
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceAmide linked chalcone derivatives, a promising class of compounds with versatile biological effects
Omar Alshazly *ab,
Gamal El-Din A. Abuo-Rahma*bc,
Mamdouh F. A. Mohamedad and
Mohamed Abdel-Azizb
*ab,
Gamal El-Din A. Abuo-Rahma*bc,
Mamdouh F. A. Mohamedad and
Mohamed Abdel-Azizb
aDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Sohag University, 82524 Sohag, Egypt
bDepartment of Medicinal Chemistry, Faculty of Pharmacy, Minia University, 61519 Minia, Egypt. E-mail: gamalaburahma@yahoo.com; Tel: +20-1003069431
cDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, Deraya University, New-Minia, Egypt
dDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, New Valley University, New Valley, 72511, Egypt
First published on 5th June 2025
Abstract
Chalcone-linked acetamide derivatives represent a unique class of compounds with a broad spectrum of biological activities. Various synthetic methods for chalcones are reviewed, emphasizing their efficiency. These derivatives exhibited potent antiproliferative properties, functioning as inhibitors of key targets such as EGFR, topoisomerase I and II, ABCG2, caspase proteins, and histone deacetylase (HDAC), as well as inhibiting tubulin polymerization. The structure–activity relationships (SAR) pertinent to their anticancer activity are elucidated. In addition to their antiproliferative effects, these compounds display significant antimicrobial activities against a variety of bacterial and fungal pathogens. Their antiviral potential is also highlighted, with capabilities to inhibit critical viral enzymes and pathways. The antiprotozoal properties of chalcone-linked acetamide derivatives underscore their efficacy against protozoan infections. Furthermore, these derivatives possess strong anti-inflammatory and antioxidant activities, contributing to their overall therapeutic potential. By exploring these diverse biological activities, these compounds present significant opportunities for the development of novel therapeutic agents overcoming various medical challenges.
1 Introduction
Chalcones, also known as 1,3-diphenylpropan-1-ones, serve as the initial intermediate structures in the biosynthesis of all flavonoids. These open-chain flavonoids consist of two aromatic rings (A and B) connected by a three-carbon α, β-unsaturated carbonyl system (Fig. 1).1,2 | ||
| Fig. 1 Chalcones scaffold. | ||
Many natural chalcones exhibit multiple hydroxyl groups on their aromatic rings. Typically, hydroxyl groups are found at the C2′, C4′, and/or C6′ positions in the A-ring. The B-ring usually contains a hydroxyl group at the C4 position. Chalcones lacking an oxygen at the 2′-position are known as retrochalcones. Prenyl and methoxyl groups are commonly found as substituents in natural chalcones.1,3
These small molecular structures possess Michael acceptor features, enabling chalcones to interact with various biological molecules, facilitating their binding and reactivity. Consequently, chalcones demonstrate a wide range of biological activities encompassing anticancer, anti-inflammatory, antibacterial, antituberculosis, antidiabetic effects, antioxidant capabilities, antimicrobial and antiviral, antimalarial, neuroprotective effects, among others.4–13 Interestingly, a single chalcone compound can manifest multiple types of bioactivity. For instance, isoliquiritigenin (Fig. 2a) exhibits anticancer, cancer-chemo-preventive, antioxidant, and anti-inflammatory activities.14,15 Similarly, xanthohumol (Fig. 2b) demonstrates anti-HIV-1, antibacterial, and anticancer effects.16 It is worth noting that alongside the therapeutic potentials of chalcones, researchers have also evaluated their potential side effects.17,18 Notably, Xing et al.19 discovered a hepatotoxic risk associated with a specific type of chalcone, emphasizing the importance of further comprehensive investigation in this area.
 | ||
| Fig. 2 Examples of natural bioactive chalcones; (a) isoliquiritigenin, (b) xanthohumol. | ||
Amide group are not only common structural motifs in biologically active molecules but also play a critical role in modulating physicochemical properties such as solubility, lipophilicity, and metabolic stability.20,21 More importantly, the amide moiety contributes significantly to molecular recognition through various non-covalent interactions, including hydrogen bonding and dipole–dipole interactions, which are essential for enhancing binding affinity and specificity toward biological targets.22
By integrating an amide functionality into the chalcone structure, it is possible to synergistically combine the electrophilic Michael acceptor character of chalcones with the favorable binding properties of amides.10 This hybrid design strategy is expected to enhance target interaction, improve bioavailability, and potentially lead to compounds with superior pharmacological profiles.23 Thus, chalcone-linked acetamide derivatives represent a promising class of compounds for drug discovery and development.24
This article present various synthetic approaches for chalcone derivatives and comprehensively review the specific biological activities associated with chalcones incorporating amide functionalities, with a particular focus on recent advancements and current research developments in this field.
2 Methods of synthesis of chalcones
Many methods have been developed for synthesizing chalcones, which are a significant class of natural products possessing diverse biological activities. Moreover, chalcones serve as precursors for the synthesis of numerous compounds. Furthermore, there has been a considerable amount of research focused on developing synthetic strategies to produce chalcones that mimic their natural counterparts, as well as synthesis of novel molecules. The following sub-sections provide an overview of the primary methodologies employed for this purpose (Scheme 1).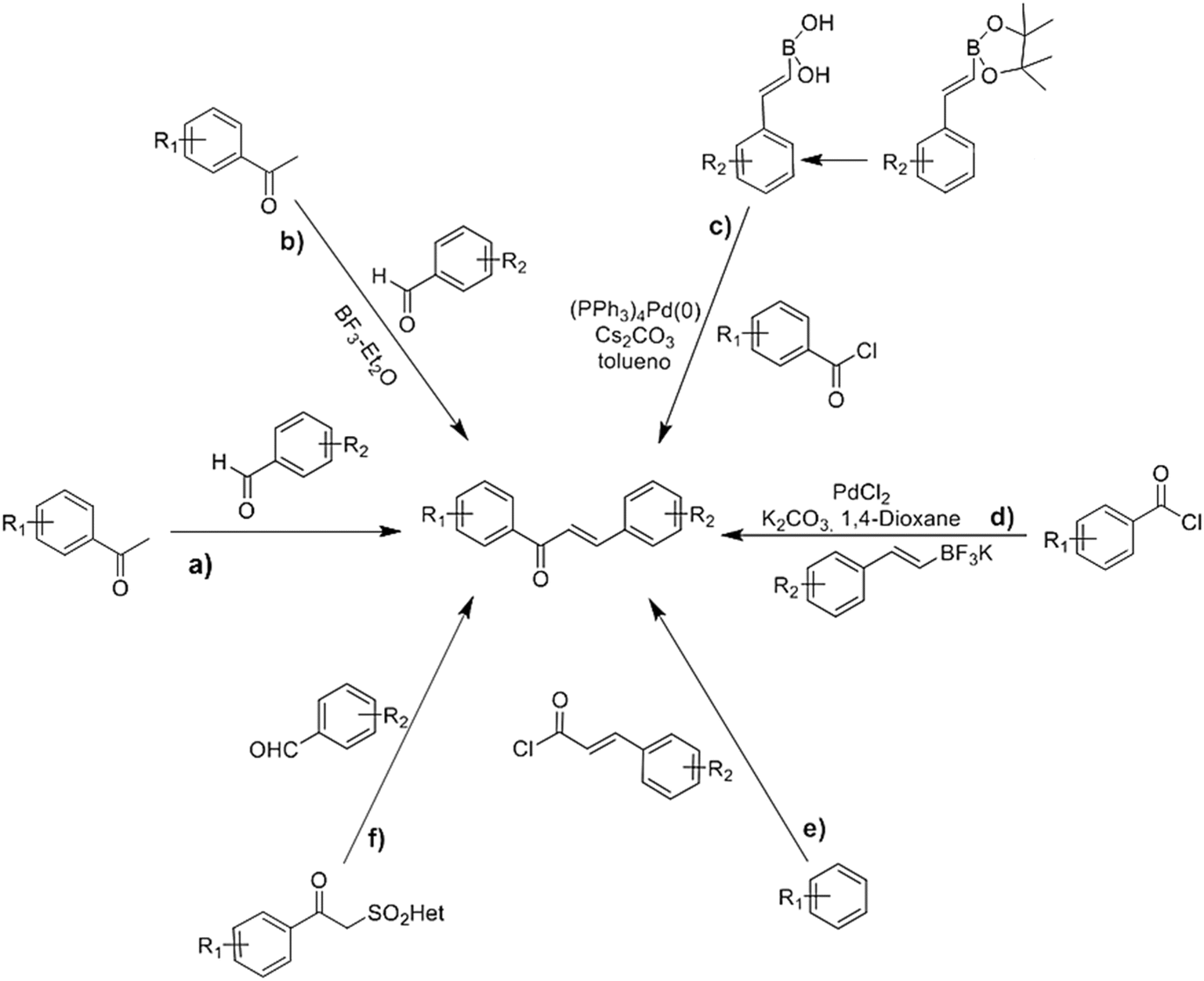 | ||
| Scheme 1 Different methods of chalcones synthesis. (a) Claisen–Schmidt reaction; (b) using borontrifluoride-etherate; (c) via Suzuki coupling reaction; (d) direct cross-coupling reaction; (e) Friedel–Crafts acylation; (f) Julia–Kocienski olefination. R1 and R2 = electron donating or electron withdrawing groups. | ||
2.1 Synthesis of chalcones by Claisen–Schmidt reaction
The Claisen–Schmidt condensation is a widely used method for synthesizing chalcones, involving the condensation of acetophenone and benzaldehyde derivatives to form α,β-unsaturated ketones, linking aromatic rings A and B (Scheme 1a).25 The reaction proceeds through the nucleophilic addition of the carbanion, derived from the aryl ketones to the carbonyl carbon of the aromatic aldehydes, the Claisen–Schmidt reaction is typically carried out in the presence of an aqueous, methanolic or ethanolic alkaline solution, at room temperature or at reflux for several hours.26 Moreover, microwave (MW) irradiation has been effectively employed. This method involves the condensation of acetophenone and benzaldehyde derivatives in the presence of anhydrous potassium carbonate under free-solvent conditions. Anhydrous K2CO3 is a cost-effective, non-toxic, and user-friendly alternative to stronger bases like NaOH or KOH, which are typically used in traditional Claisen–Schmidt reactions but can be harmful, toxic, and environmentally polluting. Khan, S. A., et al.27 successfully applied this methodology to synthesize various chalcone derivatives with high yields.2.2 Synthesis of chalcones using borontrifluoride etherate
In 2007, Narender and Reddy developed a method utilizing borontrifluoride etherate for the synthesis of diverse chalcones. Instead of the traditional base-catalyzed Claisen–Schmidt reaction, this method involves the reaction between various substituted acetophenones and aromatic aldehydes using BF3–Et2O as a catalyst, yielding chalcones within a timeframe of 15–150 minutes (Scheme 1b).28 Compared to the conventional Claisen–Schmidt condensation reactions employing KOH or NaOH, this method offers several advantages. These include high yields, simplified work-up procedures, shorter reaction times, absence of side reactions, and compatibility with sensitive functional groups such as amides and esters.282.3 Synthesis of chalcones by direct cross-coupling reaction
The direct cross-coupling reaction involves the coupling of benzoyl chlorides and potassium styryltrifluoroborates using a palladium-catalyzed system under microwave (MW) irradiation. This method enables the one-pot synthesis of α,β-unsaturated aromatic ketones. Al-Masum et al.29 were the first to utilize this approach for the synthesis of several chalcones, employing 1,4-dioxane as a solvent and K2CO3 as a base (Scheme 1d). Despite 1,4-dioxane being a non-polar solvent typically not used in MW irradiation reactions, its polarizability due to the presence of two oxygen atoms and relatively high boiling point is advantageous for facilitating the desired cross-coupling reaction. This process offers notable advantages, including the non-toxicity, ease of preparation, and ease of removal of potassium styryltrifluoroborates.2.4 Synthesis of chalcones by Friedel–Crafts acylation
Chalcones can be synthesized through a direct Friedel–Crafts acylation reaction involving a phenol. In this reaction, the phenol serves as the precursor for the formation of the chalcone's A-ring, while the acylating agent contributes to the formation of both the B-ring and the three-carbon bridge connecting rings A and B (Scheme 1e). Various catalysts have been employed for this reaction, including anhydrous aluminum chloride,30 aluminum bromide,31 and chiral phosphoric acids.322.5 Synthesis of chalcones by Julia–Kocienski olefination
The approach described in this method plays a pioneering role in the synthesis of chalcones and flavanones. It involves the condensation of aldehydes with 2-(benzo[d]thiazol-2-ylsulfonyl)-1-phenylethanones, which are reagents developed for Julia–Kocienski olefination. This reaction is carried out in the presence of a base (Scheme 1f). The resulting product is obtained in good yields.332.6 Synthesis of chalcones by Suzuki coupling reaction
The Suzuki reaction is a valuable tool utilized for the formation of carbon–carbon (C–C) bonds. It involves the coupling of organoboron compounds with organic halides or triflates, catalyzed by palladium in the presence of a base (Scheme 1c). This reaction has found widespread application in the synthesis of various compounds, including chalcones.34In 2003, Eddarir et al.35 introduced a method for synthesizing chalcones through the Suzuki coupling reaction. This method involves the coupling of cinnamoyl chlorides with phenylboronic acids (Scheme 2a) or the coupling of benzoyl chlorides with phenylvinylboronic acid (Scheme 2b).
 | ||
| Scheme 2 Method of synthesis of chalcones via Suzuki coupling reaction. | ||
2.7 Synthesis of chalcones by grinding technique
By utilizing the grinding technique, certain chalcone derivatives have been successfully synthesized. This method involves the reaction between substituted 2-acetyl-1-naphthol and various substituted benzaldehydes in the presence of a base. Notably, this reaction does not require a catalyst, making it non-hazardous and environmentally safer. Furthermore, it offers the advantage of yielding excellent results in a short reaction time, leading to high product yields.263 Antiproliferative properties
The documentation of the antiproliferative activity of acetamido chalcones is increasing. Many research groups synthesized or modified natural chalcones that possess antiproliferative activity. Chalcones showed an ability of targeting multiple cellular molecules, such as mouse double minute 2 MDM2/p53,36 tubulin,37 nuclear factor kappa-light-chain-enhancer of activated B cells NF-kappa B,38 vascular endothelial factor VEGF, VEGFR-2 kinase,39 hypoxia-inducible factor-1 HIF-1,40 matrix metalloproteinase MMP-2/9 (ref. 41) and P-glycoprotein P-gp42/multidrug resistance-associated protein MRP1 (ref. 43)/breast cancer-resistance protein BCRP.44 Which indicates that chalcones can act as an anticancer via tumor cell apoptosis induction, microtubule polymerization, anti-inflammatory, antiangiogenesis.45 These characters made chalcones highly desirable as fundamental constituents for developing agents that target cancer molecules.3.1 Chalcones as EGFR inhibitors
EGFR (epidermal growth factor receptor) is a protein involved in cell growth and survival. However, in certain cancers, EGFR becomes overactive or mutated, leading to uncontrolled cell growth.46 Targeting EGFR has emerged as an important strategy in anticancer treatment. EGFR inhibitors, such as small molecule tyrosine kinase inhibitors (TKIs) or monoclonal antibodies, are designed to block EGFR activity and its downstream signaling pathways.47 Small molecule TKIs, like erlotinib or osimertinib (Fig. 3), inhibit the intracellular domain of EGFR, preventing the phosphorylation of downstream molecules.48 Monoclonal antibodies, such as cetuximab or panitumumab, bind to the extracellular domain, blocking ligand binding and receptor activation.49 By inhibiting EGFR, these drugs suppress cell proliferation and survival, induce apoptosis, and inhibit tumor angiogenesis. EGFR inhibitors can also modulate the immune response against cancer cells, enhancing immune recognition and promoting antitumor immune responses.50 However, the effectiveness of EGFR-targeted therapies can vary depending on cancer type and the presence of specific EGFR mutations. Resistance to EGFR inhibitors can also develop, necessitating combination therapies and personalized treatment approaches.51 | ||
| Fig. 3 EGFR inhibitors – erlotinib and osimertinib. | ||
A series of quinoline/chalcone hybrids containing 1,2,4-triazole moiety was tested by ref. 52, for its antiproliferative activities, All of the selected compounds were tested in four cancer cell lines: the pancreatic cancer cell line Panc-1, the breast cancer cell line MCF-7, the colon cancer cell line HT-29, and the epithelial line cancer cell A-549. The five most active compounds were 1, 2, 3, 4, and 5 (Fig. 4). Among these compounds, 2 exhibited the highest activity against cancer cell growth, with a GI50 of 3.325 μM. Compound 2, which has an allyl-triazole backbone, displayed the highest anticancer potential. On the other hand, compound 1, which has the same substitution pattern as 2 but a different phenyltriazole moiety, showed almost 2.5 times lower activity than 2. These findings suggest that all compounds with an allyl-triazole backbone were better inhibitors of cancer cell growth compared to derivatives with the same substitutions but a different phenyltriazole backbone, and the presence of trimethoxy phenyl at the chalcone moiety showed increase in the antiproliferative activity. All investigated compounds 1, 2, 3, 4, and 5 exhibited inhibitions of EGFR with IC50 ranging from 1.3 to 4.8 μM in comparison to the positive control erlotinib (IC50 = 0.08 ± 0.04 μM). This study demonstrates that these compounds are effective EGFR inhibitors.
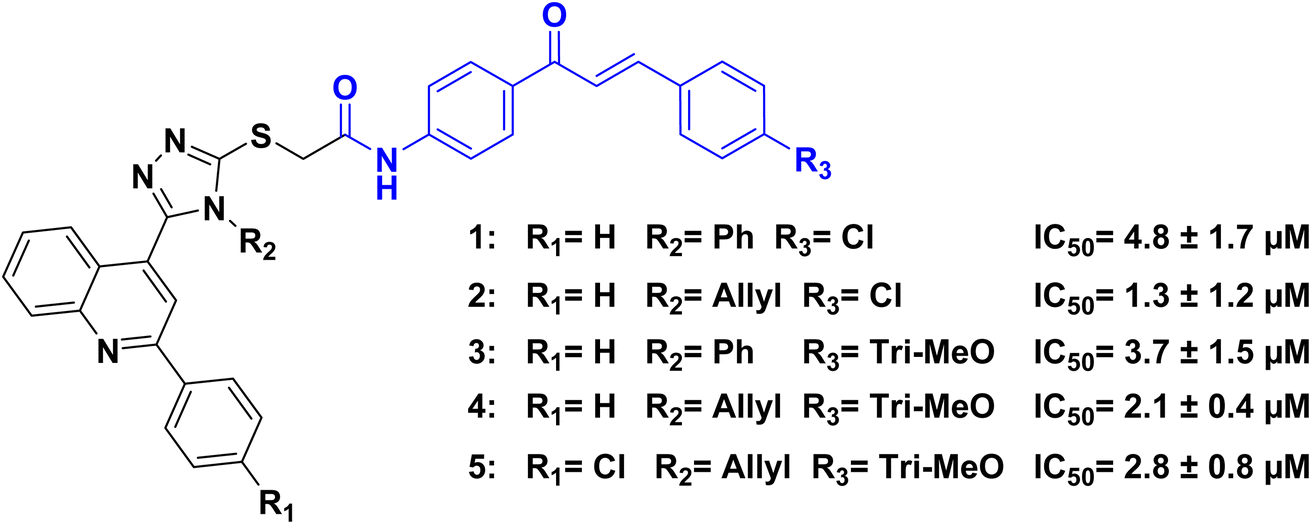 | ||
| Fig. 4 Quinoline/chalcone hybrids 1–5 as EGFR inhibitors. | ||
Fathi and coworkers53 synthesized series of chalcone/1,3,4-oxadiazole derivatives in order to study the effect of these derivatives against different cancer cell lines. The compounds exhibited promising anticancer activities particularly leukemia cell lines compounds, 6, 7, 8, and 9 (Fig. 5) were the most potent (mean growth inhibition% = 37.77, 27.29, 28.20, and 132.29; respectively). Compound 9 exhibited potent cytotoxic activity against most of the tested cell lines. Compounds 6, 7, 8, and 9 displayed the highest inhibitory activities against EGFR (IC50 = 0.24–2.35 μM) as well as Src kinase (IC50 = 0.96–6.24 μM). Out of all the derivatives, 6, 7, and 9 demonstrated the highest effectiveness in inhibiting the activity of signal transducer and activator of transcription 3 (STAT3), which indicate that the substitution of phenyl ring carrying the oxadiazole moiety showed the highest activity with the 3,4,5-trimethoxy < H < p-methoxy, and the most effective substitution of the chalcone phenyl ring was p-methoxy, as a result they found that to achieve the best possible activity, it is necessary for R1 to consist of 3,4,5-trimethoxy groups and for R2 to contain a methoxy group in the p-position. Therefore, in order to exhibit effective anti-cancer properties, it is ideal for either both phenyl rings to be unsubstituted or for one of the rings to contain a p-methoxy group, while the other ring should have 3,4,5-trimethoxy groups.
 | ||
| Fig. 5 Chalcone/1,3,4-oxadiazole hybrids 6–9 as EGFR inhibitors. | ||
Abou-Zied, et al.54 tested a series of xanthine/chalcone derivatives for possible antiproliferative activity. Compounds 10, 11, 12, 13, and 14 (Fig. 6), demonstrated strong inhibition of cancer cell growth, with IC50 values ranging from 1.0 ± 0.1 to 3.5 ± 0.4 μM against four cancer cell lines A-549 (epithelial cancer cell line), MCF-7 (breast cancer cell line), Panc-1 (pancreas cancer cell line), and HT-29 (colon cancer cell line),. In comparison, doxorubicin exhibited IC50 values ranging from 0.90 ± 0.62 to 1.41 ± 0.58 μM. Compounds 11 and 14 showed the highest effectiveness in the study. To understand their anticancer mechanism, compounds 10, 11, 12, 13, and 14 were tested for their inhibition of EGFR. Compound 11 demonstrated the strongest inhibition with an IC50 value of 0.3 μM, which is more potent than the reference drug staurosporine (IC50 = 0.4 μM). The activity of the compounds can be attributed to the substitutions (R) at position 4 of the chalcone phenyl ring. Notably, among the 1,3-dimethyl xanthine series, compounds with NO2, Cl, and OCH3 groups on the chalcone phenyl ring exhibited the highest inhibitory activity across various cell lines. Despite the differing chemical nature of the substituents, this enhanced activity may be attributed to different physicochemical nature of the chalcone/xanthine hybrids which could result in improved cell membrane permeability of these hybrids and lead to this enhanced antiproliferative activity.
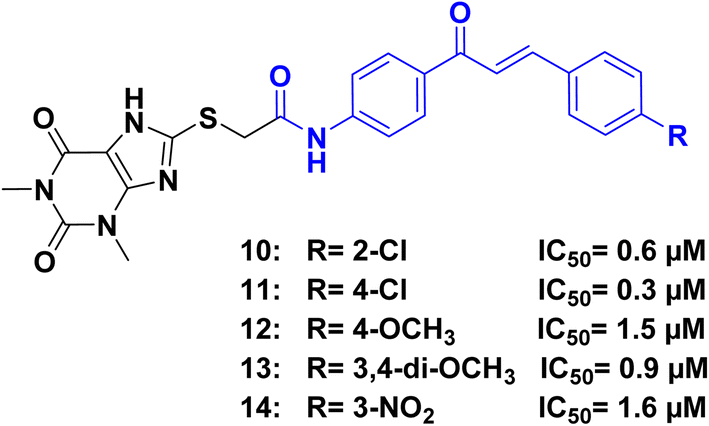 | ||
| Fig. 6 Xanthine/chalcone hybrids 10–14 as EGFR inhibitors. | ||
Hisham et al.,55 synthesized a series of hybrid molecules consisting of quinazoline-4-one and chalcone parts as EGFR enzyme inhibitors with antiproliferative activity against cancer cells. The target compounds were synthesized and tested in vitro against various cancer cell lines, the EGFR enzyme, and the BRAF enzyme which is a serine/threonine-protein kinase involved in the MAPK signaling pathway, regulating cell growth and differentiation. Out of the synthesized compounds, three showed the greatest antiproliferative activity (15, 16, and 17) and were the most potent inhibitors of the EGFR enzyme with IC50 = 0.11, 0.90, and 0.56 μM, respectively comparable to the positive control erlotinib (IC50 = 0.08 μM). The order of effectiveness for the (R2) substituent was 4-methoxy > 4-Br > 4-Cl > 3,4-dimethoxy > H > CH3 (Fig. 7).
 | ||
| Fig. 7 Structure–activity relationship of the target compounds (15–17). | ||
Hybrids of cyanopyridine/chalcone acetamide derivatives were synthesized by Abou-Zied et al.56 These derivatives were designed to inhibit EGFR enzyme. Compounds 18, and 19 (Fig. 8) exhibited strong inhibition of cancer cell growth in multiple cell lines, outperforming the reference compound doxorubicin. These compounds were found to have a dual inhibitory effect on both EGFR and BRAFV600E (which is a mutant form of the BRAF enzyme with a V600E substitution) based on in vitro studies. Compounds 8, 12, and 13 demonstrated significant inhibition of EGFR protein, with IC50 values of 110 and 94 nM, respectively, in the synthesized compounds. The study revealed that the specific atoms or arrangements in the aryl R distal chalcone' group affected potency, with 3,4-dimethoxy being more potent than hydrogen, 4-bromo, 4-chloro, and 3,4,5-trimethoxy substitutions for compounds with the 4,6-bis(3,4-dimethoxyphenyl)pyridine core structure.
 | ||
| Fig. 8 Cyanopyridine/chalcone hybrids 18, and 19 as EGFR inhibitors. | ||
Hagar et al.,57 designed and synthesized a series of 1,3,4-oxadiazole chalcone/benzimidazole hybrids. These hybrids showed notably strong abilities to inhibit the growth of different types of cancer cells in an initial screening test. The abilities of the synthesized compounds to inhibit the growth of cancer cells were evaluated against four human cancer cell lines: lung cancer cells (A-549), breast cancer cells (MCF-7), pancreatic cancer cells (Panc-1), and colon cancer cells (HT-29). Acetamido compounds and their acetate isostere compounds, showed promising abilities to inhibit cancer cell growth with IC50 values ranging from 0.80 to 2.27 μM compared to doxorubicin (IC50 values ranging from 0.90 to 1.41 μM). The most effective compound was compound 20, with an IC50 value of 1.8 ± 0.7 μM against EGFR. This compound had the following substituents: R1 = methoxy, R2 = chlorine, and X = nitrogen. The second most effective compound was 21, with an IC50 of 1.9 ± 0.8 μM against EGFR. This compound had the substituents: R1 = methoxy, R2 = chlorine, and X = oxygen. The study found that the acetamide molecular group connecting the benzimidazole, oxadiazole unit, and chalcone showed better activity than the acetate linker. Regarding the substitution pattern on the phenyl ring of the chalcone unit, in both the acetamide and ester derivatives, the order of preference was: 4-methoxy > 4-chlorine > 3,4-dimethoxy > 3,4,5-trimethoxy > unsubstituted hydrogen, as shown in (Fig. 9).
 | ||
| Fig. 9 Structure–activity relationship of the target compounds (20–21). | ||
Abdelbaset et al.,58 synthesized thienoquinoline carboxamide–chalcone derivatives by cyclizing acylated chalcones with 2-mercaptoquinoline-3-carbaldehyde. These thienoquinolines demonstrated promising antiproliferative effects across all tested cell lines and exhibited significant activity as inhibitors of EGFR. Among the series, compound 22 (Fig. 10) exhibited significant antiproliferative activities with IC50 values of 1.0, 0.9, 0.9 and 1.2 μM against the colon cancer cell line HT-29, pancreatic carcinoma cell line paca-2, lung cancer cell line H-460 and human pancreatic cancer cell line Panc-1, respectively. Compound 23 exhibited the highest antiproliferative activity among all the tested thienoquinolines, and its activities were comparable to those of erlotinib against all the tested cancer cell lines. Moreover, thienoquinoline 23 showed IC50 values of 0.5, 0.8, 0.2 and 1.0 μM against the colon cancer cell line HT-29, pancreatic carcinoma cell line paca-2, lung cancer cell line H-460 and human pancreas cancer cell line Panc-1, respectively. Furthermore, compound 23 was found to influence pre G1 apoptosis and induce cell cycle arrest at the G2/M phase.
 | ||
| Fig. 10 Thienoquinoline/chalcone hybrids 22, and 23 as EGFR inhibitors. | ||
Maghraby et al.,59 synthesized a series of 1,2,3-triazole/chalcone hybrids, and evaluated for the antiproliferative activity of these hybrids against four different cancer cell lines A-549 (epithelial cancer cell line), MCF-7 (breast cancer cell line), Panc-1 (pancreas cancer cell line), and HT-29 (colon cancer cell line), with doxorubicin used as a reference compound. Among the hybrids tested, multiple compounds demonstrated the remarkable antiproliferative activity, with IC50 values ranging from 0.95 to 1.80 μM, more potent than doxorubicin (IC50 1.14 μM). Specifically, compound 24 (Fig. 11) showed the most potent antiproliferative effect and was also a highly effective inhibitor of EGFR, with an IC50 of 0.09 ± 0.05 μM, comparable to the reference drug erlotinib (IC50 = 0.05 ± 0.03 μM). Furthermore, compound 24 exhibited modest inhibitory activity against BRAF, with an IC50 of 0.90 ± 0.10 μM. Molecular docking studies provided insights into the strong interactions of the inhibitors with the EGFR-TK domain. Additionally, cell cycle analysis revealed that compound 24 induced cell cycle arrest at the G1 transition phase.
 | ||
| Fig. 11 1,2,3-Triazole/chalcone acetamides 24 as EGFR inhibitor. | ||
The studies on amide linked chalcone based EGFR inhibitors reveal several common trends that enhance their potency. Substitutions on the chalcone phenyl ring, particularly with methoxy groups (especially in the 4-position), significantly improve EGFR inhibition, with 3,4,5-trimethoxy or p-methoxy substitutions proving most effective. Additionally, the inclusion of heterocyclic moieties, such as triazole, oxadiazole, quinazoline, xanthine, and thienoquinoline, boosts binding affinity and enhances potency, with quinoline/chalcone hybrids particularly showing strong activity. The allyl-triazole backbone also stands out as a key feature, offering higher efficacy compared to the phenyltriazole variant, emphasizing the importance of linker structure in overall compound effectiveness. These compounds demonstrated broad cell line selectivity, inhibiting cancer cell growth in pancreatic, lung, colon, and breast cancer cells, with IC50 values comparable to the standard EGFR inhibitor erlotinib (∼0.08 μM), suggesting their potential as viable candidates. Moreover, several compounds exhibited dual inhibition of EGFR alongside other targets, such as BRAF and STAT3, offering a promising strategy to overcome resistance mechanisms and enhance therapeutic outcomes.
3.2 Topoisomerase I and II inhibitors
Certain molecules can interfere with the function of topoisomerase enzymes, which are proteins that modify DNA strands during cell activities like copying and expressing genes. There are two main topoisomerase types: topoisomerase I and II. Inhibiting topoisomerase activity can stop cell growth by disrupting DNA handling, making topoisomerase inhibitors a group of substances that prevent proliferation. Some structures exhibit antiproliferative properties through their ability to block one or both topoisomerase types, thereby hindering cell division. Researchers examine the capacity of novel molecules to inhibit topoisomerase I and II as a means of evaluating their potential antiproliferative and anticancer effects.60,61A series of urea-linked ciprofloxacin–chalcone hybrid compounds were synthesized by ref. 62 and compounds 25 and 26 (Fig. 12), demonstrated remarkable abilities to inhibit cell growth in both colon HCT-116 and leukemia SR cancer cell lines, outperforming the reference compounds camptothecin, topotecan, and staurosporine. Specifically, compounds 25 and 26 exhibited IC50 values of 2.53 and 2.01 μM, respectively, against the HCT-116 cell line, and 0.73 and 0.64 μM, respectively, against the leukemia SR cell line. These values were significantly lower than those of the reference compounds. In addition to their potent antiproliferative effects, compounds 25 and 26 also showed inhibitory activity against topoisomerase I and II β. Compound 26 exhibited 56.72% inhibition of topoisomerase I and 60.06% inhibition of topoisomerase II β, compared to 60.05% and 71.09% inhibition by the reference compounds camptothecin and topotecan, respectively. Regarding the structure–activity relationship, the study found that replacing three hydrogen atoms on the phenyl ring of the chalcone moiety with methoxy groups resulted in the most active compound. For compounds with a single substituent, a chlorine atom in the para position produced the highest activity compared to other halogen or electron-donating groups.
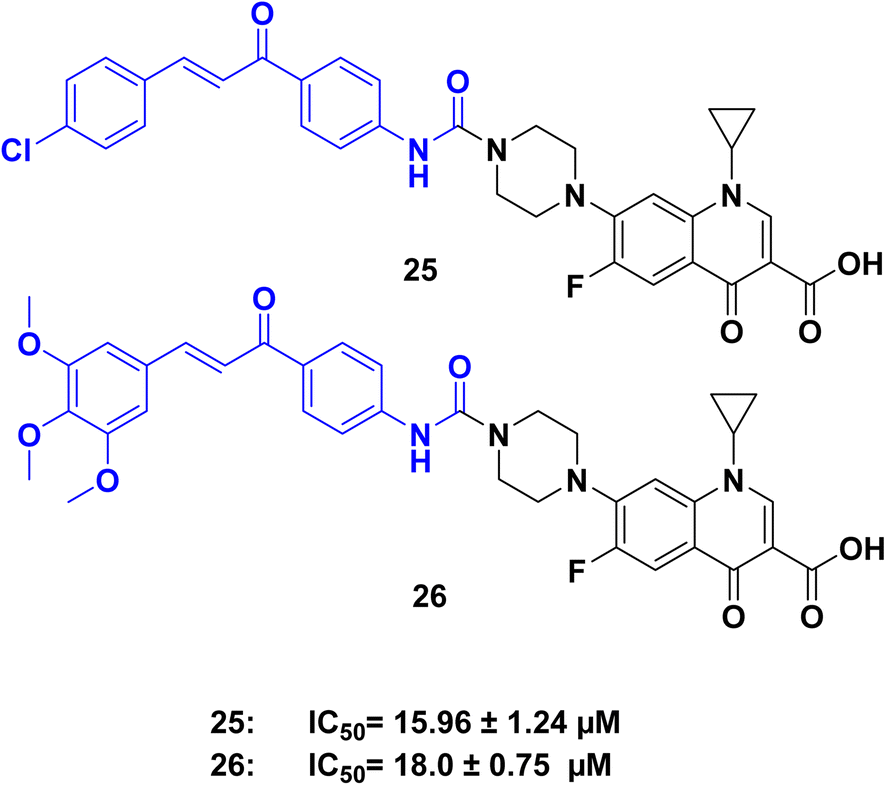 | ||
| Fig. 12 Ciprofloxacin/chalcone hybrids 25, and 26 as topoisomerase inhibitors. | ||
Kim et al.,63 conducted a study in which they synthesized a series of chalcone derivatives. The aim of the study was to identify compounds that could inhibit the topoisomerase enzyme and exhibit antiproliferative activity against cancer cells. The synthesized compounds were tested in vitro against various cancer cell lines and the topoisomerase enzyme. Compound 27 (Fig. 13), exhibited 100% inhibition of the topoisomerase I enzyme at a concentration of 100 μM, comparable to the positive control camptothecin, which showed 70.5% inhibition at the same concentration. Compound 27 also showed moderate cytotoxic activity against human breast ductal carcinoma cell line (T47D) with an IC50 of 12.33 μM and human gastric cancer cell line (SNU638) with an IC50 of 17.41 μM.
 | ||
| Fig. 13 Chalcone hybrid 27 as topoisomerase inhibitor. | ||
Abdel-Aziz et al.,64 prepared N-4-piperazinyl-ciprofloxacin–chalcone derivatives. The results of a test on a single dose of anticancer compounds showed that compounds 28 and 29 (Fig. 14) were the most effective at inhibiting the growth of various cancer cell lines. In an in vitro five-dose test on the full NCI 60 cell panel, compound 28 demonstrated a broad-spectrum antitumor activity against all nine tumor subpanels tested, without significant selectivity. Compound 29, on the other hand, exhibited high selectivity towards the leukemia subpanel. Most of the tested compounds showed good inhibitory activity against both topoisomerase I and topoisomerase II at 100 μM. The study found that the presence of three OCH3 groups is more effective than the presence of one OCH3 group. Compound 29, with three OCH3 groups, showed superior anticancer activity against various cancer cell lines compared to compound had one OCH3 group. Additionally, the presence of an electron-donating group (OCH3) in compound 29 was more effective against cancer cells than an electron-withdrawing group.
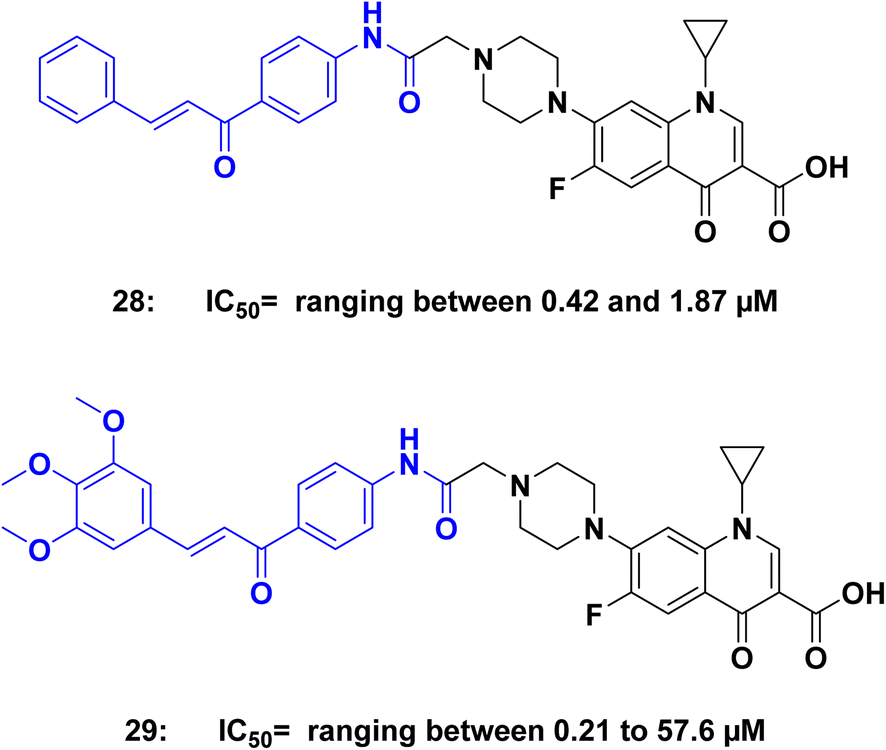 | ||
| Fig. 14 Ciprofloxacin/chalcone hybrids 28, and 29 as Topoisomerase inhibitors. | ||
The studies on chalcone derivatives as topoisomerase I and II inhibitors highlight key structural trends that enhance potency. Methoxy groups, especially in the para position, and halogen substitutions, such as chlorine, on the phenyl ring, significantly boost enzyme inhibition. Compounds with three methoxy groups showed stronger antiproliferative effects, suggesting that electron-donating groups improve enzyme inhibition.
3.3 ABCG2 modulators
ABCG2 modulators represent a promising pathway in the development of anticancer drugs. ABCG2, also known as breast cancer resistance protein (BCRP), is a member of the ATP-binding cassette (ABC) transporter family.65 It is highly expressed in various tissues, including the intestines, liver, kidney, and blood–brain barrier. The role of ABCG2 in cancer is significant, as it acts as a multidrug efflux pump, pumping out a wide range of chemotherapeutic agents from cancer cells. This efflux activity contributes to drug resistance, limiting the effectiveness of chemotherapy and leading to treatment failure.66 In recent years, many researches have focused on developing ABCG2 modulators to overcome drug resistance and enhance the efficacy of anticancer drugs.67 These modulators are compounds that can either inhibit or enhance the activity of ABCG2, thereby influencing its efflux function. Inhibitors of ABCG2 can block the pump's activity, preventing the efflux of anticancer drugs and increasing their intracellular concentration within cancer cells.68 This approach can sensitize cancer cells to chemotherapy and improve treatment outcomes. On the other hand, enhancers of ABCG2 can stimulate the pump's activity, promoting the efflux of toxic metabolites and protecting normal tissues from drug-induced toxicity.69Kraege et al.,70 synthesized a series of 35 compounds by combining a typical chalcone structure with different acid chlorides. An amide linker was introduced at positions 2′, 3′, or 4′ on ring A of the chalcone. These compounds exhibited a diverse range of substitution patterns, which facilitated the establishment of structure–activity relationships and identification of optimal structural characteristics for further investigation. The inhibitory activity against ABCG2 and intrinsic cytotoxicity of the synthesized acryloylphenylcarboxamides were examined. The study revealed that the ortho position of the amide linker and the presence of 3,4-dimethoxy groups on the distal phenyl ring of the chalcone were crucial for inhibitory activity. Additionally, the highest potency was observed with unsubstituted phenyl, thiophene, and quinoline rings linked to the amide group. Compound 30 (Fig. 15), exhibited the most significant activity, with an IC50 of 0.600 μM against MDCK II BCRP cells by using pheophorbide A assay.
 | ||
| Fig. 15 Chalcone hybrids 30, and 31 as ABCG2 inhibitors. | ||
Again Kraege et al.,71 synthesized and examined a series of chalcones combined with an additional aromatic residue to evaluate their inhibitory effects on ABC transporters. In their previous mentioned article, they determined that the ortho position on the chalcone A-ring was the preferred location for the amide linker, and they discovered various substitution patterns on the additional ring that enhanced potency. In this study, they investigated whether introducing a methoxy group, known to improve the inhibitory activity of chalcones, would also be advantageous for the acryloylphenylcarboxamide scaffold. Remarkably, this modification resulted in highly potent ABCG2 inhibitors. To further support the hypothesis regarding the beneficial impact of the amide linker, six acryloylphenylcarboxylates were synthesized and examined for their inhibitory activity. Substituting the amide linker with an ester group led to decreased inhibition. Among the derivatives tested, compound 31 (Fig. 15), exhibited the highest level of activity with IC50 = 0.211 μM against ABCG2. These include the amide linker positioned ortho, a 4′-methoxy substitution on ring A, 3,4-dimethoxy groups on ring B, and an unsubstituted phenyl ring connected to the amide linker.
In conclusion, the findings of this study can be summarized as follows: the 3,4-dimethoxy substitution on ring B was found to be beneficial for inhibiting ABCG2, based on previous studies.72 A 4′-methoxy substitution on ring A of the chalcone moiety was identified as a beneficial structural feature, leading to increased inhibitory activity against ABCG2. However, 4′,5′-dimethoxy substitution resulted in slightly decreased potency. There was no significant difference between chalcones substituted with 3,4- or 3,5-dimethoxy groups on ring B, as well as comparable 4′-methoxy-substituted acryloylphenylcarboxamides and benzoates. The replacement of the amide linker with an ester function in the acryloylphenylcarboxylates resulted in decreased inhibitory effects towards ABCG2. This could be attributed to the loss of donor–acceptor behavior or differences in preferred conformation. These findings provide valuable insights into the structure–activity relationships and the potential for developing selective ABCG2 inhibitors. Further studies and optimization of compound design could lead to the development of more potent and selective inhibitors for therapeutic applications.
3.4 Caspase protein inhibitors
Caspases are a family of proteins that play a crucial role in the regulation of programmed cell death, known as apoptosis. These proteins act as key players in the intricate network of signaling pathways responsible for maintaining cellular homeostasis.73 Dysregulation of apoptosis has been implicated in various diseases, including cancer, making caspases attractive targets for the development of anticancer agents.74 In cancer cells, the dysregulation of apoptotic pathways often leads to uncontrolled cell proliferation and resistance to cell death, contributing to tumor development and progression.75 Targeting caspases and modulating their activity has emerged as a promising strategy for developing novel anticancer agents.76 By promoting apoptosis in cancer cells, caspase-targeted therapies aim to induce selective cell death, inhibit tumor growth, and overcome drug resistance.77 Targeting caspase proteins and their associated apoptotic pathways holds great potential for the development of more effective and selective cancer therapies.75,78Tok et al.,79 synthesized a series of chalcone derivatives and assessed it's in vitro antiproliferative activities against HeLa, MCF-7, MKN-45 cancer cell lines, as well as the NIH-3T3 cell line using the MTT assay. Notably, compound 32 (Fig. 16), exhibited significant cytotoxic effects on all three cancer cells while demonstrating no cytotoxicity towards NIH-3T3 normal cells. The IC50 values for compound 32 were 26.66 μM on HeLa, 9.41 μM on MCF-7, and 5.17 μM on MKN-45. Furthermore, compound 32 exhibited the ability to upregulate the protein expression of Bax while downregulating the protein expression of Bcl-2 in cells. Additionally, these compounds increased caspase-3 activity in the cells. Based on these findings, it can be concluded that 32 activated apoptosis by inducing mitochondrial apoptotic proteins in HeLa, MCF-7, and MKN-45.
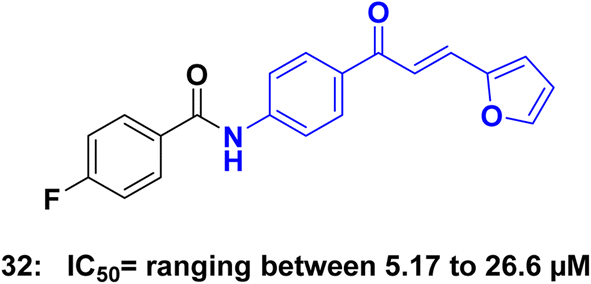 | ||
| Fig. 16 Chalcone hybrid 32 as caspase inhibitor. | ||
Ahmed et al.,80 synthesized a series of hybrids combining 1,2,4-triazole and chalcone. The synthesized compounds demonstrated remarkable cytotoxic activity against various cancer cell lines. Among the tested compounds, compounds 33, 34, 35, 36, 37, and 38 (Fig. 17), exhibited the highest cytotoxicity against human lung adenocarcinoma A549 cells, with IC50 values 19.43, 6.06, 4.4, 7.55, 16.04 and 8.04 μM, respectively. In comparison, cisplatin had an IC50 of 15.3 μM. Further investigation into the mechanism of action revealed that the 1,2,4-triazole-chalcone hybrids induced apoptosis by increasing the level of proapoptotic protein Bax, releasing cytochrome c from mitochondria, and activating caspase-3/8/9 proteins.
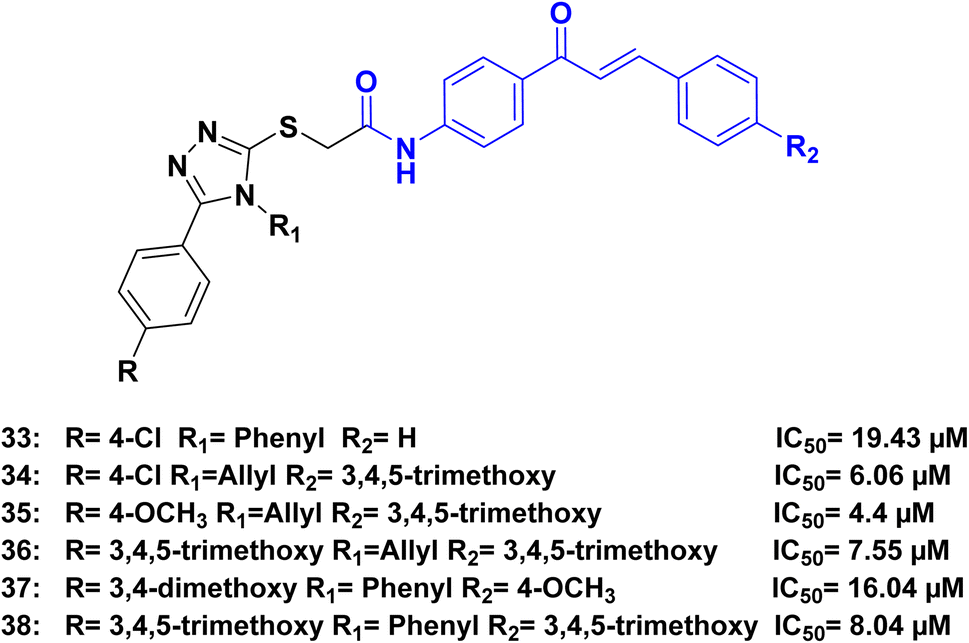 | ||
| Fig. 17 1,2,4-Triazole/chalcone hybrids 33–38 as caspase inhibitors. | ||
In the analysis of the structure–activity relationship (SAR), we found that the presence of N-4-allyl triazole in the hybrids resulted in higher activity compared to N-4-phenyl triazole. It was observed that for optimal activity in the allyl triazole hybrids, the presence of 3,4,5-trimethoxy groups in R2 was crucial. Additionally, the chalcone phenyl ring B in these hybrids needed to be substituted with either electron-donating or electron-withdrawing groups (such as OCH3 or Cl) preferably in the p-position. However, full substitution of the phenyl ring slightly decreased the activity, while 3,4-dimethoxy substitution greatly reduced it. Nonetheless, the activity was maintained when trimethoxy substitution was present. On the other hand, in the phenyl triazole hybrids, optimal activity was achieved when both R and R2 were 3,4,5-trimethoxy groups. The activity slightly decreased when the phenyl ring was substituted with electron-donating or electron-withdrawing groups in the p-position (Fig. 18).
 | ||
| Fig. 18 Structure–activity relationship of the compounds (34–38). | ||
Romagnoli et al.,81 synthesized a series of bromoacryloylamido/chalcones hybrids and evaluated for their ability to inhibit the growth of cancer cells in five different cell lines. These hybrid derivatives displayed significantly enhanced anti-tumor activity when compared to the corresponding amino chalcones. The most promising molecules identified as potential leads were compounds 39, 40, and 41 (Fig. 19), the antiproliferative activity of these three compounds was evaluated against five different cancer cell lines: L1210, FM3A, Molt4, CEM, and HeLa. Compound 39 exhibited remarkable activity, with IC50 values of 0.24 μM, 0.68 μM, 0.61 μM, 0.75 μM, and 0.75 μM for the respective cell lines. Similarly, compound 40 displayed notable activity, with IC50 values of 0.25 μM, 0.52 μM, 0.55 μM, 0.73 μM, and 0.34 μM. Compound 41 also demonstrated significant potency, with IC50 values of 0.63 μM, 0.53 μM, 0.68 μM, 0.84 μM, and 0.51 μM. Flow cytometry analysis conducted on K562 cells revealed that the most active compounds caused a substantial proportion of cells to enter the apoptotic sub-G0–G1 peak, indicating cell death. Compound 39 increased activated caspase-3 after 24 h of treatment, and a further increase after 48 h.
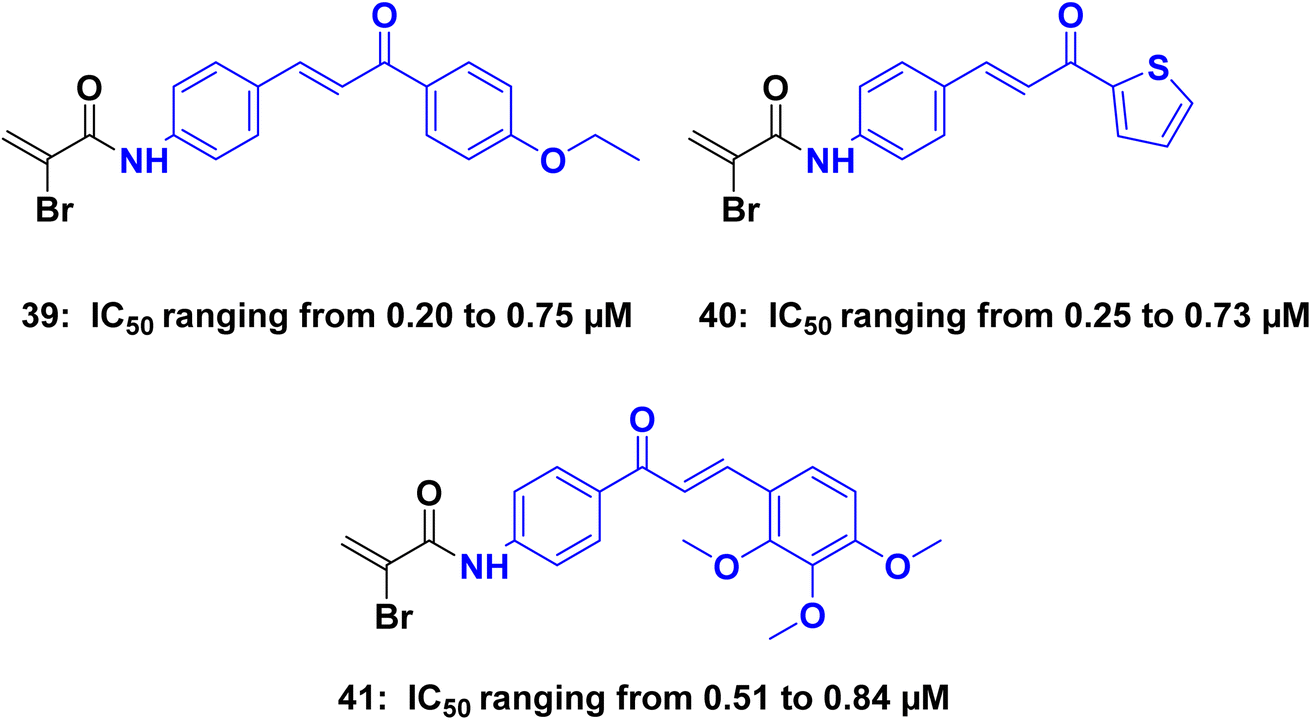 | ||
| Fig. 19 Chalcone hybrids 39–41 as caspase inhibitors. | ||
3.5 Histone deacetylase (HDAC) inhibitors
Histone deacetylase (HDAC) inhibitors have emerged as promising agents in the development of anticancer therapies. HDACs are enzymes that regulate gene expression by modifying histone proteins, which play a critical role in chromatin remodeling and gene transcription.82 Dysregulation of HDAC activity is observed in various cancers, promoting tumor growth and progression.83 HDAC inhibitors target and inhibit these enzymes, leading to the accumulation of acetyl groups on histone proteins. This alters chromatin structure and gene expression, activating tumor suppressor genes and repressing oncogenes.84 HDAC inhibitors have shown antitumor effects by inducing cell cycle arrest, differentiation, and apoptosis in cancer cells.85 They have been approved for hematological malignancies and are being investigated for solid tumors.86Valente et al.,87 synthesized compounds combining pyrrole and benzene with hydroxamate and chalcone groups. Among the synthesized compounds, 42 and 43 (Fig. 20) exhibited selective inhibition of HDAC6 at the nanomolar level 30 and 10 nM respectively. In human acute myeloid leukemia U937 cells, the other hydroxamates led to increased levels of acetyl-α-tubulin. Furthermore, in the same U937 cell line, compounds 42 and 43 demonstrated apoptotic effects of 18.4% and 21.4%, respectively, compared to 16.9% for SAHA (a reference HDAC inhibitor). The antiproliferative effects of compound 43 were also investigated across various cancer cell lines. It showed growth inhibition at sub-micromolar concentrations in neuroblastoma LAN-5 and SH-SY5Y cells, as well as chronic myeloid leukemia K562 cells. Moreover, in lung H1299 and A549, colon HCT116 and HT29 cancer cells, compound 43 exhibited growth inhibition at low-micromolar concentrations. In HT29 cells, compound 43 increased histone H3 acetylation and significantly reduced the colony forming potential of the cancer cells by up to 60%.
 | ||
| Fig. 20 Chalcone/hydroxymate hybrids 42, and 43 as HDAC inhibitors. | ||
A series of chalcone-based hybrids with α-phthalimido substituents as a dual inhibitors targeting histone deacetylases (HDACs) and tubulin for potential anticancer applications.88 The synthesized compounds were evaluated for their anticancer activity against MCF-7 and HepG2 human cancer cell lines using the MTT assay. They measured the in vitro β-tubulin polymerization and inhibitory activity against HDAC 1 and 2 for the most potent hybrids. Among the compounds tested, the trimethoxy derivative 44 (Fig. 21) exhibited the highest anticancer activity. It demonstrated potent β-tubulin polymerase and HDAC 1 and 2 inhibitory activity with IC50 of 0.22 ± 0.01 μM against HDAC 1. Additionally, compound 44 effectively induced cell cycle arrest at both the G2/M and preG1 phases in the MCF-7 cell line.
 | ||
| Fig. 21 Chalcone hybrid 44 as HDAC inhibitor. | ||
3.6 Tubulin polymerization inhibitors
Tubulin polymerization inhibitors are a class of compounds that play a crucial role in the field of medicinal chemistry. These inhibitors target the process of microtubule assembly and disassembly,89 which is essential for cellular functions such as cell division, intracellular transport, and maintenance of cell shape. By interfering with tubulin polymerization, these inhibitors disrupt the formation of microtubules, leading to cell cycle arrest and inhibition of cell proliferation.90 They have shown significant promise as anticancer agents, as they selectively target rapidly dividing cancer cells. Tubulin polymerization inhibitors exhibit various mechanisms of action, including binding to tubulin subunits, preventing their assembly into microtubules, or destabilizing existing microtubule structures. This disruption of microtubule dynamics ultimately leads to cell death through apoptosis or mitotic catastrophe.91 Ongoing research aims to discover and optimize novel tubulin polymerization inhibitors with improved efficacy, selectivity, and reduced toxicity, paving the way for the development of more effective treatments against cancer and other diseases characterized by uncontrolled cell growth.92Amin et al.,93 Synthesized a series of 8-hydroxyquinoline/chalcone hybrids, and investigated for their potential anticancer activity. Notably, compounds 45 and 46 (Fig. 22) demonstrated exceptional activity against both HCT116 and MCF7 cells, while also showing improved safety towards normal WI-38 cells when compared to staurosporine. Further enzymatic assays indicated that compounds 45 and 46, displayed effective inhibition of tubulin polymerization, with IC50 values of 5.3 and 8.5 μM respectively, in comparison to the reference compound combretastatin A4 (IC50 = 2.15 μM). Additionally, compounds 45, and 46 demonstrated inhibition of EGFR (epidermal growth factor receptor), with IC50 values of 0.097 μM, and 0.334 μM, respectively, in contrast to erlotinib (IC50 = 0.056 μM). Compounds 45 and 46 were further investigated to assess their effects on the cell cycle, induction of apoptosis, and suppression of the wnt1/β-catenin gene. Western blot analysis was conducted to detect the apoptosis markers Bax, Bcl2, Casp3, Casp9, PARP1, and β-actin.
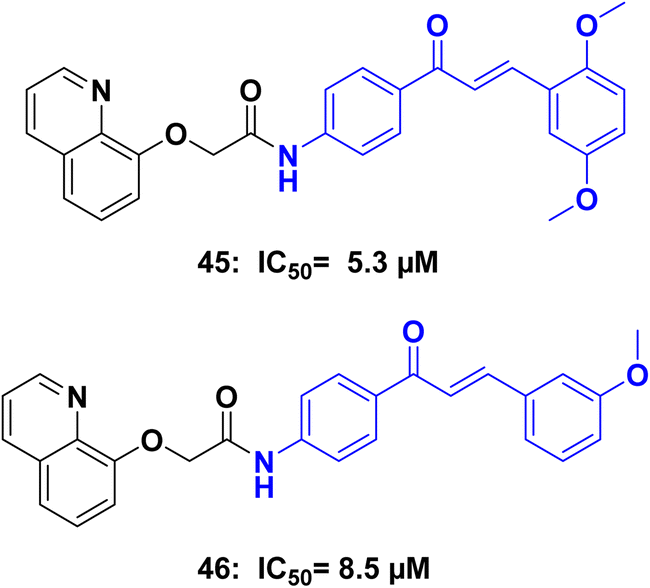 | ||
| Fig. 22 Chalcone/quinoline hybrids 45, and 46 as tubulin inhibitors. | ||
A series of chalcone derivatives synthesized and their antiproliferative activities were assessed against five human tumor cells.94 These chalcone derivatives contain an additional aromatic or heterocyclic ring connected via an ether or an ester or an amide functional group. Furthermore, the impact of the presence of one or three methoxy groups or a 2,4-dimethoxy-3-methyl system on the B ring of the chalcone structure on cytotoxicity was investigated. The findings demonstrated that the most cytotoxic chalcones contained a furoyl substituent connected through an ester or an amide to the 2′-hydroxy or 2′-amino group of the A ring in the chalcone skeleton. These compounds exhibited IC50 values ranging from 0.2 μM to 1.3 μM against human leukemia cells. Specifically, the synthetic compound 47 (Fig. 23) exhibited at least ten-fold greater potency than the antineoplastic agent etoposide against U-937 cells, while showing lower cytotoxicity against human peripheral blood mononuclear cells. Treatment of U-937 and HL-60 cells with FMC resulted in cell cycle arrest at the G2–M phase, an increase in the percentage of sub-G1 and annexin-V positive cells, release of mitochondrial cytochrome c, activation of caspase, and cleavage of poly(ADP-ribose) polymerase.
 | ||
| Fig. 23 Chalcone hybrid 47 as tubulin inhibitor. | ||
3.7 Miscellaneous anticancer chalcone acetamides
Histone lysine specific demethylase 1 (LSD1) has gained significant attention as a promising molecular target for the development of potent anticancer drugs to combat leukemia. Li, Y., et al.,95 synthesized a series of chalcone derivatives and assessed their inhibitory activity against LSD1. All chalcone–dithiocarbamate hybrids demonstrated considerable inhibitory activity against LSD1. Notably, chalcone 48 (Fig. 24), exhibited the most potent LSD1 inhibitory activity, with an IC50 value of 0.14 μM. Furthermore, compound 48 displayed inhibitory effects on cell proliferation, with IC50 values of 1.10 μM, 3.64 μM, 3.85 μM, 1.87 μM, 0.87 μM, and 2.73 μM against HAL-01, KE-37, P30-OHK, SUP-B15, MOLT-4, and LC4-1 leukemia cells, respectively. Additional investigations revealed that compound 48 selectively and reversibly inhibited LSD1 in a time-dependent manner. It also up-regulated the expression levels of H3K9me1 and H3K9me2 in MOLT-4 cells. Notably, in an in vivo xenograft model, chalcone 48 inhibited tumor growth without causing apparent toxicity. | ||
| Fig. 24 Miscellaneous anticancer chalcone hybrids 48–59. | ||
A series of chalcone–dithiocarbamate hybrids and assessed their ability to inhibit cell proliferation in various cancer cell lines, including MGC803, MCF7, and PC3.96 Among these hybrids, compound 49 (Fig. 24), exhibited the most potent inhibitory activity against PC3 cells, with an IC50 value of 1.05 μM. Preliminary investigations into the biological mechanisms of compound 49 revealed several effects. It was found that 49 could reduce colony formation, induce DNA damage, and arrest the cell cycle at the G2/M phase. This effect was associated with the upregulation of P21 and the downregulation of CDK1 and cyclinB1, and it occurred in a concentration-dependent manner. Notably, 49 also induced apoptosis in PC3 cells by reducing the mitochondrial membrane potential and altering the expression levels of BCl-2 family proteins. Additionally, as an apoptosis inducer, 49 triggered the accumulation of intracellular reactive oxygen species (ROS) in PC3 cells by inhibiting the activity of the catalase enzyme.
Lamie & Philoppes,97 synthesized a group of hybrid compounds combining 2-thiopyrimidine and chalcone, and its cytotoxic activities were assessed against three different cell lines: K-562, MCF-7, and HT-29. Most derivatives exhibited significant activity against the K-562 cell line, with IC50 values ranging from 0.77 to 1.74 m μM. Against the MCF-7 cell line, the IC50 values ranged from 1.37 to 3.56 μM, while against the HT-29 cell line, the IC50 values ranged from 2.10 to 2.37 μM. Furthermore, five derivatives were selected for further evaluation of their cytotoxicity against the normal fibroblast cell line WI38. Additionally, the inhibitory activities against STAT3 and STAT5a were determined for the most active derivatives. Notably, compound 50 (Fig. 24), exhibited dual inhibitory activity, with IC50 values of 113.31 and 50.75 μM against STAT3 and STAT5a, respectively.
Ren et al.,98 synthesized homoserine lactones/chalcone hybrids. These compounds were then tested in vitro to evaluate their cytotoxic activity against four different human cancer cell lines. Compounds with the chalcone scaffold exhibited significantly increased cytotoxicity compared to compounds with hydrophobic side chains. Additionally, certain compounds showed higher potency than the commonly used drug 5-Fu and a natural compound called OdDHL. Among the compounds tested, those with the 4-amino chalcone scaffold demonstrated excellent inhibition against all cancer cell lines, surpassing the effectiveness of 5-Fu. Compound 51(Fig. 24), which contained a 3,4,5-trimethoxy group with IC50 1.32 μM, proved to be the most potent against all cancer cell lines. Further analysis using flow cytometry revealed that analog 52 with IC50 of 5.57 μM induced cellular apoptosis and cell cycle arrest at the G2/M phase of MCF-7 cells in a concentration- and time-dependent manner.
M. A. Mourad,99 synthesized a group of nitric oxide (NO) releasing chalcones by combining amino chalcones with different NO-releasing moieties such as nitrate esters, oximes, and furoxans. The compounds were then screened to evaluate their potential as anticancer agents, and the results demonstrated a range of cytotoxic activity, varying from mild to strong. Among the NO-releasing compounds tested, compounds 53 and 54 (Fig. 24), displayed notable cytotoxic activity against a diverse set of cancer cell lines, including leukemia, lung, renal, breast, colon, melanoma, ovarian, and skin cancer cells. The nitrate ester compound 53 exhibited the highest level of inhibitory activity. Additionally, compound 53 exhibited moderate selectivity specifically towards colon cancer, with a selectivity ratio of 5.87 at the TGI (total growth inhibition) level.
Cao et al.,100 designed and synthesized Pt(IV) complexes that were combined with P-glycoprotein (P-gp) inhibitors to overcome resistance (MDR) and improve the effectiveness of anticancer treatments. Among these complexes, complex 55 (Fig. 24), demonstrated notable abilities: it efficiently reversed cisplatin resistance in the SGC-7901/CDDP cell line, with an IC50 value of 3.37 μM, and exhibited an increased selectivity index of 6.9 against the normal HL-7702 cell line. Detailed investigations conducted on SGC-7901/CDDP cells provided insights into the mechanisms behind the efficacy of complex 55. It was found that the complex induced apoptosis effectively by suppressing the expression of P-gp, leading to improved cellular uptake of platinum. Moreover, it arrested cells at the G2/M phase, induced DNA damage, and initiated the mitochondrial apoptosis pathway. Furthermore, in vivo studies demonstrated that the enhanced accumulation of complex 55 resulted in a tumor inhibition rate of 75.6% in SGC-7901/CDDP xenografts, surpassing the rates achieved by cisplatin (25.9%) and oxaliplatin (43%). Additionally, the low systemic toxicity of complex 55 indicated its potential as a P-gp-mediated MDR modulator.
Patel et al.,101 synthesized a series of amides combining deoxycholic acid and chalcone synthesized and evaluated against two cancer cell lines: A549 (human lung cancer) and SiHa (cervical cancer). Among the synthesized deoxycholic acid–chalcone conjugates, several compounds exhibited promising anticancer activity in vitro. Specifically, conjugates 56 (IC50: 0.51 μM) and 58 (IC50: 0.84 μM) (Fig. 24), containing 2-nitrophenyl and 3,4,5-trimethoxyphenyl groups respectively, demonstrated good activity against the SiHa cell line. Similarly, compounds 57 (IC50: 0.25 μM) and 56 (IC50: 1.71 μM) showed improved activity against the A549 lung cancer cell line compared to deoxycholic acid and chalcones alone. The conjugation of chalcones with deoxycholic acid resulted in enhanced anticancer activity compared to the individual components. This suggests that employing a bile acid conjugate strategy could be advantageous in enhancing the biological effectiveness of chalcone derivatives. The increased activity observed in certain compounds might be attributed to their improved bioavailability.
Lu et al.,102 synthesized Amino chalcone derivatives and investigated for their antiproliferative activity in vitro. Several compounds exhibited moderate to good activity against three human cancer cell lines: MGC-803, HCT-116, and MCF-7 cells. Notably, compound 59 (Fig. 24), displayed the most potent antiproliferative activity, with IC50 values of 1.52 μM (MGC-803), 1.83 μM (HCT-116), and 2.54 μM (MCF-7), surpassing the efficacy of the positive control (5-Fu). Further investigations were conducted to understand the mechanisms involved. The colony formation assay indicated that compound 59 inhibited the formation of cell colonies in MGC-803 cells. DAPI fluorescent staining and flow cytometry analysis revealed that compound 59 induced apoptosis in MGC-803 cells. Additionally, western blotting experiments demonstrated that compound 13e triggered cell apoptosis through the extrinsic/intrinsic apoptosis pathway in MGC-803 cells. These findings suggest that compound 59 holds promise as a valuable lead compound for antiproliferative agents, and further efforts should be made to enhance the potency of amino chalcone derivatives.
P. N. Bandeira et al.,103 synthesized a series of acetamido-chalcone derivatives were and subjected to initial biological screening using HCT-116 cells. Among the compounds tested, chalcone 60 (Fig. 25), exhibited potent and specific activity against HCT-116 cells, with an IC50 value of 2.37 ± 0.73 μM. Preliminary analysis of the structure–activity relationship suggested that the cytotoxic effects of these compounds might be attributed to the combined action of two electron-withdrawing groups: the nitro group (NO2) located at the meta-position of ring B and the acetyl group positioned at the para-position of ring A. Furthermore, chalcone 60 demonstrated the ability to induce G2/M cell cycle arrest and apoptosis when incubated at a concentration of 10 μM for 24 hours.
 | ||
| Fig. 25 Chalcone hybrids 60–71 with different targets. | ||
Durgapal et al.,104 synthesized a series of chalcone derivatives containing 3-aminomethyl pyridine synthesized and evaluated for their anticancer activity and DNA binding affinity in vitro. Most of the compounds exhibited significant antimitotic activity against the A549 cell line, surpassing the effectiveness of fluorouracil. Compounds 61 and 62 (Fig. 25), were specifically chosen for DNA-binding studies due to their excellent activity against cancer cell lines in the MTT assay. The binding affinity of compounds 61 and 62 to CT-DNA was investigated using UV-based DNA titration and fluorescence emission studies in the presence of DNA–EtBr complex. Notably, compound 62 demonstrated remarkable antiproliferative activity, with an IC50 value of 0.0067 ± 0.0002 μM, against the MCF-7 cell line. Cytotoxicity studies using MTT, LDH, as well as EtBr/AO assay revealed that compound 62 induced apoptosis in the cancerous cell line.
El-Sherief et al.,105 constructed a study with an objective to explore a synthesis approach utilizing a molecular hybridization strategy by incorporating a nitric oxide-releasing moiety, oxime, into coumarin–chalcone hybrids. Multiple derivatives were synthesized, and their in vitro anti-proliferative activity was evaluated. Moderate activity was observed for some of the prepared compounds, with growth inhibition values of 40.86, and 39.25 for compound against leukemia, central nervous system, and breast cancer cells, respectively. Additionally, compounds 63 and 64 (Fig. 25), exhibited IC50 values of 9.62 and 14.40, respectively, against breast Michigan Cancer Foundation-7 cell lines.
Farrag et al.,106 synthesized a series of indomethacin analogs with the aim of investigating their effects on three human colon cancer cell lines: HCT-116, HT-29, and Caco-2. The MTT assay was employed to assess the cytotoxicity of these derivatives. The results from the cytotoxicity assay revealed that the indomethacin amide analog 65 (Fig. 25), displayed remarkable antitumor activity specifically against Caco-2 cells, with an IC50 value of 1.5 μM.
Rodrigues et al.,107 synthesized a series of chalcone derivatives hybridized with 4-maleamic acid and 4-maleamide peptidyl groups and evaluated in vivo and in vitro for their antitumor activity against human prostate cancer. A total of twenty-one compounds were tested, and three of them 66, 67, and 68 (Fig. 25), exhibited activities against both androgen-independent (PC-3) and androgen-sensitive (LNCaP) human prostate cancer cells. Of particular interest was compound 67, an analog of 4-maleamide peptidyl chalcone, which incorporated a leucine amino acid as a substituent group. This compound demonstrated the highest cytotoxicity among the tested compounds in both tumor cell lines, with an IC50 value of 29.5 μg mL−1 for PC-3 cells and 21.4 μg mL−1 for LNCaP cells.
Bhojwani et al.,108 synthesized chalcone/benzamide hybrids, aiming to investigate their effectiveness as inhibitors of c-Met kinase. Chalcone analogs containing 4-methylbenzamide and 4-chlorobenzamide were synthesized, characterized, and subsequently assessed for their antiproliferative activity using the sulforhodamine-B stain (SRB) assay on various cell lines, including Michigan Cancer Foundation-7 (MCF-7), HT-29, MDA-MB-231, COLO-205, and A549. Compound 70 and compound 71 (Fig. 25), showed GI50 15.6, 6.05 μM respectively which demonstrated better activity than sorafenib (GI50 = 10.79 μM) in the breast cancer cell line MCF-7, indicating their potential as anticancer agents for breast cancer treatment. Furthermore, all tested compounds, including 69, 70, and 71 exhibited excellent activity in the colon cancer cell line, suggesting their effectiveness in colon cancer treatment. In the lung cancer cell line, compound 69 showed better activity than sorafenib, making it a potential candidate for lung cancer treatment. These promising results highlight the potential of these compounds as anticancer agents.
Lakshmanan et al.,109 synthesized a series of paracetamol chalcone derivatives and evaluated for their cytotoxic activity. The synthesized compounds 72, 73, and 74 (Fig. 26), have been examined and it is confirmed that the most promising cytotoxicity and also cell morphology analysis exhibited good apoptotic activity against lung cancer A549 cell.
 | ||
| Fig. 26 Anticancer chalcone acetamide hybrids 72–79 with different targets. | ||
Um, Y., et al.110 synthesized unsymmetrical curcuminoids with various amide groups and evaluated for their ability to reverse multidrug resistance (MDR). The MDR reversal activity was assessed using the potent anticancer agents, vincristine and paclitaxel, against P-gp non-expressing KB cells and P-gp expressing KBV20C cells. Verapamil was used as a positive control for comparison. Among the compounds tested, 75, 76, and 77 (Fig. 26), exhibited potent MDR reversal activity by inhibiting the drug efflux function of P-gp. The remaining compounds showed moderate potency. Based on the preliminary structure–activity relationship analysis, it was observed that the curcumin half-structure, feruloyl benzamidobenzene, holds promise as a lead structure for developing MDR reversal agents. Furthermore, the presence of one or two chloride groups at the meta- or para-position on benzamide was found to enhance the MDR reversal activity.
A set of chalcone/sulfonylurea hybrids was synthesized by Avupati, V. R.,111 and compounds were subjected to in vitro testing to assess their cytotoxicity and antimicrobial activities. In terms of cytotoxicity, compounds 78 and 79 (Fig. 26), demonstrated significant activity in the brine shrimp lethality assay, with ED50 values of 3.96 ± 0.21 and 4.02 ± 0.19 lg mL−1, respectively. These values were comparable to the reference drug podophyllotoxin, which had an ED50 value of 3.61 ± 0.17 lg mL−1. The findings suggest that compounds 78 and 79 hold potential as starting points for the development of lead molecules with strong cytotoxic effects.
3.8 Structure–activity relationship of chalcone amide as anticancer
To conclude the SAR analysis, systematic examination of the structural features of the most potent and promising anticancer chalcone derivatives identified in the reviewed literature were obtained. Statistical evaluation was performed on key structural components, including the chalcone phenyl rings A and B, the chalcone ketone bridge, and the amide group attached to the chalcone moiety. By analyzing the relationship between these structural features and the anticancer activity, the most influential modifications that enhance potency were identified. The introduction of an electron-donating group to the phenyl ring, in certain instances, results in an increase in activity compared to an unsubstituted ring, most likely: 3,4,5-trimethoxy > 4-methoxy > 3,4-dimethoxy > unsubstituted > 4-methyl. The substitution of a phenyl ring with an electron-withdrawing group, in certain cases, may result in comparable activity (in the case of fluorine isostere to hydrogen) or a moderate increase in activity compared to an unsubstituted ring. The suggested order of activity, which is subject to variation, is as follows: 4-chloro > 4-fluoro > 4-bromo > unsubstituted > 4-nitro. However, it is important to note that these observations are speculative and may not hold true universally (Fig. 27). | ||
| Fig. 27 Suggested structure–activity relationship of amide linked chalcone as anticancer. | ||
4 Antimicrobial properties
Antimicrobial resistance has become a global health concern, necessitating the development of effective antimicrobial agents.112 Chalcones offer promising opportunities in this context, as they have exhibited antimicrobial activity against a broad spectrum of pathogens, including bacteria, and fungi.113 Multiple mechanisms have been proposed to explain their antimicrobial action, such as inhibiting essential enzymes, disrupting microbial membranes, and interfering with cellular signaling pathways.114 The antimicrobial potential of chalcones has been evaluated against various drug-resistant strains, including methicillin-resistant Staphylococcus aureus (MRSA) and multidrug-resistant Escherichia coli (MDR E. coli).115 Additionally, chalcones have demonstrated antifungal activity against clinically relevant fungi, such as Candida species.116 Furthermore, the structural versatility of chalcones allows for the synthesis of numerous analogs with modified functional groups, leading to enhanced antimicrobial activity and selectivity.117H. H. Mohammed et al.,118 synthesized a series of hybrids combining ciprofloxacin with chalcones, linked by urea group. The antibacterial activity of these compounds was evaluated against S. aureus, P. aeruginosa, E. coli, and C. albicans strains. The ciprofloxacin derivatives 2a–j exhibited broad-spectrum antibacterial activity, with MIC values ranging from 0.06 to 42.23 μg mL−1, compared to ciprofloxacin alone, which had an MIC range of 0.15 to 3.25 μg mL−1. Notably, hybrids 80, and 81 (Fig. 28), demonstrated significant antibacterial activity, with MIC values ranging from 0.06 to 1.03 μg mL−1 against the tested bacterial strains. Moreover, compounds 82, 81, exhibited antifungal activity against Candida albicans comparable to that of ketoconazole, with MIC values 2.03 to 3.89 μg mL−1 respectively. Further investigations revealed that some of the ciprofloxacin hybrids showed inhibitory activity against DNA gyrase, a potential molecular target, with IC50 values ranging from 0.231 ± 0.01 to 7.592 ± 0.40 μM, compared to ciprofloxacin with an IC50 value of 0.323 ± 0.02 μM.
 | ||
| Fig. 28 Chalcone acetamides 80–91 with antibacterial activity. | ||
W.-C. Chu et al.,119 synthesized a set of chalcone derivatives to mimic the characteristics of cationic antimicrobial peptides. The antibacterial activities of these compounds were assessed against various drug-sensitive bacteria, including Staphylococcus aureus, Enterococcus faecalis, Escherichia coli, and Salmonella enterica. Additionally, clinical isolates of methicillin-resistant S. aureus (MRSA), as well as carbapenem-resistant Enterobacteriaceae producing KPC-2 and NDM-1, were evaluated. Among the compounds tested, representative molecules 83 and 84 (Fig. 28), demonstrated potent bactericidal activity against both Gram-positive and Gram-negative bacteria, including drug-resistant strains such as MRSA, KPC, and NDM. Compound 83 exhibited a minimum inhibitory concentration (MIC) of 1 mg mL−1 against S. aureus and 0.5 mg mL−1 against MRSA, while compound 83 showed an MIC of 0.5 μg mL−1 against S. aureus and 0.25 μg mL−1 against MRSA. These membrane-active antibacterial compounds effectively reduced the viable cell counts in bacterial biofilms and did not promote the development of bacterial resistance. Furthermore, these representative molecules exhibited minimal toxicity to mammalian cells at appropriate concentrations.
Another series of chalcones containing amide groups and conjugated with various secondary amines were synthesized and assessed for their antibacterial in vitro activity.120 Among the series of compounds, 85, 86, and 87 (Fig. 28), exhibited notable activity against different bacterial strains, with MIC values ranging from 2.0 to 10.0 μg mL−1. Compound 85 showed comparable potency to the standard drug Ampicillin, with an MBC value of 2.0 μg mL−1 against the bacterial strain Staphylococcus aureus. Furthermore, the compounds were evaluated for their anti-biofilm activity. Compounds 85, 86, and 87 demonstrated promising anti-biofilm effects, with IC50 values ranging from 2.4 to 8.6 μg. Molecular modeling studies were conducted to explore the potential anti-biofilm mechanisms. The results suggested that the AspB327 and HisB340 residues, involved in arene–arene interactions, played a crucial role in inhibiting c-di-GMP. Additionally, the hydrophobic nature of the compounds seemed to be important for their activity. ADME calculations were performed, indicating that compounds 85, 86, and 87 have the potential to be orally absorbed as effective anti-biofilm agents.
Joshi et al.,121 synthesized a series of chalcones containing 1,3,4-oxadiazole derivatives as antimicrobial agents targeting multidrug-resistant bacteria and fungi. The antimicrobial efficacy of these compounds was assessed against a wide range of bacteria and fungi. Notably, derivatives 88, and 89 (Fig. 28), exhibited significant antibacterial and antifungal properties, with more than half of the derivatives displaying superior minimum inhibitory concentration (MIC) values compared to the standard drugs used. Based on the structure–activity relationship (SAR) study, it is suggested that these bio-active molecules can be further improved as potent lead compounds by incorporating additional electron-donating groups into the core structure. The presence of electron-donating groups such as hydroxy (–OH) and methoxy (–OCH3) in the synthesized derivatives resulted in enhanced electron density, leading to the development of heterocyclic scaffolds with potent antibacterial and antifungal properties.
In an effort to enhance the antibacterial effects of chalcones and rhodanine-3-acetic acid, a series of hybrid compounds containing both chalcone and rhodanine-3-acetic acid components were synthesized by Z.-H. Chen et al.,122 and evaluated for their antibacterial activity. The antibacterial activities of these compounds were tested against both Gram-positive and Gram-negative bacteria, with a particular focus on multidrug-resistant strains of clinical isolates. Most of the synthesized compounds exhibited promising antibacterial activities, particularly against Gram-positive bacteria, including multidrug-resistant strains. Among the compounds tested, compound 90 (Fig. 28), demonstrated potent inhibitory capacity, with a minimum inhibitory concentration (MIC) of 2 μg mL−1. This activity was comparable to that of the standard drug norfloxacin and slightly lower than that of oxacillin. These findings suggest that the hybrid compounds incorporating chalcone and rhodanine-3-acetic acid moieties may possess enhanced antibacterial properties while introduction of the acetamide group (compound 91) in place of the halogens group significantly decreased the activity.
The analysis of chalcone derivatives highlights their promising antimicrobial properties, particularly against multidrug-resistant bacteria and fungi. The incorporation of ciprofloxacin into chalcone hybrids enhances their antibacterial activity, as evidenced by the reduced MIC values compared to ciprofloxacin alone. This suggests that the synergistic effects of chalcone and ciprofloxacin may offer a new approach for combating resistant bacterial strains. Additionally, the ability of these compounds to inhibit DNA gyrase further supports their potential as dual-action antibacterial agents. The derivatives mimicking cationic antimicrobial peptides show potent activity against a broad range of pathogens, including drug-resistant strains like MRSA. This is significant because it points to the ability of chalcones to bypass some resistance mechanisms that hinder traditional antibiotics. Furthermore, the incorporation of electron-donating substituents like methoxy and hydroxyl groups increases the antibacterial and anti-biofilm activity of chalcone derivatives. These findings indicate that careful structural modifications can enhance the potency and selectivity of chalcones. The hybridization approach, combining chalcones with other bioactive compounds such as rhodanine-3-acetic acid, demonstrates how combining different chemical entities can further improve antimicrobial activity.
5 Antiviral properties
The chalcone antiviral properties have also been investigated, showing potential in inhibiting the replication of certain viruses,123 and in the search for development of antiviral agents, a series of chalcone derivatives containing a purine component were designed and synthesized by Gan, X., et al.,124 via combining bioactive substructures. These derivatives were evaluated for their antiviral activity against tobacco mosaic virus (TMV) and cucumber mosaic virus (CMV). The results revealed that most of the derivatives exhibited antiviral properties. Notably, compounds 92 and 93 (Fig. 29), demonstrated excellent curative, protective, and inactivation activities against TMV. Their effective concentration values (EC50) were 452.4, 416.2, 241.2, and 438.7, 418.6, 261.7 μg mL−1, respectively, surpassing those of the reference drug ribavirin (585.8, 436.0, and 268.7 μg mL−1). Furthermore, compounds 92 and 93 displayed remarkable curative and protective activities against CMV. Compound 92 exhibited moderate affinity for the TMV coat protein, with binding constants (Ka) and dissociation constants (Kd) of 1.5 × 104 L mol−1 and 79.8 μmol L−1, respectively. These findings not only provide valuable insights into the structural aspects for the design of highly potent chalcone derivatives. | ||
| Fig. 29 Chalcone acetamides 92–96 with antiviral activity. | ||
Al-Hazam et al.,125 synthesized a series of substituted chalcone-incorporated amide derivatives of mefenamic acid through the Claisen–Schmidt reaction with the aim of developing HIV non-nucleoside reverse transcriptase inhibitors. Additionally, two thiopyrimidine analogues were prepared. The compounds were evaluated for their inhibitory activity against HIV-1 and HIV-2. The synthesis involved the coupling of mefenamic acid with 4-amino-acetophenone, resulting in the formation of 4-(acetylphenyl)-2-((2,3-dimethylphenyl)amino)benzamide. Further condensation reactions with various substituted benzaldehydes led to the synthesis of substituted chalcone-incorporated amide derivatives. The synthesized compounds were then tested for their inhibitory activity against HIV-1 and HIV-2 using MT-4 cells. Compounds 94 and 95 (Fig. 29), exhibited cytotoxicity with IC50 values of 2.17 and 2.06 μM, respectively, against mock-infected MT-4 cells. These findings suggest that compounds 94 and 95 hold promise as potential antileukemic agents.
A series of caffeoyl–anilide derivatives were synthesized by Bodiwala et al.,126 as dual inhibitors targeting both HIV-1 integrase (IN) and cellular CCR5. The intermediate compound, 4-amino-3,4-dihydroxychalcone. In cell-based anti-HIV assays, compound 96 (Fig. 29), demonstrated a lower EC50 = 0.9 μM and a better therapeutic index (TI) compared to AZT, a standard antiretroviral drug. Mechanistic investigations indicated that the synthesized compounds inhibited both the entry of CCR5-tropic viruses and the integration step of the virus life cycle. Hence, the anti-HIV activity of these inhibitors likely stems from their specificity towards both targets. Molecular modeling studies provided insights into the predicted conformations of the compounds and their interactions with key amino acid residues (HIV-1 IN: Thr66, His67, Gln148, Lys159, and Mg2+; CCR5: Ser160, Lys197, and Phe245) within the active sites. These interactions help explain the observed inhibitory effects of the compounds.
6 Antiprotozoal properties
With the aim of developing potent antileishmanial compounds, a medicinal chemistry-driven approach was employed by ref. 127 to synthesize scaffolds with common pharmacophoric features of dihydropyrimidine and chalcone. The design of these compounds was guided by the X-ray structure of pteridine reductase 1 (PTR1) from Leishmania major. Several dihydropyrimidine-based derivatives were synthesized, targeting specific.Several derivatives of bis-chalcone were synthesized by Domínguez et al.,128 and characterized, and their antimalarial activities were evaluated. Most of the synthesized compounds showed mild to moderate susceptibilities against Plasmodium berghei, with compound 1,1-bis-[(3′,4′-N-(urenylphenyl)-3-(3′′,4′′,5′′-trimethoxyphenyl))]-2-propen-1-one 97 exhibiting the highest activity. Compound 97 (Fig. 30), inhibited heme polymerization by 87.05 ± 0.77% and demonstrated increased survival time, reduced parasitemia, and delayed progression of malaria in infected mice. Compound 97 was further evaluated for its inhibitory effects on globin hydrolysis using a trophozoite-rich extract. The compound effectively inhibited the degradation of hemoglobin, surpassing the control groups (pepstatin and leupeptin). In an in vivo study using mice infected with P. berghei, compound 97 (administered at 20 μg kg−1, ip once daily) and chloroquine (administered at 25 mg kg−1, ip once daily) were compared to a saline control group. Compound 97 significantly reduced parasitemia at the 4th day after infection compared to the control group. Moreover, compound 97 increased the survival time of infected mice, with deaths occurring between days 6 and 19 after infection, compared to the control group, where deaths occurred between days 9 and 11. While compound 97 showed the ability to reduce parasitemia and delay malaria progression, it did not completely eradicate the infection.
 | ||
| Fig. 30 Chalcone acetamides 97–105 with antiprotozoal activity. | ||
León, C., et al.129 synthesized a series of sulfonylureas and evaluated for their antimalarial activities against Plasmodium falciparum. Compound 98 (Fig. 30), exhibited the highest activity with an IC50 of 1.2 μM against cultured P. falciparum parasites. The results suggest potent antimalarial activity for this compound, possibly through unknown mechanisms. Additionally, compound 99 also showed significant antimalarial effects with an IC50 value of 2.1 μM and demonstrated activity in a P. berghei mouse model, reducing parasitemia by 77% and increasing survival. The presence of 2,4-difluoro substitution in the aromatic ring of the α,β-unsaturated ketone system was found to be important for mediating activity against P. falciparum. The fluorine substitution likely enhances chemical interactions with the biological substrate. The results indicate that compound (98) inhibits hemoglobin degradation and hemozoin formation, contributing to its antimalarial activity in vitro. The findings also suggest that sulfonylurea derivatives with fluorine substitution in the aromatic ring maintain antimalarial efficacy in vitro and moderate activity in vivo, possibly due to electronic effects. The synthesized sulfonylurea compounds demonstrated antimalarial activity against P. falciparum. Compound (98) exhibited the highest potency, inhibiting parasite growth in cultured cells and reducing parasitemia in a mouse model. The presence of 2,4-difluoro substitution in the aromatic ring was crucial for mediating activity against P. falciparum. These findings contribute to the understanding of the antimalarial potential of sulfonylurea derivatives and suggest that compound (98) holds promise as a potential antimalarial compound.
Domínguez et al.,130 synthesized phenylurenyl chalcone derivatives and evaluated for their antimalarial properties against Plasmodium falciparum. Compound 100 (Fig. 30), showed the highest activity, with an IC50 of 1.76 μM against cultured P. falciparum parasites. Several other chalcone derivatives also exhibited significant antimalarial effects, both in vitro and in a murine malaria model. Compound 101 demonstrated good antimalarial activity in vitro and in vivo. It exhibited inhibitory effects on heme detoxification (89.2% inhibition) and globin hydrolysis (98.64% inhibition). This compound contained an aromatic pyridinyl group, resembling the nitrogen of chloroquine, which may concentrate in the food vacuoles of malaria parasites. The relationship between antiparasitic activity, hemozoin formation inhibition, and hemoglobin hydrolysis was observed, indicating that potent inhibition of heme formation and hemoglobin hydrolysis does not guarantee antimalarial activity. Based on the data, compound 100 had the best antimalarial properties, inhibiting the development of P. falciparum in culture (IC50 of 1.76 μM) and reducing parasitemia in P. berghei infected mice (from 23% to 6.8%). However, its mechanism of action does not appear to be related to hemoglobin hydrolysis or heme detoxification.
Jiang et al.,131 synthesized chalcone derivatives and investigated as potential anti-Toxoplasma agents. The compounds and evaluated for their anti-Toxoplasma activity in vitro and in vivo. The results demonstrated that several chalcone derivatives exhibited potent anti-Toxoplasma activity with low toxicity. Using the drug-food-homologous chalcone skeleton as a starting point, six series of chalcone derivatives were designed and synthesized. Approximately half of these compounds showed good anti-Toxoplasma activity in vitro. A quantitative structure–activity relationship (QSAR) model was established, indicating the significance of the Michael receptor in the chalcone molecular skeleton for enhancing activity. Four chalcone derivatives displayed potent anti-T. gondii activity and low cytotoxicity in vitro. In vivo studies revealed that three of these compounds 102, 103, and 105 (Fig. 30), effectively inhibited the proliferation of Toxoplasma tachyzoites. Moreover, these compounds exhibited liver-protecting effects, as evidenced by reduced liver and spleen index, as well as decreased levels of biochemical parameters associated with liver function. These findings suggest that these chalcone derivatives have protective effects on the liver of mice infected with Toxoplasma tachyzoites. The second study further supported the anti-Toxoplasma activity of chalcone derivatives. A QSAR model was established, confirming the importance of the Michael acceptor for their anti-Toxoplasma activity. Four compounds (102, 103, 104, and 105) demonstrated potent activity against T. gondii in vitro while exhibiting low toxicity towards host cells.
7 Anti-inflammatory and antioxidant activity
The anti-inflammatory properties of chalcones arise from their ability to modulate key molecular targets and signaling pathways involved in the inflammatory response. Through various mechanisms, chalcones exhibit remarkable anti-inflammatory effects, making them promising candidates for the development of anti-inflammatory agents.132 One of the primary mechanisms through which chalcones exert their anti-inflammatory effects is by inhibiting the activity of enzymes responsible for the production of inflammatory mediators. By targeting cyclooxygenase (COX) and lipoxygenase (LOX) enzymes, chalcones can impede the synthesis of prostaglandins and leukotrienes, respectively, thereby reducing the production of these pro-inflammatory mediators. Furthermore, chalcones have been shown to suppress the expression and release of pro-inflammatory cytokines. These small proteins, such as tumor necrosis factor-alpha (TNF-α), interleukin-1 beta (IL-1β), and interleukin-6 (IL-6), play critical roles in orchestrating the inflammatory response. Chalcones can modulate the transcriptional activity of key transcription factors, such as nuclear factor-kappa B (NF-κB) and activator protein-1 (AP-1), leading to the downregulation of pro-inflammatory cytokine production. In addition to their impact on enzymes and cytokines, chalcones exhibit antioxidant properties.133 By scavenging reactive oxygen species (ROS), chalcones can mitigate oxidative stress, a hallmark of inflammation. Oxidative stress can propagate inflammatory damage, and the antioxidant activity of chalcones helps to counteract this phenomenon, protecting cells from oxidative injury.134 Moreover, chalcones modulate various signaling pathways implicated in inflammation, they can interfere with the activation of mitogen-activated protein kinases (MAPKs) and the phosphoinositide 3-kinase (PI3K)/Akt pathway, thereby regulating the expression of inflammatory genes. Through these actions, chalcones attenuate the inflammatory response at the molecular level.135A series of trimethoxy phenyl-containing chalcone derivatives, pyrazoline derivatives, and pyrazole derivatives were synthesized by Abdelall, Lamie et al.,136 and evaluated for their COX-2 inhibitory activity, anti-inflammatory activity, histopathological effects. The compounds exhibited promising results in various assays. The synthesized compounds demonstrated selective COX-2 inhibitory activity, with higher potency against COX-2 compared to COX-1. Compound 106 (Fig. 31), displayed the most potent COX-2 inhibitory activity (IC50 = 0.039 μM), surpassing that of the reference drug celecoxib (IC50 = 0.045 μM), and exhibited a selectivity index value of 321.28, similar to celecoxib (S.I. = 326.66). Additionally, derivatives 107, exhibited excellent COX-2 inhibitory activity (IC50 = 0.041–0.049 μM) compared to celecoxib, with selectivity index values ranging from 230.61 to 278.05. In terms of anti-inflammatory activity, several compounds demonstrated prolonged inhibition of inflammation in an in vivo model, with inhibition percentages ranging from 33.21% to 44.52% after 7 hours following carrageenan injection.
 | ||
| Fig. 31 Chalcone acetamides 106–113 with anti-inflammatory activity. | ||
A hit-to-lead effort was conducted by Zhang et al.,137 to discover and optimize NLRP3 inflammasome inhibitors. The research led to the identification of a potent lead compound 108 (Fig. 31), which exhibited improved inhibitory potency and minimal toxicity. Mechanistic investigations revealed that compound 108 suppressed NLRP3 inflammasome activation by inhibiting ROS production. Furthermore, treatment with compound 108 showed remarkable therapeutic effects in sepsis and colitis models induced by LPS and DSS, respectively. These findings encourage further development of more potent inhibitors based on the chemical scaffold of compound 108 and provide a valuable chemical tool for identifying its cellular binding target. During the study, a clinical compound library screen was utilized to identify elafibranor (GFT505) as a NLRP3 inflammasome inhibitor with good safety. The chalcone pharmacophore of GFT505 was subjected to optimization through structure–activity relationship (SAR) studies. By employing both simplification and replacement strategies guided by SAR and structure information, the researchers achieved enhancements in inhibitory activity. Notably, modifications that included the removal of a carboxyisopropanyl moiety and the presence of suitable hydrogen bond donors at specific positions significantly improved the potency of the compound. Further exploration revealed that the 3,5-dimethyl moiety in the phenol group was crucial for potent inhibition of NLRP3 inflammasome activation. Subsequent modifications on the methylthio moiety resulted in the discovery of compound 108, which exhibited approximately 30 times greater potency against NLRP3 inflammasome activation and lower toxicity compared to GFT505 at the cellular level. Compound 108 demonstrated favorable drug-like properties and showed efficacy in sepsis and colitis models. Ongoing chemoproteomic studies are aimed at identifying the cellular binding target of compound 108. Collectively, these findings from a single study support the potential of compound 108 as a promising lead compound for NLRP3 inhibitors. The research highlights its therapeutic potential for inflammation and related diseases, while also providing insights into its mechanism of action and opportunities for further chemical development and investigation of its pharmacological properties.
A group of chalcone derivatives with nitric oxide (NO) donating properties were synthesized by Abuo-Rahma et al.,138 and evaluated for their anti-inflammatory and gastroprotective activities. The compounds were prepared by incorporating different NO donating moieties, including nitrate ester, oximes, and furoxans, into amino chalcones. The synthesized compounds demonstrated significant anti-inflammatory activity when tested using the carrageenan-induced rat paw edema method. Compared to the reference drug indomethacin, most of the prepared compounds exhibited higher levels of protection against inflammation. Moreover, the compounds showed a decreased incidence of gastric toxicity compared to indomethacin, as confirmed by histopathological investigation, which revealed reduced ulcer formation. The incorporation of NO-donating groups into the parent chalcone derivatives resulted in a moderate increase in anti-inflammatory activity and a marked decrease in gastric ulcerations compared to their parent chalcone derivatives. Notably, compounds, 109, and 110 (Fig. 31), demonstrated the strongest anti-inflammatory activity among the synthesized compounds, with edema inhibition percentages of, 75%, and 72%, respectively, after 4 hours. These values corresponded to, 91%, and 87% of the activity exhibited by indomethacin. Additionally, the NO-donating furoxans exhibited the highest amount of NO release among the synthesized NO-donating hybrids. The prepared chalcone/NO hybrids, regardless of the NO donating moiety (nitrate ester, oxime, or furoxan), demonstrated pronounced gastroprotective activity, likely attributed to the release of NO. Histopathological examination further supported the reduction in gastric ulceration incidence due to the presence of the NO donating moiety.
J. Chu et al.,139 synthesized a group of chalcone derivatives and were evaluated for their antioxidant and anti-inflammatory activities, specifically targeting hepatic fibrosis patients. In terms of antioxidant activity, the chalcone derivatives demonstrated significant radical scavenging abilities, as determined by various methods such as H2O2, DPPH, ferrous reducing power, and nitric oxide assays. These compounds exhibited considerable efficacy in scavenging free radicals, indicating their potential as antioxidant agents. Furthermore, the synthesized chalcone derivatives were assessed for their anti-inflammatory activity by evaluating their inhibitory potency against NF-kB activation induced by LPS. The results revealed that all of the compounds efficiently inhibited the NF-kB activation provoked by LPS. Among the series, compound 111 (Fig. 31), was identified as the most potent inhibitor of NF-kB, displaying a relative NF-kB activity of 1.12 ± 0.53. Additionally, this compound exhibited inhibitory effects on various inflammatory mediators, including TNF-a, IL-1b, IL-6, and PGE2. The developed chalcone derivatives not only exhibited excellent antioxidant activity but also demonstrated considerable inhibitory activity against NF-kB, a key regulator of inflammation. Combined with their favorable dock score, these compounds hold promise as lead molecules for future drug discovery initiatives aimed at treating liver cirrhosis and hepatic fibrosis patients.
A series of quinoline linked chalcones derivatives were synthesized by Polo et al.,140 and evaluated for their inhibitory activity against acetylcholinesterase (AChE) and their antioxidative properties. Several compounds demonstrated selectivity against AChE, with compounds 112, and 113 (Fig. 31), exhibiting significant in vitro activity, displaying IC50 values of 7.50, and 12.58 μM, respectively. Compound 112 was identified as the most potent AChE inhibitor, with an IC50 value of 7.50 μM.
8 Conclusion and future directions
Chalcone-linked acetamide derivatives exhibit a remarkable array of biological activities, positioning them as promising candidates for therapeutic applications. The detailed exploration of their synthesis methods reveals innovative approaches that enhance the efficiency and scalability of these compounds. Through extensive analysis, it is evident that these derivatives possess potent antiproliferative properties, targeting critical pathways such as EGFR, topoisomerase I and II, ABCG2, caspase proteins, and histone deacetylase (HDAC), alongside their ability to inhibit tubulin polymerization. The elucidation of structure–activity relationships further underscores their potential as anticancer agents. In addition to their antiproliferative effects, chalcone-linked acetamide derivatives demonstrate significant antimicrobial, antiviral, and antiprotozoal properties, highlighting their versatility in combating a wide range of pathogens. Their robust anti-inflammatory and antioxidant activities further contribute to their therapeutic promise. The comprehensive evaluation of these bioactivities suggests that chalcone-linked acetamide derivatives are valuable candidates for drug discovery and development.Data availability
No new data were created or analyzed during this study. Data sharing is not applicable to this article.Conflicts of interest
The authors declare that there is no conflict of interest.References
- S.-X. Lin, J. Chen, M. Mazumdar, D. Poirier, C. Wang, A. Azzi and M. Zhou, Nat. Rev. Endocrinol., 2010, 6, 485–493 CrossRef CAS PubMed
.
- D. K. Mahapatra, S. K. Bharti and V. Asati, Eur. J. Med. Chem., 2015, 98, 69–114 CrossRef CAS PubMed
- L. H. Cazarolli, V. D. Kappel, A. P. Zanatta, D. O. H. Suzuki, R. A. Yunes, R. J. Nunes, M. G. Pizzolatti and F. R. M. B. Silva, Stud. Nat. Prod. Chem., 2013, 39, 47–89 CAS
- B. Zhou and C. Xing, Med. Chem., 2015, 5, 388 Search PubMed
- C. Karthikeyan, N. S. H. Narayana Moorthy, S. Ramasamy, U. Vanam, E. Manivannan, D. Karunagaran and P. Trivedi, Recent Pat. Anti-Cancer Drug Discovery, 2015, 10, 97–115 CrossRef CAS PubMed
- A. Boumendjel, X. Ronot and J. Boutonnat, Curr. Drug Targets, 2009, 10, 363–371 CrossRef PubMed
- A. J. Leon-Gonzalez, N. Acero, D. Muñoz-Mingarro, I. Navarro and C. Martín-Cordero, Curr. Med. Chem., 2015, 22, 3407–3425 CrossRef CAS PubMed
- D. K. Mahapatra, V. Asati and S. K. Bharti, Eur. J. Med. Chem., 2015, 92, 839–865 CrossRef CAS PubMed
- D. K. Mahapatra and S. K. Bharti, Life Sci., 2016, 148, 154–172 CrossRef CAS PubMed
- C. Zhuang, W. Zhang, C. Sheng, W. Zhang, C. Xing and Z. Miao, Chem. Rev., 2017, 117, 7762–7810 CrossRef CAS PubMed
- D. Kumar, M. Kumar, A. Kumar and S. Kumar Singh, Mini-Rev. Med. Chem., 2013, 13, 2116–2133 CrossRef CAS PubMed
- A. Hameed, M. I. Abdullah, E. Ahmed, A. Sharif, A. Irfan and S. Masood, Bioorg. Chem., 2016, 65, 175–182 CrossRef CAS PubMed
- Z. Wan, D. Hu, P. Li, D. Xie and X. Gan, Molecules, 2015, 20, 11861–11874 CrossRef CAS PubMed
- F. Peng, Q. Du, C. Peng, N. Wang, H. Tang, X. Xie, J. Shen and J. Chen, Phytother. Res., 2015, 29, 969–977 CrossRef CAS PubMed
- J.-Y. Kim, S. J. Park, K.-J. Yun, Y.-W. Cho, H.-J. Park and K.-T. Lee, Eur. J. Pharmacol., 2008, 584, 175–184 CrossRef CAS PubMed
- E. Oledzka, Int. J. Mol. Sci., 2024, 25, 3398 CrossRef CAS PubMed
- M. Yang, N. Li, F. Li, Q. Zhu, X. Liu, Q. Han, Y. Wang, Y. Chen, X. Zeng and Y. Lv, Int. Immunopharmacol., 2013, 16, 466–474 CrossRef CAS PubMed
- M. Liu, P. E. Hansen, G. Wang, L. Qiu, J. Dong, H. Yin, Z. Qian, M. Yang and J. Miao, Molecules, 2015, 20, 754–779 CrossRef PubMed
- S. C. Narayanapillai, P. Leitzman, M. G. O'Sullivan and C. Xing, Chem. Res. Toxicol., 2014, 27, 1871–1876 Search PubMed
- B. Das, A. T. Baidya, A. T. Mathew, A. K. Yadav and R. Kumar, Bioorg. Med. Chem., 2022, 56, 116614 CrossRef CAS PubMed
- S. Mahesh, K.-C. Tang and M. Raj, Molecules, 2018, 23, 2615 CrossRef PubMed
- Z. Chen, H. Luo, A. Gubu, S. Yu, H. Zhang, H. Dai, Y. Zhang, B. Zhang, Y. Ma and A. Lu, Front. Cell Dev. Biol., 2023, 11, 1091809 CrossRef PubMed
- V. S. Gontijo, F. P. D. Viegas, C. J. Ortiz, M. de Freitas Silva, C. M. Damasio, M. C. Rosa, T. G. Campos, D. S. Couto, K. S. Tranches Dias and C. Viegas, Curr. Neuropharmacol., 2020, 18, 348–407 CrossRef CAS PubMed
- S. Shukla, A. K. Sood, K. Goyal, A. Singh, V. Sharma, N. Guliya, S. Gulati and S. Kumar, Anti-Cancer Agents Med. Chem., 2021, 21, 1650–1670 CrossRef CAS PubMed
- G. B. Souza, T. A. Santos, A. P. Silva, A. L. S. Barreiros, V. B. Nardelli, I. B. Siqueira, S. S. Dolabella, E. V. Costa, P. B. Alves and R. Scher, Nat. Prod. Res., 2022, 1–8 Search PubMed
- S. Nasir Abbas Bukhari, M. Jasamai, I. Jantan and W. Ahmad, Mini-Rev. Org. Chem., 2013, 10, 73–83 CrossRef
- S. A. Khan, A. M. Asiri, N. S. M. Al-Ghamdi, M. Asad, M. E. Zayed, S. A. Elroby, F. M. Aqlan, M. Y. Wani and K. Sharma, J. Mol. Struct., 2019, 1190, 77–85 CrossRef CAS
- T. Narender and K. P. Reddy, Tetrahedron Lett., 2007, 48, 3177–3180 CrossRef CAS
- M. Al-Masum, E. Ng and M. C. Wai, Tetrahedron Lett., 2011, 52, 1008–1010 CrossRef CAS
- S. Nasir Abbas Bukhari, M. Jasamai and I. Jantan, Mini-Rev. Med. Chem., 2012, 12, 1394–1403 Search PubMed
- M. M. Heravi, V. Zadsirjan, P. Saedi and T. Momeni, RSC Adv., 2018, 8, 40061–40163 RSC
- A. Scettri, R. Villano and M. R. Acocella, Molecules, 2009, 14, 3030–3036 CrossRef CAS PubMed
- A. Kumar, S. Sharma, V. D. Tripathi and S. Srivastava, Tetrahedron, 2010, 66, 9445–9449 CrossRef CAS
- M. Haddach and J. R. McCarthy, Tetrahedron Lett., 1999, 40, 3109–3112 CrossRef CAS
- S. Eddarir, N. Cotelle, Y. Bakkour and C. Rolando, Tetrahedron Lett., 2003, 44, 5359–5363 CrossRef CAS
- D. Wu and C. Prives, Cell Death Differ., 2018, 25, 169–179 CrossRef CAS PubMed
- A. Kamal, G. B. Kumar, M. Vishnuvardhan, A. B. Shaik, V. S. Reddy, R. Mahesh, I. B. Sayeeda and J. S. Kapure, Org. Biomol. Chem., 2015, 13, 3963–3981 RSC
- V. R. Yadav, S. Prasad, B. Sung and B. B. Aggarwal, Int. Immunopharmacol., 2011, 11, 295–309 CrossRef CAS PubMed
- M. J. Mphahlele, M. M. Maluleka, N. Parbhoo and S. T. Malindisa, Int. J. Mol. Sci., 2018, 19, 2552 CrossRef PubMed
- L. Wang, G. Chen, X. Lu, S. Wang, S. Han, Y. Li, G. Ping, X. Jiang, H. Li and J. Yang, Eur. J. Med. Chem., 2015, 89, 88–97 CrossRef CAS PubMed
- J. H. Jeong, H. J. Jang, S. Kwak, G. J. Sung, S. H. Park, J. H. Song, H. Kim, Y. Na and K. C. Choi, J. Cell. Biochem., 2019, 120, 977–987 CrossRef CAS PubMed
- Z. Parveen, G. Brunhofer, I. Jabeen, T. Erker, P. Chiba and G. F. Ecker, Bioorg. Med. Chem., 2014, 22, 2311–2319 CrossRef CAS PubMed
- I. K. Lindamulage, H.-Y. Vu, C. Karthikeyan, J. Knockleby, Y.-F. Lee, P. Trivedi and H. Lee, Sci. Rep., 2017, 7, 10298 CrossRef PubMed
- E. Winter, P. c. Devantier Neuenfeldt, L. D. Chiaradia-Delatorre, C. Gauthier, R. A. Yunes, R. J. Nunes, T. n. B. Creczynski-Pasa and A. Di Pietro, J. Med. Chem., 2014, 57, 2930–2941 CrossRef CAS PubMed
- Y. Ouyang, J. Li, X. Chen, X. Fu, S. Sun and Q. Wu, Biomolecules, 2021, 11, 894 CrossRef CAS PubMed
- T. Zubair and D. Bandyopadhyay, Int. J. Mol. Sci., 2023, 24, 2651 CrossRef CAS PubMed
- P. Wee and Z. Wang, Cancers, 2017, 9, 52 CrossRef PubMed
- P. Seshacharyulu, M. P. Ponnusamy, D. Haridas, M. Jain, A. K. Ganti and S. K. Batra, Expert Opin. Ther. Targets, 2012, 16, 15–31 CrossRef CAS PubMed
- W.-Q. Cai, L.-S. Zeng, L.-F. Wang, Y.-Y. Wang, J.-T. Cheng, Y. Zhang, Z.-W. Han, Y. Zhou, S.-L. Huang and X.-W. Wang, Front. Oncol., 2020, 10, 1249 CrossRef PubMed
- S. Kumagai, S. Koyama and H. Nishikawa, Nat. Rev. Cancer, 2021, 21, 181–197 CrossRef CAS PubMed
- X. Du, B. Yang, Q. An, Y. G. Assaraf, X. Cao and J. Xia, Innovation, 2021, 2(2), 100103 CAS
- A. M. Mohassab, H. A. Hassan, D. Abdelhamid, A. M. Gouda, B. G. Youssif, H. Tateishi, M. Fujita, M. Otsuka and M. Abdel-Aziz, Bioorg. Chem., 2021, 106, 104510 CrossRef CAS PubMed
- M. A. A. Fathi, A. A. Abd El-Hafeez, D. Abdelhamid, S. H. Abbas, M. M. Montano and M. Abdel-Aziz, Bioorg. Chem., 2019, 84, 150–163 CrossRef CAS PubMed
- H. A. Abou-Zied, B. G. Youssif, M. F. Mohamed, A. M. Hayallah and M. Abdel-Aziz, Bioorg. Chem., 2019, 89, 102997 CrossRef CAS PubMed
- M. Hisham, H. A. Hassan, H. A. Gomaa, B. G. Youssif, A. M. Hayallah and M. Abdel-Aziz, J. Mol. Struct., 2022, 1254, 132422 CrossRef CAS
- H. A. Abou-Zied, E. A. Beshr, H. A. Gomaa, Y. A. Mostafa, B. G. Youssif, A. M. Hayallah and M. Abdel-Aziz, Arch. Pharm., 2022, e2200464 Search PubMed
- F. F. Hagar, S. H. Abbas, D. Abdelhamid, H. A. Gomaa, B. G. Youssif and M. Abdel-Aziz, Arch. Pharm., 2023, 356, 2200357 CrossRef CAS PubMed
- M. S. Abdelbaset, M. Abdel-Aziz, M. Ramadan, M. H. Abdelrahman, S. N. A. Bukhari, T. F. Ali and G. E.-D. A. Abuo-Rahma, Bioorg. Med. Chem., 2019, 27, 1076–1086 CrossRef CAS PubMed
- M. T. E. Maghraby, O. I. Salem, B. G. Youssif and M. M. Sheha, Chem. Biol. Drug Des., 2023, 101, 749–759 CrossRef CAS PubMed
- X. Liang, Q. Wu, S. Luan, Z. Yin, C. He, L. Yin, Y. Zou, Z. Yuan, L. Li and X. Song, Eur. J. Med. Chem., 2019, 171, 129–168 CrossRef CAS PubMed
- W. A. Denny, Expert Opin. Emerging Drugs, 2004, 9, 105–133 CAS
- H. H. Mohammed, S. H. Abbas, A. M. Hayallah, G. E.-D. A. Abuo-Rahma and Y. A. Mostafa, Bioorg. Chem., 2021, 106, 104422 CrossRef CAS PubMed
- S.-H. Kim, E. Lee, K. H. Baek, H. B. Kwon, H. Woo, E.-S. Lee, Y. Kwon and Y. Na, Bioorg. Med. Chem. Lett., 2013, 23, 3320–3324 CrossRef CAS PubMed
- M. Abdel-Aziz, S.-E. Park, G. E.-D. A. Abuo-Rahma, M. A. Sayed and Y. Kwon, Eur. J. Med. Chem., 2013, 69, 427–438 Search PubMed
- K. P. Locher, Nat. Struct. Mol. Biol., 2016, 23, 487–493 CrossRef CAS PubMed
- Q. Mao and J. D. Unadkat, AAPS J., 2015, 17, 65–82 Search PubMed
- I. F. Zattoni, L. C. Delabio, J. de Paula Dutra, D. H. Kita, G. Scheiffer, M. Hembecker, G. da Silva Pereira, V. R. Moure and G. Valdameri, Eur. J. Med. Chem., 2022, 237, 114346 CrossRef CAS PubMed
- Y. Toyoda, T. Takada and H. Suzuki, Front. Pharmacol., 2019, 10, 208 CrossRef CAS PubMed
- S. Kukal, D. Guin, C. Rawat, S. Bora, M. K. Mishra, P. Sharma, P. R. Paul, N. Kanojia, G. K. Grewal and S. Kukreti, Cell. Mol. Life Sci., 2021, 1–53 Search PubMed
- S. Kraege, S. C. Köhler and M. Wiese, ChemMedChem, 2016, 11, 2422–2435 CrossRef CAS PubMed
- S. Kraege, K. Stefan, S. C. Köhler and M. Wiese, ChemMedChem, 2016, 11, 2547–2558 CrossRef CAS PubMed
- K. Juvale, V. F. Pape and M. Wiese, Bioorg. Med. Chem., 2012, 20, 346–355 CrossRef CAS PubMed
- A. B. Parrish, C. D. Freel and S. Kornbluth, Cold Spring Harbor Perspect. Biol., 2013, 5, a008672 Search PubMed
- G. Pistritto, D. Trisciuoglio, C. Ceci, A. Garufi and G. D'Orazi, Aging, 2016, 8, 603 CrossRef CAS PubMed
- S. H. MacKenzie, J. L. Schipper and A. C. Clark, Curr. Opin. Drug Discovery Dev., 2010, 13, 568 CAS
- S. Hashem, T. A. Ali, S. Akhtar, S. Nisar, G. Sageena, S. Ali, S. Al-Mannai, L. Therachiyil, R. Mir and I. Elfaki, Biomed. Pharmacother., 2022, 150, 113054 CrossRef CAS PubMed
- M. Vizovisek, D. Ristanovic, S. Menghini, M. G. Christiansen and S. Schuerle, Int. J. Mol. Sci., 2021, 22, 2514 CrossRef CAS PubMed
- J. Murray and A. R. Renslo, Curr. Opin. Struct. Biol., 2013, 23, 812–819 CrossRef CAS PubMed
- F. Tok, B. İ. Abas, Ö. Çevik and B. Koçyiğit-Kaymakçıoğlu, Bioorg. Chem., 2020, 102, 104063 CrossRef CAS PubMed
- F. F. Ahmed, A. A. Abd El-Hafeez, S. H. Abbas, D. Abdelhamid and M. Abdel-Aziz, Eur. J. Med. Chem., 2018, 151, 705–722 CrossRef CAS PubMed
- R. Romagnoli, P. G. Baraldi, M. D. Carrion, O. Cruz-Lopez, C. L. Cara, J. Balzarini, E. Hamel, A. Canella, E. Fabbri and R. Gambari, Bioorg. Med. Chem. Lett., 2009, 19, 2022–2028 CrossRef CAS PubMed
- Y. Li and E. Seto, Cold Spring Harbor Perspect. Med., 2016, 6, a026831 CrossRef PubMed
- S. Patra, D. P. Panigrahi, P. P. Praharaj, C. S. Bhol, K. K. Mahapatra, S. R. Mishra, B. P. Behera, M. Jena and S. K. Bhutia, Cell. Mol. Life Sci., 2019, 76, 3263–3282 CrossRef CAS PubMed
- H. P. Chen, Y. T. Zhao and T. C. Zhao, Crit. Rev. Oncog., 2015, 20(1–2), 35–47 CrossRef PubMed
- M. Dickinson, R. W. Johnstone and H. M. Prince, Invest. New Drugs, 2010, 28, 3–20 CrossRef CAS PubMed
- P. Chun, Arch. Pharmacal Res., 2015, 38, 933–949 CrossRef CAS PubMed
- S. Valente, D. Trisciuoglio, M. Tardugno, R. Benedetti, D. Labella, D. Secci, C. Mercurio, R. Boggio, S. Tomassi and S. Di Maro, ChemMedChem, 2013, 8, 800–811 Search PubMed
- A. A. Mourad, M. A. Mourad and P. G. Jones, Drug Des., Dev. Ther., 2020, 3111–3130 CrossRef CAS PubMed
- M. Knossow, V. Campanacci, L. A. Khodja and B. Gigant, iScience, 2020, 23(9), 101511 CrossRef CAS PubMed
- W. Liu, M. He, Y. Li, Z. Peng and G. Wang, J. Enzyme Inhib. Med. Chem., 2022, 37, 9–38 CrossRef CAS PubMed
- E. Mukhtar, V. M. Adhami and H. Mukhtar, Mol. Cancer Ther., 2014, 13, 275–284 CrossRef CAS PubMed
- G. Ş. Karatoprak, E. Küpeli Akkol, Y. Genç, H. Bardakcı, Ç. Yücel and E. Sobarzo-Sánchez, Molecules, 2020, 25, 2560 CrossRef CAS PubMed
- M. M. Amin, G. E.-D. A. Abuo-Rahma, M. S. A. Shaykoon, A. A. Marzouk, M. A. Abourehab, R. E. Saraya, M. Badr, A. M. Sayed and E. A. Beshr, Bioorg. Chem., 2023, 134, 106444 CrossRef CAS PubMed
- H. Del Rosario, E. Saavedra, I. Brouard, D. González-Santana, C. García, E. Spínola-Lasso, C. Tabraue, J. Quintana and F. Estévez, Bioorg. Chem., 2022, 127, 105926 CrossRef CAS PubMed
- Y. Li, Y. Sun, Y. Zhou, X. Li, H. Zhang and G. Zhang, J. Enzyme Inhib. Med. Chem., 2021, 36, 207–217 CrossRef CAS PubMed
- D.-J. Fu, J.-H. Li, J.-J. Yang, P. Li, Y.-B. Zhang, S. Liu, Z.-R. Li and S.-Y. Zhang, Bioorg. Chem., 2019, 86, 375–385 Search PubMed
- P. F. Lamie and J. N. Philoppes, J. Enzyme Inhib. Med. Chem., 2020, 35, 864–879 CrossRef CAS PubMed
- J.-L. Ren, X.-Y. Zhang, B. Yu, X.-X. Wang, K.-P. Shao, X.-G. Zhu and H.-M. Liu, Eur. J. Med. Chem., 2015, 93, 321–329 CrossRef CAS PubMed
- M. A. Mourad, M. Abdel-Aziz, G. E.-D. A. Abuo-Rahma and H. H. Farag, Eur. J. Med. Chem., 2012, 54, 907–913 CrossRef CAS PubMed
- X. Cao, R. Li, H. Xiong, J. Su, C. Guo, T. An, H. Zong and R. Zhao, Eur. J. Med. Chem., 2021, 221, 113520 CrossRef CAS PubMed
- S. Patel, N. Challagundla, R. A. Rajput and S. Mishra, Bioorg. Chem., 2022, 127, 106036 CrossRef CAS PubMed
- C.-F. Lu, S.-H. Wang, X.-J. Pang, T. Zhu, H.-L. Li, Q.-R. Li, Q.-Y. Li, Y.-F. Gu, Z.-Y. Mu and M.-J. Jin, Molecules, 2020, 25, 5530 CrossRef CAS PubMed
- P. N. Bandeira, T. L. G. Lemos, H. S. Santos, M. C. S. de Carvalho, D. P. Pinheiro, M. O. de Moraes Filho, C. Pessoa, F. W. A. Barros-Nepomuceno, T. H. S. Rodrigues and P. R. V. Ribeiro, Med. Chem. Res., 2019, 28, 2037–2049 Search PubMed
- S. D. Durgapal, R. Soni, S. Umar, B. Suresh and S. S. Soman, Chem. Biol. Drug Des., 2018, 92, 1279–1287 CrossRef CAS PubMed
- H. A. El-Sherief, G. E.-D. A. Abuo-Rahma, M. E. Shoman, E. A. Beshr and R. M. Abdel-baky, Med. Chem. Res., 2017, 26, 3077–3090 Search PubMed
- A. M. Farrag, Arch. Pharm., 2016, 349, 904–914 CrossRef CAS PubMed
- J. Rodrigues, C. Abramjuk, L. Vásquez, N. Gamboa, J. Domínguez, B. Nitzsche, M. Höpfner, R. Georgieva, H. Bäumler and C. Stephan, Pharm. Res., 2011, 28, 907–919 Search PubMed
- H. Bhojwani, K. Begwani, V. Bhor, P. Bedi, N. Balasinor, S. Raut and U. Joshi, Arch. Pharm., 2023, e2200405 CrossRef PubMed
- S. Lakshmanan, D. Govindaraj, K. Mahalakshmi, K. Thirumurugan, N. Ramalakshmi and S. A. Antony, Struct. Chem., 2021, 32, 1597–1609 CrossRef CAS
- Y. Um, S. Cho, H. B. Woo, Y. K. Kim, H. Kim, J. Ham, S.-N. Kim, C. M. Ahn and S. Lee, Bioorg. Med. Chem., 2008, 16, 3608–3615 CrossRef CAS PubMed
- V. R. Avupati, R. P. Yejella, G. Guntuku and P. Gunta, Bioorg. Med. Chem. Lett., 2012, 22, 1031–1035 Search PubMed
- D. Chinemerem Nwobodo, M. C. Ugwu, C. Oliseloke Anie, M. T. Al-Ouqaili, J. Chinedu Ikem, U. Victor Chigozie and M. Saki, J. Clin. Lab. Anal., 2022, 36, e24655 Search PubMed
- M. Ritter, R. Mastelari Martins, D. Dias and C. MP Pereira, Lett. Org. Chem., 2014, 11, 498–508 CrossRef CAS
- S. Wang, C. Li, L. Zhang, B. Sun, Y. Cui and F. Sang, Bioorg. Med. Chem., 2023, 117454 Search PubMed
- F. Farhadi, B. Khameneh, M. Iranshahi and M. Iranshahy, Phytother. Res., 2019, 33, 13–40 CrossRef CAS PubMed
- D. Gupta and D. Jain, J. Adv. Pharm. Technol. Res., 2015, 6, 114 CrossRef CAS PubMed
- R. Irfan, S. Mousavi, M. Alazmi and R. S. Z. Saleem, Molecules, 2020, 25, 5381 CrossRef CAS PubMed
- H. H. Mohammed, D. M. E. Ali, M. Badr, A. G. Habib, A. M. Mahmoud, S. M. Farhan, S. S. H. A. E. Gany, S. A. Mohamad, A. M. Hayallah and S. H. Abbas, Mol. Diversity, 2023, 27, 1751–1765 CrossRef CAS PubMed
- W.-C. Chu, P.-Y. Bai, Z.-Q. Yang, D.-Y. Cui, Y.-G. Hua, Y. Yang, Q.-Q. Yang, E. Zhang and S. Qin, Eur. J. Med. Chem., 2018, 143, 905–921 CrossRef CAS PubMed
- S. M. El-Messery, E.-S. E. Habib, S. T. Al-Rashood and G. S. Hassan, J. Enzyme Inhib. Med. Chem., 2018, 33, 818–832 CrossRef CAS PubMed
- D. Joshi and K. S. Parikh, Med. Chem. Res., 2014, 23, 1855–1864 CrossRef CAS
- Z.-H. Chen, C.-J. Zheng, L.-P. Sun and H.-R. Piao, Eur. J. Med. Chem., 2010, 45, 5739–5743 CrossRef CAS PubMed
- D. Elkhalifa, I. Al-Hashimi, A.-E. Al Moustafa and A. Khalil, J. Drug Targeting, 2021, 29, 403–419 Search PubMed
- X. Gan, Y. Wang, D. Hu and B. Song, Chin. J. Chem., 2017, 35, 665–672 CrossRef CAS
- H. A. Al-Hazam, Z. A. Al-Shamkani, N. A. Al-Masoudi, B. A. Saeed and C. Pannecouque, Z. Naturforsch., B, 2017, 72, 249–256 CrossRef CAS
- H. S. Bodiwala, S. Sabde, P. Gupta, R. Mukherjee, R. Kumar, P. Garg, K. K. Bhutani, D. Mitra and I. P. Singh, Bioorg. Med. Chem., 2011, 19, 1256–1263 CrossRef CAS PubMed
- U. Rashid, R. Sultana, N. Shaheen, S. F. Hassan, F. Yaqoob, M. J. Ahmad, F. Iftikhar, N. Sultana, S. Asghar and M. Yasinzai, Eur. J. Med. Chem., 2016, 115, 230–244 CrossRef CAS PubMed
- J. N. Domínguez, N. Gamboa de Dominguez, J. Rodrigues, M. E. Acosta, N. Caraballo and C. León, J. Enzyme Inhib. Med. Chem., 2013, 28, 1267–1273 Search PubMed
- C. León, J. Rodrigues, N. G. de Domínguez, J. Charris, J. Gut, P. J. Rosenthal and J. N. Domínguez, Eur. J. Med. Chem., 2007, 42, 735–742 CrossRef PubMed
- J. N. Domínguez, C. León, J. Rodrigues, N. Gamboa de Domínguez, J. Gut and P. J. Rosenthal, J. Med. Chem., 2005, 48, 3654–3658 Search PubMed
- L. Jiang, B. Liu, S. Hou, T. Su, Q. Fan, E. Alyafeai, Y. Tang, M. Wu, X. Liu and J. Li, Eur. J. Med. Chem., 2022, 234, 114244 CrossRef CAS PubMed
- J. Wu, J. Li, Y. Cai, Y. Pan, F. Ye, Y. Zhang, Y. Zhao, S. Yang, X. Li and G. Liang, J. Med. Chem., 2011, 54, 8110–8123 Search PubMed
- H. Inoue and R. Nakata, Endocr., Metab. Immune Disord.: Drug Targets, 2015, 15, 186–195 CrossRef CAS PubMed
- S. Dhakal, P. A. Ramsland, B. Adhikari and I. Macreadie, Int. J. Mol. Sci., 2021, 22, 9456 CrossRef CAS PubMed
- Q. Fang, L. Deng, L. Wang, Y. Zhang, Q. Weng, H. Yin, Y. Pan, C. Tong, J. Wang and G. Liang, J. Pharmacol. Exp. Ther., 2015, 355, 235–246 Search PubMed
- E. K. Abdelall, P. F. Lamie, L. S. Aboelnaga and R. M. Hassan, Bioorg. Chem., 2022, 124, 105806 CrossRef CAS PubMed
- C. Zhang, H. Yue, P. Sun, L. Hua, S. Liang, Y. Ou, D. Wu, X. Wu, H. Chen and Y. Hao, Eur. J. Med. Chem., 2021, 219, 113417 CrossRef CAS PubMed
- G. E.-D. A. Abuo-Rahma, M. Abdel-Aziz, M. A. Mourad and H. H. Farag, Bioorg. Med. Chem., 2012, 20, 195–206 CrossRef CAS PubMed
- J. Chu and C. L. Guo, Arch. Pharm., 2016, 349, 63–70 CrossRef CAS PubMed
- E. Polo, N. Ibarra-Arellano, L. Prent-Peñaloza, A. Morales-Bayuelo, J. Henao, A. Galdámez and M. Gutiérrez, Bioorg. Chem., 2019, 90, 103034 CrossRef CAS PubMed
| This journal is © The Royal Society of Chemistry 2025 |