
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePreparation of ruthenium electrode materials and their application to the bactericidal properties of acidic electrolyzed oxidizing water†
Yanxue Li,
Wei Zong,
Hao Zhang* and
Dawei Lou *
*
College of Chemical and Pharmaceutical Engineering, Jilin Institute of Chemical Technology, Jilin 132022, People's Republic of China. E-mail: dwlou@hotmail.com; zhangh_0915@126.com; Tel: +86-432-62185559
First published on 22nd April 2025
Abstract
The anode chlorine evolution electrode materials used for producing acidic electrolyzed oxidizing water (AEOW) typically requires platinum, iridium, ruthenium, and other expensive and non-renewable precious metals. This not only results in high production costs but also hinders the development of the industry. To reduce the economic cost of the electrode and obtain better chlorine evolution anode materials, the effects of ruthenium electrode materials doped with different elements, ruthenium–tin doping ratio, and electrolytic process parameters on the AEOW physicochemical parameter of the electrode production were studied. The findings indicated that the novel SnO2/RuO2 electrode exhibited better catalytic performance, especially the electrode with a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :3 ruthenium–tin doping ratio (SnO2/RuO2-3), the active chlorine content (ACC) was 123 mg L−1, and the oxidation–reduction potential (ORP) was 1381 mV, exhibiting higher ACC and ORP values. In addition, when the current density was 50 mA cm−2, the chlorine evolution reaction potential of the SnO2/RuO2-3 electrode decreased to 55 mV, the oxygen evolution reaction potential increased to 155 mV, and the difference in potential between the CER and OER enhanced to 446 mV relative to the RuO2 electrode. The CER selectivity of the SnO2/RuO2 electrode was significantly improved, which was approximately twice that of the RuO2 electrode. Furthermore, the effects of electrolysis voltage, time, and concentration on AEOW were investigated. AEOW with an ACC content of 120 mg L−1 killed more than 99.9% of Escherichia coli within 60 seconds.
:3 ruthenium–tin doping ratio (SnO2/RuO2-3), the active chlorine content (ACC) was 123 mg L−1, and the oxidation–reduction potential (ORP) was 1381 mV, exhibiting higher ACC and ORP values. In addition, when the current density was 50 mA cm−2, the chlorine evolution reaction potential of the SnO2/RuO2-3 electrode decreased to 55 mV, the oxygen evolution reaction potential increased to 155 mV, and the difference in potential between the CER and OER enhanced to 446 mV relative to the RuO2 electrode. The CER selectivity of the SnO2/RuO2 electrode was significantly improved, which was approximately twice that of the RuO2 electrode. Furthermore, the effects of electrolysis voltage, time, and concentration on AEOW were investigated. AEOW with an ACC content of 120 mg L−1 killed more than 99.9% of Escherichia coli within 60 seconds.
1 Introduction
There are a variety of pathogens in the environment that can spread widely through direct or indirect contact, such as person-to-person or person-to-object contact.1 Therefore, sterilization is critical for human survival as it effectively eliminates pathogens and thus protects the public from infectious diseases.2,3 With the improvement of human biosecurity awareness, especially in recent years, increasing number of people are now aware of the importance of environmental health and sterilization. Electrolyzed oxidizing water (EOW), which is a bactericidal water containing active chlorine content (ACC) prepared through the electrolysis of low-concentration sodium chloride solutions, has been widely used as a disinfection technique in Japanese hospitals and other institutions since the 1980s.4–6 Electrochemical sterilization is an indirect sterilization technology and also a new type of environmentally friendly physicochemical disinfection method,7 which is now widely used in the fields of personal protection, food sanitation,8–11 medical sterilization, agriculture, fisheries,12–16 and forestry.17,18 According to its physicochemical properties, EOW is categorized into acidic electrolyzed oxidizing water (AEOW) and slightly acidic electrolyzed water (SAEW). AEOW generally possesses a low pH (pH 2–3) coupled with a high oxidation–reduction potential (ORP > 1100 mV), whereas SAEW possesses a pH range of 5–6 and an oxidation–reduction potential ranging from 700 to 1000 mV.19,20 EOW, particularly AEOW, exhibits advantages including rapid disinfection, high efficiency, broad-spectrum antimicrobial activity, and the absence of secondary pollution.21 EOW effectively disrupts the cytoplasm, cell wall, and cytoplasmic membrane of microbial cells. This disruption increases membrane permeability, resulting in the leakage of intracellular components such as Mg2+, K+, DNA, and proteins.22,23 EOW also effectively kills pathogenic microorganisms by disrupting the pathways such as protein synthesis activity and adenosine triphosphate synthesis.24,25 The highly effective sterilizing action is mainly owing to its physical (ORP, pH) and chemical factors (active chlorine and active oxygen content). In addition, research has proven that ACC is a crucial bactericidal component in acidic electrolytic oxidized water.26 Over the years, electrolytic water disinfection technology has been proven to be a green, safe, and scientifically validated method with no secondary pollution. It has the advantages of user convenience, low cost, non-toxicity, and residue-free properties, making it indispensable in both industrial applications and daily life.27–30The core of the acidic electrolyzed water production equipment lies in the performance of the anodic chlorine evolution reaction (CER) electrode within the electrolysis tank. The electrode performance is influenced by many factors, such as elemental composition, structure, catalytic activity, and other factors, thus affecting the active chlorine content in the solution.31,32 Initially, low-cost graphite materials were used as the anode of CER in the industry. However, it has been demonstrated that the graphite anodes used for the CER suffer from significant drawbacks. The primary issues include being prone to dissolution and detachment, which drastically reduce their service life, as well as inefficient catalytic performance for the CER. Consequently, graphite anodes were phased out in the 1970s.33,34 In the meantime, researchers have proposed the concept of dimensionally stable anodes (DSA), i.e., Ti metal electrodes coated with mixed metal oxides.35,36 DSA is a new type of insoluble anode, which has been extensively utilized in the electrolysis industry due to its outstanding anode properties. Compared to traditional graphite electrodes, DSA exhibits superior performance, including dimensional stability, lower operating voltage, reduced power consumption, enhanced catalytic efficiency, and prolonged service life. Furthermore, DSAs effectively solve the problem of graphite electrode dissolution, prevent electrolyte contamination, and improve product quality.37 In the next half-century, DSA was widely used in the field of electrochemical chlorine evolution reaction and was also an important equipment for AEOW production simultaneously. Nevertheless, the fabrication of DSA predominantly depends on noble metals such as platinum (Pt), iridium (Ir), and ruthenium (Ru). These elements are scarce, costly, and non-renewable, which substantially limits the large-scale industrial adoption and commercialization of DSA technology.38–41 Even today, Ru and Ir remain regarded as ideal materials for CER electrodes.42,43 Researchers have tried various ways to reduce the use of noble metal elements over the past few decades. Research showed that RuO2 material doped with transition metals could be used to enhance the catalytic activity and stability of the electrodes; for instance, Zhang et al. incorporated tantalum (Ta) doping into RuO2 electrodes, which enhanced the corrosion resistance of RuO2 and thereby improved material stability. In industrial demonstrations, the Ta–RuO2 catalytic electrode exhibited stability comparable to IrO2 while achieving a lower overpotential at identical current densities.44–46 The RuO2/TiO2 electrode fabricated by Wang et al. improved the chlorine evolution selectivity.47
This study systematically investigated electrode materials, composition ratios, and process parameters (time, voltage) to enhance the electrocatalytic efficiency of acidic electrolyzed water production, aiming to obtain acidic electrolytic oxidized water with higher ACC, ORP and lower pH values and higher bactericidal efficiency. In the experiment, the electrode was fabricated through the thermal decomposition method, utilizing a titanium plate as the substrate. A new solvent system was used to prepare the precursor solution. Ruthenium was first doped with different metal elements to prepare the electrodes, and water electrolysis experiments were conducted. The experimental results indicated that the catalytic performance of ruthenium doped with tin element was better; when the ruthenium–tin molar ratio was 1:3, the catalytic performance reached the effect of commercial electrodes (RuO2–IrO2/Ti, 20 × 20 × 1 mm). On this basis, the material characterization and electrochemical testing of novel SnO2/RuO2 electrodes were carried out. It was shown that the SnO2/RuO2 electrode had higher CER activity and lower OER activity than the RuO2 electrode, and the significantly increased potential difference (ΔECER–OER) was beneficial in improving the selectivity of SnO2/RuO2 electrodes. Finally, the process conditions were optimized with SnO2/RuO2-3 electrodes to prepare AEOW and the bactericidal effect of Escherichia coli was tested. Compared with prior studies,48 the SnO2/RuO2-3 electrode developed in this work demonstrated superior performance in generating AEOW. Specifically, the produced AEOW achieved an ORP exceeding 1300 mV and a higher ACC than commercial electrodes, indicating its significant advantages for industrial-scale AEOW production.
2 Experimental
2.1 Materials
Ruthenium trichloride trihydrate (RuCl3·3H2O), stannous fluoride (SnF2), nickel chloride hexahydrate (NiCl2·6H2O), zinc sulfate heptahydrate (ZnSO4·7H2O), manganese sulfate monohydrate (MnSO4·H2O), cobalt sulfate heptahydrate (CoSO4·7H2O), tetra butyl titanate (C16H36O4Ti), acetone (CH3COCH3), sulfuric acid (H2SO4), nitric acid (HNO3), oxalic acid (H2C2O4), N,N-dimethylformamide (C3H7NO, DMF), ethanol (C2H5OH), starch indicator (C12H22O11), potassium iodide (KI), sodium thiosulfate (Na2S2O3), and sodium chloride (NaCl) were purchased from Annaiji, China. All reagents were analytical grade and used without further purification.2.2 Electrode preparation
Electrodes were fabricated using RuCl3·3H2O-based precursor solutions doped with different elements through thermal decomposition and impregnation-pulling method, including titanium plate pretreatment and electrode coating preparation. Titanium plates (20 × 20 × 1 mm) were ultrasonically cleaned sequentially with a certain concentration of detergent and acetone. Subsequently, they were immersed for 3 h in a mixture of concentrated nitric acid and concentrated sulfuric acid. Following this, the plates were treated in 10 wt% boiling oxalic acid for 3 h to achieve a uniformly rough surface on its exterior. Finally, the samples were preserved with anhydrous ethanol.The precursor RuCl3·3H2O was dissolved with SnF2, NiCl2·6H2O, ZnSO4·7H2O, MnSO4·H2O, and CoSO4·7H2O in a mixture of DMF and ethanol with a volume ratio of 1:1 respectively, and the metal doping ratio was maintained at 1:1, and the total metal concentration of the precursor solution was kept at 0.2 M. The pre-treated titanium plates were immersed in the mixed solution for 1 minute and then gradually withdrawn to form a uniform liquid film on their surface. The titanium plates were dried at 170 °C for 15 minutes to evaporate the surface solvent, subsequently transferred to a muffle furnace, and heated at 450 °C for 10 minutes. Finally, they were cooled to ambient temperature. The coating process was repeated for 10 cycles to ensure uniform electrode coverage. The final reaction duration was extended to 60 minutes to enhance the layer adhesion. RuO2 and SnO2/RuO2 electrodes were prepared using the same approach. The electrode doping ratios were 1:0 (RuO2), 3:1 (SnO2/RuO2-1), 1:1 (SnO2/RuO2-2), and 1:3 (SnO2/RuO2-3) respectively.
The remaining precursor liquid from the experiment was dried at 170 °C for 150 minutes to completely evaporate the solvent and form a solid block. The solid block was ground into a powder, which was then transferred to a muffle furnace. The powder was heated at 450 °C for 150 minutes and cooled to ambient temperature. A titanium-plate-free powder sample was prepared for XRD measurements.
2.3 Electrochemical measurements
Electrochemical-related measurements were conducted at ambient temperature using a CHI760E electrochemical workstation (Shanghai CH Instruments) configured with a three-electrode system. A graphite sheet served as the counter electrode and Ag/AgCl electrode as the reference electrode. The working electrodes were RuO2, SnO2/RuO2 electrodes, and commercial electrodes, respectively. The double-layer currents were acquired by performing cyclic voltammetry (CV) scans at different scanning rates. The j–v curve can be obtained by plotting the association between the scan rate (v) and the double-layer current density (j). The double-layer capacitance (Cdl), represented by the slope of the j–v curve during plotting, is divided by 60 mF cm−2 (60 mF cm−2—the reference capacitance per unit area) to calculate the ECSA value.49 The potential range of double-layer capacitance scanning was 1.15–1.25 V vs. RHE in 0.5 mol per L H2SO4 solution. The CER activity was evaluated using linear sweep voltammetry (LSV) in a 4.0 mol per L NaCl and 0.5 mol per L H2SO4 (pH = 1) at a scan rate of 5 mV s−1, with a potential range of 1.2 to 1.8 V vs. RHE. The OER activity was evaluated using the same method in a mixed solution of 1.33 mol per L Na2SO4 and 0.5 mol per L H2SO4 (pH = 1) at a scan rate of 5 mV s−1, with a potential range of 1.2 to 2 V. RHE. The electrode potential vs. RHE is calculated using the formula: ERHE = EAg/AgCl + 0.1971 V + 0.0592 pH.2.4 Sterilization validation test
The lyophilized Escherichia coli powder (BNCC185254), obtained from Beina Chuanglian Biotechnology Institute, was activated and cultured. Bacteria from third-passage cultures were then used for sterility validation. A bacterial suspension with an initial concentration of 1 × 108 CFU mL−1 was prepared. Subsequently, 1 mL of AEOW containing 120 mg per L ACC was mixed with 1 mL of the suspension, and the mixture was subjected to controlled sterilization durations. After each experiment interval, 1 mL of the sterilized solution was mixed with 9 mL of neutralizer (0.5% Na2S2O3 + 0.85% NaCl) to terminate the sterilization process by neutralizing residual ACC. The sterilization rate of AEOW was calculated using the plate counting method.3 Results and discussion
3.1 Optimization of the preparation conditions for electrode materials
Electrode material is a key factor that impacts the production of AEOW and its associated physical and chemical characteristics. Electrode materials with different elements have different electrolytic catalytic activities due to their different structures. A custom-designed intermittent diaphragm electrolysis cell was employed for the electrolysis of 300 mL NaCl solution. The synthetic electrode served as the anode and a pure titanium plate as the cathode. The electrolysis experiments were conducted under constant voltage mode using a DC-regulated power supply (MS-3610DS). The preparation conditions of AEOW were an electrolytic voltage of 15 V, electrolyte concentration of 1 mg L−1, electrolytic time of 30 minutes, and electrode distance of 6 cm. The ORP and pH values were measured by a portable redox potentiostat (Shanghai Leimagnet, YHBJ-262) and pH meter. The doping electrodes with the best catalytic effect were optimized in different proportions, and the better catalytic performance electrodes (SnO2/RuO2-3) were selected for characterization measurements, process optimization and sterilization experiments.The ACC content was determined via iodometric titration35 using the following protocol: a mixture of 10.00 mL AEOW, 10.00 mL of 10% (w/v) KI, and 10.00 mL of 20% (v/v) H2SO4 was prepared in a 250 mL conical flask. The solution was protected from light and allowed to react for 10 min, and 20.00 mL ultrapure water was subsequently added. The solution was titrated with 0.01 mol per L Na2S2O3 until pale yellow endpoint, and then 1.0 mL of 10 g per L starch indicator was introduced, resulting in a dark blue solution. Titration was continued until the complete colour faded. Triplicate measurements were performed per sample, with results averaged, and blank controls substituted 10.00 mL AEOW with ultrapure water. ACC concentration is calculated as follows:
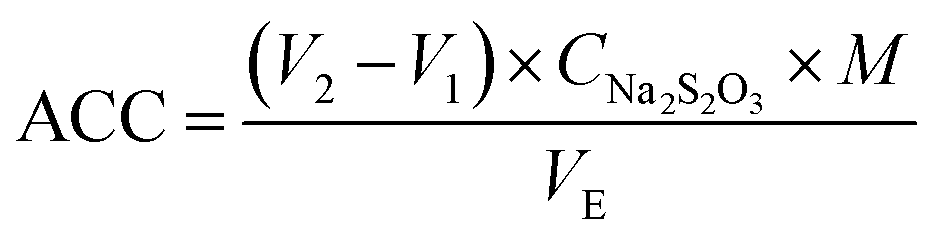 | (1) |
453 mg mol−1).
The current efficiency (CE) of CER19 was measured in a 300 mL NaCl solution (1 mg L−1). According to Faraday's law, the CE is defined as the ratio of actual active chlorine productivity to theoretical chlorine productivity:
 | (2) |
500 C mol−1), C is the active chlorine concentration (g L−1), V is the solution volume (L), M is the molecular weight of chlorine (g mol−1), i is the current (A), and t is the electrolysis time (s).
As shown in Fig. 1, under identical preparation conditions, the Sn-doped synthetic electrodes exhibited superior chlorine catalytic activity and higher ACC and ORP values in the generated AEOW compared to Ru-doped other electrodes. The pH values of AEOW produced by the Sn-doped element electrodes were nearly identical to those of Ni-, Mn-, and Ti-doped electrodes, slightly higher than those of Zn- and Co-doped electrodes, but overall showed minimal variation. All values met the requirements for AEOW applications. In addition, the synthetic electrode doped with tin element produced AEOW; the ACC, ORP and pH values were 109 mg L−1, 1382 mV, and 2.62, respectively, while the commercial electrode was 90 mg L−1, 1324 mV, and 2.65, with better physical and chemical parameters, therefore, different ratios of ruthenium tin electrodes were prepared for the following experimental studies.
 | ||
| Fig. 1 ACC, ORP and pH values of AEOW prepared using different doped element electrodes (preparation conditions: U = 15 V, V = 300 mL, t = 30 minutes, d = 6 cm, CNaCl = 1 g L−1). | ||
Fig. 2 illustrates that relative to the RuO2 electrode, the SnO2/RuO2 electrode with varying doping ratios significantly improved the physicochemical parameters of AEOW. Notably, the SnO2/RuO2-3 electrode (doping ratio 1:3) demonstrated superior performance, yielding AEOW with the most optimal physicochemical properties. As the proportion of tin in the electrode rises, the ACC content gradually grows. Fig. 2(b and c) shows how the ORP and pH of the acidic electrolytic water produced by different electrodes change with time. The ORP of the electrodes shows an overall increasing trend. In the first 10 minutes, ORP for the SnO2/RuO2 electrode quickly increased by about 500 mV, while that of the RuO2 electrode was about 430 mV. Subsequently, the rate of rise slowed until it remained essentially constant, and ORP changed by about 100 mV in 20 minutes. The SnO2/RuO2 electrode proved both a faster rate of ORP increase and a higher final ORP value than the RuO2 electrode during the 30 minute measurement period, while the performance of the SnO2/RuO2-3 electrode was more obvious. The pH also exhibited a similar trend as ORP. In the initial 5 minutes, pH decreased more rapidly, then the descent speed slowed down, finally remained stable, and the change was no longer obvious. The current efficiencies of the RuO2 and SnO2/RuO2-(1,2,3) electrodes were 50.48%, 53.25%, 65.06%, 82.06%, respectively. According to the four parameters, it is demonstrated that the ruthenium tin electrode has better catalytic performance than the RuO2 electrode, with the SnO2/RuO2-3 electrode having the best performance.
 | ||
| Fig. 2 ACC (a), ORP (b), and pH (c) values of AEOW prepared using RuO2 and SnO2/RuO2-(1,2,3) electrodes (preparation conditions: U = 15 V, V = 300 mL, t = 30 minutes, d = 6 cm, CNaCl = 1 g L−1). | ||
3.2 Material characterization
The microscopic morphology and surface characteristics of the electrode were analyzed using field emission scanning electron microscopy (FESEM, JSM-7610F Plus, JEOL), while the chemical compositions were examined through energy dispersive spectroscopy (EDS, XFlash, Bruker). The crystalline structure was characterized using X-ray diffraction (XRD, Empyrean), and the elemental valence states were determined by X-ray photoelectron spectrometry (XPS, Thermo Scientific K-Alpha).Through screening and comparison, the SnO2/RuO2-3 electrode showed superior catalytic performance. Fig. 3a shows that the surface of the RuO2 electrode had obvious cracks, the flat cracked coating completely covered the Ti substrate, and in Fig. 3b, the RuO2 crystal shows a regular cubic structure at 100 nm. The SnO2/RuO2-3 electrode (Fig. 3c) exhibited a crack-free and rough, honeycomb-like morphology. It was observed that the SnO2 crystal was smaller and covered the Ti matrix as well as the surface of RuO2 at 100 nm (Fig. 3d). The ruthenium tin synergistic modification of the electrode surface fills the cracks on the electrode surface to a certain extent and forms a loosened honeycomb structure, thereby enlarging the electrode surface area, which enhanced the interfacial contact area between the NaCl electrolyte and the electrode, thereby boosting the overall electrocatalytic performance.
 | ||
| Fig. 3 SEM images of the RuO2 (a and b) and SnO2/RuO2-3 electrodes (c and d) with a doping ratio of 1:3. | ||
Fig. 4 presents the surface elemental distribution and atomic percentage content of the electrode. During the thermal decomposition cycling oxidation process, ruthenium, tin, and oxygen elements on the surface of the SnO2/RuO2-3 electrode interacted, resulting in the formation of a ruthenium-tin mixed metal oxide coating with relatively uniform distribution across the Ti substrate surface. Through EDS analysis, the atomic percentage of Ru and Sn was found to be 1:3, which is consistent with the doping ratio set.
 | ||
| Fig. 4 Elemental mapping images (a–e) and EDS of SnO2/RuO2-3 electrode (f). | ||
Fig. 5a presents the X-ray diffraction pattern of the titanium plate, RuO2, and SnO2/RuO2-(1,2,3) electrodes, and the SnO2/RuO2-(1,2,3) powder sample without the titanium substrate. The primary diffraction peaks of RuO2 at 28.0°, 35.0°, 40.5°, and 69.5° correspond to the (110), (101), (111), and (311) planes (PDF # 88-0323), respectively.50 As the ruthenium content decreased, the RuO2 diffraction peaks progressively weakened while the SnO2 diffraction peaks emerged. The diffraction peaks of SnO2 and RuO2 were not obvious because the diffraction peaks of the titanium plate were too strong. The samples were synthesized in powder form and characterized by XRD without the influence of the titanium matrix (Fig. 5b), and the presence of characteristic peaks of SnO2 (PDF # 99-0024) and RuO2 (PDF # 88-0323) can be clearly observed. It was also observed that at higher ruthenium contents, the ruthenium did not fully oxidize, resulting in detectable ruthenium diffraction peaks (PDF # 89-4903). As the ruthenium content decreased, these peaks gradually diminished and eventually disappeared.
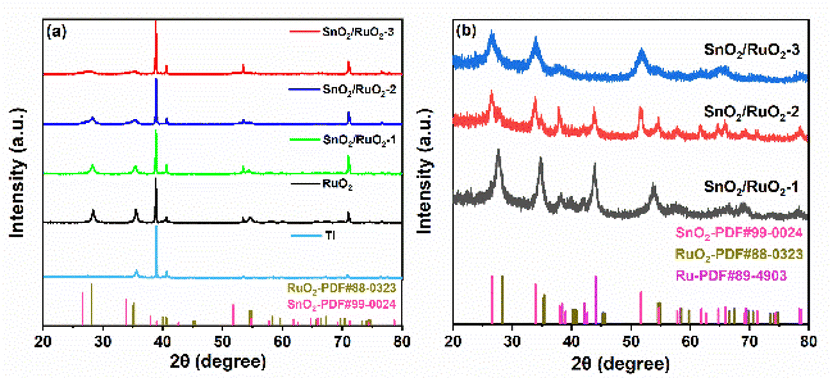 | ||
| Fig. 5 X-ray diffraction diagram of titanium plate, RuO2 electrode, and SnO2/RuO2-(1,2,3) electrodes (a). Powder X-ray diffraction diagram of SnO2/RuO2-(1,2,3) without titanium substrate (b). | ||
The elemental states of the SnO2/RuO2-3 electrode were determined using spectroscopy (XPS) measurements. Ru 3d, Sn 3d, and O 1s were observed at binding energies of around 284, 487, 494, and 530 eV in the full spectrum (Fig. 6a).51 The peaks at 280.9 and 285.1 eV correspond to the 3d5/2 and 3d3/2 orbitals of Ru4+, respectively, while 282.5 and 286.9 eV are their corresponding satellite peaks (Fig. 6b).52 The peaks at 487 and 495 eV correspond to the 3d5/2 and 3d3/2 orbitals of Sn4+ (Fig. 6c).53 XPS analysis confirmed SnO2 as the predominant species on the electrode surface.
 | ||
| Fig. 6 Survey (a), Ru 3d (b), and Sn 3d (c) XPS spectra of SnO2/RuO2-3 electrode. | ||
3.3 Electrochemical measurements
Experiments, including ECSA, CER, OER, ΔECER–OER, and Tafel, were conducted to evaluate the electrochemical performance of Ru-doped SnO2 electrodes synthesized with different doping ratios and were compared with RuO2 and commercial electrodes. The specific results of the experiment are as follows.ECSA is important for exploring electrochemistry because it reflects the total surface area of electrocatalytic reactions. It is generally believed that the electrochemical active centers are positively correlated with ECSA, and the more the electrochemical active centers, the better the electrocatalytic activity and the larger the ECSA. The ECSA of different synthetic electrodes was measured using the double-layer capacitance method. The CV profiles of the electrode at varying sweep rates and those of different electrodes at a fixed sweep rate of 10 mV s−1 are presented in Fig. S1.† The double-layer capacitance of the electrode increased with higher tin content, with the most pronounced enhancement observed in the SnO2/RuO2-3 electrode. Fig. 7a shows that the double-layer capacitance (Cdl) increased with increasing tin content, and the Cdl of the SnO2/RuO2-3 electrode was slightly higher than that of the commercial electrode and 2.57 times higher than that of the RuO2 electrode. Fig. 7b illustrates that with the increased tin content, ECSA had a significant increasing trend; especially for the SnO2/RuO2-3 electrode, the ECSA of the RuO2 electrode was 232 mA cm2, and that of the SnO2/Ruo2-3 electrode reached 593 mA cm2. Tin doping increased the ECSA value and helped to enhance the catalytic performance to a certain extent.
 | ||
| Fig. 7 Double-layer capacitances (Cdl) of RuO2, SnO2/RuO2-(1,2,3), and commercial electrodes (a). ECSA of RuO2, SnO2/RuO2-(1,2,3), and commercial electrodes (b). | ||
The chlorine evolution performance is significantly affected by the CER and OER activities, as demonstrated in Fig. 8. E0(CER) is the standard potential of CER (1.36 V) in Fig. 8a. With the adjustment of the ruthenium–tin ratio when the current density was 10 mA cm−2, the CER potential decreased from 1.476 V (RuO2) to 1.448 V (SnO2/RuO2-3), with a difference (ΔECER) of 28 mV. At the current density of 50 mA cm−2, the potential decreased from 1.653 V to 1.598 V, and the ΔECER became 55 mV (from 1.653 to 1.598 V). It is worth noting that the SnO2/RuO2-3 electrode exhibited a lower chlorine evolution potential compared to the commercial electrode. It can be seen that the doped electrode can reduce the chlorine evolution potential and make the CER occur more easily in the electrolytic process. E0(OER) is the standard potential of OER (1.23 V), as shown in Fig. 8c. By adjusting the ruthenium–tin ratio, the OER potential increased from 1.628 V to 1.646 V at a current density of 10 mA cm−2, and from 1.89 V (RuO2) to 2.045 V (SnO2/RuO2-3) at 50 mA cm−2. This indicated that the doped electrode increased the oxygen evolution potential, suppressed the OER activity, and thereby hindered the occurrence of the oxygen evolution reaction. Appropriate adjustment of the ruthenium–tin composition can enhance the activity of the chlorine evolution reaction while suppressing the activity of the oxygen evolution reaction, thereby improving the selectivity of CER and the overall catalytic performance of the electrode.
 | ||
| Fig. 8 Apparent CER activities (electrolysis: 4.0 mol per L NaCl and 0.5 mol per L H2SO4 (pH = 1), scan rate: 5 mV s−1) (a and b) and OER activities (electrolysis: 1.33 mol per L Na2SO4 and 0.5 mol per L H2SO4 (pH = 1), scan rate: 5 mV s−1) (c and d) of RuO2, SnO2/RuO2-(1,2,3), and commercial electrodes. | ||
As shown in Fig. 9, when the current density was 50 mA cm−2, the ΔECER–OER of the electrodes was 237 mV (RuO2), 298 mV (SnO2/RuO2-1), 349 mV (SnO2/RuO2-2), and 446 mV (SnO2/RuO2-3), while that of the commercial electrode was 357 mV. The ΔECER–OER of the SnO2/RuO2-3 electrode was approximately twice that of RuO2 and also surpassed that of the commercial electrode, indicating that the SnO2/RuO2-3 electrode exhibited outstanding selectivity for the CER. Compared with the previous literature, the SnO2/RuO2-3 electrode exhibited relatively high chlorine evolution activity. Qi et al. prepared the CuO/RuO2 electrode, which the ΔECER–OER was 105 mV,50 Zhang et al. developed TiO2-NTs/Sb–SnO2/PbO2 electrode, the ΔECER–OER was about 0.45 V.54 The larger ΔECER–OER value suppressed the OER activity, thereby enhancing the selectivity of the CER and overall electrode performance, ultimately increasing the ACC content in the AEOW.
 | ||
| Fig. 9 Difference in potential between CER (electrolysis: 4.0 mol per L NaCl and 0.5 mol per L H2SO4 (pH = 1), scan rate: 5 mV s−1) and OER (electrolysis: 1.33 mol per L Na2SO4 and 0.5 mol per L H2SO4 (pH = 1), scan rate: 5 mV s−1) for RuO2, SnO2/RuO2-(1,2,3), and commercial electrodes. | ||
The reaction mechanism of CER can be elucidated from the slope of the Tafel plot, which is typically associated with an unconventional electrochemical desorption process, primarily comprising three steps. The ‘‘S’’ indicates the active site in steps (3)–(5). A Tafel slope of 120 mV dec−1 indicates that the formation of adsorbed Cl (step (3)) is the rate-determining step (RDS). A Tafel slope of 40 mV dec−1 suggests that the second electron transfer (step (4)) is the rate-determining step (RDS), while the 30 mV dec−1 indicates that the electrochemical desorption process is the RDS.49 In Fig. 10a, the Tafel slopes of RuO2, SnO2/RuO2-(1,2,3), and the commercial electrode were 65.31, 56.71, 60.80, 65.25, and 72.98 mV dec−1, respectively, with all values corresponding to the second electron transfer step of the reaction mechanism. This suggested that the process was predominantly governed by the second electron transfer step (step (4)).
| S + Cl− → S–Cl + e | (3) |
| S–Cl → S–Cl+ + e | (4) |
| S–Cl+ + Cl− → S + Cl2 | (5) |
 | ||
| Fig. 10 Tafel curves of CER for RuO2, SnO2/RuO2-(1,2,3) and commercial electrodes (a). Stability testing of RuO2 and SnO2/RuO2-3 electrodes (b). | ||
Fig. 10b demonstrates that the SnO2/RuO2-3 electrode operated stably at 10 mA cm−2 in 0.5 M H2SO4 for nearly 90 h with a minimal voltage change of 90 mV, outperforming the RuO2 electrode in stability by a significant margin. This instability arises because RuO2 undergoes dissolution in highly oxidative acidic environments due to the formation of acid-soluble RuO4 with a higher oxidation state, leading to structural distortion, increased RuO2 electrode potential, and decreased stability.55 This indicated that tin doping markedly improved the stability of the electrode, enhancing its potential for practical applications.
3.4 Applications
The optimization of the preparation of acidic oxidation potential water experiment was carried out by exploring different electrolysis voltages (5, 15, 25, and 35 V), electrolysis times (10, 20, 30, 40, 50, 60, 70, 80, 90, and 100 minutes), and electrolyte concentrations (0.5, 1, 2, 3, and 4 g L−1) to explore the best electrolysis conditions for the preparation of AEOW.As shown in Fig. 11, the ACC initially increased with higher applied voltage, peaking at 25 V before decreasing at elevated potentials. The ORP increased progressively with higher electrolytic voltages, exhibiting a pronounced upward trend. After 25 minutes, the ORP stabilized at approximately 1400 mV under all tested voltages except 5 V. The pH value decreased progressively with increasing electrolytic voltage and stabilized at 2.6 under all tested voltages except 5 V. Therefore, AEOW had better physical and chemical properties when the electrolysis voltage was 25 V.
 | ||
| Fig. 11 ACC, ORP and pH values of AEOW prepared by the SnO2/RuO2-3 electrode under different electrolytic voltages (preparation conditions: V = 300 mL, t = 30 minutes, d = 6 cm, CNaCl = 1 g L−1). | ||
As illustrated in Fig. 12, the ACC, ORP, and pH values of AEOW underwent significant changes within the first 60 minutes, followed by a marked deceleration in their rates of change between 60 and 100 minutes. During the 20 to 60 minutes electrolysis period, the ACC value increased rapidly. However, beyond 60 minutes, the rate of ACC increases significantly slowed while both ORP and pH values stabilized, indicating that an electrolytic time of 60 minutes was optimal for maintaining stable conditions.
 | ||
| Fig. 12 ACC (a) and ORP and pH values (b) of AEOW prepared using the SnO2/RuO2-3 electrode at different electrolytic times (preparation conditions: U = 15 V, V = 300 mL, d = 6 cm, CNaCl = 1 g L−1). | ||
As shown in Fig. 13, within a certain range, the concentration of electrolytes increased, and the ACC content continued to increase. When the concentration was in the range of 1–2 g L−1, the rate of ascent of the ACC content was significantly faster, and ORP and pH were the best values. However, increasing the electrolyte concentration not only raises costs but also leads to chlorine gas release during electrolysis, which accelerates equipment corrosion. The generation of chlorine gas poses significant risks to human health and the environment, and in severe cases, exposure to it can be life-threatening. The current industry usually uses 1–1.5 g L−1 electrolytic concentration to produce AEOW.
 | ||
| Fig. 13 ACC (a) and ORP and pH values (b) of AEOW prepared using the SnO2/RuO2-3 electrode at different electrolytic concentrations (preparation conditions: U = 15 V, V = 300 mL, t = 30 minutes, d = 6 cm). | ||
3.5 Sterilization validation test
Fig. 14 shows the bactericidal effect against Escherichia coli when the ACC was 120 mg L−1. The AEOW prepared by the SnO2/RuO2-3 electrode had a significant bactericidal effect. The sterilization rate reached 90% within 10 s and exceeded 99.9% after 60 s, with the efficacy dependence on sterilization time detailed in Fig. S2.† | ||
| Fig. 14 Effect of AEOW prepared using the SnO2/RuO2-3 electrode on the sterilization rate of Escherichia coli at different action times. | ||
4 Conclusions
In this study, the effects of element-doped ruthenium electrodes, Ru–Sn doping ratios, and electrolytic process parameters on the physicochemical properties of AEOW electrodes were investigated. The results showed that the SnO2/RuO2-3 electrode exhibited the highest ACC and ORP values, maintained pH levels comparable with other electrodes, and displayed superior catalytic performance. In comparison to the undoped RuO2 electrode, the CER potential of the SnO2/RuO2-3 electrode decreased to 55 mV, the OER potential increased to 155 mV at a current density of 50 mA cm−2 and the ΔECER–OER potential increased to 446 mV; accordingly, the selectivity and catalytic performance of the SnO2/RuO2-3 electrode was significantly improved. Optimizing the electrolytic parameters (voltage, time, and concentration) enhanced the AEOW production quality. The ACC concentration served as the critical determinant for Escherichia coli bactericidal efficacy. The higher the ACC content, the shorter the bactericidal action time and the higher the bactericidal effect. When the ACC value was 120 mg L−1, the sterilization rate reached more than 99.9% within 1 minute.Data availability
The data will be made available on reasonable request.Author contributions
Formal analysis, Y. L.; writing-original draft, Y. L.; data curation, Y. L., and W. Z.; investigation, Y. L., and W. Z.; validation, Y. L., and W. Z.; resources, D. L., and H. Z.; supervision, D. L., and H. Z.; writing-review & editing, D. L., and H. Z.; methodology, Y. L., W. Z., D. L., and H. Z.; project administration, D. L.; funding acquisition, D. L. All authors have read and agreed to the published version of the manuscript.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Jilin Province Science and Technology Development Plan Project (20230201147GX). The financial support from the Key Laboratory of Fine Chemicals of Jilin Province is also acknowledged.Notes and references
- Y.-L. Oon, Y.-S. Oon, M. Ayaz, M. Deng, L. Li and K. Song, Front. Microbiol., 2023, 14, 1286923–1286952 CrossRef PubMed.
- Q. Zhang, R. Ma, Y. Tian, B. Su, K. Wang, S. Yu, J. Zhang and J. Fang, Environ. Sci. Technol., 2016, 50, 3184–3192 CrossRef CAS PubMed.
- X. Li, Z. Li, D. Yang, S. Li, L. Yang, W. Yan, W. Xiang, L. Pei, Z. Ye and H. Xu, J. Environ. Chem. Eng., 2024, 12, 114073–114090 CrossRef CAS.
- Z. Li, D. Yang, S. Li, L. Yang, W. Yan and H. Xu, Sci. Total Environ., 2024, 912, 169043–169062 CrossRef CAS PubMed.
- S. M. E. Rahman, I. Khan and D. H. Oh, Compr. Rev. Food Sci. Food Saf., 2016, 15, 471–490 CrossRef PubMed.
- O. Habimana, M. Kurahashi, T. Ito and A. Naka, PLoS One, 2021, 16, 0253595–0253605 Search PubMed.
- D. Alwazeer, Food Chem.:X, 2024, 24, 101973–101982 CAS.
- Y. Du, Q. Tian, G. Li, J. Yi, X. Hu and Y. Jiang, Food Res. Int., 2024, 195, 114996–115009 CrossRef CAS PubMed.
- L. Lin, Y. Lin, B. Chen, X. Li, L. Zhu, Z. Huang, Y. Liu, Z. He and X. Lin, Postharvest Biol. Technol., 2025, 222, 113402–113412 CrossRef CAS.
- M. Rebezov, K. Saeed, A. Khaliq, S. J. U. Rahman, N. Sameed, A. Semenova, M. Khayrullin, A. Dydykin, Y. Abramov, M. Thiruvengadam, M. A. Shariati, S. P. Bangar and J. M. Lorenzo, Appl. Sci., 2022, 12, 6639–6654 CrossRef CAS.
- W. Zhang, J. Cao and W. Jiang, Trends Food Sci. Technol., 2021, 114, 599–607 CrossRef CAS.
- H. Yang, J. Li, H. Xu, C. Peng, J. Cui, S. Hu, L. Xia and Y. Zhang, Aquacult. Int., 2022, 31, 957–973 CrossRef.
- H. Huang, J. He, X. Gao, J. Lei, Y. Zhang, Y. Wang, X. Liu and J. Hao, J. Proteomics, 2023, 286, 104952–104963 CrossRef CAS PubMed.
- Y.-J. Kim, K.-D. Choi and I.-S. Shin, J. Korean Soc. Food Sci. Nutr., 2010, 39, 776–781 CrossRef CAS.
- Y. Lin, H. Chen, S. Dong, Y. Chen, X. Jiang and Y. Chen, Foods, 2024, 13, 1556–1570 CrossRef CAS PubMed.
- C. Liu, W. Zheng, Z. Li, L. Zhou, Y. Sun and S. Han, Poult. Sci., 2022, 101, 101643–101651 CrossRef CAS PubMed.
- P. Yan, R. Chelliah, K. h. Jo and D. H. Oh, Processes, 2021, 9, 2240–2260 CrossRef CAS.
- J. Hao, H. Li, Y. Wan and H. Liu, Food Control, 2015, 50, 699–704 CrossRef CAS.
- D. Shao, W. Yan, L. Cao, X. Li and H. Xu, J. Hazard. Mater., 2014, 267, 238–244 CrossRef CAS PubMed.
- S. Wei, L. Shi, Z. Ren, A. Zhang, R. Ming, B. Chai and Y. Zhu, RSC Adv., 2017, 7, 45377–45384 RSC.
- J. Yan and J. Xie, LWT--Food Sci. Technol., 2021, 151, 112044–112051 CrossRef CAS.
- Z. Ye, S. Wang, T. Chen, W. Gao, S. Zhu, J. He and Z. Han, Sci. Rep., 2017, 7, 6279–6289 CrossRef PubMed.
- Q. Liu, L. Chen, A. K. C. Laserna, Y. He, X. Feng and H. Yang, Food Control, 2020, 110, 107026–107070 CrossRef CAS.
- B.-K. Chen and C.-K. Wang, Molecules, 2022, 27, 1222–1237 CrossRef CAS PubMed.
- Y. Shimamura, Y. Oura, M. Tsuchiya, Y. Yamanashi, A. Ogasawara, M. Oishi, M. Komuro, K. Sasaki and S. Masuda, Front. Microbiol., 2024, 14, 1328055–1328068 CrossRef PubMed.
- R. Ming, Y. Zhu, L. Deng, A. Zhang, J. Wang, Y. Han, B. Chai and Z. Ren, New J. Chem., 2018, 42, 12143–12151 RSC.
- P. Yan, E. B.-M. Daliri and D.-H. Oh, Microorganisms, 2021, 9, 136–155 CrossRef CAS PubMed.
- L. Zhao, S. Li and H. Yang, Curr. Opin. Food Sci., 2021, 41, 180–188 CrossRef CAS.
- S. Adhikari, S. Mandal and D. H. Kim, Small, 2022, 19, 2206003–2206041 CrossRef PubMed.
- H. Bergmann, Curr. Opin. Electrochem., 2021, 28, 100694–110080 CrossRef CAS.
- Z. Deng, S. Xu, C. Liu, X. Zhang, M. Li and Z. Zhao, Nano Res., 2023, 17, 949–959 CrossRef.
- H. Dong, W. Yu and M. R. Hoffmann, J. Phys. Chem. C, 2021, 125, 20745–20761 CrossRef CAS.
- H. L. Andersen, L. Djuandhi, U. Mittal and N. Sharma, Adv. Energy Mater., 2021, 11, 2102693–2102714 CrossRef CAS.
- J. Ji, J. Liu, L. Shi, S. Guo, N. Cheng, P. Liu, Y. Gu, H. Yin, H. Zhang and H. Zhao, Small Struct., 2023, 5, 2300240–2300251 CrossRef.
- W. Tang, Y. Li, W. Li, X. Chen and X. Zeng, LWT--Food Sci. Technol., 2016, 66, 606–614 CrossRef CAS.
- C. Orha, C. Bandas, C. Lazau, M. I. Popescu, A. Baciu and F. Manea, Water, 2022, 14, 821–837 CrossRef CAS.
- J. Hao, H. Li, Y. Wan and H. Liu, RSC Adv., 2015, 50, 699–704 CAS.
- S. Han, S. Kim, S. Kwak, C. Lee, D. H. Jeong, C. Kim and J. Yoon, J. Ind. Eng. Chem., 2022, 108, 514–521 CrossRef CAS.
- H. Liu, M. Li, Z. Zhang, Y. Li, Q. Hao, L. Liang and W. Zhang, Dalton Trans., 2025, 54, 2922–2929 RSC.
- Z.-T. Yu, Dalton Trans., 2025, 54, 2718–2736 RSC.
- T. Zhao, Y. Jia, Q. Fang, R. Du, G. Hao, W. Sun, G. Liu, D. Zhong, J. Li and Q. Zhao, J. Energy Chem., 2025, 104, 414–421 CrossRef CAS.
- B. Liu and J. Zhang, RSC Adv., 2020, 10, 25952–25957 RSC.
- S. Ding, C. Yang, J. Yuan, H. Li, X. Yuan and M. Li, RSC Adv., 2023, 13, 12309–12319 RSC.
- C. Zhu, L. Xiao, Y. Zhang, Y. Jiang, X. Zhao, Q. Liu, A. Abdukayum and G. Hu, Mater. Today Energy, 2025, 48, 101796–101825 CrossRef CAS.
- T. Kavinkumar, T. R. N. Kumar, C. Sengottaiyan, A. T. Sivagurunathan, A. Thirumurugan and D.-H. Kim, Int. J. Hydrogen Energy, 2025, 102, 321–331 CrossRef CAS.
- J. Zhang, X. Fu, S. Kwon, K. Chen, X. Liu, J. Yang, H. Sun, Y. Wang, T. Uchiyama, Y. Uchimoto, S. Li, Y. Li, X. Fan, G. Chen, F. Xia, J. Wu, Y. Li, Q. Yue, L. Qiao, D. Su, H. Zhou, W. A, G. III and Y. Kang, Science, 2025, 387, 48–55 CrossRef CAS PubMed.
- D. Wang, T. Dong, Y. Heng, Z. Xie, H. Jiang, M. Tian, H. Jiang, Z. Zhang, Z. Ren and Y. Zhu, ACS Omega, 2022, 7, 23170–23178 CrossRef CAS PubMed.
- X. Song, H. Zhao, K. Fang, Y. Lou, Z. Liu, C. Liu, Z. Ren, X. Zhou, H. Fang and Y. Zhu, RSC Adv., 2019, 9, 3113–3119 RSC.
- L. Liu, M. Wang, Y. Zhan, Z. Lin, S. Xiong, H. Ye, Y. Luo, F. Fu, Z. Ren and Y. Zhu, New J. Chem., 2024, 48, 13048–13057 RSC.
- H. Qi, Y. Heng, J. Liu, W. Yi, J. Zou, Z. Ren, X. Zhou, M. Wang and Y. Zhu, J. Electroanal. Chem., 2024, 963, 118297–118305 CrossRef CAS.
- S. S. P. Rahardjo, Y.-J. Shih and C.-S. Fan, J. Hazard. Mater., 2024, 469, 134042–134055 CrossRef CAS PubMed.
- D. B. Adam, M.-C. Tsai, Y. A. Awoke, W.-H. Huang, Y.-W. Yang, C.-W. Pao, W.-N. Su and B. J. Hwang, ACS Sustainable Chem. Eng., 2021, 9, 8803–8812 CrossRef CAS.
- D. Wang, J. Wang, S. Lu and S. P. Jiang, J. Electroanal. Chem., 2014, 712, 55–61 CrossRef CAS.
- Z. Zhang, J. Liu, H. Ai, A. Chen, L. Xu, L. Labiadh, M.-L. Fu and B. Yuan, J. Environ. Chem. Eng., 2023, 11, 109834–109844 CrossRef CAS.
- J. Cao, D. Zhang, B. Ren, P. Song and W. Xu, Chin. Chem. Lett., 2024, 35, 109863–109870 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01122a |
| This journal is © The Royal Society of Chemistry 2025 |