DOI:
10.1039/D5RA00523J
(Paper)
RSC Adv., 2025,
15, 13497-13504
Forensic electrochemical sensor for fentanyl and morphine detection using an Au–NiOx-electrodeposited carbon electrode†
Received
22nd January 2025
, Accepted 18th April 2025
First published on 28th April 2025
Abstract
The overuse and abuse of narcotics, such as fentanyl and morphine, has resulted in serious threats to human health. Although current detection methods are generally effective, they rely on specialized laboratory equipment. Herein, a forensic electrochemical sensor was developed for the on-site detection of trace quantities of fentanyl and morphine. A screen-printed carbon electrode (SPCE) was modified via the electrodeposition of Au and nickel oxide (NiOx) to enhance its electrochemical activity. Au electrodeposition was performed using a multi-potential step method that alternated between deposition and resting potentials (−0.2 and 0.7 V, respectively) with pulse durations of 1 s and 500 ms, respectively. Subsequently, NiOx deposited onto the Au–SPCE by applying a constant potential of −1.0 V for 1 h resulted in uniform NiOx nanofilms on the three-dimensional leaf-shaped Au structure, as observed by focused ion beam scanning electron microscopy. In addition, X-ray diffraction and photoelectron spectroscopy analyses confirmed the presence and chemical states of Au and NiOx on the SPCE surface. The electrochemical sensing performance of the modified SPCE was evaluated using linear sweep voltammetry, which revealed detectable signals at 0.85 and 0.5 V for fentanyl and morphine, respectively. Moreover, the calibration curves exhibited a linear relationship between concentration and current density, thereby confirming the sensitivity of the sensor electrode. The distinct oxidation peak potentials enabled the simultaneous detection of fentanyl and morphine in mixed solutions, confirming its selectivity for target molecules. The findings of this study demonstrate the potential of Au–NiOx-modified SPCEs as a practical tool for rapid and selective electrochemical detection of narcotic substances.
Introduction
The growing prevalence of drug addiction and the misuse of narcotics and psychotropic substances have increased drug-related incidents worldwide, posing significant threats to public health and safety. In particular, fentanyl and morphine, which are synthetic and natural opioids, respectively, are frequently abused owing to their potent analgesic effects and contribute to overdoses and fatal incidents.1–3 Fentanyl is often illicitly manufactured and mixed with other substances, making it difficult to control and monitor. Owing to the serious health risks and rapid onset of effects associated with these opioids, effective and rapid detection methods are urgently required for patient safety in clinical settings and to support public health interventions and law enforcement efforts addressing the opioid crisis. However, the rapid detection of various narcotics, including opioids and stimulants, remains challenging with conventional analytical methods, such as chromatography and immunoassays, which are time-consuming and require specialized laboratory equipment.3–6 These limitations potentially delay timely interventions and hinder real-time, on-site analyses. Therefore, portable diagnostic tools that can rapidly identify substances on-site without requiring transportation to a testing facility have been investigated.
Biosensors can translate biochemical information from target analytes into electrical signals. They have become pivotal to recent advancements in portable diagnostic technology with the potential for real-time, location-independent detection.3,7 In point-of-care testing and in vitro diagnostics, sensors must offer rapid testing, high accuracy, and user-friendliness.2,6,8 Among the various types of biosensors, electrochemical biosensors, which detect and quantify target substances by converting chemical or biological signals into electrical signals, are actively researched as a technology that meets these essential requirements.
Electrochemical sensors offer several advantages, including miniaturization and portability, ease of use, rapid operation, high measurement reliability, and cost-effectiveness.9–11 Therefore, they have been applied to on-site diagnostic analysis devices in various industrial fields. Moreover, they enable numerical monitoring beyond the level of qualitative analysis, which only determines the presence or absence of target substances. Electrochemical sensors, capable of quantitative analysis at low detection limits and high operating speeds, have been researched and developed as forensic tools for sensing narcotic substances.12–15 The performance and electrochemical properties of these sensors can be further improved through electrode modifications employing functional materials and advanced processing techniques.16–18 Among various electrode materials, carbon-based materials, such as glassy carbon, graphite, and screen-printed carbon, are widely utilized in forensic electrochemical sensor research due to their excellent durability, wide potential window range, and low background current.19–22 Modifying carbon electrode surfaces with various materials, such as metals, metal oxides, conductive polymers, and nanomaterials, can enhance their electrochemical catalytic properties and signal amplification, thereby improving the critical performance metrics of the electrochemical sensor, including the limit of detection (LOD), sensitivity, and selectivity.23–25
In this study, we aimed to improve the electrochemical properties of a forensic electrochemical sensor electrode by selectively modifying the carbon electrode surface via the electrochemical deposition of Au and nickel oxides (NiOx, nickel oxide, or nickel hydroxide). Au has demonstrated exceptional electrical conductivity, high chemical stability, excellent biocompatibility, and versatile surface modification capabilities, making it widely applicable in electrochemical biosensors and chemical sensors.26,27 Moreover, NiOx is a promising candidate for electrochemical sensors owing to its excellent electrochemical catalytic properties, good chemical stability, and low cost.28–30 Au nanoparticles were electrochemically deposited onto the electrode surface, thereby increasing the surface area and reducing the charge transfer resistance at the electrode interface to improve the signal-to-noise ratio and signal amplification. Subsequently, NiOx formed a nanofilm on the electrode surface via electrodeposition, further enhancing the electrode's electrochemical catalytic properties.31–33 In addition, we focused on using the Au–NiOx-modified electrode to detect narcotic/psychotropic substances, such as fentanyl and morphine, which are critical targets for forensic analysis due to their widespread misuse and significant impact on public health. The possibility of simultaneous detection of both fentanyl and morphine was also investigated to explore its potential as a dual forensic electrochemical sensor.
Experimental
Materials
Sodium tetrachloroaurate(III) dihydrate (NaAuCl4·2H2O), sodium perchlorate (NaClO4), sodium acetate (NaAc), acetic acid (HAc), potassium phosphate monobasic (KH2PO4), potassium phosphate dibasic (K2HPO4), sodium hydroxide (NaOH), potassium hexacyanoferrate(II) trihydrate (K4Fe(CN)6·3H2O), and potassium hexacyanoferrate(III) (K3Fe(CN)6) were purchased from Sigma-Aldrich (Merck). Nickel(II) nitrate hexahydrate (Ni(NO3)2·6H2O) was purchased from Alfa Aesar (Thermo Fisher Scientific). Sulfuric acid (H2SO4) and potassium chloride (KCl) were purchased from Daejung Chemicals & Metals Co., Ltd. Fentanyl citrate was supplied by Lipomed, and morphine sulfate hydrate was supplied by Cayman Chemical. Both compounds were acquired from Sam Eung Industrial Co., Ltd. We obtained approval from the Gyeongin Regional Office of Food and Drug Safety, part of the Ministry of Food and Drug Safety, to handle narcotics in this study as academic researchers.
A 0.1 M acetate buffer (AB) solution (pH 4.0) was prepared by dissolving appropriate proportions of the supporting electrolytes NaAc and HAc for nickel oxide (NiOx) electrodeposition. A 0.1 M phosphate buffer (PB) solution (pH 7.0) was prepared by dissolving appropriate amounts of the electrolytes KH2PO4 and K2HPO4 for the passivation step of the NiOx-electrodeposited electrode. All chemicals and reagents were of analytical grade and used directly without modifications, and all aqueous solutions were prepared in deionized water (18 MΩ cm).
Surface modification of the carbon electrode via the electrodeposition of Au and NiOx
A screen-printed carbon electrode (SPCE; thick-film carbon electrode by Micrux Technologies) was used as the working electrode for the forensic electrochemical sensors. An electrochemical cell with a three-electrode system was used, and Ag/AgCl (in 3 M NaCl) and Pt-coiled wire were employed as the reference and counter electrodes, respectively. The electrochemical experiments were performed using a Multi AutoLab M204 electrochemical workstation (Metrohm AG, Switzerland). First, the carbon surface of the SPCE was modified via Au electrodeposition using a precursor solution containing 2 mM NaAuCl4 in 0.2 M NaClO4. The deposition was conducted using a potential pulse, applied through a multi-potential step method, with a holding potential of −0.2 V for 1 s and stepping to 0.7 V for 500 ms. The potential pulse was repeated 2000 times. Cyclic voltammetry (CV) was performed in 0.5 M H2SO4 to examine the electrodeposited Au on the electrode surface. The cathodic electrodeposition of NiOx onto the Au–SPCE surface was performed in a 0.1 M AB solution containing 4 mM Ni(NO3)2·6H2O by applying a constant potential of −0.1 V for 3600 s. Subsequently, the metallic Ni on the electrode surface was passivated in a 0.1 M PB solution by cycling the potential between −0.5 and 0.8 V for 10 cycles at a scan rate of 200 mV s−1. Finally, the Au–NiOx-modified electrode was electrochemically activated in a 0.5 M NaOH solution for 100 cycles, with a potential range from −0.2 to 0.6 V at a scan rate of 100 mV s−1.31,32
Electrochemical characterizations of the Au–NiOx-modified SPCEs
The electrochemical properties of Au–NiOx-modified SPCEs were characterized by CV and electrochemical impedance spectroscopy (EIS) using a potentiostat (Multi AutoLab M204, Metrohm AG, Switzerland). All electrochemical analyses were performed using three-electrode cells. The working, reference, and counter electrodes were the Au–NiOx-modified SPCE, Ag/AgCl (in 3 M NaCl), and coiled Pt wire, respectively. A ferricyanide/ferrocyanide ([Fe(CN)6]3−/4−) redox couple was prepared as an electron transfer mediator, and CV profiles and Nyquist plots were obtained in 0.1 M KCl containing 1 mM [Fe(CN)6]3−/4−. CV was performed by cycling the potential from −0.1 to 0.5 V at a scan rate of 50 mV s−1, and EIS was conducted in a frequency range of 10 kHz to 0.1 Hz at a potential of 0.25 V.
Characterization of the Au–NiOx-modified SPCE surface
The Au–NiOx-modified SPCEs were characterized using the analytical equipment at the Gyeonggi Semiconductor Innovation Center analysis laboratory (Advanced Institute of Convergence Technology). The surface and cross-sectional morphologies of the Au–NiOx-modified SPCEs were investigated using focused ion beam field-emission scanning electron microscopy (FIB/FE-SEM; Helios 5 UC, FEI Company, USA) operating at 2.0 kV. The images were obtained at appropriate magnifications for comparison. The crystal structure of NiOx on Au–SPCEs was characterized by X-ray diffraction (XRD; D8 Discover, Bruker, Germany) in a 2θ range of 5–90°. The chemical compositions of NiOx on the Au–SPCEs were determined using high-performance X-ray photoelectron spectroscopy (XPS; AXIS Supra+, Kratos Analytical Ltd, UK).
Electrochemical determination of narcotic substances using the Au–NiOx-modified SPCE
To confirm the electrochemical detection of fentanyl and morphine, CV profiles were measured in a 0.1 M PB solution containing 1 mM of fentanyl citrate or morphine sulfate hydrate in the potential range of 0.1–1.0 V at a scan rate of 100 mV s−1. Linear sweep voltammetry (LSV) was performed to monitor the current change in response to the concentration gradients of fentanyl or morphine from low (0.25 μM) to high (500 μM) by sweeping the potential from 0.2 to 1.0 V at a scan rate of 100 mV s−1. The calibration curve was acquired by obtaining the current values at specific voltages for each concentration of fentanyl and morphine at potentials of 0.85 and 0.50 V, respectively. To evaluate the feasibility of simultaneous sensing of fentanyl and morphine, three types of fentanyl/morphine mixed samples were prepared: samples A (0.25 mM fentanyl + 0.5 mM morphine), B (0.5 mM fentanyl + 0.25 mM morphine), and C (1 mM each of fentanyl and morphine). These samples were electrochemically detected using LSV, and the difference in current values obtained at the low and high concentrations was investigated at potentials of 0.85 and 0.50 V for fentanyl and morphine, respectively.
Results and discussion
Surface modification of the SPCE via the electrodeposition of Au and NiOx
The SPCE surface was electrodeposited with Au and NiOx to improve its electrochemical activity for detecting trace amounts of narcotic substances. An electrodeposition method was employed to selectively and uniformly deposit Au and NiOx onto the SPCE surface with high reproducibility. First, Au was electrodeposited using a multi-potential step method with pulsed deposition (−0.2 V) and resting (0.7 V; open circuit) potentials applied in an Au precursor solution of 2 mM NaAuCl4 in 0.2 M NaClO4 (Fig. 1a). By repeatedly applying pulsed deposition and resting potentials for 1 s and 500 ms, respectively, the Au precursor was rapidly diffused and uniformly deposited onto the electrode surface. The cathodic and anodic current densities gradually increased with successive pulsed potentials, indicating that Au was seeded and grew on the electrode, thereby increasing the electrochemically active surface area. To deposit NiOx onto the Au–SPCE, a constant cathodic potential (−1.0 V) was applied (Fig. 1b), and the reduction current of NiOx was continuously monitored throughout the reaction.
 |
| | Fig. 1 Electrodeposition of Au and NiOx onto the surface of SPCE and subsequent electrochemical confirmation. (a) Au electrodeposition onto SPCE. The top image shows the profile diagram of pulse potentials applied for Au electrodeposition, and the bottom image shows the corresponding current–time (i–t) curve for Au electrodeposition. The inset graph in the lower image of (a) shows the zoomed in i–t curve according to the applied pulse potentials. The blue area represents an applied potential of −0.2 V for 1 s, and the gray area represents an applied potential of 0.7 V for 500 ms. (b) NiOx electrodeposition onto Au-electrodeposited SPCE. The top image shows the profile diagram of the constant potential applied for NiOx electrodeposition, and the bottom image shows the corresponding i–t curve for NiOx electrodeposition onto the electrode. | |
The electrodeposited Au on SPCE was confirmed by observing the oxidation and reduction peaks using CV in 0.5 M H2SO4 (Fig. S1a†). These peaks indicated the formation and removal of an oxide film on the Au surface. NiOx on the Au–SPCE was activated by performing CV in 0.5 M NaOH, and the redox peak of NiOx was observed.28,32 Specifically, oxidation–reduction reactions occurred through the conversion of Ni(II) (NiO or Ni(OH)2) to Ni(III) (NiOOH) (Fig. S1b†). The electrodeposited NiO was transformed into Ni(OH)2, and OH− ions infiltrated Ni(OH)2 to form NiOOH via electrochemical activation in an alkaline environment:32–34
| Ni(OH)2 + OH− ↔ NiOOH + H2O + e− |
Electrochemical characterization of the Au–NiOx-modified SPCE
The electrochemical properties of the Au–NiOx-modified SPCEs were analyzed using the ferricyanide/ferrocyanide redox couple ([Fe(CN)6]3−/4−) as an electron transfer mediator. The Au–NiOx-modified SPCEs exhibited improved electrochemical activity and catalytic properties compared to bare SPCEs. Fig. 2a shows the CV profiles obtained at a concentration of 1 mM ([Fe(CN)6]3−/4−) in 0.1 M KCl. The Au–NiOx-modified SPCEs exhibited larger peak currents (ipa/ipc) and narrower redox peak-to-peak potentials (Epa/Epc) compared to the bare SPCE. Owing to the excellent electrical conductivity of the electrodeposited Au, the surface area of the electrode contributing to the electrochemical reaction was increased, thereby leading to signal amplification. In addition, the electrodeposited NiOx possesses good electrocatalytic properties, enabling a rapid electron transfer at the electrode interface. Thus, the electrochemical properties of the electrode were improved, which directly enhanced the performance of the electrochemical sensor.
 |
| | Fig. 2 Electrochemical characterizations of Au–NiOx-modified SPCE. (a) Comparison of the cyclic voltammograms for (1) bare SPCE versus (2) Au–NiOx-modified SPCE at a scan rate of 50 mV s−1. (b) Comparison of the EIS profiles for (1) bare SPCE versus (2) Au–NiOx-modified SPCE from 0.1 Hz to 10 kHz with an amplitude of 0.005 V. (c) CV profiles at different scan rates: 0.005, 0.01, 0.02, 0.05, 0.1, 0.2, and 0.5 V s−1. (d) Plot of the oxidation (Ox) and reduction (Red) peak currents of the Au–NiOx-modified SPCE vs. scan rate in the CV profiles. The black line is from the fitting analysis of the plot. All experiments were performed in a 0.1 M KCl solution containing 1 mM [Fe(CN)6]3−/4−. | |
EIS was employed to compare the interfacial properties of the SPCEs before and after Au–NiOx modification, with the results displayed in the Nyquist plot shown in Fig. 2b. Compared to the bare SPCE, the semicircular portion decreased for the Au–NiOx-modified SPCE, resulting in a lower interfacial resistance. Furthermore, CV was performed at different scan rates to investigate the electrochemical behavior at the interface between the Au–NiOx-modified SPCE and the electrolyte. The CV profiles of the modified SPCE at different scan rates of 5–500 mV s−1 are illustrated in Fig. 2c. The anodic and cathodic peak potentials (Epa and Epc, respectively) gradually increased with increasing scan rates. As shown in Fig. 2d, the anodic and cathodic peak currents (ipa and ipc) were linearly proportional to the square root of the scan rate. Thus, the electrochemical behavior on the surface-modified SPCE surface was reversible. Both ipa and ipc followed linear relationships with v, and the regression equations are ipa = 0.04433v − 0.03836 (R2 = 0.999) and ipc = −0.03546v + 0.00606 (R2 = 0.999). These results indicated that redox reactions occurred as a diffusion-controlled process on the modified SPCE surface.
Multifaceted characterization of the Au–NiOx-modified SPCEs
The surface and cross-sectional morphologies of the Au–NiOx-modified SPCEs were investigated using FIB/FE-SEM. To examine the formation of Au and NiOx on the SPCE surface via electrodeposition, a comparative analysis was performed using bare SPCE as the control. Fig. 3a shows the relatively smooth surface of the bare SPCE, whereas Fig. 3b clearly demonstrates that the sequential electrochemical deposition of Au and NiOx markedly roughens the electrode. When Au was electrodeposited onto the bare SPCE under the conditions shown in Fig. 1a, Au was initially seeded on the electrode surface and subsequently grew uniformly into nanoparticles with an average size of approximately 226 nm. In addition, regions of excessive Au growth were observed, forming leaf-shaped particles with branches extending up to several micrometers (Fig. S2†). Following Au modification, NiOx was electrodeposited under the conditions presented in Fig. 1b. Nanograins accumulated across the entire Au surface, as shown in Fig. 3b and S3,† confirming the successful deposition of NiOx on Au–SPCE. Notably, a uniform NiOx nanofilm was formed even on the three-dimensional, leaf-shaped, overgrown Au structures (Fig. S3†). A direct comparison between the Au surface obtained by Au-only deposition (Fig. S2d†) and that obtained by sequential Au and NiOx deposition (Fig. S3b†) further reveals that the originally smooth Au surface becomes rough due to the coverage of NiOx nanograins. Moreover, a cross-sectional analysis of the Au–NiOx-modified SPCE (Fig. 3d) and the bare SPCE (Fig. 3c) reveals a distinct difference: a uniform NiOx nanofilm with a thickness of approximately 60 nm is clearly shown on the surface of the electrodeposited Au nanoparticles. In contrast, when NiOx was deposited alone onto the bare SPCE under the same conditions, the electrode surface was also covered with nanograins; however, the thickness of the nanofilm was approximately 150 nm (Fig. S4†). Thus, the NiOx layer on the unmodified electrode is roughly twice as thick as that on the Au–SPCE, possibly due to the interference caused by the three-dimensional morphology of Au during NiOx deposition. Despite the differences in NiOx thickness, electrochemical characterization confirmed that the Au–NiOx-modified SPCE exhibited stable electrochemical activity, which was attributed to the uniform NiOx nanofilm covering the Au surface.
 |
| | Fig. 3 FIB/FE-SEM images of the Au–NiOx-modified SPCE compared to a bare SPCE. (a) and (b) Surface morphology images of the bare and Au–NiOx-modified SPCEs, respectively. (c) and (d) Cross-sectional images of the bare and Au–NiOx-modified SPCEs, respectively. | |
The surface of the Au–NiOx-modified SPCE was analyzed using XRD and XPS to confirm the presence of Au and Ni and their chemical states. The XRD patterns of the Au–NiOx-modified and bare SPCEs are shown in Fig. 4a. The XRD pattern of the SPCE, revealed strong substrate peaks (below 2θ of 30°), along with several additional peaks, indicating the presence of unknown substances. This complexity makes it difficult to clearly assign peaks to the deposited materials. To clarify this ambiguity, XRD measurements were conducted on glassy carbon plate (GCP) electrodes, which provide a clean baseline due to their simple carbon composition. Au and NiOx layers were electrodeposited individually (Au–GCP, NiOx–GCP) and sequentially (Au–NiOx–GCP) on the GCP under the same conditions used for the SPCEs. The XRD patterns of the bare GCP confirmed the absence of interfering substrate peaks, enabling the identification of peaks originating from the deposited layers. Then, the peaks observed in the Au–NiOx-modified SPCE were compared with those from the Au–NiOx–GCP (Fig. S5†), allowing for the proper assignment of each peak to its corresponding chemical species. On the GCP electrode, the electrodeposited Au layer was identified as a cubic Au phase (JCPDS No. 66-0091), with 2θ values of 38.12°, 44.30°, 64.58°, 77.63°, and 81.80° corresponding to the (111), (200), (220), (311), and (222) crystalline planes, respectively. The subsequent NiOx layer covered the surface of the Au-modified GCP electrode. The XRD patterns of Au–NiOx–GCP showed peaks corresponding to cubic NiO (JCPDS No. 04-0835), hexagonal β-phase Ni(OH)2 (JCPDS No. 14-0117), and hexagonal γ-phase NiOOH (JCPDS No. 06-0075). This finding was consistent with previous studies showing that NiOx electrodes fabricated via electrodeposition initially comprise metallic Ni, which undergoes passivation and alkaline treatment to form Ni(OH)2 and NiOOH.32,35–37 For Au–NiOx-modified SPCEs, despite peak overlapping, specific peaks related to the NiOx layer were identified. For example, the peaks at 44.30° and 75.26° were assigned to the (200) and (311) planes of NiO, respectively. The peaks at 38.12°, 39.92°, and 52.10° corresponded to the (101), (002), and (102) planes of Ni(OH)2, respectively. The peak at 44.30° was attributed to the (105) plane of NiOOH. These results confirmed the successful deposition of NiOx phases onto the SPCE. The chemical states of the elements present on the surface of the Au–NiOx-modified SPCE were analyzed using XPS. As shown in the survey scan (Fig. 4b), peaks corresponding to carbon from the SPCE substrate, along with Au, Ni, and O from the modification, were observed at their respective binding energies. To analyze the chemical states of Ni and O on the surface of the modified electrode, high-resolution XPS spectra were obtained and deconvoluted using Gaussian–Lorentzian fitting (Fig. 4c and d). As shown in Fig. 4c, the Ni 2p spectrum exhibited two major peaks at 873.20 and 854.40 eV, corresponding to Ni 2p3/2 and Ni 2p1/2, respectively. The Ni 2p3/2 profile was further deconvoluted into three peaks, corresponding to Ni2+ from NiO (852.37 eV), Ni3+ from NiOOH (854.87 eV), and Ni2+ from Ni(OH)2 (855.84 eV). Similarly, the Ni 2p1/2 profile was deconvoluted to peaks at 870.67, 872.69, and 873.99 eV, corresponding to NiO, NiOOH, and Ni(OH)2, respectively.32,38–40 The O 1s spectrum shown in Fig. 4d presented a dominant peak at 531.10 eV. After deconvolution, three peaks at binding energies of 528.92, 530.75, and 531.61 eV were assigned to O2−, defective oxygen (Odef), and OH−, respectively. In this study, the oxygen peaks do not correspond to a single NiOx species but rather reflect the complex results of all three forms of NiOx, as shown in Fig. 4d. NiO generally shows an O2− peak; however, in environments where Ni3+ is present (such as NaOOH), an Odef peak also appears. The oxygen species in Ni(OH)2 are represented by the OH− peak, while NiOOH exhibits both O2− and OH− oxygen peaks, which is consistent with previous studies.40–43 Therefore, the peaks in the O 1s spectrum are the result of interactions between the three NiOx species. In particular, the O2− and Odef peaks are related to the interactions between Ni2+ and Ni3+ in NiO and NiOOH, while the OH− peak is influenced by Ni(OH)2 and NiOOH.
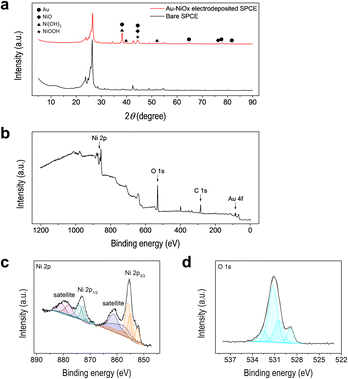 |
| | Fig. 4 Surface characteristics of the electrochemically modified electrode. (a) XRD patterns of the bare SPCE (black line) and Au–NiOx-modified SPCE (red line). (b) XPS survey scan of the Au–NiOx-modified SPCE. (c) High resolution Ni 2p spectrum of the Au–NiOx-modified SPCE fitted with Ni2+ and Ni3+. (d) High resolution O 1s spectrum of the Au–NiOx-modified SPCE fitted with three O species: O2−, Odef, and OH−. | |
Electrochemical sensing of narcotic substances using Au–NiOx-modified SPCE
The electrochemical detection capability of the developed electrode for target narcotic substances was evaluated at high concentrations (1 mM) of fentanyl citrate or morphine sulfate hydrate in 0.1 M PBS (pH 7.0) using CV (Fig. 5a and b). In contrast to the CV profile of 0.1 M PBS without narcotic substances (black line), oxidation reactions were observed on the surface of the modified electrode. For fentanyl, the current increased from 0.7 V, and the oxidation peak appeared at 0.85 V. The CV profile of morphine showed an increasing current from 0.3 V with an oxidation peak at 0.5 V. Thus, the proposed Au–NiOx-modified SPCE demonstrated its potential as an electrochemical sensor for detecting fentanyl and morphine. Moreover, the irreversible electrochemical behaviors of fentanyl and morphine have been reported in previous studies,15,44–48 and the possible routes for the electrooxidation processes on the modified electrode are as follows:
| Fentanyl + H2O → Norfentanyl + Phenylacetaldehyde + 2H+ + 2e− |
| Morphine → Pseudomorphine + 2H+ + 2e− |
 |
| | Fig. 5 Electrochemical analysis of target narcotic substances using the Au–NiOx-modified SPCE. CV results of high concentration narcotics in 0.1 M PB solution at a scan rate of 100 mV s−1: (a) 1 mM fentanyl (red line) and (b) 1 mM morphine (red line). The CV results of the 0.1 M PB solution without target substances are co-plotted as a black line. LSV profiles at a scan rate of 50 mV s−1 for the concentrations of 0.25, 2.5, 25, 250, and 500 μM for (c) fentanyl and (d) morphine, and calibration curves for (e) fentanyl and (f) morphine (n = 3). | |
To evaluate the sensing performance of the modified electrode, LSV measurements were conducted for fentanyl (Fig. 5c) and morphine (Fig. 5d) in the concentration range of 0.25–500 μM. As shown in Fig. 5c and d, the peak current increased with the concentration of the narcotic substance, with peak potentials of 0.85 and 0.5 V for fentanyl and morphine, respectively. Calibration curves were plotted to show the relationship between the peak current measured at the peak potential and the concentration of each analyte. As shown in Fig. 5e and f, both calibration curves exhibited linearity over a low-concentration range from 0.25 to 25 μM and a high-concentration range from 25 to 500 μM. To determine the sensitivity and the LODs of the fentanyl and morphine sensors based on the Au–NiOx modified SPCE, linear regression analysis was performed on the low-concentration range. The resulting calibration equations were as follows: jp,fentanyl = 0.02931Cfentanyl + 0.23775 (R2 = 0.969) and jp,morphine = 0.00367Cmorphine + 0.06214 (R2 = 0.874). Accordingly, the sensitivities of the sensors were calculated to be 2.0718 μA μM−1 for fentanyl and 0.25942 μA μM−1 for morphine, while the corresponding LOD values were determined to be 0.01088 μM and 0.03601 μM, respectively. In addition, the sensing performance of the Au–NiOx-modified SPCE was compared with that of previously reported sensors for the detection of fentanyl and morphine. As summarized in Table 1, the developed sensor based on Au–NiOx-modified SPCE exhibits comparable or better analytical performance compared to other reported electrochemical and non-electrochemical sensing platforms. The enhancement in electrochemical performance and sensing sensitivity is attributed to the uniform and homogeneous modification of the SPCE surface with Au–NiOx via an electrochemical deposition method, which provides a synergistic effect by combining the excellent conductivity of Au and the strong electrocatalytic activity of NiOx.
Table 1 Performance comparison of different sensors for determination of fentanyl and morphine
| Analyte |
Sensing method |
Modified electrode or sensing platform |
Linear range |
LOD |
Ref. |
| Fentanyl |
Electrochemistry |
Au–NiOx–SPCE |
0.25–500 μM |
0.01 μM |
This work |
| NiO@CNTs/GCE |
1–160 μM |
0.01 μM |
14 |
| MWCNTs/GCE |
0.5–100 μM |
0.1 μM |
45 |
| Colorimetry |
CB[7]-FCA |
0.1–10 mM |
5 mM |
49 |
| Raman (SERS) |
MOF-Au-nanostructure |
5–200 ng mL−1 |
0.46 ng mL−1 |
50 |
| Morphine |
Electrochemistry |
Au–NiOx–SPCE |
0.25–500 μM |
0.036 μM |
This work |
| NiO/SWCNTs/DDPM/CPE |
0.9–400 μM |
0.4 μM |
51 |
| NiO/MWCNT paste electrode |
0.34–12 μM |
0.14 μM |
52 |
| Photochemistry |
Cr-MOF-NPs |
0.1–350 nM |
0.167 nM |
53 |
| D-cys-GQDs |
0–88 μM |
1.9 μM |
54 |
As shown in Fig. 5, fentanyl and morphine exhibited oxidation peaks at distinct potentials; therefore, it is possible to detect the two substances in mixed solutions simultaneously. To confirm this, three mixed solutions (A, B, and C) were prepared with fentanyl/morphine concentrations of 0.25 mM/0.5 mM, 0.5 mM/0.25 mM, and 1 mM/1 mM, respectively, for LSV analysis. Fig. 6a shows the LSV profiles of the mixed solutions, and Fig. 6b illustrates the current densities at the peak potential for both substances. The current density increased with increasing concentrations of fentanyl and morphine in the mixed solutions. For fentanyl, both the concentration and its current density increased in the order A < B < C, while for morphine, the order was B < A < C. These findings indicated that the developed sensor based on Au–NiOx-modified SPCE demonstrated great potential to detect different narcotic substances simultaneously as long as their peak potentials during the electrochemical reaction were separated.
 |
| | Fig. 6 (a) LSV profiles of a mixed samples of fentanyl and morphine at the following concentrations: (A) 0.25 mM fentanyl and 0.5 mM morphine, (B) 0.5 mM fentanyl and 0.25 mM morphine, and (C) 1 mM fentanyl and 1 mM morphine. (b) Current densities at the peak potentials of each substance: fentanyl at 0.8 V and morphine at 0.5 V. | |
Conclusions
This study successfully developed a forensic electrochemical sensor that could detect narcotic substances (fentanyl and morphine). To construct the sensor, the SPCE surface was selectively electrodeposited with Au of high conductivity and NiOx, which possesses excellent electrocatalytic properties. The electrochemically deposited Au nanoparticles and NiOx nanofilms improved the electrochemical properties of the sensor electrode. In addition, the chemical state and structure of the NiOx nanofilm, which was the outmost layer of the modified electrode, were analyzed in detail. This layer exhibited electrochemical catalytic properties; therefore, it participated in the direct electrochemical reaction with fentanyl/morphine. The surface-modified electrodes could generate signals at specific potentials (0.85 and 0.5 V for fentanyl and morphine, respectively), and the signals show a linear relationship with the concentration of the analytes. Moreover, the electrode can simultaneously detect fentanyl and morphine in their mixed solutions. This study contributes to the future development of forensic electrochemical sensors that can be used directly in real world to ensure reliable detection in samples, such as biological fluids or food and beverages.
Data availability
The data supporting the findings of this study are available within the article and its ESI.†
Author contributions
Eun Joong Kim conceptualization, data curation, funding acquisition, investigation, supervision, writing – original draft, writing – review & editing. Yekyung Kim conceptualization, data curation, investigation, supervision, writing – original draft, writing – review & editing. Soyeon Kwon formal analysis. Sungho Kang writing – review & editing. Tae Hoon Park writing – review & editing.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by a grant (2023-008) from the Gyeonggi Research Development Program for Women in Science, Engineering and Technology funded by Gyeonggi Province. This work was supported by the National Research Foundation of Korea (NRF) grant funded by the Korean government (MSIT) (No. 2021R1A2C1093427, No. 2021R1F1A1060062).
References
- R. A. Rudd, N. Aleshire, J. E. Zibbell and R. M. Gladden, Morb. Mortal. Wkly. Rep., 2016, 64, 1378–1382 CrossRef PubMed.
- Z. Li and P. Wang, Arch. Pathol. Lab. Med., 2020, 144, 1325–1334 CrossRef CAS PubMed.
- S. O. Fakayode, P. N. Brady, C. Grant, V. Fernand Narcisse, P. Rosado Flores, C. H. Lisse and D. K. Bwambok, Chemosensors, 2024, 12, 58 CrossRef CAS.
- N. K. Singh, G. K. Sidhu and K. Gupta, Biomedicines, 2022, 10, 743 CrossRef CAS PubMed.
- M. Yence, A. Cetinkaya, S. I. Kaya and S. A. Ozkan, Crit. Rev. Anal. Chem., 2024, 54, 882–895 CrossRef CAS PubMed.
- N. Anzar, S. Suleman, S. Parvez and J. Narang, Process Biochem., 2022, 113, 113–124 CrossRef CAS.
- M. Razlansari, F. Ulucan-Karnak, M. Kahrizi, S. Mirinejad, S. Sargazi, S. Mishra, A. Rahdar and A. M. Díez-Pascual, Eur. J. Pharm. Biopharm., 2022, 179, 79–94 CrossRef CAS PubMed.
- S. R. Ahmed, R. Chand, S. Kumar, N. Mittal, S. Srinivasan and A. R. Rajabzadeh, TrAC, Trends Anal. Chem., 2020, 131, 116006 CrossRef CAS.
- X. Li, B. Luo, M. Liao and A. Mohamed, Front. Chem., 2022, 10, 1–12 Search PubMed.
- R. Zhang, K. Fu, F. Zou, H. Bai, G. Zhang, F. Liang and Q. Liu, Electrochim. Acta, 2021, 370, 137803 CrossRef CAS.
- G. B. Slepchenko, T. M. Gindullina and S. V. Nekhoroshev, J. Anal. Chem., 2017, 72, 703–709 CrossRef CAS.
- H. M. Elbardisy, A. García-Miranda Ferrari, C. W. Foster, O. B. Sutcliffe, D. A. C. Brownson, T. S. Belal, W. Talaat, H. G. Daabees and C. E. Banks, ACS Omega, 2019, 4, 1947–1954 CrossRef CAS.
- L. Shaw and L. Dennany, Curr. Opin. Electrochem., 2017, 3, 23–28 CrossRef CAS.
- H. Ding and W. Tao, Int. J. Electrochem. Sci., 2021, 16, 1–12 Search PubMed.
- M. W. Glasscott, K. J. Vannoy, P. U. A. Iresh Fernando, G. K. Kosgei, L. C. Moores and J. E. Dick, TrAC, Trends Anal. Chem., 2020, 132, 116037 CrossRef CAS.
- P. Abraham, S. Renjini, P. Vijayan, V. Nisha, K. Sreevalsan and V. Anithakumary, J. Electrochem. Soc., 2020, 167, 037559 CrossRef CAS.
- J. Yang, D. He, N. Zhang and C. Hu, J. Electroanal. Chem., 2022, 905, 115997 CrossRef CAS.
- H. Rui, Y. Ting and M. Y. Yan, New J. Chem., 2022, 47, 2161–2172 RSC.
- C. Gibi, C. H. Liu, S. C. Barton, S. Anandan and J. J. Wu, J. Taiwan Inst. Chem. Eng., 2024, 154, 1–7 Search PubMed.
- W. Zhang, S. Zhu, R. Luque, S. Han, L. Hu and G. Xu, Chem. Soc. Rev., 2016, 45, 715–752 RSC.
- R. Van Echelpoel, J. Schram, M. Parrilla, D. Daems, A. Slosse, F. Van Durme and K. De Wael, Sensors & Diagnostics, 2022, 1, 793–802 Search PubMed.
- A. M. Arjun, P. H. Krishna, A. R. Nath and P. A. Rasheed, Anal. Methods, 2022, 14, 4040–4052 RSC.
- V. Kumar, P. Kumar, A. Pournara, K. Vellingiri and K. H. Kim, TrAC, Trends Anal. Chem., 2018, 106, 84–115 CrossRef CAS.
- S. Kempahanumakkagari, A. Deep, K. H. Kim, S. Kumar Kailasa and H. O. Yoon, Biosens. Bioelectron., 2017, 95, 106–116 CrossRef CAS PubMed.
- H. Karimi-Maleh, F. Karimi, A. FallahShojaei, K. Tabatabaeian, M. Arshadi and M. Rezapour, Curr. Anal. Chem., 2019, 15, 136–142 CrossRef CAS.
- P. V. Dudin, M. E. Snowden, J. V. MacPherson and P. R. Unwin, ACS Nano, 2011, 5, 10017–10025 CrossRef CAS PubMed.
- P. V. Dudin, P. R. Unwin and J. V. Macpherson, J. Phys. Chem. C, 2010, 114, 13241–13248 CrossRef CAS.
- A. M. Ghonim, B. E. El-Anadouli and M. M. Saleh, Int. J. Electrochem. Sci., 2016, 11, 621–639 CrossRef CAS.
- E. Sharel Pei, D. Liu, R. A. Lazenby, J. Sloan, M. Vidotti, P. R. Unwin and J. V. Macpherson, J. Phys. Chem. C, 2016, 120, 16059–16068 CrossRef.
- N. S. Pham, P. T. Q. Phan and V. X. Le, J. Appl. Electrochem., 2022, 52, 1343–1351 CrossRef CAS.
- S. Park, H. Kang, C. M. Kang, Y. Kim and E. J. Kim, Electron. Lett., 2023, 59, e12761 CrossRef CAS.
- E. J. Kim, C. M. Kang and J. H. Han, Electrochem. Commun., 2024, 162, 107701 CrossRef CAS.
- I. Sohail, Z. Hussain, A. N. Khan and K. Yaqoob, Mater. Res. Express, 2017, 4, 116412 CrossRef.
- R. R. Poolakkandy, N. Ar, K. A. Padmalayam, G. K. Rajanikant and M. M. Menamparambath, ACS Appl. Nano Mater., 2023, 6, 1347–1359 CrossRef CAS.
- A. Salimi, A. Noorbakhsh, H. A. Rafiee-Pour and H. Ghourchian, Electroanalysis, 2011, 23, 683–691 CrossRef CAS.
- K. Wang, L. Li and T. Zhang, Int. J. Electrochem. Sci., 2013, 8, 6252–6257 CrossRef CAS.
- M. Jamal, S. Chakrabarty, H. Shao, D. McNulty, M. A. Yousuf, H. Furukawa, A. Khosla and K. M. Razeeb, Microsyst. Technol., 2018, 24, 4217–4223 CrossRef CAS.
- S. R. Ede, S. Anantharaj, K. T. Kumaran, S. Mishra and S. Kundu, RSC Adv., 2017, 7, 5898–5911 RSC.
- N. Weidler, J. Schuch, F. Knaus, P. Stenner, S. Hoch, A. Maljusch, R. Schäfer, B. Kaiser and W. Jaegermann, J. Phys. Chem. C, 2017, 121, 6455–6463 CrossRef CAS.
- B. P. Payne, A. P. Grosvenor, M. C. Biesinger, B. A. Kobe and N. S. McIntyre, Surf. Interface Anal., 2007, 39, 582–592 Search PubMed.
- B. P. Payne, M. C. Biesinger and N. S. McIntyre, J. Electron Spectrosc. Relat. Phenom., 2012, 185, 159–166 CrossRef CAS.
- B. P. Payne, M. C. Biesinger and N. S. McIntyre, J. Electron Spectrosc. Relat. Phenom., 2009, 175, 55–65 CrossRef CAS.
- A. F. Carley, P. R. Chalker and M. W. Roberts, Proc. R. Soc. London, Ser. A, 1985, 399, 167–179 Search PubMed.
- A. L. Sanati, H. Karimi-Maleh, A. Badiei, P. Biparva and A. A. Ensafi, Mater. Sci. Eng., C, 2014, 35, 379–385 CrossRef CAS PubMed.
- M. Najafi, E. Sohouli and F. Mousavi, J. Anal. Chem., 2020, 75, 1209–1217 CrossRef.
- F. Li, J. Song, C. Shan, D. Gao, X. Xu and L. Niu, Biosens. Bioelectron., 2010, 25, 1408–1413 CrossRef CAS PubMed.
- N. F. Atta, H. K. Hassan and A. Galal, Anal. Bioanal. Chem., 2014, 406, 6933–6942 Search PubMed.
- D. Giovanelli, N. S. Lawrence, L. Jiang, T. G. J. Jones and R. G. Compton, Sens. Actuators, B, 2003, 88, 320–328 CrossRef CAS.
- A. C. Mora, M. Vara, P. Reust, A. Code, P. Oliver and C. R. Mace, ACS Sens., 2024, 9, 3198–3204 CrossRef CAS PubMed.
- X. Li, L. Sun, B. Xu, L. Dai, Y. Xiao, Y. Ding, Q. Liu, M. Meng, R. Xi, L. Guo and Y. Yin, Sens. Actuators, B, 2023, 385, 133710 Search PubMed.
- Y. Akbarian, M. Shabani-Nooshabadi and H. Karimi-Maleh, Sens. Actuators, B, 2018, 273, 228–233 CrossRef CAS.
- A. L. Sanati, H. Karimi-Maleh, A. Badiei, P. Biparva and A. A. Ensafi, Mater. Sci. Eng., C, 2014, 35, 379–385 CrossRef CAS PubMed.
- M. Alhaddad and S. M. Sheta, ACS Omega, 2020, 5, 28296–28304 CrossRef CAS PubMed.
- P. Mohammadian, M. Masteri-Farahani and N. Mosleh, J. Photochem. Photobiol., A, 2025, 458, 115970 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *,
Yekyung Kim
*,
Yekyung Kim




