DOI:
10.1039/D5RA00657K
(Paper)
RSC Adv., 2025,
15, 21838-21858
Harnessing thiazole chemistry for antifungal strategies through an experimental and computational chemistry approach: anti-biofilm, molecular docking, dynamics, and DFT analysis†
Received
28th January 2025
, Accepted 13th June 2025
First published on 26th June 2025
Abstract
This study reports the design, synthesis, and evaluation of four novel (E)-2-(2-(1-(5-chlorothiophen-2-yl)ethylidene)hydrazineyl)-4-(aryl)thiazole derivatives (4a–4d) as potential anti-biofilm agents against Candida albicans. The compounds were structurally characterized by FT-IR, 1H NMR, 13C NMR, and HRMS spectral techniques. Biofilm inhibition assays revealed that derivatives 4a–4c suppressed over 50% of biofilm formation at a concentration of 12.5 μg mL−1, although exopolysaccharide production remained largely unaffected. Molecular docking indicated strong binding affinities toward lanosterol 14α-demethylase, with 4a achieving the highest docking score (−8.715 kcal mol−1) through hydrogen bonding and π–π stacking interactions. Stability of the 4c–protein complex was confirmed by molecular dynamics simulations, supported by RMSD and flexibility analyses. An in-depth computational analysis was also performed on the most active thiazole derivative, compound 4c. DFT and NBO analyses of 4c indicated favourable geometry and key electron delocalization, while ELF, LOL, NCI, and RDG studies highlighted the role of non-covalent interactions in stabilizing the molecular framework. Additionally, the ADME profile of 4c demonstrated favourable pharmacokinetic properties, including high gastrointestinal absorption and a moderate lipophilicity index, highlighting its potential as a lead antifungal scaffold.
1 Introduction
Antimicrobial resistance has become more prevalent, especially in fungal infections, which has increased the need for innovative treatment using pharmaceuticals.1,2 Because of their potential to be effective biofilm inhibitors, the synthesis of hybrid heterocyclic compounds containing the thiazole moiety, has attracted a lot of attention in this regard.3 When compared to their mono-heterocyclic counterparts, these compounds which combine several pharmacophores into a single chemical framework have demonstrated increased biological activity.4–6 Potential therapeutic candidates containing thiophene and thiazole are recognised for their varied pharmacological actions, which include antibacterial,7,8 anti-inflammatory,9,10 anticancer characteristics11,12 and many others.13–15 Particularly thiophene has been shown to increase the antimicrobial effectiveness of synthetic compounds, making it an important ingredient in the development of new antifungal agents.16
One common fungal pathogen that can be quite dangerous to people with weakened immune systems is Candida albicans.17 Being able to build biofilms on a variety of surfaces, including host tissues and medical equipment, is a crucial component of its pathogenesis.18 The formation of biofilms complicates treatment of C. albicans infections by conferring resistance to antifungal therapies and the immune response of the host.19 Antifungal medicines are unable to penetrate and effectively treat biofilms due to their thick extracellular matrix and modified microenvironment, which increases the possibility of recurrent infections as well as morbidity and mortality. Therefore, developing compounds that inhibit the formation of biofilms is crucial for enhancing the effectiveness of treatment.20–22 One innovative way of developing new antifungal agents is to synthesise hybrid heterocyclic compounds with potent activity.23,24 Through exploiting the combined advantages of thiophene and thiazole moieties, these compounds have significant antifungal properties.25–27 Some biologically potent anti-biofilm thiazole derivatives (I to V) are depicted in Fig. 1. Gondru et al. synthesized pyrazole–thiazole hybrids,28 Bhosale et al. focused on Schiff base derivatives containing 4,5-disubstituted thiazole,29 Kassab et al. reported on isatin-decorated thiazole derivatives,30 Gondru et al. studied 1,2,3-triazole–thiazole hybrids,31 and Dhonnar et al. investigated 2-(2-hydrazineyl)thiazole derivatives32 which showed good biofilm inhibition and notable antifungal activity. In view of this, molecular hybridization of thiazole scaffolds has proven to be a powerful strategy in enhancing anti-biofilm activity by combining complementary pharmacophores to target microbial resistance and disrupt biofilm formation effectively.
 |
| | Fig. 1 Antibiofilm activity of some thiazole-based hybrid compounds. | |
Computer-aided drug design and molecular docking simulations provide valuable insights into the molecular interactions between potential drug candidates and their biological targets.33,34 Molecular docking enables the prediction of binding affinities and the identification of critical interactions that contribute to the biological effectiveness of synthesized compounds.35,36 Integrating computational analyses with experimental evaluations allows researchers to more efficiently identify effective biofilm inhibitors, minimize the time and resources needed for developing new therapies, and accelerate the drug discovery process.37 This work is noteworthy because it addresses the critical need for additional antifungal medications that can effectively treat C. albicans-caused biofilm-associated conditions. Through the synthesis, evaluation, and extensive in silico studies of (E)-2-(2-(1-(5-chlorothiophen-2-yl)ethylidene)hydrazineyl)-4-(aryl)thiazoles (4a–4d), this research expands the comprehension of the molecular foundations of biofilm inhibition and establishes the groundwork for the development of more potent antifungal treatments. The results of this investigation could widen up the possibilities to innovative therapeutic approaches that can get around the shortcomings of the antifungal drugs while improving outcomes.
2 Materials and methods
2.1. General remarks
All the chemicals used in this study were sourced from reputable suppliers, including Sigma-Aldrich, Avra Synthesis, and HiMedia, ensuring high-quality reagents for reliable experimental outcomes. The glassware utilized throughout the research was procured from Borosil Glass Works Ltd. (India). Additionally, plastic ware employed in the experiments was obtained from Tarson Products Pvt Ltd. Fourier-transform infrared (FT-IR) spectroscopy was conducted using a Shimadzu spectrophotometer to identify functional groups and analyze the molecular structure of the compounds. Nuclear magnetic resonance (NMR) spectroscopy, an essential technique for elucidating chemical structures, was performed on a Bruker Avance III HD NMR 500 MHz instrument. Thin-layer chromatography (TLC) was used to monitor the progress of chemical reactions and evaluate the purity of synthesized compounds. The TLC analyses were carried out on Merck TLC plates. These advanced tools and high-quality materials collectively ensured the accuracy and reproducibility of the experimental results.
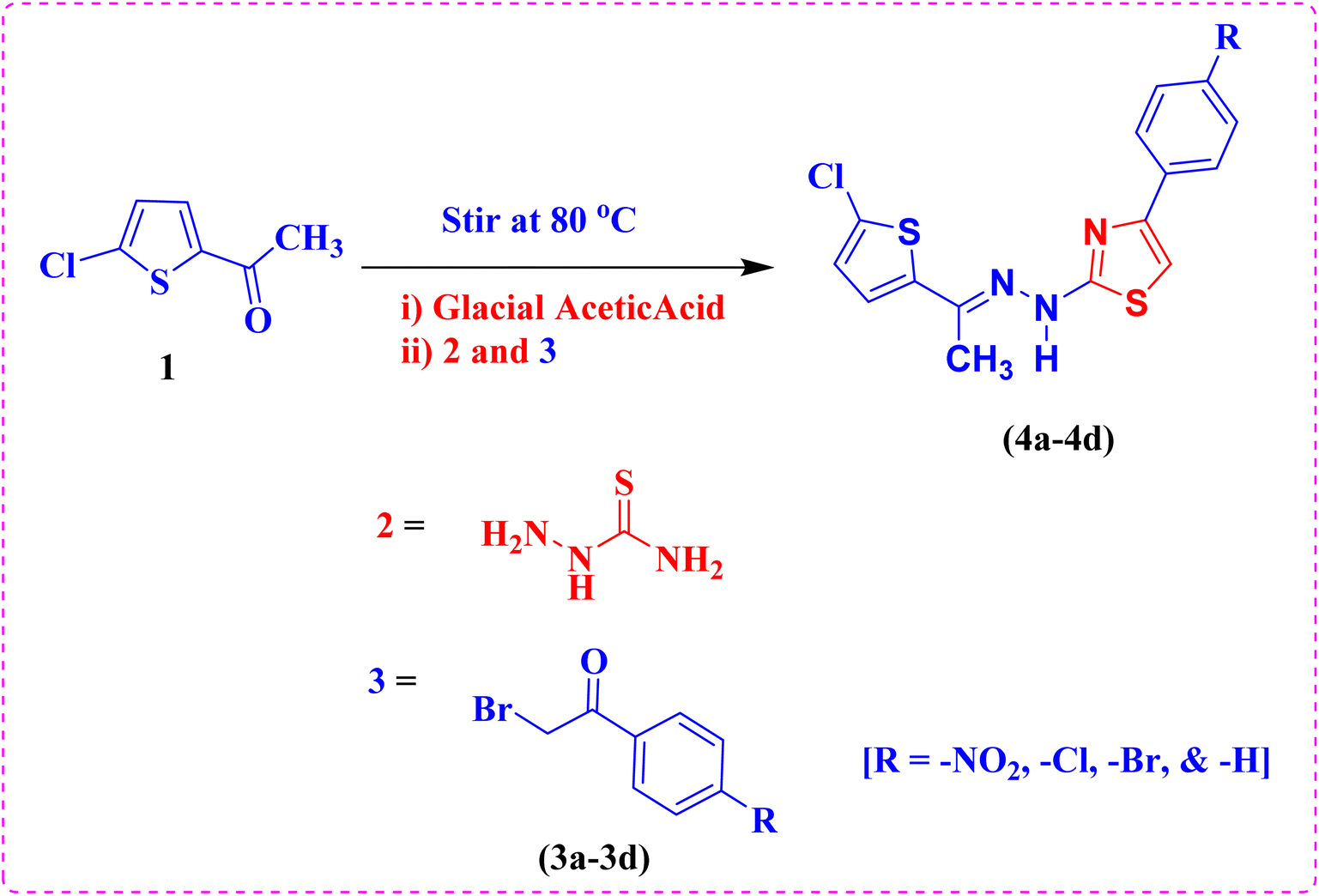
2.2. Synthesis of (E)-2-(2-(1-(5-chlorothiophen-2-yl)ethylidene)hydrazineyl)-4-(aryl)thiazoles (4a–4d)
To synthesize (E)-2-(2-(1-(5-chlorothiophen-2-yl)ethylidene)hydrazineyl)-4-(aryl)thiazoles, a precise stoichiometric reaction was employed. A solution comprising 5-chloro-2-acetyl thiophene (1, 0.01 mol) and thiosemicarbazide (2, 0.01 mol) was prepared in 20 mL of ethanol, contained within a conical flask. To catalyze the reaction, 2–3 drops of glacial acetic acid were added to the reaction mixture, which was then subjected to continuous stirring and maintained at a temperature of 80 °C for 45 minutes. Subsequently, phenacyl bromide derivatives (3a–3d, 0.01 mol) were incrementally introduced to the reaction system. The reaction mass was stirred further for 10–15 minutes to ensure completion. The progression of the reaction was meticulously monitored using TLC. Upon confirming the reaction's completion via TLC, the reaction mixture was allowed to cool to ambient temperature. The resultant precipitate was filtered using a Büchner funnel under vacuum, ensuring efficient recovery of the product. The obtained solid was dried under vacuum conditions and characterized extensively using FT-IR, NMR and HRMS methods to confirm the structural integrity of the synthesized compounds. The synthetic route and chemical transformations involved are illustrated in Scheme 1.
 |
| | Scheme 1 Synthesis of (E)-2-(2-(1-(5-chlorothiophen-2-yl)ethylidene)hydrazineyl)-4-(4-aryl)thiazoles (4a–4d). | |
2.3. Spectral data
(E)-2-(2-(1-(5-Chlorothiophen-2-yl)ethylidene)hydrazineyl)-4-(4-nitrophenyl)thiazole (4a): yield: 94% (3.56 g); mp 256–258 °C.
FT-IR (cm−1): 3340.71, 3107.32, 2918.30, 2814.14, 1564.27, 1500.62, 1448.54, 1330.88, 1273.02, 1209.37, 1107.14, 1043.49, 1002.98, 844.82, 779.24, 707.88, 653.87, 524.64, 474.49, 1H NMR (500 MHz, DMSO-d6) δ: 11.51 (s, 1H), 8.29 (d, J = 9.0 Hz, 2H), 8.12 (d, J = 9.0 Hz, 2H), 7.74 (s, 1H), 7.27 (d, J = 4.0 Hz, 1H), 7.09 (d, J = 4.0 Hz, 1H), 2.30 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ: 169.42, 148.49, 146.08, 142.86, 142.20, 140.59, 129.70, 127.39, 126.21, 126.05, 124.03, 109.12, 13.51; HRMS: (M + H) calculated for molecular formula C15H11ClN4O2S2 379.0090, found: 379.0085.
(E)-4-(4-Chlorophenyl)-2-(2-(1-(5-chlorothiophen-2-yl)ethylidene)hydrazineyl)thiazole (4b): yield: 90% (3.31 g); mp 214–216 °C.
FT-IR (cm−1): 3313.35, 3033.58, 2914.42, 1617.13, 1489.07, 1430.44, 1364.17, 1292.99, 1189.89, 1093.21, 1055.63, 1004.18, 910.61, 807.63, 763.96, 726.71, 711.44, 660.02; 1H NMR (500 MHz, DMSO-d6) δ: 11.33 (s, 1H), 7.88 (d, J = 8.6 Hz, 2H), 7.47 (d, J = 8.6 Hz, 2H), 7.40 (s, 1H), 7.26 (d, J = 4.0 Hz, 1H), 7.09 (d, J = 4.0 Hz, 1H), 2.29 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ: 169.14, 142.72, 142.29, 141.85, 133.31, 131.85, 129.56, 129.46, 128.55, 128.33, 127.38, 127.12, 125.94, 104.99, 13.48; HRMS: (M + H) calculated for molecular formula C15H11Cl2N3S2-367.9849, found: 367.9849.
(E)-4-(4-Bromophenyl)-2-(2-(1-(5-chlorothiophen-2-yl)ethylidene)hydrazineyl)thiazole (4c): yield 88% (3.64 g); mp: 228–230 °C.
FT-IR (cm−1): 3317.76, 3033.83, 2915.74, 1616.89, 1571.86, 1483.11, 1428.73, 1363.57, 1282.08, 1187.99, 1108.28, 1071.01, 1003.50, 909.48, 807.62, 760.25, 724.92, 694.83; 1H NMR (500 MHz, DMSO-d6) δ: 11.65 (s, 1H), 7.81 (d, J = 8.5 Hz, 2H), 7.60 (d, J = 8.6 Hz, 2H), 7.41 (s, 1H), 7.26 (d, J = 4.0 Hz, 1H), 7.08 (d, J = 4.0 Hz, 1H), 2.29 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ: 169.71, 143.27, 142.87, 134.25, 132.03, 131.83, 130.30, 130.13, 128.00, 127.96, 126.51, 121.01, 105.65, 14.05; HRMS: (M + H + 2) calculated for molecular formula C15H11BrClN3S2-413.9501, found: 413.9325.
(E)-2-(2-(1-(5-Chlorothiophen-2-yl)ethylidene)hydrazineyl)-4-phenylthiazole (4d): yield: 84% (2.81 g); mp 223–225 °C.
FT-IR (cm−1): 3315.75, 3032.10, 2929.87, 2738.92, 1612.49, 1494.83, 1435.04, 1365.60, 1286.52, 1213.23, 1112.93, 1002.98, 810.10, 752.24, 711.73, 580.57, 536.21, 472.56; 1H NMR (500 MHz, DMSO-d6) δ: 11.29 (s, 1H), 7.86 (d, J = 7.2 Hz, 2H), 7.44–7.39 (m, J = 7.7 Hz, 2H), 7.34–7.28 (m, 2H), 7.26 (d, J = 4.0 Hz, 1H), 7.09 (d, J = 4.0 Hz, 1H), 2.29 (s, 3H); 13C NMR (126 MHz, DMSO-d6) δ: 169.04, 142.73, 142.37, 134.32, 129.52, 128.53, 127.77, 127.49, 127.38, 125.87, 125.41, 104.10, 13.48; HRMS: (M + H) calculated for molecular formula C15H12ClN3S2-334.02394, found: 334.0236.
2.4. Anti-biofilm study
2.4.1. Bacterial strain and growth medium. A standard culture of Candida albicans ATCC 227 was a kind gift from DR Zore. Overnight culture of C. albicans in YPD (yeast, peptone, dextrose) broth is used for each experiment.
2.4.2. Biofilm inhibition assay. The biofilm inhibition assay was conducted using 96-well polystyrene plates designed for tissue culture. Each well was seeded with 100 μL of a cell suspension (1 × 107 cells per mL) and incubated at 37 °C for 1.5 hours to allow cell adhesion to the polystyrene surface. Following the adhesion phase, wells were rinsed with sterile distilled water. A series of compounds (4a–4d) at concentrations ranging from 12.5 to 100 μg mL−1 were prepared in RPMI-1640 medium containing 1% dimethyl sulfoxide (DMSO). Subsequently, 100 μL of these solutions were added to the pre-seeded wells, and the plates were incubated at 37 °C for 24 hours. After the incubation period, non-adherent cells were removed, and the wells were gently rinsed to eliminate residual unadhered cells. A 25 μL (1000 μg mL−1) of 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide was added to each well, followed by incubation in the dark for 3 hours. To solubilize the formed formazan crystals, 100 μL of DMSO was added to each well. The optical density was measured at 570 nm using a microplate reader (Hidex, Germany). All experiments were performed in quadruplicate. Apart from quantitative measure of biofilm, the structural feature of biofilm was observed under microscope at sub-MIC value.
2.4.3. Effect of thiazole derivatives on hyphae formation. The hyphae formation in biofilm structure was prepared on glass slide in 24-well plates. Firstly, biofilm was formed in presence of sub-MIC concentrations of thiazole derivatives. Instead of subjecting the formed biofilm to MTT assay, the biofilm was subjected to series of alcohol dehydration and then observed under optical microscope (Cell discoverer, Ziess).
2.4.4. Quantitative measurement of bound exopolysaccharides. To quantify bound exopolysaccharides (EPS), an overnight culture of C. albicans was used. A total of 300 μL of the cell suspension was seeded into each well of a 24-well plate and incubated at 37 °C for 1.5 hours to facilitate cell adhesion. Post-adhesion, the wells were rinsed with sterile distilled water, and 750 μL of RPMI medium containing 25 μg mL−1 of compounds (4a–4d) was added to each well. The plates were incubated in the dark at 37 °C for 24 hours. After incubation, the wells were washed twice with 0.9% NaCl, and 1 mL of 0.9% NaCl was added to each well. The biofilm cells were scraped off, collected in microcentrifuge tubes, and centrifuged at 10![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 rpm for 5 minutes. The supernatant was then transferred to a new tube, and two volumes of chilled ethanol were added, followed by overnight incubation at 4 °C. The tubes were centrifuged again at 10000 rpm for 30 minutes, and the precipitate was collected and resuspended in sterile distilled water. The phenol–sulfuric acid method was employed for EPS quantification. A 200 μL sample was combined with 100 μL of 5% phenol and 500 μL of concentrated sulphuric acid, and then kept in the dark for 30 minutes. The EPS content was measured at 490 nm using a multimode plate reader (Hidex Sense, Hidex).
000 rpm for 5 minutes. The supernatant was then transferred to a new tube, and two volumes of chilled ethanol were added, followed by overnight incubation at 4 °C. The tubes were centrifuged again at 10000 rpm for 30 minutes, and the precipitate was collected and resuspended in sterile distilled water. The phenol–sulfuric acid method was employed for EPS quantification. A 200 μL sample was combined with 100 μL of 5% phenol and 500 μL of concentrated sulphuric acid, and then kept in the dark for 30 minutes. The EPS content was measured at 490 nm using a multimode plate reader (Hidex Sense, Hidex).
2.4.5. Molecular docking. Molecular docking studies were performed using the GLIDE module in Schrödinger Maestro to evaluate the interactions between ligands (4a–4d) and lanosterol 14-alpha demethylase (PDB ID 5EQB). The 2D structures of the ligands (4a–4d) were designed in ChemDraw (2022) and saved in SDF format. Ligand preparation was carried out using the Maestro Build Panel, followed by optimization with LigPrep 2.2. This process generated low-energy conformers using the OPLS 2005 force field, ensuring that the ligands were in their most stable conformations prior to docking.38 The X-ray crystal structure of the 5EQB protein was retrieved from the RCSB Protein Data Bank. Protein preparation was conducted using the Protein Preparation Wizard in Maestro 8.0.39 During this step, bond orders were assigned, water molecules were removed, and hydrogens were added to maintain a physiological pH of 7.0.39,40 Missing side chains and loops were reconstructed using Epik Prime, and terminal regions were capped. The protein structure was minimized using the OPLS 2005 force field to optimize geometry.41 The binding site of the protein was defined based on the co-crystallized ligand, and a receptor grid was generated. Docking simulations were carried out in Standard Precision (SP) mode using the Glide tool, and the results were analyzed to assess the ligand–protein binding interactions.42
2.4.6. Molecular dynamics. Molecular dynamics (MD) simulations were conducted for the protein–ligand complexes involving the 4c compound in complex with lanosterol 14-alpha demethylase (5EQB) using the Desmond program, an explicit solvent MD package, along with the OPLS_2005 force field.43 The protein–ligand complex was prepared using the Protein Preparation Wizard, employing the predefined SPC (simple point charge) water model and an orthorhombic box with dimensions of 3 Å × 3 Å × 3 Å. Sodium chloride, at a physiological concentration of 0.15 M, was introduced into 10 Å buffer region between the protein atoms and the simulation box to maintain ionic strength, using the system-builder option.44 To relax the system into a local energy minimum, energy minimization was performed, followed by a 100 ns MD simulation using the OPLS_2005 force field. The Nose–Hoover chain thermostat was used to maintain the temperature at 300 K, while the Martyna–Tobias–Klein barostat was applied at a pressure of 1.01325 bar, with isotropic coupling.45,46 A coulombic cutoff of 0.9 nm was employed, while other parameters were kept at default settings.47 The MD simulation trajectories were analyzed using the Simulation Interaction Diagram (SID) tool to assess ligand–receptor interactions.48,49 From the results of the MD simulation, we evaluated the various parameters, including the root mean square deviation (RMSD), radius of gyration (Rg), root mean square fluctuation (RMSF), and the number of hydrogen bonds (H-bonds). We also created a plot illustrating ligand interactions within the binding cavity, developed a stacked bar chart for protein–ligand contact analysis, and performed a total contacts timeline analysis of the MD trajectory. Additionally, we examined the torsional degrees of freedom for the rotatable bonds of the selected ligand over the 100 ns simulation of the docked protein–ligand complex.
2.4.7. Free energy landscape. The free energy landscape (FEL) of the 4c compound in complex with the lanosterol 14-alpha demethylase during protein folding was analyzed. The FEL provides valuable insights into the dynamics of biological processes by examining its minima, which represent stable states of the system, and its barriers, which represent transient states.50 This analysis offers a comprehensive understanding of protein folding behavior. The MD trajectory was used to generate a 3-dimensional energy profile plot, with RMSD and radius of gyration (Rg) as variables, using the Schrödinger Python script python3 Fel.py.
2.4.8. Computational details. Molecular docking and MD simulation study was performed by Glide and Desmond module of Schrodinger, respectively. All Density Functional Theory (DFT) simulations were conducted at B3LYP functional and 6-311++G(d,p) basis set using the Gaussian 9W software package.51–57 Results were visualized with GaussView 6 and Chemcraft software.58,59 For topological analysis, Multiwfn software and the https://Atomistica.online web tool were utilized, with visual representations prepared in VMD.60–63
3 Results and discussion
3.1. Chemistry
The synthesis of (E)-2-(2-(1-(5-chlorothiophen-2-yl)ethylidene)hydrazineyl)-4-(aryl)thiazoles (4a–4d) involve a one pot reaction process. Initially, equimolar amounts of 5-chloro-2-acetyl thiophene (1) and thiosemicarbazide (2) were dissolved in ethanol. To this reaction mixture, 2–3 drops of glacial acetic acid were added as a catalyst. The mixture was then stirred and heated at 80 °C for 45 minutes to facilitate the formation of an intermediate thiosemicarbazone. Subsequently, phenacyl bromide (3a–3d) was added to the reaction mixture. This step aims to introduce the phenacyl group to form the desired thiazole ring. The resulting mixture was stirred for an additional 10–15 minutes to ensure the completion of the reaction. The synthesized compounds were characterized using FT-IR, 1H NMR, and 13C NMR and mass spectral methods. IR spectroscopy provided information about the functional groups present in the molecule, while NMR spectroscopy offered detailed insights into the molecular structure and the environment of specific atoms within the compound. These characterization techniques confirmed the successful synthesis of the target thiazole derivatives and provided essential data for further analysis and application of these compounds.
The IR spectroscopic analysis of (E)-2-(2-(1-(5-chlorothiophen-2-yl)ethylidene)hydrazineyl)-4-(4-aryl)thiazoles (4a–4d) revealed several characteristic peaks corresponding to various functional groups and structural features in the compounds. The broad absorption peak at 3310–3340 cm−1 can be attributed to the N–H stretching vibration, indicating the presence of a hydrazineyl group. The peak around 3000–3100 cm−1 corresponds to the aromatic C–H stretching, confirming the aromatic nature of the thiophene and phenyl rings. The peaks between 2920 cm−1 and 2810 cm−1 are indicative of aliphatic C–H stretching vibrations. The strong absorption at 1564.27 cm−1 in compound 4a is characteristic of the nitro group's (NO2) asymmetric stretching vibration, while the peak at 1500.62 cm−1 is due to the symmetric stretching of the nitro group. The strong absorption around 1620 cm−1 is characteristic of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N stretching vibration. The peaks between 1600 and 1450 cm−1 corresponds to CC stretching vibrations in the aromatic rings. The absorptions at 1300–1250 cm−1 are attributed to C–N stretching vibrations, which are consistent with the thiazole and hydrazone structures. The peak around 1200 cm−1 is assigned to C–S stretching vibrations of the thiazole ring. The peak around 1000 cm−1 may correspond to out-of-plane C–H bending vibrations in the aromatic rings. The peaks at 850–750 cm−1 are indicative of aromatic C–H out-of-plane bending vibrations. The absorption bands at 650–720 cm−1 can be assigned to the C–Cl stretching vibration in the chlorothiophene moiety. Finally, the peaks at 450–450 cm−1 and correspond to skeletal vibrations involving the thiazole ring and other molecular framework vibrations. The singlet nearly at 12 ppm is assigned to the NH proton of the hydrazineyl group. The two doublets in the aromatic region, i.e. 8–7.5 with J = 8.0 to 8.9 Hz correspond to the aromatic protons of the p-di-substituted benzene ring in the compounds 4a–4c. The singlet at 7.40 to 7.75 ppm (1H) in all four compounds is assigned to the proton on the thiazole ring, which is deshielded due to the electron-withdrawing effects of the adjacent nitrogen and sulfur atoms. The doublets around 7.27 ppm (1H, J = 4.0 Hz) and 7.10 ppm (1H, J = 4.0 Hz) correspond to the protons on the 5-chlorothiophene ring. Finally, the singlet around 2.30 ppm (3H) is attributed to the methyl protons (–CH3) attached to the ethylidene group, reflecting the typical chemical shift for aliphatic protons adjacent to an electron-withdrawing group. The 13C NMR spectroscopic analysis of revealed a series of carbon signals that provide insights into the molecular structure and environment of the carbon atoms within the compound. The signal at 170 ppm is assigned to the carbon in the ethylidene group adjacent to the hydrazineyl nitrogen, which is highly deshielded due to the electron-withdrawing effects of the adjacent nitrogen and the delocalization of electrons. The signals between 110 and 150 ppm are attributed to the benzene, thiazole and 5-chlorothiophene ring. The signal at 110 ppm is attributed to the carbon in the 5-chlorothiophene ring that is ortho to the chlorine substituent, indicating a relatively shielded environment. Finally, the peak around 14 ppm is assigned to the methyl carbon (CH3) in the ethylidene group, reflecting the typical chemical shift for aliphatic carbons in such a position. High-resolution mass spectrometry (HRMS) was employed to confirm the molecular mass of compounds 4a–4d. The molecular formula of the compound, C15H12ClN4O2S2, corresponds to a calculated monoisotopic mass of 378.0085 Da. In the positive ion mode, a prominent peak was observed at m/z 379.0085, which corresponds to the protonated molecular ion [M + H]+. This closely matches the theoretical value. A second significant peak appeared at m/z 381.0056, consistent with the [M + H + 2]+ ion. This is attributable to the natural isotopic abundance of chlorine, which exists as two isotopes: 35Cl (∼75.8%) and 37Cl (∼24.2%), creating the characteristic +2 isotope pattern. Compound 4b exhibits a triplet isotopic pattern with an M:M + 2:M + 4 ratio of approximately 9:6:1, indicative of two chlorine atoms. This matches the molecular structure which includes both a chlorothiophene and a 4-chlorophenyl group. The precise mass of m/z 367.9849 confirms the monoisotopic [M + H]+ peak, and the isotopic spacing further validates the structure. The HRMS data of compound 4c, confirms its molecular composition. The spectrum exhibits a prominent molecular ion peak at m/z 413.9325, which corresponds to the protonated molecular ion [M + H + 2]+ of the expected molecular formula C15H10BrClN3S2. The isotopic distribution, characterized by a pattern of M, M + 2, and M + 4 peaks, further supports the presence of these halogens due to their natural isotopic abundances (79Br/81Br and 35Cl/37Cl). The HRMS of compound 4d, provides strong evidence for its molecular identity. The spectrum displays a dominant molecular ion peak at m/z 334.0236, which corresponds to the protonated molecular ion [M + H]+. This value is consistent with the calculated monoisotopic mass for the molecular formula C15H11ClN3S2, confirming the presence of a chlorine atom and two sulfur atoms, characteristic of the thiazole and chlorothiophene moieties. The accompanying isotopic peak at m/z 336.0204 reflects the presence of the 37Cl isotope, further validating the incorporation of chlorine. The high intensity and resolution of the peak, along with the clear isotopic pattern, demonstrate the compound's purity and accurate molecular structure, affirming the successful synthesis of compound 4d.
N stretching vibration. The peaks between 1600 and 1450 cm−1 corresponds to CC stretching vibrations in the aromatic rings. The absorptions at 1300–1250 cm−1 are attributed to C–N stretching vibrations, which are consistent with the thiazole and hydrazone structures. The peak around 1200 cm−1 is assigned to C–S stretching vibrations of the thiazole ring. The peak around 1000 cm−1 may correspond to out-of-plane C–H bending vibrations in the aromatic rings. The peaks at 850–750 cm−1 are indicative of aromatic C–H out-of-plane bending vibrations. The absorption bands at 650–720 cm−1 can be assigned to the C–Cl stretching vibration in the chlorothiophene moiety. Finally, the peaks at 450–450 cm−1 and correspond to skeletal vibrations involving the thiazole ring and other molecular framework vibrations. The singlet nearly at 12 ppm is assigned to the NH proton of the hydrazineyl group. The two doublets in the aromatic region, i.e. 8–7.5 with J = 8.0 to 8.9 Hz correspond to the aromatic protons of the p-di-substituted benzene ring in the compounds 4a–4c. The singlet at 7.40 to 7.75 ppm (1H) in all four compounds is assigned to the proton on the thiazole ring, which is deshielded due to the electron-withdrawing effects of the adjacent nitrogen and sulfur atoms. The doublets around 7.27 ppm (1H, J = 4.0 Hz) and 7.10 ppm (1H, J = 4.0 Hz) correspond to the protons on the 5-chlorothiophene ring. Finally, the singlet around 2.30 ppm (3H) is attributed to the methyl protons (–CH3) attached to the ethylidene group, reflecting the typical chemical shift for aliphatic protons adjacent to an electron-withdrawing group. The 13C NMR spectroscopic analysis of revealed a series of carbon signals that provide insights into the molecular structure and environment of the carbon atoms within the compound. The signal at 170 ppm is assigned to the carbon in the ethylidene group adjacent to the hydrazineyl nitrogen, which is highly deshielded due to the electron-withdrawing effects of the adjacent nitrogen and the delocalization of electrons. The signals between 110 and 150 ppm are attributed to the benzene, thiazole and 5-chlorothiophene ring. The signal at 110 ppm is attributed to the carbon in the 5-chlorothiophene ring that is ortho to the chlorine substituent, indicating a relatively shielded environment. Finally, the peak around 14 ppm is assigned to the methyl carbon (CH3) in the ethylidene group, reflecting the typical chemical shift for aliphatic carbons in such a position. High-resolution mass spectrometry (HRMS) was employed to confirm the molecular mass of compounds 4a–4d. The molecular formula of the compound, C15H12ClN4O2S2, corresponds to a calculated monoisotopic mass of 378.0085 Da. In the positive ion mode, a prominent peak was observed at m/z 379.0085, which corresponds to the protonated molecular ion [M + H]+. This closely matches the theoretical value. A second significant peak appeared at m/z 381.0056, consistent with the [M + H + 2]+ ion. This is attributable to the natural isotopic abundance of chlorine, which exists as two isotopes: 35Cl (∼75.8%) and 37Cl (∼24.2%), creating the characteristic +2 isotope pattern. Compound 4b exhibits a triplet isotopic pattern with an M:M + 2:M + 4 ratio of approximately 9:6:1, indicative of two chlorine atoms. This matches the molecular structure which includes both a chlorothiophene and a 4-chlorophenyl group. The precise mass of m/z 367.9849 confirms the monoisotopic [M + H]+ peak, and the isotopic spacing further validates the structure. The HRMS data of compound 4c, confirms its molecular composition. The spectrum exhibits a prominent molecular ion peak at m/z 413.9325, which corresponds to the protonated molecular ion [M + H + 2]+ of the expected molecular formula C15H10BrClN3S2. The isotopic distribution, characterized by a pattern of M, M + 2, and M + 4 peaks, further supports the presence of these halogens due to their natural isotopic abundances (79Br/81Br and 35Cl/37Cl). The HRMS of compound 4d, provides strong evidence for its molecular identity. The spectrum displays a dominant molecular ion peak at m/z 334.0236, which corresponds to the protonated molecular ion [M + H]+. This value is consistent with the calculated monoisotopic mass for the molecular formula C15H11ClN3S2, confirming the presence of a chlorine atom and two sulfur atoms, characteristic of the thiazole and chlorothiophene moieties. The accompanying isotopic peak at m/z 336.0204 reflects the presence of the 37Cl isotope, further validating the incorporation of chlorine. The high intensity and resolution of the peak, along with the clear isotopic pattern, demonstrate the compound's purity and accurate molecular structure, affirming the successful synthesis of compound 4d.
3.2. Effect of anti-biofilm activity of thiazole derivatives
A major virulence attribute of C. albicans is its ability to form biofilms, densely packed communities of cells adhered to a surface. These biofilms are intrinsically resistant to conventional antifungal therapeutics, the host immune system, and other environmental factors, making biofilm-associated infections a significant clinical challenge. Thiazole derivatives 4a–4c inhibited more than 50% of biofilm formation at low concentration of 12.5 μg mL−1 (Fig. 2). The anti-biofilm activity revealed that low doses of thiazole are sufficient to inhibit the biofilm formation in C. albicans. The standard fluconazole showed only 20% inhibition at 12.5 μg mL−1, and amphotericin B showed more than 90% biofilm inhibition at 12.5 μg mL−1 (lab observations).
 |
| | Fig. 2 Effect of anti-biofilm activity of thiazole derivatives. | |
3.3. Effect on exopolysaccharides quantification
The exopolysaccharide is secreted by microorganism itself, which helps in the biofilm formation. In the disrupt biofilm, the thiazole compounds do not show prominent effect on the EPS production. The thiazoles derivatives (4a–4d) have not shown any inhibition in the EPS production in C. albicans (Fig. 3). The EPS production plays a more prominent role in the maturation and stability of the biofilm matrix. This process is regulated by a distinct set of genetic pathways, including ZAP1, FKS1, and PHR1. In our study, the thiazole derivatives did not lead to a notable reduction in EPS production, suggesting that these biosynthetic routes remain largely unaffected. Interestingly, even when hyphal development is inhibited, yeast-form cells can still produce and secrete EPS. However, in the absence of hyphal structures, this EPS lacks the architectural framework needed to form a robust and mature biofilm, limiting its functional integrity.
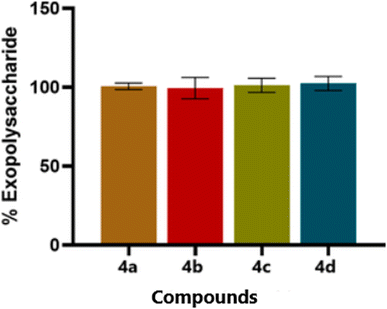 |
| | Fig. 3 Effect on exopolysaccharides quantification. | |
3.4. Effect of thiazole derivatives on hyphal inhibition in C. albicans
The ability of thiazole derivatives to inhibit hyphae formation in the biofilm structure was shown in Fig. 4. Thiazole derivatives 4b, 4c and 4d showed hyphal inhibition in C. albicans. The formation of hyphae is a critical early step in the development of biofilms by Candida albicans. Hyphal filaments enhance the organism's ability to adhere to surfaces, invade host tissues, and establish the structural foundation of mature biofilms. In our study, we observed that thiazole derivatives effectively inhibit hyphal development. This effect is likely mediated through interference with key regulatory pathways, such as the Ras1–cAMP–PKA and MAPK signalling cascades, which are known to govern hyphal induction and play a central role in initiating biofilm formation.
 |
| | Fig. 4 Effect of thiazole derivatives on biofilm structure in C. albicans. | |
In our study, thiazole derivatives exhibit significant anti-biofilm activity at low concentrations (12.5 μg mL−1), inhibiting over 50% of biofilm formation. This suggests their potential as effective agents against biofilm-associated infections. Furthermore, these compounds were found to notably suppress hyphae formation, which is a crucial step in tissue invasion and systemic infections caused by C. albicans. Among the compounds tested, derivative 4a showed higher antifungal activity, although it did not inhibit hyphal formation as effectively as derivatives 4b, 4c, and 4d, which were able to inhibit approximately 40–60% of hyphal formation under serum-stimulated conditions. Interestingly, despite the marked inhibition of biofilm formation and hyphal growth, the production of exopolysaccharides (EPS) remained unaffected. This indicates that the thiazole derivatives primarily target the hyphal development and structural development rather than the biosynthesis of the biofilm matrix.
3.5. Molecular docking
The molecular docking study, conducted using the Schrödinger Maestro suite, evaluated the binding interactions of compounds (4a–4d) with the lanosterol 14-alpha demethylase. Compound 4a exhibited the highest docking score of −8.715 kcal mol−1, indicating a strong binding affinity. It formed hydrogen bond interactions with Met509 and aromatic hydrogen bond interaction with Phe506 and Tyr140 of lanosterol 14-alpha demethylase. Compound 4a also showed π–π stacking interaction with Phe241 residue as shown in Fig. 5A with lanosterol 14-alpha demethylase. Compound 4b had the second-highest score among all complexes, with a docking score of −8.027 kcal mol−1, forming aromatic hydrogen bond interaction with Ser382 and π–π stacking interactions with Phe241, as well as halogen bond interaction is seen with Arg98 as demonstrate in Fig. 5B. In contrast, compound 4c displayed a docking score of −7.791 kcal mol−1, forming aromatic hydrogen bond interactions with Ser382 and π–π stacking interaction with Hie381 of lanosterol 14-alpha demethylase. It also showed the halogen bond interaction with Arg98 as depicted in Fig. 5C. Compound 4d exhibited the docking score of −7.293 kcal mol−1, forming π–π stacking interactions with Tyr126, Phe236, and Hie381. It also exhibited the aromatic hydrogen bond interaction with Gly314, Met509, and Ser508, as well as halogen bond interactions with Hie381 of lanosterol 14-alpha demethylase as shown in Fig. 5D. Overall, compound 4a showed the highest docking score, indicating the strongest binding affinity among all the complexes (Table 1).
 |
| | Fig. 5 3D representations of molecular interactions of compounds (A) 4a, (B) 4b, (C) 4c, (D) 4d with the lanosterol 14-alpha demethylase (5EQB). | |
Table 1 Evaluation of docking scores and glide energies for compounds 4a–4d with the lanosterol 14-alpha demethylase
| Compounds |
Docking score (kcal mol−1) |
Glide energy (kcal mol−1) |
| 4a |
−8.715 |
−55.236 |
| 4b |
−8.027 |
−46.581 |
| 4c |
−7.791 |
−45.961 |
| 4d |
−7.293 |
−45.509 |
3.6. Molecular dynamics
Molecular dynamics (MD) simulations were performed to investigate the interactions between compound 4c and the lanosterol 14-alpha demethylase. The structural dynamics of the protein–ligand complex were evaluated over a 100 ns simulation, focusing on the root mean square deviation (RMSD) of the Cα atoms to assess conformational stability and deviations from the starting structure. At the start of the simulation (0 ns), the Cα RMSD value was 1.71 Å, indicating minimal deviation from the initial configuration. A rapid increase in RMSD was observed from 1.71 Å, reaching to 6.54 Å by 15 ns due to initial system fluctuation. From 15 ns onward the protein structure was equilibrated until the end of simulation with minor fluctuation in RMSD. For the ligand RMSD, the simulation began at 1.48 Å and the ligand's RMSD initially fluctuate from 1.48 Å to 4.57 Å until 21 ns followed by the phase of stability and equilibration. A significant peak was observed at 59 ns, where the ligand RMSD reached 5.31 Å, before stabilizing around 4.7 Å until 60 ns. Another marked spike was observed at 70 ns, with the RMSD rising to 5.98 Å, followed by stabilization between 4.34 and 4.84 Å until the end of the simulation at 100 ns. The observed equilibrium phases, interspersed with spikes in both Cα and ligand RMSD values, highlight regions of conformational flexibility and stability throughout the simulation, providing insight into the dynamic interactions between compound 4c and the lanosterol 14-alpha demethylase enzyme as shown in Fig. 6A.
 |
| | Fig. 6 MD simulation analysis of the 4c compound in complex with the lanosterol 14-alpha demethylase: (A) RMSD analysis, where the protein's RMSD is depicted in grey, and the RMSD of the 4c compound is shown in red; (B) protein RMSF analysis; (C) 2D interaction diagram; and (D) protein–ligand contact analysis throughout the MD trajectory. | |
3.7. RMSF
The RMSF values for each amino acid residue in the protein backbone are illustrated in Fig. 6B. Peaks in the graph represent the fluctuation of individual residues throughout the simulation. Higher RMSF values signify greater flexibility, while lower values suggest reduced mobility and increased system stability. The secondary structural elements α-helices and β-strands are indicated in red and blue, respectively, while loop regions are shown in white as the plot's background. Typically, α-helical and β-strand regions are more rigid compared to unstructured protein regions, thus exhibiting lower fluctuations, while loop areas tend to show greater movement. A slight fluctuation observed in the residues within the active site, along with minor fluctuations in the main chain, suggests minimal conformational shifts, indicating that the lead compound remains tightly bound within the binding pocket throughout the simulation. In the 100 ns molecular dynamics (MD) simulation of the compound 4c–lanosterol 14-alpha demethylase complex, the RMSF values for the protein backbone residues in the active site of the enzyme varies from 0.5 Å to 4.0 Å. Notable peaks were observed at residue indices around 50, 100, 200, and 400, corresponding to regions with higher flexibility, likely representing loop regions, surface-exposed areas, or flexible linkers. During the simulation, compound 4c formed interactions with 21 amino acid residues in the 5EQB protein, including TYR72 (RMSF: 1.13 Å), VAL94 (1.22 Å), LEU95 (1.15 Å), MET100 (1.11 Å), TYR126 (0.86 Å), LEU129 (0.97 Å), GLY235 (1.14 Å), PRO238 (1.21 Å), PHE241 (1.05 Å), VAL242 (1.23 Å), MET313 (1.36 Å), HIS317 (1.26 Å), PRO379 (0.80 Å), LEU380 (0.96 Å), HIS381 (1.41 Å), SER282 (0.92 Å), LEU383 (0.80 Å), PHE384 (0.86 Å), TYR404 (1.22 Å), TYR507 (1.72 Å), and SER508 (1.44 Å). These interactions are crucial in maintaining the stability of the compound 4c–lanosterol 14-alpha demethylase complex within the binding pocket of the 5EQB protein.
3.8. Protein–ligand contact analysis
The stability of the compound 4c–lanosterol 14-alpha demethylase protein–ligand complex is largely driven by the interactions between the ligands and key amino acid residues within the binding pocket. Predominantly, hydrophobic interactions play a critical role, with Phe384 showing 100% involvement, Phe241 at 95%, and Tyr126 contributing 75% (Fig. 6C and D). Additionally, His381 participates in forming hydrogen bonds, hydrophobic interactions, and water bridges. These interactions are further supported by water bridges with Ser382, Ser508, and Met509, as depicted in (Fig. 6C and D).
3.9. Free energy landscape (FEL)
The free energy landscape analysis was performed to assess the structural dynamics of the system, as shown in Fig. 7. FEL was utilized to identify the lowest free energy states of the Cα backbone atoms of the proteins, using “root mean square deviation (RMSD)” and the “radius of gyration (Rg)” as key descriptors. In the case of the 4c–lanosterol 14-alpha demethylase complex, the system reached its lowest free energy at frame 209, with an RMSD of 6.869 Å and an Rg of 24.734 nm. These findings indicate that the protein undergoes conformational adjustments, likely involving folding or structural rearrangements, to reach its most energetically favourable state. The binding of the ligand appears to facilitate these conformational changes, stabilizing the protein in its lowest energy state. The FEL of the 4c–lanosterol 14-alpha demethylase complex displayed distinct regions of varying free energy. Yellow regions in the FEL corresponded to higher free energy states, indicating less stable conformations, where the protein is more flexible or disordered. In contrast, deep blue regions represented areas of low free energy, suggesting highly stable conformations where the protein adopts a more rigid, energetically favourable structure. These stable regions reflect the lowest energy states of the system, where the protein–ligand complex is likely in its most functional or biologically relevant form.
 |
| | Fig. 7 The 2D and 3D free energy landscapes generated from MD trajectories depicting the interaction of the 4c compound with the lanosterol 14-alpha demethylase. | |
3.10. Inhibition pathways in fungal sterol synthesis
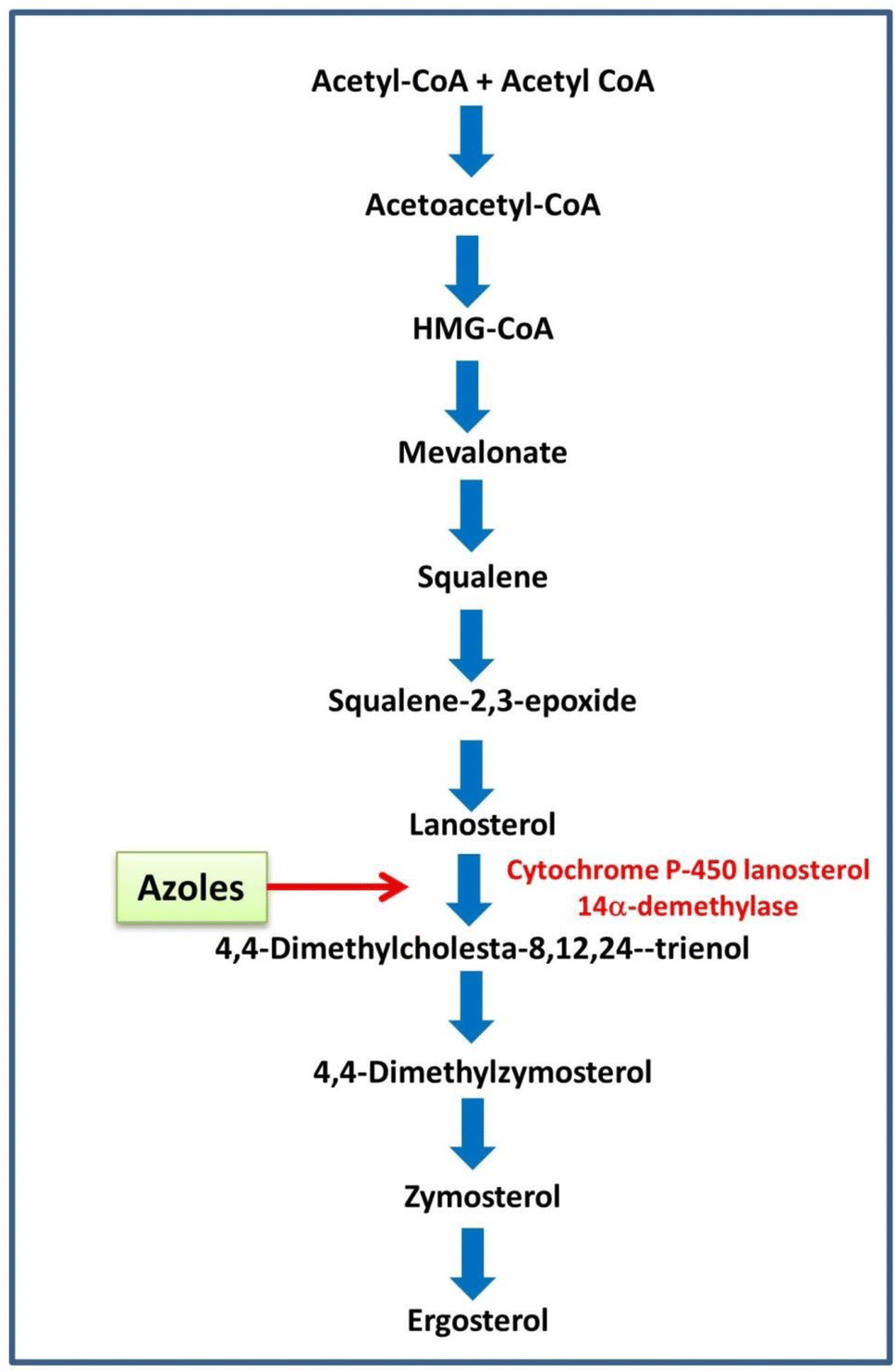
The Fig. 8 illustrates the ergosterol biosynthesis pathway in fungi, a critical target for antifungal therapy.64,65 Ergosterol, a major sterol in fungal membranes, is analogous to cholesterol in mammalian cells and is essential for maintaining fungal cell membrane integrity and function.64,65 The pathway begins with the condensation of two acetyl-CoA molecules, proceeding through several intermediates including HMG-CoA, mevalonate, and squalene, eventually leading to the synthesis of lanosterol. Lanosterol undergoes a series of demethylation, isomerization, and reduction steps to finally form ergosterol.64,65 Azole antifungals inhibit the cytochrome P450 enzyme lanosterol 14α-demethylase (CYP51), which is essential for converting lanosterol into downstream sterol intermediates.64 As shown in the diagram, this inhibition blocks the pathway at the lanosterol stage, thereby depleting ergosterol and accumulating toxic sterol intermediates, which ultimately compromise membrane integrity and inhibit fungal growth.64
 |
| | Fig. 8 Schematic representation of the ergosterol biosynthetic pathway in fungi, highlighting the key biochemical pathways targeted for inhibition. The enzyme lanosterol-14α-demethylase (CYP51) is emphasized as a crucial target for antifungal intervention by azole compounds. | |
3.11. DFT study
3.11.1. Optimized geometry. The optimized structural parameters of compound 4c have been simulated through the DFT calculations along with B3LYP functional and 6-311++(d,p) basis set was presented in Table 2 and optimized structure is given in Fig. 9. The optimized structure comprises one (Br–C, Cl–C, N–N, C–N, and N–H), two (N–C), four (C–S), fifteen (C–C), and nine (C–H) bond distances. In addition, one (Cl–C–C, Cl–C–S, S–C–H, N–C–N, N–C–S, C–S–C, S–C–N, C–N–H, and N–N–H), two (C–N–N, C–N–C, and Br–C–C), fifteen (C–C–C), sixteen (C–C–H), and three (H–C–H, C–C–S, and N–C–C) bond angles. In the structure of compound 4c, the bond distances of Br1–C4, Cl17–C18, N13–N14, C12–N13, and N13–H28 were simulated at 1.918, 1.731, 1.351, 1.376, and 1.012 Å, respectively. The simulated bond distances for C18–S22 and C21–S22 in the chlorothiophene ring were found to be 1.737 and 1.757 Å. In contrast, the bond distances of S11–C10 and S11–C12 in the thiazole ring were calculated as 1.742 and 1.773 Å, respectively. The bond lengths for C2–H23, C3–H24, C5–H25, and C6–H26 in phenyl ring were calculated at 1.084, 1.082, 1.085, and 1.082 Å, whereas the bond distances of C10–H27, C19–H32, an-Cd C20–H33 in thiazole and chlorothiophene structure were calculated at 1.077, 1.081, and 1.081 Å. In contrast, the bond distances of C15–H29, C15–H30, and C15–H31 in methyl group were calculated at 1.096, 1.088, and 1.096 Å, respectively. In addition, the bond distances of C–C and N–C were simulated between 1.291–1.511 and 1.290–1.385 Å, respectively. The bond length of Br1–C4 was observed to be longer than C–C and C–H bonds, likely due to the bromine's higher electronegativity and electron-withdrawing nature. These factors reduce the electron density between bromine and carbon, resulting in a weaker bond within the crystal structure. The bond angles of Cl17–C18–C19, Cl17–C18–S22, S11–C10–H27, N8–C12–N13, N8–C12–S11, C10–S11–C12, S11–C12–N13, C12–N13–H28, and N13–N14–H28 were calculated at 127.2, 120.3, 119.6, 126.3, 115.0, 87.70, 118.60, 118.10, and 121.2°, respectively. The bond angles of C–C–C and C–C–H were simulated between 112.2–126.4 and 110.9–129.7°. The bond angles of Br–C–C in the phenyl ring were simulated within 119.5–119.7°. The bond angles of C–N–N, C–N–C, and N–C–C were calculated between 118.5–120.8, 111.4–115.1, and 115.7–123.5°, respectively. In addition, the bond angles of C–S–C and C–C–S were simulated within 87.70–91.0 and 110.4–121.8°, respectively. The bond angle of C9–C10–H27 salvaged between nitrogen and sulfur atoms was simulated at 129.7° has been the highest as compared to other bond angles due to lone pairs in electronegative atoms, which occupy additional space and exert repulsive interactions on the bonding electrons of adjacent atoms.
Table 2 Simulated structural parameters of compound 4c
| Bond |
Bond lengths (Å) |
Bond |
Bond lengths (Å) |
Bond |
Bond lengths (Å) |
| Br1–C4 |
1.918 |
N8–C9 |
1.385 |
C15–H30 |
1.088 |
| C2–C3 |
1.389 |
N8–C12 |
1.29 |
C15–H31 |
1.096 |
| C2–C7 |
1.402 |
C9–C10 |
1.366 |
C16–C21 |
1.463 |
| C2–H23 |
1.084 |
C10–S11 |
1.742 |
Cl17–C18 |
1.731 |
| C3–C4 |
1.392 |
C10–C27 |
1.077 |
C18–C19 |
1.365 |
| C3–H24 |
1.082 |
S11–C12 |
1.773 |
C18–S22 |
1.737 |
| C4–C5 |
1.39 |
C12–N13 |
1.376 |
C19–C20 |
1.417 |
| C5–C6 |
1.391 |
N13–N14 |
1.351 |
C19–H32 |
1.081 |
| C5–H25 |
1.082 |
N13–H28 |
1.012 |
C20–C21 |
1.375 |
| C6–C7 |
1.401 |
C14–C16 |
1.291 |
C20–H33 |
1.081 |
| C6–H26 |
1.082 |
C15–C16 |
1.511 |
C21–S22 |
1.757 |
| C7–C9 |
1.474 |
C15–H29 |
1.096 |
— |
— |
| Bond |
Bond angles (°) |
Bond |
Bond angles (°) |
Bond |
Bond angles (°) |
| Br1–C4–C3 |
119.5 |
C9–N8–C12 |
111.4 |
C15–C16–C21 |
120.9 |
| Br1–C4–C5 |
119.7 |
C8–N9–C10 |
115.1 |
H29–C15–H30 |
107.2 |
| C3–C2–C7 |
121.2 |
N8–C12–S11 |
115.0 |
H29–C15–H31 |
108.0 |
| C3–C2–H23 |
118.4 |
N8–C12–N13 |
126.3 |
H30–C15–H31 |
107.2 |
| C2–C3–C4 |
119.2 |
C9–C10–S11 |
110.7 |
C16–C21–C20 |
127.8 |
| C2–C3–H24 |
120.4 |
C9–C10–H27 |
129.7 |
C16–C21–S22 |
121.8 |
| C7–C2–H23 |
120.4 |
S11–C10–H27 |
119.6 |
Cl17–C18–C19 |
127.2 |
| C2–C7–C6 |
118.2 |
C10–S11–C12 |
87.7 |
Cl17–C18–S22 |
120.3 |
| C2–C7–C9 |
122.1 |
S11–C12–N13 |
118.6 |
C19–C18–C22 |
112.5 |
| C4–C3–H24 |
120.4 |
C12–N13–N14 |
120.8 |
C18–C19–C20 |
112.2 |
| C3–C4–C5 |
120.9 |
C12–N13–H28 |
118.1 |
C18–C19–H32 |
123.2 |
| C4–C5–C6 |
119.3 |
N14–N13–H28 |
121.2 |
C18–S22–C21 |
91.0 |
| C4–C5–H25 |
120.4 |
N13–N14–C16 |
118.5 |
C20–C19–H32 |
124.7 |
| C6–C5–H25 |
120.3 |
N14–C16–C15 |
123.5 |
C19–C20–C21 |
114.0 |
| C5–C6–C7 |
121.2 |
N14–C16–C21 |
115.7 |
C19–C20–H33 |
124.4 |
| C5–C6–H26 |
120.2 |
C16–C15–H29 |
110.9 |
C21–C20–H33 |
121.6 |
| C7–C6–H26 |
118.6 |
C16–C15–H30 |
112.4 |
C20–C21–S22 |
110.4 |
| C6–C7–C9 |
119.7 |
C16–C15–H31 |
110.9 |
C7–C9–N8 |
118.5 |
| C7–C9–C10 |
126.4 |
— |
— |
— |
— |
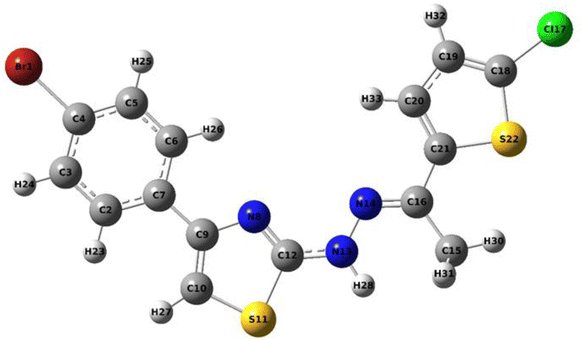 |
| | Fig. 9 Optimized molecular structure of 4c. | |
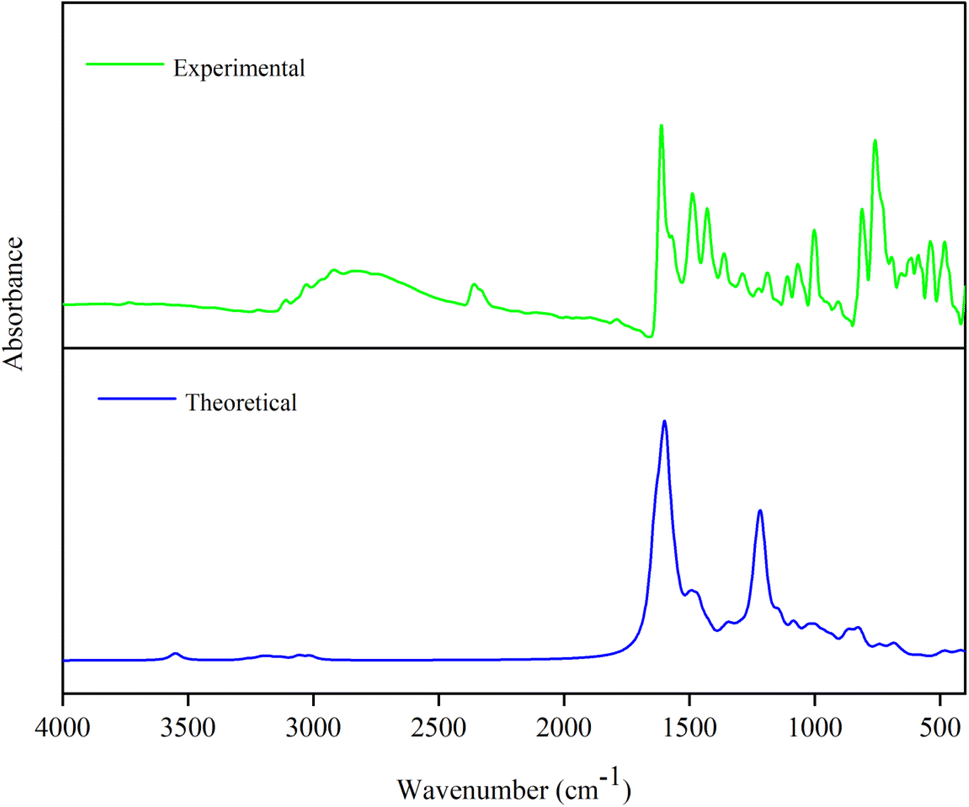
3.11.2. Vibrational analysis. The optimized molecular structure of compound 4c comprises 33 atoms and 206 electrons give rise to 93 vibrational modes according with (3N-6) normal modes assuming C1 point symmetry. The simulated and experimental FT-IR wavenumbers are presented in Table 3, with the agreeing vibrational FT-IR spectrum demonstrated in Fig. 10 highlights significant bands corresponding to C–Br, C–Cl, C–S, C–N, N–N, C–H, and C–C stretching and bending modes. The structure of the synthesized compound comprises a single bromine atom linked with C4 carbon atom in the phenyl ring, giving rise to C–Br stretching modes. In literature, the C–Br stretching modes were simulated at 593, 463, and 569 cm−1 and observed at 595 and 457 cm−1, respectively.66,67 In the present investigation, the stretching modes of C–Br were simulated at 402 and 184 cm−1 with a PED value of 37 and 13%, respectively. These values have an agreeing correlation with the aforementioned literature values. In nature, the C–Cl stretching modes are noted as solid peaks between 760 and 505 cm−1.68 The chlorine atom (Cl17) linked with the carbon atom (C18) in the chlorothiophene ring gives rise to C–Cl stretching mode was simulated at 477 cm−1 and observed at 480 cm−1 has significant coincidence with the literature values simulated at 550, 472 cm−1 and observed at 552 cm−1.69 In heteroaromatic compounds, the N–H and C–H stretching modes were observed between 3500–3220 and 3000–3100 cm−1, respectively.70,71 In the present investigation, the N–H stretching mode was simulated at 3408 cm−1 with a PED contribution of 100%. Similarly, the C–H stretching modes were simulated between 3132–2936 cm−1 and observed at 3109 cm−1. The stretching modes of C–S are observed as moderate bands within 630–930 cm−1.72 The C–S stretching bands were simulated at 708, 792, 838, and 956 cm−1. In addition, the symmetric and asymmetric stretching bands of the methyl group were simulated at 2892 and 3007 cm−1 and observed at 2918 and 3028 cm−1 respectively. The hydrogen atoms linked with carbon atoms in the thiazole, bromophenyl, and chlorothiophene rings give rise to C–H in-plane and out-of-plane deformations. The bands corresponding to C–H deformations were observed between 1450–1000 cm−1 (in-plane) and 1000–750 cm−1 (out-of-plane) deformations.73 In the present endeavor, the bands simulated between 1445–1048 cm−1 and 956–713 cm−1 for in-plane and out-of-plane deformations and observed at 1361–1066 and 810–758 cm−1. The carbon atom linked with the halogenated atom Br gives rise to C–Br deformations. In literature, C–Br deformations were simulated at 334 cm−1 (in-plane) and 102 cm−1 (out-of-plane). In the present work, the C–Br deformations were simulated at 268, 227, 195, and 132 cm−1. The C–S deformations were expected to fall between 600–400 cm−1 (in-plane) and 420–320 cm−1 (out-of-plane).72 In the present work, the bands simulated at 559, 477 cm−1 and 315, 290 cm−1 corresponds to in-plane and out-of-plane deformations and observed at 480 cm−1 (in-plane) deformation.
Table 3 Simulated and experimental wavenumbers of compound 4ca
| Mode |
Experimental |
Theoretical wave numbers (cm−1) |
Vibrational assignments |
| FT-IR |
Unscaled |
Scaled |
IIR |
| υs – symmetric stretching; υas – asymmetric stretching; β – deformation; τ – torsion; scaling factor 0.96 for all vibrations.36 |
| 1 |
— |
3550 |
3408 |
23.73 |
υ NH (100) |
| 2 |
— |
3262 |
3132 |
4.14 |
υ CH (99) |
| 3 |
3109 |
3230 |
3101 |
0.31 |
υ CH (99) |
| 4 |
— |
3216 |
3087 |
2.70 |
υ CH (100) |
| 5 |
— |
3212 |
3084 |
1.08 |
υ CH (99) |
| 6 |
— |
3203 |
3075 |
4.72 |
υ CH (99) |
| 7 |
— |
3196 |
3068 |
1.11 |
υ CH (99) |
| 8 |
— |
3177 |
3050 |
7.54 |
υ CH (99) |
| 9 |
3028 |
3132 |
3007 |
7.80 |
υas CH3 (93) |
| 10 |
— |
3058 |
2936 |
13.01 |
υ CH (100) |
| 11 |
2918 |
3012 |
2892 |
12.26 |
υs CH3 (93) |
| 12 |
1612 |
1636 |
1571 |
287.01 |
υ NC (61) |
| 13 |
— |
1632 |
1567 |
0.27 |
υ CC (29), β CCC (11) |
| 14 |
— |
1610 |
1546 |
120.31 |
υ NC (49), υ CC (34) |
| 15 |
— |
1596 |
1532 |
548.03 |
υ NC (20) |
| 16 |
— |
1569 |
1506 |
34.09 |
υ CC (38), β HNN (14) |
| 17 |
— |
1562 |
1500 |
66.97 |
υ CC (22), β HNN (37) |
| 18 |
1487 |
1547 |
1485 |
24.78 |
υ CC (45), β HNN (10) |
| 19 |
— |
1505 |
1445 |
46.96 |
υ CC (21), β HCC (40) |
| 20 |
1429 |
1491 |
1431 |
52.84 |
β HCH (48) |
| 21 |
— |
1488 |
1428 |
8.88 |
β HCH (80), τ HCCC (11) |
| 22 |
— |
1465 |
1406 |
104.26 |
υ CC (62) |
| 23 |
1361 |
1429 |
1372 |
32.61 |
υ CC (35), β HCC (32) |
| 24 |
— |
1416 |
1359 |
16.53 |
β HCH (90) |
| 25 |
— |
1369 |
1314 |
19.46 |
υ CC (50), β HCC (14) |
| 26 |
— |
1346 |
1292 |
49.99 |
υ CC (10), υ NC (23), β HCC (15) |
| 27 |
1288 |
1321 |
1268 |
15.17 |
υ CC (35), β HCC (52) |
| 28 |
— |
1307 |
1255 |
10.97 |
υ CC (57), υ NC (11) |
| 29 |
1224 |
1293 |
1241 |
24.54 |
υ CC (28), β HCC (12) |
| 30 |
1188 |
1235 |
1186 |
131.67 |
υ NN (12), β HCC (45) |
| 31 |
— |
1222 |
1173 |
5.52 |
υ CC (15), β HCS (42) |
| 32 |
— |
1216 |
1167 |
313.70 |
υ NC (19), β HCC (13) |
| 33 |
— |
1204 |
1156 |
74.72 |
β HCC (71) |
| 34 |
1109 |
1144 |
1098 |
76.26 |
υ NN (36) |
| 35 |
— |
1131 |
1086 |
9.93 |
υ CC (21), β HCC (52) |
| 36 |
1066 |
1092 |
1048 |
1.20 |
υ CC (36), β HCC (37) |
| 37 |
— |
1086 |
1043 |
33.18 |
υ CC (56) |
| 38 |
— |
1080 |
1037 |
37.58 |
υ NC (13), β HCS (23) |
| 39 |
— |
1045 |
1003 |
1.14 |
β HCH (26), τ HCCC (55), τ CCNC (13) |
| 40 |
1001 |
1043 |
1001 |
14.66 |
β CCC (19) |
| 41 |
— |
1024 |
983 |
45.37 |
β CCC (82) |
| 42 |
— |
996 |
956 |
50.02 |
υ SC (14), β CCC (11), τ HCCC (14) |
| 43 |
— |
993 |
953 |
0.41 |
τ CCCC (23), τ HCCC (69) |
| 44 |
— |
964 |
925 |
36.79 |
υ CC (28) |
| 45 |
— |
957 |
919 |
0.0004 |
τ CCCC (18), τ HCCC (69) |
| 46 |
906 |
931 |
894 |
35.71 |
β SCC (11), β CNC (14) |
| 47 |
— |
913 |
876 |
0.51 |
τ HCCC (89) |
| 48 |
— |
873 |
838 |
44.30 |
υ SC (13), β CCN (30) |
| 49 |
810 |
855 |
820 |
24.77 |
τ HCCC (89) |
| 50 |
— |
829 |
796 |
9.29 |
τ HCCC (78) |
| 51 |
— |
825 |
792 |
27.48 |
υ SC (47) |
| 52 |
758 |
818 |
785 |
39.03 |
τ HCCC (88) |
| 53 |
— |
743 |
713 |
26.26 |
τ CCCC (37), τ HCCC (14), τ CCNC (20) |
| 54 |
— |
737 |
708 |
2.02 |
υ SC (27), β CCC (60) |
| 55 |
694 |
712 |
684 |
3.91 |
β CCC (20), β CCN (16) |
| 56 |
— |
693 |
665 |
9.91 |
υ CC (16), β SCC (17) |
| 57 |
— |
681 |
654 |
27.87 |
τ CCCC (36), τ CCNC (20), τ HCSC (25) |
| 58 |
— |
672 |
645 |
3.24 |
τ CCNC (25), τ HCSC (48) |
| 59 |
— |
652 |
626 |
6.72 |
υ SC (25), β SCC (16) |
| 60 |
615 |
645 |
619 |
0.34 |
β CCC (55) |
| 61 |
586 |
597 |
573 |
1.26 |
τ CCCC (24), τ CCNC (17) |
| 62 |
— |
582 |
559 |
5.65 |
β SCC (38), β CNC (11) |
| 63 |
538 |
571 |
548 |
0.66 |
τ CCCC (15), τ NCNC (12), τ CNCC (34), τ HCSC (10) |
| 64 |
— |
545 |
523 |
0.43 |
τ CCCC (28), τ CCNC (29) |
| 65 |
480 |
497 |
477 |
2.43 |
υ CCl (22), β SCC (12) |
| 66 |
— |
495 |
475 |
5.25 |
τ CCCC (45), τ BrCCC (21) |
| 67 |
— |
480 |
461 |
8.94 |
β CCN (27), τ SCCC (49) |
| 68 |
— |
479 |
460 |
6.59 |
β CCN (23) |
| 69 |
— |
446 |
428 |
4.07 |
β CCN (11), β CCC (20), β NCC (20) |
| 70 |
— |
419 |
402 |
17.49 |
υ CC (18), υ CBr (37) |
| 71 |
— |
414 |
397 |
0.005 |
τ CCCC (57), τ HCCC (38) |
| 72 |
— |
368 |
353 |
22.48 |
υ CCl (14), β CCC (12) |
| 73 |
— |
328 |
315 |
0.53 |
τ ClCSC (14), τ CCNN (50), β SCC (16) |
| 74 |
— |
315 |
302 |
20.68 |
τ CCCC (11), τ SCCC (20), τ BrCCC (26), τ CNCC (11), τ HNNC (15) |
| 75 |
— |
302 |
290 |
0.028 |
β CCC (10), β ClCS (28), β SCC (28) |
| 76 |
— |
292 |
280 |
36.39 |
τ SCCC (17), τ HNNC (54) |
| 77 |
— |
279 |
268 |
0.58 |
β BrCC (31), β ClCS (19) |
| 78 |
— |
244 |
234 |
13.10 |
τ ClCSC (10), τ NCNC (45), τ HNNC (14) |
| 79 |
— |
236 |
227 |
2.64 |
β BrCC (18), β NCC (10), β CNN (14) |
| 80 |
— |
214 |
205 |
0.0029 |
τ SCCC (10), τ ClCSC (32), τ CCNN (16), τ NCNC (14) |
| 81 |
— |
203 |
195 |
1.21 |
β BrCC (10), β ClCS (14) |
| 82 |
— |
192 |
184 |
0.05 |
υ CBr (13), β CNN (10) |
| 83 |
— |
154 |
148 |
0.04 |
τ SCCC (24), τ BrCCC (24), τ CNCC (17) |
| 84 |
— |
137 |
132 |
0.29 |
β NCN (10), β CCC (32), β BrCC (13), β ClCS (10), β NNC (12) |
| 85 |
— |
127 |
122 |
3.01 |
τ HCCC (77) |
| 86 |
— |
89 |
85 |
0.91 |
τ CCCC (34), τ SCCC (12), τ ClCSC (15), τ NNCN (14) |
| 87 |
— |
77 |
74 |
0.83 |
β CCN (15), β CCC (37), β NCC (21) |
| 88 |
— |
68 |
65 |
7.45 |
τ CCCN (14), τ CNNC (63) |
| 89 |
— |
48 |
46 |
2.30 |
τ CCCC (48), τ SCCC (11) |
| 90 |
— |
30 |
29 |
0.0033 |
τ NCCC (82) |
| 91 |
— |
25 |
24 |
0.015 |
τ CCCC (15) |
| 92 |
— |
21 |
20 |
0.06 |
β CCN (10), β NCN (21), τ NNCN (48), τ CCCN (14), β NNC (28), β NCC (11), β CNN (15) |
| 93 |
— |
14 |
13 |
0.13 |
τ CCNN (10), τ CCCN (53), τ CNNC (18) |
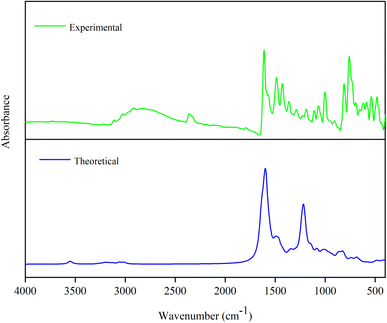 |
| | Fig. 10 Simulated and experimental FT-IR spectrum of compound 4c. | |
3.11.3. NBO analysis. In NBO analysis, the second-order Fock matrix was employed to evaluate donor and acceptor interactions within the crystal structures, revealing a transfer of electron clouds from localized NBOs of the idealized Lewis structure to vacant non-Lewis orbitals.72 The NBO analysis was performed using DFT/B3LYP method along with 6-311++G(d,p) basis set was tabulated in Table 4.
Table 4 Second order perturbation theory of Fock matrix in selected NBO basis for compound 4c
| Donor (i) |
Type |
Acceptor (j) |
Type |
Type of transition |
E(2)a (kJ mol−1) |
E(j) − E(i)b (a.u.) |
F(i,j)c (a.u.) |
| E(2) means energy of hyperconjucative interactions (stabilization energy). Energy difference between donor (i) and acceptor (j) NBO orbitals. F(i,j) is the Fock matrix element between i and j NBO orbital. |
| C2–C3 |
π |
C4–C5 |
π* |
π–π* |
20.22 |
0.28 |
0.068 |
| C2–C3 |
π |
C6–C7 |
π* |
π–π* |
19.04 |
0.29 |
0.068 |
| C4–C5 |
π |
C2–C3 |
π* |
π–π* |
19.96 |
0.30 |
0.069 |
| C4–C5 |
π |
C6–C7 |
π* |
π–π* |
18.33 |
0.30 |
0.068 |
| C6–C7 |
π |
C2–C3 |
π* |
π–π* |
20.82 |
0.28 |
0.069 |
| C6–C7 |
π |
C4–C5 |
π* |
π–π* |
22.88 |
0.27 |
0.070 |
| C6–C7 |
π |
C9–C10 |
π* |
π–π* |
19.94 |
0.26 |
0.066 |
| N8–C12 |
π |
C9–C10 |
π* |
π–π* |
19.56 |
0.35 |
0.078 |
| C9–C10 |
π |
C6–C7 |
π* |
π–π* |
10.19 |
0.32 |
0.055 |
| C18–C19 |
π |
C20–C21 |
π* |
π–π* |
13.96 |
0.32 |
0.063 |
| C20–C21 |
π |
N14–C16 |
π* |
π–π* |
16.81 |
0.29 |
0.063 |
| C20–C21 |
π |
C18–C19 |
π* |
π–π* |
16.78 |
0.28 |
0.064 |
| N8 |
LP(1) |
S11–C12 |
σ* |
LP(1)–σ* |
17.34 |
0.51 |
0.085 |
| S11 |
LP(2) |
N8–C12 |
π* |
LP(2)–π* |
26.15 |
0.26 |
0.075 |
| S11 |
LP(2) |
C9–C10 |
π* |
LP(2)–π* |
17.75 |
0.28 |
0.063 |
| N13 |
LP(1) |
N8–C12 |
π* |
LP(1)–π* |
47.92 |
0.28 |
0.106 |
| N13 |
LP(1) |
N14–C16 |
π* |
LP(1)–π* |
30.03 |
0.30 |
0.086 |
| S22 |
LP(2) |
C18–C19 |
π* |
LP(2)–π* |
22.50 |
0.26 |
0.068 |
| S22 |
LP(2) |
C20–C21 |
π* |
LP(2)–π* |
20.53 |
0.28 |
0.068 |
In the structure of compound 4c, a significant delocalization was observed from the lone pair on nitrogen (N13) to the anti-bonding orbital of N8–C12. This interaction, represented by the LP(1) → π* transition, provides a stabilizing energy of 47.92 kJ mol−1. The second significant delocalization was observed from the lone pair nitrogen (N13) to the anti-bonding orbital N14–C16 with a stabilizing energy of 30.03 kJ mol−1 by LP(1)–π* transition. In addition, the lone pairs nitrogen (N8) and sulfur (S11 and S22) create delocalization to anti-bonding S11–C12, N8–C12, C9–C10, C18–C19, and C20–C21 with stabilizing energy of 17.34, 26.16, 17.75, 22.50, and 20.53 kJ mol−1 by LP(1) → σ* and LP(1)–π* transitions, respectively. Similarly, π–π* transitions were also noticed between electron-donating from C2–C3, C4–C5, C6–C7, N8–C12, C9–C10, C18–C19, and C20–C21 to antibonding C4–C5, C6–C7, C2–C3, C6–C7, C2–C3, C4–C5, C9–C10, C6–C7. C20–C21, N14–C16, and C18–C19 with a stabilizing energy of 20.22, 19.04, 19.96, 18.33, 20.82, 22.88, 19.94, 19.56, 10.19, 13.96, 16.81, and 16.78 kJ mol−1.
3.11.4. ELF and LOL. In quantum chemistry, ELF and LOL have been used to analyze the localization and distribution of electrons within molecular structures. This offers a better insight into the understanding of chemical reactivity, bonding, stability, and delocalization of electrons.74 In compound 4c, the ELF and LOL projection, color map, and contour have been demonstrated in Fig. 11 and 12 simulated between 0.00–1.00 and 0.00–0.800, respectively. In the ELF color-filled map, the red regions at hydrogen atoms H23 and H24 in the bromo phenyl ring and H32 and H33 in the chlorothiophene ring emphasize the electrons were highly localized. Similarly, the carbon and nitrogen atoms C2 and C3 (bromophenyl ring), C18, C19, C20, and C21 (chlorothiophene ring), and N8, N13, N14, C12, C15, and C16 (aliphatic chain) encircled with blue color emphasize the electrons were highly delocalized. In contrast, the blue and red regions over the chlorothiophene and bromophenyl rings signify the delocalized and localized areas within the crystal structure.
 |
| | Fig. 11 ELF projection, color map, and contour map of compound 4c. | |
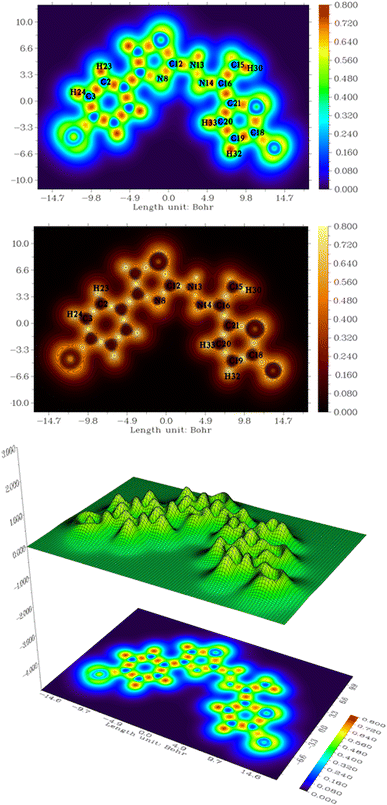 |
| | Fig. 12 LOL projection, color map, and contour map of compound 4c. | |
3.11.5. RDG and NCI. NCI and RDG analyses are significant tools for understanding the non-covalent interactions (van der Waals, hydrogen bonding, and steric repulsions) within crystal structures using isosurface and scattered plots.74 The RDG scattered plot and NCI isosurface plot of compound 4c have been simulated using Atomistica, an online molecular modeling platform. This has been demonstrated in Fig. 13. In the simulated NCI isosurface graph, red spheres within the thiazole, bromophenyl, and chlorothiophene rings highlight the steric effects of electron delocalization. Meanwhile, the green spheres at the boundaries of hydrogen atoms highlight the van der Waals interactions. In the RDG scattered plot, distinct spikes were observed between 0.012 and 0.05 a.u. were noticed (red) and −0.02 to −0.018 (green) highlights van der Waals interaction and steric effect. These interactions play a crucial role in stabilizing the crystal structure.
 |
| | Fig. 13 RDG scatter graph (top) and NCI diagram (bottom) of compound 4c. | |
3.12. ADME study
The ADME profile of the compound 4c is illustrated in Table 5. The compound 4c, with a molecular weight of 412.75 g mol−1 and moderate lipophilicity (iLOGP of 3.62), show pharmacologically suitable properties for absorption. It has high gastrointestinal absorption and is not a P-gp substrate, which suggests it can effectively reach systemic circulation. Despite its poor water solubility (logS = −6.74), its moderate lipophilicity supports membrane permeability, making it a candidate for oral administration. Pharmacologically, its inhibition of key cytochrome P450 enzymes (CYP1A2, CYP2C19, and CYP2C9) could make it useful for modulating drug metabolism, though it avoids potential interactions with CYP2D6 and CYP3A4. Additionally, it meets Lipinski's rule of five, ensuring good druglikeness. The bioavailability score of 0.55 indicates moderate oral bioavailability, enhancing its suitability as a pharmacologically relevant compound.
Table 5 ADME profile of compound 4c
| Category |
Property |
Result |
| Physicochemical properties |
Molecular weight |
412.75 g mol−1 |
| Number of heavy atoms |
22 |
| Number of aromatic heavy atoms |
16 |
| Fraction Csp3 |
0.07 |
| Number of rotatable bonds |
4 |
| Number of H-bond acceptors |
2 |
| Number of H-bond donors |
1 |
| TPSA (topological polar surface area) |
93.76 Å2 |
| Lipophilicity |
logPo/w (iLOGP) |
3.62 |
| Consensus logPo/w |
5.34 |
| Water solubility |
logS (ESOL) |
−6.74 |
| Pharmacokinetics |
GI absorption |
High |
| BBB Permeant |
No |
| P-gp substrate |
No |
| CYP1A2 inhibitor |
Yes |
| CYP2C19 inhibitor |
Yes |
| CYP2C9 inhibitor |
Yes |
| CYP2D6 inhibitor |
No |
| CYP3A4 inhibitor |
No |
| Druglikeness |
Lipinski |
Yes |
| Ghose |
No |
| Veber |
Yes |
| Egan |
No |
| Muegge |
No |
| Bioavailability score |
0.55 |
4 Conclusion
This study underscores the potential of thiazole derivatives as promising candidates for combating drug-resistant fungal infections, particularly those caused by Candida albicans. The synthesized (E)-2-(2-(1-(5-chlorothiophen-2-yl)ethylidene)hydrazineyl)-4-(aryl)thiazole derivatives (4a–4d) exhibit promising anti-biofilm activity against Candida albicans, with compounds 4a–4c inhibiting biofilm formation by over 50% at 12.5 μg mL−1. Molecular docking and dynamics analyses confirmed strong and stable binding, particularly for 4a and 4c, with compound 4a showing the highest affinity (−8.715 kcal mol−1) and 4c demonstrating complex stability via RMSD and FEL evaluations. DFT-based electronic and NBO analyses revealed favorable geometry and electron delocalization in 4c, with ELF, LOL, NCI, and RDG results confirming stabilizing non-covalent interactions. Additionally, ADME profiling indicated good oral bioavailability and drug-like properties for 4c. These findings highlight the therapeutic potential of thiazole-based scaffolds in developing novel antifungal agents targeting drug-resistant strains.
Data availability
The data supporting the findings of this study are available within the article and its ESI files.† Additional data that support the findings of this study are available from the corresponding author upon reasonable request.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was funded by the Ongoing Research Funding program, (ORF-2025-1100), King Saud University, Riyadh, Saudi Arabia. Authors gratefully acknowledge CIF, Savitribai Phule Pune University, Pune for NMR and HRMS characterization of samples. Authors are also thankful to RAP Analytical Instrumentation Laboratory, Nashik FT-IR characterization facilities. The authors also would like to thank Research Centre in Chemistry, Loknete Vyankatrao Hiray Arts, Science and Commerce College Panchavati, Nashik and MSG Arts, Science and Commerce College, Malegaon Camp, Malegaon.
References
- H. D. Marston, D. M. Dixon, J. M. Knisely, T. N. Palmore and A. S. Fauci, Antimicrobial resistance, JAMA, 2016, 316(11), 1193–1204 CrossRef CAS PubMed.
- M. C. Fisher, N. J. Hawkins, D. Sanglard and S. J. Gurr, Worldwide emergence of resistance to antifungal drugs challenges human health and food security, Science, 2018, 360(6390), 739–742 CrossRef CAS PubMed.
- A. Y. Alzahrani, Y. A. Ammar, M. Abu-Elghait, M. A. Salem, M. A. Assiri, T. E. Ali and A. Ragab, Development of novel indolin-2-one derivative incorporating thiazole moiety as DHFR and quorum sensing inhibitors: synthesis, antimicrobial, and antibiofilm activities with molecular modelling study, Bioorg. Chem., 2022, 119, 105571 CrossRef CAS PubMed.
- S. Ceylan, H. Bektas, H. Bayrak, N. Demirbas, S. Alpay-Karaoglu and S. Ülker, Syntheses and biological activities of new hybrid molecules containing different heterocyclic moieties, Arch. Pharm., 2013, 346(10), 743–756 CrossRef CAS PubMed.
- J. Baranwal, S. Kushwaha, S. Singh and A. Jyoti, A review on the synthesis and pharmacological activity of heterocyclic compounds, Curr. Phys. Chem., 2023, 13(1), 2–19 CrossRef CAS.
- N. C. Desai and D. V. Vaja, Hybrid Approach for Synthesis and Antimicrobial Activity of Heterocyclic Compounds, Int. Lett. Chem., Phys. Astron., 2018, 77, 35–52 Search PubMed.
- Y. N. Mabkhot, F. Alatibi, N. N. El-Sayed, N. A. Kheder and S. S. Al-Showiman, Synthesis and structure-activity relationship of some new thiophene-based heterocycles as potential antimicrobial agents, Molecules, 2016, 21(8), 1036 CrossRef PubMed.
- S. Abu-Melha, M. M. Edrees, H. H. Salem, N. A. Kheder, S. M. Gomha and M. R. Abdelaziz, Synthesis and biological evaluation of some novel thiazole-based heterocycles as potential anticancer and antimicrobial agents, Molecules, 2019, 24(3), 539 CrossRef PubMed.
- R. M. da Cruz, F. J. Mendonça-Junior, N. B. de Mélo, L. Scotti, R. S. de Araújo, R. N. de Almeida and R. O. de Moura, Thiophene-based compounds with potential anti-inflammatory activity, Pharmaceuticals, 2021, 14(7), 692 CrossRef PubMed.
- A. S. Tapkir, S. S. Chitlange and R. P. Bhole, Discovery of thiazole based bis heterocyclic system for anti-inflammatory potential, Antiinflamm. Antiallergy Agents Med. Chem., 2017, 16(3), 175–192 CrossRef CAS PubMed.
- S. Pathania and P. A. Chawla, Thiophene-based derivatives as anticancer agents: an overview on decade's work, Bioorg. Chem., 2020, 101, 104026 Search PubMed.
- K. K. Sahil and V. Jaitak, Thiazole and related heterocyclic systems as anticancer agents: a review on synthetic strategies, mechanisms of action and SAR studies, Curr. Med. Chem., 2022, 29(29), 4958–5009 Search PubMed.
- R. Shah and P. K. Verma, Therapeutic importance of synthetic thiophene, Chem. Cent. J., 2018, 12, 1–22 Search PubMed.
- P. M. Jadhav, S. Kantevari, A. B. Tekale, S. V. Bhosale, R. P. Pawar and S. U. Tekale, A review on biological and medicinal significance of thiazoles, Phosphorus, Sulfur Silicon Relat. Elem., 2021, 196(10), 879–895 Search PubMed.
- V. Chugh, G. Pandey, R. Rautela and C. Mohan, Heterocyclic compounds containing thiazole ring as important material in medicinal chemistry, Mater. Today: Proc., 2022, 69, 478–481 Search PubMed.
- S. Chawla, S. Sharma, S. Kashid, P. K. Verma and A. Sapra, Therapeutic potential of thiophene compounds: a mini-review, Mini-Rev. Med. Chem., 2023, 23(15), 1514–1534 Search PubMed.
- J. P. Lopes and M. S. Lionakis, Pathogenesis and virulence of Candida albicans, Virulence, 2022, 13(1), 89–121 Search PubMed.
- R. Gaziano, S. Sabbatini and C. Monari, The interplay between Candida albicans, vaginal mucosa,
host immunity and resident microbiota in health and disease: an overview and future perspectives, Microorganisms, 2023, 11(5), 1211 Search PubMed.
- S. Shahabudin, N. S. Azmi, M. N. Lani, M. Mukhtar and M. S. Hossain, Candida albicans skin infection in diabetic patients: an updated review of pathogenesis and management, Mycoses, 2024, 67(6), e13753 CrossRef CAS PubMed.
- J. Stefanska, G. Nowicka, M. Struga, D. Szulczyk, A. E. Koziol, E. Augustynowicz-Kopec, A. Napiorkowska, A. Bielenica, W. Filipowski, A. Filipowska and A. Drzewiecka, Antimicrobial and anti-biofilm activity of thiourea derivatives incorporating a 2-aminothiazole scaffold, Chem. Pharm. Bull., 2015, 63(3), 225–236 CrossRef CAS PubMed.
- H. Mohammad, H. E. Eldesouky, T. Hazbun, A. S. Mayhoub and M. N. Seleem, Identification of a phenylthiazole small molecule with dual antifungal and antibiofilm activity against Candida albicans and Candida auris, Sci. Rep., 2019, 9(1), 18941 CrossRef CAS.
- A. Biernasiuk, A. Berecka-Rycerz, A. Gumieniczek, M. Malm, K. Z. Łączkowski, J. Szymańska and A. Malm, The newly synthesized thiazole derivatives as potential antifungal compounds against Candida albicans, Appl. Microbiol. Biotechnol., 2021, 105(16), 6355–6367 CrossRef CAS PubMed.
- S. Dawbaa, A. E. Evren, Z. Cantürk and L. Yurttaş, Synthesis of new thiazole derivatives and evaluation of their antimicrobial and cytotoxic activities, Phosphorus, Sulfur Silicon Relat. Elem., 2021, 196(12), 1093–1102 CrossRef CAS.
- A. Singh, D. Malhotra, K. Singh, R. Chadha and P. M. Bedi, Thiazole derivatives in medicinal chemistry: recent advancements in synthetic strategies, structure activity relationship and pharmacological outcomes, J. Mol. Struct., 2022, 1266, 133479 CrossRef CAS.
- Y. N. Mabkhot, F. Alatibi, N. N. El-Sayed, N. A. Kheder and S. S. Al-Showiman, Synthesis and structure-activity relationship of some new thiophene-based heterocycles as potential antimicrobial agents, Molecules, 2016, 21(8), 1036 CrossRef PubMed.
- Y. N. Mabkhot, N. A. Kaal, S. Alterary, M. S. Mubarak, A. Alsayari and A. Bin Muhsinah, New thiophene derivatives as antimicrobial agents, J. Heterocycl. Chem., 2019, 56(10), 2845–2953 CrossRef CAS.
- I. Mishra, V. Sharma, N. Kumar, G. Krishna, V. A. Sethi, R. Mittal, P. K. Dhakad and R. Mishra, Exploring Thiophene Derivatives: Synthesis Strategies and Biological Significance, Med. Chem., 2025, 21(1), 11–31 CrossRef CAS PubMed.
- R. Gondru, K. Sirisha, S. Raj, S. K. Gunda, C. G. Kumar, M. Pasupuleti and R. Bavantula, Design, Synthesis, In Vitro Evaluation and Docking Studies of Pyrazole–Thiazole Hybrids as Antimicrobial and Antibiofilm Agents, ChemistrySelect, 2018, 3(28), 8270–8276 CrossRef CAS.
- D. Bhosale, A. Narale, P. Hadimani, M. Kokane, M. Mali, S. Shringare, D. Raut, M. Bamankar, G. Kathwate, M. Damale and A. Lawand, Novel Schiff base derivatives containing 4,5-disubstituted thiazole as potential antibiofilm, anti-inflammatory and antioxidant agents: green synthesis, molecular docking and ADME analysis, J. Mol. Struct., 2024, 1311, 138401 CrossRef CAS.
- R. M. Kassab, S. A. Al-Hussain, N. S. Elleboudy, A. Albohy, M. E. Zaki, K. A. Abouzid and Z. A. Muhammad, Tackling microbial resistance with isatin-decorated thiazole derivatives: design, synthesis, and in vitro evaluation of antimicrobial and antibiofilm activity, Drug Des., Dev. Ther., 2023, 2817–2832 Search PubMed.
- R. Gondru, S. Kanugala, S. Raj, C. G. Kumar, M. Pasupuleti, J. Banothu and R. Bavantula, 1,2,3-Triazole-thiazole hybrids: synthesis, in vitro antimicrobial activity and antibiofilm studies, Bioorg. Med. Chem. Lett., 2021, 33, 127746 CrossRef CAS PubMed.
- S. L. Dhonnar, V. A. Adole, R. H. Patil, B. B. Khairnar, T. B. Pawar, N. V. Sadgir and B. S. Jagdale, Synthesis, spectral, antifungal, in silico molecular docking, ADME and DFT studies of some 2-(2-hydrazineyl) thiazole derivatives: computational and experimental perspective, J. Mol. Struct., 2023, 1294, 136411 CrossRef CAS.
- S. K. Niazi and Z. Mariam, Computer-aided drug design and drug discovery: a prospective analysis, Pharmaceuticals, 2023, 17(1), 22 CrossRef PubMed.
- T. I. Adelusi, A. Q. Oyedele, I. D. Boyenle, A. T. Ogunlana, R. O. Adeyemi, C. D. Ukachi, M. O. Idris, O. T. Olaoba, I. O. Adedotun, O. E. Kolawole and Y. Xiaoxing, Molecular modeling in drug discovery, Inform. Med. Unlocked, 2022, 29, 100880 CrossRef.
- N. Patel, M. Patel, A. Patel, S. Patel, D. Sakariya, A. Parmar, R. Sarkar, M. Patel, S. Rohit, S. Patel and N. Solanki, Investigating the role of natural flavonoids in VEGFR inhibition: molecular modelling and biological activity in A549 lung cancer cells, J. Mol. Struct., 2025, 1322, 140392 CrossRef CAS.
- R. A. Shinde, V. A. Adole, R. A. More, B. S. Jagdale, R. H. Waghchaure, S. S. Gurav and S. N. Mali, Synthesis, spectroscopic characterization, DFT analysis, antibacterial, antifungal, antioxidant, molecular docking, and ADME study of 3,4-dihydro-2H-napthalen-1-one tagged chalcone derivatives, J. Mol. Struct., 2025, 1322, 140313 CrossRef CAS.
- S. K. Pathan, A. Shelar, S. Deshmukh, F. A. Kalam Khan, S. A. Ansari, I. A. Ansari, R. B. Patil, R. Arote, O. Bhusnure, R. H. Patil and J. N. Sangshetti, Exploring antibiofilm potential of some new imidazole analogs against C. albicans: synthesis, antifungal activity, molecular docking and molecular dynamics studies, J. Biomol. Struct. Dyn., 2025, 43(6), 3099–3115 CrossRef CAS.
- N. R. Tawari, S. Bag and M. S. Degani, Pharmacophore mapping of a series of pyrrolopyrimidines, indolopyrimidines and their congeners as multidrug-resistance-associated protein (MRP1) modulators, J. Mol. Model., 2008, 14, 911–921 CrossRef CAS.
- K. S. Bhadoriya, M. C. Sharma and S. V. Jain, Pharmacophore modeling and atom-based 3D-QSAR studies on amino derivatives of indole as potent isoprenylcysteine carboxyl methyltransferase (Icmt) inhibitors, J. Mol. Struct., 2015, 1081, 466–476 CrossRef CAS.
- M. M. Mysinger, M. Carchia, J. J. Irwin and B. K. Shoichet, Directory of useful decoys, enhanced (DUD-E): better ligands and decoys for better benchmarking, J. Med. Chem., 2012, 55(14), 6582–6594 CrossRef CAS.
- K. Roy, S. Kar and P. Ambure, On a simple approach for determining applicability domain of QSAR models, Chemom. Intell. Lab. Syst., 2015, 145, 22–29 CrossRef CAS.
- H. M. Patel, M. Shaikh, I. Ahmad, D. Lokwani and S. J. Surana, BREED based de novo hybridization approach: Generating novel T790M/C797S-EGFR tyrosine kinase inhibitors to overcome the problem of mutation and resistance in non small cell lung cancer (NSCLC), J. Biomol. Struct. Dyn., 2021, 39(8), 2838–2856 CrossRef CAS PubMed.
- R. Kleerebezem, L. W. Pol and G. Lettinga, Anaerobic degradation of phthalate isomers by methanogenic consortia, Appl. Environ. Microbiol., 1999, 65(3), 1152–1160 CrossRef CAS PubMed.
- B. K. Kumar, N. Faheem, K. V. Sekhar, R. Ojha, V. K. Prajapati, A. Pai and S. Murugesan, Pharmacophore based virtual screening, molecular docking, molecular dynamics and MM-GBSA approach for identification of prospective SARS-CoV-2 inhibitor from natural product databases, J. Biomol. Struct. Dyn., 2022, 40(3), 1363–1386 CrossRef CAS PubMed.
- P. Mark and L. Nilsson, Structure and dynamics of the TIP3P, SPC, and SPC/E water models at 298 K, J. Phys. Chem. A, 2001, 105(43), 9954–9960 CrossRef CAS.
- D. J. Evans and B. L. Holian, The Nose–Hoover thermostat, J. Chem. Phys., 1985, 83(8), 4069–4074 CrossRef CAS.
- W. L. Jorgensen, D. S. Maxwell and J. Tirado-Rives, Development and testing of the OPLS all-atom force field on conformational energetics and properties of organic liquids, J. Am. Chem. Soc., 1996, 118(45), 11225–11236 CrossRef CAS.
- G. Kalibaeva, M. Ferrario and G. Ciccotti, Constant pressure-constant temperature molecular dynamics: a correct constrained NPT ensemble using the molecular virial, Mol. Phys., 2003, 101(6), 765–778 CrossRef CAS.
- G. J. Martyna, Remarks on “Constant-temperature molecular dynamics with momentum conservation”, Phys. Rev. E: Stat. Phys., Plasmas, Fluids, Relat. Interdiscip. Top., 1994, 50(4), 3234 CrossRef CAS.
- T. X. Hoang, A. Trovato, F. Seno, J. R. Banavar and A. Maritan, Geometry and symmetry presculpt the free-energy landscape of proteins, Proc. Natl. Acad. Sci. U. S. A., 2004, 101(21), 7960–7964 CrossRef CAS.
- M. J. Frisch, Gaussian 09, Revision D. 01, Gaussian Inc., Wallingford CT, 2009, vol. 201 Search PubMed.
- A. D. Becke, Density-functional thermochemistry. I. The effect of the exchange-only gradient correction, J. Chem. Phys., 1992, 96(3), 2155–2160 CrossRef CAS.
- C. Lee, W. Yang and R. G. Parr, Development of the Colle-Salvetti correlation-energy formula into a functional of the electron density, Phys. Rev. B: Condens. Matter Mater. Phys., 1988, 37(2), 785 CrossRef CAS PubMed.
- B. Miehlich, A. Savin, H. Stoll and H. Preuss, Results obtained with the correlation energy density functionals of Becke and Lee, Yang and Parr, Chem. Phys. Lett., 1989, 157(3), 200–206 CrossRef CAS.
- W. Kohn and L. J. Sham, Self-consistent equations including exchange and correlation effects, Phys. Rev., 1965, 140(4A), A1133 CrossRef.
- R. G. Parr and W. Yang, Density-functional Theory of Atoms and Molecules, Oxford Univ. Press, New York, 1989 Search PubMed.
- P. Hohenberg and W. Kohn, Inhomogeneous electron gas, Phys. Rev., 1964, 136(3B), B864 CrossRef.
- R. Dennington, T. A. Keith and J. M. Millam. GaussView 6.0. 16, Semichem Inc., Shawnee Mission, KS, USA, 2016, pp. 143–150 Search PubMed.
- G. A. Zhurko and D. A. Zhurko, Chemcraft Program Version 1.6 (Build 315), 2009 Search PubMed.
- T. Lu and F. Chen, Multiwfn: a multifunctional wavefunction analyzer, J. Comput. Chem., 2012, 33(5), 580–592 CrossRef CAS PubMed.
- S. Armaković and S. J. Armaković, Atomistica.online – web application for generating input files for ORCA molecular modelling package made with the Anvil platform, Mol. Simul., 2023, 49(1), 117–123 CrossRef.
- S. Armaković and S. J. Armaković, Online and desktop graphical user interfaces for xtb programme from atomistica.online platform, Mol. Simul., 2024, 50(7–9), 560–570 CrossRef.
- W. Humphrey, A. Dalke and K. Schulten, VMD: visual molecular dynamics, J. Mol. Graphics, 1996, 14(1), 33–38 CrossRef CAS PubMed.
- A. Singh, K. Singh, A. Sharma, K. Kaur, R. Chadha and P. M. Bedi, Recent advances in antifungal drug development targeting lanosterol 14α-demethylase (CYP51): a comprehensive review with structural and molecular insights, Chem. Biol. Drug Des., 2023, 102(3), 606–639 Search PubMed.
- L. Song, S. Wang, H. Zou, X. Yi, S. Jia, R. Li and J. Song, Regulation of Ergosterol Biosynthesis in Pathogenic Fungi: Opportunities for Therapeutic Development, Microorganisms, 2025, 13(4), 862 CrossRef PubMed.
- S. Jeyavijayan, Spectroscopic (FTIR, FT-Raman), molecular electrostatic potential, NBO and HOMO–LUMO analysis of P-bromobenzene sulfonyl chloride based on DFT calculations, Spectrochim. Acta, Part A, 2015, 136, 890–899 CrossRef CAS PubMed.
- C. Arunagiri, M. Arivazhagan and A. Subashini, Vibrational spectroscopic (FT-IR and FT-Raman), first-order hyperpolarizability, HOMO, LUMO, NBO, Mulliken charges and structure determination of 2-bromo-4-chlorotoluene, Spectrochim. Acta, Part A, 2011, 79(5), 1747–1756 CrossRef CAS PubMed.
- E. B. Sas, M. Kurt, M. Karabacak, A. Poiyamozhi and N. Sundaraganesan, FT-IR, FT-Raman, dispersive Raman, NMR spectroscopic studies and NBO analysis of 2-bromo-1H-benzimidazol by density functional method, J. Mol. Struct., 2015, 1081, 506–518 CrossRef CAS.
- S. Selvaraj, A. R. Kumar, T. Ahilan, M. Kesavan, S. Gunasekaran and S. Kumaresan, Multi spectroscopic and computational investigations on the electronic structure of oxyclozanide, J. Indian Chem. Soc., 2022, 99(10), 100676 CrossRef CAS.
- M. Kurt, P. C. Babu, N. Sundaraganesan, M. Cinar and M. Karabacak, Molecular structure, vibrational, UV and NBO analysis of 4-chloro-7-nitrobenzofurazan by DFT calculations, Spectrochim. Acta, Part A, 2011, 79(5), 1162–1170 CrossRef CAS PubMed.
- S. Gunasekaran, S. Kumaresan, R. Arun Balaji, G. Anand and S. Seshadri, Vibrational spectra and normal coordinate analysis on structure of chlorambucil and thioguanine, Pramana, 2008, 71, 1291–1300 CrossRef CAS.
- A. Ram Kumar, S. Selvaraj, M. Azam, G. P. Sheeja Mol, N. Kanagathara, M. Alam and P. Jayaprakash, Spectroscopic, biological, and topological insights on lemonol as a potential anticancer agent, ACS Omega, 2023, 8(34), 31548–31566 CrossRef CAS PubMed.
- R. Gayathri and M. Arivazhagan, Experimental (FT-IR and FT-Raman) and theoretical (HF and DFT) investigation, NMR, NBO, electronic properties and frequency estimation analyses on 2,4,5-trichlorobenzene sulfonyl chloride, Spectrochim. Acta, Part A, 2012, 97, 311–325 CrossRef CAS PubMed.
- P. Rajkumar, S. Selvaraj, P. Anthoniammal, A. R. Kumar, K. Kasthuri and S. Kumaresan, Structural (monomer and dimer), spectroscopic (FT-IR, FT-Raman, UV-Vis and NMR) and solvent effect (polar and nonpolar) studies of 2-methoxy-4-vinyl phenol, Chem. Phys. Impact, 2023, 7, 100257 CrossRef.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *a,
Vishnu A. Adole
*a,
Vishnu A. Adole