
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRepurposing the phenylthiazole scaffold with 1,3,4-oxadiazole for selective, potent and well-tolerated antifungal activity†
Mohamed Hagras *a,
Hany G. Ezzata,
Abdelrahman A. Abuelkhira and
Abdelrahman S. Mayhoubab
*a,
Hany G. Ezzata,
Abdelrahman A. Abuelkhira and
Abdelrahman S. Mayhoubab
aDepartment of Pharmaceutical Organic Chemistry, College of Pharmacy, Al-Azhar University, Cairo, 11884, Egypt. E-mail: m.hagrs@azhar.edu.eg
bNanoscience Program, University of Science and Technology, Zewail City of Science and Technology, October Gardens, 6th of October, Giza 12578, Egypt
First published on 27th March 2025
Abstract
Invasive fungal infections (IFIs) represent a critical health threat, particularly among immunocompromised individuals, with mortality rates reaching up to 50%. The growing resistance to existing antifungal therapies necessitates the development of novel agents. Here, we rationally designed phenylthiazole-based oxadiazole derivatives to enhance selectivity and potency against resistant fungal strains. Among the tested compounds, compound 35 (which emerged as a lead candidate) demonstrated potent activity against Candida albicans (MIC = 1–2 μg mL−1), Candida glabrata (MIC = 0.5–1 μg mL−1), and multidrug-resistant Candida auris (MIC = 2–4 μg mL−1), outperforming fluconazole and matching amphotericin B. Additionally, compound 35 showed minimal cytotoxicity (88% cell viability at 16 μg mL−1) and negligible hemolytic activity, indicating a superior safety profile.
1 Introduction
Invasive fungal infections (IFIs) are a significant global health burden, causing roughly 1.5 to 2 million deaths annually.1,2 These life-threatening infections predominantly strike immunocompromised individuals, including organ transplant recipients, AIDS patients, and cancer patients undergoing immunosuppressive therapy. Additionally, patients hospitalized with severe underlying infections are also susceptible to IFIs.3,4 Candida, Aspergillus, and Cryptococcus species are the most frequent fungal culprits responsible for IFIs.5 Candida, particularly in intensive care unit (ICU) patients, is the leading cause of invasive fungal infections (invasive candidiasis). Notably, it ranks as the fourth most common bloodstream infection within the United States. Mortality rates associated with candidemia can be as high as 50%, while invasive aspergillosis carries a mortality rate ranging from 30–50%.6–8For the treatment of IFIs, clinicians primarily rely on three drug classes: polyenes, echinocandins, and azoles.9,10 Polyenes, such as amphotericin B and mycostatin, demonstrate potent antifungal activity against a wide range of pathogens. However, their use is often limited by the potential for moderate to severe nephrotoxicity and allergic contact dermatitis.11,12 Echinocandins offer a favorable safety profile compared to other antifungals and exhibit fungicidal activity against Candida species. Nevertheless, their efficacy is diminished against Aspergillus, the most common fungal culprit in pneumonia.13 Azoles (Fig. 1A), owing to their high therapeutic index, have emerged as the mainstay of antifungal therapy. Their broad-spectrum activity and generally well-tolerated nature make them the preferred choice for many IFIs.14,15
 | ||
| Fig. 1 (A) Structures of a typical azole antifungal drugs. (B) Rational design of this work. | ||
The widespread use of azoles, particularly fluconazole, for treating fungal infections has unfortunately led to the emergence of multidrug-resistant fungal strains. These strains exhibit resistance not only to fluconazole but also to other azole antifungals like itraconazole and voriconazole.16 Notably, Candida krusei has been shown to possess innate resistance to fluconazole, meaning it is resistant without prior exposure to the drug.17 Furthermore, a concerning number of Candida and Aspergillus species isolates, responsible for an estimated 1.4 million deaths annually, have developed significant resistance to fluconazole.18
The alarming rise of antifungal resistance, coupled with the inherent toxicity and adverse effects associated with existing antifungals, presents a significant unmet medical need. This challenge is further compounded by the difficulties in registering new antifungal compounds to combat resistant fungal strains. Consequently, the development of novel, potent antifungals is of paramount importance.
Azole antifungal drugs, such as fluconazole, voriconazole, isavuconazole, ketoconazole, and itraconazole, rely heavily on 5-membered nitrogenous heterocycles like triazoles, imidazoles, tetrazoles, and thiazoles for their antifungal activity (Fig. 1A). 1,3,4-Oxadiazoles share structural similarity with commonly used azole antifungals, such as triazoles and imidazoles, enabling them to target the same fungal enzymes. For example, replacing a portion of the fluconazole molecule with a 1,3,4-oxadiazole moiety has been shown to directly enhance its antifungal activity.19 On the other hand, the phenylthiazole scaffold, well-established for its antimicrobial activity against multi-drug resistant pathogens, shows mixed activity (antibacterial and antifungal) in its lead compound 1a. This compound possesses two key pharmacophoric features: an aminoguanidine head and an n-butyl tail. Notably, altering the n-butyl tail by replacing it with either a longer or shorter alkyl chain resulted in a decline in antimicrobial activity.20,21 Optimizing 1a by rigidifying the aminoguanidine head with a pyrimidine ring (a 6-membered ring) yielded selective antibacterial hits (Fig. 1B).21,22 Herein, we aimed to design selective antifungal compounds. Therefore, we propose inserting a 1,3,4-oxadiazole ring into the phenylthiazole scaffold (Fig. 1B). We hypothesize that incorporating this ring may enhance selectivity and potency towards resistant fungal strains, leading to the development of novel and selective antifungal lead compounds (Fig. 1B).
2 Results and discussion
2.1. Chemistry
4-Butylbenzoyl chloride 2 was added portion-wise to an ammonia solution while keeping the reaction mixture stirred at 0–5 °C to produce 4-butylbenzamide 3. The product was dried and dissolved in dry THF, and the flask was charged with Lawesson's reagent to obtain 4-butylbenzthioamide 4. Ester 5 was prepared in high yield using thioamide 4 and ethyl α-chloroacetoacetate. The key intermediate acid hydrazide 6 was then obtained via treatment of the obtained ethyl ester 5 with hydrazine hydrate (Scheme 1). Adding a carbon disulfide to 6 in the presence of potassium hydroxide followed by methylation of the free thiol group afforded the 5-methylmercapto derivative 8. The later was oxidized by m-chloroperbenzoic acid (m-CPBA) to yield methylsulfone analogue 9, the key intermediate of this scheme, which was utilized to access nucleophilic substitution reaction. So that, treatment of methylsulfone 9 with thirty-five different nitrogenous nucleophiles include primary and secondary amines, hydrazine, diamines, aminoalcohols, guanidines and carboximidines afforded the final products 10–44 (Scheme 1).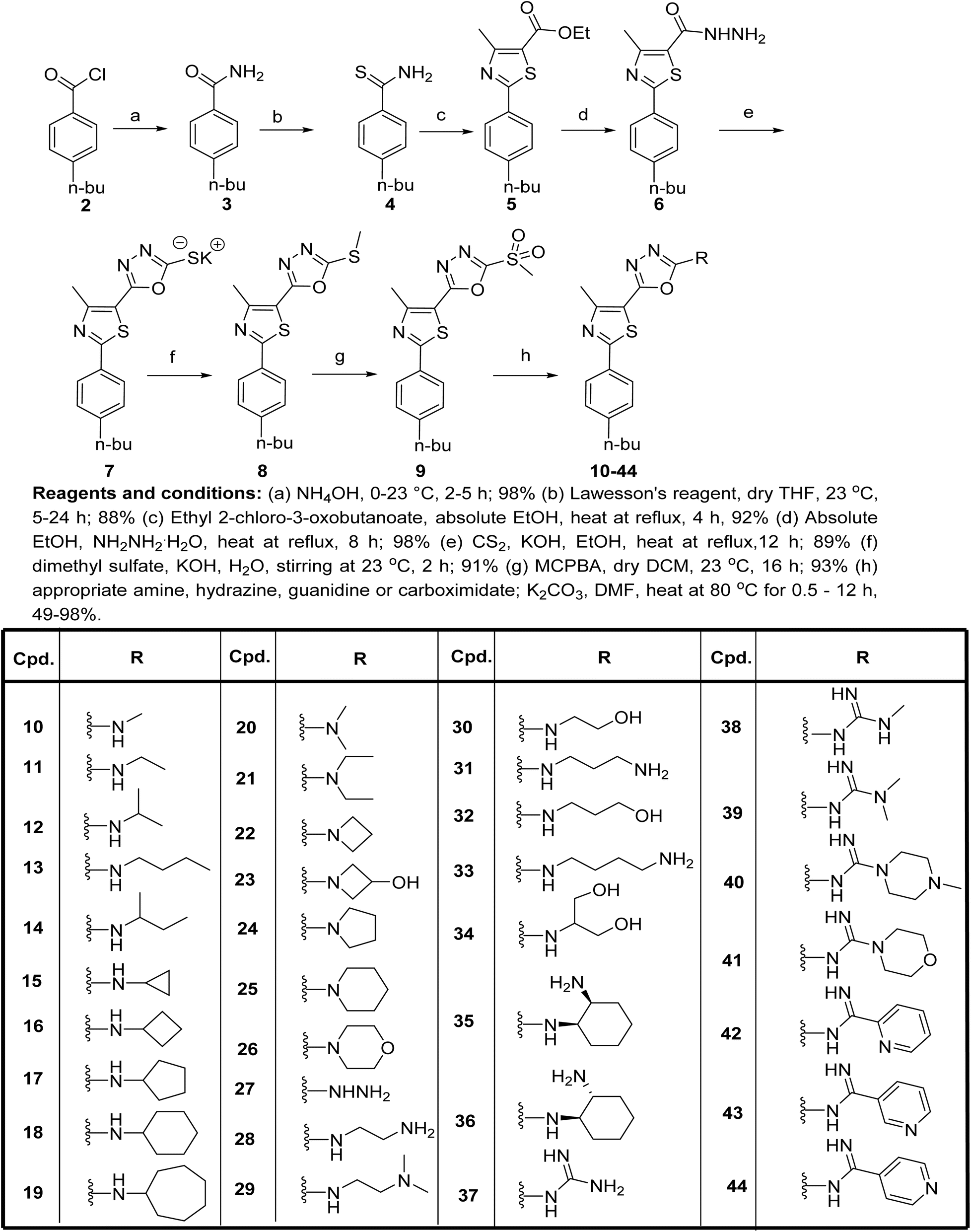 | ||
| Scheme 1 | ||
2.2. Biological evaluation
The antimicrobial results that have been presented in (Table 1) indicated that oxadiazole derivatives with aliphatic side chains, primary or secondary amines (compounds 10–26), are void from any antimicrobial activity, with the exceptions of ethylamine 11 and azetidine 22 derivatives. Compound 11 exhibited mild antimicrobial activity against all five tested microbes, with MIC values ranging from 8 to 32 μg mL−1. In contrast, azetidine derivative 22 demonstrated moderate antibacterial activity against MRSA (MIC = 16 μg mL−1) and promising antifungal activity against C. albicans (MIC = 8 μg mL−1). These results collectively suggested that the presence of a hydrophobic region around the oxadiazole position-2 is unfavorable and highlights the crucial role of the terminal heteroatom as a hydrogen bond donor HBD and/or acceptor HBA.
|
||||||
|---|---|---|---|---|---|---|
| Cpd | Side chain | MRSA NRS384 (MRSA USA300) | C. difficile ATCC BAA 1870 | E. coli JW55031 (TolC Mutant) | N. gonorrhoeae 181 | C. albicans SS5314 (wild-type) |
| a NT; not tested. | ||||||
| 10 |  |
>64 | >64 | >64 | >64 | >64 |
| 11 | 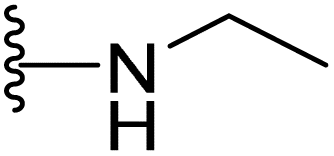 |
16 | 8 | 16 | 32 | 8 |
| 12 |  |
>64 | >64 | >64 | >64 | >64 |
| 13 |  |
>64 | >64 | >64 | >64 | >64 |
| 14 | 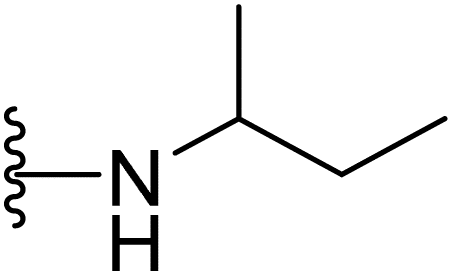 |
>64 | 64 | >64 | 64 | >64 |
| 15 |  |
>64 | >64 | >64 | >64 | >64 |
| 16 |  |
>64 | >64 | >64 | >64 | >64 |
| 17 |  |
>64 | 64 | >64 | 64 | >64 |
| 18 |  |
32 | 32 | >64 | >64 | >64 |
| 19 |  |
>64 | 64 | >64 | 64 | >64 |
| 20 |  |
64 | 32 | >64 | >64 | >64 |
| 21 |  |
>64 | >64 | >64 | 64 | >64 |
| 22 |  |
16 | >64 | 32 | 64 | 8 |
| 23 |  |
>64 | >64 | >64 | >64 | >64 |
| 24 |  |
>64 | >64 | >64 | >64 | >64 |
| 25 |  |
>64 | 64 | >64 | >64 | >64 |
| 26 |  |
>64 | 32 | >64 | >64 | >64 |
| 27 |  |
64 | 8 | 64 | 64 | 64 |
| 28 |  |
>64 | 32 | >64 | 64 | 64 |
| 29 |  |
16 | 8 | 16 | 64 | 16 |
| 30 |  |
>64 | >64 | >64 | >64 | >64 |
| 31 |  |
16 | 8 | 32 | 64 | 16 |
| 32 |  |
>64 | >64 | >64 | 64 | >64 |
| 33 |  |
32 | 16 | 32 | 64 | 64 |
| 34 |  |
>64 | 16 | >64 | 32 | 32 |
| 35 |  |
>64 | 32 | >64 | 32 | 4 |
| 36 |  |
64 | 32 | 64 | >64 | 8 |
| 37 |  |
>64 | >64 | >64 | 32 | >64 |
| 38 |  |
>64 | >64 | >64 | 64 | >64 |
| 39 |  |
>64 | 64 | >64 | >64 | >64 |
| 40 |  |
8 | 16 | >64 | 64 | 8 |
| 41 |  |
>64 | >64 | >64 | >64 | >64 |
| 42 | 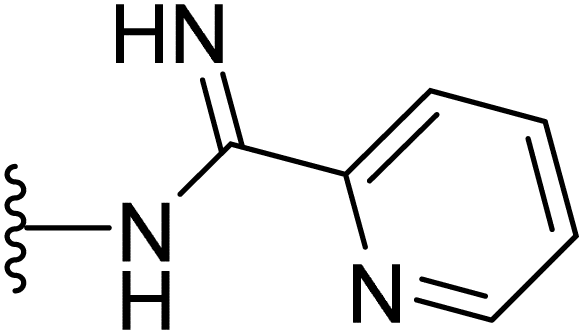 |
>64 | >64 | >64 | >64 | >64 |
| 43 |  |
>64 | >64 | >64 | >64 | >64 |
| 44 | 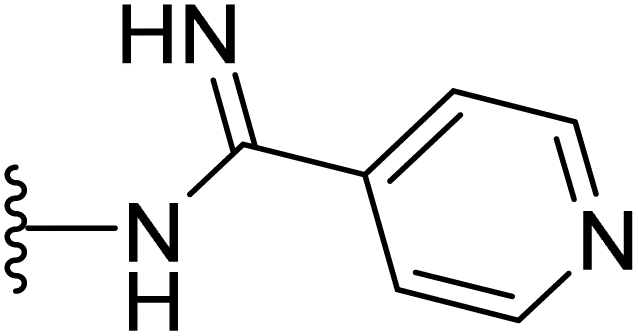 |
>64 | >64 | >64 | > 64 | >64 |
| Linezolid | 1 | >64 | 8 | NT | NT | |
| Vancomycin | 1 | NT | NT | NT | NT | |
| Gentamicin | NT | ≤0.5 | ≤0.5 | NT | NT | |
| Cefixime | NT | NT | NT | 1 | NT | |
| Amphotericin B | NT | NT | NT | NT | 1 | |
| Fluconazole | NTa | NT | NT | NT | >64 | |
Adding another polar heteroatom led to a new set of structures. The oxadiazole 27 bearing the hydrazine nitrogenous side chain was first synthesized and tested in this set. Compound 27 was limited moderate activity against C. difficile with MIC of 8 μg mL−1. Unfortunately, 27 was inactive against all other tested microorganisms (Table 1).
Increasing the distance between the two heteroatoms resulted in the synthesis of diamines and amino alcohols (compounds 28–36), which showed a slight improvement in anti-MRSA activity. Notably, the N,N-dimethyl ethylenediamine derivative 29 and the propylenediamine derivative 31 exhibited MIC values of 16 μg mL−1 against MRSA and 8 μg mL−1 against C. difficile. Additionally, both compounds demonstrated promising antifungal activity against C. albicans (MIC = 8 μg mL−1). Interestingly, the 1,2-diaminocyclohexane derivatives (35 and 36) exhibited selective antifungal activity against C. albicans with MIC values of 4 and 8 μg mL−1, respectively (Table 1).
Further expansion of the nitrogenous side chain provided the guanidine compounds 37–39, which were void from any antimicrobial activity. The antibacterial activity of guanidine-containing compound 37 was not improved even by adding one or two methyl groups to the terminal amino group (compounds 38 and 39). However, further increasing the number of carbon units around the terminal amine (compounds 40) notably enhanced both anti-MRSA and antifungal activity, with MIC value improved from above 64 to 8 μg mL−1. Finally, replacing the n-methyl piperazine side chain with a morpholine ring (compound 41) or aromatic pyridines (compounds 42–44) had a negative impact on the antibacterial activity of the tested compounds, as these compounds were void of any antimicrobial activity (Table 1).
The SAR analysis for antifungal activity indicates that oxadiazole derivatives with hydrophobic regions and simple aliphatic side chains, such as compounds 10–26, lack significant activity, except for ethylamine 11 and azetidine 22, which exhibit mild antifungal effects (MIC = 8 μg mL−1). Introducing additional heteroatoms, as in compound 27, or extending the distance between heteroatoms, as in compounds 28–36, improved antifungal activity, with N,N-dimethyl ethylenediamine 29 and propylenediamine 31 active against C. albicans (MIC = 8 μg mL−1). Notably, selective antifungal activity was observed only in the 1,2-diaminocyclohexane derivatives 35 and 36, which exhibited MIC values of 4 and 8 μg mL−1, respectively, with no significant antibacterial activity, underscoring the importance of a rigid cyclic diamine structure. Guanidine-containing compounds 37–39 were inactive, while increasing carbon units in compound 40 restored antifungal activity (MIC = 8 μg mL−1). Substitutions with morpholine 41 or pyridines 42–44 eliminated activity, highlighting the importance of specific side chains for selectivity (Fig. 2).
 | ||
| Fig. 2 SAR study of antifungal activity. | ||
| No. | Fungal strain | MICs (μg mL−1) | |||
|---|---|---|---|---|---|
| 35 | Amphotericin | Fluconazole | Itraconazole | ||
| a NT; not tested. | |||||
| 1 | C. albicans NR-29448 | 2 | 1 | >128 | NTa |
| 2 | C. albicans ATCC 10231 | 1 | 0.5 | 0.25 | NT |
| 3 | C. albicans ATCC 64124 | 1 | 2 | 128 | NT |
| 4 | C. glabrata ATCC MYA-2950 | 0.5 | 0.5 | 16 | NT |
| 5 | C. glabrata ATCC 15126 | 1 | 1 | 4 | NT |
| 6 | C. glabrata ATCC 66032 | 0.5 | 1 | 8 | NT |
| 7 | C. glabrata ATCC 2001 | 1 | 1 | 8 | NT |
| 8 | C. glabrata CAB 524041 | 1 | 1 | 8 | NT |
| 9 | C. krusei ATCC 14243 | 1 | 1 | 16 | NT |
| 10 | C. krusei ATCC 34135 | 2 | 2 | 16 | NT |
| 11 | C. krusei CAB 396420 | 1 | 1 | 16 | NT |
| 12 | C. auris AR0385 | 2 | 2 | >128 | NT |
| 13 | C. auris AR0389 | 4 | 2 | >128 | NT |
| 14 | C. auris AR0390 | 2 | 2 | >128 | NT |
| 15 | Cryptococcus gattii NR-43208 | 1 | 1 | 2 | NT |
| 16 | Cryptococcus gattii NR-43210 | 1 | 1 | 2 | NT |
| 17 | Cryptococcus gattii NR-43209 | 0.5 | 1 | 4 | NT |
| 18 | Cryptococcus neoformans NR-41298 | 1 | 1 | 2 | NT |
| 19 | Cryptococcus neoformans NR-41300 | 2 | 1 | 2 | NT |
| 20 | Cryptococcus neoformans NR-48770 | 1 | 1 | 2 | NT |
| 21 | Aspergillus fumigatus 303 | >16 | 1 | NT | 0.5 |
| 22 | Aspergillus fumigatus 304 | >16 | 1 | NT | 0.5 |
Notably, compound 35 showed efficacy against Candida auris, a multidrug-resistant pathogen of critical clinical concern. It achieved MICs of 2–4 μg mL−1, whereas fluconazole was ineffective (MICs >128 μg mL−1). The compound also displayed potent activity against Cryptococcus gattii and Cryptococcus neoformans, with MICs of 0.5–2 μg mL−1, aligning closely with amphotericin B and outperforming fluconazole, which exhibited reduced activity in some strains. However, the efficacy of compound 35 was less pronounced against Aspergillus fumigatus, where MICs exceeded 16 μg mL−1.
In the cytotoxicity assay (Fig. 3), compound 35 demonstrated minimal impact on cell viability across all tested concentrations. At 8 μg mL−1 and 16 μg mL−1, compound 35 maintained a high cell viability of 88%, in stark contrast to amphotericin B, which exhibited significant cytotoxicity, with cell viabilities of 40% and 18%, respectively. This steep decline in cell viability for amphotericin B underscores its well-documented narrow therapeutic window and high off-target toxicity. Compound 35's gentle dose–response curve highlights its potential as a safer antifungal agent for systemic use.
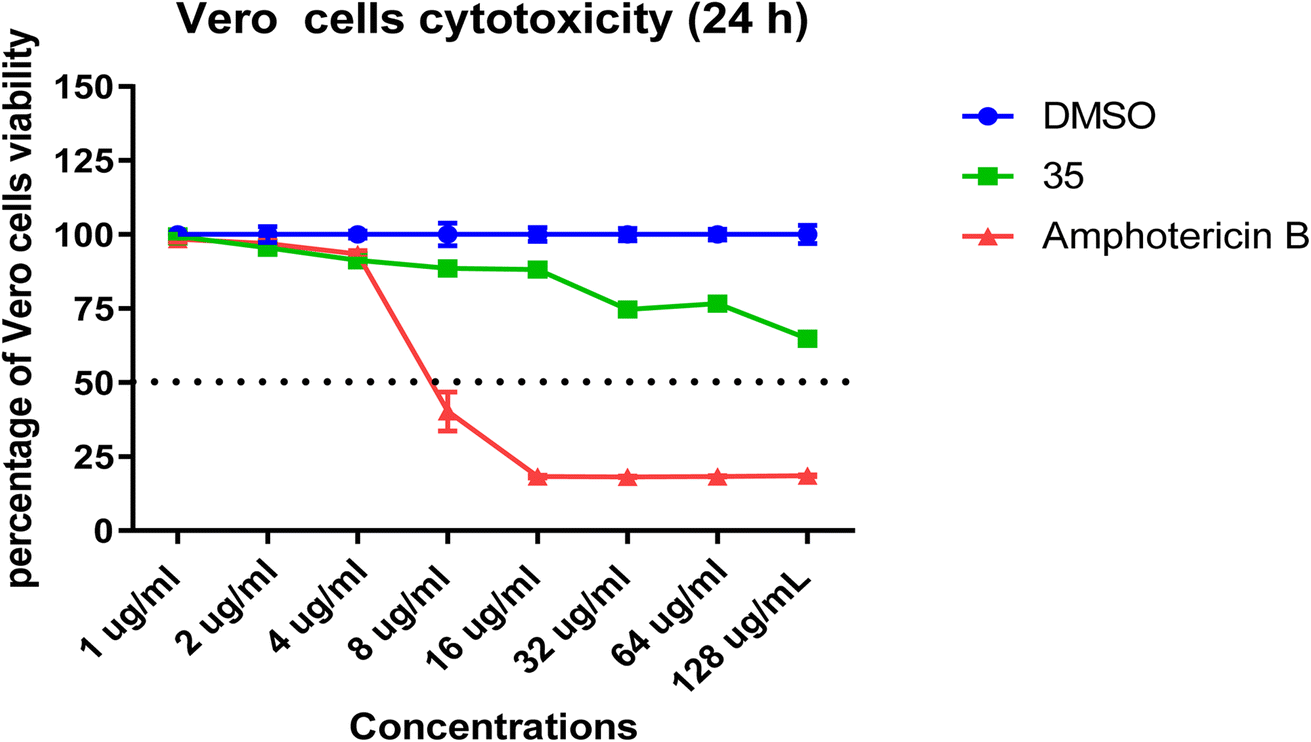 | ||
| Fig. 3 In vitro cytotoxicity assay of different concentrations of 35 and amphotericin-B against monkey kidney epithelial (Vero) cells. | ||
In the hemolysis assay (Fig. 4), amphotericin B exhibited a pronounced dose-dependent hemolytic activity, causing substantial hemolysis at higher concentrations, with levels exceeding 100% at 128 μg mL−1. In contrast, compound 35 showed negligible hemolytic activity, comparable to that of fluconazole, across all tested concentrations. The absence of significant red blood cell lysis even at elevated concentrations reinforces the favorable safety profile of compound 35 and indicates its reduced risk of causing hemolytic toxicity during systemic administration.
 | ||
| Fig. 4 RBCs hemolysis assay of different concentrations of 35 and antifungal drugs. | ||
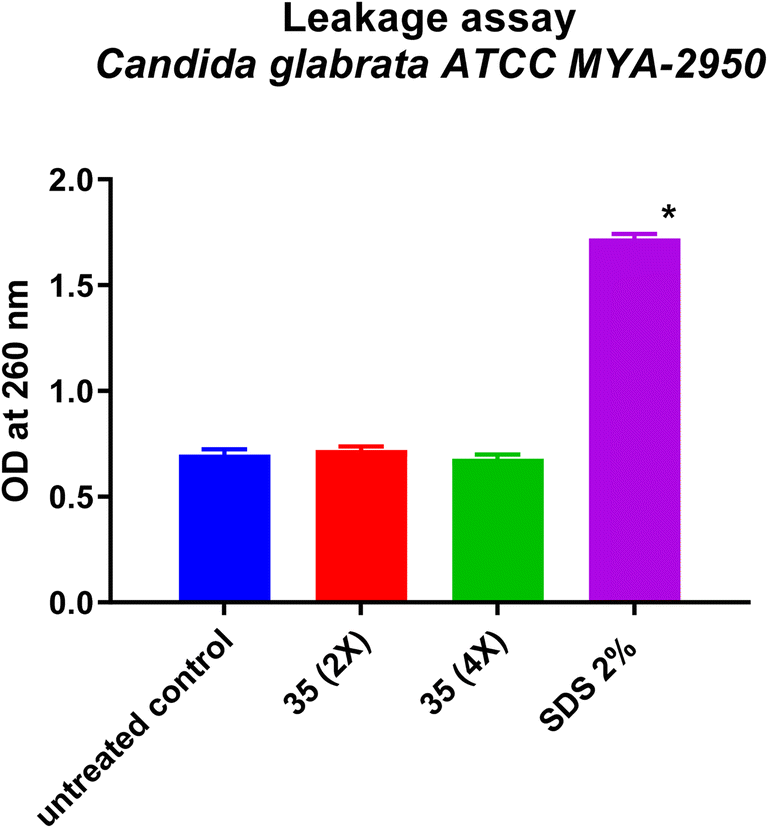 | ||
| Fig. 5 Leakage assay of Candida glabrata ATCC MYA-2950 after 4 h incubation with 2× and 4× MIC of 35 compound and 2% SDS. | ||
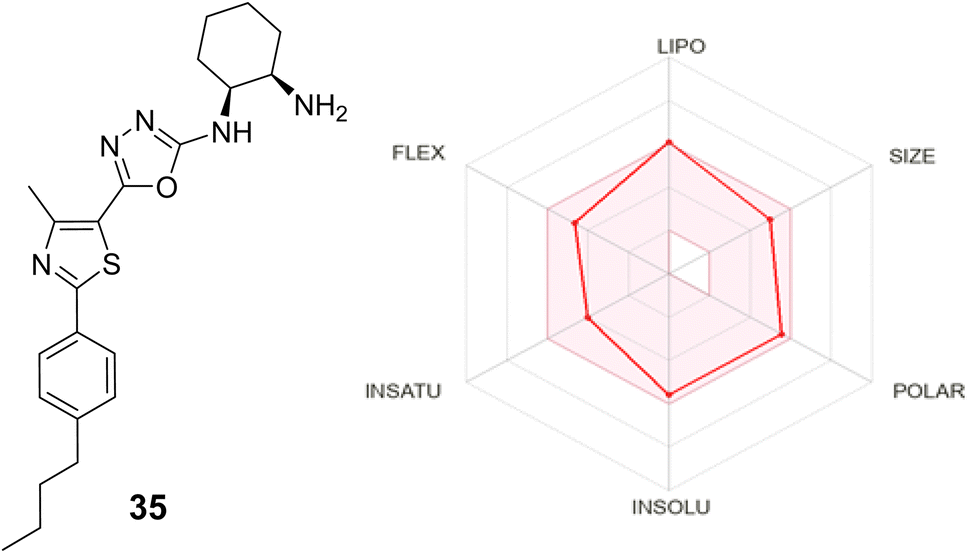 | ||
| Fig. 6 In silico ADME profile of 35. | ||
2.3. In silico ADME profile
The ADME (absorption, distribution, metabolism, and excretion) properties of the most active compound 35 were evaluated using the SwissADME online tool. Based on Lipinski's rule of five, a compound is considered to have acceptable drug-like ADME characteristics if it meets no more than one of the following criteria: molecular weight below 500 daltons, fewer than 5 hydrogen bond donors (n-OHNH), fewer than 10 hydrogen bond acceptors (n-ON), an octanol–water partition coefficient (MLogP) below 4.5, and a total polar surface area (TPSA) of 140 or less. As summarized in (Table 3), the analysis revealed that compound 35 complies with all these criteria, indicating it fully adheres to Lipinski's rule. Furthermore, the predicted solubility class suggests that compound 35 is moderately soluble. These results highlight that compound 35 possesses favorable ADME properties, supporting its potential as a therapeutic candidate.| Compound | M![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) logPa logPa |
TPSA | n-ON | n-OHNH | NRB | L.Vio. | LogS (ESOL) |
Solubility class |
|---|---|---|---|---|---|---|---|---|
| a The logarithm of partition coefficient between n-octanol and water (MlogP); topological polar surface area (TPSA); number of hydrogen bond acceptors (n-ON); number of hydrogen bond donors (n-OHNH); number of rotatable bonds (NRB); Lipinski's violation (L.Vio.); the logarithm of solubility (logS) = log(solubility measured in mol L−1). |
||||||||
| 35 | 0.21 | 118.10 Å2 | 6 | 2 | 7 | 0 | −5.57 | Moderately soluble |
3 Conclusion
The rational design of phenylthiazole-based oxadiazole derivatives led to the identification of compound 35, a promising antifungal agent with potent activity against a wide spectrum of fungal species, including resistant strains. Its excellent safety profile, as evidenced by low cytotoxicity and hemolytic activity, and its non-disruptive mechanism highlight its therapeutic potential. These results demonstrate the value of legand-based drug design in addressing the limitations of current antifungal treatments and support further development of compound 35 as a novel antifungal therapy.4 Experimental section
4.1. Chemistry
4.1.1.1. Synthesis of 2-(4-butylphenyl)-4-methylthiazole-5-carbohydrazide (6). To a solution of ester derivative 5 (1.29 g, 4 mmol) in ethanol (15 mL), hydrazine hydrate (99%, 1 mL, 20 mmol) was added dropwise. The reaction mixture was heated at reflux for 6 h then allowed to cool down to room temperature. The formed solid was separated by filtration and crystallized from ethanol to provide the desired product as white crystals (1.1 g, 93%); mp = 133–135 °C; IR (KBr) cm−1: 3289, 3185 (NH2), 3160 (NH), 3026 (C–H aromatic), 2987, 2943 (C–H aliphatic), 1668 (C
![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O amide); 1H NMR (DMSO-d6) δ: 9.66 (brs, 1H), 7.90 (d, J = 8.4 Hz, 2H), 7.37 (d, J = 8.4 Hz, 2H), 4.53 (brs, 2H), 2.78 (s, 3H), 2.68:2.65 (m, 2H), 1.58:1.56 (m, 2H), 1.38:1.30 (m, 2H), 0.93–0.89 (m, 3H); MS (m/z) 389 (M+, 100%).
O amide); 1H NMR (DMSO-d6) δ: 9.66 (brs, 1H), 7.90 (d, J = 8.4 Hz, 2H), 7.37 (d, J = 8.4 Hz, 2H), 4.53 (brs, 2H), 2.78 (s, 3H), 2.68:2.65 (m, 2H), 1.58:1.56 (m, 2H), 1.38:1.30 (m, 2H), 0.93–0.89 (m, 3H); MS (m/z) 389 (M+, 100%).
4.1.1.2. Synthesis of 5-(2-(4-butylphenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazole-2-thiolate (7). Potassium hydroxide (0.4 g, 10 mmol) was added to a solution of 6 (3 g, 10 mmol) in ethanol (15 mL), followed by drop-wise addition of carbon disulphide (3 mL, 110 mmol) over 0.5 h. The reaction mixture was stirred at room temperature for an additional 15 min and then heated to reflux until the evolution of hydrogen sulfide gas ceased. After completion of the reaction, as monitored by TLC, the obtained product was poured on cold water (50 mL), washed with water, dried and crystallized from ethanol to provide thiol derivative as yellow crystals (3.3 g, 89%); mp > 300 °C.
4.1.1.3. Synthesis of 2-(2-(4-butylphenyl)-4-methylthiazol-5-yl)-5-(methylthio)-1,3,4-oxadiazole (8). The obtained yellow solid 7 (0.25 g, 4.2 mmol) was dissolved in water (15 mL). Then, dimethyl sulfate (0.5 mL, 4 mmol) was added dropwise with vigorous stirring. After 2 h, the formed solid was filtered and washed with copious amounts of water to yield the titled compound as a yellowish white solid (0.68 g, 91%); mp = 154 °C; 1H NMR (DMSO-d6) δ: 7.90 (d, J = 8.4 Hz, 2H), 7.35 (d, J = 8.4 Hz, 2H), 2.77 (s, 3H), 2.71 (s, 3H), 2.64
:2.62 (m, 2H), 1.62:1.56 (m, 2H), 1.37–1.31 (m, 2H), 0.93:0.89 (m, 3H); MS (m/z) 345 (M+, 14%).
4.1.1.4. Synthesis of 2-(2-(4-butylphenyl)-4-methylthiazol-5-yl)-5-(methylsulfonyl)-1,3,4 oxadiazole (9). To a solution of S-methyl derivative 8 (0.5 g, 1.3 mmol) in dry DCM (5 mL), m-CPBA (0.514 g, 2.9 mmol) diluted with DCM (5 mL) was added portion-wise with continuous stirring. Afterward, the reaction mixture was kept at room temperature for 4 h, additional DCM (10 mL) was added and the reaction mixture was washed with 25 mL of 5% aqueous solution of sodium metabisulfite, and 25 mL of 5% aqueous sodium carbonate. The organic layer was separated, dried and concentrated under reduced pressure to give the desired product as yellow crystals (0.5 g, 93%) mp = 126 °C; 1H NMR (DMSO-d6) δ: 7.92 (d, J = 8.4 Hz, 2H), 7.37 (d, J = 8.4 Hz, 2H), 3.83 (s, 3H), 2.78 (s, 3H), 2.74
:2.65 (m, 2H), 1.58:1.57 (m, 2H), 1.38:1.36 (m, 2H), 0.93:0.89 (m, 3H); MS (m/z) 377 (M+, 100%).
4.1.2.1. General procedure A. To a solution of 9 (0.1 g, 0.25 mmol) in dry DMF (5 mL), appropriate amine, diamine, aminoalcohol, hydrazine, guanidine, guanidine analogue or carboximidate (0.4 mmol) was added. The reaction mixture was heated at 80 °C for 0.5–12 h, and then poured over ice water (50 mL). The formed solid was extracted with ethyl acetate (10 mL). The organic layer was evaporated under reduced pressure. The obtained crude material was then purified by crystallization or column chromatography. Physical properties and spectral analysis of isolated products are listed below.
4.1.2.2. 5-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)-N-methyl-1,3,4-oxadiazol-2-amine (10). Following the general procedure (A), and using methylamine (13 μL, 0.4 mmol), compound 10 was obtained as yellowish white solid (0.078 g, 91%); mp = 170 °C; 1H NMR (DMSO-d6) δ: 7.89 (d, J = 8.4 Hz, 2H), 7.77 (brs, 1H), 7.36 (d, J = 8.4 Hz, 2H), 2.87 (s, 3H), 2.65 (s, 3H), 2.63
:2.56 (m, 2H), 1.61:1.55 (m, 2H), 1.37:1.32 (m, 2H), 0.92:0.89 (m, 3H); 13C NMR (DMSO-d6) δ: 166.6, 164.2, 153.8, 153, 146.3, 130.2, 129.7, 126.7, 114.9, 35.1, 33.2, 29.5, 22.2, 17.4, 14.2; MS (m/z) 328 (M+, 100%); anal. calc. for: (C17H20N4OS, Mwt = 328): C, 62.17; H, 6.14; N, 17.06%; found: C, 62.22; H, 6.19; N, 17.09%.
4.1.2.3. 5-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)-N-ethyl-1,3,4-oxadiazol-2-amine (11). Following the general procedure (A), and using ethylamine (18 μL, 0.4 mmol), compound 11 was obtained as white solid (0.085 g, 95%); mp = 115 °C; 1H NMR (DMSO-d6) δ: 7.90 (d, J = 8.4 Hz, 2H), 7.77 (brs, 1H), 7.35 (d, J = 8.4 Hz, 2H), 3.26
:3.24 (m, 2H), 2.67 (s, 3H), 2.63:2.60 (m, 2H), 1.62:1.55 (m, 2H), 1.26:1.22 (m, 2H), 1.18:1.16 (m, 3H), 0.93:0.89 (m, 3H); 13C NMR (DMSO-d6) δ: 166.6, 164.7, 153.7, 152.9, 146.1, 130.2, 129.6, 126.9, 114.9, 37.9, 35.1, 33.2, 22.2, 17.4, 14.4, 13.7; MS (m/z) 342 (M+, 100%); anal. calc. for: (C18H22N4OS, Mwt = 342): C, 63.13; H, 6.48; N, 16.36%; found: C, 63.19; H, 6.52; N, 16.40%.
4.1.2.4. 5-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)-N-isopropyl-1,3,4-oxadiazol-2-amine (12). Following the general procedure (A), and using isopropylamine (23 μL, 0.4 mmol), compound 12 was obtained as white solid (0.085 g, 92%); mp = 135 °C; 1H NMR (DMSO-d6) δ: 8.05 (brs, 1H), 7.90 (d, J = 8.4 Hz, 2H), 7.37 (d, J = 8.4 Hz, 2H), 3.78
:3.71 (m, 1H), 2.84 (s, 3H), 2.68:2.54 (m, 2H), 1.58:1.57 (m, 2H), 1.33:1.28 (m, 2H), 1.23:1.21 (m, 6H), 0.93:0.89 (m, 3H); 13C NMR (DMSO-d6) δ: 166.6, 162.9, 153.7, 152.8, 146.1, 130.2, 129.6, 126.9, 115, 45.5, 35.1, 33.2, 22.7, 22, 17.4, 14; MS (m/z) 356 (M+, 68%); anal. calc. for: (C19H24N4OS, Mwt = 356): C, 64.02; H, 6.79; N, 15.72%; found: C, 64.07; H, 6.84; N, 15.77%.
4.1.2.5. N-Butyl-5-(2-(4-butylphenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-amine (13). Following the general procedure (A), and using butylamine (25 μL, 0.4 mmol), compound 13 was obtained as buff solid (0.09 g, 93%); mp = 175 °C; 1H NMR (DMSO-d6) δ: 7.90 (d, J = 8.4 Hz, 2H), 7.77 (brs, 1H), 7.38 (d, J = 8.4 Hz, 2H), 3.26
:3.21 (m, 2H), 2.68 (s, 3H), 2.62:2.51 (m, 2H), 1.62:1.58 (m, 4H), 1.39:1.31 (m, 4H), 0.98:0.91 (m, 6H); 13C NMR (DMSO-d6) δ: 166.6, 163.7, 153.7, 152.8, 146.2, 130.2, 129.7, 126.6, 115, 42.7, 35.1, 33.2, 31.3, 22.1, 19.8, 17.3, 14.2, 14; MS (m/z) 370 (M+, 100%); anal. calc. for: (C20H26N4OS, Mwt = 370): C, 64.83; H, 7.07; N, 15.12%; found: C, 64.88; H, 7.11; N, 15.15%.
4.1.2.6. N-(sec-Butyl)-5-(2-(4-butylphenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-amine (14). Following the general procedure (A), and using sec-butylamine (28 μL, 0.4 mmol), compound 14 was obtained as yellow solid (0.089 g, 92%); mp = 120 °C; 1H NMR (DMSO-d6) δ: 7.93 (d, J = 8.4 Hz, 2H), 7.81 (brs, 1H), 7.33 (d, J = 8.4 Hz, 2H), 3.58
:3.55 (m, 1H), 2.78 (s, 3H), 2.68:2.51 (m, 4H), 1.60:1.57 (m, 2H), 1.37:1.33 (m, 2H), 1.22:1.20 (m, 3H), 0.94:0.89 (m, 6H); 13C NMR (DMSO-d6) δ: 166.5, 163.2, 153.6, 152.7, 146.1, 130.2, 129.6, 126.6, 115, 51, 35.1, 33.2, 29.1, 22.1, 20.3, 17.4, 14.2, 10.8; MS (m/z) 370 (M+, 48%); anal. calc. for: (C20H26N4OS, Mwt = 370): C, 64.83; H, 7.07; N, 15.12%; found: C, 64.87; H, 7.11; N, 15.17%.
4.1.2.7. 5-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)-N-cyclopropyl-1,3,4-oxadiazol-2-amine (15). Following the general procedure (A), and using cyclopropylamine (18 μL, 0.4 mmol), compound 15 was obtained as yellow solid (0.06 g, 65%); mp = 165 °C; 1H NMR (DMSO-d6) δ: 8.20 (brs, 1H), 7.87 (d, J = 8.4 Hz, 2H), 7.35 (d, J = 8.4 Hz, 2H), 3.09
:3.07 (m, 1H), 2.68 (s, 3H), 2.64:2.62 (m, 2H), 1.58:1.56 (m, 2H), 1.37:1.31 (m, 2H), 0.93:0.89 (m, 3H), 0.75:0.72 (m, 2H), 0.59:0.53 (m, 2H); 13C NMR (DMSO-d6) δ: 166.7, 164.1, 153.8, 153.3, 146.3, 130.2, 129.7, 126.7, 114.9, 35.1, 33.2, 24.6, 22.1, 17.3, 14.2, 6.8; MS (m/z) 354.3 (M+, 100%); anal. calc. for: (C19H22N4OS, Mwt = 354): C, 64.38; H, 6.26; N, 15.81%; found: C, 64.44; H, 6.29; N, 15.85%.
4.1.2.8. 5-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)-N-cyclobutyl-1,3,4-oxadiazol-2-amine (16). Following the general procedure (A), and using cyclobutylamine (20 μL, 0.4 mmol), compound 16 was obtained as white solid (0.091 g, 95%); mp = 155 °C; 1H NMR (DMSO-d6) δ: 8.19 (brs, 1H), 7.90 (d, J = 8.4 Hz, 2H), 7.37 (d, J = 8.4 Hz, 2H), 4.07
:4.03 (m, 1H), 2.78 (s, 3H), 2.70:2.64 (m, 2H), 2.31:2.26 (m, 2H), 2.08:2.03 (m, 2H), 1.73:1.70 (m, 2H), 1.58:1.56 (m, 2H), 1.33:1.28 (m, 2H), 0.93:0.89 (m, 3H); 13C NMR (DMSO-d6) δ: 166.7, 162.5, 153.8, 152.9, 146.3, 130.2, 129.6, 127.2, 114.9, 48.2, 35.1, 33.2, 30.4, 22.1, 17, 14.8, 14.2; MS (m/z) 368 (M+, 100%); anal. calc. for: (C20H24N4OS, Mwt = 368): C, 65.19; H, 6.57; N, 15.20%; found: C, 65.26; H, 6.62; N, 15.26%.
4.1.2.9. 5-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)-N-cyclopentyl-1,3,4-oxadiazol-2-amine (17). Following the general procedure (A), and using cyclopentylamine (34 μL, 0.4 mmol), compound 17 was obtained as brown solid (0.091 g, 91%); mp = 165 °C; 1H NMR (DMSO-d6) δ: 8.16 (brs, 1H), 7.90 (d, J = 8.4 Hz, 2H), 7.35 (d, J = 8.4 Hz, 2H), 3.93
:3.88 (m, 1H), 2.68 (s, 3H), 2.57:2.53 (m, 2H), 1.92:1.90 (m, 2H), 1.72:1.70 (m, 2H), 1.58:1.52 (m, 4H), 1.33:1.23 (m, 4H), 0.93:0.87 (m, 3H); 13C NMR (DMSO-d6) δ: 166.6, 163.2, 153.7, 152.9, 146.2, 130.2, 129.7, 126.3, 115, 54.8, 35.1, 33.2, 32.6, 23.7, 22.1, 17.3, 14.2; MS (m/z) 382 (M+, 100%); anal. calc. for: (C21H26N4OS, Mwt = 382): C, 65.94; H, 6.85; N, 14.65%; found: C, 65.98; H, 6.89; N, 14.69%.
4.1.2.10. 5-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)-N-cyclohexyl-1,3,4-oxadiazol-2-amine (18). Following the general procedure (A), and using cyclohexylamine (40 μL, 0.4 mmol), compound 18 was obtained as brown solid (0.08 g, 78%); mp = 176 °C; 1H NMR (DMSO-d6) δ: 8.02 (brs, 1H), 7.90 (d, J = 8.4 Hz, 2H), 7.37 (d, J = 8.4 Hz, 2H), 3.88
:3.82 (m, 1H), 2.81 (s, 3H), 2.68:2.56 (m, 4H), 2.01:1.96 (m, 2H), 1.73:1.67 (m, 2H), 1.56:1.52 (m, 4H), 1.33:1.30 (m, 4H), 0.95:0.89 (m, 3H); 13C NMR (DMSO-d6) δ: 166.5, 162.9, 153.6, 152.7, 146.2, 130.2, 129.3, 126.3, 115, 52.3, 35.1, 33.3, 32.6, 25.3, 24.8, 22.2, 17, 14.2; MS (m/z) 396 (M+, 100%); anal. calc. for: (C22H28N4OS, Mwt = 396): C, 66.63; H, 7.12; N, 14.13%; found: C, 66.66; H, 7.15; N, 14.17%.
4.1.2.11. 5-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)-N-cycloheptyl-1,3,4-oxadiazol-2-amine (19). Following the general procedure (A), and using cycloheptylamine (50 μL, 0.4 mmol), compound 19 was obtained as orange solid (0.091 g, 85%); mp = 188 °C; 1H NMR (DMSO-d6) δ: 8.13 (brs, 1H), 7.91 (d, J = 8.4 Hz, 2H), 7.37 (d, J = 8.4 Hz, 2H), 3.65
:3.63 (m, 1H), 2.78 (s, 3H), 2.73:2.68 (m, 2H), 1.99:1.95 (m, 2H), 1.59:1.26 (m, 14H), 0.93:0.91 (m, 3H); 13C NMR (DMSO-d6) δ: 166.6, 162.8, 153.6, 152.8, 146.1, 130.9, 129.6, 127.2, 115, 54.5, 35.1, 34.4, 33.2, 29.3, 23.9, 22.1, 17.4, 14.2; MS (m/z) 410 (M+, 100%); anal. calc. for: (C23H30N4OS, Mwt = 410): C, 67.28; H, 7.37; N, 13.65%; found: C, 67.34; H, 7.41; N, 13.67%.
4.1.2.12. 5-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)-N,N-dimethyl-1,3,4-oxadiazol-2-amine (20). Following the general procedure (A), and using dimethylamine (18 μL, 0.4 mmol), compound 20 was obtained as brown solid (0.07 g, 79%); mp = 110 °C; 1H NMR (DMSO-d6) δ: 7.89 (d, J = 8.4 Hz, 2H), 7.35 (d, J = 8.4 Hz, 2H), 3.07 (s, 6H), 2.68 (s, 3H), 2.64
:2.62 (m, 2H), 1.62:1.55 (m, 2H), 1.33:1.28 (m, 2H), 0.93:0.91 (m, 3H); 13C NMR (DMSO-d6) δ: 166.7, 164.6, 153.8, 153.5, 146.3, 130.2, 129.7, 126.7, 114.8, 38.1, 35.1, 33.2, 22.2, 17.3, 14.2; MS (m/z) 342 (M+, 100%); anal. calc. for: (C18H22N4OS, Mwt = 342): C, 63.13; H, 6.48; N, 16.36%; found: C, 63.15; H, 6.51; N, 16.39%.
4.1.2.13. 5-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)-N,N-diethyl-1,3,4-oxadiazol-2-amine (21). Following the general procedure (A), and using diethylamine (25 μL, 0.4 mmol), compound 21 was obtained as yellow solid (0.068 g, 70%); mp = 158 °C; 1H NMR (DMSO-d6) δ: 7.87 (d, J = 8.4 Hz, 2H), 7.33 (d, J = 8.4 Hz, 2H), 3.49
:3.44 (m, 4H), 2.67 (s, 3H), 2.62:2.51 (m, 2H), 1.59:1.57 (m, 2H), 1.30:1.29 (m, 2H), 1.18:1.12 (m, 6H), 0.92:0.90 (m, 3H); 13C NMR (DMSO-d6) δ: 166.6, 163.4, 153.6, 153.2, 146.2, 130.2, 129.6, 126.7, 114.9, 43.5, 35.1, 33.2, 22.2, 17.3, 14.2, 13.4; MS (m/z) 370 (M+, 100%); anal. calc. for: (C20H26N4OS, Mwt = 370): C, 64.83; H, 7.07; N, 15.12%; found: C, 64.87; H, 7.11; N, 15.14%.
4.1.2.14. 2-(Azetidin-1-yl)-5-(2-(4-butylphenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazole (22). Following the general procedure (A), and using azetidine (40 mg, 0.4 mmol), compound 22 was obtained as yellow solid (0.07 g, 76%); mp = 230 °C; 1H NMR (DMSO-d6) δ: 7.90 (d, J = 8.4 Hz, 2H), 7.36 (d, J = 8.4 Hz, 2H), 4.18
:4.13 (m, 4H), 2.68 (s, 3H), 2.63:2.61 (m, 2H), 2.45:2.42 (m, 2H), 1.61:1.59 (m, 2H), 1.31:1.28 (m, 2H), 0.93:0.91 (m, 3H); 13C NMR (DMSO-d6) δ: 167.1, 164.7, 154.3, 153.4, 146.4, 130.2, 129.7, 126.8, 114.6, 52.5, 35.3, 33.2, 22.1, 17.8, 17.4, 14.2; MS (m/z) 354 (M+, 100%); anal. calc. for: (C19H22N4OS, Mwt = 354): C, 64.38; H, 6.26; N, 15.81%; found: C, 64.43; H, 6.29; N, 15.85%.
4.1.2.15. 1-(5-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl)azetidin-3-ol (23). Following the general procedure (A), and using 3-hydroxyazetidine (25 mg, 0.4 mmol), compound 23 was obtained as white solid (0.09 g, 93%); mp = 190 °C; 1H NMR (DMSO-d6) δ: 7.88 (d, J = 8.4 Hz, 2H), 7.38 (d, J = 8.4 Hz, 2H), 5.85 (brs, 1H), 4.67
:4.65 (m, 1H), 4.32 (dd, J = 4.6 Hz, J = 9.2 Hz, 2H), 3.96 (dd, J = 4.8 Hz, J = 9.2 Hz, 2H), 2.68 (s, 3H), 2.63:2.59 (m, 2H), 1.61:1.59 (m, 2H), 1.37:1.33 (m, 2H), 0.93:0.91 (m, 3H); 13C NMR (DMSO-d6) δ: 167.1, 164.8, 154.5, 154.4, 146.4, 130.2, 129.7, 126.8, 114.5, 62.4, 62.2, 35.1, 33.2, 22.1, 17.4, 14.2; MS (m/z) 370 (M+, 97%); anal. calc. for: (C19H22N4O2S, Mwt = 370): C, 61.60; H, 5.99; N, 15.12%; found: C, 61.66; H, 6.04; N, 15.16%.
4.1.2.16. 2-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)-5-(pyrrolidin-1-yl)-1,3,4-oxadiazole (24). Following the general procedure (A), and using pyrrolidine (28 μL, 0.4 mmol), compound 24 was obtained as yellow solid (0.086 g, 90%); mp = 115 °C; 1H NMR (DMSO-d6) δ: 7.89 (d, J = 8.4 Hz, 2H), 7.35 (d, J = 8.4 Hz, 2H), 3.50
:3.48 (m, 4H), 2.68 (s, 3H), 2.64:2.61 (m, 2H), 2.00:1.98 (m, 4H), 1.59:1.57 (m, 2H), 1.37:1.35 (m, 2H), 0.93:0.91 (m, 3H); 13C NMR (DMSO-d6) δ: 166.6, 162.4, 153.7, 153.4, 146.2, 130.2, 129.7, 126.6, 114.9, 48, 35.1, 33.2, 25.5, 22.2, 17.3, 14.2; MS (m/z) 368 (M+, 100%); anal. calc. for: (C20H24N4OS, Mwt = 368): C, 65.19; H, 6.57; N, 15.20%; found: C, 65.22; H, 6.59; N, 15.23%.
4.1.2.17. 2-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)-5-(piperidin-1-yl)-1,3,4-oxadiazole (25). Following the general procedure (A), and using piperidine (34 μL, 0.4 mmol), compound 25 was obtained as yellow solid (0.092 g, 93%); mp = 123 °C; 1H NMR (DMSO-d6) δ: 7.93 (d, J = 8.4 Hz, 2H), 7.41 (d, J = 8.4 Hz, 2H), 3.48
:3.44 (m, 4H), 2.69 (s, 3H), 2.65:2.62 (m, 2H), 1.62:1.53 (m, 6H), 1.34:1.28 (m, 2H), 0.91:0.85 (m, 5H); 13C NMR (DMSO-d6) δ: 166.5, 163.5, 153.7, 152.9, 146.5, 130.4, 130.3, 126.6, 113.3, 64.6, 36.9, 35.1, 33.5., 32.9, 21.9, 18.9, 13.9; MS (m/z) 382 (M+, 100%); anal. calc. for: (C21H26N4OS, Mwt = 382): C, 65.94; H, 6.85; N, 14.65%; Found: C, 65.97; H, 6.85; N, 14.66%.
4.1.2.18. 4-(5-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl)morpholine (26). Following the general procedure (A), and using morpholine (35 μL, 0.4 mmol), compound 26 was obtained as greenish yellow solid (0.048 g, 49%); mp = 143 °C; 1H NMR (DMSO-d6) δ: 7.89 (d, J = 8.4 Hz, 2H), 7.36 (d, J = 8.4 Hz, 2H), 3.75
:3.73 (m, 4H), 3.48:3.46 (m, 4H), 2.69 (s, 3H), 2.68:2.65 (m, 2H), 1.62:1.59 (m, 2H), 1.32:1.26 (m, 2H), 0.91:0.86 (m, 3H); 13C NMR (DMSO-d6) δ: 166.6, 165.9, 154.3, 153, 146.5, 130, 126.8, 126.5, 115.3, 54.5, 44.3, 35.2, 33.1, 22.3, 17.5, 14.5; MS (m/z) 384 (M+, 100%); anal. calc. for: (C20H24N4O2S, Mwt = 384): C, 62.48; H, 6.29; N, 14.57%; found: C, 62.52; H, 6.33; N, 14.60%.
4.1.2.19. 2-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)-5-hydrazinyl-1,3,4-oxadiazole (27). Following the general procedure (A), and using and using hydrazine hydrate (5 mL), compound 27 was obtained as grayish black solid (0.072 g, 85%); mp = 170 °C; 1H NMR (DMSO-d6) δ: 8.74 (brs, 1H), 7.90 (d, J = 8.4 Hz, 2H), 7.41 (d, J = 8.4 Hz, 2H), 4.56 (brs, 2H), 2.77 (s, 3H), 2.69
:2.58 (m, 2H), 1.61:1.55 (m, 2H), 1.37:1.32 (m, 2H), 0.91:0.89 (m, 3H); 13C NMR (DMSO-d6) δ: 166.5, 163.5, 153.7, 152.9, 146.5, 130, 126.8, 126.6, 115, 35.9, 33.6, 22.5, 17.4, 14.9; MS (m/z) 329 (M+, 100%); anal. calc. for: (C16H19N5OS, Mwt = 329): C, 58.34; H, 5.81; N, 21.26%; found: C, 58.38; H, 5.85; N, 21.28%.
4.1.2.20. N-(5-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl)ethane-1,2-diamine (28). Following the general procedure (A), and using ethane-1,2-diamine (20 μL, 0.4 mmol), compound 28 was obtained as yellowish brown solid (0.085 g, 92%); mp = 230 °C; 1H NMR (DMSO-d6) δ: 8.35 (brs, 1H), 7.78 (d, J = 8.4 Hz, 2H), 7.34 (d, J = 8.4 Hz, 2H), 3.74
:3.72 (m, 2H), 2.69 (s, 3H), 2.61:2.57 (m, 2H), 2.45:2.43 (m, 2H), 1.75 (brs, 2H), 1.59:1.57 (m, 2H), 1.34:1.31 (m, 2H), 0.93:0.90 (m, 3H); 13C NMR (DMSO-d6) δ: 166.7, 162.4, 153.7, 153.4, 146.2, 130.6, 126.5, 126.4, 115, 55.5, 36.9, 35.1, 31.3, 22.7, 18.5, 13.9; MS (m/z) 357 (M+, 46%); anal. calc. for: (C18H23N5OS, Mwt = 357): C, 60.48; H, 6.49; N, 19.59%; found: C, 60.52; H, 6.53; N, 19.59%.
4.1.2.21. N-(5-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl)-N,N-dimethylethane-1,2-diamine (29). Following the general procedure (A), and using N,N-Dimethylethylenediamine (30 μL, 0.4 mmol), compound 29 was obtained as yellow solid (0.095 g, 95%); mp = 179 °C; 1H NMR (DMSO-d6) δ: 7.93 (brs, 1H), 7.89 (d, J = 8.4 Hz, 2H), 7.33 (d, J = 8.4 Hz, 2H), 3.84
:3.82 (m, 2H), 2.77 (s, 3H), 2.66:2.62 (m, 2H), 2.45:2.42 (m, 2H), 2.18 (s, 6H), 1.59:1.57 (m, 2H), 1.34:1.32 (m, 2H), 0.90:0.88 (m, 3H); 13C NMR (DMSO-d6) δ: 166.6, 164.7, 153.7, 152.9, 146.2, 130.2, 129.3, 126.2, 114, 58.2, 45.6, 35.1, 33, 22.2, 17.4, 14.7, 14.2; MS (m/z) 385 (M+, 71%); anal. calc. for: (C20H27N5OS, Mwt = 385): C, 62.31; H, 7.06; N, 18.17%; found: C, 62.34; H, 7.08; N, 18.20%.
4.1.2.22. 2-((5-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl)amino)ethan-1-ol (30). Following the general procedure (A), and using ethanolamine (20 μL, 0.4 mmol), compound 30 was obtained as white solid (0.092 g, 98%); mp = 184 °C; 1H NMR (DMSO-d6) δ: 7.93 (brs, 1H), 7.85 (d, J = 8.4 Hz, 2H), 7.35 (d, J = 8.4 Hz, 2H), 4.81 (brs, 1H), 4.62
:4.60 (m, 2H), 3.32:3.26 (m, 2H), 2.76 (s, 3H), 2.68:2.65 (m, 2H), 1.59:1.57 (m, 2H), 1.34:1.32 (m, 2H), 0.92:0.90 (m, 3H); 13C NMR (DMSO-d6) δ: 168.6, 167, 155.2, 154, 146.9, 130.9, 126.9, 126.1, 115.5, 65.6, 64.6, 35.3, 33.1, 22.3, 16.5, 13.9; MS (m/z) 358 (M+, 100%); anal. calc. for: (C18H22N4O2S, Mwt = 358): C, 60.31; H, 6.19; N, 15.63%; found: C, 60.35; H, 6.22; N, 15.66%.
4.1.2.23. N1-(5-(2-(4-butylphenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl)propane-1,3-diamine (31). Following the general procedure (A), and using propane-1,3-diamine (28 μL, 0.4 mmol), compound 31 was obtained as yellow solid (0.08 g, 83%); mp = 252 °C; 1H NMR (DMSO-d6) δ: 7.89 (d, J = 8.4 Hz, 2H), 7.76 (brs, 1H), 7.35 (d, J = 8.4 Hz, 2H), 3.45
:3.42 (m, 2H), 2.68 (s, 3H), 2.62:2.59 (m, 2H), 2.43:2.41 (m, 2H), 1.79 (brs, 2H), 1.58:1.56 (m, 2H), 1.33:1.29 (m, 2H), 1.09:1.06 (m, 2H), 0.92:0.89 (m, 3H); 13C NMR (DMSO-d6) δ: 166.8, 163.6, 153.8, 152.9, 146.3, 130.2, 126.7, 126, 114.9, 46.6, 37, 35.1, 33.3, 33.2, 22.1, 17.4, 14.2; MS (m/z) 371 (M+, 75%); anal. calc. for: (C19H25N5OS, Mwt = 371): C, 61.43; H, 6.78; N, 18.85%; found: C, 61.45; H, 6.81; N, 18.89%.
4.1.2.24. 3-((5-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl)amino)propan-1-ol (32). Following the general procedure (A), and using 3-aminopropan-1-ol (28 μL, 0.4 mmol), compound 32 was obtained as yellow solid (0.091 g, 94%); mp = 249 °C; 1H NMR (DMSO-d6) δ: 7.93 (brs, 1H), 7.88 (d, J = 8.4 Hz, 2H), 7.34 (d, J = 8.4 Hz, 2H), 4.57 (brs, 1H), 3.52
:3.50 (m, 2H), 3.31:3.29 (m, 2H), 2.77 (s, 3H), 2.63:2.60 (m, 2H), 1.76:1.74 (m, 2H), 1.61:1.56 (m, 2H), 1.31:1.26 (m, 2H), 0.92:0.89 (m, 3H); 13C NMR (DMSO-d6) δ: 166.9, 161.5, 154.3, 153.8, 146.4, 130.9, 126.3, 124.1, 115.9, 64.6, 38.3, 37.9, 35.1, 34.1, 22.3, 17.5, 14.9; MS (m/z) 372 (M+, 100%); anal. calc. for: (C19H24N4O2S, Mwt = 372): C, 61.27; H, 6.49; N, 15.04%; found: C, 61.33; H, 6.53; N, 15.07%.
4.1.2.25. N-(5-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl)butane-1,4-diamine (33). Following the general procedure (A), and using butane-1,4-diamine (34 μL, 0.4 mmol), compound 33 was obtained as yellow solid (0.096 g, 96%); mp = 209 °C; 1H NMR (DMSO-d6) δ: 7.92 (brs, 1H), 7.85 (d, J = 8.4 Hz, 2H), 7.37 (d, J = 8.4 Hz, 2H), 3.87
:3.82 (m, 2H), 3.26:3.23 (m, 2H), 2.78:2.72 (m, 2H), 2.68 (s, 3H), 2.64:2.62 (m, 2H), 1.60:1.58 (m, 2H), 1.48:1.46 (m, 2H), 1.33:1.31 (m, 2H), 0.93:0.89 (m, 3H); 13C NMR (DMSO-d6) δ: 166.5, 164.9, 154, 152.8, 145.4, 130.6, 126.9, 126.3, 115.4, 64.6, 37.9, 35.1, 34.6, 31.3, 26.5, 26.4, 17.5, 14.9; MS (m/z) 385 (M+, 68%); anal. calc. for: (C20H27N5OS, Mwt = 385): C, 62.31; H, 7.06; N, 18.17%; found: C, 62.33; H, 7.08; N, 18.19%.
4.1.2.26. 2-((5-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl)amino)propane-1,3-diol (34). Following the general procedure (A), and using 2-aminopropane-1,3-diol (30 μL, 0.4 mmol), compound 34 was obtained as red solid (0.093 g, 93%); mp = 126 °C; 1H NMR (DMSO-d6) δ: 7.90 (d, J = 8.4 Hz, 2H), 7.76 (brs, 1H), 7.35 (d, J = 8.4 Hz, 2H), 4.79 (brs, 2H), 3.84
:3.79 (m, 4H), 3.59:3.57 (m, 1H), 2.78 (s, 3H), 2.70:2.64 (m, 2H), 1.58:1.55 (m, 2H), 1.33:1.31 (m, 2H), 0.93:0.89 (m, 3H); 13C NMR (DMSO-d6) δ: 168.6, 164.7, 160.6, 156.3, 146.7, 130.2, 129.9, 126.2, 113.7, 60.5, 58, 35.1, 33.2, 22.1, 17.4, 14.2; MS (m/z) 388 (M+, 100%); anal. calc. for: (C19H24N4O3S, Mwt = 388): C, 58.74; H, 6.26; N, 14.42%; found: C, 58.77; H, 6.29; N, 14.45%.
4.1.2.27. N-{5-[2-(4-Butylphenyl)-4-methylthiazol-5-yl]-1,3,4-oxadiazol-2-yl}cyclohexane-cis-1,2-diamine (35). Following the general procedure (A), and using cis-1,2-Diaminocyclohexane (0.045 g, 0.4 mmol), compound 35 was obtained as brown solid (0.09 g, 84%); mp = 83 °C; 1H NMR (DMSO-d6) δ: 7.85 (d, J = 8.4 Hz, 2H), 7.33 (d, J = 8.4 Hz, 2H), 7.11 (brs, 1H), 3.90
:3.88 (m, 1H), 3.54:3.51 (m, 1H), 3.14:3.08 (m, 2H), 2.70 (s, 3H), 2.61:2.59 (m, 2H), 1.58:1.52 (m, 2H), 1.30:1.20 (m, 8H), 1.05 (brs, 2H), 0.85:0.82 (m, 3H); 13C NMR (DMSO-d6) δ: 166.7, 161.4, 157.6, 153.4, 145.9, 130.8, 126.9, 126.5, 115.5, 49.3, 43.9, 39.9, 36.9, 35, 33.2, 26.7, 22.7, 22.1, 18.6, 14.2; MS (m/z) 411 (M+, 60%); anal. calc. for: (C22H29N5OS, Mwt = 411): C, 64.20; H, 7.10; N, 17.02%; found: C, 64.26; H, 7.15; N, 17.07%.
4.1.2.28. N-{5-[2-(4-Butylphenyl)-4-methylthiazol-5-yl]-1,3,4-oxadiazol-2-yl}cyclohexane-trans-1,2-diamine (36). Following the general procedure (A), and using trans-1,2-diaminocyclohexane (0.045 g, 0.4 mmol), compound 36 was obtained as brown solid (0.088 g, 81%); mp = 99 °C; 1H NMR (DMSO-d6) δ: 7.88 (d, J = 8.4 Hz, 2H), 7.34 (d, J = 8.4 Hz, 2H), 7.05 (brs, 1H), 3.86
:3.78 (m, 2H), 2.86:2.80 (m, 2H), 2.68 (s, 3H), 2.62:2.57 (m, 2H), 1.58:1.53 (m, 2H), 1.30:1.11 (m, 8H), 1.03 (brs, 2H), 0.93:0.87 (m, 3H); 13C NMR (DMSO-d6) δ: 166.3, 161.5, 157.3, 153.5, 145.7, 130.9, 127.1, 126.6, 115.6, 40.5, 39.9, 39.4, 36.8, 35.1, 33.3, 25.4, 22.5, 22.2, 18.4, 14.2; MS (m/z) 411 (M+, 62%); anal. calc. for: (C24H31N5S, Mwt = 411): C, 64.20; H, 7.10; N, 17.02%; found: C, 64.27; H, 7.16; N, 17.05%.
4.1.2.29. 1-(5-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl)guanidine (37). Following the general procedure (A), and using guanidine hydrochloride (0.05 g, 0.5 mmol) and potassium carbonate anhydrous (0.1 g, 0.7 mmol), compound 37 was obtained as yellow solid (0.07 g, 76%); mp = 140 °C; 1H NMR (DMSO-d6) δ: 8.19 (brs, 2H), 7.93 (d, J = 8.4 Hz, 2H), 7.36 (d, J = 8.4 Hz, 2H), 6.92 (brs, 2H), 2.73 (s, 3H), 2.65
:2.63 (m, 2H), 1.59:1.57 (m, 2H), 1.34:1.32 (m, 2H), 0.93:0.91 (m, 3H); 13C NMR (DMSO-d6) δ: 166.5, 163.5, 154.3, 153.7, 146.9, 145, 130, 126.8, 126.6, 115, 35.1, 31.3, 22.4, 17.4, 14.9; MS (m/z) 356 (M+, 30%); anal. calc. for: (C17H20N6OS, Mwt = 356): C, 57.28; H, 5.66; N, 23.58%; found: C, 57.34; H, 5.70; N, 23.62%.
4.1.2.30. 1-(5-(2-(4-butylphenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl)-3-methylguanidine (38). Following the general procedure (A), and using methylguanidine hydrochloride (0.06 g, 0.5 mmol) and potassium carbonate anhydrous (0.1 g, 0.7 mmol), compound 38 was obtained as brown solid (0.09 g, 93%); mp = 160 °C; 1H NMR (DMSO-d6) δ: 7.88 (d, J = 8.4 Hz, 2H), 7.77 (brs, 2H), 7.34 (d, J = 8.4 Hz, 2H), 2.87 (brs, 1H), 2.78 (s, 3H), 2.67 (s, 3H), 2.61
:2.59 (m, 2H), 1.61:1.59 (m, 2H), 1.35:1.29 (m, 2H), 0.91:0.89 (m, 3H); 13C NMR (DMSO-d6) δ: 166.4, 164.2, 153.7, 153, 146.2, 146, 130.2, 129.9, 126.3, 114.7, 58, 35.1, 33.2, 22.2, 17, 14.2; MS (m/z) 370 (M+, 41%); anal. calc. for: (C18H22N6OS, Mwt = 370): C, 58.36; H, 5.99; N, 22.68%; found: C, 58.38; H, 6.01; N, 22.69%.
4.1.2.31. 3-(5-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl)-1,1-dimethylguanidine (39). Following the general procedure (A), and using 1,1-dimethylguanidine hydrochloride (0.06 g, 0.5 mmol) and potassium carbonate anhydrous (0.1 g, 0.7 mmol), compound 39 was obtained as yellow solid (0.06 g, 60%); mp = 175 °C; 1H NMR (DMSO-d6) δ: 7.90 (d, J = 8.4 Hz, 2H), 7.69 (brs, 2H), 7.36 (d, J = 8.4 Hz, 2H), 3.03 (s, 6H), 2.71 (s, 3H), 2.63
:2.60 (m, 2H), 1.59:1.57 (m, 2H), 1.34:1.32 (m, 2H), 0.91:0.87 (m, 3H); 13C NMR (DMSO-d6) δ: 166.5, 163.7, 154.3, 153.7, 152.9, 146.4, 130, 126.8, 126.6, 115, 63.6, 35.1, 34.6, 22.3, 17.5, 14.9; MS (m/z) 384 (M+, 37%); anal. calc. for: (C19H24N6OS, Mwt = 384): C, 59.35; H, 6.29; N, 21.86%; found: C, 59.39; H, 6.32; N, 21.89%.
4.1.2.32. N-(5-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl)-4-methylpiperazine-1-carboximidamide (40). Following the general procedure (A), and and using 4-methylpiperazine-1-carboximidamide hydroiodide (0.11 g, 0.4 mmol) and potassium carbonate anhydrous (0.1 g, 0.7 mmol), compound 40 was obtained as orange solid (0.110 g, 92%); mp = 155 °C; 1H NMR (DMSO-d6) δ: 7.96 (brs, 2H), 7.89 (d, J = 8.4 Hz, 2H), 7.35 (d, J = 8.4 Hz, 2H), 3.62
:3.57 (m, 4H), 2.70 (s, 3H), 2.62:2.59 (m, 2H), 2.34:2.32 (m, 4H), 2.21 (s, 3H), 1.62:1.55 (m, 2H), 1.33:1.28 (m, 2H), 0.93:0.87 (m, 3H); 13C NMR (DMSO-d6) δ: 166.9, 166.8, 156.9, 154, 153, 146.2, 130.3, 129.7, 126.5, 115.4, 54.6, 46, 44.4, 35.1, 33.2, 22.2, 17.5, 14.2; MS (m/z) 459 (M+, 42%); anal. calc. for: (C22H29N7O2S, Mwt = 439): C, 60.11; H, 6.65; N, 22.31%; found: C, 60.15; H, 6.68; N, 22.34%.
4.1.2.33. N-(5-(2-(4-butylphenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl)morpholine-4-carboximidamide (41). Following the general procedure (A), and using morpholine-4-carboximidamide hydroiodide (0.1 g, 0.4 mmol) and potassium carbonate anhydrous (0.1 g, 0.7 mmol), compound 41 was obtained as buff solid (0.07 g, 63%); mp = 200 °C; 1H NMR (DMSO-d6) δ: 7.97 (brs, 2H), 7.87 (d, J = 8.4 Hz, 2H), 7.36 (d, J = 8.4 Hz, 2H), 3.64
:3.62 (m, 4H), 3.55:3.51 (m, 4H), 2.70 (s, 3H), 2.62:2.60 (m, 2H), 1.60:1.58 (m, 2H), 1.33:1.31 (m, 2H), 0.93:0.89 (m, 3H); 13C NMR (DMSO-d6) δ: 169.3, 166.6, 164.2, 153.8, 153, 146.3, 130.3, 129.7, 126.7, 114, 66.1, 44.8, 35.1, 33.2, 22.1, 17.5, 14.2; MS (m/z) 426 (M+, 32%); anal. calc. for: (C21H26N6O2S, Mwt = 426): C, 59.13; H, 6.14; N, 19.70%; found: C, 59.15; H, 6.16; N, 19.73%.
4.1.2.34. N-(5-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl)picolinimidamide (42). Following the general procedure (A), and using picolinmidamide hydrochloride (0.06 g, 0.4 mmol) and potassium carbonate anhydrous (0.1 g, 0.7 mmol), compound 42 was obtained as yellow solid (0.1 g, 92%); mp = 188 °C; 1H NMR (DMSO-d6) δ: 9.47 (brs, 1H), 8.91 (brs, 1H), 8.78
:8.72 (m, 1H), 8.41:8.38 (m, 1H), 8.07:8.05 (m, 1H), 7.93 (d, J = 8.4 Hz, 2H), 7.69:7.66 (m, 1H), 7.36 (d, J = 8.4 Hz, 2H), 2.78 (s, 3H), 2.68:2.66 (m, 2H), 1.59:1.53 (m, 2H), 1.33:1.31 (m, 2H), 0.92:0.90 (m, 3H); 13C NMR (DMSO-d6) δ: 167.8, 161.9, 159.2, 158.7, 158, 157.9, 155.1, 154.3, 154, 153.5, 146.5, 130.5, 130.3, 126.6, 113.3, 35.1, 31.3, 22.2, 18.9, 13.9; MS (m/z) 418 (M+, 100%); anal. calc. for: (C22H22N6OS, Mwt = 418): C, 63.14; H, 5.30; N, 20.08%; found: C, 63.18; H, 5.33; N, 20.11%.
4.1.2.35. N-(5-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl)nicotinimidamide (43). Following the general procedure (A), and using nicotinimidamide hydrochloride (0.06 g, 0.4 mmol) and potassium carbonate anhydrous (0.1 g, 0.7 mmol), compound 43 was obtained as yellow solid (0.1 g, 92%); mp = 139 °C; 1H NMR (DMSO-d6) δ: 9.61 (brs, 1H), 9.22 (brs, 1H), 9.06 (s, 1H), 8.82
:8.79 (m, 1H), 8.44:8.41 (m, 1H), 7.94 (d, J = 8.4 Hz, 2H), 7.60:7.58 (m, 1H), 7.33 (d, J = 8.4 Hz, 2H), 2.78 (s, 3H), 2.68:2.63 (m, 2H), 1.61:1.60 (m, 2H), 1.36:1.34 (m, 2H), 0.93:0.90 (m, 3H); 13C NMR (DMSO-d6) δ: 166.4, 163.8, 160.4, 159.1, 157.8, 156.1, 154.5, 153.4, 152.8, 146.8, 135.9, 129.7, 126.6, 126.3, 115, 34.9, 33.2, 22.1, 17.4, 14.2; MS (m/z) 418 (M+, 100%); anal. calc. for: (C22H22N6OS, Mwt = 418): C, 63.14; H, 5.30; N, 20.08%; found: C, 63.18; H, 5.33; N, 20.09%.
4.1.2.36. N-(5-(2-(4-Butylphenyl)-4-methylthiazol-5-yl)-1,3,4-oxadiazol-2-yl)isonicotinimidamide (44). Following the general procedure (A), and using isonicotinimidamide hydrochloride (0.06 g, 0.4 mmol) and potassium carbonate anhydrous (0.1 g, 0.7 mmol), compound 44 was obtained as yellow solid (0.07 g, 64%); mp = 127 °C; 1H NMR (DMSO-d6) δ: 9.67 (brs, 1H), 9.07 (brs, 1H), 8.97 (d, J = 6 Hz, 2H), 8.00 (d, J = 6 Hz, 2H), 7.90 (d, J = 8.4 Hz, 2H), 7.34 (d, J = 8.4 Hz, 2H), 2.77 (s, 3H), 2.65
:2.63 (m, 2H), 1.59:1.57 (m, 2H), 1.34:1.29 (m, 2H), 0.90:0.89 (m, 3H); 13C NMR (DMSO-d6) δ: 167.9, 166.3, 160.3, 155.5, 155.3, 150.8, 146.5, 141.6, 130.1, 129.6, 126.8, 121.9, 114.7, 35.1, 33.1, 22.2, 17.6, 14.2; MS (m/z) 418 (M+, 100%); anal. calc. for: (C22H22N6OS, Mwt = 418): C, 63.14; H, 5.30; N, 20.08%; found: C, 63.17; H, 5.34; N, 20.13%.
4.2 Biology
000 rpm for 10 min and spectrophotometric measurements of intracellular components at 260 nm were determined in the cell supernatant.29,304.3 ADME prediction
ADME parameters were predicted using the Swiss ADME open-access web tool (http://www.swissadme.ch/).Ethical approval
All animal procedures were performed in accordance with the Guidelines for Care and Use of Laboratory Animals of Al-Azhar University and Experiments were approved by the Animal Ethics Committee of Animal Care and Use of Faculty of Medicine Al-Azhar University.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
All the authors declare that they have no conflict of interest.References
- S. Emami, A. Foroumadi, M. Falahati, E. Lotfali, S. Rajabalian, S.-A. Ebrahimi, S. Farahyar and A. Shafiee, Bioorg. Med. Chem. Lett., 2008, 18, 141–146 Search PubMed.
- W. Jiang, M. Zhou, Z. Cong, J. Xie, W. Zhang, S. Chen, J. Zou, Z. Ji, N. Shao and X. Chen, Angew. Chem., 2022, 134, e202200778 CrossRef.
- I. Kato, Y. Ukai, N. Kondo, K. Nozu, C. Kimura, K. Hashimoto, E. Mizusawa, H. Maki, A. Naito and M. Kawai, J. Med. Chem., 2021, 64, 10482–10496 CrossRef CAS PubMed.
- M. S. Al Aboody and S. Mickymaray, Antibiotics, 2020, 9, 45 CrossRef CAS PubMed.
- M. D. Altıntop, Ö. Atlı, S. Ilgın, R. Demirel, A. Özdemir and Z. A. Kaplancıklı, Eur. J. Med. Chem., 2016, 108, 406–414 CrossRef PubMed.
- E. L. Berkow and S. R. Lockhart, Infect. Drug Resist., 2017, 237–245 CrossRef CAS PubMed.
- R. Traboulsi and M. A. Ghannoum, in Antifungal Therapy, CRC Press, 2016, pp. 288–302 Search PubMed.
- S. S. Magill, E. O'Leary, S. J. Janelle, D. L. Thompson, G. Dumyati, J. Nadle, L. E. Wilson, M. A. Kainer, R. Lynfield and S. Greissman, N. Engl. J. Med., 2018, 379, 1732–1744 CrossRef PubMed.
- M. C. Fisher, N. J. Hawkins, D. Sanglard and S. J. Gurr, Science, 2018, 360, 739–742 CrossRef CAS PubMed.
- M. Spitzer, N. Robbins and G. Wright, Virulence, 2017, 6, 169–185 CrossRef PubMed.
- K. J. Shaw and A. S. Ibrahim, J. Fungi, 2020, 6, 239 CrossRef CAS PubMed.
- M. H. Chowdhury, L. K. Ryan, K. Cherabuddi, K. B. Freeman, D. G. Weaver, J. C. Pelletier, R. W. Scott and G. Diamond, J. Fungi, 2018, 4, 30 CrossRef PubMed.
- A. Arastehfar, C. Lass-Flörl, R. Garcia-Rubio, F. Daneshnia, M. Ilkit, T. Boekhout, T. Gabaldon and D. S. Perlin, J. Fungi, 2020, 6, 138 CrossRef CAS PubMed.
- F. Xie, T. Ni, J. Zhao, L. Pang, R. Li, Z. Cai, Z. Ding, T. Wang, S. Yu and Y. Jin, Bioorg. Med. Chem. Lett., 2017, 27, 2171–2173 Search PubMed.
- J. Zhang, Z. Wang, C. Gai, F. Yang, X. Yun, B. Jiang, Y. Zou, Q. Meng, Q. Zhao and X. Chai, Bioorg. Med. Chem., 2024, 97, 117543 Search PubMed.
- Z. A. Kanafani and J. R. Perfect, Clin. Infect. Dis., 2008, 46, 120–128 CrossRef PubMed.
- P. Marichal, J. Gorrens, M. C. Coene, L. L. Jeune and H. V. Bossche, Mycoses, 1995, 38, 111–117 CrossRef CAS PubMed.
- G. D. Brown, D. W. Denning, N. A. Gow, S. M. Levitz, M. G. Netea and T. C. White, Sci. Transl. Med., 2012, 4, 165rv113 Search PubMed.
- J. Liao, F. Yang, L. Zhang, X. Chai, Q. Zhao, S. Yu, Y. Zou, Q. Meng and Q. Wu, Arch. Pharmacal Res., 2015, 38, 470–479 CrossRef CAS PubMed.
- H. Mohammad, A. S. Mayhoub, A. Ghafoor, M. Soofi, R. A. Alajlouni, M. Cushman and M. N. Seleem, J. Med. Chem., 2014, 57, 1609–1615 CrossRef CAS PubMed.
- H. Mohammad, H. E. Eldesouky, T. Hazbun, A. S. Mayhoub and M. N. Seleem, Sci. Rep., 2019, 9, 18941 Search PubMed.
- M. A. Seleem, A. M. Disouky, H. Mohammad, T. M. Abdelghany, A. S. Mancy, S. A. Bayoumi, A. Elshafeey, A. El-Morsy, M. N. Seleem and A. S. Mayhoub, J. Med. Chem., 2016, 59, 4900–4912 CrossRef CAS PubMed.
- M. Hagras, et al., PLoS One, 2021, 16, 0258465 CrossRef PubMed.
- M. M. Seif El-Din, M. Hagras and A. S. Mayhoub, RSC Med. Chem., 2024, 15, 1991–2001 RSC.
- M. Hagras, N. S. Abutaleb, H. G. Ezzat, E. A. Salama, M. N. Seleem and A. S. Mayhoub, RSC Med. Chem., 2023, 14, 2089–2099 RSC.
- A. A. Abuelkhir, M. Omara, Y. I. Nagy, A. E. Gouda, A. S. Attia, A. S. Mayhoub and M. Hagras, Med. Chem. Res., 2024, 33, 1178–1194 CrossRef CAS.
- M. M. Elsebaie, H. T. Nour El-Din, N. S. Abutaleb, A. A. Abuelkhir, H.-W. Liang, A. S. Attia, M. N. Seleem and A. S. Mayhoub, Eur. J. Med. Chem., 2022, 234, 114204 CrossRef CAS.
- M. Hagras, E. A. Salama, A. M. Sayed, N. S. Abutaleb, A. Kotb, M. N. Seleem and A. S. Mayhoub, Eur. J. Med. Chem., 2020, 189, 112046 Search PubMed.
- E. Mani-López, O. Cortés-Zavaleta and A. López-Malo, SN Appl. Sci., 2021, 3, 44 Search PubMed.
- A. Salama Ehab, Y. Elgammal, M. Utturkar Sagar, A. Lanman Nadia, R. Hazbun Tony and N. Seleem Mohamed, Antimicrob. Agents Chemother., 2024, 68, e00556 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra00499c |
| This journal is © The Royal Society of Chemistry 2025 |