DOI:
10.1039/D5RA01257K
(Paper)
RSC Adv., 2025,
15, 20495-20512
Synthesis, cytotoxic activity, molecular docking, molecular dynamics simulations, and ADMET studies of novel spiropyrazoline oxindoles based on domino Knoevenagel–Michael cyclization as potent non-toxic anticancer agents targeting β-tubulin and EGFR, with anti-MRSA activity†
Received
20th February 2025
, Accepted 3rd June 2025
First published on 19th June 2025
Abstract
The search for novel potent anticancer and antimicrobial agents is considered a rapidly advancing field and viewed as a constantly evolving area within medicinal chemistry. In this work, a series of novel spiropyrazoline oxindole scaffolds were synthesized based on domino Knoevenagel–Michael cyclization reactions. These compounds were tested for their in vitro cytotoxic activity against HePG-2 [human hepatocellular carcinoma cell line] and MCF-7 [breast cancer cell line]. Compounds 5a, 5b, and 5d showed potent activity against the HePG-2 cell line. Compound 5a was the most potent one and showed activity against both cell lines, with IC50 (μg mL−1) values of 12.3 and 17.3, respectively, compared to adriamycin, with IC50 (μg mL−1) values of 21.6 and 25.5, respectively. An in silico study, encompassing both molecular docking and MD simulations, highlighted the potential of compounds 5a and 5b as potent therapeutic agents targeting the 4I4T protein, compared to three commercially available drugs, namely, adriamycin, sunitinib, and spirobrassinin. This study demonstrates the importance of spiropyrazoline oxindoles for the development of new and potent cancer treatments. The MD simulations confirmed that compound 5a has a more stable and stronger interaction with the 4I4T protein, making it a promising candidate for further development. An in silico study was conducted to support the experimental results, and another one to show the binding affinity with the PBP2a receptor protein of S. aureus for future research. Compound 4a showed a binding affinity energy of −7.9 kcal mol−1, compared to −6.5 kcal mol−1 for linezolid and −6.3 kcal mol−1 for penicillin.
1 Introduction
Cancer remains a significant health concern in both developing and underdeveloped nations.1–3 The development of therapeutic agents with high potency against cancer cells is a key focus point in drug research. Researchers have made great efforts over the past decades to create new analogues with potent and selective antitumor activity.4 Tubulin-microtubule synthesis plays a crucial role in cellular processes, particularly in maintaining cell division, making it a promising molecular target for chemotherapy.5,6 Spiroheterocyclic compounds represent a significant class of heterocycles that have attracted substantial interest due to their occurrence in many bioactive, natural, and synthetic products.7 Spiro-indolinones, commonly referred to as spirooxindoles, are among the most significant spiranic structures in medicinal chemistry. Spirooxindoles can attach to different heterocycle rings at the C3 position (Fig. 1), making them a promising candidate in drug discovery.8,9
 |
| | Fig. 1 Scaffold of spirocyclic oxindoles. | |
The 2-pyrazoline moiety is a very important five-membered ring heterocycle. It has many applications as an active therapeutic agent in industry. Several compounds with the 2-pyrazoline core structure have been recently used as lead compounds in drug discovery and the identification of promising medicinal candidates. These compounds exhibit a broad spectrum of different biological activities, including anticancer,10,11 analgesic12 anti-inflammatory,13 anti-diabetic,14 antidepressant,15 antibacterial,16 antifungal,17 and antiamoebic activities.18 Spiropyrazolines represent another class of spiro compounds that serve as the core structure of various synthetic molecules, possessing antimicrobial and anticancer properties.19 Spirocyclic compounds containing indolin-2-one and pyrazoline nuclei have demonstrated significant bioactivity against cancer cell lines.20 However, spiropyrazoline indolin-3-ones have not been extensively studied or utilized in medicinal chemistry. This motivated us to synthesize these compounds to assess their biological activity. Based on previous research, we found that spiropyrazoline oxindole scaffolds would be promising therapeutic agents. In this research, we report on the synthetic method and the cytotoxic activity of this novel scaffold. Computational studies, including molecular docking, molecular dynamic simulations and ADMET, were conducted to evaluate these novel spiropyrazoline compounds.
2 Result and discussion
2.1. Chemistry
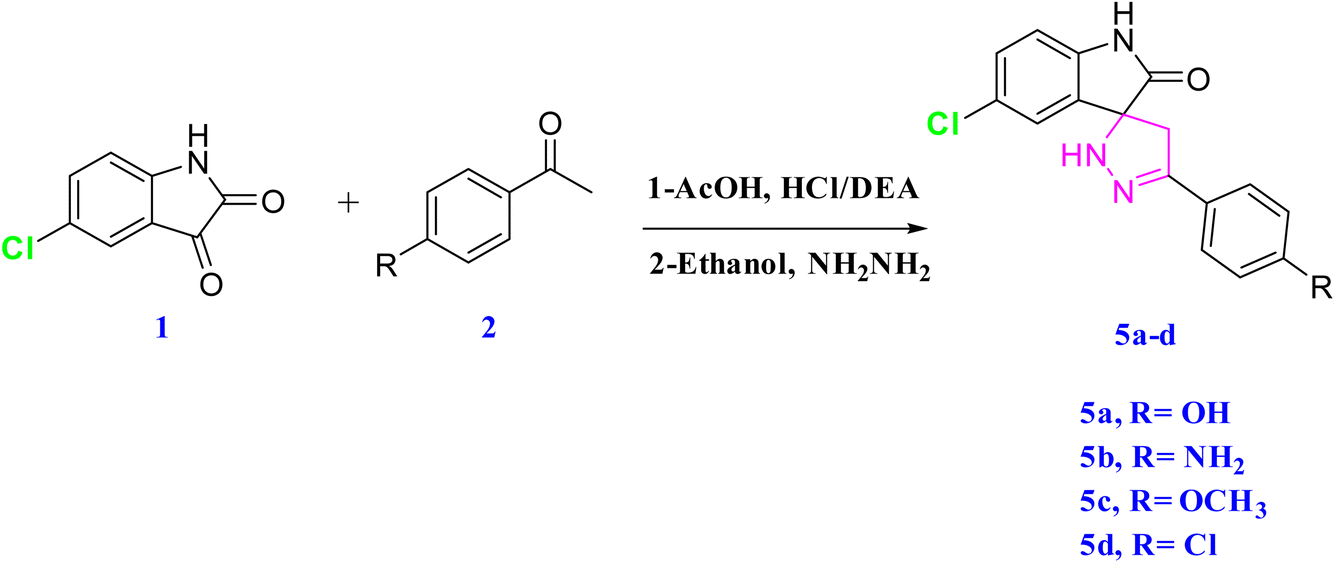
The targeted spiropyrazoline oxindole scaffolds were synthesized based on a domino Knoevenagel–Michael cyclization reaction. All the synthesized compounds were characterized by spectroscopic techniques.
2.1.1 Synthesis of isatin-based chalcone derivatives 4a,b (ref. 21). The aldol condensation reaction was used to synthesize the isatin-based chalcone compounds 4a,b. 5-Chloro isatin was used as a starting material. A carbanion was formed in basic media using DEA (diethyl amine). This carbanion is relatively stable because it can conjugate to produce enolate ions. Isatin acts as an electrophile and was attacked at c3, which led to the formation of 3a,b. This reaction was carried out in absolute methanol under basic conditions. The synthesis of 4a,b was carried out under acidic conditions in absolute ethanol, with the reaction carried out under reflux in a water bath for 1 h (Scheme 1).
 |
| | Scheme 1 Synthesis of isatin-based chalcone derivatives 4a,b. | |
2.1.2 Elucidation of the structure and configuration of the target compound 4a. Isatin stereoselectively22 reacted with acetophenone as the E-diastereomer was the sole product in this reaction, see Fig. 2. Accordingly, in the current work, the configuration around the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bond was confirmed based on 2D 1H NMR.
C bond was confirmed based on 2D 1H NMR.
 |
| | Fig. 2 1H 1H–COSY spectra of compound 4a. | |
2.1.3 Synthesis of spiropyrazoline oxindoles23. This cyclization reaction occurs through a domino Knoevenagel–Michael mechanism24 (Scheme 2). The nucleophilic Michael addition of hydrazine led to the formation of the spiropyrazoline oxindoles 5a–d (Scheme 3).
 |
| | Scheme 2 Mechanism for the synthesis of spiropyrazoline oxindoles. | |
 |
| | Scheme 3 Synthesis of spiropyrazoline oxindoles. | |
2.1.4 Optimization of the reaction conditions for the synthesis of spiropyrazoline using NH2NH2 in ethanol. To optimize the spiropyrazoline synthesis via the cyclocondensation of hydrazine hydrate with reactive intermediate chalcone derivatives, a series of reactions were performed under catalyst-free conditions, with varying the solvent systems, temperature, and reaction time. The results are summarized in Table 1.
Table 1 Optimization of reaction conditions for the synthesis of spiropyrazoline
| Entry |
Solvent |
Temperature (°C) |
Yield (%) |
| 1 |
Ethanol |
RT |
70 |
| 2 |
Ethanol |
60 |
75 |
| 3 |
Ethanol |
70 |
80 |
| 4 |
Ethanol (reflux) |
80 |
85 |
| 5 |
EtOH/H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) :1) |
78 |
82 |
| 6 |
Water |
80 |
72 |
The synthesis of spiropyrazoline via the reaction with hydrazine hydrate was investigated under green, catalyst-free conditions. The effect of temperature and the solvent system was found to significantly influence the reaction yield. At room temperature (RT) in ethanol (Entry 1), a 70% yield was achieved after an extended reaction time. Increasing the temperature to 60 °C and 70 °C (Entries 2–3) progressively improved the yields to 75% and 80%, respectively. A mixed solvent system of ethanol and water (Entry 5) slightly enhanced the yield to 82%. Performing the reaction under reflux conditions (Entry 4) provided the highest yield of 85%. However, when water was used as the sole solvent (Entry 6), the yield dropped slightly to 72%, likely due to the solubility limitations of the reactants.25 Based on these results, the optimal catalyst-free reaction conditions for the synthesis of spiropyrazoline were identified as using ethanol at 80 °C, yielding up to 85%. These conditions align well with green chemistry principles, offering a simple and sustainable synthetic approach.
2.2 Justification for protein selection based on the pharmacophore characteristics26,27
In this study, we synthesized spiropyrazoline derivatives through domino Knoevenagel–Michaele cyclization reactions, aiming to explore their potential as microtubule-stabilizing agents. To evaluate their interaction with tubulin, we selected the crystal structure of the tubulin-RB3-TTL-Zampanolide complex (PDB ID: 4I4T) as our molecular target. The 4I4T structure provides a high-resolution (1.8 Å) model of αβ-tubulin in complex with zampanolide, a potent microtubule-stabilizing agent. Zampanolide binds covalently to the taxane site on β-tubulin, promoting microtubule assembly and stability. This binding induces a helical conformation in the M-loop of β-tubulin,28 a structural change critical for microtubule stabilization. Our synthesized spiropyrazoline compounds featured pharmacophoric elements conducive to binding at the taxane site:
• Hydrophobic and aromatic moieties that can engage in van der Waals interactions within the hydrophobic pocket of the taxane site;
• Hydrogen bond donors and acceptors capable of forming interactions with key residues such as βHis229 and βLeu275;
• Rigid spirocyclic frameworks, which may favor the induction of conformational changes in the M-loop, similar to those induced by zampanolide.
By utilizing the 4I4T structure, we aimed to model the interactions of our compounds within a biologically relevant binding site, providing insights into their potential as microtubule-stabilizing agents.
The 4I4T protein is derived from Escherichia coli (E. coli) and is often used as a structural model to investigate potential interactions with synthesized compounds. Although the HePG-2 cell line is derived from Homo sapiens (human liver cancer cells), the use of the 4I4T structure for computational docking studies is justified by the high degree of structural conservation between human tubulin and tubulin from bacterial sources like E. coli.
Several studies have demonstrated that tubulin's core structure remains highly conserved across species, including between human and bacterial systems. For example, research by Geyer et al. showed that tubulin from non-human sources like E. coli exhibits similar binding properties to human tubulin,29 allowing for the use of E. coli-derived tubulin structures in docking studies for human-targeted therapies.30
Furthermore, structural studies have shown that the tubulin-binding sites for many known microtubule-targeting agents, such as taxanes and zampanolide, are almost identical between species. This conservation makes the 4I4T structure a valuable tool for identifying and characterizing potential ligands that can bind to the taxane site of β-tubulin, which is critical for the stabilization of microtubules and the inhibition of cell division.28
In summary, the use of the 4I4T protein in docking studies is scientifically supported by the structural conservation of tubulin across species, and the information gained can be translated to human systems, such as HePG-2 cells, for further biological validation.
In the case of EGFR, the protein was selected due to the increasing evidence linking EGFR overexpression to hepatocellular carcinoma, the disease model relevant to the HePG-2 cell line. EGFR inhibitors, such as gefitinib and erlotinib, are FDA-approved for various cancers, and structural analogs of our compounds have been reported to exhibit kinase inhibition.29 AQ4 exhibited a binding affinity of −7.7 kcal mol−1 indicating weaker interaction compared to our synthesized compounds.31 This comparative approach not only justifies the use of EGFR as a target but also strengthens the hypothesis that our synthesized compounds may exert dual activity via tubulin and kinase inhibition pathways.32
2.2.1 Scientific justification for selecting MRSA (1MWT) as a target. MRSA is a significant clinical concern due to its resistance to most β-lactam antibiotics, which arises from the expression of PBP2a, encoded by the mecA gene. PBP2a maintains peptidoglycan synthesis even in the presence of β-lactams, making it a critical target for antimicrobial drug development. Literature supports PBP2a as a valid and essential target in the development of anti-MRSA agents.33,34
2.2.2 Rationale for using linezolid and penicillin as reference drugs. • Penicillin was chosen because it is the prototype β-lactam, and its resistance mechanism is directly linked to PBP2a.• Linezolid was selected as a comparator because it is an FDA-approved oxazolidinone antibiotic with proven activity against MRSA, although it acts through a different mechanism (protein synthesis inhibition). Including it enabled us to assess whether our compounds offer an alternative or complementary mechanism of action with potential anti-MRSA activity.
2.2.3 Key justification for choosing spirobrassinin in our study. The justification for choosing spirobrassinin lies in its structural features, which include:• Hydrophobic/aromatic moieties, which are crucial for binding to the hydrophobic pocket of tubulin,
• A rigid spirocyclic structure, which enhances the molecule's ability to fit within the binding site of β-tubulin,
• Functional groups capable of forming interactions with important residues within the tubulin-binding site, as observed in zampanolide and other microtubule-targeting agents.29
The scientific literature28 has demonstrated that compounds with similar structural features to spirobrassinin show promising activity in stabilizing microtubules and inhibiting cell division. The spirocyclic framework, although unique, contributes to the rigidity and specificity needed for interaction with the tubulin-binding site, which we believe is a relevant characteristic for our compounds as well.
2.2.4 Criteria for defining a HIT compound in our study. The criteria were based on a combination of the binding affinity, interaction with key residues, and pharmacophoric compatibility with known active compounds against the selected target (tubulin, PDB ID: 4I4T). In this work, compound 5a was considered a HIT as it met the following criteria:• Docking score: The compound demonstrated a binding energy equal to −8.3 kcal mol−1, which is within the range typically reported for strong tubulin-binding agents.35
• Binding mode: The compound formed interactions with key amino acid residues in the taxane binding site of β-tubulin consistent with the known pharmacophoric interactions of microtubule-stabilizing agents.31
• Pharmacophore fit: The compound exhibited structural and electronic features aligned with known microtubule-stabilizing pharmacophores (e.g., hydrophobic/aromatic centers, hydrogen bond donors/acceptors).
2.3. Cytotoxic activity
The MTT assay was used to assess cell viability36 [against HePG-2 (liver), and MCF7 (breast) cancer cell lines].
IC90 values were determined, and a probit analysis was obtained (Fig. 3–5).
 |
| | Fig. 3 Probit analysis obtained from the MTT assay results against the MCF7 cancer cell line (5a). | |
 |
| | Fig. 4 Probit analysis obtained from the MTT assay results against the MCF7 cancer cell line (doxorubicin). | |
 |
| | Fig. 5 Probit analysis obtained from the MTT assay results against the HePG-2 cancer cell line. A. Doxorubicin, B. 5a, C. 5c, and D. 5d. | |
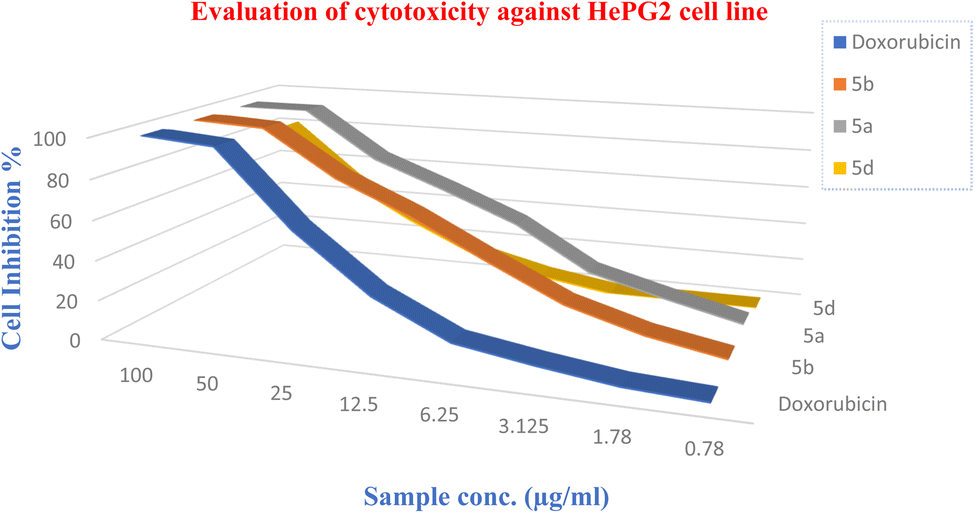
2.3.1 HePG-2 cell line. All the tested compounds showed significant antitumor effects. Compounds 5a and 5b revealed the highest potencies of the synthesized agents with efficacies higher than the standard reference (doxorubicin) (2 times potency; IC50 = 10.87, 12.3 and 21.6 μg mL−1 for 1, 2 and doxorubicin, respectively) (Tables 2–4).
Table 2 Cytotoxic activity test results of the synthesized agents and standard reference
| Entry |
Compound |
IC50 (μg mL−1) |
IC90 (μg mL−1) |
Remarks |
| HePG2 |
MCF7 |
HePG2 |
MCF7 |
HePG2 |
MCF7 |
| 4 |
5a |
12.3 ± 0.002 |
17.3 ± 0.002 |
28.7 ± 0.085 |
58.9 ± 0.065 |
100% |
75.6% |
| 5 |
5b |
13.4 ± 0.006 |
27.6 ± 0.05 |
35.1 ± 0.078 |
65.3 ± 0.079 |
100% |
71.8% |
| 6 |
5c |
57.4 ± 0.07 |
˃100 |
101.8 ± 0.080 |
˃100 |
71.6% |
26.3% |
| 7 |
5d |
23.5 ± 0.04 |
˃100 |
48.7 ± 0.079 |
˃100 |
100% |
34.2% |
| 8 |
Doxorubicin |
21.6 ± 0.05 |
25.5 ± 0.004 |
38.1 ± 0.079 |
48.6 ± 0.084 |
100% |
100% |
| 9 |
DMSO |
— |
— |
— |
— |
1% |
3% |
Table 3 Parameter estimates in the cytotoxic activity tests for the synthesized agents and standard reference (HePG 2)
| Compound |
Parameter |
Estimate |
Z |
Sig |
95% Confidence interval |
| Lower bound |
Upper bound |
| PROBIT model: PROBIT(p) = Intercept + BX. |
| 5a |
PROBITa C |
0.078 |
11.907 |
0.000 |
0.025 |
0.033 |
| Intercept |
−0.946 |
−12.103 |
0.000 |
−1.744 |
−1.573 |
| 5c |
PROBITa C |
0.078 |
11.907 |
0.000 |
0.065 |
0.091 |
| Intercept |
−0.964 |
−12.103 |
0.000 |
−1.044 |
−0.844 |
| 5d |
PROBITa C |
0.053 |
14.271 |
0.000 |
0.046 |
0.060 |
| Intercept |
−1.250 |
−15.819 |
0.000 |
−1.329 |
−1.171 |
| Doxorubicin |
PROBITa C |
0.078 |
14.247 |
0.000 |
0.067 |
0.088 |
| Intercept |
−1.680 |
−17.405 |
0.000 |
−1.777 |
−1.584 |
Table 4 Chi-square tests results
| Compound |
Chi-square |
dfa |
| HePG2 |
MCF7 |
| 5a |
32.467 |
78.020 |
6 |
| 5c |
46.508 |
— |
6 |
| 5d |
62.158 |
— |
6 |
| Doxorubicin |
10.507 |
— |
6 |
2.3.2 MCF7 cell line. Among all the tested compounds, compound 5a showed the most significant antitumor effect (Fig. 6–8).
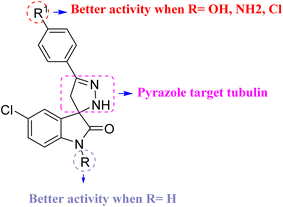 |
| | Fig. 6 Structure–activity relationship (SAR) of spiropyrazoline oxindoles with cytotoxic activity and molecular docking. | |
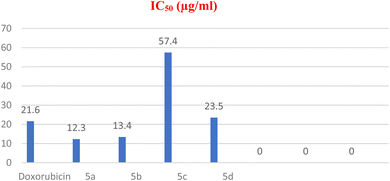 |
| | Fig. 7 Cytotoxic activities of the tested compounds against HepG-2. | |
 |
| | Fig. 8 Cytotoxic activity of new compounds compared with doxorubicin against the HePG-2 cell line. | |
2.3.3 Effect on tubulin polymerization. The most potent synthesized compounds 5a and 5b were evaluated in vitro to verify their effects on tubulin polymerization and to find out if the antitumor activity of these agents resulted from an interaction with tubulin or not (Table 5).
Table 5 Tubulin polymerization inhibition of HIT compounds 5a and 5b
| Compound |
Tubulin polymerization IC50 (μM) |
| 5a |
3.85 ± 0.09 |
| 5b |
4.35 ± 0.11 |
| Tubulin polymerization inhibitor II, referenced under CAS 1151995-69-5 |
4.5 ± 0.13 |
2.3.4 In vitro EGFR inhibition activity. Using computational studies, the enzyme activity against EGFR was evaluated. The results showed that the synthesized compounds were more potent than erlotinib (Table 6). These results further suggest that these new compounds had the correct structural features required to show significant potency against EGFR.
Table 6 IC50 values against EGFR
| Compound |
EGFR IC50 (μM)a |
| IC50 values were calculated as the mean ± SD of three separate experiments. |
| 4a |
0.10 ± 0.07 |
| 4b |
0.05 ± 0.06 |
| 5a |
0.09 ± 0.05 |
| 5b |
0.07 ± 0.09 |
| 5c |
0.08 ± 0.08 |
| 5d |
0.04 ± 0.09 |
| Erlotinib |
0.12 ± 0.04 |
2.4. Molecular docking
Molecular docking is a computational approach employed to forecast the binding behavior of a substance, such as a drug or ligand, with a certain target, usually a protein receptor. It aids in simulating the interaction between a tiny molecule and its biological target, offering valuable information on the strength of their binding and the stability of the resulting complex.37 The evaluation of the ligand's optimal positioning or orientation within the receptor's binding site is often done by determining the binding energy or affinity. The structure validation of a protein can be predicted using a Ramachandran plot (Fig. 9). As energy decreases, contact becomes more stable, indicating that the ligand has the potential to be an effective therapeutic agent.38 The molecular docking analysis of 4a, 4b, 5a, 5b, 5c and 5d compounds against the 4I4T receptor revealed varying binding affinities (Table 7). Compounds 5a and 5b exhibited the strongest binding affinities, both at −8.3 kcal mol−1, suggesting these two compounds had a potentially robust interaction with the receptor. Compound 4a followed closely with a binding affinity of −8.0 kcal mol−1, indicating a similarly strong interaction. Compound 4b showed a moderate binding affinity of −7.6 kcal mol; whereas compound 5c and 5d showed binding affinities of −7.4 and −7.7 kcal mol−1, respectively.
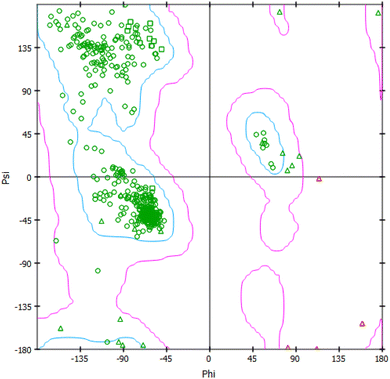 |
| | Fig. 9 Rhamachandran plot of 4I4T. | |
Table 7 Binding affinity of new compounds with protein 4I4T
| Compound |
Binding affinity with 4I4T (kcal mol−1) |
| 5a |
−8.3 |
| 5b |
−8.3 |
| 4a |
−8.0 |
| 4b |
−7.6 |
| 5c |
−7.4 |
| 5d |
−7.7 |
| Doxorubicin |
−7.8 |
| Spirobrassinin |
−5.6 |
| Sunitinib |
−7.3 |
When comparing the tested compounds with three commercially available drugs, doxorubicin displayed a binding affinity of −7.8 kcal mol−1, which was stronger than compound 4b but weaker than compounds 5a, 5b, and 4a. Sunitinib had a binding affinity of −7.3 kcal mol−1, also demonstrating a moderate interaction strength but lower than most of the tested compounds. Spirobrassinin exhibited the weakest binding affinity at −5.6 kcal mol−1. This comparative analysis highlights that compounds 5a, 5b, and 4a outperformed the reference drugs in terms of binding affinity, indicating their potential as promising candidates for further development.
Further, the interactions of the novel compounds with protein are shown in Table 8. Also, 3D and 2D poses were obtained and are presented in Fig. 10. Compound 5a formed two key hydrogen bonds with ARG 284 and GLN 281 at a distance of 2.81 Å, suggesting a strong interaction. Hydrophobic interactions were observed with LEU 286, LEU 371, PRO 274, PRO 360, PHE 272, and ALA 233 at varying distances (between 3.98 to 5.04 Å). This combination of hydrogen bonding and multiple hydrophobic contacts strengthens the binding of compound 5a to the receptor, particularly through interactions with leucine and proline residues. Similar to compound 5a, compound 5b forms hydrogen bonds with GLN 281 (2.85 Å) and ARG 284 (2.79 Å), showing strong hydrogen bonding interactions. The hydrophobic interactions were also similar, involving LEU 286, LEU 371, PRO 274, PRO 360, PHE 272, and ALA 233, with distances ranging from 3.95 to 4.97 Å.
Table 8 Interaction of new compounds with 4I4T
| Compound |
Hydrogen bonds |
Hydrophobic interactions |
| Amino acid |
Distance (Å) |
Amino acid |
Distance (Å) |
| 5a |
ARG 284 |
2.81 |
LEU 286 and 371 |
4.56 and 4.52 |
| |
GLN 281 |
2.81 |
PRO 274 and 360 |
4.20 and 4.95 |
| |
|
PHE 272 |
4.60 |
| |
|
ALA 233 |
3.98 and 5.04 |
| 5b |
GLN 281 |
2.85 |
LEU 286 and 371 |
4.62 and 4.56 |
| |
ARG 284 |
2.79 |
PRO 274 and 360 |
4.16 and 4.93 |
| |
|
PHE 272 |
4.62 |
| |
|
ALA 233 |
3.95 and 4.97 |
| 4a |
THR 276 |
2.32 |
PRO 274 and 360 |
4.83, 4.75, and 5.29 |
| |
GLN 281 |
2.94 |
LEU 371 |
5.37 and 5.31 |
| |
|
ARG 369 |
5.24 |
| |
|
VAL 23 |
4.47 |
| |
|
ALA 233 |
4.10, 4.48, and 5.10 |
| 4b |
THR 276 |
2.45, 2.47 |
LEU 275 and 371 |
5.32 and 5.46 |
| 5d |
THR 276 |
2.50 |
LEU 371 |
4.82 and 5.18 |
| |
GLN 281 |
2.19 |
PRO 360 |
4.32 |
| |
|
|
PHE 272 |
4.62 |
| |
|
|
ALA 233 |
3.50 and 4.96 |
| |
|
|
HIS 229 |
4.62 |
| |
|
|
ARG 278 |
4.22 |
| 5c |
THR 276 |
2.46 |
LEU 371 |
4.84 |
| |
GLN 281 |
2.22 |
ALA 233 |
4.91 |
| |
|
|
HIS 229 |
4.60 |
| |
|
|
LEU 217 |
3.82 |
| |
|
|
LEU 219 |
3.99 |
| |
|
|
ARG 278 |
4.28 |
| Doxorubicin |
ARG 369 |
2.25 |
LEU 371 |
3.79 and 5.18 |
| |
GLY 370 |
2.75 |
LEU 286 |
4.02 |
| |
THR 276 |
2.25, 3.07 |
PRO 274 |
5.13, 4.37, and 4.77 |
| Spirobrassinin |
PRO 274 |
2.19 |
HIS 229 |
5.42 |
| |
THR 276 |
2.53 |
LEU 217 |
3.77 and 5.03 |
| |
LEU 275 |
3.25 |
LEU 371 |
4.84 |
| Sunitinib |
|
|
VAL 23 |
5.00 and 5.02 |
| |
|
|
ALA 233 |
4.52 and 4.64 |
| |
|
|
LEU 371 |
3.88 and 5.07 |
| |
|
|
PHE 272 |
4.99 |
| |
|
|
PRO 360 |
5.45 |
| |
|
|
ARG 369 |
3.71 and 5.04 |
| |
|
|
HIS 229 |
4.34 and 4.90 |
 |
| | Fig. 10 2D and 3D poses for the molecular docking with 4I4T. | |
Compound 4a exhibited hydrogen bonds with THR 276 (2.32 Å) and GLN 281 (2.94 Å), indicating strong polar interactions. The hydrophobic interactions involved PRO 274, PRO 360, LEU 371, ARG 369, VAL 23, and ALA 233, with distances between 4.10 Å and 5.29 Å. For compound 4b, hydrogen bonds with THR 276 (2.45 Å, 2.47 Å) are critical for stabilizing the compound's binding. Hydrophobic interactions were observed with LEU 275, LEU 371, PRO 360, ARG 369, VAL 23, and ALA 233, with interaction distances ranging from 4.12 to 5.46 Å. The pattern of interactions was similar to compound 4a, but the distances were slightly longer.
Compound 5d formed hydrogen bonds with THR 276 (2.50 Å) and GLN 281 (2.19 Å), indicating strong polar interactions. Hydrophobic interactions were observed with LEU 371 (4.82 Å, 5.18 Å), PRO 360 (4.32 Å), PHE 272 (4.62 Å), ALA 233 (3.50 Å, 4.96 Å), HIS 229 (4.62 Å), and ARG 278 (4.22 Å). These interactions suggest a robust binding affinity, particularly through hydrogen bonding and multiple hydrophobic contacts. Compound 5c exhibited hydrogen bonds with THR 276 (2.46 Å) and GLN 281 (2.22 Å), showing strong polar interactions. The hydrophobic interactions involved LEU 371 (4.84 Å), ALA 233 (4.91 Å), HIS 229 (4.60 Å), LEU 217 (3.82 Å), LEU 219 (3.99 Å), and ARG 278 (4.28 Å). The combination of these interactions indicates a strong binding affinity, similar to compound 5d, with significant contributions from both hydrogen bonding and hydrophobic interactions.
Doxorubicin formed strong hydrogen bonds with ARG 369 (2.25 Å), GLY 370 (2.75 Å), and THR 276 (2.25 Å, 3.07 Å), indicating solid interactions with the receptor. The hydrophobic interactions involved LEU 371, LEU 286, and PRO 274, with distances between 3.79 and 5.18 Å. Spirobrassinin formed hydrogen bonds with PRO 274 (2.19 Å), THR 276 (2.53 Å), and LEU 275 (3.25 Å). Hydrophobic interactions with HIS 229, LEU 217, and LEU 371 occurred at distances between 3.77 and 5.42 Å, suggesting relatively strong binding. The interactions were similar in nature to those of doxorubicin but fewer in number. Sunitinib did not form any hydrogen bonds but engaged in hydrophobic interactions with VAL 23, ALA 233, LEU 371, PHE 272, PRO 360, ARG 369, and HIS 229, with distances between 3.71 and 5.45 Å.
Compounds 5a and 5b exhibited the strongest and most diverse interaction profiles, closely followed by compound 4a. Compounds 4b, 5c and 5d showed weaker interaction profiles. Among the reference drugs, doxorubicin demonstrated strong binding through both hydrogen bonding and hydrophobic interactions, while spirobrassinin and sunitinib rely more on hydrophobic interactions.
The substituent groups (–OH, –Cl, –OCH3) play a crucial role in determining the binding affinity and interaction profile of each compound. The hydroxyl group in compound 5a facilitates the formation of strong hydrogen bonds, contributing to its higher binding affinity. The chloro group in compound 5d, while still allowing for hydrogen bonding, resulted in a slightly lower binding affinity compared to compound 5a. The methoxy group in compound 5c, although capable of forming hydrogen bonds, led to the lowest binding affinity observed among the three, possibly due to steric hindrance or a less optimal interaction geometry.
2.4.1 Antitumor activity. The antitumor activity of the new compounds 4a, 4b, 5a, 5c, and 5d were predicted by its docking analysis with the EGFR protein receptor with PDB Id: 1m17;39 validation of the site of activity was done, and the RMSD was 1.057 Å, see Table 9, Fig. 11 and 12.
Table 9 Interaction analysis of new compounds and erlotinib with 1m17
| Ligands |
Binding energy (kcal mol−1) |
Amino acids |
Interactions |
| 4a |
−7.8 |
ALA: A48, VAL: A31, LEU: A23, LEU: A69, LYS: A50, THR: A79, GLU: A67, and MET: A71 |
Pi-alkyl, Conv. H-bond, Pi-sigma, Carbon-H-bond, and Pi-cation |
| 4b |
−8.3 |
ALA: A48, VAL: A31, LEU: A13, LEU: A69, LYS: A50, THR: A79, GLU: A67, and MET: A18 |
Pi-alkyl, Conv. H-bond, Pi-sigma, and Pi-cation |
| 5a |
−7.9 |
ALA: A48, VAL: A31, LEU: A23, LEU: A69, LYS: A50, and PHE: A28 |
Pi-alkyl and alkyl |
| 5b |
−8.1 |
ALA: A48, VAL: A31, LEU: A69, LYS: A50, THR: A15, GLU: A67, and MET: A18 |
Pi-alkyl, Conv. H-bond, Pi-sigma, and Pi-cation |
| 5c |
−8.1 |
ALA: A48, VAL: A31, LEU: A23, LYS: A50, THR: A79, and THR: A15 |
Pi-alkyl, Conv. H-bond, van der Waals, Pi-cation |
| 5d |
−8.3 |
ALA: A48, VAL: A31, LEU: A23, LEU: 69, LYS: A50, and THR: A15 |
Pi-alkyl, Conv. H-bond, van der Waals, and Pi-cation |
| Erlotinib |
−7.3 |
ALA: 719, LEU: 820, VAL: 702, and LEU: 694 |
Pi-sigma, Pi-alkyl, alkyl, and Pi-cation |
 |
| | Fig. 11 2D illustration of interaction between 4a, 4b and 1m17 protein receptor. | |
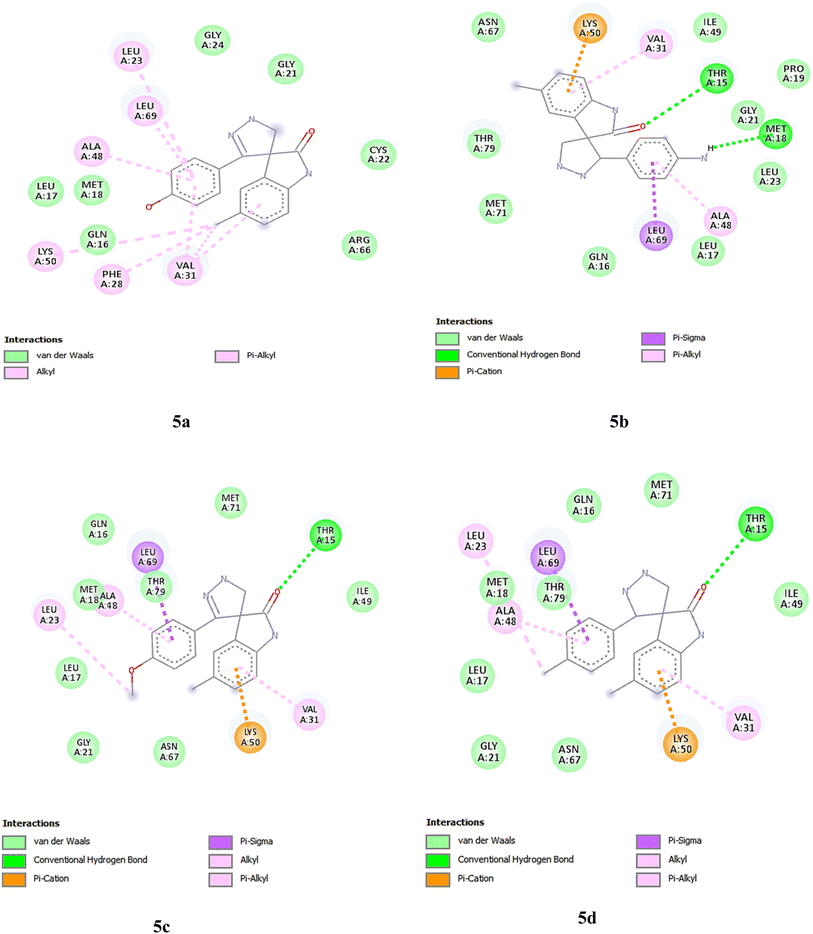 |
| | Fig. 12 2D illustration of interaction between 5a, 5b, 5c, 5d and 1m17 protein receptor. | |
2.4.2 MRSA computational studies on 1MWT. A molecular docking study of the synthesized compounds with MRSA protein (PDB ID 1MWT) was performed. Here, to analyzes the difference in binding efficiency of the newly synthesized compounds against the allosteric site of PBP2a, molecular docking analysis was carried out (Fig. 14 and 15). The site of activity was determined, and the RMSD was found to be 0.21 Å (Fig. 13 and Table 10).
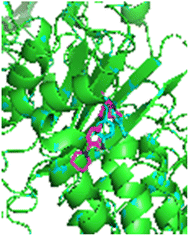 |
| | Fig. 13 3D illustration of the active site with superimposed co-crystal (cyan) and re-docked (pink) linezolid. | |
 |
| | Fig. 14 2D illustrations of interactions between 4a, 4b. 5a, 5b, 5c, 5d and the 1mwt protein receptor. | |
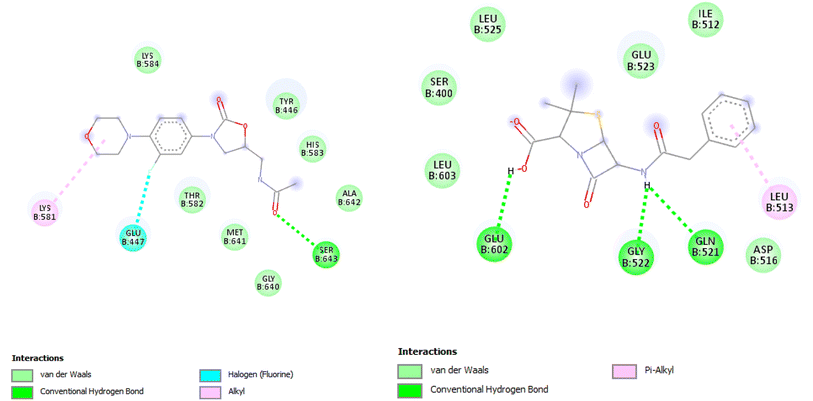 |
| | Fig. 15 2D illustration of the interaction between linezolid, penicillin and the 1mwt protein receptor. | |
Table 10 Interaction analysis of new compounds with 1mwt
| Ligands |
Binding energy (kcal mol−1) |
Amino acids |
Interactions |
| 4a |
−7.9 |
ALA: A48, VAL: A31, LEU: A23, LEU: A69, LYS: A50, THR: A79, GLU: A67, and MET: A71 |
Pi-alkyl, Conv. H-bond, Pi-sigma, carbon-H-bond, and Pi-cation |
| 4b |
−6.6 |
ALA: A48, VAL: A31, LEU: A13, LEU: A69, LYS: A50, THR: A79, GLU: A67, and MET: A18 |
Pi-alkyl, Conv. H-bond, Pi-sigma, and Pi-cation |
| 5a |
−6.4 |
ALA: A48, VAL: A31, LEU: A23, LEU: A69, LYS: A50, and PHE: A28 |
Pi-alkyl and alkyl |
| 5b |
−6.7 |
ALA: A48, VAL: A31, LEU: A69, LYS: A50, THR: A15, GLU: A67, and MET: A18 |
Pi-alkyl, Conv. H-bond, Pi-sigma and Pi-cation |
| 5c |
−6.8 |
ALA: A48, VAL: A31, LEU: A23, LYS: A50, THR: A79, and THR: A15 |
Pi-alkyl, Conv. H-bond, van der Waals, and Pi-cation |
| 5d |
−6.7 |
ALA: A48, VAL: A31, LEU: A23, LEU: 69, LYS: A50, and THR: A15 |
Pi-alkyl, Conv. H-bond, van der Waals, and Pi-cation |
| Linezolid |
−6.5 |
ALA: 719, LEU: 820, LYS: 721, VAL: 702, LEU: 694, and LEU: 764 |
Pi-alkyl, alkyl, Pi-cation, and Pi-sigma |
| Penicillin |
−6.3 |
|
|
2.4.3 Molecular dynamics. Molecular dynamics (MD) study offers a valuable understanding of the ever-changing behavior of systems. MD simulations aid in comprehending the binding mechanisms between drug candidates and their biological targets.
2.4.4 Root mean square deviation (RMSD). RMSD is a widely employed metric in MD simulations for quantifying a molecule's structural variations or conformational alterations, specifically proteins, nucleic acids, or tiny molecules, over the simulation period.40–44 It quantifies the average distance between atoms in a specific structure at different time points compared to a reference structure. RMSD gives an indication of how much the system is deviating from its starting structure. A stable system will show low RMSD values after an initial equilibration period, indicating that the structure is not undergoing large fluctuations.45,46 Fig. 16 shows the RMSD curve of the complexes formed for compounds 5a and 5b with the protein 4I4T. For both, there was a gradual increase in RMSD in the first 10 ns, suggesting that the system was undergoing equilibration, moving away from the initial configuration as it finds a more stable conformation. The RMSD for compound 5a remained relatively stable after 20 ns, indicating that it had reached equilibrium and it then stayed close to this conformation throughout the simulation. The moderate fluctuations suggest that the compound is relatively stable but undergoes some dynamic motion. For compound 5b, after 10 ns, the RMSD gradually increased to around 0.25 nm and remained fluctuating between 0.225–0.275 nm.
 |
| | Fig. 16 RMSD curve of compounds 5a and 5b with 4I4T. | |
2.4.5 Root mean square fluctuation (RMSF)47. The RMSF curve shows the flexibility of each residue in the molecular system over the course of the MD simulation for the two compounds, denoted as compound 5a (black) and compound 5b (red) (Fig. 17). The RMSF provides a measure of how much each residue fluctuates relative to its average position during the simulation. Compound 5a showed distinct peaks at several residue numbers, indicating regions of higher flexibility. Notably, residues around the positions 50, 150, and 425 displayed higher RMSF values, with fluctuations reaching up to 0.3 nm at specific points. Most of the middle regions of the system (residues between 100 and 400) exhibited relatively low fluctuations, suggesting that these regions were more rigid and stable during the simulation. For compound 5b, its general RMSF pattern was quite similar to that of compound 5b, with peaks at similar residue positions, but with some differences in magnitude. The fluctuation at residue 50 was slightly higher compared to compound 5a, reaching approximately 0.3 nm. Compound 5b showed consistently higher RMSF values between the residues, suggesting that this region may exhibit more flexibility or mobility compared to compound 5a.
 |
| | Fig. 17 RMSF curve of compounds 5a and 5b with 4I4T. | |
2.4.6 Number of hydrogen bonds (H-bonds)48. Fig. 18 shows the number of H-bonds for compounds 5a and 5b formed during the simulations with protein 4I4T. Compound 5a consistently formed a higher number of hydrogen bonds, fluctuating mostly between 3 and 5 H-bonds. This indicates that compound 5a maintained a relatively stable and strong interaction with its surrounding environment, contributing to its overall stability during the molecular dynamics (MD) simulation. Peaks reaching 6 H-bonds could be observed at various points, particularly early in the simulation. These occasional spikes suggested periods where compound 5a formed additional hydrogen bonds, possibly because of favorable conformational changes. After an initial equilibration phase, which seemed to last around the first 10 ns, compound 5a stabilized, consistently forming a robust hydrogen bond network that likely contributed to the structural rigidity observed in the earlier RMSD and RMSF analyses. Compound 5b formed fewer hydrogen bonds, fluctuating predominantly between 1 and 3 H-bonds for most of the simulation. This indicates a more dynamic or flexible interaction, with fewer stable hydrogen bond interactions compared to compound 5a. The lower number of H-bonds suggests that compound 5b may exhibit more conformational flexibility or less interaction stability.
 |
| | Fig. 18 Number of H-bonds of compounds 5a and 5b with 4I4T. | |
The MD simulations of compounds 5a and 5b with the 4I4T protein provided valuable insights into their dynamic behavior and stability. Compound 5a demonstrated a stable interaction profile, maintaining a consistent RMSD after the initial equilibration phase and forming a robust hydrogen bond network throughout the simulation. This stability was further supported by the relatively low RMSF values, indicating minimal fluctuations in the protein–ligand complex. In contrast, compound 5b exhibited higher RMSD and RMSF values, suggesting greater conformational flexibility. The lower number of hydrogen bonds formed by compound 5b also indicates its fewer stable interactions compared to compound 5a. Overall, the MD simulations confirmed that compound 5a had a more stable and stronger interaction with the 4I4T protein, making it a promising candidate for further development.
3 In silico studies
3.1. Molecular docking
The molecular docking of compounds 5a, 5b, 4a, 4b, 5c, and 5d, along with three market-available drugs, namely doxorubicin, spirobrassinin, and sunitinib, was done using Autodock Tools.49 The structures of the compounds and the drugs were drawn using ChemDraw software.50 The crystal structure of a protein with PDB ID 4I4T was downloaded from the RCSB protein data bank.51 Further, the ligands and the protein were converted to PDBQT using Autodock, and the coordinates of the active site were obtained using Discovery Studio visualizer. The size of the grid box was taken as 20 Å × 20 Å × 20 Å. Also, for further analysis to obtain 2D and 3D poses as well as the distances of the interactions taking place, Discovery Studio was used.
3.2. Molecular dynamics (MD) simulations
MD simulations for the complexes of compound 5a and 5b with 4I4T were performed using GROMACS software. The ligand was derived from the findings of the molecular docking study utilizing Discovery Studio. The protein topology was generated using the CHARMM27 force field. The Swiss Param Server produced the ligand topology and data that are compatible with GROMACS. The system was hydrated using the TIP3P water model.52,53 After applying PBC (periodic boundary condition) correction and eliminating the solvents, ions, and other particles from the 100 ns calculations, GROMACS software's built-in functions were utilized to compute essential parameters, such as the root mean square deviation (RMSD), root mean square fluctuation (RMSF), and multiple hydrogen bonds (H-bonds) formed between the protein and the ligand.54,55
3.3. Conclusion of the computational study
The comprehensive silico study, encompassing both molecular docking and MD simulations, highlights the potential of compounds 5a and 5b as effective therapeutic agents targeting the 4I4T protein. The molecular docking results identified compounds 5a and 5b as having the highest binding affinities, supported by strong hydrogen bonding and hydrophobic interactions. The subsequent MD simulations reinforced these findings, with compound 5a showing superior stability and interaction strength. The study underscores the importance of combining molecular docking with MD simulations to gain a holistic understanding of the binding mechanisms and dynamic behavior of potential drug candidates. These findings pave the way for the further experimental validation and optimization of compounds 5a and 5b, contributing to the development of novel therapeutic agents.
3.4. ADMET analysis40
The results are presented in Table 11 and Fig. 19.
Table 11 In silico physicochemical properties of compounds 4a, 4b, 5a, 5b, 5c and 5d
| Compound |
MW |
MR |
TPSA |
LOGP |
H-bond donors |
H-bond acceptors |
Rotatable bonds |
| 4a |
299.71 |
83.61 |
66.4 |
2.03 |
2 |
3 |
2 |
| 4b |
298.72 |
85.99 |
72.19 |
1.99 |
2 |
2 |
2 |
| 5a |
313.74 |
94.37 |
73.72 |
1.74 |
3 |
3 |
1 |
| 5b |
327.76 |
98.83 |
62.72 |
2.42 |
2 |
3 |
2 |
| 5c |
312.75 |
96.75 |
79.51 |
1.77 |
3 |
2 |
1 |
| 5d |
332.18 |
97.35 |
53.49 |
2.47 |
2 |
2 |
1 |
 |
| | Fig. 19 Biological activity and physicochemical parameters of 4a,4b, 5a, 5b, 5c and 5d. | |
4 Experimental section
General: The starting material, solvents, and reagents were purchased from Sigma Aldrich, Merck, Alfa Aser, and were used without further purification. The reactions were monitored by TLC using pre-coated silica gel aluminum plates (Kieselgel 60, 254, E. Merck, Germany) and visualized under a UV lamp (254 nm or 365 nm). The melting points of the compounds were determined using a Veego digital melting point apparatus and are reported uncorrected. Elemental analysis was performed on a Vario Micro cube CHNS analyzer from Elementar. The IR spectra of the synthesized compounds were recorded on a Shimadzu FTIR 8400S instrument. The 1H NMR (400 MHz) spectra were recorded on a Bruker spectrometer.
4.1. General procedure: synthesis of 5-chloro-3-hydroxy-3-(2-(4-substituted phenyl)-2-oxoethyl)indolin-2-one 3a,b
An equimolar mixture of 5-chloroindoline-2,3-dione (1) (15 mmol, 1.47 g) and acetophenone derivatives (2a,b) (15 mmol) was refluxed for 5 h in absolute ethanol (20 mL) using diethylamine. After the reaction was completed, the reaction was allowed to cool; then the obtained white crystals were separated out, filtered, and recrystallized from ethanol to give white crystals of compounds 3a,b.
4.1.1 5-Chloro-3-hydroxy-3-(2-(4-hydroxyphenyl)-2-oxoethyl)indolin-2-one 3a. White crystals, m.p 180–182 °C, yield 91%. 1H NMR (400 MHz, DMSO) δ 10.40 (s, 1H), 10.35 (s, 1H), 7.75 (d, J = 8.7 Hz, 2H), 7.35 (d, J = 2.0 Hz, 1H), 7.20 (dd, J = 8.2, 2.1 Hz, 1H), 6.87–6.75 (m, 3H), 6.13 (s, 1H), 4.02 (d, J = 17.6 Hz, 1H), 3.51 (d, J = 17.6 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 194.99, 178.62, 162.75, 142.40, 134.66, 130.97, 128.98, 128.17, 125.53, 124.31, 115.68, 111.20, 73.59, 45.61. Analysis for C16H12ClNO4 (317.72): calcd. (%) C 60.49; H 3.81; N 4.41; found C 60.72; H 4.01; N 4.54.
4.1.2 3-(2-(4-Aminophenyl)-2-oxoethyl)-5-chloro-3-hydroxyindolin-2-one 3b. White crystals, m.p 178–180 °C, yield 88%. 1H NMR (400 MHz, DMSO) δ 10.30 (s, 1H), 7.78–7.09 (m, 4H), 6.89–6.39 (m, 3H), 6.07 (s, 2H), 4.33 (s, 1H), 3.92 (s, 2H). 13C NMR (101 MHz, DMSO) δ 205.49, 193.75, 178.77, 154.35, 142.43, 134.89, 130.80, 125.46, 124.21, 112.91, 111.14, 73.70, 45.32. Analysis for C16H13ClN2O3 (316.74): calcd. (%) C 60.67; H 4.14; N 8.84; C 60.78; H 4.31; N 9.01.
4.2. General procedure: synthesis of 5-chloro-3-hydroxy-3-(2-(4-substituted phenyl)-2-oxoethyl)indolin-2-one 4a,b
Compounds 3a,b (15 mmol) were refluxed in glacial acetic acid (10 mL) and HCl (6 mL) using a water bath for 1 h. The reaction was then allowed to cool. The obtained orange-red crystals were separated and filtered to give compounds 4a,b.
4.2.2 (E)-5-Chloro-3-(2-(4-hydroxyphenyl)-2-oxoethylidene)indolin-2-one 4a. Red needles, m.p 197–199 °C, yield 85%. 1H NMR (400 MHz, DMSO) δ 10.89 (s, 1H), 10.67 (s, 1H), 8.04 (s, 1H), 7.98 (d, J = 8.4 Hz, 2H), 7.76 (s, 1H), 7.38 (d, J = 6.3 Hz, 1H), 6.93 (d, J = 8.4 Hz, 2H), 6.89 (d, J = 6.3 Hz, 1H). 13C NMR (101 MHz, DMSO) δ 189.07, 168.17, 163.37, 143.56, 134.88, 132.11, 131.71, 128.74, 128.28, 126.30, 125.66, 121.65, 116.01, 111.90. Analysis for C16H10ClNO3 (299.71): calcd. (%) C 64.12; H 3.36; N 4.67; C 63.89; H 3.71; N 4.91.
4.3. General procedure: synthesis of 5-chloro-5′-(4-substituted phenyl)-2′,4′-dihydrospiro[indoline-3,3′-pyrazol]-2-one 5a–e
Compounds 3a–e was refluxed with hydrazine hydrate in ethanol (20 mL) using diethylamine (6 drops) for 6–8 h. After cooling, acetic acid (5 mL) was added. Compounds 5a–e was precipitated, filtered, and recrystallized from ethanol.
4.3.1 5-Chloro-5′-(4-hydroxyphenyl)-2′,4′-dihydrospiro[indoline-3,3′-pyrazol]-2-one 5a. Off-white crystals, m.p 230–232 °C, yield 75%. FTIR: νmax/cm−1 3352, 3267, 3194, 3250, 3032, 2850, 1651, 1589, 1516, 1470, 825.740; 1H NMR (400 MHz, DMSO) δ 10.05 (s, 1H), 9.97 (s, 1H), 8.26 (s, 1H), 8.17 (d, J = 8.5 Hz, 2H), 8.07 (d, J = 10.2 Hz, 2H), 7.79 (d, J = 8.5 Hz, 1H), 6.93 (d, J = 8.5 Hz, 2H), 4.72 (s, 2H). 13C NMR (101 MHz, DMSO) δ 166.0, 160.05, 156.75, 146.91, 140.75, 130.98, 129.47, 129.17, 124.63, 117.77, 116.23, 111.38, 70.28, 44.86. (EI-MS) m/z: 313.55, M+2: 315.22. Analysis for C16H12ClN3O2 (313.74): calcd. (%) C 61.25; H 3.86; N 13.39; found C 61.44; H 3.91; N 13.62.
4.3.2 5′-(4-Aminophenyl)-5-chloro-2′,4′-dihydrospiro[indoline-3,3′-pyrazol]-2-one 5b. Off-white crystals, m.p 235–237 °C, yield 70%. FTIR: νmax/cm−1 3437, 3321, 3286, 3217, 3082, 2920, 2850, 1639, 1585, 1523, 1489, 1180, 883, 694; 1H NMR (400 MHz, DMSO) δ 10.02 (s, 1H), 8.21 (d, J = 2.1 Hz, 1H), 8.06–7.96 (m, 4H), 7.75 (dd, J = 8.5, 2.1 Hz, 1H), 6.69 (d, J = 8.5 Hz, 2H), 5.68 (s, 2H, exchangeable with D2O), 4.70 (s, 2H). 13C NMR (101 MHz, DMSO) δ 166.15, 157.11, 151.55, 147.01, 140.38, 130.74, 129.02, 125.27, 124.56, 117.35, 114.17, 65.36, 44.75. (EI-MS) m/z: 312.84, M+2: 314.26. Analysis for C16H13ClN4O (312.75): calcd. (%) C 61.45; H 4.19; N 17.91; found C 61.74; H 4.31; N 18.02.
4.3.3 5-Chloro-5′-(4-methoxyphenyl)-2′,4′-dihydrospiro[indoline-3,3′-pyrazol]-2-one 5c. White crystals, m.p 255–257 °C, yield 72%. FTIR: νmax/cm−1 3267, 3194, 3085, 2858, 1651, 1516, 1489, 1238, 1153, 825, 740; 1H NMR (400 MHz, DMSO) δ 10.08 (s, 1H), 8.27 (d, J = 8.5 Hz, 2H), 8.23 (s, 1H), 8.14 (s, 1H), 8.09 (d, J = 8.9 Hz, 1H), 7.81 (d, J = 8.9 Hz, 1H), 7.12 (d, J = 8.5 Hz, 2H), 4.74 (s, 2H), 3.85 (s, 3H). 13C NMR (101 MHz, DMSO) δ 165.50, 161.10, 156.01, 146.47, 140.43, 131.44, 130.61, 130.26, 128.92, 123.97, 117.48, 114.40, 64.12, 55.40, 46.70. (EI-MS) m/z: 327.50, M+2: 329.95. Analysis for C17H14ClN3O2 (327.76): calcd. (%) C 62.30; H 4.31; N 12.82; found C 62.73; H 4.51; N 12.98.
4.3.4 5-Chloro-5′-(4-chlorophenyl)-2′,4′-dihydrospiro[indoline-3,3′-pyrazol]-2-one 5d. White crystals, m.p 242–244 °C, yield 85%. FTIR: νmax/cm−1 3263, 3089, 2924, 2850, 1643, 1624, 1531, 1485, 1087, 887, 750; 1H NMR (400 MHz, DMSO) δ 10.09 (s, 1H), 8.34 (d, J = 8.5 Hz, 2H), 8.30 (s, 1H), 8.22 (s, 1H), 8.15 (d, J = 9.0 Hz, 1H), 7.86 (dd, J = 9.0, 2.2 Hz, 1H), 7.65 (d, J = 8.5 Hz, 2H), 4.75 (s, 2H). 13C NMR (101 MHz, DMSO) δ 165.70, 155.56, 146.83, 141.18, 137.09, 135.60, 132.13, 131.34, 129.50, 124.74, 118.29, 67.79, 45.52. (EI-MS) m/z: 332.16 M+2: 334.65, M+4: 336.87Analysis for C16H11Cl2N3O (332.18): calcd. (%) C 57.85; H 3.34; N 12.65; found C 58.03; H 3.51; N 12.78.
Abbreviations
| MD | Molecular dynamics |
| PBP2a | Penicillin binding protein 2a |
| MTT | 3-[4,5-Dimethylthiazol-2-yl]-2,5 diphenyl tetrazolium bromide |
| SAR | Structure–Activity relationship |
| TIP3P | Transferable intermolecular potential with 3 points |
| MW | Molecular weight |
| ADMET | Absorption distribution metabolism excretion toxicity |
| MR | Molar refractivity |
| PSA | Polar surface area |
| CYP | Cytochrome P450 |
| DEA | Diethyl amine |
Biological studies
The procedures for the biological studies are mentioned in the ESI file.†
Data availability
Data related to this research paper are included in the ESI† and within the paper.
Conflicts of interest
The authors declare that they have no conflicts of interest related to this work.
Acknowledgements
I would like to express my sincere appreciation to the editors and anonymous reviewers for their valuable time, constructive comments, and insightful suggestions, which have greatly improved the quality and clarity of this manuscript. This research was self-funded and carried out in Dr Seliem's laboratory.
References
- G. Kaur, R. Gupta, N. Hooda and N. R. Gupta, Wirel. Pers. Commun., 2022, 125, 2537–2564 CrossRef.
- T. Al-Warhi, M. Abualnaja, O. A. Abu Ali, N. M. Alyamani, F. G. Elsaid, A. A. Shati, S. Albogami, E. Fayad, A. H. Abu Almaaty, K. O. Mohamed, W. M. Alamoudi and I. Zaki, symmetry, 2022, 14(9), 1814 CrossRef CAS.
- E. A. Rakha, G. M. Tse and C. M. Quinn, Histopathology, 2023, 82, 5–16 CrossRef PubMed.
- J. S. Modica-Napolitano and V. Weissig, Treatment Strategies that Enhance the Efficacy and Selectivity of Mitochondria-Targeted Anticancer Agents, MDPI AG, 2015.
- S. Khwaja, K. Kumar, R. Das and A. S. Negi, Bioorg. Chem., 2021, 116, 105320 CrossRef CAS PubMed.
- R. Kaul, A. L. Risinger and S. L. Mooberry, J. Nat. Prod., 2019, 82, 680–685 CrossRef CAS PubMed.
- M. Chalkha, K. Chebbac, H. Nour, A. Nakkabi, A. El Moussaoui, B. Tüzün, M. Bourhia, S. Chtita, M. Bakhouch, H. Laaroussi, S. M. A. Kawsar, T. Ben Hadda, G. Al Houari, M. Augustyniak, M. A. M. Aboul-Soud and M. El Yazidi, Arabian J. Chem., 2023, 105465 Search PubMed.
- S. M. M. Kamal, A. Hossain, S. Sultana, V. Begum, N. Haque, J. Ahmed, T. M. A. Rahman, K. A. Hyder, S. Hossain, M. Rahman, R. A. Chowdhury, K. J. M. Aung, A. Islam, R. Hasan and A. Van Deun, Int. J. Tuberc. Lung Dis., 2015, 19, 151–156 CrossRef CAS PubMed.
- A. M. Al-Majid, H. M. Ghawas, M. S. Islam, S. M. Soliman, F. F. El-Senduny, F. A. Badria, M. Ali, M. R. Shaik, H. A. Ghabbour and A. Barakat, J. Mol. Struct., 2020, 1204, 127500 CrossRef CAS.
- K. Gangarapu, G. Thumma, S. Manda, A. Jallapally, R. Jarapula and S. Rekulapally, Med. Chem. Res., 2017, 26, 819–829 CrossRef CAS.
- Y. Zhang, Y. Qin, D. Tang, M. Yang, B. Li, Y. Wang, H. Cai, B. Wang and H. Zhu, ChemMedChem, 2016, 11, 1446–1458 CrossRef CAS PubMed.
- S. Sridhar and Y. Rajendraprasad, E-J. Chem., 2012, 9, 1810–1815 CAS.
- D. M. Lokeshwari, D. K. Achutha, B. Srinivasan, N. Shivalingegowda, L. N. Krishnappagowda and A. K. Kariyappa, Bioorg. Med. Chem. Lett., 2017, 27, 3806–3811 CrossRef CAS PubMed.
- Z. Uğraş, F. Tok, C. Çakir, K. Tuna, G. Tatar-Yilmaz, D. Mutlu, Y. Sicak, Ş. Arslan, M. Öztürk and B. Koçyіğіt-Kaymakçioğlu, J. Mol. Struct., 2024, 1315, 138978 CrossRef.
- Ö. D. Can, Ü. D. Özkay, Z. A. Kaplancıklı and Y. Öztürk, Arch. Pharmacal Res., 2009, 32, 1293–1299 CrossRef PubMed.
- S. Kumar, S. Bawa, S. Drabu, R. Kumar and H. Gupta, Recent Pat Antiinfect Drug Discov., 2009, 4, 154–163 CrossRef CAS PubMed.
- P. M. Sivakumar, S. Ganesan, P. Veluchamy and M. Doble, Chem. Biol. Drug Des., 2010, 76, 407–411 CrossRef CAS PubMed.
- F. Hayat, A. Salahuddin, S. Umar and A. Azam, Eur. J. Med. Chem., 2010, 45, 4669–4675 CrossRef CAS PubMed.
- T. A. Farghaly, A. M. Abo Alnaja, H. A. El-Ghamry and M. R. Shaaban, Bioorg. Chem., 2020, 102, 104103 CrossRef CAS PubMed.
- L. R. Raposo, A. Silva, D. Silva, C. Roma-Rodrigues, M. Espadinha, P. V. Baptista, M. M. M. Santos and A. R. Fernandes, Bioorg. Med. Chem., 2021, 30, 115880 CrossRef CAS PubMed.
- C. Karthikeyan, V. R. Solomon, H. Lee and P. Trivedi, BPN, 2013, 3, 325–330 Search PubMed.
- S. Pramanik, S. Ray, S. Maity, P. Ghosh and C. Mukhopadhyay, Synthesis, 2021, 53, 2240–2252 CrossRef CAS.
- M. N. Ibrahim, M. F. El-Messmary and M. G. A. Elarfi, E-J. Chem., 2010, 7, 55–58 CAS.
- B. Yu, D. Q. Yu and H. M. Liu, Rasayan J. Chem., 16, 3, 1599–1604 Search PubMed.
- C. Reichardt, Solvents and Solvent Effects in Organic Chemistry. Wiley-VCH, Weinheim/Germany, 2003 Search PubMed.
- M. J. Horgan, L. Zell, B. Siewert, H. Stuppner, D. Schuster and V. Temml, J. Chem. Inf. Model., 2023, 63, 6396–6411 CrossRef CAS PubMed.
- V. W. Y. Liao, A. Kumari, R. Narlawar, S. Vignarajan, D. E. Hibbs, D. Panda and P. W. Groundwater, Mol. Pharmacol., 2020, 97, 409–422 CrossRef CAS PubMed.
- A. E. Prota, M. M. Magiera, M. Kuijpers, K. Bargsten, D. Frey, M. Wieser, R. Jaussi, C. C. Hoogenraad, R. A. Kammerer, C. Janke and M. O. Steinmetz, J. Cell Biol., 2013, 200, 259–270 CrossRef CAS PubMed.
- P. Giannakakou, D. Sackett and T. Fojo, JNCI, J. Natl. Cancer Inst., 2000, 92, 182–183 CrossRef CAS PubMed.
- E. A. Geyer, A. Burns, B. A. Lalonde, X. Ye, F.-A. Piedra, T. C. Huffaker and L. M. Rice, Elife, 2015, 10113 CrossRef PubMed.
- S. Zhu, K. T. Mc Henry, W. S. Lane and G. Fenteany, Chem. Biol., 2005, 12, 981–991 CrossRef CAS PubMed.
- F. Ciardiello and G. Tortora, N. Engl. J. Med., 2008, 358, 1160–1174 CrossRef CAS PubMed.
- C. Fuda, D. Hesek, M. Lee, K. Morio, T. Nowak and S. Mobashery, J. Am. Chem. Soc., 2005, 127, 2056–2057 CrossRef CAS PubMed.
- C. Fuda, D. Hesek, M. Lee, K. Morio, T. Nowak and S. Mobashery, J. Am. Chem. Soc., 2005, 127, 2056–2057 CrossRef CAS PubMed.
- A. E. Prota, K. Bargsten, D. Zurwerra, J. J. Field, J. F. Díaz, K.-H. Altmann and M. O. Steinmetz, Science, 2013, 339, 587–590 CrossRef CAS PubMed.
- T. Mosmann, J. Immunol. Methods, 1983, 65, 55–63 CrossRef CAS PubMed.
- A. Rani, M. Aslam, J. Khan, G. Pandey, P. Singh, R. S. Maharia and B. N. Pant, Chem. Biodiversity, 2024, 21(10), e202400782 CrossRef PubMed.
- S. Yadav, S. Sewariya, M. Babu Singh, B. Sachdeva, P. Singh, R. Chandra, S. Kukreity, S. K. Singh and K. Kumari, ChemistrySelect, 2024, 9, e202400770 CrossRef CAS.
- I. A. Seliem, Bioorg. Chem., 2024, 150, 107601 CrossRef CAS PubMed.
- N. Mahmoodi, M. Bayat, D. Gheidari and Z. Sadeghian, J. Saudi Chem. Soc., 2024, 28, 101894 CrossRef CAS.
- D. Gheidari, M. Mehrdad and M. Bayat, PLoS One, 2024, 19, e0299301 CrossRef CAS PubMed.
- D. Gheidari, M. Mehrdad and M. Bayat, J. Biomol. Struct. Dyn., 2024, 42, 7860–7873 CrossRef CAS PubMed.
- D. Gheidari, M. Mehrdad and Z. karimelahi, Sci. Rep., 2024, 14, 26431 CrossRef CAS PubMed.
- Z. Amiri, M. Bayat and D. Gheidari, BMC Chem., 2025, 19, 90 CrossRef CAS PubMed.
- M. Babu Singh, Himani, S. Yadav and P. Singh, ChemistrySelect, 2024, 9, e202304767 CrossRef CAS.
- S. Yadav, S. Sewariya, P. Singh, R. Chandra, P. Jain and K. Kumari, Chem. Biodiversity, 2024, e202400495 CrossRef CAS PubMed.
- Z. Amiri, M. Bayat and D. Gheidari, BMC Chem., 2025, 90 CrossRef CAS PubMed.
- D. Gheidari, M. Mehrdad and Z. Karimelahi, Sci. Rep., 2024, 14, 26431 CrossRef CAS PubMed.
- G. M. Morris, R. Huey, W. Lindstrom, M. F. Sanner, R. K. Belew, D. S. Goodsell and A. J. Olson, J. Comput. Chem., 2009, 30, 2785–2791 CrossRef CAS PubMed.
- K. R. Cousins, J. Am. Chem. Soc., 2005, 127, 4115–4116 CrossRef CAS.
- H. M. Berman, J. Westbrook, Z. Feng, G. Gilliland, T. N. Bhat, H. Weissig, I. N. Shindyalov and P. E. Bourne, Nucleic Acids Res., 2000, 28, 235–242 CrossRef CAS PubMed.
- S. Boonstra, P. R. Onck and E. van der Giessen, J. Phys. Chem. B, 2016, 120, 3692–3698 CrossRef CAS PubMed.
- H. J. C. Berendsen, D. van der Spoel and R. van Drunen, Comput. Phys. Commun., 1995, 91, 43–56 CrossRef CAS.
- R. Reza, T. Dutta, N. Baildya, N. N. Ghosh, A. A. Khan and R. K. Das, Microb. Pathog., 2022, 169, 105615 CrossRef CAS PubMed.
- M. Aslam, G. Pandey, N. Deshwal, A. Kumar, K. Kumari, I. Bahadur, P. Singh, F. Mohammad and A. Abdullah Soleiman, J. Mol. Liq., 2024, 397, 124070 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*