
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceEnhanced photocatalytic performance of Ag-BiOBr nanosheets under LED with different wavelengths†
Shuqing Wanga,
Guodong Shen *abc,
Yijun Yaoa and
Runjun Suna
*abc,
Yijun Yaoa and
Runjun Suna
aSchool of Textile Science and Engineering, Xi'an Polytechnic University, Xi'an, 710048, China. E-mail: shenguodong@xpu.edu.cn
bKey Laboratory of Auxiliary Chemistry and Technology for Chemical Industry, Ministry of Education, Shaanxi University of Science and Technology, Xi'an, 710021, China
cShaanxi Collaborative Innovation Center of Industrial Auxiliary Chemistry and Technology, Shaanxi University of Science and Technology, Xi'an 710021, China
First published on 22nd April 2025
Abstract
The surface deposition of noble metals can effectively improve photocatalytic performance by the effect of electron trapping (ET) or surface plasmon resonance (SPR), but it is not clear which effect dominates the photocatalytic degradation of organic dyes under light irradiation at specific wavelengths. In this work, a series of xAg-(1−x)BiOBr (x = 0.01, 0.02, 0.03, 0.04, 0.05, and 0.06) nanosheets were synthesized via an ultrasonic-assisted chemical coprecipitation and photo-deposition reaction. The effect of light-emitting diode (LED) with different wavelengths on the photocatalytic degradation of Rhodamine B (RhB) dye was investigated. The results revealed that the hole (h+) and superoxide free radical (O2˙−) of xAg-(1−x)BiOBr were the main reactive groups contributing to the rapid decomposition of RhB dye. However, it is worth noting that the sources of the main active groups under LED with different wavelengths were different. Notably, photocatalytic performance was enhanced by the ET effect or SPR effect of Ag particles under LED of 390 nm or 570 nm, respectively. Moreover, both the ET and SPR effects synergistically enhanced the photocatalytic performance of Ag-BiOBr under full-spectrum irradiation (xenon lamp). This work provides theoretical and experimental insights into noble metal deposition modification and accelerates the application of photocatalysis technology in the degradation of printing and dyeing wastewater.
1. Introduction
Photocatalysis, as an advanced oxidation technology (AOT), has widely drawn attention owing to its promising application prospects in environmental improvement, such as wastewater degradation.1–3 BiOBr is an ideal visible-light-response semiconductor with a band gap of 2.87 eV.4 The [Bi2O2]2+ layer and double Br− layer in BiOBr with a tetragonal PbFCl crystal structure overlap with each other along the c-axis to form an open-layered crystal structure. Such a specific crystal structure enables BiOBr to exhibit stable photocatalytic performance and a two-dimensional sheet morphology. Meanwhile, BiOBr has the drawback of a high photo-generated charge-carrying recombination rate, which seriously restricts the application of BiOBr in wastewater treatment.5,6 Various modification methods (based on different mechanisms) on BiOBr photocatalysts have been reported, including element doping,7 noble metal deposition,8–10 semiconductor compounding or dye sensitization,9,10 for improving the separation efficiency of photo-generated electrons and holes. Among them, surface deposition of noble metals is one of the simplest and most efficient methods for significantly enhancing photocatalytic performance.Noble metals (Pt, Au, Ag) reduce the recombination of the electron–hole pairs via electron trapping (ET) or surface plasmon resonance (SPR).11,12 Among them, silver is the most widely used material and is applicable for industrial applications at the cheapest price.13 Yu et al. demonstrated that Ag could reduce the recombination of photo-generated charge carriers by trapping photo-generated electrons in Ag-BiOBr sheets.14 In contrast, Jiang et al. reported that the modification with 0.2–2 wt% Ag particles deposited on the surface of BiOBr sheets significantly decreased the photocatalytic performance.15 They concluded that a part of Ag easily formed AgBr with Br, while the other part of Ag was transformed into Ag+ and then converted into Ag2O. On the one hand, Ag broadened the spectral absorption intensity of BiOBr in the visible region (especially 430–800 nm). On the other hand, Ag2O and the structural defects of BiOBr caused by Br− detachment jointly reduced the photocatalytic performance. In addition, the particle size also affects the role of noble metals on the surface of BiOBr. Ye et al. investigated the effect of Au particle size on the photocatalytic performance of the BiOBr/g-C3N4/Au ternary composite photocatalyst.16 Larger Au particles played a bridging role in the electron transport medium between g-C3N4 sheets and BiOBr sheets, making holes the main active groups for degrading organic dyes. The SPR effect caused by the smaller Au particles led the free electrons to become the main active groups.
In this study, the photocatalytic performance of Ag-BiOBr under a light-emitting diode (LED) with a specific wavelength was explored to reveal the effects of Ag content and light source on the photocatalytic degradation of organic dyes. In addition, the mechanism by which Ag-BiOBr suppresses the recombination of photo-generated charge carriers under different light irradiation (LED and full spectrum light) was explained. The results could provide more experimental basis and theoretical guidance for noble-metal deposition modification and promote the application of AOT in the deep degradation of printing and dyeing wastewater.
2. Experimental
2.1 Synthesis of xAg-(1−x)BiOBr
According to our previous report,17,18 300–500 nm round BiOBr nanosheets were prepared by ultrasonic-assisted chemical coprecipitation. In general, the silver content of the noble metal-modified photocatalysts was lower for excellent ET or SPR effect.14,15,19 Meanwhile, the size of Ag particles on the surface of the photocatalysts increased with the extension of photo-deposition time.20 The Kessil PR160 LED was selected as the irradiation source for the photo-deposition synthesis to ensure that low concentrations of AgNO3 were rapidly converted into small-sized Ag particles and uniformly distributed on the surface of BiOBr.As shown in Fig. 1, BiOBr nanosheets and AgNO3 were first dispersed in a mixed solution of CH3OH and H2O (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 by volume) via ultrasonic stirring for 10 min. Then, xAg-(1−x)BiOBr (x = 0.01, 0.02, 0.03, 0.04, 0.05, and 0.06) samples were obtained by irradiation for 30 s. CH3OH acted as a reducing agent to accelerate the photo-deposition process of Ag particles by consuming ambient oxygens and holes produced by BiOBr.15
:1 by volume) via ultrasonic stirring for 10 min. Then, xAg-(1−x)BiOBr (x = 0.01, 0.02, 0.03, 0.04, 0.05, and 0.06) samples were obtained by irradiation for 30 s. CH3OH acted as a reducing agent to accelerate the photo-deposition process of Ag particles by consuming ambient oxygens and holes produced by BiOBr.15
 | ||
| Fig. 1 Schematic of the synthesis of Ag-BiOBr. | ||
2.2 Characterization and measurements
The instrument details of microstructure, crystal structure characterization and property testing are provided in the experimental section of the ESI.† The photocatalytic performance was evaluated by degrading Rhodamine B (RhB) dye solution (20 mg L−1). The photocatalyst dose was 10 mg L−1.As shown in Fig. S1,† the main wavelengths of the LED (PR160, Kessil) were 390 nm and 570 nm, respectively, with a maximum light intensity of 230 mW cm−2. A xenon lamp (190–1100 nm, 2000 mW cm−2, CEL-HXF300) was used as the simulated full-spectrum light source.
3. Results and discussion
The XRD patterns of BiOBr and xAg-(1−x)BiOBr are shown in Fig. 2a. Due to the low content of Ag, the XRD patterns of xAg-(1−x)BiOBr (x = 0.01, 0.02, 0.03, 0.04, 0.05, and 0.06) were dominated by the characteristic diffraction peaks of tetragonal BiOBr. Part of the XRD patterns (2θ = 37–46°) were amplified to observe the content change of Ag in xAg-(1−x)BiOBr. The excessively low content (about 0.4 wt%) of Ag in 0.01 Ag-0.99BiOBr results in the inability to observe the characteristic peaks of elemental silver. Except for the two characteristic diffraction peaks at 38.1° and 44.2° corresponding to the 111 and 220 crystal planes of elemental Ag, no peaks other than the tetragonal BiOBr appeared in xAg-(1−x)BiOBr (x = 0.02, 0.03, 0.04, 0.05, and 0.06) samples. The intensity of the above two characteristic diffraction peaks increased with the increase in Ag in xAg-(1−x)BiOBr samples. In addition, SEM, EDS, and TEM will be employed to confirm the morphology and size of elemental Ag in xAg-(1−x)BiOBr. In addition, as shown in Fig. 2b, spherical particles were attached to the surfaces of the BiOBr nanosheets. The EDS results present that the atomic ratio of Bi to Br is 28.77:25.79, which is close to the atomic ratio of Bi to Br in BiOBr (1:1). The presence of 4.27 mol% Ag indicates that Ag particles were successfully photo-deposited on the surface of BiOBr (Fig. 2c). As shown in Fig. 2d–f, 10–20 nm spherical Ag particles are dispersed deposited on the surface of BiOBr nanosheets without accumulation. The uniform distribution of Ag particles is advantageous for the capture of light energy by Ag and BiOBr.
 | ||
| Fig. 2 (a) XRD patterns of BiOBr and xAg-(1−x)BiOBr, (b) SEM, (c) EDS and (d–f) TEM images of 0.03Ag-0.97BiOBr. | ||
The chemical states and binding forms of BiOBr and Ag in xAg-(1−x)BiOBr were analyzed by XPS. The binding energy absorption peaks of BiOBr and xAg-(1−x)BiOBr samples are similar, except for the characteristic Ag absorption peaks at 366–378 eV (Fig. S2a†). As shown in Fig. S2b,† the binding energy absorption peak interval of Ag 3d2/3 and Ag 3d5/2 in xAg-(1−x)BiOBr samples is 6.0 eV, which is attributed to elemental Ag.20 The XPS results further demonstrate the formation of Ag particles on the surface of BiOBr nanosheets. Neither the XRD nor XPS results demonstrate the formation of Ag2O and AgBr, that is, the enhancement of the photocatalytic performance in Ag-BiOBr is only related to the photo-deposited elemental Ag particles. The atomic proportions of the selected target elements are shown in Table S1.† On the one hand, the atomic ratio of the selected target element proves that the increase in Ag content in the xAg-(1−x)BiOBr samples is consistent with the increment of x, indicating that the Ag variation law of the as-prepared Ag-BiOBr samples is consistent with the composition design. On the other hand, XPS was applied to measure the surface element content, and the oxygen element may also come from the conductive adhesive or the instrument chamber, resulting in lower contents of Bi and Br. Therefore, the atomic ratio of Bi/Ag is different from the design components of Ag-BiOBr. Overall, the XPS results verify that the increment of Ag in xAg-(1−x)BiOBr series samples is consistent with the component design.
In addition, ICP-OES (inductively coupled plasma-optical emission spectroscopy) was performed to detect the content of metallic elements (Ag and Bi). Similar to the XPS results, ICP-OES results present that the increase in Ag content is consistent with the component design of xAg-(1−x)BiOBr (Fig. S3a†). More importantly, the ICP-OES-detected contents of Ag and Bi were only slightly higher than the theoretical content of xAg-(1−x)BiOBr (Fig. S3a and b†), verifying that the Ag content conformed to the component design. All of the above characterization and test results (XRD, XPS and ICP-OES) provide a series of reliable photocatalysts with reliable photocatalytic performance and degradation mechanisms.
UV-Vis DRS treated by data normalization was applied to investigate the effect of the photo-deposited Ag particles on the spectral absorption property of Ag-BiOBr powders. There was almost no difference in spectral absorption between xAg-(1−x)BiOBr and BiOBr in the range of 250–400 nm (Fig. 3). The spectral absorption intensity of Ag-BiOBr increases significantly with a gradual increase in deposited Ag particles from 400 nm. However, BiOBr exhibits almost no spectral absorption in the range of 430–800 nm. According to the empirical formula Eg = 1240/λ,21 the bandgap width of BiOBr is approximately 2.87 eV, which is consistent with the previous results reported.22,23 In contrast, xAg-(1−x)BiOBr has a higher spectral absorption intensity in the range of 430–750 nm, which is due to the absorption difference caused by the SPR effect of Ag.12,20 The results also indicate that Ag deposition on the surface of xAg-(1−x)BiOBr triggers a significant SPR effect in the spectral range of 570–590 nm. Based on the above results, the real effect of surface-deposited Ag particles on the photocatalytic performance of BiOBr can be determined by comparing the efficiency of the photocatalytic degradation of organic dyes under specific wavelength light irradiation (such as LED). Since the spectral absorption boundary of BiOBr and the highest visible spectral absorption peak of Ag-BiOBr are about 432 and 575 nm, two LEDs with specific wavelengths of 390 or 570 nm were selected to comparatively evaluate the photocatalytic performance for degrading organic dye. The former LED (390 nm) stimulated the photo-generated charge carriers of BiOBr, but it was difficult to induce the SPR effect of Ag in xAg-(1−x)BiOBr. In contrast, the latter LED (570 nm) did not excite BiOBr to produce electron and hole pairs but was conducive to promoting the SPR effect of Ag particles.
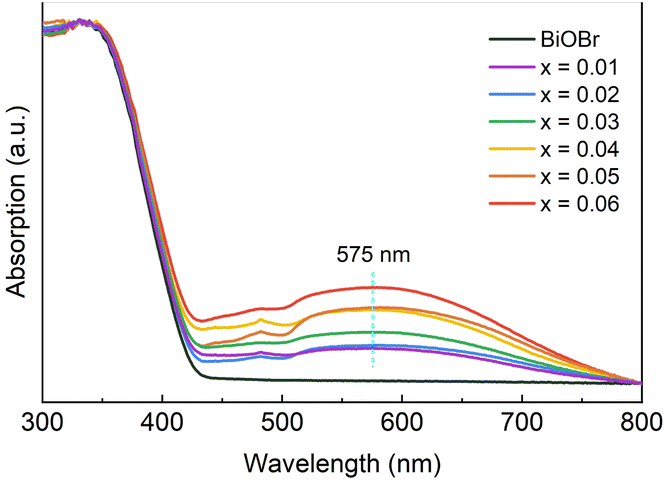 | ||
| Fig. 3 UV-Vis DRS of BiOBr and xAg-(1−x)BiOBr powders. | ||
As shown in Fig. 4a and c, the adsorption efficiency of xAg-(1−x)BiOBr and BiOBr for RhB dye is close in the dark reaction stage (−30–0 min). To explore the effect of surface-deposited Ag particles on the adsorption capacity of Ag-BiOBr, a comparison of the adsorption efficiency within 30 min is shown in Fig. S5.† Moreover, N2 adsorption–desorption isotherm curves were obtained to measure the specific surface areas of BiOBr and xAg-(1−x)BiOBr samples. As listed in Table S2,† the surface deposited Ag nanoparticles increase the specific surface area of the BiOBr sheets. The reduction in the specific surface area of xAg-(1−x)BiOBr (x = 0.04, 0.05, and 0.06) samples can be inferred from the agglomeration of Ag nanoparticles on the surface of BiOBr. On the one hand, the adsorption capacity of xAg-(1−x)BiOBr for RhB dye is higher than that of BiOBr (Fig. S4†), which is attributed to the fact that the surface morphology of the BiOBr sheets is changed by surface-deposited Ag nanoparticles. On the other hand, excessive surface-deposited Ag particles easily led to aggregation. Therefore, the adsorption performance of 0.03Ag-0.97BiOBr for RhB dye is the highest under both LED wavelengths of 390 nm and 570 nm. Both xAg-(1−x)BiOBr and BiOBr could photocatalytic degrade RhB dye under the LED (390 nm) irradiation, but the degradation efficiency of xAg-(1−x)BiOBr is significantly higher than that of BiOBr (Fig. 4a). Meanwhile, since there is no obvious SPR effect in the wavelength range of 390 nm, it can be inferred that Ag particles probably reduce the secondary recombination of photo-generated electrons and holes by acting as an electron trapping trap. The 0.03Ag-0.97BiOBr sample shows the highest degradation efficiency (95.50%) for RhB dye in 60 min.
 | ||
| Fig. 4 Photocatalytic degradation performances and pseudo-first-order reaction kinetics of BiOBr and xAg-(1−x)BiOBr over RhB dye under LED irradiation with different wavelengths: (a and c) 390 nm, (b and d) 570 nm. | ||
The LED with a wavelength of 570 nm could not excite the photo-generated electrons from the valence band to the conduction band of BiOBr (Fig. 4c), so the photocatalytic degradation efficiency of BiOBr over RhB dye does not change significantly with the extension of light exposure time (0–120 min). However, Ag-BiOBr also showed slow degradation of RhB dye, which was attributed to the SPR effect of the Ag particles under an LED wavelength of 570 nm. Meanwhile, the SPR effect gradually increased with increasing content of Ag particles. The photocatalytic degradation efficiency of 0.06Ag-0.94BiOBr over RhB dye is 50.06% after 120 min.
Fig. 4b and d display the photocatalytic degradation rate curves calculated according to the pseudo-first-order kinetic model. The reaction rate curves show a linear relationship with the reaction time under an LED of 390 nm, whereas the photocatalytic degradation rate is relatively lower under an LED of 570 nm. The photocatalytic degradation rate of 0.03Ag-0.97BiOBr for the target dye was 2.7 times that of BiOBr.
To explore the main active groups of degrading RhB dye under different LEDs. P-benzoquinone (BQ), triethanolamine (TEOA) and isopropyl alcohol (IPA) were selected as the scavengers of the superoxide free radical (O2˙−), hole (h+) and hydroxyl free radical (˙OH), respectively. As shown in Fig. 5a, the degradation efficiency of 0.03Ag-0.97BiOBr over RhB dye decreased by more than 90% upon the addition of TEOA, indicating that h+ is the main active group for oxidative decomposition of RhB dye under an LED wavelength of 390 nm. On the contrary, the degradation efficiency hardly decreased with the addition of IPA, suggesting that ˙OH is not an active group for degrading RhB dyes, while O2˙− is another active group behind h+. As shown in Fig. 5b, O2˙− is the unique active group for photocatalytic degrading RhB dye, while ˙OH hardly participates in photocatalytic degradation under an LED wavelength of 570 nm.
 | ||
| Fig. 5 Trapping experiments under LED irradiation of (a) 390 nm and (b) 570 nm. (c) Photodegradation performance of BiOBr and xAg-(1−x)BiOBr under xenon lamp irradiation and (d) recycling reaction of 0.03Ag-0.97BiOBr. | ||
The sources of the main active groups under two different LEDs with different wavelengths are completely different. Under an LED of 390 nm, the most important active group (h+) comes from the valence band of BiOBr. The noble Ag particles inject hot electrons into the conduction band of BiOBr via the SPR effect to produce enough O2˙− groups, which make O2˙− play the most important role in degrading RhB dye under an LED wavelength of 570 nm.
Subsequently, a comparative experiment using a suspension of silver nanoparticles was performed to verify the above inference that BiOBr is a carrier for receiving the hot electrons produced by Ag particles. The 20 nm silver nanoparticle dispersion solution with an initial concentration of 100 mg L−1 was added to the RhB dye solution to dilute to 10 mg L−1 Ag dispersion solution. As shown in Fig. S5,† the photocatalytic degradation efficiency of Ag nanoparticles over RhB dye is very low as to be negligible in the absence of BiOBr sheets. The results of the comparative experiments confirm that BiOBr acts as a carrier for receiving hot electrons.
Another free radical capture comparison experiment was conducted by replacing the aqueous solution with a CH3OH/H2O (1:1 by volume) solution of RhB dye. The reducing agent methanol (CH3OH) was used for the capture of h+.24 The photocatalytic degradation efficiency decreased by more than 60% while the photo-generated h+ is reduced by CH3OH under an LED of 390 nm. In contrast, no significant decline in photocatalytic degradation efficiency was observed under an LED wavelength of 570 nm. The results again confirm that h+ is the main active group for degrading RhB dye under an LED of 390 nm, while the O2˙− groups generated by the SPR effect of Ag particles have been the new dominant factor for RhB dye degradation under 570 nm LED irradiation.
To confirm the importance of O2 in the reaction mechanism, argon (Ar) gas is continuously injected into the RhB dye solution to expel O2. As shown in Fig. 5a and b, the photocatalytic degradation efficiency was reduced by continuous Ar injection. Combined with the results of trapping O2˙− experiments, these results show that O2 is an important factor in photocatalytic degradation. The lack of O2 made it difficult to produce O2˙−, which is a similar effect to that of adding BQ. The absence of the main active group (O2˙−) reduces the photocatalytic degradation efficiency, especially under the LED of 590 nm.
The most common simulation of the visible light degradation experiment using a representative 300 W xenon lamp is shown in Fig. 5c. In the dark reaction stage (−30–0 min), The amounts of RhB dye adsorbed by BiOBr and xAg-(1−x)BiOBr are equal (approximately 20%). In the photocatalytic reaction stage (0–40 min), the concentration of RhB dye solution decreases linearly with the extension of reaction time. The degradation efficiency of xAg-(1−x)BiOBr is much higher than that of BiOBr. Under full-spectrum xenon lamp irradiation, the noble metal Ag particles deposited on the surface of BiOBr not only serve as ET but also exert the SPR effect. These two effects work together to achieve a rapid degradation for the target dye. The photocatalytic degradation efficiency of 0.03Ag-0.97BiOBr over RhB dye is as high as 97.50% after 40 min.
In addition, based on the experimental results of the free radical capture experiment of CH3OH, Ag particles reduced the secondary recombination of photo-generated charge carriers of BiOBr by trapping the electrons from the conduction band of BiOBr in the full-spectrum irradiation environment, which is the main reason for significantly improving the photocatalytic degradation efficiency. 0.03Ag-0.97BiOBr has excellent cyclic photocatalytic performance under xenon lamp irradiation. The photocatalytic degradation efficiency of 5 cycles is more than 95% (Fig. 5d).
The significant enhancement of the photocatalytic activity of BiOBr modified by the surface-deposited noble Ag particles again indicates that the high-intensity ultraviolet light source (LED), low addition amount of AgNO3, continuous ultrasonic stirring, short photo-deposition time (30 s), and regular morphology and controllable size of two-dimensional BiOBr nanosheets are the key factors in ensuring the uniform deposition and fastness of small-sized Ag particles (10–20 nm) on the surface of BiOBr.
Based on the above results, Ag particles enhance the photocatalytic performance of BiOBr in different ways under LED at different wavelengths (Fig. 6). (1) The photo-generated electrons (e−) transition to the conduction band of BiOBr, while a large number of holes (h+) are generated in the valence band under an LED of 390 nm. Due to the low Fermi level of Ag, the secondary recombination of photo-generated charge carriers is effectively reduced and inhibited by trapping photo-generated electrons from the conduction band of BiOBr to Ag particles.11 Therefore, the photo-generated holes in the valence band of BiOBr are the main active groups in the oxidation decomposition of organic dyes. Furthermore, the free electrons captured by the trapping effect of Ag particles combine with ambient O2 to form more O2˙− active groups, which participate in the degradation of organic dyes. (2) Although it is difficult to excite BiOBr with a bandgap width of 2.87 eV by the LED of 570 nm, the specific light energy matches the collective oscillation frequency of the electrons inside the Ag particles. The thermal motion of electrons in Ag particles is induced by the absorption of large numbers of photons. Subsequently, hot electrons are first transferred to the conduction band of BiOBr and then form many O2˙− active groups with ambient O2. Therefore, different from the photocatalytic degradation mechanism of the oxidation decomposition of organic dyes by photo-generated holes under an LED of 390 nm, the abundant O2˙− active groups generated by Ag particles are the main forces for degrading organic dyes under an LED of 570 nm. Last but not least, the synergistic effect of electron trapping (ET) and surface plasmon resonance (SPR) ensures that the target organic dyes are rapidly photocatalytically degraded within 40 min under xenon lamp irradiation.
 | ||
| Fig. 6 Photocatalytic mechanism diagrams of Ag-BiOBr under different wavelengths of LED irradiation. | ||
4. Conclusions
In this study, a series of xAg-(1−x)BiOBr (x = 0.01, 0.02, 0.03, 0.04, 0.05, and 0.06) nanosheets were synthesized using ultrasonic-assisted chemical coprecipitation and photo-deposition reaction. The photocatalytic performance was enhanced by the ET effect or SPR effect of Ag particles under an LED irradiation at 390 nm or 570 nm, respectively. Moreover, both the ET effect and SPR effect synergistically promoted the significant enhancement of photocatalytic performance in Ag-BiOBr under full-spectrum irradiation (xenon lamp). The photocatalytic degradation efficiency of 0.03Ag-0.97BiOBr over RhB dye is 97.50% after 40 min. This study provides a theoretical and experimental basis for noble metal deposition modification and accelerates the application of photocatalysis technology in the degradation of printing and dyeing wastewater.Data availability
The data supporting this article are included in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported by Innovation Capability Support Program of Shaanxi (2024ZC-KJXX-044), Open Foundation of Key Laboratory of Auxiliary Chemistry and Technology for Chemical Industry, Ministry of Education (KFKT2022-08), Natural Science Basic Research Plan in Shaanxi Province of China (2021JQ-681).References
- Y. Guo, L. Luo, T. Liu, L. Hao, Y. Li, R. Liu and T. Zhu, J. Environ. Sci., 2024, 136, 682–697 CrossRef PubMed.
- C. Hu, J. Cao, X. Jia, H. Sun, H. Lin and S. Chen, Appl. Catal., B, 2023, 337, 122957 CrossRef CAS.
- Y. Guo, X. Tong and N. Yang, Nano-Micro Lett., 2023, 15, 77 CrossRef CAS PubMed.
- L. Zhan, J. Hu, Y. Cao, X. Ning, J. Xie, Z. Lu and A. Hao, Chem. Commun., 2024, 60, 1892–1895 RSC.
- Y. Li, J. Guan, S. Li, H. Wang, J. Liu, J. Fu and M. Yu, Sep. Purif. Technol., 2025, 360, 131312 CrossRef CAS.
- Z. Chen, Z. Lu, W. Tian, B. Liu, B. Huo, J. Zhao, M. Zou, R. Zhang, X. Yu and X. Zhang, Sep. Purif. Technol., 2025, 362, 131720 CrossRef.
- Y. Qian, Q. Zhang, L. Chen, Y. Dong, T. Song, Y. Pei, X. Wang, X. Wu, X. Zheng, W. He and B. Long, J. Colloid Interface Sci., 2024, 441, 114841 Search PubMed.
- T. Tian, X. Sun, L. Di, C. Sun, H. Li, C. Ma and H. Yang, Appl. Surf. Sci., 2025, 638, 158136 Search PubMed.
- S. Liu, Y. Li, K. Yang, X. Li, W. Jin, X. Zhong, H. Liu and R. Xie, Appl. Surf. Sci., 2025, 688, 162404 CrossRef CAS.
- H. Li, C. He, A. Xiao, Y. Hu, L. Luo and F. Jiang, J. Cleaner Prod., 2024, 469, 143239 CrossRef CAS.
- S. Pamu, K. Vashist, P. Ganesh, P. Costa and S. Singh, Catal. Today, 2024, 441, 114841 CrossRef CAS.
- M. Du, W. Lin, N. Liu, Y. Ling and S. Kang, Nano Energy, 2024, 124, 109495 CrossRef CAS.
- A. Sabir, T. Sherazi and Q. Xu, Surf. Interface, 2021, 26, 101318 CrossRef CAS.
- C. Yu, C. Fan, X. Meng, K. Yang, F. Cao and X. Li, React. Kinet., Mech. Catal., 2011, 103, 141–145 CrossRef CAS.
- L. Lu, L. Kong, Z. Jiang, H. Lai, T. Xiao and P. Edwards, Catal. Lett., 2012, 142, 771–778 CrossRef CAS.
- Y. Bai, T. Chen, P. Wang, L. Wang, L. Ye, X. Shi and W. Bai, Sol. Energy Mater. Sol. Cells, 2016, 157, 406–414 CrossRef CAS.
- G. Shen, Y. Pu, Y. Cui, P. Jing and R. Sun, New J. Chem., 2021, 45, 10496 RSC.
- G. Shen, Y. Pu, R. Sun, Y. Shi, Y. Cui and P. Jing, New J. Chem., 2019, 43, 12932 RSC.
- G. Jiang, R. Wang, X. Wang, X. Xi, R. Hu, Y. Zhou, S. Wang, T. Wang and W. Chen, ACS Appl. Mater. Interfaces, 2012, 4, 4440–4444 CrossRef CAS PubMed.
- J. Ni, D. Liu, W. Wang, A. Wang, J. Jia, J. Tian and Z. Xing, Chem. Eng. J., 2021, 419, 129969 CrossRef CAS.
- G. Shen, Y. Pu, Y. Cui and P. Jing, Ceram. Int., 2017, 43, S664–S670 CrossRef CAS.
- A. Ganose, M. Cuff, K. Butler, A. Walsh and D. Scanlon, Chem. Mater., 2016, 28, 1980–1984 CrossRef CAS PubMed.
- G. Shen, Y. Pu, Y. Cui, P. Jing, M. Yang and R. Sun, Appl. Surf. Sci., 2021, 550, 149366 CrossRef CAS.
- Y. Zhou, Z. Zhang, Z. Fang, M. Qiu, L. Ling, J. Long, L. Chen, Y. Tong, W. Su, Y. Zhang, J. Wu, J. Basset, X. Wang and G. Yu, Proc. Natl. Acad. Sci. U.S.A., 2019, 116, 10232–10237 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra01035g |
| This journal is © The Royal Society of Chemistry 2025 |