RT cells generously provided by Dr Peter Houghton from St. Jude Children's Research Hospital.32 Although both cell lines originate from the same type of pediatric brain tumor, they exhibit different levels of chemosensitivity. BT-12 cells are more responsive to chemotherapeutic drugs, such as vincristine, cisplatin, doxorubicin, and etoposide, while BT-16 cells exhibit a higher level of resistance to these treatments.33,34 These differences in chemosensitivity make the two cell lines valuable for identifying effective FDA-approved drugs and obtaining significant data for potential therapies.
Results
Forty-two non-chemotherapeutic drugs identified as effective against AT/RT cells
Many FDA-approved drugs have been successfully repurposed for various types of anticancer activities. However, there has been no report on repurposing FDA-approved drugs specifically for AT/RTs. In this study, a library of 2130 FDA-approved drugs was screened against AT/RT cells. The initial single-concentration screening identified 117 and 169 out of 2130 compounds that inhibited the viability of BT-12 and BT-16 cells by more than 80%, respectively (Fig. 1A and B). When the effective compounds for both cell lines were merged, 104 hit compounds were identified (Fig. 1C and Table S1†). These 104 hit compounds were further classified into non-chemotherapeutic drugs and chemotherapeutic drugs. Amongst them, 42 compounds were identified as non-chemotherapeutic agents. These non-chemotherapeutic agents underwent secondary screening, including dose-dependent testing, to determine their IC50 values. | ||
| Fig. 1 High-throughput screening of 2130 FDA-approved drugs against AT/RT cells. The primary screening was conducted in BT-12 cells (A) and BT-16 cells (B) using FDA-approved drugs at a concentration of 10 μM. Hit compounds were identified based on their ability to inhibit cancer cell viability by over 80% indicated by the orange dotted line. A total of 104 hit compounds were effective against BT-12 cells and BT-16 cells as shown in (C). Amongst these, 62 compounds were classified as chemotherapeutic drugs (orange dots), while 42 compounds were identified as non-chemotherapeutic drugs (blue dots) (C). Doxorubicin served as the positive control (red dot), and DMSO served as the negative control (green dot). | ||
Analysis of the dose–response relationship of three selected FDA-approved drugs
From the primary screening, 42 non-chemotherapeutic drugs (Fig. 1C, blue dot) were investigated for their potential to act as anti-AT/RT agents. These compounds were assayed in a dose-dependent manner on BT-12 and BT-16 cells. Amongst them, three FDA-approved drugs, colchicine, digoxin, and emetine, exhibited significant effects on AT/RT cell viability, with IC50 values below 0.1 μM for both cell lines (Fig. 2A and B). Colchicine, an anti-inflammatory drug, was effective with IC50 values of 0.016 and 0.056 mM for BT-12 and BT-16 cells, respectively (Fig. 2B, C, and G). Meanwhile, digoxin, a cardiovascular drug, inhibited the proliferation of BT-12 and BT-16 cells at IC50 values of 0.080 and 0.076 mM, respectively (Fig. 2B, D, and H). Emetine, an antiprotozoal drug, also showed potential effects on BT-12 and BT-16 cells at IC50 values of 0.090 and 0.093 mM, respectively (Fig. 2B, E, and I). These findings suggested that colchicine, digoxin, and emetine have strong potential as anti-AT/RT agents.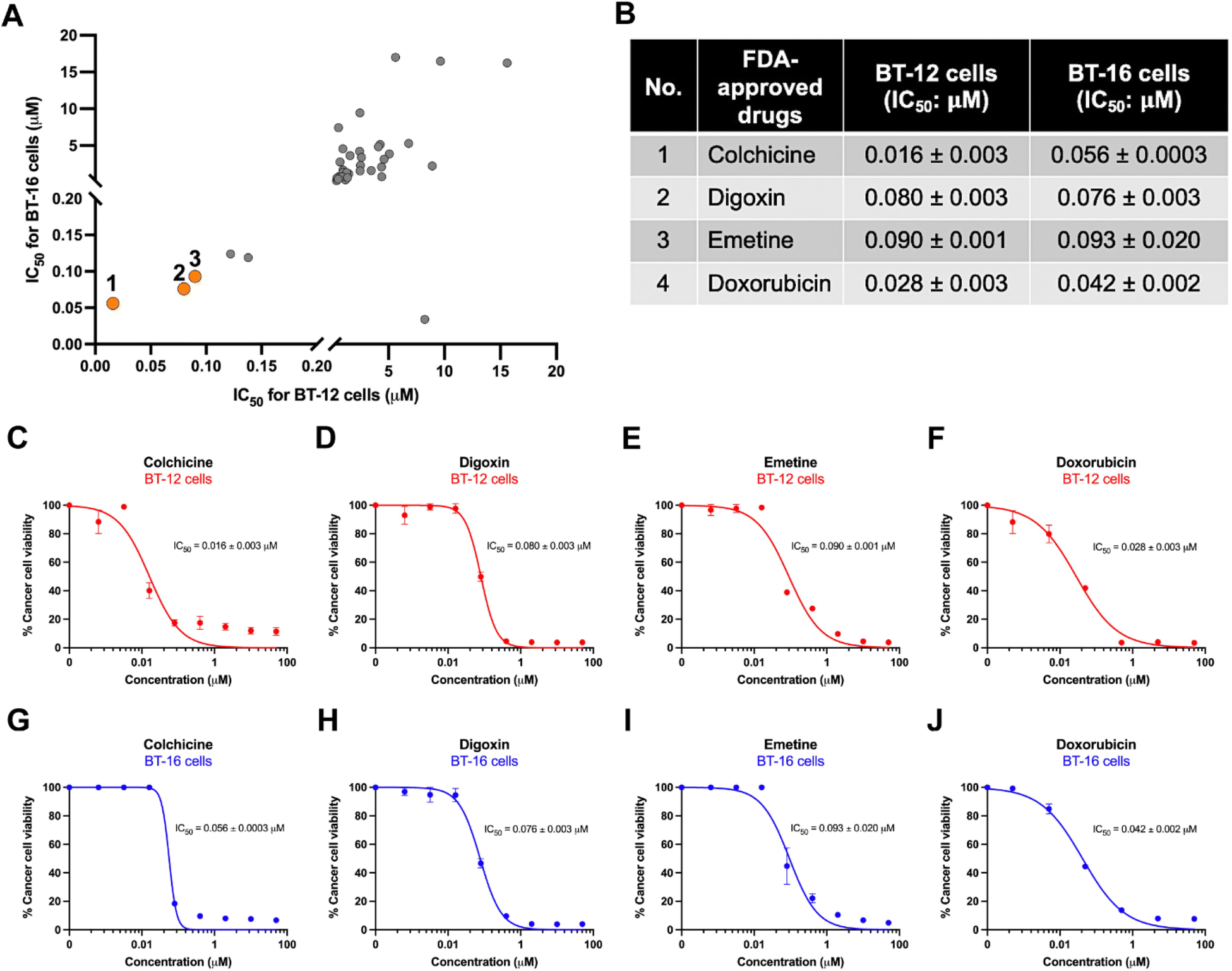 | ||
| Fig. 2 Dose–response effects of three FDA-approved drug candidates on AT/RT cells. (A) IC50 comparison graph between BT-12 cells and BT-16 cells highlighting the three drug candidates in orange dots. (B) IC50 values of three FDA-approved drugs affect AT/RT cell viability corresponding to the numbered orange dots in (A). (C–F) Dose-dependent effects on BT-12 cell viability for colchicine (C), digoxin (D), emetine (E), with doxorubicin as a control (F). (G–J) Dose-dependent effects on BT-16 cell viability for colchicine (G), digoxin (H), emetine (I), and doxorubicin (J). | ||
Colchicine demonstrates anti-AT/RT activity without cytotoxicity in brain endothelial and brain normal cells
The IC50 values of the hit compounds—colchicine, digoxin, and emetine—are critical to evaluating their potential as anti-AT/RT agents. However, assessing the CC50 of these compounds in normal cells is equally important to determine their therapeutic windows using the selectivity index (SI) criterion. The SI is calculated as the ratio of a compound's toxic concentration (CC50) to its effective bioactive concentration (IC50). Thus, cytotoxicity testing of these hit compounds in normal cells is essential to assess their safety profile. In this study, the concentration of colchicine that inhibits 50% of normal cell viability (CC50) was found to exceed 20 μM in human cerebral microvascular endothelial cells (hCMEC/D3 cells) and human normal brain cells (astrocytes). This CC50 value is over two orders of magnitude higher than its effective concentrations (IC50) against AT/RT cells, making it safer compared to other hit compounds (Fig. 3). These findings suggest that colchicine is relatively safe for normal brain cells and brain endothelial cells, which are likely to encounter the drug before its penetration into the brain. | ||
| Fig. 3 Cytotoxicity testing (CC50) of the hit compounds against the normal cells. (A–D) Dose-dependent cytotoxicity effects of colchicine (A), digoxin (B), emetine (C), and doxorubicin (D) on the viability of human cerebral microvascular endothelial cells (hCMEC/D3 cells). (E–H) Dose-dependent cytotoxicity effects of colchicine (E), digoxin (F), emetine (G), and doxorubicin (H) on the viability of primary astrocytes. | ||
The SI values of the hit compounds were evaluated and are presented in Table 1 for hCMEC/D3 cells and Table 2 for astrocytes. The analysis revealed that colchicine exhibited SI values exceeding 100 for both cell types, indicating a relatively large therapeutic window. By contrast, the SI values for digoxin and emetine were less than 100, which aligns with the SI values observed for the chemotherapeutic drug doxorubicin. These findings highlight colchicine as a safe candidate for further research in the treatment of AT/RTs.
| No. | FDA-approved drugs | BT-12 cells (IC50: μM) | BT-16 cells (IC50: μM) | hCMEC/D3 cells (CC50: μM) | SI (CC50/IC50) | |
|---|---|---|---|---|---|---|
| hCMEC/D3: BT-12 | hCMEC/D3: BT-16 | |||||
| 1 | Colchicine | 0.016 | 0.056 | >20 | >1250 | >357.143 |
| 2 | Digoxin | 0.080 | 0.076 | 0.101 | 1.263 | 1.329 |
| 3 | Emetine | 0.090 | 0.093 | 2.697 | 29.267 | 29 |
| 4 | Doxorubicin | 0.028 | 0.042 | 0.699 | 24.964 | 16.64 |
| No. | FDA-approved drugs | BT-12 cells (IC50: μM) | BT-16 cells (IC50: μM) | Astrocytes (CC50: μM) | SI (CC50/IC50) | |
|---|---|---|---|---|---|---|
| Astrocytes: BT-12 | Astrocytes: BT-16 | |||||
| 1 | Colchicine | 0.016 | 0.056 | >20 | >1250 | >357.143 |
| 2 | Digoxin | 0.080 | 0.076 | 0.102 | 1.275 | 1.342 |
| 3 | Emetine | 0.090 | 0.093 | 1.238 | 13.756 | 13.312 |
| 4 | Doxorubicin | 0.028 | 0.042 | 0.131 | 4.679 | 3.119 |
Colchicine inhibits cell growth in three-dimensional (3D) AT/RT spheroid models
In addition to its effects in 2D conventional cancer cell cultures, the effects of colchicine were also evaluated in 3D cancer spheroid models, which better mimic the behavior of cancer cells in the human body. The results of this study found that colchicine effectively inhibited the viability of 3D AT/RT spheroids at nanomolar concentrations. Colchicine demonstrated EC50 values of 14 and 37 nM for BT-12 and BT-16 spheroids, respectively (Fig. 4). Additionally, the EC50 value of colchicine in the BT-12 spheroid showed greater efficacy compared to doxorubicin (Fig. 4C). Meanwhile, the EC50 value of colchicine in the BT-16 spheroid displayed a similar effectiveness to that of doxorubicin (Fig. 4F). These findings suggest that colchicine exhibits significant efficacy in 3D AT/RT spheroid models, supporting its potential as a therapeutic agent for AT/RT treatment. | ||
| Fig. 4 Effect of colchicine on three-dimensional (3D) AT/RT spheroid growth models. BT-12 spheroids were treated with colchicine (A) or doxorubicin (B) at varying concentrations. The percentage effect of colchicine (red line) and doxorubicin (dark line) on BT-12 spheroid viability is shown in (C). Similarly, BT-16 spheroids were exposed to colchicine (D) or doxorubicin (E) at the stated concentrations. The percentage effect of colchicine (red line) and doxorubicin (black line) on BT-16 spheroid viability is presented in (F). Hoechst (green) and ethidium homodimer (orange) staining were used to distinguish live and dead cells, respectively. | ||
Discussion
The primary objective of this study was to repurpose a non-chemotherapeutic FDA-approved drug for anti-AT/RT activity. This study revealed that colchicine, an established anti-gout and anti-familial Mediterranean fever (FMF) drug, exhibits promising anti-AT/RT activity by inhibiting AT/RT cell proliferation. Colchicine, an anti-mitotic agent, demonstrated an IC50 value significantly lower than that of doxorubicin, a commonly used chemotherapeutic agent for AT/RT treatment. Colchicine, derived from Colchicum autumnale, exerts its anticancer activity by binding to microtubular ends and inhibiting the polymerization of microtubules, thus disrupting cell division. It is well-established that colchicine prevents mitotic progression by arresting cells in the metaphase. Colchicine's ability to attach to tubulins is one of its common modes of action in therapeutic activities.35 Microtubules are composed of α- and β-tubulin heterodimers, which are important cytoskeleton components. Colchicine binds to soluble tubulin to form the tubulin-colchicine (TC) complex, which subsequently binds to microtubular ends and disturbs tubulin lattice dynamics.36–38 Depending on its concentration, colchicine can either promote or inhibit microtubule polymerization. At lower concentrations, colchicine specifically inhibits microtubule polymerization, ultimately leading to cell cycle arrest and reduced tumor cell viability.35In this study, BT-12 cells were found to be more sensitive to colchicine than BT-16 cells. This observation is consistent with findings for another mitotic inhibitor, paclitaxel, which has demonstrated effectiveness against both AT/RT cell lines.33 Additionally, BT-12 cells showed significantly higher sensitivity to cisplatin than BT-16 cells,34 suggesting that BT-16 cells may exhibit a desensitized response to certain drugs relative to BT-12 cells. Higher proliferative activity has also been observed in BT-12 cells, as indicated by increased Ki-67 protein expression,39 a widely used marker for cell proliferation in human tumors.40 The process of cell proliferation depends heavily on microtubule polymerization, which plays a central role in maintaining the intracellular cytoskeletal framework. The dynamic nature of microtubule polymerization is particularly crucial for numerous cellular functions, especially in tumor cells.41 These factors likely contribute to the enhanced sensitivity of BT-12 cells to anti-mitotic agents such as colchicine and paclitaxel compared to BT-16 cells.
Additionally, colchicine was evaluated for its cytotoxicity in human brain endothelial cells and normal human astrocytes, and the results showed a CC50 value higher than 20 μM, which is more than two orders of magnitude higher than its IC50. This indicates that colchicine is relatively safe for normal brain cells and brain endothelial cells, critical components of the blood–brain barrier, which are among the first cell types exposed to drugs prior to their penetration into the brain. By contrast, doxorubicin displayed higher cytotoxicity with a considerably narrower therapeutic window compared to colchicine. These results strongly support the further investigation of colchicine as a safer and more effective anti-AT/RT agent. Beyond traditional in vitro 2D cell cultures, the anticancer activity of colchicine was also assessed in 3D cancer spheroid models, which more closely mimic the in vivo tumor microenvironment. These spheroid models are composed of heterogeneous cell populations that exhibit varying behavior depending on their levels of exposure to oxygen and nutrients. These include proliferating cells, quiescent cells, and hypoxic cells, which alter drug responsiveness, leading to changes in IC50 values that differ from those in 2D culture.42 This study demonstrated that colchicine effectively inhibited 3D AT/RT spheroid viability at nanomolar concentrations, 14 nM for BT-12 spheroid and 37 nM for BT-16 spheroid. These findings further validate colchicine's potential as a repurposed therapeutic agent for AT/RT treatment.
Studies on the effects of colchicine on 3D AT/RT spheroids have revealed that, even at higher concentrations, colchicine fails to completely eradicate cancer cells within the spheroid. This limitation is likely due to the enhanced drug resistance of cancer cells residing in the deeper layers of the spheroid. These cells may adapt by upregulating efflux transporter proteins, which protect them from the cytotoxic effects of drugs and other harmful agents.43–45 To address this issue, a co-treatment strategy combining colchicine with an efflux transporter inhibitor, such as verapamil, is proposed to enhance therapeutic efficacy. Furthermore, cancer cells located in the inner regions of the spheroid often transition from a highly proliferative state to a quiescent state, reducing ATP consumption required for processes such as cell cycle progression.46 This adaptation further limits the effectiveness of colchicine, which acts as an antimitotic agent by binding to tubulin heterodimers near the exchangeable GTP-binding site, disrupting microtubule polymerization. This destabilization ultimately induces programmed cell death, or apoptosis.47
Colchicine is well known for its therapeutic applications in treating gout48–50 and familial Mediterranean fever (FMF).51–53 Additionally, colchicine has been used to manage other conditions such as Behcet's disease (BD),54–56 pericarditis,57–59 coronary artery disease,60,61 and various inflammatory and fibrotic disorders.62,63 When colchicine is prescribed daily and long-term for FMF, the most common side effects (affecting up to 20% of patients) include abdominal pain, diarrhea, nausea, and vomiting. These symptoms are typically mild, temporary, and resolve when the dose is reduced. When used acutely for gout flares at doses up to 1.8 mg within 2 hours, diarrhea (23%) is the most frequently reported side effect.48 However, colchicine is considered safe at a dosage of 1.5 mg, although it was not effective in halting disease progression or reducing mortality in patients with severe COVID-19.64 For children under 5 years of age with FMF or other autoinflammatory diseases, a daily colchicine dose of 0.5 mg is considered safe.65,66 According to the 2007 guidelines, the recommended initial dosage varies by age: 0.5 mg per day for children aged under 5 years, 1.0 mg per day for those aged 5–10 years, and 1.5 mg per day for those aged over 10 years.66 Based on this dosing approach, colchicine may be a potential alternative treatment option for pediatric patients with AT/RT.
Numerous studies have highlighted the anticancer properties of colchicine across a variety of cancers. One such study investigated the effects of colchicine on human breast adenocarcinoma MCF-7 cells, showing that it inhibited MMP-2 mRNA expression and caused cell cycle arrest at the G2/M stage of mitosis, which led to reduced cell proliferation.67 Colchicine's efficacy against hypopharyngeal carcinoma has also been reported. Colchicine inhibited the formation and proliferation of human hypopharyngeal carcinoma cells in a dose-dependent manner. This was achieved through the suppression of key pathways involved in cancer progression.
Colchicine downregulated MMP9, μPA, and FAK/SRC signaling pathways, effectively reducing cell invasion, migration, and adhesion. Furthermore, colchicine induced apoptosis by activating caspase-3,68 preventing metastasis and promoting cancer cell death. In addition to breast and hypopharyngeal carcinoma, colchicine has shown a significant anticancer effect on gastric carcinoma cells in a dose-dependent manner. Colchicine promoted apoptosis in gastric carcinoma cells through the activation of caspase-3. This was mediated via the suppression of the PI3K/Akt/mTOR signaling pathway.69 Colchicine has also been studied against colorectal cancer cells, where it exhibits dose-dependent cytotoxic effects. Its administration led to an increase in ROS generation, activation of caspase-3, upregulation of pro-apoptotic protein BAX, and downregulation of the anti-apoptotic protein Bcl-2. These findings indicate that colchicine induces apoptosis, possibly via the intrinsic apoptotic signaling route, in colorectal cancer cells.70 These studies collectively highlight colchicine's ability to target multiple mechanisms involved in cancer progression, including inhibition of proliferation, migration, and invasion, as well as the induction of apoptosis.
While colchicine has been extensively studied in various cancer types, its potential as an anti-AT/RT agent remains unexplored. The evidence of its anticancer effectiveness strongly supports its capacity to act as a potential candidate for AT/RT treatment. By targeting key signaling pathways, inhibiting metastasis, and promoting apoptosis, colchicine could offer a novel therapeutic approach for AT/RTs. Furthermore, this study suggests a potential mechanism for colchicine's effect on AT/RTs. Colchicine binds to tubulin, forming a tubulin-colchicine (TC) complex, which interferes with cell cycle progression by arresting mitotic cells at the G2/M phase. This disruption ultimately inhibits AT/RT cell proliferation, particularly within the proliferative zone of 3D AT/RT spheroids (Fig. 5). Additionally, colchicine's FDA-approved status makes it a particularly attractive candidate for drug repurposing. Unlike conventional chemotherapeutic agents, colchicine demonstrates a lower risk of adverse effects as evidenced by its selective toxicity and favourable therapeutic window in normal brain and endothelial cells.
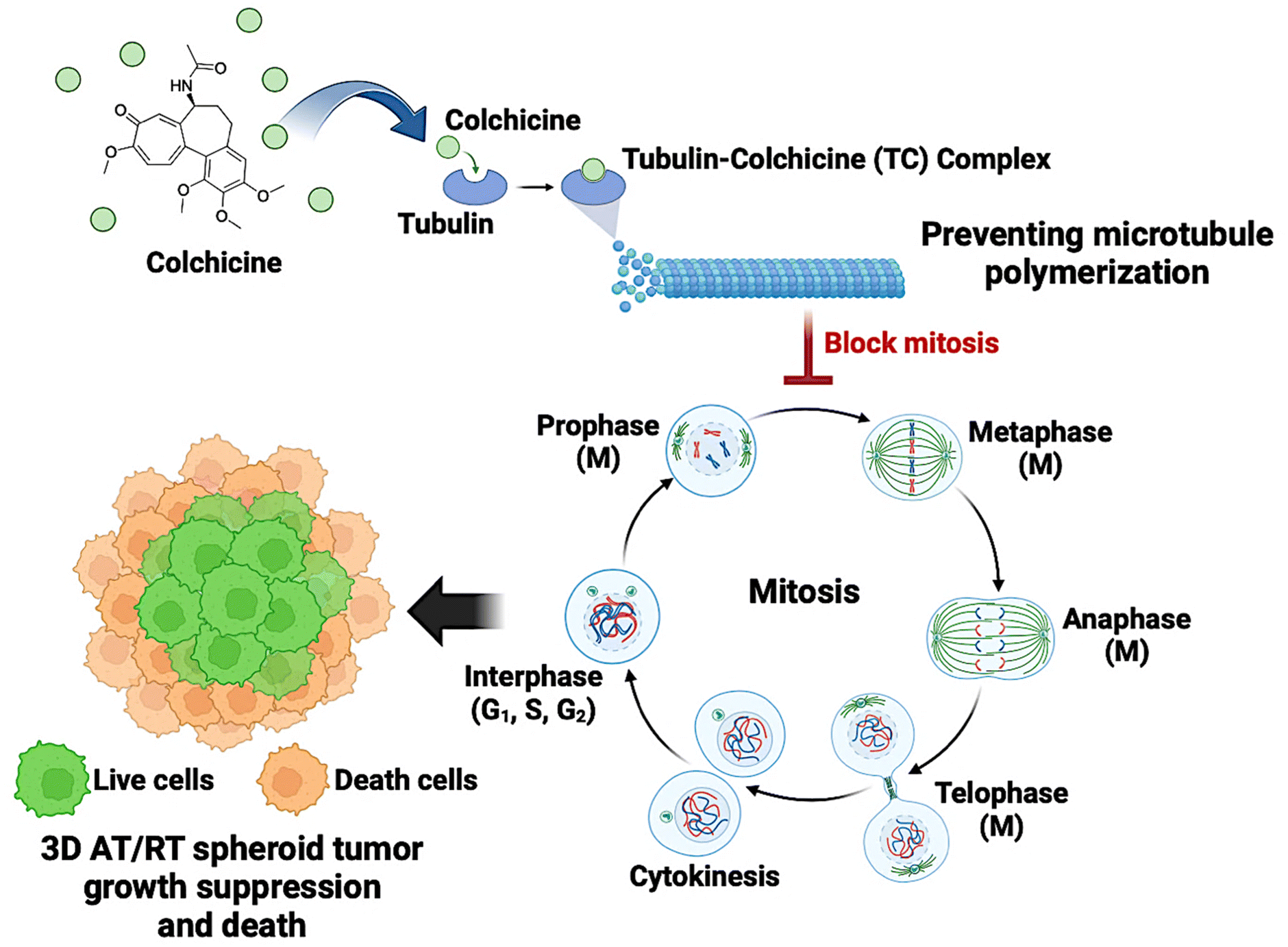 | ||
| Fig. 5 Proposed antimitotic mechanism of colchicine. Colchicine disrupts microtubule stability by binding to two tubulin heterodimers near the exchangeable GTP-binding site, forming a tubulin-colchicine (TC) complex. This interaction destabilizes tubulin, preventing proper microtubule assembly and arresting mitotic cells at the G2/M phase. As a result, AT/RT cell proliferation is inhibited, particularly in the proliferative zone of 3D AT/RT spheroids, ultimately triggering programmed cell death, often known as apoptosis.47 The cell cycle consists of four main phases: G1 (first growth phase), where the cell grows and carries out normal metabolic functions; S (synthesis phase), during which DNA replication occurs; G2 (second growth phase), preparing the cell for mitosis; and M (mitotic phase), where nuclear division results in two genetically identical daughter cells. The M phase is further divided into prophase, metaphase, anaphase, and telophase. (Created by http://BioRender.com/Mahidol University.) | ||
Collectively, colchicine has the potential to be successfully repurposed as an AT/RT treatment, and combining colchicine with other drugs to overcome resistance may offer a potential strategy for improving treatment outcomes. Further investigation into the combination treatment of colchicine and other efflux transporter inhibitors and the mechanistic study of colchicine are also underway to confirm its therapeutic value. These findings will contribute to our understanding of colchicine as a possible new treatment for AT/RTs, offering a safer and potentially more effective alternative to traditional chemotherapy.
Methods
Cell culture
The human atypical teratoid/rhabdoid tumor cell lines (BT-12 and BT-16 cells) were generously provided by Peter Houghton from the St. Jude Children's Research Hospital (Memphis, TN, USA) through Professor Suradej Hongeng. BT-12 and BT-16 cells were cultured in RPMI 1640 medium, which was supplemented with 10% FBS and 1% P/S (Penicillin/Streptomycin). The human blood–brain barrier cell line (hCMEC/D3 cells) was purchased from Merck (Schuchardt, Darmstadt, Germany). hCMEC/D3 cells were cultured in EndoGRO™-MV complete medium, which was supplemented with 1 ng mL−1 FGF-2. The primary human astrocytes (HA) were purchased from ScienCell™ Research Laboratories (Carlsbad, CA, USA). Astrocytes were cultured in astrocyte medium (AM), which was supplemented with astrocyte growth supplement (AGS) and 2% FBS.FDA-approved drug library preparation
A library of 2130 FDA-approved drugs was purchased from Selleck Chemicals (Houston, TX, USA). All FDA-approved drugs were dissolved in DMSO at 10 mM concentration. For the compound screening, 2130 FDA-approved drugs were diluted in an assay medium at the final concentration of 10 μM.High-throughput screening of 2130 FDA-approved drugs for anti-AT/RT cells in 2D traditional culture
Atypical teratoid/rhabdoid tumor cell lines were seeded into 96-well clear plates (Corning Acton, MA, USA) at a density of 2 × 103 cells per well. The cells were cultured in RPMI 1640 medium supplemented with 10% FBS and 1% P/S (Penicillin/Streptomycin) and incubated at 37 °C with 5% CO2 for 24 hours. Following this incubation period, the screening of compounds was carried out using a high-throughput liquid handling system (JANUS) (PerkinElmer, Waltham, MA, USA). For primary screening, the compounds were added into the cell plates at a final concentration of 10 μM and incubated for 72 hours under the same conditions. After 72 hours of compound treatment, the culture media containing the compounds was removed, and serum-free media containing MTT was added to each well. The cells were incubated with the MTT solution for 3 hours at 37 °C with 5% CO2. After the 3 hours incubation period, the serum-free medium containing MTT was removed, and DMSO was added to each well and the absorbance of resulting-colored solution was measured at 570 nm by Multi-Mode Microplate Reader (ENVISION) (PerkinElmer, Waltham, MA, USA). The cell viability was quantified as a percentage of the untreated control. The compounds were selected for further investigation if they reduced cancer cell viability to 20% or less.Secondary screening (dose-dependent manner) for IC50 of anti-AT/RT cells
Atypical teratoid/rhabdoid tumor cell lines were seeded into 96-well clear plates at a density of 2 × 103 cells per well. The cells were cultured in RPMI 1640 medium supplemented with 10% FBS and 1% P/S and incubated at 37 °C with 5% CO2 for 24 hours. After 24 hours of incubation, the test compounds were added to the wells in a dose-dependent manner with the final concentrations of 20, 2, 0.2, 0.02, 0.002, and 0.0002 μM. The plates were then incubated for 72 hours at 37 °C, 5% CO2. After the 72-hour treatment, the culture media containing the compounds was removed, and serum-free media containing MTT was added to each well. The cells were incubated for 3 hours at 37 °C with 5% CO2. Following this incubation, the serum-free media containing MTT was removed, and DMSO was added to the wells and the absorbance of resulting-colored solution was measured at 570 nm by Multi-Mode Microplate Reader (ENVISION). The calculation of IC50 was performed using GraphPad Prism 9 (GraphPad Software, Inc., San Diego, CA, USA).High-content imaging analysis of hit compounds for anti-AT/RT cells in 3D spheroid culture
Atypical teratoid/rhabdoid tumor cell lines were seeded into 96-well ultra-low attachment clear plates (Corning (Acton, MA, USA)) at a density of 5 × 103 cells per well. The cells were cultured in RPMI 1640 medium supplemented with 10% FBS and 1% P/S. The plates were incubated at 37 °C with 5% CO2 for 72 hours to allow the formation of 3D spheroids. After this incubation period, compounds were added to the plates in a dose-dependent manner and further incubated for an additional 72 hours at 37 °C with 5% CO2. Following this treatment period, the 3D spheroids were stained with Hoechst (live cell staining) and ethidium homodimer (dead cell staining) at a 1:1000 dilution ratios of each dye reagent to the culture medium. The 3D spheroids were incubated with the dye reagents for 2 hours. The stained spheroids were then analyzed using a high-content imaging system (Operetta) (PerkinElmer (Waltham, MA, USA)). The calculation of IC50 was performed using GraphPad Prism 9.
Cytotoxicity test in BBB and astrocytes
The hCMEC/D3 cells representing the BBB were seeded into 96-well clear plates at a density of 2 × 104 cells per well. The cells were cultured in EndoGRO™-MV complete medium supplemented with 1 ng mL−1 FGF-2 and incubated at 37 °C with 5% CO2 for 24 hours. Alternatively, human astrocytes were seeded into 96-well clear plates at a density of 5 × 103 cells per well and cultured in astrocyte medium (AM) supplemented with astrocyte growth supplement (AGS) and 2% FBS under the same condition. After 24 hours of incubation, selected compounds were added to the wells of both BBB and astrocytes plates in a dose-dependent manner with final concentrations of 20, 5, 1.25, 0.313, 0.078, 0.020, and 0.005 μM. The plates were then incubated for 72 hours at 37 °C with 5% CO2. Following this period, the culture media containing the compound was removed, and serum-free media containing MTT was added to each well. The cells were incubated with the MTT solution for 3 hours under the same conditions.After the 3-hour incubation, the solution was removed, and DMSO was added to each well and the absorbance of the resulting-colored solution was measured at 570 nm by Multi-Mode Microplate Reader (ENVISION). The calculation of CC50 was performed using GraphPad Prism 9.
Statistical analysis
All data were analyzed using GraphPad Prism 9 (GraphPad Software, Inc., San Diego, CA, USA). The Z-factor is a measure of statistical effect size and an attempt to quantify the suitability of a particular assay and assess the quality of an assay for high throughput screening.Abbreviations
| AT/RT | Atypical teratoid/rhabdoid tumor |
| AKT | Ak strain transforming/protein kinase B (PKB) |
| BAX | Bcl2 associated X-protein |
| Bcl2 | B-cell lymphoma 2 |
| CNS | Central nervous system |
| FAK/SRC | Focal adhesion kinase/sarcoma non-receptor tyrosine kinase |
| FDA | Food and Drug Administration |
| GTP | Guanosine triphosphate |
| hCMEC/D3 | Human cerebral microvascular endothelial cell/clone D3 |
| HIV | Human immunodeficiency virus |
| MMP-2 | Matrix metalloproteinase-2 |
| MMP-9 | Matrix metalloproteinase-9 |
| mTOR | Mammalian target of rapamycin |
| PI3K | Phosphoinositide 3-kinases |
| ROS | Reactive oxygen species |
| SMARCB1 | SWI/SNF-related matrix-associated actin-dependent regulator of chromatin subfamily B member 1 |
| VEGFR2 | Vascular endothelial growth factor 2 |
Data availability
The data supporting the findings of this article have been included as part of the ESI.†Author contributions
P. K., K. J., and S. S. conducted the experiments and analyzed the data; P. K., S. B., P. W., S. H. and S. C. conceptualized the idea; S. B., P. W., S. H. and S. C. provided materials and resources; S. B., S. H. and S. C. provided funding; S. C. supervised all research; and P. K. and S. C. wrote, reviewed, and edited the manuscript. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This research was partially supported by the Ramathibodi Foundation and Thailand Center of Excellence for Life Sciences (TCELS), and also by the Faculty of Science, Mahidol University via the Reinventing University Project. The authors would like to thank Dr Witchuda Saengsawang, Dr Jiraporn Panmanee and Dr Sujira Mukda for discussions.References
- M. Bhattacharjee, J. Hicks, L. Langford, R. Dauser, D. Strother, M. Chintagumpala, M. Horowitz, L. Cooley and H. Vogel, Ultrastruct. Pathol., 1997, 21, 369–378 CrossRef CAS PubMed.
- N. Gökden, O. Nappi, P. E. Swanson, J. D. Pfeifer, R. T. Vollmer, M. R. Wick and P. A. Humphrey, Am. J. Surg. Pathol., 2000, 24, 1329–1338 CrossRef PubMed.
- I. Versteege, N. Sévenet, J. Lange, M.-F. Rousseau-Merck, P. Ambros, R. Handgretinger, A. Aurias and O. Delattre, Nature, 1998, 394, 203–206 CrossRef CAS PubMed.
- D. N. Louis, A. Perry, G. Reifenberger, A. von Deimling, D. Figarella-Branger, W. K. Cavenee, H. Ohgaki, O. D. Wiestler, P. Kleihues and D. W. Ellison, Acta Neuropathol., 2016, 131, 803–820 CrossRef PubMed.
- Y. S. Dho, S. K. Kim, J. E. Cheon, S. H. Park, K. C. Wang, J. Y. Lee and J. H. Phi, Childs Nerv. Syst., 2015, 31, 1305–1311 CrossRef PubMed.
- T. Tamiya, H. Nakashima, Y. Ono, S. Kawada, S. Hamazaki, T. Furuta, K. Matsumoto and T. Ohmoto, Pediatr. Neurosurg., 2000, 32, 145–149 CrossRef CAS PubMed.
- Q. T. Ostrom, Y. Chen, P. M de Blank, A. Ondracek, P. Farah, H. Gittleman, Y. Wolinsky, C. Kruchko, M. L. Cohen, D. J. Brat and J. S. Barnholtz-Sloan, Neuro-Oncology, 2014, 16, 1392–1399 CrossRef PubMed.
- A. T. Reddy, D. R. Strother, A. R. Judkins, P. C. Burger, I. F. Pollack, M. D. Krailo, A. B. Buxton, C. Williams-Hughes, M. Fouladi, A. Mahajan, T. E. Merchant, B. Ho, C. M. Mazewski, V. A. Lewis, A. Gajjar, L. G. Vezina, T. N. Booth, K. W. Parsons, V. L. Poss, T. Zhou, J. A. Biegel and A. Huang, J. Clin. Oncol., 2020, 38, 1175–1185 CrossRef CAS PubMed.
- L. Lafay-Cousin, T. Fay-McClymont, D. Johnston, C. Fryer, K. Scheinemann, A. Fleming, J. Hukin, L. Janzen, S. Guger, D. Strother, D. Mabbott, A. Huang and E. Bouffet, Pediatr. Blood Cancer, 2015, 62, 1265–1269 CrossRef PubMed.
- M. C. Frühwald, J. A. Biegel, F. Bourdeaut, C. W. Roberts and S. N. Chi, Neuro-Oncology, 2016, 18, 764–778 CrossRef.
- M. Park, J. W. Han, S. M. Hahn, J. A. Lee, J. Y. Kim, S. H. Shin, D. S. Kim, H. I. Yoon, K. T. Hong, J. Y. Choi, H. J. Kang, H. Y. Shin, J. H. Phi, S. K. Kim, J. W. Lee, K. H. Yoo, K. W. Sung, H. H. Koo, D. H. Lim, H. J. Shin, H. Kim, K. N. Koh, H. J. Im, S. D. Ahn, Y. S. Ra, H. J. Baek, H. Kook, T. Y. Jung, H. S. Choi, C. Y. Kim, H. J. Park and C. J. Lyu, Cancer Res. Treat., 2021, 53, 378–388 CrossRef PubMed.
- J. M. Hilden, S. Meerbaum, P. Burger, J. Finlay, A. Janss, B. W. Scheithauer, A. W. Walter, L. B. Rorke and J. A. Biegel, J. Clin. Oncol., 2004, 22, 2877–2884 CrossRef PubMed.
- J. S. Shim and J. O. Liu, Int. J. Biol. Sci., 2014, 10, 654–663 CrossRef CAS PubMed.
- N. Nosengo, Nature, 2016, 534, 314–316 CrossRef PubMed.
- C. M. Rudin, J. R. Brahmer, R. A. Juergens, C. L. Hann, D. S. Ettinger, R. Sebree, R. Smith, B. T. Aftab, P. Huang and J. O. Liu, J. Thorac. Oncol., 2013, 8, 619–623 CrossRef CAS PubMed.
- E. S. Antonarakis, E. I. Heath, D. C. Smith, D. Rathkopf, A. L. Blackford, D. C. Danila, S. King, A. Frost, A. S. Ajiboye, M. Zhao, J. Mendonca, S. K. Kachhap, M. A. Rudek and M. A. Carducci, Oncologist, 2013, 18, 163–173 CrossRef CAS PubMed.
- J. Xu, Y. Dang, Y. R. Ren and J. O. Liu, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 4764–4769 CrossRef CAS PubMed.
- B. A. Nacev, P. Grassi, A. Dell, S. M. Haslam and J. O. Liu, J. Biol. Chem., 2011, 286, 44045–44056 CrossRef CAS PubMed.
- T. B. Brunner, M. Geiger, G. G. Grabenbauer, M. Lang-Welzenbach, T. S. Mantoni, A. Cavallaro, R. Sauer, W. Hohenberger and W. G. McKenna, J. Clin. Oncol., 2008, 26, 2699–2706 CrossRef CAS PubMed.
- E. C. Costa, V. M. Gaspar, P. Coutinho and I. J. Correia, Biotechnol. Bioeng., 2014, 111, 1672–1685 CrossRef CAS PubMed.
- K. Ludwig, E. S. Tse and J. Y. Wang, BMC Cancer, 2013, 13, 221 CrossRef CAS PubMed.
- A. Amann, M. Zwierzina, G. Gamerith, M. Bitsche, J. M. Huber, G. F. Vogel, M. Blumer, S. Koeck, E. J. Pechriggl, J. M. Kelm, W. Hilbe and H. Zwierzina, PLoS One, 2014, 9, e92511 CrossRef PubMed.
- I. Dufau, C. Frongia, F. Sicard, L. Dedieu, P. Cordelier, F. Ausseil, B. Ducommun and A. Valette, BMC Cancer, 2012, 12, 15 CrossRef CAS PubMed.
- S. Shankar, D. Nall, S. N. Tang, D. Meeker, J. Passarini, J. Sharma and R. K. Srivastava, PLoS One, 2011, 6, e16530 CrossRef CAS PubMed.
- A. Takagi, M. Watanabe, Y. Ishii, J. Morita, Y. Hirokawa, T. Matsuzaki and T. Shiraishi, Anticancer Res., 2007, 27, 45–53 CAS.
- M. Wartenberg, S. Gronczynska, M. M. Bekhite, T. Saric, W. Niedermeier, J. Hescheler and H. Sauer, Int. J. Cancer, 2005, 113, 229–240 CrossRef CAS PubMed.
- M. Wartenberg, E. Hoffmann, H. Schwindt, F. Grünheck, J. Petros, J. R. Arnold, J. Hescheler and H. Sauer, FEBS Lett., 2005, 579, 4541–4549 CrossRef CAS PubMed.
- E. C. Costa, A. F. Moreira, D. de Melo-Diogo, V. M. Gaspar, M. P. Carvalho and I. J. Correia, Biotechnol. Adv., 2016, 34, 1427–1441 CrossRef PubMed.
- A. McIntyre, S. Patiar, S. Wigfield, J. L. Li, I. Ledaki, H. Turley, R. Leek, C. Snell, K. Gatter, W. S. Sly, R. D. Vaughan-Jones, P. Swietach and A. L. Harris, Clin. Cancer Res., 2012, 18, 3100–3111 CrossRef CAS PubMed.
- M. P. Valley, K. R. Kupcho, C. A. Zimprich, A. L. Niles, J. J. Cali, J. M. Kelm, W. Moritz and D. F. Lazar, Cancer Res., 2014, 74, 3731 CrossRef.
- I. Ayvaz, D. Sunay, E. Sariyar, E. Erdal and Z. F. Karagonlar, J. Gastrointest. Cancer, 2021, 52, 1294–1308 CrossRef PubMed.
- I. Alimova, D. K. Birks, P. S. Harris, J. A. Knipstein, S. Venkataraman, V. E. Marquez, N. K. Foreman and R. Vibhakar, Neuro-Oncology, 2013, 15, 149–160 CrossRef CAS PubMed.
- A. Morin, C. Soane, A. Pierce, B. Sanford, K. L. Jones, M. Crespo, S. Zahedi, R. Vibhakar and J. M. Mulcahy Levy, Neuro-Oncology Adv., 2020, 2, vdaa051 CrossRef PubMed.
- P. Hannon Barroeta, S. Magnano, M. J. O'Sullivan and D. M. Zisterer, Cancer Treat. Res. Commun., 2022, 32, 100584 CrossRef PubMed.
- Y. Y. Leung, L. L. Yao Hui and V. B. Kraus, Semin. Arthritis Rheum., 2015, 45, 341–350 CrossRef CAS PubMed.
- M. A. Jordan, Curr. Med. Chem.:Anti-Cancer Agents, 2002, 2, 1–17 CrossRef CAS PubMed.
- B. Karahalil, S. Yardım-Akaydin and S. Nacak Baytas, Arh. Hig. Rada Toksikol., 2019, 70, 160–172 CrossRef CAS PubMed.
- Z. Cheng, X. Lu and B. Feng, Transl. Cancer Res., 2020, 9, 4020–4027 CrossRef CAS PubMed.
- J. Casaos, S. Huq, T. Lott, R. Felder, J. Choi, N. Gorelick, M. Peters, Y. Xia, R. Maxwell, T. Zhao, C. Ji, T. Simon, J. Sesen, S. J. Scotland, R. E. Kast, J. Rubens, E. Raabe, C. G. Eberhart, E. M. Jackson, H. Brem, B. Tyler and N. Skuli, Oncotarget, 2018, 9, 8054–8067 CrossRef PubMed.
- S. Uxa, P. Castillo-Binder, R. Kohler, K. Stangner, G. A. Müller and K. Engeland, Cell Death Differ., 2021, 28, 3357–3370 CrossRef CAS PubMed.
- F. Mollinedo and C. Gajate, Apoptosis, 2003, 8, 413–450 CrossRef CAS PubMed.
- M. Kapałczyńska, T. Kolenda, W. Przybyła, M. Zajączkowska, A. Teresiak, V. Filas, M. Ibbs, R. Bliźniak, Ł. Łuczewski and K. Lamperska, Arch. Med. Sci., 2018, 14, 910–919 Search PubMed.
- A. Filipiak-Duliban, K. Brodaczewska, A. Kajdasz and C. Kieda, Int. J. Mol. Sci., 2022, 23, 1166 CrossRef CAS PubMed.
- M. Świerczewska, K. Sterzyńska, M. Ruciński, M. Andrzejewska, M. Nowicki and R. Januchowski, Biomed. Pharmacother., 2023, 165, 115152 CrossRef PubMed.
- T. M. Achilli, S. McCalla, J. Meyer, A. Tripathi and J. R. Morgan, Mol. Pharm., 2014, 11, 2071–2081 CrossRef CAS PubMed.
- M. Zanoni, F. Piccinini, C. Arienti, A. Zamagni, S. Santi, R. Polico, A. Bevilacqua and A. Tesei, Sci. Rep., 2016, 6, 19103 CrossRef CAS PubMed.
- P. Dhyani, C. Quispe, E. Sharma, A. Bahukhandi, P. Sati, D. C. Attri, A. Szopa, J. Sharifi-Rad, A. O. Docea, I. Mardare, D. Calina and W. C. Cho, Cancer Cell Int., 2022, 22, 206 CrossRef CAS PubMed.
- R. A. Terkeltaub, D. E. Furst, K. Bennett, K. A. Kook, R. S. Crockett and M. W. Davis, Arthritis Rheum., 2010, 62, 1060–1068 CrossRef CAS PubMed.
- M. Hamburger, H. S. Baraf, T. C. Adamson III, J. Basile, L. Bass, B. Cole, P. P. Doghramji, G. A. Guadagnoli, F. Hamburger, R. Harford, J. A. Lieberman III, D. R. Mandel, D. A. Mandelbrot, B. P. McClain, E. Mizuno, A. H. Morton, D. B. Mount, R. S. Pope, K. G. Rosenthal, K. Setoodeh, J. L. Skosey and N. L. Edwards, Postgrad. Med., 2011, 123, 3–36 CrossRef PubMed.
- D. Khanna, P. P. Khanna, J. D. Fitzgerald, M. K. Singh, S. Bae, T. Neogi, M. H. Pillinger, J. Merill, S. Lee, S. Prakash, M. Kaldas, M. Gogia, F. Perez-Ruiz, W. Taylor, F. Lioté, H. Choi, J. A. Singh, N. Dalbeth, S. Kaplan, V. Niyyar, D. Jones, S. A. Yarows, B. Roessler, G. Kerr, C. King, G. Levy, D. E. Furst, N. L. Edwards, B. Mandell, H. R. Schumacher, M. Robbins, N. Wenger and R. Terkeltaub, Arthritis Care Res., 2012, 64, 1447–1461 CrossRef CAS PubMed.
- C. A. Dinarello, S. M. Wolff, S. E. Goldfinger, D. C. Dale and D. W. Alling, N. Engl. J. Med., 1974, 291, 934–937 CrossRef CAS PubMed.
- E. Ben-Chetrit, J. Nephrol., 2003, 16, 431–434 CAS.
- T. Kallinich, D. Haffner, T. Niehues, K. Huss, E. Lainka, U. Neudorf, C. Schaefer, S. Stojanov, C. Timmann, R. Keitzer, H. Ozdogan and S. Ozen, Pediatrics, 2007, 119, e474–e483 CrossRef PubMed.
- M. Takeuchi, Y. Asukata, T. Kawagoe, N. Ito, T. Nishide and N. Mizuki, Ocul. Immunol. Inflammation, 2012, 20, 193–197 CrossRef CAS PubMed.
- F. Davatchi, B. Sadeghi Abdollahi, A. Tehrani Banihashemi, F. Shahram, A. Nadji, H. Shams and C. Chams-Davatchi, Mod. Rheumatol., 2009, 19, 542–549 CrossRef CAS PubMed.
- A. Sun, Y. P. Wang, J. S. Chia, B. Y. Liu and C. P. Chiang, J. Oral Pathol. Med., 2009, 38, 401–405 CrossRef CAS PubMed.
- M. Imazio, A. Brucato, R. Cemin, S. Ferrua, R. Belli, S. Maestroni, R. Trinchero, D. H. Spodick and Y. Adler, Ann. Intern. Med., 2011, 155, 409–414 CrossRef PubMed.
- M. Imazio, R. Belli, A. Brucato, R. Cemin, S. Ferrua, F. Beqaraj, D. Demarie, S. Ferro, D. Forno, S. Maestroni, D. Cumetti, F. Varbella, R. Trinchero, D. H. Spodick and Y. Adler, Lancet, 2014, 383, 2232–2237 CrossRef CAS PubMed.
- M. Imazio, A. Brucato, R. Cemin, S. Ferrua, S. Maggiolini, F. Beqaraj, D. Demarie, D. Forno, S. Ferro, S. Maestroni, R. Belli, R. Trinchero, D. H. Spodick and Y. Adler, N. Engl. J. Med., 2013, 369, 1522–1528 CrossRef CAS PubMed.
- S. M. Nidorf, J. W. Eikelboom, C. A. Budgeon and P. L. Thompson, J. Am. Coll. Cardiol., 2013, 61, 404–410 CrossRef CAS PubMed.
- S. Deftereos, G. Giannopoulos, K. Raisakis, C. Kossyvakis, A. Kaoukis, V. Panagopoulou, M. Driva, G. Hahalis, V. Pyrgakis, D. Alexopoulos, A. S. Manolis, C. Stefanadis and M. W. Cleman, J. Am. Coll. Cardiol., 2013, 61, 1679–1685 CrossRef CAS PubMed.
- V. Fontes, L. Machet, B. Huttenberger, G. Lorette and L. Vaillant, Ann. Dermatol. Venereol., 2002, 129, 1365–1369 CAS.
- E. Fiorucci, G. Lucantoni, G. Paone, M. Zotti, B. E. Li, M. Serpilli, P. Regimenti, I. Cammarella, G. Puglisi and G. Schmid, Eur. Rev. Med. Pharmacol. Sci., 2008, 12, 105–111 CAS.
- A. Absalón-Aguilar, M. Rull-Gabayet, A. Pérez-Fragoso, N. R. Mejía-Domínguez, C. Núñez-Álvarez, D. Kershenobich-Stalnikowitz, J. Sifuentes-Osornio, A. Ponce-de-León, F. González-Lara, E. Martín-Nares, S. Montesinos-Ramírez, M. Ramírez-Alemón, P. Ramírez-Rangel, M. F. Márquez, J. C. Plata-Corona, G. Juárez-Vega, D. Gómez-Martín and J. Torres-Ruiz, J. Gen. Intern. Med., 2022, 37, 4–14 CrossRef PubMed.
- T. Welzel, A. L. Wildermuth, N. Deschner, S. M. Benseler and J. B. Kuemmerle-Deschner, Pediatr. Rheumatol., 2021, 19, 142 CrossRef PubMed.
- A. M. Knieper, J. Klotsche, D. Föll, H. Wittkowski, E. Lainka and T. Kallinich, Pediatr. Rheumatol., 2015, 13, O44 CrossRef.
- F. Bakar Ateş, N. Özmen, E. Kaya Sezginer and E. E. Kurt, Turk Hij. Deneysel Biyol. Derg., 2018, 75, 239–244 CrossRef.
- J. H. Cho, Y. H. Joo, E. Y. Shin, E. J. Park and M. S. Kim, Anticancer Res., 2017, 37, 6269–6280 CrossRef CAS PubMed.
- T. Zhang, W. Chen, X. Jiang, L. Liu, K. Wei, H. Du, H. Wang and J. Li, Biosci. Rep., 2019, 39, BSR20181802 CrossRef PubMed.
- Z. Huang, Y. Xu and W. Peng, Mol. Med. Rep., 2015, 12, 5939–5944 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01341k |
| This journal is © The Royal Society of Chemistry 2025 |