
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioanalytical method development and validation for determination of olutasidenib and its application to pharmacokinetic studies
Madhusudhana Reddy Nimmakayalaa,
Kuruva Rangamunia,
Jasti Surendrab,
Jatla Murali Prakasha and
Deepti Kolli *a
*a
aDepartment of Chemistry, Koneru Lakshmaiah Education Foundation, Green Fields, Vaddeswaram, Guntur, Andhra Pradesh 522302, India. E-mail: atluri.deepti1984@gmail.com; Tel: +91 9490494699
bDepartment of Mechanical Engineering, Prasad V Potluri Siddhartha Institute of Technology, Kanuru, Vijayawada, Andhra Pradesh, India
First published on 8th July 2025
Abstract
Olutasidenib is an inhibitor licensed by the FDA, indicated against mutations in isocitrate dehydrogenase-1 (IDH1). For individuals with vulnerable IDH1 mutations, it has been demonstrated to be a very effective therapy for recurrent or refractory acute myeloid leukemia (AML). After a long review procedure, olutasidenib was finally given an approval by the FDA in December of 2022. To determine the concentration of olutasidenib in rat plasma, an LC-MS/MS approach was applied. The drug ibrutinib serves as a standard for comparison. Inertsil ODS, 150 mm × 4.6 mm, 3.5 μm, mobile phase was Acetonitrile (ACN), and Ammonium formate buffer (AmF), pH 3.0 (50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 v/v) at 1.0 ml min−1 was used for the separation process. Liquid–liquid extraction (LLE) was adopted for both olutasidenib and the Internal standard (IS). Proton adducts of olutasidenib and ibrutinib were observed at m/z 354.8589 and 239.8107 and m/z 441.573 and 372.1236 in MRM positive mode, correspondingly. The approach was shown accurate throughout a range of 3.0–60.0 ng ml−1 and correlation values of (r2) ≥ 0.999.6 replicates including olutasidenib at 4 distinct QC levels were analyzed to determine intra-assay precision and accuracy; the Coefficient of variations (CV) were reported to be 3.41% to 0.58% to 0.31% to 0.36, and the accuracy ranged from 97.40, 99.69, 99.4, and 99.16%, respectively, for LOQQC, LQC, MQC, and HQC. In a pharmacokinetic investigation using rat plasma, this strategy has proven effective.
:50 v/v) at 1.0 ml min−1 was used for the separation process. Liquid–liquid extraction (LLE) was adopted for both olutasidenib and the Internal standard (IS). Proton adducts of olutasidenib and ibrutinib were observed at m/z 354.8589 and 239.8107 and m/z 441.573 and 372.1236 in MRM positive mode, correspondingly. The approach was shown accurate throughout a range of 3.0–60.0 ng ml−1 and correlation values of (r2) ≥ 0.999.6 replicates including olutasidenib at 4 distinct QC levels were analyzed to determine intra-assay precision and accuracy; the Coefficient of variations (CV) were reported to be 3.41% to 0.58% to 0.31% to 0.36, and the accuracy ranged from 97.40, 99.69, 99.4, and 99.16%, respectively, for LOQQC, LQC, MQC, and HQC. In a pharmacokinetic investigation using rat plasma, this strategy has proven effective.
Introduction
USFDA has given approval to the use of olutasidenib, a novel medication, to treat recurring or refractory acute myeloid leukemia (AML) in adults who are sensitive to IDH1 mutation.1,2 Rezlidhia is the commercial name for the medicine olutasidenib, which is the active component in this medication.3,4 Individuals with AML who have a poor prognosis often have mutant IDH1 enzymes, which this medication works to combat. The acceptance of olutasidenib as a therapy for AML is a major step forward. The therapy choices for individuals with recurrent or refractory AML were severely restricted prior to the approval of olutasidenib. Nausea, tiredness, aches and pains, constipation, white blood cell count increase, difficulty breathing, temperature increase, rash, mucositis, diarrhoea, and transaminitis are among the most prevalent unwanted effects.5 Clinical studies of olutasidenib, however, have shown encouraging results, and the drug was officially approved by the FDA in December 2022. Isocitrate dehydrogenase (IDH) inhibitors, such as olutasidenib, are a kind of medication. The mutant version of IDH1, which is prevalent in individuals with AML, is the primary target of IDH inhibitors. It is believed that 2-hydroxyglutarate buildup, caused by the IDH1 mutation, is essential in the initiation and advancement of AML. AML Patients have benefited greatly from the usage of IDH inhibitors like olutasidenib. Therapeutic studies of individuals with IDH1-mutated AML and myelodysplastic syndrome demonstrated that olutasidenib was effective and exhibited good therapeutic efficacy.The science of pharmacokinetics investigates how medications are taken in, transported across the body, broken down, and removed. The therapeutic effectiveness, toxicity, and dosage schedule of a medicine are all heavily influenced by its pharmacokinetics.6,7 Thus, it is crucial to learn the pharmacokinetics of olutasidenib to create a successful treatment plan for individuals with AML. There is a lack of data about olutasidenib's pharmacokinetics.
The recent approval by the FDA for the use of olutasidenib represents a significant milestone in the treatment of AML in adults.8 Olutasidenib, marketed under the name Rezlidhia, is a ground-breaking medication designed to target and address a specific genetic mutation known as Isocitrate dehydrogenase-1 (IDH1). This approval marks a crucial advancement in the field of oncology.9
Patients suffering from recurrent or refractory AML, particularly those with mutant IDH1 enzymes, often face limited treatment options and poor prognoses. Olutasidenib's mechanism of action, which specifically targets mutant IDH1 enzymes, offers newfound hope for these individuals. By inhibiting these aberrant enzymes, olutasidenib has the potential to disrupt the growth and proliferation of leukemia cells, improving the prognosis for patients.10
The FDA's decision to greenlight olutasidenib for AML underscores its commitment to advancing innovative therapies and improving outcomes for patients facing this challenging disease. This approval paves the way for healthcare professionals to incorporate olutasidenib into their treatment protocols, providing a more tailored and effective approach for eligible AML patients, ultimately enhancing the quality of care and patient outcomes in the fight against this devastating form of leukemia.11
Before olutasidenib's approval, the therapeutic options available for individuals grappling with recurrent or refractory Acute Myeloid Leukemia (AML) were notably limited. This dearth of effective treatments left patients and healthcare providers with few choices for managing this aggressive disease. However, olutasidenib's approval represents a pivotal moment in AML treatment.12
This calls for an analysis of olutasidenib's pharmacokinetics and a determination of its concentration by means of LC-MS/MS in rat plasma. A reliable and accurate approach for determining drug levels in biological matrices, LC-MS/MS, was used for measuring olutasidenib. LC-MS/MS is a very effective analytical method for separating and detecting analytes in complicated matrices. Quantifying olutasidenib by means of LC-MS/MS is a crucial step in elucidating the drug's metabolism and distribution, leading to a better overall knowledge of its pharmacokinetics. Fig. 1 shows the structure of olutasidenib.
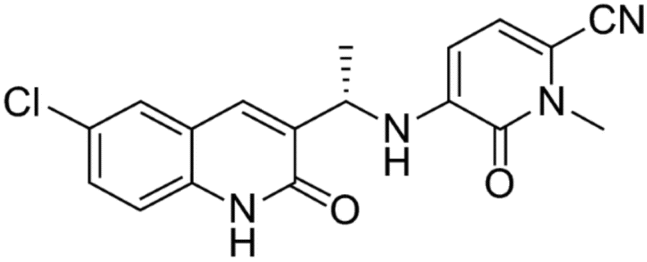 | ||
| Fig. 1 Structure of olutasidenib. | ||
So far, LC-MS/MS techniques have not been developed for the purpose of quantifying olutasidenib. Olutasidenib quantitation using RP-HPLC has been developed,13 however this approach is only useful for bulk pharmaceutical formulations. A more targeted and sensitive analytical method is necessary for accurate pharmacokinetic assessment. LC-MS/MS has several benefits over RP-HPLC, such as more selectivity in intricate biological matrices, lower detection limits, and enhanced resilience for pharmacokinetic research. In order to guarantee accurate pharmacokinetic assessment as well as therapeutic monitoring, this work focuses on establishing and verifying an LC-MS/MS approach for precise evaluation of olutasidenib in plasma.
Methods
Chemicals and reagents
Moltus Research Laboratories Tarapur, M.I.D.C. Boisar, Maharashtra, kindly supplied the reference sample of olutasidenib. All chemicals, including HPLC-grade acetonitrile (ACN) and methanol (MeOH), were acquired from Merck chemical division, Mumbai. Water purification system by Milli-Q was supplied for the experiment. Manisha Analytical Laboratories in Mumbai, Maharashtra, India, was the source for the rat plasma used in this study.Instrumentation
The Waters alliance e-2695 model HPLC system, having a column oven, auto sampler, and degasser, was utilized for the purpose of analysis. The HPLC equipment was connected to a SCIEX QTRAP 5500 mass spectrometer that was outfitted with an electrospray ionisation interface. The data from the chromatogram was analysed using SCIEX software.Olutasidenib stock solution (120 ng ml−1)
A quantity of 6 mg of olutasidenib working standard was measured and thereafter put into a 10 ml volumetric flask. The flask was then filled to its full volume with a suitable diluent. Sonication and subsequent vortexing was performed to produce a dissolved and uniform solution. The initial volume of 0.1 ml was further diluted with diluent to a final volume of 10 ml. Transfer 0.2 millilitres of the aforementioned solution into a 10 millilitres volumetric flask, and thereafter fill the flask to the desired volume with a suitable diluent.Preparation of internal standard stock solution (120 ng ml−1)
A 6 mg quantity of ibrutinib working standard was measured and thereafter put into a 10 ml volumetric flask. The flask was then filled to its full volume with a diluent. The initial volume of 0.1 ml was then diluted to a final volume of 10 ml using a diluent. Transfer 0.2 ml of the aforementioned solution into a volumetric flask (10 ml), and afterwards fill the flask to the desired volume with a suitable diluent.Preparation of standard solution (30 ng ml−1 of olutasidenib)
The standard stock solution was transferred into a 2 ml centrifuge tube, using a volume of 500 μl. To the mixture, include 200 μl of plasma, 500 μl of IS, 300 μl of ACN, and 500 μl of diluent. Subject the sample to centrifugation for a duration of 20 minutes. The supernatant liquid should be filtered and afterwards transferred into a container suitable for High Performance Liquid Chromatography (HPLC).Buffer preparation
The procedure involves precisely measuring and transferring 6.3 grams of ammonium formate into a 1 liter volume of HPLC water. The mixture should be thoroughly stirred and its pH should be adjusted to a value of 3.0 using formic acid. The sample was passed through a filter paper of 0.22μ.Preparation of mobile phase
Combine equal volumes of acetonitrile and ammonium formate buffer in a 1:1 ratio. The solution was passed through a membrane filter paper of 0.45μ.
Detection
Olutasidenib (m/z, 239.8107) and ibrutinib (m/z, 372.1236); collision energy: 15 V; optimized mass parameters yielded product ions of m/z, 354.8589 and m/z, 441.5732; drying gas and source temperatures range from 120–250 and 550 °C, correspondingly, while the ion spray voltage is 5500 volts. Nitrogen for use as collision gas, the following parameters were determined for the drying gas; flow stream: 5 l min−1, declustering potential 40 V, entrance potential 10 V, exit potential 7 V, and dwell duration 1 s.Chromatographic conditions
At room temp., 1.0 ml min−1 of a mobile phase having ACN and AmF, pH 3.0 (50:50 v/v) was used for separation on an analytical column of Intertsil ODS, 150 mm × 4.6 mm, 3.5 μm. As an internal reference for chromatography and extractability, ibrutinib was analyzed. With an overall run duration of 5 minutes, the drug and IS were extracted at 2.965 and 2.046 minutes, correspondingly.
Standards and QC samples preparation
Both drug and IS were produced as 30.0 ng ml−1 stock solutions in a solvent. Freezer settings (2 °C to 8 °C) were maintained for the storage of these solutions and internal standard spiking solutions prior to analysis. Quality control (QC) standards were prepared by adding olutasidenib standard stock solution to evaluate clean rat plasma at concentrations of 3.0, 7.5, 15.00, 22.50, 30.00, 45.00, and 60.0 ng ml; these were then frozen at 30 °C until investigation. ACN was used to generate aqueous standards, which were then maintained in fridge at 2–8 degrees Celsius until investigation.Sample preparation
The drug and IS were extracted from rat plasma by LLE. Because of its better sensitivity, enhanced analyte recovery, and reduced matrix effects, Liquid–liquid extraction (LLE) was selected over Solid phase extraction (SPE) and Protein precipitation (PPT).14–17 In order to do this, 500 μL of dilutant, standard (appropriate concentration), and IS were introduced to marked polypropylene tubes, along with 200 μL of plasma sample, and the tubes were quickly vortexed. Then, after adding and vortexing for 10 minutes, a total of 300 μL of extracting solvent (ACN) was introduced. The materials went through a centrifuge for 15–20 minutes at 20 °C and 4000 rpm. After that, we placed the supernate from each sample into separate polypropylene tubes and gave them a quick swirling. After extraction, the material was placed in auto-sampler vials and then fed into the column.Bioanalytical method validation18–33
:1.LOQ is minimum detectable level of a drug in any sample that yields reliable results. The LOQ was determined by studying olutasidenib concentrations in both mobile phase and plasma standards.
At both the low and high quality control levels, duplicate extractions of six different batches of blank biological matrices were performed and then post-spiked with the aqueous standard. The coefficient of variation (% CV) provides a measure of accuracy for the matrix factor and a value of <15% is considered acceptable.
6 Male Sprague-Dawley rats (SD rats) were given olutasidenib (6 mg g−1 b.w.) orally through a BD syringe linked to an oral gavage needle (size 18), and the devised LC-MS/MS technology was effectively adopted for the pharmacokinetic investigation. Each rat was given isoflurane anesthesia, and blood samples were taken via retro-orbital sampling and placed into tubes carrying 10% of K2EDTA anticoagulant (20 μL) at regular intervals. Samples were taken at 0, 1, 2, 4, 8, 16, 32, 64, and 128 hours after dosing. Every rat's blood sample was between 1.9 and 2.2 ml, which is much below the 20% maximum that is advised. Approximately ten to fifteen minutes of centrifugation at a speed of three thousand revolutions per minute were required to separate plasma from whole blood. The collected samples were placed in microcentrifuge tubes with labels and frozen at 30 °C. The developed approach was applied to all samples. Non-compartmental analysis was used to determine pharmacokinetic parameters with the help of the WinNonlin (version 5.2) program. The proposed approach was employed to produce concentration data (FDA 2002; FDA 2003). Incurred sample reanalysis (ISR) verified the consistency of the study's samples. Every subject had two samples taken at different times throughout the pharmacokinetic profile's Cmax and clearance phase for ISR. Not exceeding a twenty percent discrepancy was allowed between samples, at which point they were deemed stable.
Results and discussion
Method development and validation
Due to its specificity, sensitivity, and consistency, LC-MS/MS has proven to be among the useful analytical approaches in clinical pharmacokinetics. The purpose of this research was to design and verify a straightforward assay for the quantitative assessment of olutasidenib in rat plasma. Multiple studies were performed to find the optimal chromatographic conditions for maximizing the resolution and boosting the detection of olutasidenib and IS. These included the makeup and properties of the mobile phase, the use of distinct columns, and various techniques like SPPE and LLE for sample extraction. Directly injecting olutasidenib and IS solutions into the spectroscope's ESI pump was used to achieve optimal MS performance. Improved ionizing of the protonated ionic olutasidenib and IS was achieved by optimizing the ESI source's vital factors, including the needle and capillary voltages, source temperature, heater, nebulizer, and desolvation gases. Olutasidenib and IS both had abundant fragment ions at m/z 354.8589 and m/z 441.5732 in the corresponding product ion spectra (see Fig. 2 and 3, accordingly). To get a quick and selective LC approach, we first tuned the MS settings, and then we adjusted the chromatographic conditions, including the mobile phase, column, and extraction procedure. At 1.0 ml min−1 with an injection volume of 10 μL, a mobile phase of ACN and AmF was found to be optimal for extraction and recovery. Fig. 4 shows the optimal chromatographic conditions: an Intertsil ODS column (150 mm × 4.6 mm, 3.5 m) and a LLE procedure. The new approach was shown to be accurate throughout a linear range of concentrations, from 3.00 to 60.0 ng ml−1. Selectivity, LOD, LOQ, matrix effect, accuracy, reliability, recovery, and stability were all assessed as part of the validation process. | ||
| Fig. 2 MRM of olutasidenib parent and product ions. | ||
 | ||
| Fig. 3 MRM of ibrutinib parent ion and product ion. | ||
 | ||
| Fig. 4 Optimized chromatogram. | ||
Selectivity and specificity
There were no interfering chemicals in the MRM study of olutasidenib and IS. Fig. 5 and 6 displays chromatograms of olutasidenib and ibrutinib-spiked plasma. The drug's retention time of 2.965 minutes and the internal standard's retention time of 2.046 minutes demonstrate that he approach is highly selective and can efficiently separate the two compounds. When the stationary phase interacts with it, separation takes place without interference or peak overlap. This separation, along with the absence of peak overlap, confirms that the method is highly specific, ensuring interference-free identification and measurement. Therefore, the results indicate that the olutasidenib analysis using the proposed LC-MS/MS method is both highly selective and specific.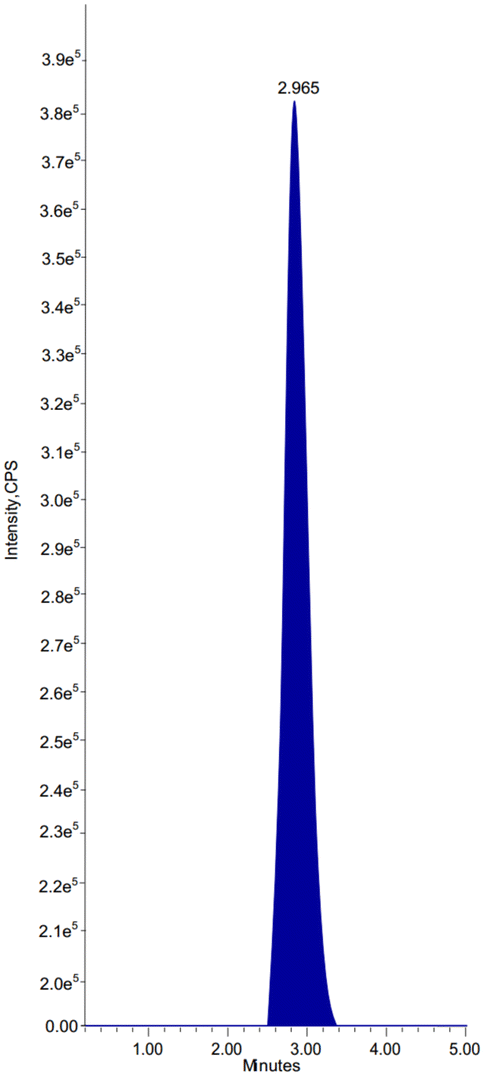 | ||
| Fig. 5 Chromatogram of olutasidenib. | ||
 | ||
| Fig. 6 Chromatogram of ibrutinib. | ||
Limit of detection and quantification
Based on the response standard deviation (σ) and the calibration curve slope (S), the LOD and LOQ values were calculated (in accordance with ICH recommendations). The LOD was calculated as . Similarly, the LOQ was calculated as
. Similarly, the LOQ was calculated as 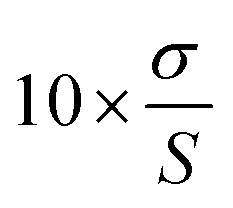 . Table 1 shows the instrument detection values for olutasidenib at very low doses based on the LOD. The LOD for the analyte was determined to be 3 ng ml−1, demonstrating that the approach is sensitive enough to identify analyte concentrations as low as 3 ng ml−1. Additionally, the analyte's LOQ was found to be 10 ng ml−1, indicating that the approach can accurately and consistently measure analyte concentrations as low as 10 ng ml−1.
. Table 1 shows the instrument detection values for olutasidenib at very low doses based on the LOD. The LOD for the analyte was determined to be 3 ng ml−1, demonstrating that the approach is sensitive enough to identify analyte concentrations as low as 3 ng ml−1. Additionally, the analyte's LOQ was found to be 10 ng ml−1, indicating that the approach can accurately and consistently measure analyte concentrations as low as 10 ng ml−1.
| Drug name and details | LOD (ng ml−1) | LOQ (ng ml−1) |
|---|---|---|
| Olutasidenib (S/B) value | 3 | 10 |
Matrix effect
For accuracy, selectivity, and sensitivity to remain unaffected, it is necessary to assess the matrix effect. The aqueous standard was post-spiked into six batches of blank biological mediums at the intermediate QC level, and the samples were assessed against neat standards at identical levels in alternating injections. Olutasidenib has a matrix factor of 0.9973, which indicates a high degree of accuracy. This further indicates that the matrix does not significantly impact the measurement of the analyte. Additionally, the effects of IS and the drug on ion suppression/enhancement were not seen at their expected retention durations.Ion suppression or enhancement in LC-MS/MS analysis can be greatly affected by species-specific differences in protein binding, enzyme activity, and phospholipid composition. In particular, human plasma is known to have a higher protein content and different metabolic enzyme profiles than rat plasma, which could result in distinct matrix effects.15 Therefore, it is important for subsequent studies to evaluate and take into consideration matrix effects in human plasma to guarantee the reliability and accuracy of the technique in clinical contexts. This will allow for a smooth transition from preclinical to clinical trials.
Linearity
The linearity of the standard curves was seen across 3–60 ng ml−1 of olutasidenib, this means that the approach can reliably quantify olutasidenib in biological samples across this concentration range (Table 2). The calculated average correlation coefficient was determined to be 0.999. It demonstrates that, across the measured range, the analytical response (peak area ratio) is directly proportional to the analyte concentration. The quantification of samples was performed by calculating the ratio between the peak area of the analyte and the peak area of the IS. The graphic Fig. 7 depicted the relationship between peak area ratios and plasma concentrations.| Final conc. (ng ml−1) | Res | Area response ratio |
|---|---|---|
| 0 | 0 | 0.0 |
| 3.00 | 0.392 × 105 | 0.102 |
| 7.50 | 0.936 × 105 | 0.245 |
| 15.00 | 1.927 × 105 | 0.500 |
| 22.50 | 2.934 × 105 | 0.760 |
| 30.00 | 3.852 × 105 | 0.992 |
| 37.50 | 4.761 × 105 | 1.228 |
| 45.00 | 5.689 × 105 | 1.486 |
| 60.00 | 7.584 × 105 | 1.990 |
| Slope | 0.0330 | |
| Intercept | 0.00206 | |
| R2 value | 0.99985 | |
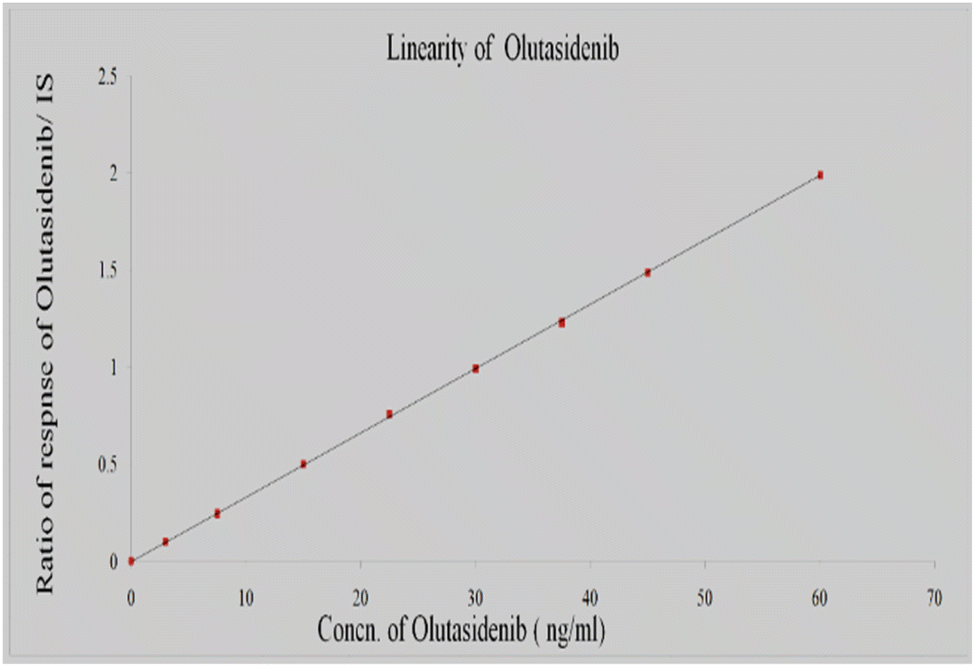 | ||
| Fig. 7 Calibration plot for concentration vs. area ratio of olutasidenib. | ||
Precision and accuracy
The estimation of intra-assay precision and accuracy was conducted by analysing six duplicates consisting of olutasidenib at four distinct quality control (QC) levels. The assessment of inter-assay precision involved the analysis of four tiers of quality control (QC) samples across four distinct runs. The acceptance standards for the data encompass two key aspects: accuracy and precision. Accuracy is defined as the data falling within a range of 85–115% of the actual values. Precision, on the other hand, is determined by the relative standard deviation (RSD) being within ±15%, except for the LLQC. For LLQC, the accuracy should be within 80–120% and the RSD should be less than 20%. The accuracies for LLQC, LQC, MQC, and HQC were 97.40%, 99.69%, 99.40%, and 99.16%, respectively, while the coefficient of variation (% CV) ranged from 0.31% to 3.41%. If the method's mean% accuracy is between 85% and 115%, and its mean% CV is less than 15% across all QC levels, it is considered both accurate and precise. The low % CV demonstrates the method's precision and consistency, while the accuracy is satisfactory. These results (Table 3) indicate that the approach is accurate and can be reliably used for bioanalytical or clinical analyte quantification.| Parameter | HQC | MQC | LQC | LLQC |
|---|---|---|---|---|
| Nominal conc. (ng ml−1) | ||||
| 45.01 | 30.0 | 15.0 | 3.0 | |
| n | 6 | 6 | 6 | 6 |
| Mean | 5.669 × 105 | 3.827 × 105 | 1.919 × 105 | 0.375 × 105 |
| SD | 0.021 | 0.012 | 0.011 | 0.013 |
| % CV | 0.36 | 0.31 | 0.58 | 3.41 |
| % Mean accuracy | 98.16% | 99.40% | 99.69% | 97.40% |
Recovery of analyte
The evaluation of olutasidenib and IS recovery was conducted at three distinct concentration levels, specifically LQC, MQC, and HQC. The calculation of recovery involved the comparison of the response of duplicate samples with the response of the neat reference solution.The recovery of an analyte from a sample matrix, also known as extraction efficiency, involves comparing the analytical response obtained from a certain analyte quantity added to the response obtained from the sample matrix. The extraction process was conducted utilizing a mobile phase solvent due to the fundamental characteristics of olutasidenib. Table 4 presents the mean recovery at each QC level. The recovery rates for LQC, MQC, and HQC are 99.38%, 99.32%, and 98.22%, respectively. With recoveries so close to 100%, these results demonstrate the efficiency of the extraction procedure. The % CV values across all QC levels are very low, ranging from 0.21% to 0.67%. This indicates that the recovery procedure is highly precise, with minimal variation across the replicates. By averaging the recoveries at the LQC, MQC, and HQC concentrations, the overall recovery is 99.10%. This confirms that the technique is capable of consistently and reliably quantifying analytes across different concentrations, without significant variation due to analyte-related factors or sample preparation.
| — | Extracted LQC | Unextracted LQC | Extracted MQC | Unextracted MQC | Extracted HQC | Unextracted HQC |
|---|---|---|---|---|---|---|
| 1 | 1.916 × 105 | 1.924 × 105 | 3.811 × 105 | 3.824 × 105 | 5.674 × 105 | 5.686 × 105 |
| 2 | 1.897 × 105 | 1.907 × 105 | 3.829 × 105 | 3.836 × 105 | 5.683 × 105 | 5.694 × 105 |
| 3 | 1.903 × 105 | 1.912 × 105 | 3.814 × 105 | 3.819 × 105 | 5.688 × 105 | 5.693 × 105 |
| 4 | 1.908 × 105 | 1.915 × 105 | 3.826 × 105 | 3.832 × 105 | 5.649 × 105 | 5.662 × 105 |
| 5 | 1.919 × 105 | 1.926 × 105 | 3.839 × 105 | 3.845 × 105 | 5.675 × 105 | 5.684 × 105 |
| 6 | 1.933 × 105 | 1.941 × 105 | 3.827 × 105 | 3.837 × 105 | 5.663 × 105 | 5.679 × 105 |
| Mean | 1.913 × 105 | 1.921 × 105 | 3.824 × 105 | 3.832 × 105 | 5.672 × 105 | 5.683 × 105 |
| SD | 0.013 | 0.012 | 0.010 | 0.009 | 0.014 | 0.012 |
| % CV | 0.67 | 0.64 | 0.27 | 0.25 | 0.25 | 0.21 |
| % Mean recovery | 99.38% | 99.79% | 99.32% | 99.53% | 98.22% | 98.41% |
| Overall recovery | 99.10% | |||||
Stability
Table 5 describe the short-term stability of olutasidenib in plasma after it has been exposed to 3 cycles of freezing/thawing (−30 °C to ambient temp.), an autosampler, and benchtop. The medication passed the stability tests, as the percentage CV of the freeze-thaw, autosampler, and benchtop samples was less than 15%. The CV% results ranged from 0.20% to 0.77%, indicating excellent accuracy and negligible degradation. These stability tests demonstrate that the drug's concentration remains relatively constant, suggesting that it remains stable under varying conditions suitable for sample analysis and storage. Stability tests under these conditions are recommended by current regulatory guidelines, with variable degrees of accuracy. For example, stability on a benchtop for 4–24 hours and at least 3 freeze/thaw cycles are required. From a scientific perspective, however, it is of utmost importance to address the application-specific maximum storage duration and number of cycles.| Stability | Spiked plasma conc. (ng mL−1) | Conc. (ng mL−1) (mean ± SD; n = 6) | (CV%) (n = 6) |
|---|---|---|---|
| Freeze–thaw | 15.0 | 1.924 × 105 ± 0.007 | 0.37 |
| 45.0 | 5.670 × 105 ± 0.018 | 0.32 | |
| Autosampler | 1.911 × 105 ± 0.015 | 0.77 | |
| 5.665 × 105 ± 0.018 | 0.32 | ||
| Benchtop | 1.1916 × 105 ± 0.014 | 0.73 | |
| 5.657 × 105 ± 0.012 | 0.22 | ||
| Short term stability | 1.881 × 105 ± 0.009 | 0.20 | |
| 5.626 × 105 ± 0.011 | 0.5 |
Table 6 shows results from long-term stability experiments, demonstrating that the medication has strong plasma stability, as indicated by the low CV% in the observed concentrations over time. Although there is a slight drop in concentration over the 28 days period, this is typical of drugs tested for long-term stability and suggests that the drug remains mostly unchanged during this time. This type of stability research may help better predict the drug's storage conditions and shelf life.
| Days | Spiked plasma conc. (ng mL−1) | Conc. (ng mL−1) (mean ± SD; n = 6) | (CV%) (n = 6) |
|---|---|---|---|
| Day 1 | 15.0 | 1.918 × 105 ± 0.013 | 0.69 |
| 45.0 | 5.683 × 105 ± 0.012 | 0.22 | |
| Day 7 | 1.878 × 105 ± 0.007 | 0.35 | |
| 5.618 × 105 ± 0.011 | 0.20 | ||
| Day 14 | 1.842 × 105 ± 0.010 | 0.55 | |
| 5.546 × 105 ± 0.009 | 0.17 | ||
| Day 21 | 1.800 × 105 ± 0.011 | 0.55 | |
| 5.486 × 105 ± 0.009 | 0.16 | ||
| Day 28 | 1.688 × 105 ± 0.008 | 0.46 | |
| 5.367 × 105 ± 0.011 | 0.24 |
Pharmacokinetics and statistical analysis
The internal standard (IS) and olutasidenib were both extracted using liquid–liquid extraction (LLE). To measure olutasidenib concentrations in the plasma of SD rats, the optimized and validated LC-MS/MS technique was used. Six separate rats were dosed with the analyte, and blood samples were collected at various intervals: 0, 1, 2, 4, 8, 16-, 32-, 64-, and 128-hours post-dosing. Once the samples were prepared, they were injected into the chromatographic system, and the results were recorded according to the test method. Olutasidenib concentrations were effectively quantified into a single dosage (6 mg/200 g b.w.) orally through a BD syringe linked to an oral gavage needle (size 18). Maximum drug concentration (Cmax), the area under the curve (AUC0–128) (24 hours trapezoid rule), time to achieve Cmax (Tmax), elimination rate constant (Kel) (established using a semi-log graph of the plasma level-time plot (by applying the least-squares method)), and half-life (T1/2; calculated by quotient 0.693/Kel) were the variables assessed. Table 7 provides information on pharmacokinetics. Fig. 8 depicts the average olutasidenib plasma concentration vs. time profile in rats.| Time intervals (hours) | Olutasidenib (ng ml−1) |
|---|---|
| 1 | 9.765 |
| 2 | 18.317 |
| 4 | 27.012 |
| 8 | 22.190 |
| 16 | 15.210 |
| 32 | 7.298 |
| 64 | 2.379 |
| 128 | 0 |
 | ||
| Fig. 8 Recovery plot for olutasidenib in rat plasma. | ||
A concentration of 9.765 ng ml−1 at 1 hour indicates rapid drug absorption into the circulation. After two hours, the concentration increases to 18.317 ng ml−1, and after four hours, it rises further to 27.012 ng ml−1. This shows that the drug is entering the bloodstream and continues to circulate. After 8 hours, the concentration begins to decrease to 22.190 ng ml−1, suggesting that the medication is being metabolized or eliminated from the body. The concentration continues to fall, reaching 15.210 ng ml−1 after 16 hours. It decreases further to 7.298 ng ml−1 at the 32nd hour, and drops to 2.379 ng ml−1 after 64 hours. The medication is almost entirely eliminated from the body by 128 hours, as the concentration reaches 0 ng ml−1. Olutasidenib appears to have a half-life characterized by a slow decline in concentration until it is fully excreted from the body. When the drug's clearance rate is directly proportional to its concentration, it likely follows first-order kinetics.
Conclusion
The decision was made to employ ibrutinib as the IS. To isolate the drugs, a mobile phase consisting of 50:50 ACN and ammonium formate buffer was used, with a flow rate of 1 ml min−1. The mobile phase was paired with an Inertsil ODS column measuring 150 mm × 4.6 mm and having a 3.5 μm particle size. The drug and the IS both exhibited proton adducts at approximately m/z 354.8589–239.8107 and m/z 441.573–372.1236–143.7695–76.7964, respectively, which could be detected via MRM positive modalities. The method demonstrated a linearity range of 3.00–60.00 ng ml−1 with an r2 value of 0.9996. Intraday precision and accuracy for this approach were 96.40% and 99.59%, respectively.
According to the results of the stability studies, benchtop testing, and three freeze-thaw cycles, olutasidenib remained stable. The goal of this study was to investigate the oral pharmacokinetics of olutasidenib by quantifying the drug in rat plasma using a newly developed and well-established LC-MS/MS analytical method. The pharmacokinetic (PK) properties of olutasidenib demonstrated an immediate onset of action followed by absorption. The collected data have the potential to pave the way for olutasidenib's clinical and preclinical studies while also providing benchmarks and valuable information for the continued development of new medications and pharmacology pathways.
Data availability
All pertinent data supporting the conclusions of this study are presented within the article. Certain data access is restricted due to privacy or ethical considerations.Author contributions
All authors have made equal contributions to the research findings and writing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors gratefully acknowledge the support provided by the Koneru Lakshmaiah Education Foundation, Vaddeswaram, Guntur, Andhra Pradesh. Authors like to acknowledge the DST through technical support from SR/PURSE/2023/196.Notes and references
- Rigel Announces U.S., FDA Approval of Rezlidhia (olutasidenib) for the Treatment of Adult Patients with Relapsed or Refractory Acute Myeloid Leukemia with a Susceptible IDH1 Mutation, Rigel Pharmaceuticals, Inc., 1 December 2022, Retrieved 2 December, 2022.
- Rigel Announces U.S., FDA Approval of Rezlidhia (olutasidenib) for the Treatment of Adult Patients with Relapsed or Refractory Acute Myeloid Leukemia with a Susceptible IDH1 Mutation. Rigel Pharmaceuticals, 1 December 2022, Retrieved 2 December, 2022 – via PR Newswire.
- Rezlidhia-olutasidenib Capsule, DailyMed, 13 December 2022, Retrieved 21 January, 2023 Search PubMed.
- https://www.accessdata.fda.gov/drugsatfda_docs/appletter/2022/215814Orig1s000ltr.pdf, This article incorporates text from this source, which is in the public domain.
- U.S. Food and Drug Administration (FDA), FDA Approves Olutasidenib for Relapsed or Refractory Acute Myeloid Leukemia with a Susceptible IDH1 Mutation, 1 December, 2022, Retrieved 20 December, 2022 Search PubMed.
- World Health Organization, International Nonproprietary Names for Pharmaceutical Substances (INN): Recommended INN: List 82, WHO Drug Information, 2019, vol. 33(3), hdl:10665/330879 Search PubMed.
- M. Thompson, S. L. Ellison and R. Wood, Pure Appl. Chem., 2008, 74(5), 835–855 CrossRef.
- J. E. Megías-Vericat, O. Ballesta-López, E. Barragán and P. Montesinos, IDH1-mutated relapsed or refractory AML: current challenges and future prospects, Blood Lymphatic Cancer, 2019, 9, 19–32, DOI:10.2147/BLCTT.S177913.
- REZLIDHIA (olutasidenib), FDA-approved for treatment of adult patients with relapsed or refractory acute myeloid leukemia, available at biologics by McKesson, December 19, 2022, Accessed December 21, 2022.
- The American Cancer Society, Key Statistics for Acute Myeloid Leukemia (AML), Revised January 12, 2022, Accessed Aug. 1, 2022 at https://www.cancer.org/cancer/acute-myeloid-leukemia/about/key-statistics.html.
- Leukaemia Care, Relapse in Acute Myeloid Leukaemia (AML), Version 3. Reviewed October 2021, 2019, Accessed Dec 2, 2021 at https://media.leukaemiacare.org.uk/wpcontent/uploads/Relapse-in-Acute-Myeloid-Leukaemia-AML-Web-Version.pdf.
- F. Thol, R. F. Schlenk, M. Heuser and A. Ganser, How I treat refractory and early relapsed acute myeloid leukemia, Blood, 2015, 126(3), 319–327, DOI:10.1182/blood-2014-10-551911.
- P. R. Sankar, K. H. Sri, C. V. P. Rao and G. Jhansi, Stability Indicating RP-HPLC Method for the Estimation of Olutasidenib in Bulk Pharmaceutical Formulations, Pharmaceutical Science: New Insights and Developments, 2025, pp. 35–69, DOI:10.9734/bpi/psnid/v2/4023.
- H. Trufelli, P. Palma, G. Famiglini and A. Cappiello, Matrix effects in liquid chromatography-mass spectrometry, J. Mass Spectrom., 2011, 46(3), 309–321 Search PubMed.
- P. Panuwet, R. E. Hunter, P. E. D'Souza, X. Chen, S. A. Radford, J. R. Cohen and D. B. Barr, Biological matrix effects in quantitative tandem mass spectrometry-based analytical methods: Advancing biomonitoring, Crit. Rev. Anal. Chem., 2016, 46(2), 93–105 CrossRef CAS PubMed.
- A. R. Chaves, P. Fernández and M. P. Lorenzo, The role of liquid-liquid extraction in bioanalysis: Applications in LC-MS/MS drug quantification, Biomed. Chromatogr., 2020, 34(12), e4956 CrossRef PubMed.
- G. Vandermeer, H. Lauwers and R. T'Kindt, Comparison of extraction techniques in LC-MS bioanalysis: LLE vs. SPE vs. PPT, Anal. Bioanal. Chem., 2019, 411(17), 4267–4275 Search PubMed.
- U.S. Department of Health and Human Services, Food and Drug Admnistration and Center for Drug Evaluation and Research, Guidance for industry: Bioanalytical method validation, 2018, https://www.fda.gov/regulatory-information/search-fda-guidance-documents/bioanalytical-method-validation-guidance-industry.
- N. Sakurai, Y. Nakamura, H. Kawagachi, J. Abe, K. Yamada, K. Nagayama and H. Kakeya, Chem. Pharm. Bull., 2019, 67, 439–444 CrossRef CAS PubMed.
- T. Belabbas, T. Yamada, Y. Tsuchiya, K. Suetsugu, N. Egashira and I. Ieiri, Chem. Pharm. Bull., 2021, 69, 646–651 CrossRef CAS PubMed.
- G. Srinubabu, B. Veera Venkata Ratnam, A. Appa Rao and M. N. Rao, Chem. Pharm. Bull., 2008, 56, 28–33 CrossRef CAS PubMed.
- W.-S. Ku, H.-J. Cho, In-S. Yoon, J. H. Kim, B.-J. Cha, S. K. Jung, K.-M. Kim, S.-K. Kang, S.-J. Chung, C.-K. Shim and D.-D. Kim, Chem. Pharm. Bull., 2011, 59, 1083–1088 CrossRef CAS PubMed.
- B. Narasimhan, A. Khan and K. Srinivas, Chem. Pharm. Bull., 2008, 56, 413–417 CrossRef CAS PubMed.
- R. Wood, Trends Anal. Chem., 2005, 18, 624–632 CrossRef.
- A. Thulaseedhar, M. Karunasree, N. Amar Babu LA and K. Aravind, Int. J. Appl. Pharm. Sci., 2022, 5(14), 53–61 Search PubMed.
- M. L. Chiu, W. Lawi, S. T. Snyder, P. K. Wong, J. C. Liao and V. Gau, J. Assoc. Lab. Autom., 2010, 15, 233–242 CrossRef CAS.
- International Conference on Harmonization (ICH), Validation of Analytical Methods: Methodology, ICH Q2B, 1996 Search PubMed.
- EURACHEM, The Fitness for Purpose of Analytical Methods—A Laboratory Guide to Method Validation and Related Topics, 1998 Search PubMed.
- V. S. Lalit, N. P. Bhagwat, V. U. Sharad, V. W. Pradeepkumar and H. S. Laxman, Pharm. Anal. Acta, 2014, 5, 3 Search PubMed.
- M. Thompson, S. L. R. Ellison and R. Wood, Pure Appl. Chem., 2002, 74, 835–855 CrossRef CAS.
- C. Anhang, Anforderungen an die Durchfuhrung von Analysen, Validierung, Toxichem Krimtech, 2004, 74, 146–154 Search PubMed.
- L. A. N. Amar Babu, K. Kalyani, P. Babji, S. V. K. Srinivas and R. K. Prabhakara, Int. J. Res. Pharm. Sci., 2020, 11(2), 2210–2220 CrossRef.
- K. Prabhakara Rao, L. A. N. Amar Babu, K. Kalyani, P. Babji and S. V. K. Srinivas, SN Appl. Sci., 2021, 3, 321 CrossRef.
| This journal is © The Royal Society of Chemistry 2025 |