DOI:
10.1039/D5RA00779H
(Review Article)
RSC Adv., 2025,
15, 12494-12527
Comprehensive methodologies for synthesizing tricyclic fused pyrimidoquinolines of biological relevance: a review
Received
3rd February 2025
, Accepted 14th March 2025
First published on 22nd April 2025
Abstract
Among quinoline-fused heterocycles, tricyclic pyrimidoquinoline nuclei have received considerable attention from synthetic chemists and medicinal and materials scientists over many years because they occur commonly in various biologically important natural products and potent drugs that exhibit anticancer, antibacterial, anti-inflammatory, antilipidemic, antioxidant and antimalarial activities. This study will be beneficial for medicinal chemists in the field of drug discovery to synthesize new fused tricyclic pyrimidoquinolines as potent therapeutic agents. This review provides a comprehensive compilation of the methodologies developed for the synthesis of all six known types of pyrimidoquinolines reported thus far. This article includes synthesis via solvent-free reactions, Vilsmeier–Haack reaction, Lewis and Brønsted acid catalysis, Pictet–Spengler reaction, the use of metal oxide nanoparticles as a green catalyst, multicomponent reactions (MCR), the use of L-proline as an environmentally friendly organocatalyst, aza-Wittig reaction, the use of β-cyclodextrin (β-CD) as a supramolecular catalyst, ultrasound irradiation, microwave-assisted reaction and ultraviolet light (UV365) irradiation. To the best of our knowledge, this is the first review that focuses on the synthesis of all six types of pyrimidoquinolines along with mechanistic aspects. Some medicinal applications are also mentioned.
1. Introduction
Pyrimidine and its derivatives have been studied for over a century due to their chemical and biological significance. They occur widely in nature1 as substituted and ring-fused compounds and derivatives, including nucleotides, alloxan and thiamine (vitamin B1). They are also found in many synthetic compounds, such as zidovudine and barbiturates. In medicinal chemistry, pyrimidines are well known for their therapeutic applications. One possible explanation for this activity is the presence of a pyrimidine base in uracil, cytosine, and thymine, which are essential binding blocks for nucleic acids, DNA, and RNA. The literature indicates that pyrimidines and heterocyclic annulated pyrimidines have a broad range of fascinating biological and pharmacological properties, such as antiproliferative,2 antitumor,3,4 antibacterial,5 anti-inflammatory,6 antimycobacterial,7,8 antifungal,9 anticancer,10 sedative,11 anti-HIV,12,13 antimicrobial and antitubercular,14 antimalarial,15 antineoplastic,16 and antibiotic17,18 activities.
Quinoline derivatives are an important class of N-heteroaromatic compounds used in the development of new drugs. Many theoretical and experimental studies have shown that the quinoline ring system is an important structural unit widely found in natural products, pharmaceuticals, dyestuffs, materials, agrochemicals and synthetic analogues. Furthermore, many quinolines have been shown to have various useful pharmacological and biological activities, such as antileishmanial activity,19 antifungal activity,20,21 antidiabetic activity22 anti-Alzheimer activity,23 antiasthmatic activity,24 antipsychotic activity,25 antibiotic activity,26 the presence of potent melaninconcentrating hormone 1 receptor (MCH1R) antagonists 1,27–31 antiprotozoal activity,32–37 the potential to treat lupus38,39 and neurodegenerative diseases,36 Src kinase inhibition activity,40 and antihypertensive activity.41
In the last few decades, the chemistry of fused heterocycles has remained a promising area in organic synthesis owing to their abundance in various biologically important natural products and synthetic molecules with wide applications for various purposes, such as biological materials, potent drugs, chemosensors, agrochemicals and pharmaceuticals, polymers and ligands.42–48 In particular, pyrimidine-fused quinoline derivatives are found in several drugs and bioactive natural products.49,50 Hybrid molecules having a pyrimidine ring fused with quinoline, as shown in Fig. 1, are also known as 5-deazaisoalloxazines or deazaflavins. The N-5 analogues of these molecules are known as flavins and are available in the biomolecule riboflavin and flavin adenine dinucleotide (FAD). Considering their structural resemblance to flavins, they are very useful molecules in medicinal chemistry. The compounds with a pyrimidoquinoline core demonstrate several significant and therapeutically useful biological activities, such as anti-allergic,51,52 antifolate,53 radioprotective,54 antimitotic,55 antioxidant,56,57 antiproliferative,58 anticancer,59,60 antimicrobial,61,62 antitumor,63,64 antiviral,65 analgesic66 and antimalarial67 activities. Given their tremendous applications, the design and development of new and efficient protocols for the synthesis of pyrimidoquinoline derivatives remains an important topic.
 |
| | Fig. 1 Names and structures of representative-fused pyrimidoquinolines and their naturally available analogues. | |
The six known types of pyrimidine fused to quinoline according to the sites of fusion at the quinoline substrate are pyrimido[1,2-a]quinoline, pyrimido[1,6-a]quinoline, pyrimido[4,5-b]quinoline, pyrimido[4,5-c]quinoline, pyrimido[5,4-b]quinoline and pyrimido[5,4-c]quinoline, as shown in Fig. 2. To the best of our knowledge, there is no review article about the synthetic procedures of the reported 6 types of pyrimidoquinoline derivatives, and only a few review articles highlighting the synthesis of pyrimido[5,4-c]quinolines and pyrimido-[4,5-b]quinoline have been published very recently.68–72 Therefore, we here wish to report, for the first time, this review to present a comprehensive survey of the literature on the synthetic approaches employed for the synthesis of all 6 types of pyrimidoquinolines.
 |
| | Fig. 2 Six most known types of pyrimidoquinolines ring system. | |
2. Synthesis of pyrimidoquinolines
2.1. Synthesis of pyrimido[4,5-b]quinolines
Pyrimido[4,5-b]quinolines are considered an important class of heterocyclic compounds because the pharmacological and biological properties displayed by these compounds mainly depend on the position and nature of substituents, and they also possess antiallergic,52 antimicrobial,73 anti-inflammatory74 and antitumor75 activities.
In 1982, Yamazaki and coworkers76 developed an efficient synthesis of 2,4-dioxo-pyrimido[4,5-b]-quinolines 2,3 by Vilsmeier–Haack cyclization of 6-(arylamino)uracil 1. The reactions were carried out by treating 6-(arylamino)uracil 1 with a mixture of dimethylformamide (DMF) and phosphorus oxychloride (POCl3) at room temperature under an argon atmosphere for 60 min. The desired 2,4-dioxopyrimido[4,5-b]quinolines 2a,b and 3a,b were obtained in very good to excellent yields (Scheme 1).
 |
| | Scheme 1 Synthesis of 2,4-dioxopyrimido[4,5-b]quinolines 2a,b and 3a,b from 6-(arylamino)-uracil 1. | |
On heating iminophosphorane 4 with N,N′-dialkylbarbituric acids 5a–e in pyridine under reflux for 48 h, pyrimido[4,5-b]quinoline derivatives 6a–e were obtained in 35–53% yields (Scheme 2).77
 |
| | Scheme 2 Synthesis of pyrimido[4,5-b]quinolines 6a–e via the reaction of iminophosphorane 4 with N,N′-dialkylbarbituric acids 5a–e. | |
Selvi et al.61 developed eco-friendly, solvent free and microwave-induced techniques for the synthesis of a series of 2-oxopyrimido[4,5-b]- 8a–g and 2-thioxopyrimido[4,5-b]quinolines 9a–g as antibacterial and antifungal agents. The condensation of 2-chloro-3-formylquinolines 7a–g with urea (or thiourea) in the presence of p-toluenesulfonic acid (PTSA) as a catalyst under microwave heating for 5 min. Afforded the required 2-oxopyrimido[4,5-b]- 8a–g and 2-thioxo-pyrimido[4,5-b]quinolines 9a–g in good to excellent yields (Scheme 3).
 |
| | Scheme 3 Green method for the synthesis of 2-oxopyrimido[4,5-b]- 8a–g and 2-thioxo-pyrimido[4,5-b]quinolines 9a–g under microwave heating. | |
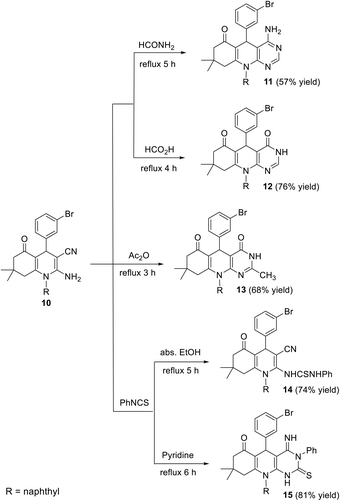
The synthesis of a new series of pyrimido[4,5-b]quinolines 11–15 was reported, starting from 2-amino-4-(3-bromophenyl)-7,7-dimethyl-1-(naphthalen-1-yl)-5-oxo-1,4,5,6,7,8-hexa-hydroquinoline-3-carbonitrile (10).78 When compound 10 was heated with formamide at reflux temperature for 5 h, 4-amino-5-(3-bromophenyl)-8,8-dimethyl-10-(naphthalen-1-yl)-5,8,9,10-tetrahydropyrimido[4,5-b]quinolin-6(7H)-one (11) was obtained in 57% yield. Heating compound 10 with formic acid for 4 h caused intramolecular cyclization to give the corresponding 5-(3-bromophenyl)-8,8-dimethyl-10-(naphthalen-1-yl)-5,8,9,10-tetrahydro-pyrimido[4,5-b]quinoline-4,6(3H,7H)-dione (12) in 76% yield. When compound 10 was made to reflux with acetic anhydride for 3 h, the 5-(3-bromo-phenyl)-2,8,8-trimethyl-10-(naphthalen-1-yl)-5,8,9,10-tetrahydropyrimido[4,5-b]quinoline-4,6(3H,7H)-dione (13) was isolated in 68% yield. In addition, the behavior of 10 towards phenyl isothiocyanate under different conditions was investigated. Thus, the reaction of equimolar amounts of 10 and phenyl isothiocyanate in boiling absolute ethanol for 5 h afforded 1-(4-(3-bromophenyl)-3-cyano-7,7-dimethyl-1-(naphthalen-1-yl)-5-oxo-1,4,5,6,7,8-hexahydroquinolin-2-yl)-3-phenylthiourea (14) in 74% yield. However, the reaction of 10 with phenyl isothiocyanate in absolute pyridine under reflux conditions for 6 h led to the formation of 5-(3-bromophenyl)-4-imino-8,8-dimethyl-10-(naphthalen-1-yl)-3-phenyl-2-thioxo-2,3,4,5,7,8,9,10-octahydropyrimido[4,5-b]quinolin-6(1H)-one (15) in 81% yield (Scheme 4).
 |
| | Scheme 4 Synthesis of new derivatives of tetrahydropyrimido[4,5-b]quinolines 11–15. | |
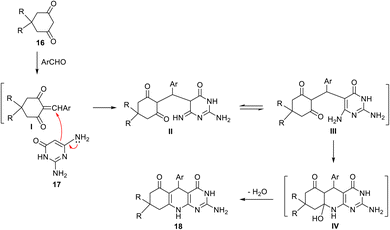
In 2005, a clean and expeditious microwave-mediated one-pot methodology for the synthesis of a new series of pyrimido[4,5-b]quinolines was reported by Tu and his coworkers.79 A mixture of aromatic aldehyde, cyclic 1,3-dicarbonyl compound 16a,b and 2,6-diamino-pyrimidin-4(3H)-one (17) in glycol was irradiated in a microwave at 198 °C (300 W) for 4–7 min to provide different linear 2-amino-5-aryl-8-substituted-5,8,9,10-tetrahydropyrimido[4,5-b]-quinoline-4,6(3H,7H)-diones 18a–l (Scheme 5). The protocol in the absence of a catalyst has the advantage of short reaction time, excellent yield (90–95%) and an environmentally friendly technique. A plausible mechanism for the formation of 18 is given in Scheme 6. The reaction may occur via a condensation, addition, cyclization, or elimination mechanism. The initial condensation between cyclic 1,3-dicarbonyl compound 16 and aldehyde afforded the corresponding 2-arylidene-5,5-dimethyl-1,3-cyclohexane-dione I. Then, Michael addition between I and 2,6-diaminopyrimidin-4-one 17 furnished the intermediate II, which isomerized to III. Intramolecular dehydration of IV yielded the desired tricyclic product 18.
 |
| | Scheme 5 Microwave-assisted synthesis of 2-amino-5-aryl-8-substituted-5,8,9,10-tetrahydro-pyrimido[4,5-b]quinoline-4,6(3H,7H)-diones 18a–l. | |
 |
| | Scheme 6 Plausible mechanism for the formation of 2-amino-5-aryl-8-substituted-5,8,9,10-tetrahydropyrimido[4,5-b]quinoline-4,6(3H,7H)-diones 18a–l. | |
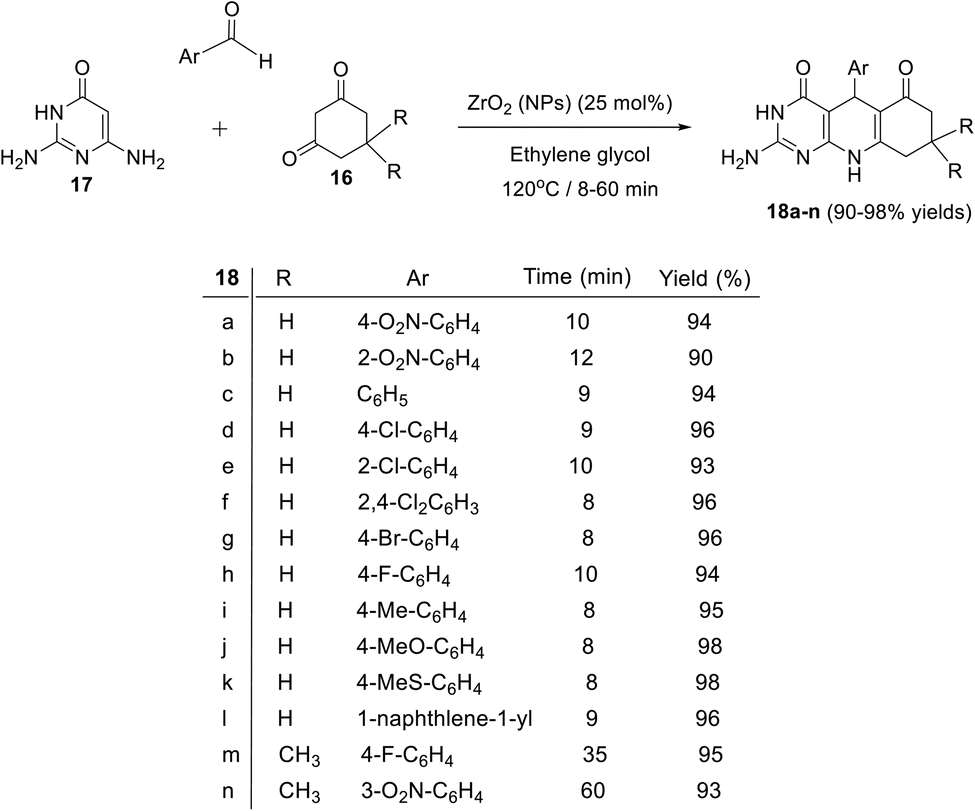
In 2017, Mamaghani et al.80 described an ecofriendly and efficient multi-component reaction for the green synthesis of 2-amino-5-aryl-pyrimido[4,5-b]quinolinediones using metal oxide nanoparticles [ZrO2 (NPs)] as a green catalyst. On heating equimolar amounts of 2,6-diamino-pyrimidin-4(1H)-one (17), aromatic aldehydes and 1,3-cyclohexane-dione or 5,5-dimethyl-1,3-cyclohexanedione (16) in the presence of ZrO2 (NPs) (25 mol%) in ethylene glycol at 120 °C for 8–60 min, the desired 2-amino-5-aryl-pyrimido[4,5-b]quinoline-4,6(1H,5H,7H,10H)-diones 18a–n were obtained in excellent yields (90–98%) (Scheme 7).
 |
| | Scheme 7 Synthesis of 2-amino-5-aryl-pyrimido[4,5-b]quinolinediones 18a–n via ZrO2 (NPs) catalyzed reaction. | |
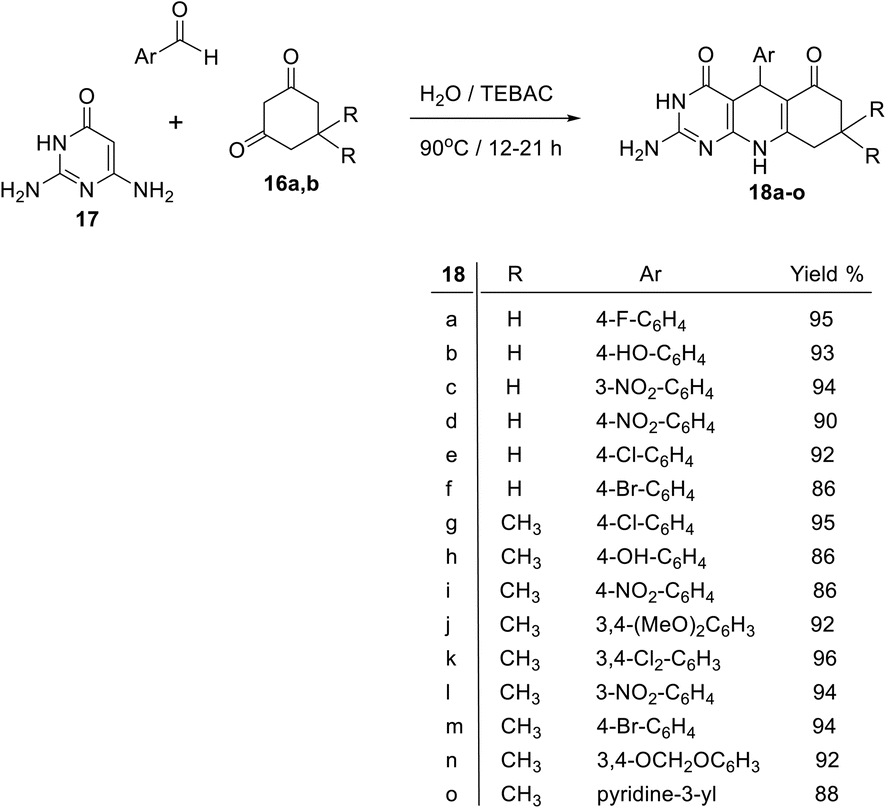
An efficient, clean, one-pot, three-component reaction of 2,6-diaminopyrimidin-4(3H)-one (17), aromatic aldehyde and 1,3-cyclohexanedione (16a) or 5,5-dimethyl-1,3-cyclo-hexanedione (16b) in water in the presence of triethylbenzylammonium chloride (TEBAC) as a catalyst under conventional heating conditions at 90 °C for 12–21 h produced the pyrimidine fused quinoline (PFQ), namely 2-amino-5-aryl-8,9-dihydropyrimidino[4,5-b]quinoline-4,6-(1H,3H,5H,10H)-diones 18a–o, in high yields (Scheme 8). This new method has the advantages of mild reaction conditions, the use of inexpensive reagents, easy work-up, high yields, and environmentally friendly procedures.81
 |
| | Scheme 8 Clean and one-pot synthesis of 2-amino-5-aryl-8,9-dihydropyrimidino[4,5-b]-quinoline-4,6-(1H, 3H, 5H, and 10H)-diones 18a–o. | |
In 2007, Wilson et al.82 described a short and efficient synthesis of 10-aryl-7-nitro-pyrimido[4,5-b]quinoline-2,4(3H,10H)-diones 23a–c. The starting 6-chlorouracil (20) was synthesized by heating 2,4,6-trichloropyrimidine (19) with a solution of sodium hydroxide under reflux for 1 h. The next stage involves a two-step convergent approach where 6-chloro-uracil (20) was fused at the melt temperature (170 °C for 20 min) with the appropriate arylamine, followed by heating the resulting 6-N-aryl-aminouracils 21 with 2-chloro-5-nitrobenzaldehyde (22) in DMF at 110 °C for 90 min to give 10-(3-chlorophenyl)-7-nitro-10H-pyrimido[4,5-b]-quinoline-2,4-dione (23a), 10-(4-chlorophenyl)-7-nitro-10H-pyrimido[4,5-b]quinoline-2,4-dione (23b) and 10-(4-methylphenyl)-7-nitro-10H-pyrimido-[4,5-b]quinoline-2,4-dione (23c) in 26%, 22% and 79% yields, respectively, in two steps (Scheme 9).
 |
| | Scheme 9 Efficient synthesis of 10-aryl-7-nitro-pyrimido[4,5-b]quinoline-2,4(3H and 10H)-diones 23a–c. | |
A facile synthesis of new 6,7,8,9-tetrahydropyrimido[4,5-b]quinoline derivatives 28–30, a family of new pyrimido[4,5-b]quinolines with potential antifungal activity, was developed by Elkholy and Morsy in 2006.83 Refluxing a solution of cyclohexanone (24) and 2-benzylidenemalononitrile (25) in absolute ethanol containing an excess of ammonium acetate for 3 h yielded the 2-amino-4-phenyl-5,6,7,8-tetrahydroquinoline-3-carbonitrile (26). Heating 26 with dimethylformamide dimethylacetal (DMF–DMA) in dioxane at reflux temperature for 4 h afforded 2-dimethylaminomethelenimino-4-phenyl-5,6,7,8-tetrahydroquinoline-3-carbonitrile (27). Reacting 27 with hydrazine hydrate in refluxing absolute ethanol for 4 h produced 3-amino-4(3H)-imino-5-phenyl-6,7,8,9-tetrahydropyrimido[4,5-b]quinoline (28). The reactivity of compound 26 towards urea, thiourea and carbon disulfide was also investigated. Thus, heating a mixture of 26 with urea (or thiourea) and sodium ethoxide in absolute EtOH at reflux temperature for 6 h gave 4-amino-5-phenyl-6,7,8,9-tetrahydro-pyrimido[4,5-b]quinoline-2(1H)-one/thione derivatives 29a,b. However, reacting 26 with carbon disulphide in dry pyridine under reflux conditions for 6 h afforded the corresponding 5-phenyl-6,7,8,9-tetrahydropyrimido[4,5-b]quinoline-2,4(1H,3H)-dithione (30) (Scheme 10). The yields of the products were not reported.
 |
| | Scheme 10 Facile synthesis of new 6,7,8,9-tetrahydropyrimido[4,5-b]quinolines 28–30. | |
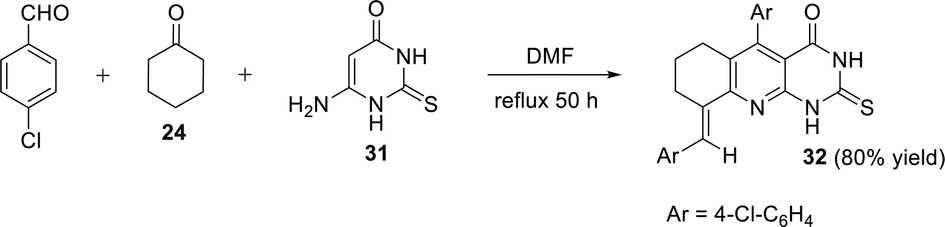
A one-pot synthesis of 5-(4-chlorophenyl)-9-(4-chlorophenyl-methylene)-2-thioxo-6,7,8,9-tetrahydropyrimido[4,5-b]quinolin-4-one (32) was reported by El-Gazzar et al. in 2009.84 This synthetic approach proceeded by heating a mixture of 4-chlorobenzaldehyde, cyclohexanone (24) and 6-amino-2-thioxo-2,3-dihydropyrimidin-4(1H)-one (31) in DMF at reflux temperature for 50 h to give 32 in 80% yield (Scheme 11). In the same year, they developed a new synthetic strategy for the preparation of 32 by refluxing a solution of 6-aminothiouracil (31) and α,β-unsaturated ketone 33 in DMF for 30 h. The respective tricyclic 32 was obtained in 83% yield (Scheme 12).85
 |
| | Scheme 11 One-pot synthesis of 5-(4-chlorophenyl)-9-(4-chlorophenyl-methylene)-2-thioxo-6,7,8,9-tetrahydropyrimido[4,5-b]quinolin-4-one (32). | |
 |
| | Scheme 12 New synthetic strategy for the synthesis of 9-(4-chlorobenzylidene)-5-(4-chloro-phenyl)-2-thioxo-2,3,6,7,8,9-hexahydropyrimido[4,5-b]quinolin-4(1H)-one (32). | |
Dow et al. described a general synthetic route for the synthesis of pyrimido[4,5-b]quinolin-4(3H)-ones 36a–e and 38a,b, which are potent and selective inhibitors of the tyrosine-specific kinase activity associated with pp6Oc-src.86 The reactions were performed in three steps, starting with o-nitrobenzaldehyde 34. First, condensation of o-nitrobenzaldehyde 34 with ethyl cyanoacetate or cyanoacetamide under basic conditions was followed by reductive cyclization, which gave the corresponding 2-aminoqninolines 35a,b. Reaction of ethyl 2-amino-quinoline-3-carboxylates 35a with carboxamides at elevated temperatures; alternatively, treatment of carboxamides 35b with an ortho ester in the presence of an acid catalyst provided fully-aromatized tricyclic pyrimido[4,5-b]quinolin-4(3H)-ones 36a–e. Synthesis of the corresponding N-3 functionalized analogs was performed by the condensation of 35a with dimethylformamide dimethylacetal (MDF–DMA) to give the corresponding ethyl 2-(((dimethylamino)methylene)amino)quinoline-3-carboxylate 37. When compound 37 was reacted with the appropriate amine, it underwent intramolecular cyclization to give N-3 substituted pyrimido[4,5-b]quinoline-4-ones 38a,b (Scheme 13). The yields of the products were not reported.
 |
| | Scheme 13 General synthetic route for the synthesis of 5,10-dihydropyrimido[4,5-b]quinolin-4(3H)-ones 36a–e and 38a,b. Reagents and reaction conditions: (a) CNCH2CO2Et or CNCH2CONH2/piperdine/EtOH, reflux; (b) zinc or iron dust AcOH/reflux; (c) R3-CONH2/over 150 °C or R3-CH(OEt)3/PTSA/reflux; (d) (MeO)2CHNMe2/PTSA/toluene/reflux; (e) R4-NH2/EtOH/reflux. | |
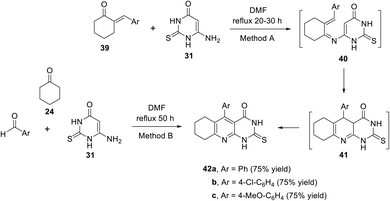
In 2008, El-Gazzar and his coworkers66 developed a new approach for the synthesis of a novel series of pyrimido[4,5-b]quinolines 42a–c in good yields. The reaction was accomplished by heating a mixture of arylidene cyclohexanone 39 and 6-amino-thiouracil 31 in DMF under reflux for 20–30 h to give 5-aryl-2-thioxo-2,3,6,7,8,9-hexahydro-1H,4H-pyrimido-[4,5-b]quinoline-4-ones 42a–c via intermediacies 40 and 41 (Method A). Alternatively, compound 42 could also be obtained by a one-pot synthesis by refluxing a solution of 6-aminothiouracil (31), cyclohexanone (24) and aromatic aldehydes in DMF for 50 h (Method B) (Scheme 14).
 |
| | Scheme 14 Synthesis of a novel series of pyrimido[4,5-b]quinolines 42a–c. | |
In 2010, Alqasoumi and his workers63 investigated the reaction of 2-amino-quinoline-3-carbonitrile derivatives 43, bearing biologically active sulfonamide with isothiocyanates, and they found that the type of products depends on the reaction conditions. Thus, the nucleophilic reaction of compound 43 on the highly positive carbon of the isothiocyanates (RNCS) in dry pyridine under reflux conditions for 1 h afforded the corresponding thioureido derivatives 44 (Scheme 15), while a 28 h reaction time gave the novel tricyclic system pyrimido[4,5-b]quinoline derivatives 45a–e in one step. Alternatively, compound 45 could also be obtained by boiling compound 44 in dry pyridine for 24 h (Scheme 15). Product 45 exhibited higher activity with IC50 values (5.5, 6.9, and 7 mg ml−1) when compared with doxorubicin as a reference drug (IC50 value of 38 mg ml−1).
 |
| | Scheme 15 Synthesis of novel pyrimido[4,5-b]quinoline derivatives 45a–e. | |
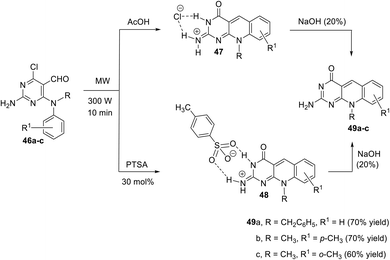
An efficient microwave-assisted synthesis of a series of pyrimido[4,5-b]quinolines 49a–c, flavin analogues, via intramolecular cyclization of 2,4-diamino-6-chloro-pyrimidine-5-carbaldehydes 46a–c, was reported by Trilleras et al.87 When 2,4-diamino-6-chloro-pyrimidine-5-carbaldehydes 47a–c were heated with an excess of acetic acid under microwave irradiation (maximum power 300 W for 10 min at a controlled temperature of 300 °C) using a focused microwave reactor, they underwent intramolecular cyclo-condensation to furnish the 4-oxo-4,10-dihydropyrimido[4,5-b]quinolin-2(3H)-iminium chlorides 47. To avoid substituting the chloro atom to maintain the possibility of adding molecular diversity and complexity to the molecule, the same reaction was carried out using an excess of 4-toluenesulfonic acid (PTSA). Thus, compounds 46a–c (1 mmol) were reacted with an excess of PTSA monohydrate (1.3 mmol) under the same conditions described above. Reaction products were characterized from the spectroscopic data and X-ray analysis as 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 salt 2-amino-pyrimido[4,5-b]quinolin-4(10H)-one:PTSA 48. The treatment of salts 47 and 48 with aqueous NaOH (20%) was carried out to directly give the neutral tricyclic ring system 2-amino-pyrimido[4,5-b]quinolin-4(10H)-one derivatives 49a–c in good yields (Scheme 16).
:1 salt 2-amino-pyrimido[4,5-b]quinolin-4(10H)-one:PTSA 48. The treatment of salts 47 and 48 with aqueous NaOH (20%) was carried out to directly give the neutral tricyclic ring system 2-amino-pyrimido[4,5-b]quinolin-4(10H)-one derivatives 49a–c in good yields (Scheme 16).
 |
| | Scheme 16 Efficient microwave-assisted synthesis of a series of 2-amino-pyrimido[4,5-b]-quinolin-4(10H)-ones 49a–c. | |
A new synthetic approach to polyfunctionally substituted pyrimido[4,5-b]quinoline-2,4(1H,3H)-diones 52 via a three-component one-pot reaction of aromatic amines 50, barbituric acid (51) and aromatic aldehydes is reported.88 The use of commercially available aniline derivatives allowed the facile syntheses of pyrimido[4,5-b]quinolinediones 52 to be substituted in all the positions on the benzene ring with electron donor or electron withdrawing groups. On heating an equimolar mixture of aniline 50, compound 51 and aromatic aldehydes in AcOH at reflux temperature, a wide range of the desired tricyclic pyrimido[4,5-b]quinoline-2,4(1H,3H)-diones 52a–m were obtained in 25–77% yields (Scheme 17).
 |
| | Scheme 17 One-pot procedure for the synthesis of polyfunctionally substituted pyrimido-[4,5-b]quinoline-2,4(1H,3H)-diones 52a–m. | |
Ghorab et al.89 reported the synthesis of new pyrimido[4,5-b]quinoline derivatives 54, 56, 57 using 4-(2-amino-3-cyano-4-(4-fluorophenyl)-7,7-dimethyl-5-oxo-5,6,7,8-tetrahydroquinolin-1(4H)-yl)benzenesulfonamide (53) as the starting material. Refluxing a solution of 53 in formic acid for 5 h furnished 4-(5-(4-fluorophenyl)-8,8-dimethyl-4,6-dioxo-3,4,6,7,8,9-hexahydropyrimido[4,5-b]quinolin-10(5H)-yl)benzenesulfonamide (54) in 79% yield. However, heating 53 with acetic anhydride and formamide at reflux temperature gave the new fused pyrimido[4,5-b]quinolines 56 (via the intermediacy of 55) and 57 in 75% and 45% yields, respectively (Scheme 18). The products showed significant anticancer activity.
 |
| | Scheme 18 Synthesis of new pyrimido[4,5-b]quinoline derivatives 54, 56, and 57. | |
In 2011, Chandra and his coworkers90 developed a simple and rapid synthesis of 2-amino-3H-pyrimido[4,5-b]quinolin-4(3H)-ones 60a–i via t-BuOK-catalyzed cyclization of 2-chloroquinoline-3-carbonitriles 58 with guanidine hydrochloride (59) in a very short reaction time in good yields. On heating 2-chloroquinoline-3-carbonitriles 58 (1 equiv.) with guanidine hydrochloride (59) (1 equiv.) in the presence of t-BuOK (0.5 equiv.) in EtOH at 90 °C for 5 min, the cyclized products 60a–i were obtained in 80–90% yields (Scheme 19). The electron-donating and -withdrawing substituents at the benzene ring of the quinoline moiety show better yields of the cyclized products.
 |
| | Scheme 19 Simple and rapid synthesis of 2-amino-3H-pyrimido[4,5-b]quinolin-4(3H)-ones 60a–i. | |
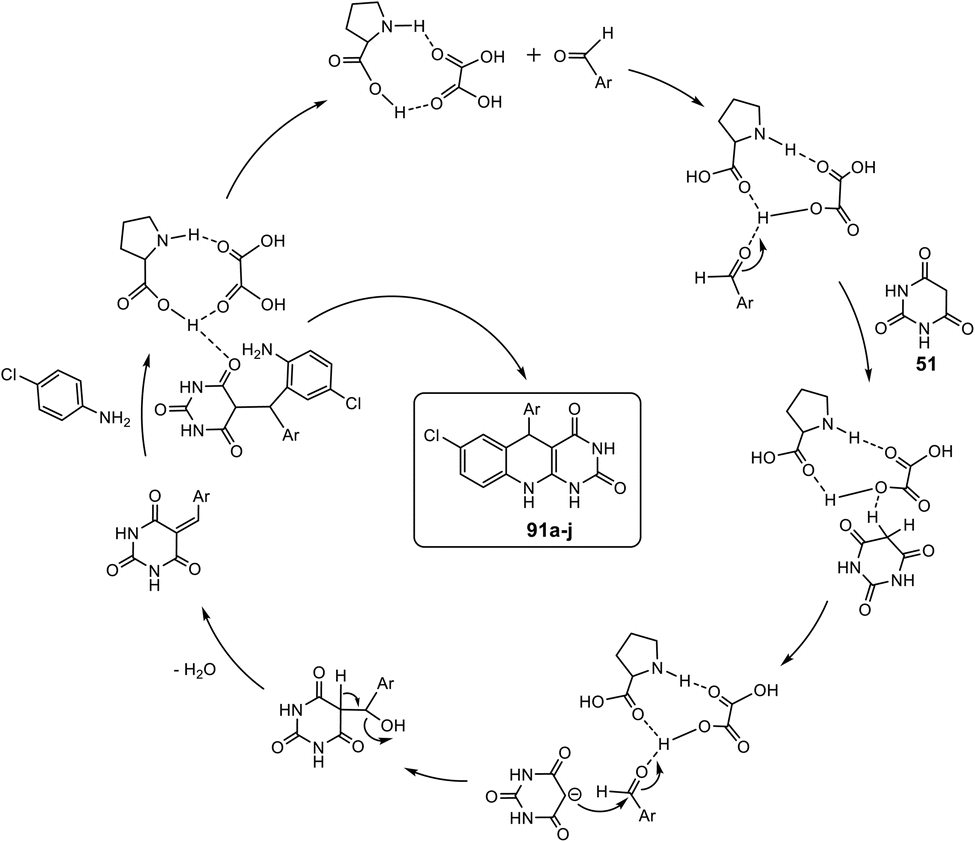
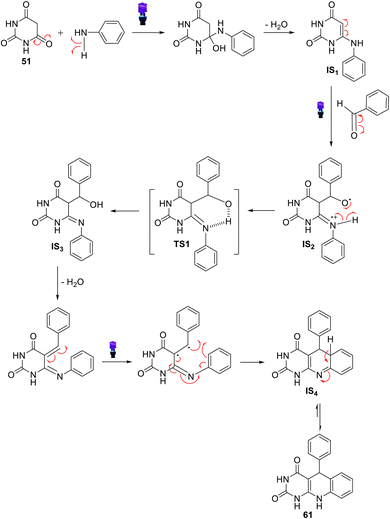
A green, convenient and efficient procedure for the regioselective synthesis of 5-aryl-pyrimido-[4,5-b]quinoline-diones 61a–k using the three-component coupling reaction involving aromatic amines, aldehydes, and barbituric acids 52a,b was reported by Khalafi-Nezhad et al. in 2012.91 This protocol was accomplished efficiently using L-proline, an environmentally friendly organocatalyst, in an aqueous medium to produce the desired tricyclic products in high yields. The MCRs showed good regioselectivity, and computational studies were used to investigate selectivity. Thus, when a mixture of aromatic amines, barbituric acid (51) or thiobarbituric acid (51a), aromatic aldehydes and L-proline (20 mol%), as catalysts, in refluxing H2O was stirred for 10–20 h, the 5-aryl-pyrimido[4,5-b]quinolines 61a–k were obtained in high yields (82–92%) (Scheme 20). As is clearly shown in Scheme 20, this multicomponent route can be used for both aromatic aldehydes with electron-donating and electron-withdrawing groups. Similarly, heterocyclic aldehydes can be used under optimized conditions. Furthermore, a wide range of aromatic amines were applied successfully in this reaction with excellent results.
 |
| | Scheme 20 Green, convenient and efficient synthesis of 5-aryl-pyrimido[4,5-b]quinoline-diones 61a–k. | |
The proposed mechanism to explain the formation of 61 is shown in Scheme 21. First, the aldehyde is activated by L-proline. Simultaneously, L-proline acts as a Brønsted acid/base, assisting the enolization of barbituric acids 51, which is subsequently reacted with adduct I to generate intermediate II. Intermediate II can lose one molecule of L-proline, so barbiturate III, as an unsaturated carbonyl compound, is formed. However, L-proline can activate adduct III to produce intermediate IV, which undergoes a reaction with aniline derivatives to afford intermediate V. Then, intermediate V undergoes an intramolecular cyclization reaction to form the desired product 61. Although L-proline plays a key role in this reaction, it does not affect the formation of a chiral center, so stereoselectivity does not occur.
 |
| | Scheme 21 Plausible mechanism for the one-pot three-component synthesis of 5-aryl-pyrimido[4,5-b]quinoline-diones 61a–k using L-proline as a catalyst. | |
In 2014, Mosslemina and his coworkers92 reported a green and efficient synthesis of 5-aryl-(1H,3H,5H,10H)-pyrimido[4,5-b]quinoline-2,4-diones 61a–l via a one-pot, three-component reaction of anilines, barbituric acids 51 and aldehydes catalyzed by 1,4-diaza-bicyclo[2.2.2]octane (DABCO) in H2O. This synthetic approach proceeded by heating a mixture of equimolar amounts of anilines, (thio)barbituric acids 51 and aldehydes in the presence of a catalytic amount of DABCO (15 mol%) in H2O at a reflux temperature for 12 h to give the new derivatives of 5-aryl-(1H,3H,5H,10H)-pyrimido[4,5-b]quinoline-2,4-diones 61a–l in 83–96% yields (Scheme 22). Using microwave heating (at 90 °C, 400 W), reaction times were shortened from 12 h to under a minute (30 s) and yields were generally higher (Scheme 22). A suggested mechanism for the formation of 61 is illustrated in Scheme 23. DABCO initially activated the aldehyde. Simultaneously, DABCO as Brønsted acid/base assists the enolization of the barbituric acids 51, which is subsequently reacted with adduct I to give intermediate II. The latter intermediate can lose one molecule of H2O, so barbiturate III, as an unsaturated carbonyl compound, is formed. However, DABCO can activate adduct III to generate intermediate IV to undergo a reaction with aniline, resulting in the production of adduct V. DABCO could act as a nucleophilic catalyst, reacting with adduct V to produce intermediate VI. Subsequently, intermediate VI undergoes an intramolecular cyclization reaction to afford intermediate VII. DABCO can assist VII to lose one molecule of H2O and give the final products 61a–l.
 |
| | Scheme 22 Green and efficient synthesis of new 5-aryl-(1H,3H,5H,10H)-pyrimido[4,5-b]-quinoline-2,4-diones 61a–l. | |
 |
| | Scheme 23 Plausible reaction mechanism for DABCO-catalyzed synthesis of 5-aryl-pyrimido[4,5-b]quinoline-diones 61. | |
Recently, an efficient, fast and straightforward protocol towards the construction of various new derivatives of 5-aryl-pyrimido[4,5-b]quinolinedione 61 via a three-component condensation of electronically different aromatic amines, aromatic aldehydes and barbituric acid (51) promoted by β-cyclodextrin (β-CD), as a supramolecular catalyst, in water has been developed, for the first time, by Reddy and his group.93 This strategy provides a benign method for building pyrimido-[4,5-b]quinolinediones in an environmentally safer reaction medium. The reactions were carried out by heating a mixture of equimolar amounts of aromatic amines, aromatic aldehydes and barbituric acid (51) in H2O in the presence of β-cyclodextrin (β-CD), as a supramolecular catalyst, at 80 °C for 4–5 h to afford new 5-aryl-pyrimido[4,5-b]quinoline-diones 61a–l in good to excellent yields (Scheme 24). A plausible mechanism to account for the formation of 61 is suggested in Scheme 25. A hydrophobic environment of the catalyst (β-CD facilitated the reaction by forming the β-CD-aldehyde complex). This complex reacts with barbituric acid (51) to give enone I. The latter reacts with aromatic amines via the Michael addition reaction, followed by intramolecular cyclization, giving the corresponding 5-aryl-pyrimido[4,5-b]quinolinedione 61.
 |
| | Scheme 24 Green approach for the synthesis of 5-aryl-pyrimido[4,5-b]quinolinediones 61a–l. | |
 |
| | Scheme 25 Plausible mechanism for the β-CD catalyzed synthesis of pyrimido[4,5-b]-quinoline-diones 61a–l. | |
In 2018, Nongthombam and his coworkers94 developed a green, highly efficient and environmentally benign UV365 light-mediated synthesis of several biologically important pyrimido[4,5-b]quinoline-2,4-diones 61 from barbituric acid (51), aromatic amines and aromatic aldehyde. Thus, when a mixture of barbituric acid (51), aryl amines and aryl aldehydes in water–glycerol (1:1) was irradiated by long ultraviolet light (UV365) for 60–90 min, the desired pyrimido[4,5-b]quinoline-2,4-diones 61a–x were obtained in 85–98% yields (Scheme 26). This synthetic approach operates at room temperature under direct irradiation from a UV365 light source in a water–glycerol medium and in the absence of a photocatalyst. This reported method shows several merits, such as clean reaction conditions, chromatography-free synthesis, and the use of an inexpensive water–glycerol solvent system, which is also environmentally friendly and results in high yields. The proposed mechanism to explain the formation of 61a–x is shown in Scheme 27.
 |
| | Scheme 26 Synthesis of pyrimido[4,5-b]quinoline-2,4-diones 61a–x under UV365 irradiation. | |
 |
| | Scheme 27 Suggested mechanism for UV365-aided synthesis of 61a–x via a free radical pathway. | |
In the same year, Nongthombam and Nongkhlaw95 reported an efficient, economical and environment benign protocol for the synthesis of 5-aryl-2-thioxo-2,3,5,10-tetrahydropyrimido[4,5-b]quinolin-4(1H)-ones 61a–g utilizing glutathione on super-paramagnetic iron-oxide nanoparticle (SPION) (SPION@glutathione) as a nano-organo-catalyst and ultrasound irradiation as an energy source. When a mixture of thiobarbituric acid (51a), aniline derivatives, aryl aldehyde, and SPION@glutathione (10 mg) in H2O was ultrasonicated for 15 min, the desired tricyclic 5-aryl-2-thioxo-2,3,5,10-tetrahydropyrimido[4,5-b]quinolin-4(1H)-ones 61a–g were obtained in 92–98% yields (Scheme 28). In this strategy, the nano-organocatalyst (SPION@glutathione) was successfully recovered and reused without any loss in its activity.
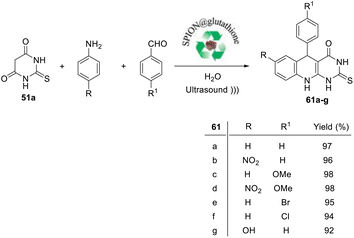 |
| | Scheme 28 Synthesis of 5-aryl-2-thioxo-2,3,5,10-tetrahydropyrimido[4,5-b]quinolin-4(1H)-ones 61a–g using SPION@ glutathione as a nano-organocatalyst under ultrasonic conditions. | |
In 2023, Dai and his group96 reported a one-pot alcohol oxidation/three-component green synthesis of new 5-aryl-pyrimido[4,5-b]quinolinedione derivatives 61, for the first time, utilizing a bifunctional nanomolecular catalyst L-Pro-Mn-Anderson by grafting L-proline onto an Mn-Anderson POM (polyoxometalate). This synthetic method involves heating a mixture of alcohol (1 mmol), catalyst (L-Mn-Anderson POM, 0.5 mol%) and aqueous H2O2 (30%, 3 mmol) in H2O at 90 °C for 12–15 h to give the corresponding aldehyde, followed by in situ reaction with aromatic amine compound (1 mmol) and barbituric acid (51) (1 mmol) in H2O at a reflux temperature for 12–15 h to afford 61a–d in excellent yields (85–93%) (Scheme 29). A postulated reaction mechanism for the formation of 61 is presented, as shown in Scheme 30. First, the Mn-Anderson skeleton of the L-Pro modified compound catalyzes the oxidation of alcohol to aldehyde. The pyrrolidine grafted on the POM may activate the aldehyde to form intermediate I. Meanwhile, L-Pro-Mn-Anderson Brønsted acid/base helps the enolization of barbituric acid 51 and then reacts enolate with intermediate I to give intermediate II. 5-(4-Fluorobenzylidene)-pyrimidine-2,4,6-trione (61A) is generated (the compound was successfully isolated) after removing L-Pro-Mn-Anderson from intermediate II. Subsequently, L-Pro-Mn-Anderson can activate 61A to generate intermediate III to facilitate the reaction with 4-fluoroaniline, resulting in the formation of intermediate IV. Finally, the latter intermediate IV undergoes an intramolecular cyclization reaction to afford the target product 61.
 |
| | Scheme 29 L-Pro-Mn-Anderson catalyzed a one-pot alcohol oxidation/three-component condensation reaction from alcohols to 5-aryl-pyrimido[4,5-b]quinoline-diones 61. | |
 |
| | Scheme 30 Proposed mechanism for the synthesis of 5-aryl-pyrimido[4,5-b]quinoline-diones 61 via a one-pot alcohol oxidation/three-component condensation reaction. | |
Khurana et al.97 described a new, simple, environmentally benign one-pot and three-component protocol for the synthesis of novel pyrimido[4,5-b]quinolines 63 using indium trichloride (InCl3) as a catalyst in water. The condensation of 6-amino-1,3-dimethyluracil (62), 5,5-dimethylcyclohexane-1,3-dione (16b) and various aromatic aldehydes in water in the presence of InCl3 (20 mol%), as a catalyst, at reflux temperature for 60–90 min afforded a series of novel pyrimido[4,5-b]quinolines 63a–f in excellent yields (Scheme 31). The advantages of this method include operational simplicity, the reusability of the catalyst and high yields.
 |
| | Scheme 31 Indium trichloride catalyzed the synthesis of pyrimido[4,5-b]quinolines 63a–f. | |
In 2014, Tabatabaeian and his group98 described a novel, convenient, efficient and environmentally benign one-pot three-component coupling reaction of 6-amino-1,3-dimethyluracil (62), aromatic aldehydes and cyclic 1,3-diketones 16a,b for the synthesis of bioactive pyrimido[4,5-b]quinoline derivatives 63 utilizing RuCl3·xH2O as a reusable homogenous catalyst in the absence of any organic solvents and H2O as a green solvent. The main advantages of this route are (i) high atom economy of the reaction by avoiding the use of toxic organic solvents, (ii) clean and simple work-up for the isolation and purification of products using non-chromatographic methods, (iii) short reaction time, (iv) energy saving by employing multicomponent reactions, (iv) excellent yields, (v) environmentally benign procedures, and (vi) reusability of the catalyst. Several derivatives of pyrimido[4,5-b]quinolines 63 showed extremely high levels of antibacterial activity.
In this investigation, 6-amino-1,3-dimethyluracil (62) was used as an important partner in the synthesis of tricyclic fused rings. This compound provided C5 and C6 carbons in products 63. 1,3-Cyclohexanedione (16a) or dimedone (16b) as a cyclic ketone with strong nucleophilic properties provided C2 and C3 carbons in products 63. The reaction was carried out by heating equimolar amounts of 6-amino-1,3-dimethyluracil (62), aromatic aldehydes and cyclic 1,3-diketones 16a,b in deionized H2O in the presence of a catalytic amount of RuCl3·xH2O (3 mol%) either via long reflux or by short time ultrasound (US) irradiations (40 kHz, 40 °C) to furnish the desired tricyclic pyrimido[4,5-b]quinolines 63a–r. This reaction under ultrasound (US) irradiation gave an excellent yield of products and increased the reaction rate (Scheme 32). It was found that the electronic nature of the substituents on the phenyl ring of the applied aromatic aldehydes significantly affected this reaction. Aromatic aldehydes with electron-withdrawing groups (EWG) (such as nitro and halide groups) reacted at a faster rate and in better yields compared to electron-donating groups (EDG) (such as methoxy and methyl groups). When an aliphatic aldehyde, such as acetaldehyde, reacted with cyclic ketone and 6-amino-1,3-dimethyluracil under the same reaction conditions mentioned above, the desired product, pyrimido[4,5-b]quinoline 63, was not obtained.
 |
| | Scheme 32 RuCl3·xH2O catalyzed the synthesis of pyrimido[4,5-b]quinolines 63a–r. | |
In 2015, Mohammadi et al.99 described a green, efficient and convenient method for the synthesis of pyrimido[4,5-b]quinolines via a three-component one-pot cyclo-condensation of 6-amino-1,3-dimethyluracil (62), aromatic aldehydes and cyclic 1,3-dicarbonyl compounds 16a,b in the presence of a catalytic amount of 1,3-disulfonic acid imidazolium hydrogen sulfate [dsim]HSO4 as an environmentally benign and reusable catalyst. The reactions were carried out by heating a mixture of 6-amino-1,3-dimethyluracil (62), aromatic aldehydes and cyclic 1,3-dicarbonyl compounds 16a,b in EtOH in the presence of a catalytic amount of [dsim]HSO4 at 70 °C for 15–35 min to afford the polyfunctionalized pyrimido[4,5-b]quinolines 63a–q in 85–92% yields (Scheme 33). The notable advantages of the present methodology are mild conditions, excellent yields of the products, efficiency, short reaction times, easy work-up procedures and non-chromatographic purification of the products, making this method an attractive and useful process for the synthesis of pyrimido[4,5-b]quinolines as biologically interesting compounds. Moreover, the catalyst is recyclable and can be reused several times without a significant loss of activity. A probable mechanism for the formation of pyrimido-[4,5-b]quinolines 63a–q is outlined in Scheme 34. Initially, the [dsim]HSO4-catalyzed Knoevenagel condensation between the aldehyde and cyclic 1,3-diketone gave adduct I. Then, the 6-amino-1,3-dimethyluracil (62) attacks adduct I through a Michael addition to provide an open chain intermediate II. Subsequently, the latter intermediate II undergoes intramolecular cyclization by the reaction of nucleophilic amino function (NH2) to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O group, followed by dehydration, to produce pyrimido[4,5-b]quinolines 63.
O group, followed by dehydration, to produce pyrimido[4,5-b]quinolines 63.
 |
| | Scheme 33 [dsim]HSO4 catalyzed the synthesis of polyfunctionalized pyrimido[4,5-b]-quinolines 63a–q. | |
 |
| | Scheme 34 Proposed mechanistic pathway for the [dsim]HSO4 catalyzed formation of polyfunctionalized pyrimido[4,5-b]quinolines 63a–q. | |
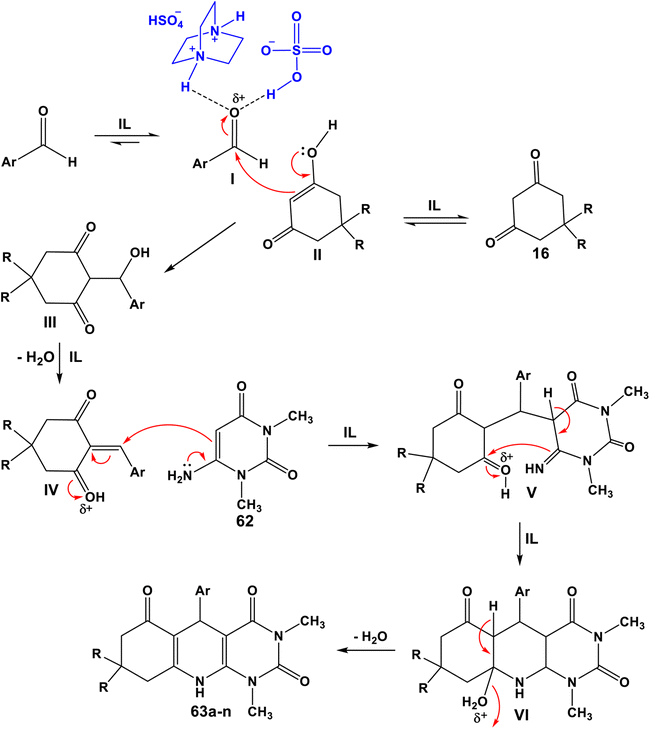
A novel methodology for the synthesis of pyrimido[4,5-b]quinolines 63 utilizing a new ionic liquid (IL), [H2-DABCO][HSO4]2 from the reaction of 1,4-diazabicyclo[2.2.2]octane (DABCO) and H2SO4, as a catalyst, was developed by Shirini et al. in 2017.100 The results show the applicability of the prepared ionic liquid as a reusable catalyst without losing its activity. This protocol has some advantages such as short reaction times, excellent yields and use of non-toxic and affordable catalyst. This synthetic procedure proceeded by stirring a mixture of the 6-amino-1,3-dimethyluracil (62), aromatic aldehyde and 1,3-diketone 16 and in the presence of a catalytic amount of [H2-DABCO][HSO4]2 in EtOH–H2O (1:2) at 75 °C for 60–150 min to give the tricyclic pyrimido[4,5-b]quinolines 63a–n in 85–95% yields (Scheme 35). A plausible mechanism to account for the formation of 63a–n is suggested in Scheme 36. The ionic liquid [H2-DABCO] [HSO4]2 may activate the aldehyde via hydrogen bonding formation. Then, 6-amino-1,3-dimethyluracil (62) was engaged in a Michael addition with intermediate IV to afford intermediate V. Subsequently, the ionic liquid promotes intramolecular cyclization by removing a hydrogen proton (H+) from intermediate V, resulting in intermediate VI. Finally, the latter intermediate VI loses one molecule of H2O to afford the desired products 63a–n.
 |
| | Scheme 35 Synthesis of pyrimido[4,5-b]quinolines 63a–n utilizing [H2-DABCO][HSO4]2 as the catalyst. | |
 |
| | Scheme 36 Suggested mechanism for the synthesis of pyrimido[4,5-b]quinolines 63a–n catalyzed by [H2-DABCO][HSO4]2. | |
Very recently, Gholami and his group101 reported a one-pot, three-component synthesis of novel azo and sulfonated pyrimido[4,5-b]quinoline derivatives 63 by the reaction of azo or sulfonated aldehydes, 6-amino-1,3-dimethyluracil (62) and 1,3-cyclohexadione (16a) or dimedone (16b). The reaction occurred in choline chloride/oxalic acid (ChCl:Oxa), as a green solvent and catalyst, at 80 °C for 40–60 min, delivering pyrimido[4,5-b]quinolines 63a–h (Scheme 37). This approach has several advantages, including high efficiency, excellent yields over short reaction times and a low cost. The catalytic role of ChCl:Oxa in the synthesis of pyrimido[4,5-b]quinolines 63 is indicated by the mechanistic pathway (Scheme 38). Hydrogen bonding with the acidic hydrogen of oxalic acid increases the electrophilicity of the carbonyl group of aldehydes. It is supposed that the reaction may proceed at first by the reaction of 1,3-diketone 16a,b with aldehyde by Knoevenagel condensation to produce the required intermediate I. The 6-amino-1,3-dimethyluracil (62) then attacks intermediate I in a Michael-type reaction to form intermediate II. Finally, the latter intermediate II underwent intramolecular cyclization via attack of the NH group to a carbonyl group, followed by dehydration to form novel pyrimido[4,5-b]quinoline 63.
 |
| | Scheme 37 Synthesis of novel azo and sulfonated pyrimido[4,5-b]quinolines 63a–h catalyzed by ChCl:Oxa. | |
 |
| | Scheme 38 Proposed mechanism for the synthesis of azo and sulfonated pyrimido[4,5-b]-quinolines 63 by ChCl:Oxa. | |
In 2012, Singh and his coworkers102 developed a two-step synthesis of pyrimido[4,5-b]quinoline-4-ones 65a–h from 2-chloroquinoline-3-carbonitriles 58 via amination and cyclization reactions. The amination reactions proceeded much faster in water via the simple SNAr displacement reactions of chlorine atoms at C-2 in 58. The cyclization reactions using the Vilsmeier reagent at lower temperatures gave the best yield of the products. When a mixture of 2-chloroquinoline-3-carbonitriles 58 (1 equiv.) and benzylamine (3 equiv.) was heated in water at 90 °C for 10–90 min, the corresponding 2-benzylamino-quinoline-3-carbonitriles 64 were formed. The authors examined the scope of the Vilsmeier reagent for the cyclization of 2-benzylaminoquinoline-3-carbonitriles 64, and they found that the Vilsmeier reaction with 1:3 molar ratios of DMF and POCl3 at 60 °C was the best optimal reaction conditions for cyclization to afford excellent yields of the desired products pyrimido-[4,5-b]quinoline-4-ones 65a–h (Scheme 39). The electron donating substituents at position 7 afforded better yields of the products than the substituents at position 6. Notably, the ethyl group gave a better product yield than the CH3 group at position 8. However, the electron-withdrawing substituent at position-6/7 afforded a better yield of the product. Notably, the faster reaction rates with the methoxy group could be attributed to the resonance effect of the group. A plausible mechanism for the formation of 65 is depicted in Scheme 40.
 |
| | Scheme 39 Two-step synthesis of 1-benzyl-pyrimido[4,5-b]quinoline-4-ones 65a–h. | |
 |
| | Scheme 40 Plausible mechanism for the formation of 1-benzyl-pyrimido[4,5-b]quinoline-4-ones 65a–m. | |
The synthesis of new pyrimido[4,5-b]quinoline-2,4(3H,10H)-diones 68a–u was developed by Dickens et al.103 in 2013. Reactions were carried out by heating 6-chlorouracil (20) (1 equiv.) with a wide variety of anilines (6 equiv.) at 180–200 °C for 1.5–3 h to afford the corresponding 6-anilinouracils 66, which were then refluxed with 2-halo-benzaldehydes 67 in DMF for 4 h to give the desired pyrimido-[4,5-b]quinoline-2,4(3H,10H)-diones 68a–u in 11–79% yields (Scheme 41).
 |
| | Scheme 41 Synthesis of new pyrimido[4,5-b]quinoline-2,4(3H,10H)-diones 68a–u. | |
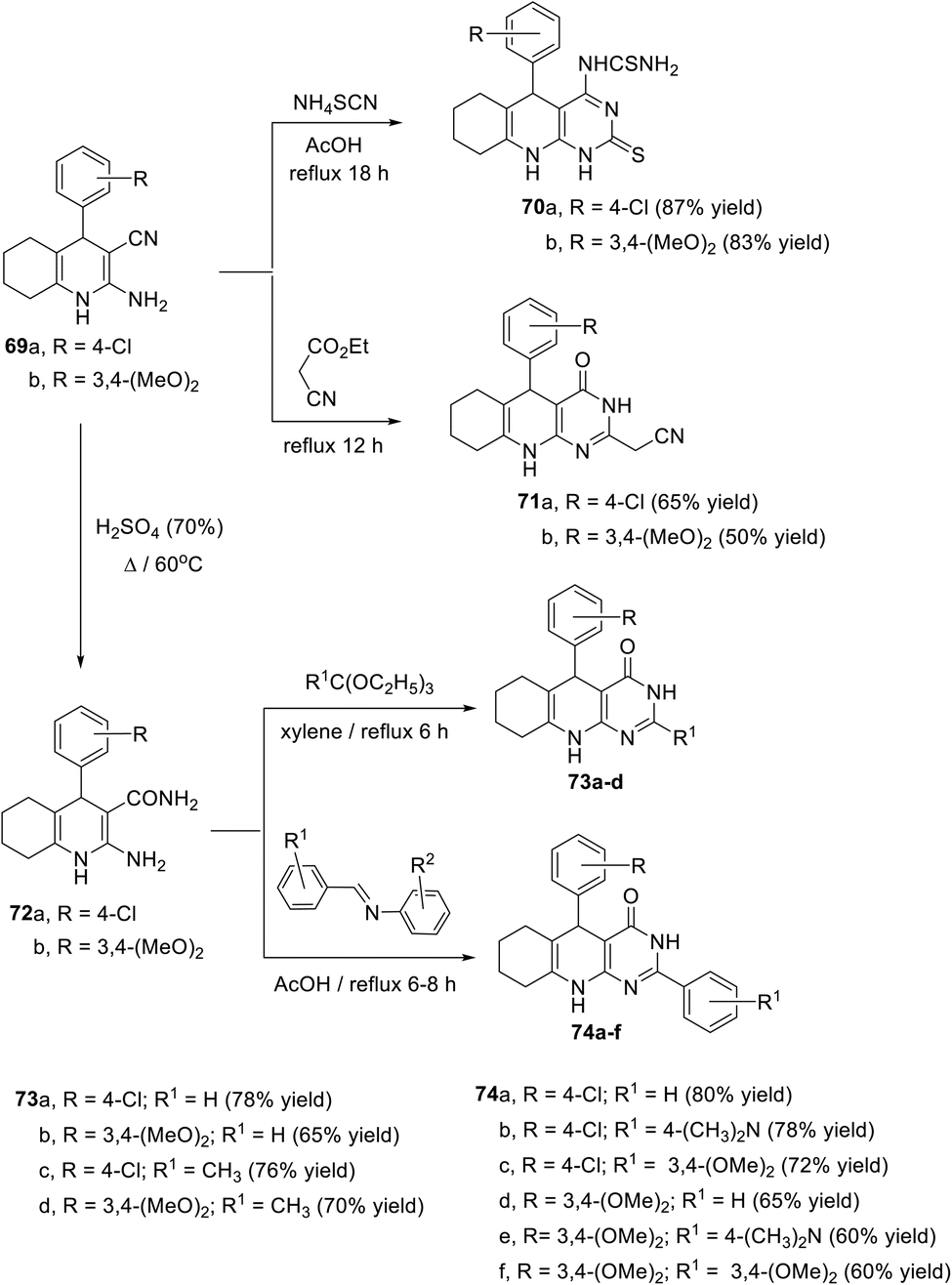
In 2013, El-Gohary104 described the synthesis of a series of new pyrimido[4,5-b]quinolines as potential antitumor agents. Reactions were carried out by heating the key 2-amino-4-aryl-1,4,5,6,7,8-hexahydroquinoline-3-carbonitriles 69a,b with ammonium thiocyanate (NH4SCN) in glacial acetic acid at reflux temperature for 18 h to give 5-aryl-1,2,5,6,7,8,9,10-octahydro-2-thioxopyrimido[4,5-b]quinolins 70a,b in very good yields. When compounds 69a,b were refluxed with an excess of ethyl cyanoacetate for 12 h, new 5-aryl-2-(cyanomethyl)-6,7,8,9-tetrahydropyrimido[4,5-b]quinolin-4(3H,5H,10H)-ones 71a,b were formed in moderate yields (Scheme 42). However, the hydrolysis of compounds 69a,b using H2SO4 (70%) at 60 °C gave the corresponding 2-amino-quinoline-3-carboxamides 72a,b (Scheme 42). Refluxing 72a,b with an appropriate triethyl orthoester in xylene for 6 h gave 5-aryl-6,7,8,9-tetrahydro-2-(unsubstituted or methyl)pyrimido[4,5-b]quinolin-4(3H,5H,10H)-ones 73a–d in 65–78% yields. However, when 72a,b (1 equiv.) were refluxed with the appropriate benzylideneaniline (2 equiv.) in glacial AcOH for 6–8 h, tricyclic products 74a–f were obtained in 60–80% yields, as a new derivative of pyrimido[4,5-b]quinolin-4-ones (Scheme 42).
 |
| | Scheme 42 Synthesis of 2-thioxopyrimido[4,5-b]quinolins 70a,b and pyrimido[4,5-b]-quinolin-4-ones 71a,b, 73a–d and 74a–f. | |
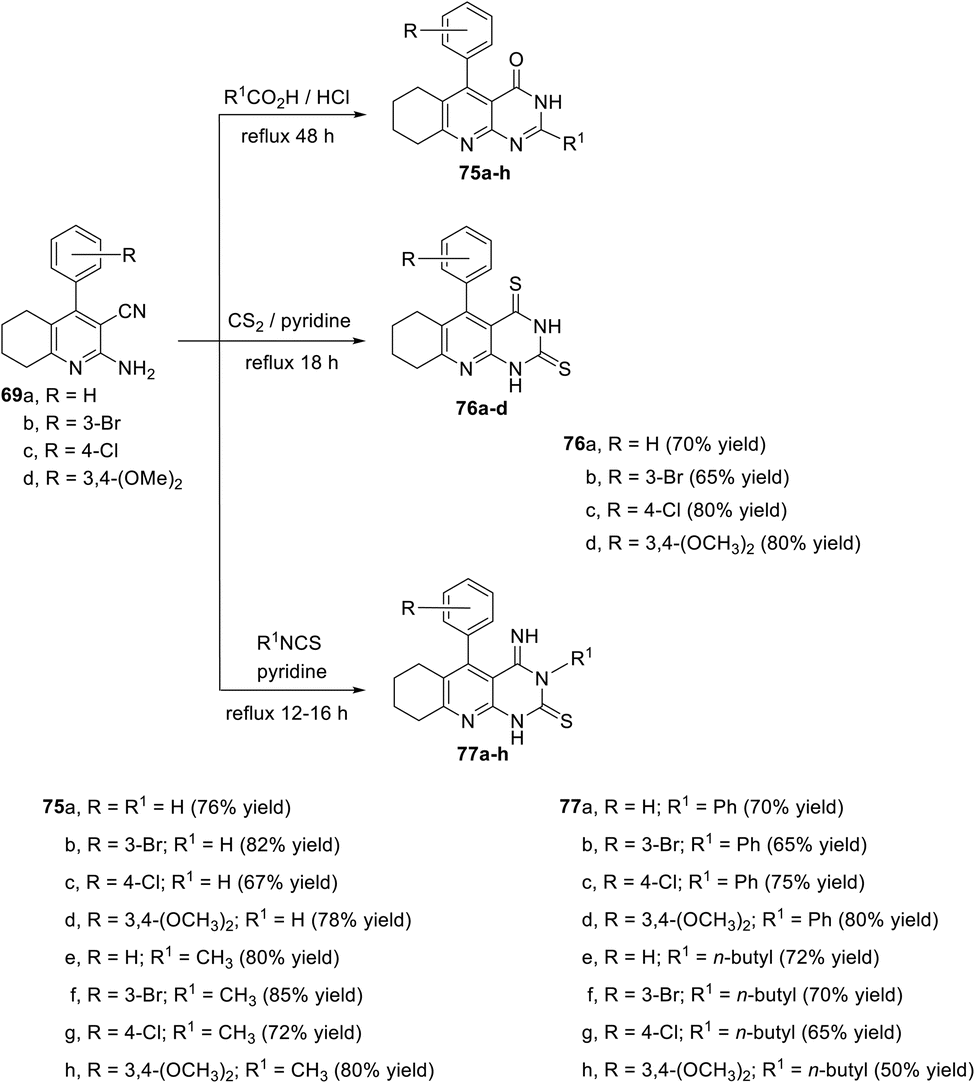
In another report, El-Gohary and his coworkers105 described the synthesis of a new series of tricyclic pyrimido[4,5-b]quinolines via a reaction of 2-amino-quinoline-3-carbonitriles 69a–d with different reagents, as shown in Scheme 38. Thus, heating compounds 69a–d with aliphatic acids (HCO2H and CH3CO2H) in the presence of a catalytic amount of conc. HCl at reflux temperature for 48 h gave 5-aryl-6,7,8,9-tetrahydro-2-(unsubstituted or methyl)-pyrimido[4,5-b]quinolin-4(3H,5H,10H)-ones 75a–h in 67–85% yields. When compounds 69a–d (1 equiv.) were refluxed with carbon disulfide (CS2) (1 equiv.) in pyridine on a water bath (80 °C) for 18 h, they underwent a cyclo-condensation reaction to afford 5-aryl-5,6,7,8,9,10-hexahydropyrimido[4,5-b]quinoline-2,4(1H,3H)-dithiones 76a–d in 65–80% yields. On heating compounds 69a–d with isothiocyanates in pyridine at reflux temperature for 12–16 h, 5-aryl-3,4,5,6,7,8,9,10-octahydro-4-imino-3-(n-butyl or phenyl)-pyrimido[4,5-b]-quinoline-2(1H)-thiones 77a–h were obtained in 50–80% yields (Scheme 43).
 |
| | Scheme 43 Synthesis of new series of pyrimido[4,5-b]quinolin-4-ones 75a–h, pyrimido-[4,5-b]quinoline-2,4-dithiones 76a–d and 4-imino-pyrimido[4,5-b]quinoline-2-thiones 77a–h. | |
In the same year, Faidallah and Rostom75 reported the synthesis of some new derivatives of tetrahydropyrimido[4,5-b]quinolines utilizing 2-amino-8-methyl-4-substituted-5,6,7,8-tetrahydro-quinoline-3-carbonitriles 78a,b as the key precursors. The reaction of 78a,b with phenyl isothiocyanate in pyridine under reflux for 2 h afforded the corresponding substituted tricyclic thiones 79a,b. The cyclization of compound 78a,b with formamide led to the formation of the required 4-amino-9-methyl-5-substituted-6,7,8,9-tetrahydropyrimido[4,5-b]quinolines 80a,b. Moreover, the fusion of 78a,b (1 equiv.) either with urea or thiourea (5 equiv.) at 260–300 °C using a sand bath for 1 h was used as a fruitful way for a one-step synthesis of the target tricyclic compounds 81a,b. However, reacting compounds 78a,b with either formic acid or acetic anhydride at 80 °C for 30 min gave the targeted tetrahydropyrimido-[4,5-b]quinolin-4-ones 82a,b and their 2-methyl analogs 82c,d (Scheme 44).
 |
| | Scheme 44 Synthesis of some new derivatives of tetrahydropyrimido[4,5-b]quinolines 79–82. | |
Dudkin et al.106 developed a new, simple and general methodology for the synthesis of novel 1,3-dimethyl-5-(polyfluoroalkyl)pyrimido[4,5-b]quinoline-2,4(1H,3H)-diones 84. This approach was based on the intramolecular cyclization reaction of 6-anilino-5-(polyfluoroacyl)-1,3-dimethyluracils 83 under acidic conditions. Thus, when uracils 83 were dissolved in conc. H2SO4 and allowed to stand at room temperature for 3 h, the tricyclic 1,3-dimethyl-5-(polyfluoroalkyl)pyrimido[4,5-b]quinoline-2,4(1H,3H)-diones 84a–t were obtained in good to excellent yields (49–92%) (Scheme 45).
 |
| | Scheme 45 Synthetic route to 1,3-dialkyl-5-(polyfluoroalkyl)pyrimido[4,5-b]quinoline-2,4(1H,3H)-diones 84a–t. | |
Cyclocondensation of 6-N-(3-hydroxypropyl)aminouracil (85) with 2-fluoro-benzaldehyde in DMF at reflux temperature for 6 h provided the desired 10-(3-hydroxypropyl)-pyrimido[4,5-b]quinoline-2,4(3H,10H)dione (86) in 83% yield. However, the reaction of 85 with 2,3-dimethoxybenzaldehyde under the same reaction conditions mentioned above gave the corresponding 10-(3-hydroxypropyl)-9-methoxy-pyrimido[4,5-b]quinoline-2,4(3H,10H)-dione (87) in 54% yield (Scheme 46).107
 |
| | Scheme 46 Synthesis of new pyrimido[4,5-b]quinoline-2,4(3H,10H)-diones 86 and 87. | |
Naik et al.108 described a versatile and useful access to different scaffolds of biologically significant pyrimido[4,5-b]quinoline-2-ol/thiol 90a,b utilizing a simple and efficient methodology based on the microwave (MW) irradiation technique. The reaction was carried out by heating 2-chloroquinoline-3-carbaldehyde (7a) (1 equiv.) with urea or thiourea (1 equiv.) in the presence of anhydrous K2CO3 (2 equiv.) in DMF for 10 min under microwave irradiation to give pyrimido[4,5-b]quinoline-2-ol (90a) and pyrimido[4,5-b]quinoline-2-thiol (90b) via intermediacy of 88 in 75% and 73% yields, respectively (Scheme 47). The efficiency of this methodology can be explained by the fact that MW energy is much higher than the activation energy required for each reaction, so the rate of reaction increases and yields are higher. The DNA binding properties of these two newly synthesized 90a,b were investigated using viscosity, absorption spectra and thermal denaturation experiments. The results showed that sulfur-containing 90b had more interaction with CT-DNA compared to 90a. Additionally, the authors carried out DNA cleavage via an oxidative route. The cleavage study results demonstrated that sulfur-containing 90b is more nuclease than 90a.
 |
| | Scheme 47 Microwave-induced one-pot synthesis of pyrimido[4,5-b]quinoline-2-ol/thiol 90a,b. | |
Mohire et al.109 developed a new, green, highly efficient cost-effective and atom-economic approach for the synthesis of 5-aryl-7-chloro-5,10-dihydropyrimido[4,5-b]-quinoline-2,4(1H,3H)-diones 91 via one-pot three-component condensation of 4-chloroaniline, aromatic aldehyde and barbituric acid (51) utilizing oxalic acid dihydrate:proline, as a low transition temperature mixture (LTTM), as new generation and green solvents instead of the hazardous organic solvents. This methodology was accomplished using ecofriendly and recyclable reaction media, easy work-up procedures, simple methodology, high atom economy and no chromatographic purification with high yields. The reactions were carried out by heating a mixture of 4-chloroaniline, aromatic aldehydes and barbituric acid (51) in oxalic acid:proline (LTTM), as a solvent, at 80 °C for 25–45 min to afford the new tricyclic 5-aryl-7-chloro-5,10-dihydropyrimido[4,5-b]quinoline-2,4(1H,3H)-diones 91a–j in 82–92% yields (Scheme 48).
 |
| | Scheme 48 Oxalic acid dihydrate:proline (LTTM)-mediated synthesis of 5-aryl-7-chloro-5,10-dihydropyrimido[4,5-b]quinoline-2,4(1H,3H)-diones 91a–j. | |
A plausible mechanism for the formation of 5-aryl-7-chloro-5,10-dihydropyrimido-[4,5-b]quinoline-2,4(1H,3H)-dione derivatives 91 is shown in Scheme 49. Owing to the hydrogen bonding nature of LTTM, it facilitates the electrophilic activation of the carbonyl group of aromatic aldehydes. Then, Knoevenagel condensation of aromatic aldehydes and barbituric acid (51) occurs to form 5-arylidene-pyrimidine-2,4,6(1H,3H,5H)-triones. Subsequently, the aza-Michael addition forms the ring nitrogen of 4-chloroaniline, resulting in the formation of an intermediate aza-Michael adduct, which is then cyclized to the tricyclic product via intramolecular nucleophilic attack from the endo-nitrogen on the amino heterocycle species. Finally, air auto-oxidation leads to the desired products 91a–j.
 |
| | Scheme 49 Plausible mechanism for the one-pot synthesis of 5-aryl-pyrimido[4,5-b]-quinolinediones 91 using oxalic acid dihydrate:proline (LTTM) as a solvent. | |
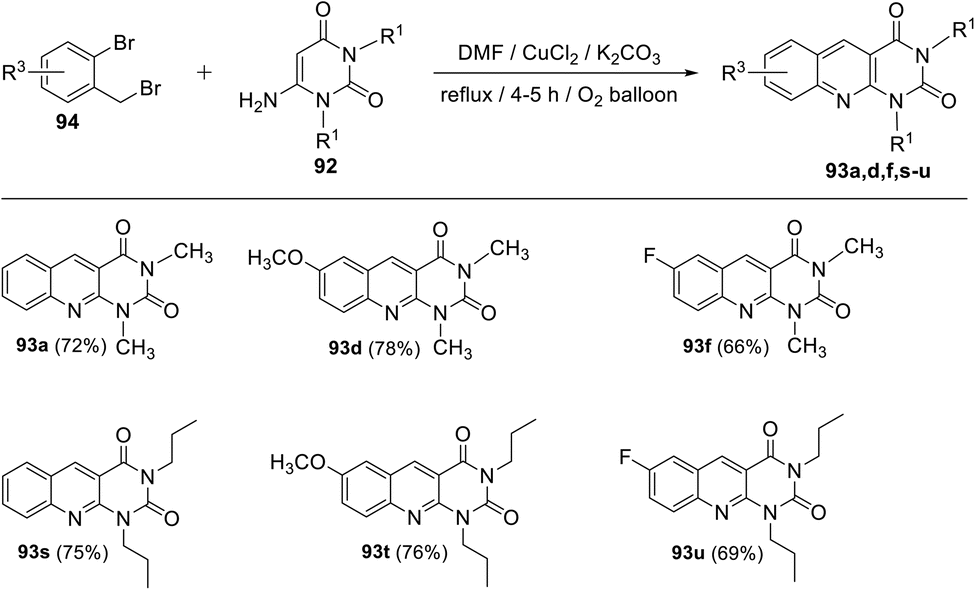
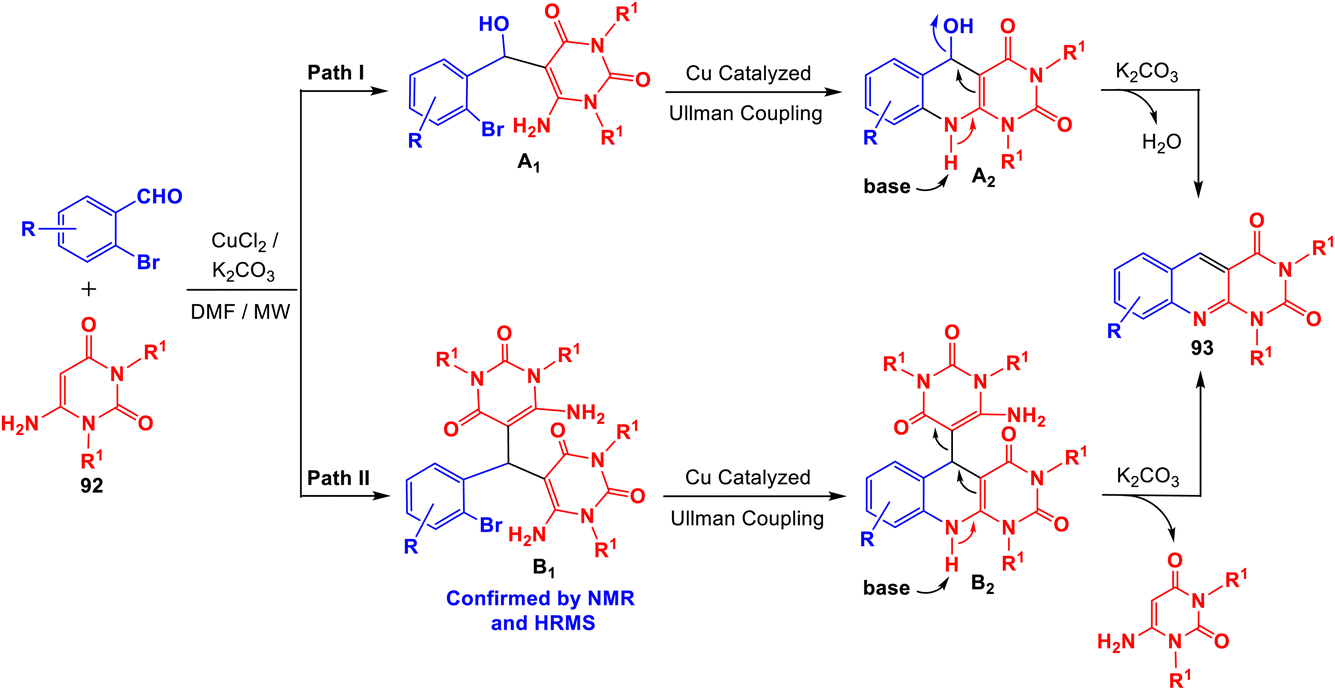
Panday and his group110 developed two efficient and convenient methodologies for the synthesis of new 1,3-dialkylpyrimido[4,5-b]quinoline-2,4(1H,3H)-diones 93 via the Cu-catalyzed coupling reaction of 6-aminouracils 92 and 2-bromobenzaldehydes/2-bromo-benzyl bromides 94. The reaction of 2-bromo-benzaldehydes with 6-aminouracils 92 in the presence of K2CO3 as base and a catalytic amount of CuCl2 (10 mol%) in DMF under microwave (MW) heating afforded the corresponding 1,3-dialkyl-pyrimido[4,5-b]quinoline-2,4(1H,3H)-diones 93a–r, in 60–86% yields, within 30 min (Scheme 50). Alternatively, 1,3-dialkyl-pyrimido[4,5-b]quinoline-2,4(1H,3H)-diones 93a,d,f,s–u were synthesized by reacting 2-bromobenzyl bromides 97 with 6-aminouracils 95 in the presence of molecular oxygen, K2CO3 as base and CuCl2 (10 mol%) in DMF at reflux temperature for 4–5 h (Scheme 51). A plausible mechanism (a) for the synthesis of 93 from 2-bromo benzaldehydes and 92 under MW heating and mechanism (b) for the formation of 93 from 94 and 92 under reflux conditions are outlined in Schemes 52 and 53, respectively.
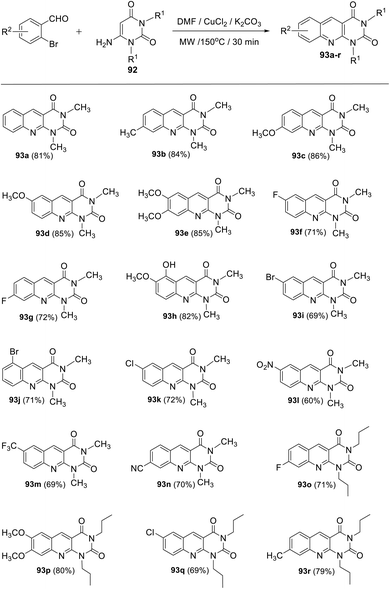 |
| | Scheme 50 Copper-catalyzed synthesis of pyrimido[4,5-b]quinolines 93a–r from the reaction of 2-bromobenzaldehyde and 6-aminouracils 92 under MW heating. | |
 |
| | Scheme 51 Copper-catalyzed synthesis of pyrimido[4,5-b]quinolines 93 from the reaction of 2-bromobenzyl bromides 94 and 6-aminouracils 92 under reflux conditions. | |
 |
| | Scheme 52 Proposed mechanism for the formation of 93 from 2-bromobenzaldehydes and 92 under MW heating. | |
 |
| | Scheme 53 Proposed mechanism for the formation of 93 from 92 and 94 under reflux conditions. | |
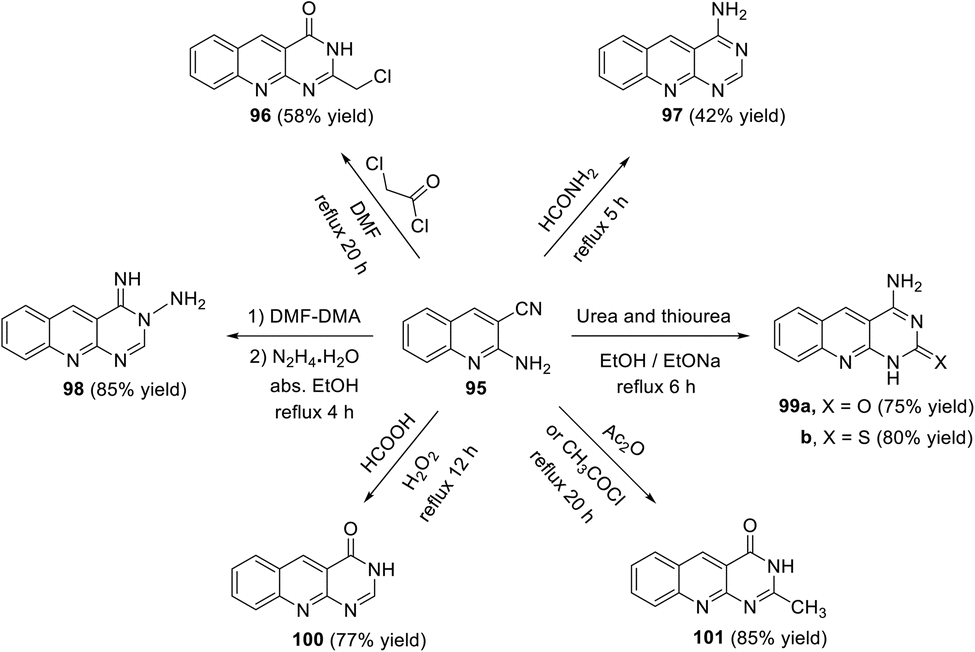
In 2017, El-Gamal111 described the synthesis of a new series of pyrimido[4,5-b]-quinolines 96–101 via the reaction of the key 2-aminoquinoline-3-carbonitrile (95) with various reagents. Thus, the reaction of 95 with chloroacetic chloride, formamide, DMF–DMA/N2H4, urea (or thiourea), formic acid and acetic anhydride (or acetyl chloride) gave the corresponding pyrimido[4,5-b]quinoline derivatives 96–101 (Scheme 54). The results of the in vitro cancer activity and docking study revealed that the synthesized compounds have potential cancer activity.
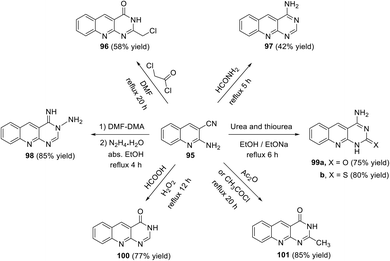 |
| | Scheme 54 Synthesis of pyrimido[4,5-b]quinolines 96–101 via the reaction of 95 with different reagents. | |
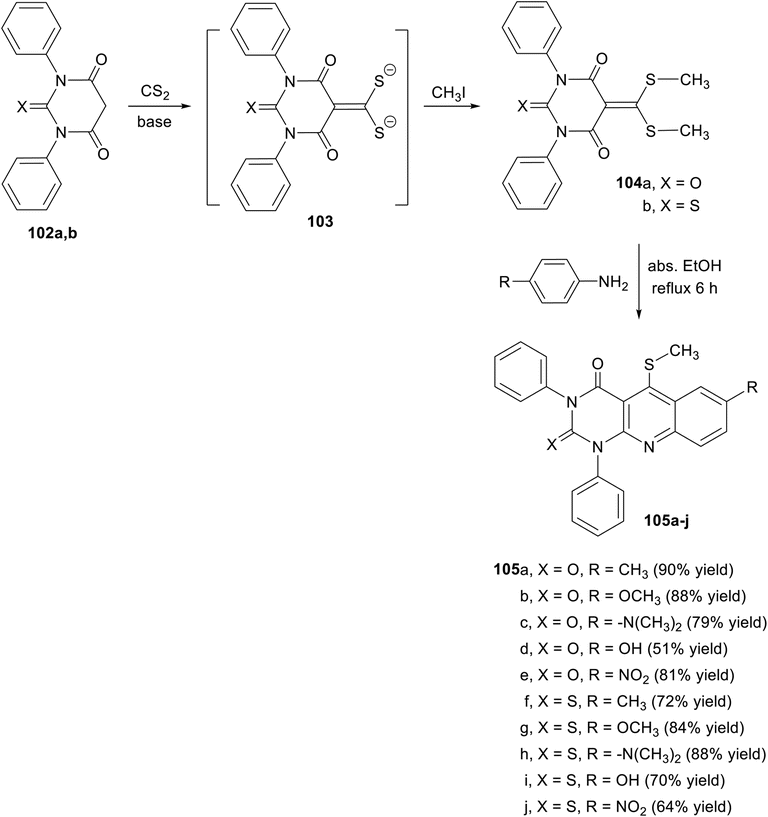
In the same year, Husain and his coworkers112 designed and synthesized a new class of pyrimido-[4,5-b]quinolines 105a–j utilizing the starting materials, 5-(bis(methylthio)-methylene)-1,3-diphenylpyrimidine-2,4,6(1H,3H,5H)-trione (104a) and 5-(bis(methylthio)-methylene)-1,3-diphenyl-2-thioxodihydropyrimidine-4,6(1H,5H)-dione (104b), as efficient α,α-ketene dithioacetals. α,α-Ketene dithioacetals 104a,b were synthesized from 1,3-diphenyl-barbituric acid (102a) and 1,3-diphenyl-2-thiobarbituric acid (102b), as shown in Scheme 55. Thus, heating the desired starting materials 104a,b with different para-substituted anilines in absolute EtOH at reflux temperature for 6 h afforded the novel tricyclic pyrimido[4,5-b]quinoline derivatives 105a–j in high yields (Scheme 55). Some of these compounds exhibited outstanding antibacterial activity (100%) against the strains E. coli and S. aureus, comparable to that of the standard drug Ciprofloxacin at the same concentration, while the others revealed significant antifungal activity.
 |
| | Scheme 55 Synthesis of some novel functionalized pyrimido[4,5-b]quinoline derivatives 105a–j from α,α-ketene dithioacetals 104a,b. | |
2.2. Synthesis of pyrimido[5,4-c]quinolines
Pyrimido[5,4-c]quinoline derivatives are found in a wide range of biologically important natural products and potent drugs. Compounds with pyrimido[5,4-c]quinoline cores exhibit various important and therapeutically useful biological activities, including antioxidant,57 antiherpetic,113 and antimalarial activities114 and potent 5-HT1A/2A and 5-HT7 receptor ligands.115 The synthesis of novel 2-imino-4-methyl-2,6-dihydropyrimido[5,4-c]quinolin-5(1H)-one (107), 4-methyl-pyrimido[5,4-c]quinoline-2,5(1H,6H)-dione (108a) and 4-methyl-2-thioxo-2,6-dihydropyrimido[5,4-c]quinolin-5(1H)-one (108b) was developed by Sankaran et al.57 utilizing 3-acetyl-4-hydroxy-quinoline-2-one (106) as the key precursor. The reaction of 106 with guanidine nitrate, urea and thiourea (as nitrogen bases) in refluxing EtOH in the presence of a catalytic amount of sodium acetate afforded the corresponding pyrimido[5,4-c]quinolines 107 and 108a,b (Scheme 56). The yields of the products and the reaction times were not reported. These compounds were screened for their in vitro antioxidant activities against Trolox equivalent antioxidant capacity (TEAC), radical scavenging capacity using DPPH., superoxide radical (O2˙−) scavenging activity, total antioxidant activity by FRAP, nitric oxide scavenging activity and metal chelating activity, and they exhibited significant antioxidant activities.
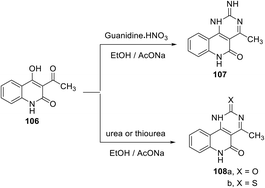 |
| | Scheme 56 Synthesis of pyrimido[5,4-c]quinolines 107 and 108a,b from 3-acyl-4-hydroxy-quinoline-2-one (106). | |
Ai and his group58,116 designed and synthesized new derivatives of 2-(amino/aroxy)-5-methyl-pyrimido[5,4-c]quinolin-4(3H)-ones 112a,b and 113a–i via an aza-Wittig reaction, starting from ethyl 4-amino-2-methyl-quinoline-3-carboxylate (109), as shown in Scheme 57. Treatment of key intermediate 109 with triphenylphosphine, hexachloroethane and triethylamine at room temperature for 24 h afforded iminophosphorane 110 in a satisfactory yield (70% yield). Aza–Wittig reaction between iminophosphorane 110 and substituted phenyl isocyanate in dry methylene chloride under a nitrogen atmosphere at 45 °C for 12 h provided the corresponding carbodiimides 111, which were used directly without further purification in the next step. Reacting 111 with alkyl amines in absolute EtOH in the presence of EtONa at room temperature for 12 h gave the desired tricyclic 2-(alkylamino)-3-aryl-5-methyl-pyrimido[5,4-c]quinoline-4(3H)-ones 112a,b. Meanwhile, the reaction of carbodiimide 111 with substituted phenols in CH3CN in the presence of a catalytic amount of K2CO3 at 75 °C for 4–6 h produced new 5-methyl-2-aryloxy-pyrimido[5,4-c]quinolin-4(3H)-ones 113a–l in 30–39% yields. The products showed potential antiproliferative activity with broad spectrum against several human cancer cell lines.
 |
| | Scheme 57 Synthesis of 2-(amino/aroxy)-5-methyl-pyrimido[5,4-c]quinolin-4(3H)-ones 112a,b and 113a–l via an aza-Wittig reaction. | |
In 2008, Ismaili et al.56 developed a simple and general methodology for the synthesis of several new hexahydropyrimido[5,4-c]quinoline-2,5-diones 117a–f and 2-thioxo-hexahydropyrimido[5,4-c]quinoline-5-ones 117g–j by Biginelli reaction in two steps from ethyl 4-aryl-6-methyl-2-oxo-tetrahydropyrimidine-5-carboxylates 116a–f or ethyl 4-aryl-6-methyl-2-thioxotetrahydropyrimidine-5-carboxylates 116g–j, as good precursors. When a mixture of 2-chlorobenzaldehyde derivatives 114, ethyl acetoacetate (115), urea or thiourea derivatives 116 and boric acid (H3BO3) in glacial CH3CO2H was heated at 100 °C for 3 h, they underwent condensation reactions to give the corresponding 117a–f and 117g–j. Intramolecular cyclization of 117 was achieved by heating in ammonia at 250 °C under 10 bars for 16 h to afford the desired tricyclic compounds 118a–j in 65–90% yields (Scheme 58).
 |
| | Scheme 58 Synthesis of new hexahydropyrimido[5,4-c]quinoline-2,5-diones 118a–f and 2-thioxohexahydropyrimido[5,4-c]quinoline-5-ones 118g–j. | |
In 2010, Rajanarendar et al.117 reported the synthesis of a novel series of 1-aryl-4-methyl-3,6-bis-(5-methylisoxazol-3-yl)-2-thioxo-2,3,6,10b-tetrahydro-1H-pyrimido[5,4-c]quinolin-5-ones 122a–h, as antibacterial and antifungal agents, utilizing ethyl 3-aryl-4-(2-chloro-phenyl)-6-methyl-1-(5-methyl-isoxazol-3-yl)-2-thioxo-1,2,3,4-tetrahydropyrimidine-5-carboxylates 120a–h as starting materials, which were obtained by Biginelli reaction, through CAN-catalyzed one-pot condensation reaction of 2-chloro-benzaldehyde (114), ethyl acetoacetate (115) and isoxazolyl thioureas 119 (Scheme 59). The compounds 120a–h when heated with 3-amino-5-methyl-isoxazole (121) in diphenyl ether under a nitrogen atmosphere at 200 °C for 10 h underwent intramolecular cyclization to give directly the new tricyclic ring system 122a–h (Scheme 59). The yields of the products were not reported.
 |
| | Scheme 59 Synthesis of new 1-aryl-4-methyl-3,6-bis-(5-methylisoxazol-3-yl)-2-thioxo-2,3,6,10b-tetrahydro-1H-pyrimido-[5,4-c]quinolin-5-ones 122a–h. | |
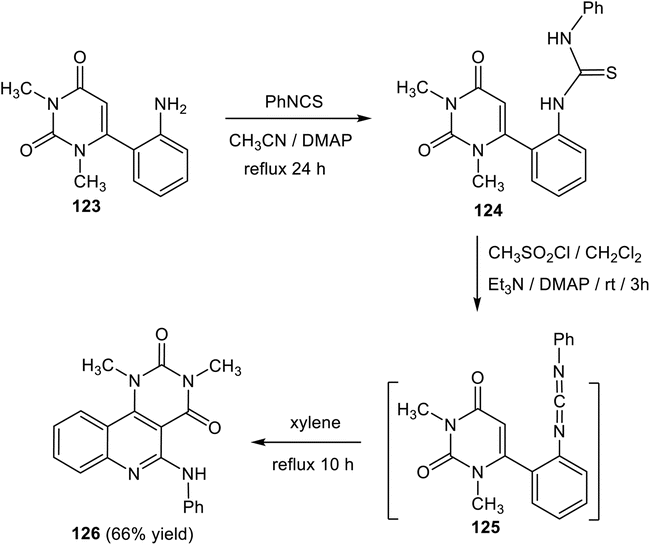
Krajsovszky and his coworkers118 described the synthesis of a new 1,3-dimethyl-5-(phenylamino)-pyrimido[5,4-c]quinoline-2,4(1H,3H)-dione (126) via a carbodiimide intermediate by electrocyclic ring closure. The reactions were carried out by refluxing 6-(2-aminophenyl)-1,3-dimethyl-pyrimidine-2,4(1H,3H)-dione (123) with phenylisothio-cyanate in acetonitrile catalyzed by 4-(dimethylamino)-pyridine (DMAP) for 24 h to produce the thiourea derivative 124. By stirring a solution of 124 with methane-sulfonylchloride in dichloromethane in the presence of triethylamine and 4-dimethylamino-pyridine (DMAP) at ambient temperature for 3 h, the corresponding carbodiimide 125 was generated in situ, which was transformed by refluxing in xylene to the cyclized product pyrimido[5,4-c]quinoline 126 (Scheme 60).
 |
| | Scheme 60 Synthesis of new 1,3-dimethyl-5-(phenylamino)-pyrimido[5,4-c]quinoline-2,4(1H,3H)-dione (126) via ring closure reaction of carbodiimide 125. | |
An efficient and straightforward synthesis of pyrimido[5,4-c]quinoline-5(1H)-ones by cyclocondensation method utilizing 1-ethyl-4-hydroxy-3-(2-nitroacetyl)quinolin-2(1H)-one (127), as a promising building block, with thiourea and cyanoguanidine was described by Ibrahim and his group in 2012.119 Thus, refluxing 127 with thiourea and cyanoguanidine in ethanolic KOH solution for 4 h afforded the corresponding 6-ethyl-4-(nitromethyl)-2-thioxo-2,6-dihydropyrimido[5,4-c]quinolin-5(1H)-one (128) and 6-ethyl-4-(nitromethyl)-pyrimido[5,4-c]-quinolin-5(6H)-one derivative 129 in 61% and 57% yields, respectively (Scheme 61).
 |
| | Scheme 61 Synthesis of 6-ethyl-4-(nitromethyl)-2-thioxo-2,6-dihydropyrimido[5,4-c]-quinolin-5(1H)-one (128) and 6-ethyl-4-(nitromethyl)-pyrimido[5,4-c]quinolin-5(6H)-one derivative 129. | |
In 2013, Ismail et al.120 developed an efficient procedure for the synthesis of a series of pyrimido-[5,4-c]quinoline-2,4-diones 131a–k with different substituents on the quinoline ring as a useful scaffold for the synthesis of antimicrobial agents via a thermolysis reaction of an equimolar ratio of 5-arylidene-1,3-dimethylbarbituric acid derivatives 130a–d with different aromatic amines at 150–180 °C for 1–2 h (Scheme 62).
 |
| | Scheme 62 Efficient procedure for the synthesis of a series of pyrimido[5,4-c]quinoline-2,4-dione derivatives 131a–k. | |
4-Aryl-6-phenyl-4,6-dihydropyrimido[5,4-c]quinoline-2,5(1H,3H)-diones 133a–d and 4-aryl-6-phenyl-2-thioxo-2,3,4,6-tetrahydropyrimido[5,4-c]quinolin-5(1H)-ones 133e–h were synthesized via Biginelli condensation reactions of 4-hydroxy-1-phenyl-quinolin-2(1H)-one (132), aromatic aldehydes and urea or thiourea in DMF under microwave irradiation for 3–5 min (Scheme 63).121
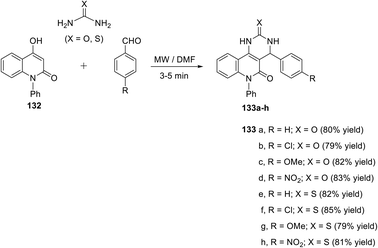 |
| | Scheme 63 Synthesis of 4-aryl-6-phenyl-4,6-dihydropyrimido[5,4-c]quinoline-2,5(1H,3H)-diones 133a–d and 4-aryl-6-phenyl-2-thioxo-2,3,4,6-tetrahydropyrimido[5,4-c]quinolin-5(1H)-ones 133e–h. | |
A simple and solvent free reaction of 5-(3-nitrobenzylidene)pyrimidine-2,4,6(1H,3H,5H)-trione (134) with various sulfanilamides to synthesize highly functionalized 5-(3-nitro-phenyl)pyrimido-[5,4-c]quinoline-2,4(1H,3H)-diones 135a–e was developed by Mubeen in 2018.122 Thus, fusion of 134 with sulfanilamide derivatives in a sealed tube at 170–212 °C for 1–2 h in an oil bath afforded the desired tricyclic pyrimido[5,4-c]quinoline-2,4(1H,3H)-diones 135a–e in 50–74% yields (Scheme 64). The synthesized compounds are potential drug candidates for the development of new antibacterial and antiviral agents.
 |
| | Scheme 64 Synthesis of highly functionalized 5-(-3-nitrophenyl)-pyrimido[5,4-c]quinoline-2,4(1H,3H)-diones 135a–e. | |
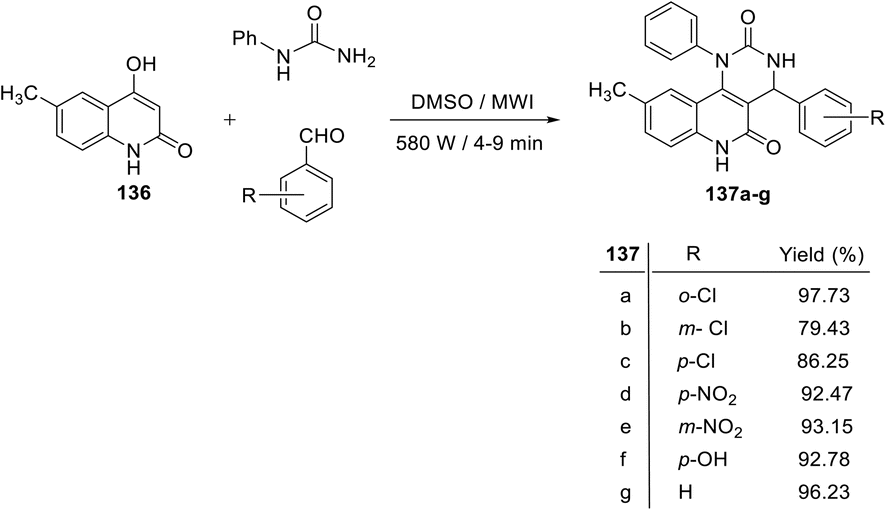
Recently, Nadaraj and his coworkers123 developed a new synthetic method for the synthesis of 4-aryl-9-methyl-1-phenyl-1,4-dihydropyrimido[5,4-c]quinolin-2,5(3H,6H)-diones 137a–g by the reaction of 6-methyl-4-hyrdoxyquinolin-2(1H)-one (136), an aromatic aldehyde, with phenyl urea via the Biginelli reaction. A mixture of 6-methyl-4-hydroxy-quinolin-2(1H)-one (136), aromatic aldehydes and phenyl urea in dimethyl sulphoxide (DMSO) was irradiated in a microwave (580 W) for 4–9 min to provide 4-aryl-9-methyl-1-phenyl-1,4-dihydro-pyrimido[5,4-c]quinolin-2,5(3H,6H)-diones 137a–g in very good to excellent yields (Scheme 65). The notable features of this protocol are mild reaction conditions, easy work of the products, excellent yields, cleaner reactions and short reaction times.
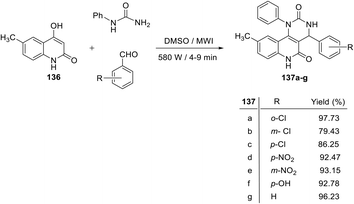 |
| | Scheme 65 Synthesis of 4-aryl-9-methyl-1-phenyl-1,4-dihydropyrimido[5,4-c]quinolin-2,5(3H,6H)-diones 137a–g. | |
The synthesis of pyrimido[5,4-c]quinolines 140, structurally analogous to biologically active benzonaphthyridines present in alkaloids, was described by Agarwal et al. in 2009.124 This synthetic strategy is based on the modified Pictet–Spengler reaction. The 2-amino-pyrimidine substrates 138a–c when heated with various aldehydes in DMF in the presence of a catalytic amount of triflic acid (1%) at 120 °C for 8 h underwent Pictet–Spengler cyclization to give the corresponding tricyclic pyrimido-[5,4-c]quinolines 140a–v in good yields via intermediacy of 139 (Scheme 66).
 |
| | Scheme 66 Synthesis of pyrimido[5,4-c]quinolines 140a–v via Pictet–Spengler reaction from 2-amino-pyrimidine substrates 138a–c. | |
A novel three-component synthesis of fluorine-containing pyrimido[5,4-c]quinolines 143 via the condensation reactions of 4-amino-3-trifluoroacetyl-quinoline (141) with different aldehydes and aq. NH3 was developed by Okada and his coworkers in 2014.125 Thus, stirring a mixture of 141 (1 mmol) with aldehydes (5 mmol) and aq. NH3 [28% (w/w)] (3 to 10 mmol) in MeCN at 50 °C for 24–96 h gave the corresponding fluorine-containing dihydropyrimido-[5,4-c]quinolines 142a–n. Treatment of 142a–n with DDQ in MeCN at room temperature for 1 h led to successful dehydrogenation to afford the desired 2-substituted-4-(trifluoromethyl)-pyrimido[5,4-c]quinolines 143a–n in 80–100% yields (Scheme 67).
 |
| | Scheme 67 Synthesis of 2-substituted-4-(trifluoromethyl)-pyrimido[5,4-c]quinolines 143 via three-component condensation reactions of 4-amino-3-trifluoroacetylquinoline (141) with different aldehydes and aq. NH3. | |
Zhang et al.126 synthesized a new series of 2-substituted-pyrimido[5,4-c]-zquinolin-5(6H)-one derivatives 148 and evaluated their cytotoxic activity against human lung carcinoma (H460), human colorectal cancer (HT-29) and human breast cancer (MDA-MB-231) cell lines. The results showed that most of these compounds exhibited stronger activity in the three selected cell lines. The condensation of ethyl 3-(2-nitrophenyl)-3-oxopropanoate (144) with dimethylformamide dimethylacetal (DMF–DMA) in toluene at reflux temperature for 10 h afforded ethyl 3-(dimethylamino)-2-(2-nitrobenzoyl)acrylate (145). When compound 145 was heated with various guanidines 146 in EtOH in the presence of K2CO3 at reflux temperature for 7 h, it underwent intermolecular cyclization to produce ethyl 2-substituted-4-(2-nitrophenyl)-pyrimidine-5-carboxylates 147a–d. Finally, the reduction of the nitro group in 147 with Fe in AcOH at 85 °C for 8 h and ring-closure in one pot gave the target tricyclic 2-substituted-pyrimido-[5,4-c]quinolin-5(6H)-ones 148a–d in 45–75% yields (Scheme 68).
 |
| | Scheme 68 Synthesis of 2-substituted-pyrimido[5,4-c]quinolin-5(6H)-ones 148a–d. | |
In 2022, Mekheimer and his group127 developed a new, affordable, simple, and one-step methodology for the construction of a novel series of pyrimido[5,4-c]quinolines variously substituted at positions 2 and 5, as potential antiproliferative agents with multitarget actions, via a rapid base-catalyzed cyclization reaction of 2,4-dichloro-quinoline-3-carbonitrile (149) with guanidine hydrochlorides 59; 150a,b. The reactions were carried out by refluxing compound 149 (1 mmol) with 59; 150a,b (4 mmol) in the presence of anhydrous K2CO3 (4 mmol) in absolute EtOH for 3–6 h, as inferred by TLC. The reaction occurred via an initial nucleophilic attack of the guanidine on the quinoline C-4, followed by 6-exo-dig cyclization to afford the new tricyclic 4-amino-5-chloro-2-substituted-pyrimido[5,4-c]quinolines 153a–c in one step via intermediacies 151 and 152 in very good to excellent yields (Scheme 69).
 |
| | Scheme 69 One-step synthesis of new 4-amino-5-chloro-2-substituted-pyrimido[5,4-c]quinolines 153a–c. | |
2.3. Synthesis of pyrimido[5,4-b]quinolines
Fenner and Teichmann128 developed a synthetic route for the synthesis of pyrimido[5,4-b]quinoline derivatives as analogues of lumichrome. The bromination of 154 in glacial AcOH gave the corresponding 5-bromo-1,3-disubstituted-6-(2-nitrobenzyl)-pyrimidine-2,4(1H,3H)-diones 155. The reduction of 155 with stannous chloride in glacial acetic acid/HCl at 10 °C for 3 days gave 6-(2-aminobenzyl)-5-bromo-1,3-disubstituted-pyrimidine-2,4(1H,3H)-dione derivatives 156. When compound 156 was warmed in a saturated NaHCO3 solution at 100 °C for 3 h, the desired tricyclic 1,3-disubstituted-pyrimido-[5,4-b]quinoline-2,4(3H)-diones 159a,b were obtained in low yields as fluorescent substances (Scheme 70).
 |
| | Scheme 70 Synthesis of 1,3-disubstituted-pyrimido[5,4-b]quinoline-2,4(3H)-diones 159a,b. | |
Fenner et al.129 described the synthesis of 1,3-dimethyl-2,4-dioxo-8-substituted-1,2,3,4-tetrahydropyrimido[5,4-b]quinoline-10-carbonitriles 163a,b by the trimethylphosphite cyclization of 2-(1,3-dimethyl-2,6-dioxo-1,2,3,6-tetrahydropyrimidin-4-yl)-2-(2-nitro-5-substituted-phenyl)acetonitrile derivatives 162. As shown in Scheme 71, the reactions were achieved in two steps. Stirring a mixture of 6-chloro-1,3-dimethyluracil (160) (1 equiv.), 2-nitro-phenylacetonitrile (161a) or 5-methoxy-2-nitro-phenylacetonitrile (161b) (1 equiv.) and NaH (5.8 equiv.) in 1,2-dimethoxyethane at room temperature for 1 h gave 162. The reductive cyclization of nitro-substituted aromatics in 162 was performed by heating 162 in trimethylphosphite at reflux temperature under an argon atmosphere for 5 h to obtain the desired tricyclic products 163a,b directly in low yields.
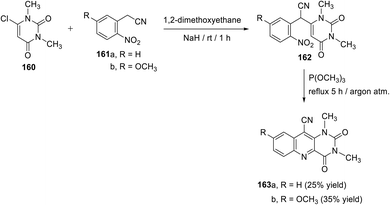 |
| | Scheme 71 Synthesis of 1,3-dimethyl-2,4-dioxo-8-substituted-1,2,3,4-tetrahydropyrimido-[5,4-b]quinoline-10-carbonitriles 163a,b. | |
2.4. Synthesis of pyrimido[4,5-c]quinolines
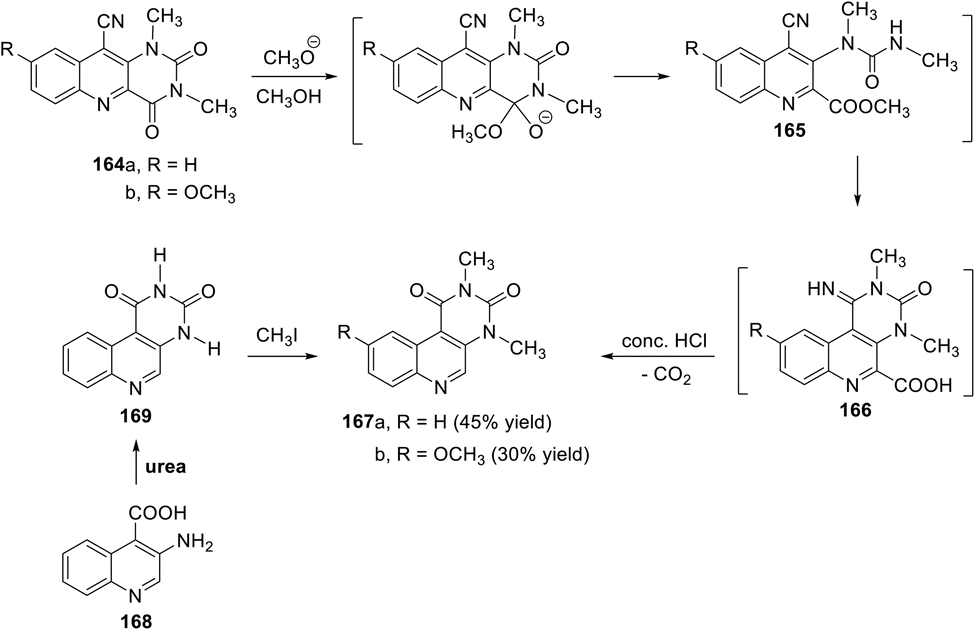
In 1986, Fenner and his coworkers130 described a facile and unexpected transformation of the pyrimido[5,4-b]quinoline ring system to pyrimido[4,5-c]quinoline derivatives. They reported that investigations of the reactivity of deazaalloxazines towards nucleophiles showed that in the case of 5-deazaalloxazine and 10-deazaalloxazine, ring-opening reactions occur at the pyrimidine-2,4-dione system and preferably occur by attack in the 2-position. Unexpectedly, with the 10-cyano substitution of the 10-deazaalloxazine system, ring opening follows exclusively under attack in the 4-position. The reaction was performed by adding sodium methoxide solution dropwise to a suspension of 1,3-dimethyl-2,4-dioxo-pyrimido[5,4-b]quinoline-10-carbonitriles 164a,b in absolute MeOH until the substances dissolved and a dark color appeared. Then, acidification with conc. HCl resulted in the formation of the tricyclic products, namely 2,4-dimethylpyrimido[4,5-c]quinoline-1,3(2H,4H)-diones 167a,b, in moderate yields via intermediates 165 and 166 (Scheme 72). The following mechanism is proposed based on the structure of the end product. First, ring opening to 165 occurs, which results in a rapid subsequent relationship action to form the angularly annulated pyrimido[4,5-c]quinoline 166. Upon acidification, the hydrolysis of imine 166 occurs, followed by decarboxylation to 167 (Scheme 72). This unexpected transformation of pyrimido[5,4-b]quinoline 164 to pyrimido[4,5-c]quinoline 167 is supported by the independent synthesis of 167a. The condensation of 3-amino-quinoline-4-carboxylic acid (168) with urea afforded pyrimido[4,5-c]quinoline-1,3(2H,4H)-dione (169), which reacted with methyl iodide to produce a product identical to 167a (Scheme 72).
 |
| | Scheme 72 Facile and unexpected transformation of pyrimido[5,4-b]quinolines 164a,b to pyrimido[4,5-c]quinolines 167a,b. | |
In 2010, Metwally et al.131 described the synthesis of a novel series of 2-amino-pyrimido[4,5-c]quinolin-1(2H)-ones 174a–n having several substituents with various electronic and steric properties at different positions of the pyrimidine and quinoline rings (positions 3,5,7 and 9) as potent cytotoxic antimitotic agents. The synthetic route to the target 2-amino-pyrimido[4,5-c]quinolin-1(2H)-ones 174 is illustrated in the general reaction sequence depicted in Scheme 73. The starting 3-amino-2-aryl-quinoline-4-carboxylic acids 172 were synthesized by refluxing isatins 170 with 4-chloro- (171a) or 4-bromo-phenacyl-amine hydrochloride (171b) in aq. NaOH for 30 min. The treatment of acids 172 with 4-chlorobenzoyl chloride in pyridine at room temperature for 36 h led to the formation of the lactones, [1,3]oxazino[4,5-c]-quinolin-1-ones 173. Finally, the hydrazinolysis of the lactones 173 by heating with hydrazine hydrate (99%) in 2-ethoxyethanol, as a high boiling solvent, at reflux temperature for 24 h afforded the desired pyrimido[4,5-c]quinolin-1(2H)-ones 174a–n in 41–54 yields.
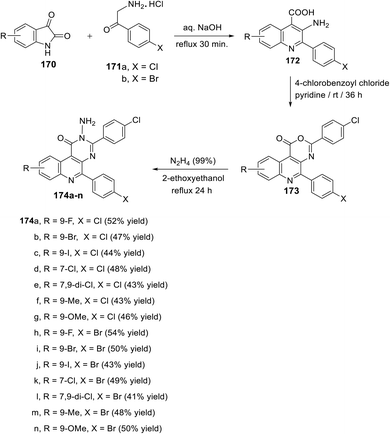 |
| | Scheme 73 Synthesis of new 2-amino-pyrimido[4,5-c]quinolin-1(2H)-ones 174a–n. | |
In 2011, Pierre et al.132 described the first one-pot synthesis of novel substituted pyrimido[4,5-c]-quinolines, which act mechanistically as ATP-competitive inhibitors of protein kinase CK2. Palladium-catalyzed coupling between methyl 5-bromo-2-substituted-pyrimidine-4-carboxylates 175a,b and 2-amino-4-(methoxycarbonyl)-phenylboronic acid hydrochloride (176) in dioxane at reflux temperature resulted in the one-pot formation of cyclized methyl 5-oxo-5,6-dihydropyrimido[4,5-c]quinoline-8-carboxylates 177a,b (Scheme 74). Reaction times were not reported.
 |
| | Scheme 74 One-pot procedure for the synthesis of novel substituted pyrimido[4,5-c]quinolines 177a,b. | |
2.5. Synthesis of pyrimido[1,2-a]quinolines
In 2007, Ukrainets and his group133 described the synthesis of 1-hydroxy-3,6-dioxo-4,6-dihydro-3H-pyrimido[1,2-a]quinoline-5-carbonitrile (181) in 76% yield via the reaction of ethyl 2-(4-oxo-4H-benzo[d][1,3]oxazin-2-yl)acetate (178) with malononitrile in dry pyridine at reflux temperature for 10 h (Scheme 75). In the proposed reaction mechanism, benzoxazinone 178 reacted with the highly nucleophilic carbanion generated from malononitrile to give the acylmalononitrile intermediate 179, which was cyclized into the corresponding aminoquinolone intermediate 180. The isolation of 180 was unsuccessful because under the reaction conditions of the synthesis, the amino group was subject to intramolecular acylation with the formation of the desired tricyclic product 1-hydroxy-3,6-dioxo-4,6-dihydro-3H-pyrimido[1,2-a]quinoline-5-carbonitrile (181) (Scheme 75).
 |
| | Scheme 75 Synthesis of 1-hydroxy-3,6-dioxo-4,6-dihydro-3H-pyrimido[1,2-a]quinoline-5-carbonitrile (181). | |
In 2011, Marjani et al.134,135 reported a short, facile and highly effective method for the synthesis of functionalized pyrimido[1,2-a]quinoline derivatives 185a–i by the rearrangement of N-quinolinyl-isoxazol-5(2H)-ones 184a–i under mild basic conditions (Scheme 76). Heating a neat mixture of ethyl 5-oxo-2,5-dihydroisoxzole-4-carboxylate (182) with 2-chloroquinolines 183a–i at 130 °C under nitrogen atmosphere for 15 min afforded the corresponding N-quinolinylisoxazolones 184a–i. When compounds 184a–i were refluxed in CHCl3 in the presence of a catalytic amount of triethylamine for 4 h, the desired ethyl 3-hydroxy-5,8,9,10-tetrasubstituted-1-oxo-1H-pyrimido[1,2-a]quinoline-2-carboxylates 185a–i were produced in 69–80% yields (Scheme 76). A mechanism for the formation of 185a–i is outlined in Scheme 77.
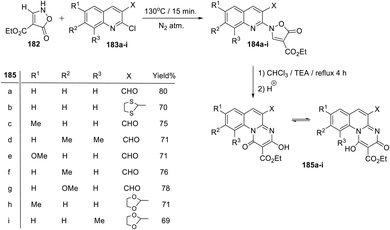 |
| | Scheme 76 Facile synthesis of a new series of ethyl 3-hydroxy-5,8,10-trisubstituted-1-oxo-1H-pyrimido[1,2-a]quinoline-2-carboxylates 185a–i. | |
 |
| | Scheme 77 Plausible mechanism for the formation of 185. | |
In 2013, Jadhav and Halikar136 reported for the first time a simple route for the synthesis of 1-imino-3-(methylthio)-1H-pyrimido[1,2-a]quinoline-2,5-dicarbonitrile (187) under mild conditions with good yield, exhibiting significant antibacterial and antifungal activities. On reacting 2-amino-quinoline-3-carbonitrile (185) with 2-(bis-(methylthio)-methylene)malononitrile (186) in N,N-dimethylformamide in the presence of a catalytic amount of anhydrous K2CO3 under reflux for 4 h, 1-imino-3-(methylthio)-1H-pyrimido[1,2-a]quinoline-2,5-dicarbonitrile (187) was obtained in 88% yield (Scheme 78).
 |
| | Scheme 78 Novel and simple route for the synthesis of 1-imino-3-(methylthio)-1H-pyrimido[1,2-a]quinoline-2,5-dicarbonitrile (187). | |
Recently, Soleimani-Amiri and co-workers137 reported a green and one-pot synthesis of pyrimido[1,2-a]-quinolin-3-ones 191a–c in high yields from the reaction of quinoline (188), dialkylacetylene-dicarboxylate or propiolate 189 and amides 190 in the presence of ZnO nanorods (ZnO-NRs) (15 mol%), as an efficient catalyst, under solvent-free conditions at room temperature (Scheme 79). The ease of use, solvent-free conditions, and reusability of the catalyst make this method an interesting alternative to others. The current method has several advantages, including high atom economy and yield, clean and mild reaction conditions, a short reaction time and low catalyst loading.
 |
| | Scheme 79 Green and one-pot synthesis of pyrimido[1,2-a]quinolin-3-ones 192a–c. | |
2.6. Synthesis of pyrimido[1,6-a]quinolines
In 2015, Ramanathan and Pitchumani138 reported for the first time an unprecedented copper(I)–Y zeolite-catalyzed tandem process involving ketenimine-based termolecular [2 + 2 + 2]/[NC + CC + NC] cycloaddition using sulfonyl azides 192, alkynes 193 and quinoline derivative 194 to synthesize highly functionalized pyrimido[1,6-a]quinolines 196a–x (Scheme 80). In this simple, highly atom- and step-economical protocol, copper(I) promotes azide–alkyne [3 + 2] cycloaddition, which is followed by ring-rearrangement/ketenimine formation/regio- and stereoselective [2 + 2 + 2] termolecular cycloaddition and dehydrogenation cascade to afford selectively the E-isomer of pyrimido[1,6-a]quinoline. The advantages of performing two cycloaddition reactions ([3 + 2] [2 + 2 + 2]) in one operation, remarkable simplicity of operation and significant molecular complexity generated render this catalytic system environmentally friendly and potentially cost-effective. The reaction was carried out by adding alkyne 193 (2 equiv.) to a mixture of Cu(I)–Y zeolite (20 mg, 10 mol%), sulfonyl azide 192 (2 equiv.), quinoline derivative 194 (1 equiv.) and Cs2CO3 (1.2 equiv.) in DCM under an open air atmosphere at room temperature. The methodology furnished the desired tricyclic 196, via intermediacy of 195 in 26–76% yields (Scheme 80). The plausible mechanistic pathway for the formation of 196 is described in Scheme 81. Sulfonyl azide 192 and alkyne 193 in the presence of Cs2CO3 and Cu(I)–Y zeolite form copper triazolyl intermediate I. This is followed by a ring opening reaction to give II, which undergoes rearrangement and extrusion of nitrogen to yield N-sulfonyl-ketenimine III, simultaneously regenerating the copper catalyst. The N-sulfonyl-ketenimine III as an energetic dipolar intermediate, reacting simultaneously as an electrophile (across CN) and nucleophile (across CC bond), undergoes addition to the CN bond of quinoline in a novel termolecular [2 + 2 + 2] cycloaddition reaction. The observed [NC + CC + NC] termolecular cycloaddition reaction occurs regioselectively and stereoselectively to form a dihydro intermediate IV in a concerted manner. The subsequent dehydrogenation of intermediate IV by air oxidation generates pyrimido[1,6-a]quinolines 196.
 |
| | Scheme 80 Synthesis of highly functionalized pyrimido[1,6-a]quinolines 196a–x. | |
 |
| | Scheme 81 Plausible mechanism for the formation of pyrimido[1,6-a]quinolines 196a–x. | |
3. Conclusion
In this review, we highlighted the synthesis of all six known types of pyrimidoquinolines with notable pharmacological and biological properties. Rapid progress in the chemistry of pyrimidoquinoline derivatives over the past two decades has led to a wide range of valuable synthetic methods for all major classes of these ring systems. Because of the established biological and pharmaceutical activities of the synthesized ring systems, we hope that this review will attract more research efforts to this field and provide a useful aid to crop protection, medicinal and other chemists dealing with heterocyclic systems daily. In addition, the lack of a comprehensive literature overview on the synthesis of all six known types of pyrimidoquinolines is behind the present attempt to provide for the first time a detailed literature survey and summarize the synthesis of these ring systems.
Data availability
No primary research results, software, or code are included, and no new data are generated or analyzed in this review.
Conflicts of interest
There are no conflicts to declare.
References
- I. M. Lagoja, Pyrimidine as constituent of natural biologically active compounds, Chem. Biodivers., 2005, 2, 1–50 CrossRef CAS PubMed.
- S. Noll, M. Kralj, L. Suman, H. Stephan and I. Piantanida, Synthesis of modified pyrimidine bases and positive impact of chemically reactive substituents on their in vitro antiproliferative activity, Eur. J. Med. Chem., 2009, 44, 1172–1179 CrossRef CAS PubMed.
- P. G. Baraldi, M. G. Pavani, M. Nunez, P. Brigidi, B. Vitali, R. Gambari and R. Romagnoli, Antimicrobial and antitumor activity of N-heteroimmine-1,2,3-dithiazoles and their transformation in triazolo-, imidazo-, and pyrazolopyrimidines, Bioorg. Med. Chem., 2002, 10, 449–456 CrossRef CAS PubMed.
- H. I. Ali, N. Ashida and T. Nagamatsu, Antitumor studies. Part 3: Design, synthesis, antitumor activity, and molecular docking study of novel 2-methylthio-, 2-amino-, and 2-(N-substituted amino)-10-alkyl-2-deoxo-5-deazaflavins, Bioorg. Med. Chem., 2007, 15, 6336–6352 CrossRef CAS PubMed.
- L. B. Narayana, R. R. A. Rao and S. P. Rao, Synthesis of new 2-substituted pyrido[2,3-d]-pyrimidin-4(1H)-ones and their antibacterial activity, Eur. J. Med. Chem., 2009, 44, 1369–1376 CrossRef PubMed.
- S. M. Sondhi, M. Johar, S. Rajvanshi, S. G. Dastidar, R. Shukla, R. Raghubir and J. W. Lown, Anticancer, anti-inflammatory and analgesic activity evaluation of heterocyclic compounds synthesized by the reaction of 4-isothiocyanato-4-methylpentan-2-one with substituted o-phenylenediamines, o-diaminopyridine and (un)substituted O, Aust. J. Chem., 2001, 54, 69–74 CrossRef CAS.
- L. Ballell, R. A. Field, G. A. C. Chung and R. J. Young, New thiopyrazolo[3,4-d]pyrimidine derivatives as anti-mycobacterial agents, Bioorg. Med. Chem. Lett., 2007, 17, 1736–1740 CrossRef CAS PubMed.
- M. T. Chhabria and M. H. Jani, Design, synthesis and anti-mycobacterial activity of some novel imidazo[1,2-c]pyrimidines, Eur. J. Med. Chem., 2009, 44, 3837–3844 CrossRef CAS PubMed.
- G. Mangalagiu, M. Ungnreanu, G. Grosu, I. Mangalagiu and M. Petrovanu, New pyrrolopyrimidine derivatives with antifungal or antibacterial properties in vitro, Ann. Pharm. Fr., 2001, 59, 139–140 CAS.
- K. Ghoshal and S. T. Jacob, An alternative molecular mechanism of action of 5-fluorouracil, a potent anticancer drug, Biochem. Pharmacol., 1997, 53, 1569–1575 CrossRef CAS PubMed.
- G. Fahron, F. Martens and U. Frei, Phenobarbital: A Good choice for long-term sedation, Crit. Care, 2001, 5, 201 CrossRef.
- N. Fujiwara, T. Nakajima, Y. Ueda, H. Fujita and H. Kawakami, Novel piperidinyl-pyrimidine derivatives as inhibitors of HIV-1 LTR activation, Bioorg. Med. Chem., 2008, 16, 9804–9816 CrossRef CAS PubMed.
- M. Hurst and S. Noble, Stavudine: An Update of its use in the treatment of HIV infection, Drugs, 1999, 58, 919–949 CrossRef CAS PubMed.
- R. B. Patel, P. S. Desai, K. R. Desai and K. H. Chikhalia, Synthesis of pyrimidine based thiazolidinones and azetidinones: antimicrobial and antitubercular agents, Indian J. Chem., 2006, 45(B), 773–778 Search PubMed.
- A. Agarwal, K. Srivastava, S. K. Puri, S. Sinha and P. M. S. Chauhan, A small library of trisubstituted pyrimidines as antimalarial and antitubercular agents, Bioorg. Med. Chem. Lett., 2005, 15, 5218–5221 Search PubMed.
- R. Isha, K. Navgeet, G. Anju and S. Manish, An Appraisal on synthetic and medicinal aspects of fused pyrimidines as anti neoplastic agents, Anticancer Agents Med. Chem., 2023, 23, 525–561 Search PubMed.
- G. Daves, R. Robins and C. Cheng, Communications-The Structure of fervenulin, a new antibiotic, J. Org. Chem., 1961, 26, 5256–5257 CrossRef CAS.
- G. D. Daves, R. K. Robins and C. C. Cheng, Antibiotics. I. Synthesis of 1,6-dimethyl-5,7-dioxo-1,5,6,7-tetrahydropyrimido[5,4-e]as-triazine (toxoflavin) and related compounds, J. Am. Chem. Soc., 1962, 84, 1724–1729 CrossRef CAS.
- J. Desrivot, C. Herrenknecht, G. Ponchel, N. Garbi, E. Prina, A. Fournet, C. Bories, B. Figadere, R. Hocquemiller and P. M. Loiseau, Antileishmanial 2-substituted quinolines: in vitro behaviour towards biological components, Biomed. Pharmacother., 2007, 61, 441–450 CrossRef CAS PubMed.
- A. R. Gholap, K. S. Toti, F. Shirazi, R. Kumari, M. K. Bhat, M. V. Deshpande and K. V. Srinivasan, Synthesis and evaluation of antifungal properties of a series of the novel 2-amino-5-oxo-4-phenyl-5,6,7,8-tetrahydroquinoline-3-carbonitrile and its analogues, Bioorg. Med. Chem., 2007, 15, 705–6715 CrossRef PubMed.
- C. M. M. Gómez, V. V. Kouznetsov, M. A. Sortino, S. L. Alvarez and S. A. Zacchino, in vitro antifungal activity of polyfunctionalized 2-(hetero)arylquinolines prepared through imino Diels-Alder reactions, Bioorg. Med. Chem., 2008, 16, 7908–7920 CrossRef PubMed.
- D. Edmont, R. Rocher, C. Plisson and J. Chenault, Synthesis and evaluation of quinoline carboxyguanidines as antidiabetic agents, Bioorg. Med. Chem. Lett., 2000, 10, 1831–1834 CrossRef CAS PubMed.
- Z. R. Wang, J. H. Hu, X. P. Yang, X. Feng, X. S. Li, L. Huang and A. S. C. Chan, Design, synthesis, and evaluation of orally bioavailable quinoline-indole derivatives as innovative multitarget-directed ligands: promotion of cell proliferation in the adult murine hippocampus for the treatment of Alzheimer's Disease, J. Med. Chem., 2018, 61, 1871–1894 CrossRef CAS PubMed.
- D. Dubé, M. Blouin, C. Brideau, C. C. Chan, S. Desmarais, D. Ethier, J. P. Falgueyret, R. W. Friesen, M. Girard, Y. Girard, J. Guay, D. Riendeau, P. Tagari and R. N. Young, Quinolines as potent 5-lipoxygenase inhibitors: Synthesis and biological profile of L-746,530, Bioorg. Med. Chem. Lett., 1998, 8, 1255–1260 CrossRef PubMed.
- P. Zajdel, A. Partyka, K. Marciniec, A. J. Bojarski, M. Pawlowski and A. Wesolowska, Quinoline- and isoquinoline-sulfonamide analogs of aripiprazole: Novel antipsychotic agents?, Future Med. Chem., 2014, 6, 57–75 CrossRef CAS PubMed.
- A. Mahamoud, J. Chevalier, A. Davin-Regli, J. Barbe and J.-M. Pages, Quinoline derivatives as promising inhibitors of antibiotic efflux pump in multidrug resistant enterobacter aerogenes isolates, Curr. Drug Targets, 2006, 7, 843–847 CrossRef CAS PubMed.
- J. Jiang, M. Hoang, J. R. Young, D. Chaung, R. Eid, C. Turner, P. Lin, X. Tong, J. Wang, C. Tan, S. Feighner, O. Palyha, D. L. Hreniuk, J. Pan, A. W. Sailer, D. J. MacNeil, A. Howard, L. Shearman, S. Stribling, R. Camacho, A. Strack, L. H. Van der Ploeg, M. T. Goulet and R. J. DeVita, 2-Aminoquinoline melanin-concentrating hormone (MCH)1R antagonists, Bioorg. Med. Chem. Lett., 2006, 16, 5270–5274 CrossRef CAS PubMed.
- T. Ulven, P. B. Little, J.-M. Receveur, T. M. Frimurer, ø. Rist, P. K. Nørregaard and T. Högberg, 6-Acylamino-2-amino-4-methylquinolines as potent melanin-concentrating hormone 1 receptor antagonists: structure-activity exploration of eastern and western parts, Bioorg. Med. Chem. Lett., 2006, 16, 1070–1075 CrossRef CAS PubMed.
- J. Jiang, P. Lin, M. Hoang, L. Chang, C. Tan, S. Feighner, O. C. Palyha, D. L. Hreniuk, J. Pan, A. W. Sailer, N. R. Morin, D. J. MacNeil, A. D. Howard, L. H. Y. Van der Ploeg, M. T. Goulet and R. J. DeVita, 4-Aminoquinoline melanin-concentrating hormone 1-receptor (MCH1R) antagonists, Bioorg. Med. Chem. Lett., 2006, 16, 5275–5279 CrossRef CAS PubMed.
- T. Ulven, T. M. Frimurer, J.-M. Receveur, P. B. Little, Ø. Rist, P. K. Nørregaard and T. Högberg, 6-Acylamino-2-aminoquinolines as potent melanin-concentrating hormone 1 receptor antagonists. identification, structure-activity relationship, and investigation of binding mode, J. Med. Chem., 2005, 48, 5684–5697 CrossRef CAS PubMed.
- R. Arienzo, D. E. Clark, S. Cramp, S. Daly, H. J. Dyke, P. Lockey, D. Norman, A. G. Roach, K. Stuttle, M. Tomlinson, M. Wong and S. P. Wren, Structure-activity relationships of a novel series of melanin-concentrating hormone (MCH) receptor antagonists, Bioorg. Med. Chem. Lett., 2004, 14, 4099–4102 CrossRef CAS PubMed.
- S. Bawa, S. Kumar, S. Drabu and R. Kumar, Structural modifications of quinoline-based antimalarial agents: Recent developments, J. Pharm. BioAllied Sci., 2010, 2, 64–71 CrossRef CAS PubMed.
- B. Gryzło and K. Kulig, Quinoline-a promising fragment in the search for new antimalarials, Mini-Rev. Med. Chem., 2014, 14, 332–344 CrossRef PubMed.
- K. Kaur, M. Jain, R. P. Reddy and R. Jain, Quinolines and structurally related heterocycles as antimalarials, Eur. J. Med. Chem., 2010, 45, 3245–3264 CrossRef CAS PubMed.
- A. P. Gorka, A. de Dios and P. D. Roepe, Quinoline drug-heme interactions and implications for antimalarial cytostatic versus cytocidal activities, J. Med. Chem., 2013, 56, 5231–5246 CrossRef CAS PubMed.
- S. Bongarzone and M. L. Bolognesi, The Concept of privileged structures in rational drug design: Focus on acridine and quinoline scaffolds in neurodegenerative and protozoan diseases, Expert Opin. Drug Discovery, 2011, 6, 251–268 CrossRef CAS PubMed.
- K. A. Reynolds, W. A. Loughlin and D. J. Young, Quinolines as chemotherapeutic agents for leishmaniasis, Mini-Rev. Med. Chem., 2013, 13, 730–743 CrossRef CAS PubMed.
- N. Costedoat-Chalumeau, B. Dunogué, N. Morel, V. Le Guern and G. Guettrot-Imbert, Hydroxychloroquine: A multifaceted treatment in lupus, Presse Med., 2014, 43, e167–e180 CrossRef PubMed.
- I. Pendrak, S. Barney, R. Wittrock, D. M. Lambert and W. D. Kingsbury, Synthesis and Anti-HSV activity of A-ring-deleted mappicine ketone analog, J. Org. Chem., 1994, 59, 2623–2625 CrossRef CAS.
- D. H. Boschelli, Y. D. Wang, F. Ye, B. Wu, N. Zhang, M. Dutia, D. W. Powell, A. Wissner, J. M. Weber and F. Boschelli, Synthesis and Src kinase inhibitory activity of a series of 4-phenylamino-3-quinolinecarbonitriles, J. Med. Chem., 2001, 44, 822–833 CrossRef CAS PubMed.
- N. Muruganantham, R. Sivakumar, N. Anbalagan, V. Gunasekaran and J. T. Leonard, Synthesis, anticonvulsant and antihypertensive activities of 8-substituted quinoline derivatives, Biol. Pharm. Bull., 2004, 27, 1683–1687 CrossRef CAS PubMed.
- Y. Engel, A. Dahan, E. Rozenshine-Kemelmakher and M. Gozin, Phenanthroline-derived ratiometric chemosensor for ureas, J. Org. Chem., 2007, 72, 2318–2328 CrossRef CAS PubMed.
- H. Deng, T. Han, E. Zhao, R. T. K. Kwok, J. W. Y. Lam and B. Z. Tang, Multicomponent click polymerization: A Facile strategy toward fused heterocyclic polymers, Macromolecules, 2016, 49, 5475–5483 CrossRef.
- V. Kavala, Z. Yang, A. Konala, C.-Y. Huang, C.-W. Kuo and C.-F. Yao, Synthesis of benzopyridoindolone derivatives via a one-pot copper catalyzed tandem reaction of 2-iodobenzamide derivatives and 2-iodobenzylcyanides, J. Org. Chem., 2017, 82, 7280–7286 CrossRef CAS PubMed.
- C.-Y. Huang, V. Kavala, C.-W. Kuo, A. Konala, T.-H. Yang and C.-F. Yao, Synthesis of biologically active indenoisoquinoline derivatives via a one-pot copper(II)-catalyzed tandem reaction, J. Org. Chem., 2017, 82, 1961–1968 CrossRef CAS PubMed.
- N. M. Evdokimov, S. Van slambrouck, P. Heffeter, L. Tu, B. Le Calvé, D. Lamoral-Theys, C. J. Hooten, P. Y. Uglinskii, S. Rogelj, R. Kiss, W. F. A. Steelant, W. Berger, J. J. Yang, C. G. Bologa, A. Kornienko and I. V. Magedov, Structural simplification of bioactive natural products with multicomponent synthesis. 3. Fused uracil-containing heterocycles as novel topoisomerase-targeting agents, J. Med. Chem., 2011, 54, 2012–2021 CrossRef CAS PubMed.
- P. Martins, J. Jesus, S. Santos, L. Raposo, C. Roma-Rodrigues, P. Baptista and A. Fernandes, Heterocyclic anticancer compounds: Recent advances and the paradigm shift towards the use of nanomedicine's tool box, Molecules, 2015, 20, 16852–16891 CrossRef CAS PubMed.
- J. F. Guastavino and R. A. Rossi, Synthesis of benzo-fused heterocycles by intramolecular α-arylation of ketone enolate anions, J. Org. Chem., 2012, 77, 460–472 CrossRef CAS PubMed.
- G. Joshi, H. Nayyar, J. M. Alex, G. S. Vishwakarma, S. Mittal and R. Kumar, Pyrimidine-fused derivatives: Synthetic strategies and medicinal attributes, Curr. Top. Med. Chem., 2016, 16, 3175–3210 CrossRef CAS PubMed.
- S. Kumar, S. Bawa and H. Gupta, Biological activities of quinoline derivatives, Mini-Rev. Med. Chem., 2010, 9, 1648–1654 CrossRef PubMed.
- T. H. Althuis, P. F. Moore and H. J. Hess, Development of ethyl 3,4-dihydro-4-oxopyrimido-[4,5-b]quinoline-2-carboxylate, a new prototype with oral antiallergy activity, J. Med. Chem., 1979, 22, 44–48 CrossRef CAS PubMed.
- T. H. Althuis, S. B. Kadin, L. J. Czuba, P. F. Moore and H. J. Hess, Structure-activity relationships in a series of novel 3,4-dihydro-4-oxopyrimido[4,5-b]quinoline-2-carboxylic acid antiallergy agents, J. Med. Chem., 1980, 23, 262–269 CrossRef CAS PubMed.
- R. J. Perner, Y.-G. Gu, C.-H. Lee, E. K. Bayburt, J. McKie, K. M. Alexander, K. L. Kohlhaas, C. T. Wismer, J. Mikusa, M. F. Jarvis, E. A. Kowaluk and S. S. Bhagwat, 5,6,7-Trisubstituted-4-aminopyrido[2,3-d]pyrimidines as novel inhibitors of adenosine kinase, J. Med. Chem., 2003, 46, 5249–5257 CrossRef CAS PubMed.
- M. M. Ghorab, F. A. Ragab, E. Noaman, H. I. Heiba and E. M. El-Hossary, Synthesis of some novel quinolines and pyrimido[4,5-b]quinolines bearing a sulfonamide moiety as potential anticancer and radioprotective agents, Arzneimittelforschung, 2007, 57, 795–803 CAS.
- K. Metwally, H. Pratsinis and D. Kletsas, Pyrimido[4,5-c]quinolin-1(2H)-ones as a novel class of antimitotic agents: Synthesis and in vitro cytotoxic activity, Eur. J. Med. Chem., 2007, 42, 344–350 CrossRef CAS PubMed.
- L. Ismaili, A. Nadaradjane, L. Nicod, C. Guyon, A. Xicluna, J. F. Robert and B. Refouvelet, Synthesis and antioxidant activity evaluation of new hexahydropyrimido[5,4-c]-quinoline-2,5-diones and 2-thioxohexahydropyrimido[5,4-c]quinoline-5-ones obtained by Biginelli reaction in two steps, Eur. J. Med. Chem., 2008, 43, 1270–1275 CrossRef CAS PubMed.
- M. Sankaran, C. Kumarasamy, U. Chokkalingam and P. S. Mohan, Synthesis, antioxidant and toxicological study of novel pyrimidoquinoline derivatives from 4-hydroxy-3-acyl quinolin-2-one, Bioorg. Med. Chem. Lett., 2010, 20, 7147–7151 CrossRef CAS PubMed.
- Y. Ai, Y.-J. Liang, J.-C. Liu, H.-W. He, Y. Chen, C. Tang, G.-Z. Yang and L.-W. Fu, Synthesis and in vitro antiproliferative evaluation of pyrimido[5,4-c]quinoline-4-(3H)-one derivatives, Eur. J. Med. Chem., 2012, 47, 206–213 CrossRef CAS PubMed.
- A. Długosz and D. Duś, Synthesis and anticancer properties of pyrimido[4,5-b]quinolines, Farmaco, 1996, 51, 367–374 Search PubMed.
- M. M. Ghorab, F. A. Ragab, H. I. Heiba, R. K. Arafa and E. M. El-Hossary, Docking study, in vitro anticancer screening and radiosensitizing evaluation of some new fluorine-containing quinoline and pyrimidoquinoline derivatives bearing a sulfonamide moiety, Med. Chem. Res., 2011, 20, 388–400 CrossRef CAS.
- S. T. Selvi, V. Nadaraj, S. Mohan, R. Sasi and M. Hema, Solvent free microwave synthesis and evaluation of antimicrobial activity of pyrimido[4,5-b]- and pyrazolo[3,4-b]-quinolines, Bioorg. Med. Chem., 2006, 14, 3896–3903 CrossRef CAS PubMed.
- O. A. El-Sayed, B. A. Al-Bassam and M. E. Hussein, Synthesis of some novel quinoline-3-carboxylic acids and pyrimidoquinoline derivatives as potential antimicrobial agents, Arch. Pharm., 2002, 335, 403–410 CrossRef CAS PubMed.
- S. I. Alqasoumi, A. M. Al-Taweel, A. M. Alafeefy, E. Noama and M. M. Ghorab, Novel quinolines and pyrimido[4,5-b]quinolines bearing biologically active sulfonamide moiety as a new class of antitumor agents, Eur. J. Med. Chem., 2010, 45, 738–744 CrossRef CAS PubMed.
- H.-A. S. Abbas, H. N. Hafez and A. B. A. El-Gazzar, Synthesis, in vitro antimicrobial and in vivo antitumor evaluation of novel pyrimidoquinolines and its nucleoside derivatives, Eur. J. Med. Chem., 2011, 46, 21–30 CrossRef CAS PubMed.
- D. Dorjsuren, A. Burnette, G. N. Gray, X. Chen, W. Zhu, P. E. Roberts, M. J. Currens, R. H. Shoemaker, R. P. Ricciardi and S. Sei, Chemical library screen for novel inhibitors of Kaposi's sarcoma-associated herpesvirus processive DNA synthesis, Antiviral Res., 2006, 69, 9–23 CrossRef CAS PubMed.
- A.-R. B. A. El-Gazzar, M. M. El-Enany and M. N. Mahmoud, Synthesis, analgesic, anti-inflammatory, and antimicrobial activity of some novel pyrimido[4,5-b]quinolin-4-ones, Bioorg. Med. Chem., 2008, 16, 3261–3273 CrossRef CAS PubMed.
- A. A. Joshi, S. S. Narkhede and C. L. Viswanathan, Design, synthesis and evaluation of 5-substituted amino-2,4-diamino-8-chloropyrimido[4,5-b]quinolines as novel antimalarials, Bioorg. Med. Chem. Lett., 2005, 15, 73–76 CrossRef CAS PubMed.
- M. A. Gouda, A. A. Abu-Hashem, T. A. Ameen, S. H. Althagafi, W. S. Hamama and A.-G. M. Khalil, Pyrimido[5,4-c]quinolines: Synthesis from 3,4-difunctionallized quinoline, reactivity and biological activities, Chem. Biodiversity, 2024, 21, e202301968 CrossRef CAS PubMed.
- H. N. Tawfeek, T. H. A. Hasanin and S. Bräse, Synthetic methodology of pyrimido[4,5-b]quinoline derivatives, J. Heterocycl. Chem., 2024, 61, 971–1008 CrossRef CAS.
- N. S. M. Ibrahim, H. H. Kadry, A. F. Zaher and K. O. Mohamed, Review: synthesis and anticancer activity of pyrimido[4,5-b]quinolines in the last twenty years, Chem. Pap., 2024, 78, 2729–2755 CrossRef CAS.
- A. A. Abu-Hashem, O. Hakami, N. Amri, T. A. Ameen, M. A. Bajaber, M. M. Youssef and M. A. Gouda, Recent routes in synthesis and biological activity of pyrimido[4,5-b]-quinoline derivatives: A review, Mini-Rev. Org. Chem., 2024, 22(3), 340–358 CrossRef.
- M. A. Gouda, A. A. Abu-Hashem, T. A. Ameen, M. A. Salem and A. Aljuhani, Recent progress in synthetic chemistry and biological activities of pyrimido[4,5-b]quinoline derivatives (Part III), Mini-Rev. Org. Chem., 2024, 21, 779–792 CrossRef CAS.
- M. A. El-Hashash, S. M. Sherif, A. A. E. Badawy and H. R. M. Rashdan, Synthesis of some new antimicrobial 5,6,7,8-tetrahydropyrimido[4,5-b]quinolone derivatives, Pharma Chem., 2014, 6, 23–29 Search PubMed.
- S. Mukherjee and M. Pal, Quinolines: A New hope against inflammation, Drug Discovery Today, 2013, 18, 389–398 CrossRef CAS.
- H. M. Faidallah and S. A. F. Rostom, Synthesis, in vitro antitumor evaluation and DNA-binding study of novel tetrahydroquinolines and some derived tricyclic and tetracyclic ring systems, Eur. J. Med. Chem., 2013, 63, 133–143 CrossRef CAS PubMed.
- S. Yamazaki, L. Tsai and T. C. Stadtman, Analogs of 8-hydroxy-5-deazaflavin cofactor: relative activity as substrates for 8-hydroxy-5-deazaflavin-dependent NADP+ reductase from methanococcus vannielii, Biochemistry, 1982, 21, 934–939 CrossRef CAS PubMed.
- P. Molina, M. J. Vilaplana and A. Pastor, A Facile entry to fused pyrimidines: Preparation of pyrimido[4,5-b]quinoline and pyrido[2,3-d:6,5-d′]dipyrimidine derivatives, Synthesis, 1992, 827–829 CrossRef CAS.
- S. M. Abdel-Gawad, M. S. A. El-Gaby, H. I. Heiba, H. M. Aly and M. M. Ghorab, Synthesis and radiation stability of some new biologically active hydroquinoline and pyrimido- [4,5-b]quinoline derivatives, J. Chin. Chem. Soc., 2005, 52, 1227–1236 CrossRef CAS.
- S. Tu, F. Fang, T. Li, S. Zhu and X. Zhang, An Efficient one-pot synthesis of novel pyrimidoquinoline derivatives under microwave irradiation without catalyst, J. Heterocycl. Chem., 2005, 42, 707–710 CrossRef CAS.
- M. Mamaghani, M. Jamali-Moghadam and R. Hossein-Nia, A Facile ZrO2 nanoparticles catalyzed synthesis of 2-amino-5-arylpyrimido[4,5-b]quinolinediones, J. Iran. Chem. Soc., 2017, 14, 395–401 CrossRef CAS.
- D.-Q. Shi, L.-H. Niu, H. Yao and H. Jiang, An Efficient synthesis of pyrimido[4,5-b]-quinoline derivatives via three-component reaction in aqueous media, J. Heterocycl. Chem., 2009, 46, 237–242 CrossRef CAS.
- J. M. Wilson, G. Henderson, F. Black, A. Sutherland, R. L. Ludwig, K. H. Vousden and D. J. Robinsa, Synthesis of 5-deazaflavin derivatives and their activation of P53 in Cells, Bioorg. Med. Chem., 2007, 15, 77–86 CrossRef CAS PubMed.
- Y. M. Elkholy and M. A. Morsy, Facile synthesis of 5,6,7,8-tetrahydropyrimido[4,5-b]-quinoline derivatives, Molecules, 2006, 11, 890–903 CrossRef CAS PubMed.
- A. B. A. El-Gazzar, M. M. Youssef, A. M. S. Youssef, A. A. Abu-Hashem and F. A. Badria, Design and synthesis of azolopyrimidoquinolines, pyrimidoquinazolines as anti-oxidant, anti-inflammatory and analgesic activities, Eur. J. Med. Chem., 2009, 44, 609–624 CrossRef CAS PubMed.
- A. B. A. El-Gazzar, H. N. Hafez, A. A. Abu-Hashem and A. S. Aly, Synthesis and antioxidant, anti-inflammatory, and analgesic activity of novel polycyclic pyrimido[4,5-b]quinolines, Phosphorus Sulfur Silicon Relat. Elem., 2009, 184, 379–405 CrossRef CAS.
- R. L. Dow, B. M. Bechle, T. T. Chou, C. Goddard and E. R. Larson, Selective inhibition of the tyrosine kinase Pp60src by analogs of 5,10-dihydropyrimido[4,5-b]quinolin-4(1H)-one, Bioorg. Med. Chem. Lett., 1995, 5, 1007–1010 CrossRef CAS.
- J. Trilleras, L. G. López, D. J. Pacheco, J. Quiroga, M. Nogueras, J. M. de la Torre and J. Cobo, Efficient microwave-assisted synthesis of 5-deazaflavine derivatives, Molecules, 2010, 15, 7227–7234 CrossRef CAS PubMed.
- K. Aknin, S. Desbène-Finck, P. Helissey and S. Giorgi-Renault, A New synthetic approach to functionalize pyrimido[4,5-b]quinoline-2,4(1H,3H)-diones via a three-component one-pot reaction, Mol. Diversity, 2010, 14, 123–130 CrossRef CAS PubMed.
- M. M. Ghorab, M. A. Shaaban, H. I. Heiba, A. Zaher and A. A. Hamed, Anticancer and radiosensitizing evaluation of novel sulfonamides with quinoline and pyrimidoquinoline groups, Res. Chem. Intermed., 2015, 41, 647–661 CrossRef CAS.
- A. Chandra, S. Upadhyay, B. Singh, N. Sharma and R. M. Singh, Base-catalyzed cyclization reaction of 2-chloroquinoline-3-carbonitriles and guanidine hydrochloride: A Rapid synthesis of 2-amino-3H-pyrimido[4,5-b]quinolin-4-ones, Tetrahedron, 2011, 67, 9219–9224 CrossRef CAS.
- A. Khalafi-Nezhad, S. Sarikhani, E. S. Shahidzadeh and F. Panahi, L-Proline-promoted three-component reaction of anilines, aldehydes and barbituric acids/malononitrile: Regioselective synthesis of 5-arylpyrimido[4,5-b]quinoline-diones and 2-amino-4-arylquinoline-3-carbonitriles in water, Green Chem., 2012, 14, 2876–2884 RSC.
- M. H. Mosslemin, E. Hzarenezhad, N. Shams, M. N. S. Rad, H. Anaraki-Ardakani and R. Fayazipoor, Green synthesis of 5-aryl-(1H,3H,5H,10H)-pyrimido[4,5-b]quinoline-2,4-diones catalysed by 1,4-diazabicyclo[2.2.2]octane in water, J. Chem. Res., 2014, 38, 169–171 CrossRef CAS.
- S. S. Reddy, M. V. K. Reddy and P. V. G. Reddy, β-Cyclodextrin in water: As an efficient green protocol for the synthesis of pyrimido[4,5-b]quinoline-diones, ChemistrySelect, 2018, 3, 4283–4288 CrossRef CAS.
- G. S. Nongthombam, G. K. Kharmawlong, J. E. Kumar and R. Nongkhlaw, UV365 Light promoted catalyst-free synthesis of pyrimido[4,5-b]quinoline-2,4-diones in aqueous-glycerol medium, New J. Chem., 2018, 42, 9436–9442 RSC.
- G. S. Nongthombam and R. Nongkhlaw, Experimental and theoretical studies on SPION@glutathione catalyzed synthesis of indolyl chromene, indolo xanthene, and pyrimido[4,5-b]quinoline, Synth. Commun., 2018, 48, 541–552 CrossRef CAS.
- G. Dai, Q. Li, D. Zang and Y. Wei, A Bifunctional molecular catalyst built up of L-proline grafted polyoxometalate for one-pot three-component green synthesis of heterocycles, Green Chem., 2023, 25, 6263–6269 RSC.
- J. M. Khurana, A. Chaudhary, B. Nand and A. Lumb, Aqua mediated indium(III) chloride catalyzed synthesis of fused pyrimidines and pyrazoles, Tetrahedron Lett., 2012, 53, 3018–3022 CrossRef CAS.
- K. Tabatabaeian, A. F. Shojaei, F. Shirini, S. Z. Hejazi and M. Rassa, A Green multi-component synthesis of bioactive pyrimido[4,5-b]quinoline derivatives as antibacterial agents in water catalyzed by RuCl3·xH2O, Chin. Chem. Lett., 2014, 25, 308–312 CrossRef CAS.
- K. Mohammadi, F. Shirini and A. Yahyazadeh, 1,3-Disulfonic acid imidazolium hydrogen sulfate: A Reusable and efficient ionic liquid for the one-pot multi-component synthesis of pyrimido[4,5-b]quinoline derivatives, RSC Adv., 2015, 5, 23586–23590 RSC.
- F. Shirini, M. S. N. Langarudi, N. Daneshvar, M. Mashhadinezhad and N. Nabinia, Preparation of a new DABCO-based ionic liquid and investigation on its application in the synthesis of benzimidazoquinazolinone and pyrimido[4,5-b]quinoline derivatives, J. Mol. Liq., 2017, 243, 302–312 CrossRef CAS.
- A. Gholami, M. Mokhtary and M. Nikpassand, Choline chloride/oxalic acid (chcl/oxa) catalyzed one-pot synthesis of novel azo and sulfonated pyrimido[4,5-b]quinoline derivatives, Dyes Pigm., 2020, 180, 108453 CrossRef CAS.
- R. M. Singh, N. Sharma, R. Kumar, M. Asthana and S. Upadhyay, An Alternative synthesis of pyrimido[4,5-b]quinoline-4-ones via metal-free amination in water and Vilsmeier-Haack cyclization, Tetrahedron, 2012, 68, 10318–10325 CrossRef CAS.
- M. P. Dickens, P. Roxburgh, A. Hock, M. Mezna, B. Kellam, K. H. Vousden and P. M. Fischer, 5-Deazaflavin derivatives as inhibitors of P53 ubiquitination by HDM2, Bioorg. Med. Chem., 2013, 21, 6868–6877 CrossRef CAS PubMed.
- N. S. El-Gohary, Synthesis and in vitro antitumor activity of new quinoline, pyrimido-[4,5-b]quinoline, [1,2,3]triazino[4,5-b]quinoline, and [1,2,4]triazolo[2′,3′:3,4]pyrimido-[6,5-b]quinoline analogs, Med. Chem. Res., 2013, 22, 5236–5247 CrossRef CAS.
- M. B. El-Ashmawy, M. A. El-Sherbeny and N. S. El-Gohary, Synthesis and antitumor screening of new series of pyrimido[4,5-b]quinolines and [1,2,4]triazolo[2′,3′:3,4]-pyrimido[6,5-b]quinolines, Med. Chem. Res., 2013, 22, 2724–2736 CrossRef CAS.
- S. Dudkin, V. O. Iaroshenko, V. Y. Sosnovskikh, A. A. Tolmachev, A. Villingera and P. Langer, Synthesis and reactivity of 5-polyfluoroalkyl-5-deazaalloxazines, Org. Biomol. Chem., 2013, 11, 5351–5361 RSC.
- R. G. Melik-Ohandzanyan, T. R. Hovsepyan, S. G. Israelyan, R. A. Tamazyan, A. G. Ayvazyan and G. A. Panosyan, Synthesis, molecular and crystal structure of new 9,10-substituted deazaflavins, Russ. J. Org. Chem., 2014, 50, 1161–1163 CrossRef CAS.
- H. R. P. Naik, H. S. Bhojya Naik, T. R. Ravikumar Naik, H. Raja Naik, D. S. Lamani and T. Aravinda, Pyrimido[4,5-b]quinoline-2-thiol/ol: Microwave-induced one-pot synthesis, DNA binding and cleavage studies, J. Sulfur Chem., 2008, 29, 583–592 CrossRef CAS.
- P. P. Mohire, R. B. Patil, D. R. Chandam, S. J. Jadhav, A. A. Patravale, D. R. Kumbhar, J. S. Ghosh and M. B. Deshmukh, Low transition temperature mixtures prompted one-pot synthesis of 5,10-dihydropyrimido[4,5-b]quinoline-2,4(1H,3H)-dione derivatives, Res. Chem. Intermed., 2017, 43, 7013–7028 CrossRef CAS.
- A. K. Panday, R. Mishra, A. Jana, T. Parvin and L. H. Choudhury, Synthesis of pyrimidine fused quinolines by ligand-free copper-catalyzed domino reactions, J. Org. Chem., 2018, 83, 3624–3632 CrossRef CAS PubMed.
- K. El-Gamal, Synthesis and anticancer screening of heterocyclic compounds bearing pyrimido[4,5-b]quinoline moiety, Int. J. Pharm. Sci., 2017, 8, 570–581 CAS.
- K. Husain, E. A. M. Saleh and I. Hassan, Synthesis, antibacterial, and antifungal evaluation of new class of pyrimido[4,5-d]pyrimidine, pyrazolo[3,4-d]pyrimidine, and pyrimido- [4,5-b]quinoline derived from α,α-ketene dithioacetals as fused five and six-membered heterocycle derivatives, Russ. J. Bioorg. Chem., 2023, 49, 1367–1380 CrossRef CAS.
- M. P. Wentland, J. A. Carlson, P. H. Dorff, S. C. Aldous, R. B. Perni, D. C. Young, M. G. Woods, S. D. Kingsley, K. A. Ryan, D. Rosi, M. L. Drozd and F. J. Dutko, Cyclic variations of 3-quinolinecarboxamides and effects on antiherpetic activity, J. Med. Chem., 1995, 38, 2541–2545 CrossRef CAS PubMed.
- M. Nasr, I. Nabih and J. H. Burckhalter, Synthesis of pyrimido[5,4-c]quinolines and related quinolines as potential antimalarials, J. Med. Chem., 1978, 21, 295–298 CrossRef CAS PubMed.
- W. Lewgowd, A. J. Bojarski, M. Szczesio, A. Olczak, M. L. Glowka, S. Mordalski and A. Stanczak, Synthesis and structural investigation of some pyrimido[5,4-c]quinolin-4(3H)-one derivatives with a long-chain arylpiperazine moiety as potent 5-HT1A/2A and 5-HT7 receptor ligands, Eur. J. Med. Chem., 2011, 46, 3348–3361 CrossRef CAS PubMed.
- Y. Ai, G. Yang, J. Liu, Y. Chen, L. Liu and X. Lei, Synthesis and structure elucidation of five new pyrimido[5,4-c]quinoline-4(3H)-one derivatives using 1D and 2D NMR spectroscopy, Magn. Reson. Chem., 2010, 48, 955–959 CrossRef CAS PubMed.
- E. Rajanarendar, M. N. Reddy, K. R. Murthy, K. G. Reddy, S. Raju, M. Srinivas, B. Praveen and M. S. Rao, Synthesis, antimicrobial, and mosquito larvicidal activity of 1-aryl-4-methyl-3,6-bis-(5-methylisoxazol-3-yl)-2-thioxo-2,3,6,10b-tetrahydro-1H-pyrimido[5,4-c]quinolin-5-ones, Bioorg. Med. Chem. Lett., 2010, 20, 6052–6055 CrossRef CAS PubMed.
- G. Krajsovszky, L. Károlyházy, P. Dunkel, S. Boros, A. Grillo and P. Mátyus, Suzuki-Aza-Wittig, Suzuki-condensation and Aza-Wittig-electrocyclic ring-closure tandem reactions for synthesis of fused nitrogen-containing ring systems, Arkivoc, 2011,(10), 229–253 Search PubMed.
- M. A. Ibrahim, H. M. Hassanin, Y. A. Gabr and Y. A. Alnamer, Studies on the chemical behavior of 3-(nitroacetyl)-1-ethyl-4-hydroxyquinolin-2(1H)-one towards some electrophilic and nucleophilic reagents, J. Braz. Chem. Soc., 2012, 23, 905–912 CrossRef CAS.
- M. A. Ismail, S. Al-Shihry, R. K. Arafa and U. El-Ayaan, Synthesis, antimicrobial activity and molecular modeling study of substituted 5-aryl-pyrimido[5,4-c]quinoline-2,4-diones, J. Enzyme Inhib. Med. Chem., 2013, 28, 530–538 CrossRef CAS PubMed.
- A.-F. E. Mourad, A. A. Amer, K. M. El-Shaieb, A. M. Ali and A. A. Aly, 4-Hydroxy-1-phenylquinolin-2(1H)-one in one-pot synthesis of pyrimidoquinolines and related compounds under microwave irradiation and conventional conditions, J. Heterocycl. Chem., 2016, 53, 383–388 CrossRef CAS.
- S. Mubeen, A. Rauf and A. M. Qureshi, Synthesis of new quinoline scaffolds via a solvent-free fusion method and their anti-microbial properties, Trop. J. Pharm. Res., 2018, 17, 1853–1858 CrossRef CAS.
- V. Nadaraj, S. Thamarai and M. Abirami, Modified Biginelli reaction: Synthesis of pyrimidoquinoline derivatives, Asian J. Chem., 2019, 31, 1243–1245 CAS.
- P. K. Agarwal, S. K. Sharma, D. Sawant and B. Kundu, Application of the Pictet-Spengler reaction to aryl amine-based substrates having pyrimidine as a π-nucleophile: Synthesis of pyrimidoquinolines with structural analogy to benzonaphthyridines present in alkaloids, Tetrahedron, 2009, 65, 1153–1161 CrossRef CAS.
- E. Okada, M. Hatakenaka, S. Nakano, T. Sakaemura, T. Mori and T. Terauchi, Efficient syntheses of fluorine-containing pyrimido[5,4-c]quinolines and benzo[h][1,6]-naphthyridines by condensation reactions of 3-trifluoroacetylquinolin-4-amine with aldehydes and ketones, Heterocycles, 2014, 89, 2303–2317 CrossRef CAS.
- F. Zhang, X. Zhai, L. J. Chen, J. G. Qi, B. Cui, Y. C. Gu and P. Gong, Synthesis and cytotoxic activity of 2,5-disubstituted pyrimido[5,4-c]quinoline derivatives, Chin. Chem. Lett., 2011, 22, 1277–1280 CrossRef CAS.
- R. A. Mekheimer, S. M. R. Allam, M. A. Al-Sheikh, M. S. Moustafa, S. M. Al-Mousawi, Y. A. Mostafa, B. G. M. Youssif, H. A. M. Gomaa, A. M. Hayallah, M. Abdelaziz and K. U. Sadek, Discovery of new pyrimido[5,4-c]quinolines as potential antiproliferative agents with multitarget actions: Rapid synthesis, docking, and ADME studies, Bioorg. Chem., 2022, 121, 105693 CrossRef CAS PubMed.
- H. Fenner and R. Teichmann, Pyrimido[5,4-b]chinoline-10-deaza-alloxazine, Arch. Pharm., 1978, 311, 115–125 CrossRef CAS PubMed.
- H. Fenner, C. Hentzsch and C. Wiegreffe, Pyrimido[5,4-b]chinoline durch trialkyl-phosphit-cyclisierung von 2-nitro-6′-benzylpyrimidindionen, Arch. Pharm., 1986, 319, 379–381 CrossRef CAS.
- H. Fenner, C. Wiegreffe and B. Iffert, Pyrimido[4,5-c]chinoline aus 10-cyanopyrimido- [5,4-b]chinolinen, Arch. Pharm., 1986, 319, 382–384 CrossRef CAS.
- K. Metwally, A. Khalil, H. Pratsinis and D. Kletsas, Synthesis, in vitro cytotoxicity, and a preliminary structure-activity relationship investigation of pyrimido[4,5-c]quinolin-1(2H)-ones, Arch. Pharm., 2010, 343, 465–472 CrossRef CAS PubMed.
- F. Pierre, S. E. O'Brien, M. Haddach, P. Bourbon, M. K. Schwaebe, E. Stefan, L. Darjania, R. Stansfield, C. Ho, A. Siddiqui-Jain, N. Streiner, W. G. Rice, K. Anderes and D. M. Ryckman, Novel potent pyrimido[4,5-c]quinoline inhibitors of protein kinase CK2: SAR and preliminary assessment of their analgesic and antiviral properties, Bioorg. Med. Chem. Lett., 2011, 21, 1687–1691 CrossRef CAS PubMed.
- I. V. Ukrainets, L. V. Sidorenko, O. V. Gorokhova and S. V. Slobodzyan, 4-Hydroxy-2-quinolones. 112. Reaction of 2-ethoxycarbonylmethyl-4H-3,1-benzoxazin-4-one with active methylene compounds, Chem. Heterocycl. Compd., 2007, 43, 63–66 CrossRef CAS.
- A. P. Marjani, J. Khalafy and A. R. M. Ebrahimlo, Facile synthesis of some new pyrimido-quinolines, Synth. Commun., 2011, 41, 2475–2482 CrossRef CAS.
- A. P. Marjani and J. Khalafy, A Short and efficient method for the synthesis of pyrimido- [1,2-a]quinolines, Chem. Heterocycl. Compd., 2011, 47, 96–100 CrossRef CAS.
- A. G. Jadhav and N. K. Halikar, Synthesis and biological activity of pyrimido[1,2-a]-quinoline moiety and its 2-substituted derivatives, J. Phys.: Conf. Ser., 2013, 423, 12007 CrossRef CAS.
- S. Soleimani-Amiri, Z. Hossaini, M. Arabkhazaeli, H. Karami and S. A. S. Abad, Green synthesis of pyrimidoisoquinolines and pyrimidoquinoline using ZnO nanorods as an efficient catalyst: study of antioxidant activity, J. Chin. Biochem. Soc., 2019, 66, 438–445 CrossRef CAS.
- D. Ramanathan and K. Pitchumani, Copper(I)-Y Zeolite-catalyzed regio- and stereoselective [2+2+2] cyclotrimerization cascade: An Atom- and step-economical synthesis of pyrimido[1,6-a]quinoline, J. Org. Chem., 2015, 80, 10299–10308 CrossRef CAS PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Samar M. R. Allamb,
Mariam A. Al-Sheikhc,
Hanadi Y. Medrasic,
Mohamed Abd-Elmonema and
Kamal U. Sadek
*a,
Samar M. R. Allamb,
Mariam A. Al-Sheikhc,
Hanadi Y. Medrasic,
Mohamed Abd-Elmonema and
Kamal U. Sadek