
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMechanistic insights from simulations of drug–drug conjugate nanoclusters for co-delivery across cancer cell membranes†
Cherdpong Choodet,
Unnop Srikulwong,
Pakawat Toomjeen,
Adulvit Chuaephon,
Witthawat Phanchai and
Theerapong Puangmali *
*
Department of Physics, Faculty of Science, Khon Kaen University, Khon Kaen 40002, Thailand. E-mail: theerapong@kku.ac.th
First published on 10th April 2025
Abstract
Amphiphilic drug–drug conjugates (ADDCs) such as gemcitabine–camptothecin (GEM–CPT) and doxorubicin–10-hydroxycamptothecin (DOX–HCPT) nanoclusters offer innovative solutions to overcome the limitations of conventional cancer therapies, including poor solubility and nonspecific targeting. Using molecular dynamics (MD) simulations, we explored the mechanisms by which these nanoclusters interact with and penetrate cancer and normal cell membranes. GEM–CPT exhibited enhanced membrane penetration in cancer cells through combined hydrophilic and hydrophobic interactions, along with its ability to extract cholesterol and induce membrane remodelling. In contrast, DOX–HCPT maintained structural integrity through stable π–π stacking interactions, showing selective binding to membrane head groups (HG) with minimal cholesterol interaction, particularly in normal membranes. The GEM–CPT nanocluster disrupted the cancer membrane by inducing asymmetric lipid distribution and facilitating water infiltration, whereas the hydrophobic DOX–HCPT repelled water, maintaining membrane stability. The size of the nanocluster further influenced the behaviour; larger clusters drove steric assembly and lipid reorganisation, while smaller clusters achieved deeper penetration at the cost of structural integrity. The contrasting behaviours of GEM–CPT and DOX–HCPT highlight the critical roles of size, charge, and amphiphilicity in membrane transport mechanisms. These findings provide valuable insights into the design of efficient and selective nanomedicines, paving the way for optimised drug delivery systems with reduced off-target effects.
1 Introduction
The high incidence and mortality rates associated with cancer have fueled the continuous innovation of new anticancer treatment methods. Despite progress in this field, the significant side effects commonly linked to conventional therapies often undermine patient results, affecting both survival rates and life quality.1 To alleviate these negative effects, combination chemotherapy, which uses multiple chemical agents to concurrently attack malignant tumours, has become a promising strategy.2 Although combination therapies offer substantial benefits over single-drug options, their clinical effectiveness remains limited when administered via traditional dosage methods due to still-incomplete data on their results.3 To overcome these challenges, amphiphilic drug conjugates (ADCs) have been developed as innovative co-delivery systems that tackle key hurdles in cancer treatment, such as multidrug resistance, inadequate bioavailability, and swift systemic elimination.4,5 These advanced ADC platforms comprise various subcategories, including polymer–drug conjugates (PDCs), phospholipid–mimetic prodrugs, peptide–drug conjugates (PepDCs), pure nanodrugs (PNDs), Janus drug–drug conjugates (JDDCs), and amphiphilic drug–drug conjugates (ADDCs).6 ADDCs represent a novel approach in cancer therapy, enhancing the effectiveness of combination chemotherapy due to their amphiphilic characteristics.7 After biodegradation in tissues or cells, these conjugates release free anticancer agents that together target affected regions, which enhances cellular drug absorption. This technique has been advanced by binding hydrophilic and hydrophobic anticancer drugs to synergistically work. Through the integration of drugs with various physicochemical characteristics, ADDCs address significant issues such as low solubility, lack of target specificity, and poor pharmacokinetics.8 These features promote efficient drug delivery and lead to better therapeutic results.An illustrative case in this drug delivery model is the gemcitabine–camptothecin (GEM–CPT) conjugate, which has recently gained attention as a promising candidate in advanced drug delivery conjugates. Gemcitabine (GEM), a nucleoside analogue, inhibits DNA synthesis and is commonly used in the treatment of cancers such as the pancreas, breast, and lung. Camptothecin (CPT), known for its strong inhibition of topoisomerase I, disrupts DNA replication and is especially effective against solid tumours. The amphiphilic properties of GEM–CPT conjugates facilitate their self-assembly into nanoparticles, improving the solubility and bioavailability. This attribute allows for the simultaneous release of drugs, achieving synergistic anticancer outcomes while reducing systemic toxicity. Due to their pharmacokinetic and pharmacodynamic benefits, GEM–CPT conjugates represent a highly effective and targeted drug delivery system, potentially overcoming the drawbacks of standard combination chemotherapies. Quantitative calculation of the combination index suggested that GEM–CPT exhibited a significant synergistic anticancer effect at elevated drug concentrations. According to research by Meili Hou et al.,9 GEM–CPT efficiently circumvented tumour cell multidrug resistance.
In addition to the ADDCs, the PND model has been introduced as a novel approach to more efficient and less toxic drug delivery. The PND strategy eliminates the need for supplementary carriers or excipients by utilizing drug–drug conjugates that deliver to target cells via the formation of nanoparticles or nanoclusters entirely made up of active pharmaceutical ingredients.10 PNDs represent a novel carrier-free drug delivery approach that incorporates multiple active drugs, enhancing anticancer effects.10–13 The high hydrophobicity of chemotherapeutic agents poses a significant obstacle in the design and clinical application of nanomedicines.14 Though various solubilizing agents and specific chemical modifications have somewhat reduced solubility issues, the dependency on additional agents such as surfactants remains an barrier to the clinical efficacy and translational prospects of these drugs. Better clinical outcomes are likely achieved by avoiding such methods of fabrication. An effective alternative is to employ the active pharmaceutical ingredients themselves as surfactants.
Doxorubicin (DOX), characterised by its hydrophobic anthracycline rings and abundant hydroxyl structure, exhibits surfactant-like properties, suggesting its role as a stabiliser for drug nanosizing.15,16 As an active pharmaceutical ingredient, DOX avoids limitations associated with inactive ingredients, enabling dual-drug combination therapy in PND. Liang et al.17 reported the development of a carrier-free PND combining 10-hydroxycamptothecin (HCPT) and DOX, referred to as doxorubicin–10-hydroxycamptothecin (DOX–HCPT), utilising a straightforward and environmentally friendly technique. The surfactant-like characteristics of DOX eliminated the need for additives, increasing both the solubility in water and the stability. Although DOX-based PNDs and GEM–CPT ADDCs hold considerable promise for enhanced drug delivery, the critical factor of their interaction with cellular membranes, which influences drug uptake and effectiveness, remains essential, particularly due to the structural and compositional variances between normal and cancerous cell membranes.
Cell membranes are vital for the absorption of drugs into cells, managing how therapeutic agents access healthy and cancerous cells. These membranes consist of a lipid bilayer with proteins embedded within, which function as selective barriers that regulate molecular entry and exit.18 Typically, healthy cell membranes maintain a well-balanced lipid composition, asymmetric arrangement, and selective permeability, all of which are crucial to maintaining cellular stability. Conversely, altered lipid compositions and symmetrical distribution are common traits of various diseases, including cancer.19 Cancer cells, known for their uncontrolled growth, demand increased biomolecule supplies, such as fatty acids and lipids.20 To facilitate their rapid expansion, tumour cells enhance fatty acid biosynthesis to produce the necessary components for new membrane formation.21–23 The profiles of lipids and phospholipids are notably altered in cancers, such as breast24,25 and colorectal cancers, as well as chemoresistant tumours.26 Notably, hepatocellular carcinoma cells display a quadrupled sphingomyelin profile and increased levels of unsaturated fatty acids compared to normal cells.
The distribution of the lipid profile within the plasma membrane, sustained by the actions of flippases and floppases, is vital for its function.27,28 The outer leaflet is predominantly composed of phosphatidylcholine (PC) and sphingomyelin (SM), while the inner leaflet primarily comprises phosphatidylserine (PS) and phosphatidylethanolamine (PE). Understanding these membrane properties and uptake mechanisms is crucial for advancing drug design and enhancing therapeutic outcomes, as the membrane penetration ability of a drug is a key factor in its efficacy. In recent years, simulations of cancerous and normal cell membranes have become a popular approach to study how drugs enter cells. These investigations offer critical insights, elucidating how structural and compositional variations between these cell types impact drug uptake.29,30
To understand the intricate interactions between drugs and cell membranes, we employed atomistic molecular dynamics (MD) simulations. This research aimed to explore the molecular processes by which amphiphilic drug conjugates interact with and integrate into cell membranes using MD simulations. The study concentrated on two model drugs: ADDCs (GEM–CPT) and PNDs (DOX–HCPT). The study assessed the process by which these drugs form nanoclusters (Fig. 1(a) and (b)) and explored their interactions with both cancerous and healthy cell membranes (Fig. 1(c)). In addition, the study analysed drug transport mechanisms across membranes, offering insights into therapeutic effectiveness and targeted delivery.
 | ||
| Fig. 1 (a) and (b) illustrate DOX–HCPT and GEM–CPT drug conjugates forming PND and ADDC nanoclusters, respectively. (c) Shows ADC nanoclusters interacting with both cancerous and normal membranes. Lipid components are colour-coded: sphingomyelin (SM) in purple, DOPE in orange, DOPS in pink, DOPC in green, and cholesterol (CHL) in yellow, with lipid head groups depicted as spheres. Histograms on the right reveal the normalized abundance of lipid species in the monolayers of the membranes. | ||
2 Methods
2.1 Molecular dynamics simulation
2.2 Analysis
The Molecular Mechanics Poisson–Boltzmann Surface Area (MMPB-SA) tool integrated with GROMACS was utilized to calculate the free binding energy between the drug and the membrane.55,56 The technique was originally developed to examine the binding interactions between proteins and ligands.57 Later it has been widely used for various biomolecules.56 Numerous studies have effectively applied this methodology to investigate the interactions among biomolecules, such as photosensitizers,58 peptides,59,60 with cell membranes. This calculation was carried out using gmx mmpbsa.57 The binding free energy (Gbinding) when the drug associates with the membrane in a solvent is determined by the following equation:61| ΔGbinding = Gcomplex − (Gmembrane + Gdrug) | (1) |
| Gx = Vbonded + VVdW + Velec + Gpolar + Gnonpolar − TS | (2) |
| ΔGbinding = VVdWcomplex + Veleccomplex + ΔGpolar + ΔGnonpolar | (3) |
| ΔGpolar = Gpolarcomplex − (Gpolardrug + Gpolarmembrane) | (4) |
| ΔGnonpolar = Gnonpolarcomplex − (Gnonpolardrug + Gnonpolarmembrane). | (5) |
3 Results and discussion
To gain a comprehensive understanding of the cellular drug uptake mechanisms, we began by studying the structure of nanoclusters formed through the self-assembly of drug–drug conjugates. We then analysed the interactions between these drug nanoclusters and the cell membrane to assess how nanocluster size and configuration affect drug penetration. Lastly, we examined how co-delivery affects membrane deformation.3.1 Structure of drug–drug conjugate nanocluster
The electrostatic distribution across the drug surface is crucial for identifying the position and intensity of both hydrophobic and hydrophilic areas, thereby aiding in the self-organization of drug molecules. DOX and GEM are abundant in functional groups that contain oxygen, such as ketones and hydroxyl groups. In contrast, CPT and HCPT have fewer oxygen-rich groups but consist of several bonded aromatic rings, as shown in their chemical structures and electrostatic potential (ESP) maps in Fig. 2. The red and blue areas on the ESP maps indicate significant polar zones, which indicate hydrophilic properties. Importantly, the joined aromatic rings present in DOX and HCPT facilitate robust π–π stacking interactions. GEM–CPT conjugates are classified as hydrophilic drug models exhibiting higher SASA, while DOX and HCPT serve as hydrophobic models with reduced SASA, as shown in Fig. S1.† Negative charges on the ESP surfaces correspond with electronegative atoms such as oxygen and nitrogen, while positive charges cluster on less electronegative atoms. | ||
| Fig. 2 Chemical structures and electrostatic potential (ESP) visualizations of pharmaceutical models. (a and b) Molecular structures of gemcitabine (GEM) and camptothecin (CPT) with corresponding ESP maps, highlighting regions of positive (blue) and negative (red) electrostatic potential. (c) Formation of GEM–CPT amphiphilic drug–drug conjugates (ADDCs), illustrating the integration of GEM and CPT via a disulfide linkage. (d and e) Molecular configurations of doxorubicin (DOX) and hydroxycamptothecin (HCPT) and the distribution of their electrostatic charge. | ||
The self-assembly of drug–drug conjugate nanoclusters in a solvent highlights the dynamic behaviour of drug molecules in an aqueous environment. Initially, amphiphilic drug–drug conjugates and pure nanodrugs were randomly distributed within the simulation box (Fig. S2 and S3†), but over time they self-organised into stable nanoclusters driven by thermodynamic forces. Fig. 3 compares the size-dependent structural effects of these nanoclusters. In smaller GEM–CPT ADDC nanoclusters, CPT molecules exhibit well-defined π–π stacking interactions with an interplanar distance of 3.56 Å, stabilised by aromatic interactions, while GEM adjusts its conformation to enhance non-covalent interactions with CPT rings (Fig. 3(a)). However, larger GEM–CPT nanoclusters show reduced stacking π–π among CPT molecules due to steric hindrance, with GEM primarily contributing to the bulk structure due to its disulfide tether, limiting its role in π–π interactions (Fig. 3(b)). In contrast, DOX–HCPT PNDs maintain consistent and highly ordered π–π stacking patterns in both small and large nanoclusters, as shown in Fig. 3(c) and (d), reflecting the structural advantage of their design. These findings underscore the crucial influence of steric effects and molecular architecture on nanocluster assembly and stability.
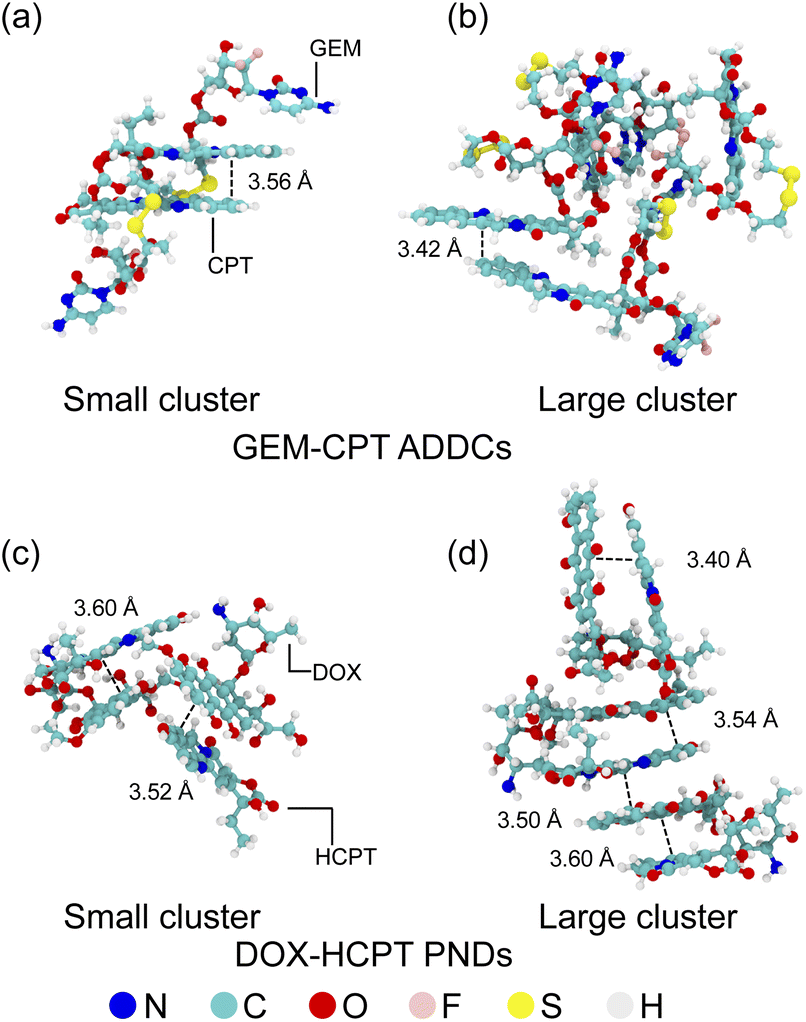 | ||
| Fig. 3 Molecular configurations and π–π interactions in small and large aggregates of GEM–CPT and DOX–HCPT. The figure illustrates the aggregation structures of GEM–CPT (a and b) and DOX–HCPT (c and d), highlighting the π–π stacking interactions that stabilize these molecular assemblies. Distinct configurations for both small and large aggregates are depicted, emphasizing the role of π–π interactions in the self-assembly process. | ||
3.2 Influence of nanocluster size in cellular uptake
The process of cellular drug uptake is affected by the interaction between drug nanoclusters and the cell membrane, primarily influenced by the strength of the binding energy. To study these interactions, MD simulations were performed on GEM–CPT and DOX–HCPT nanoclusters of varying sizes, which react with normal and cancer cell membranes in an aqueous setting, as shown in Fig. 4. The large and small nanoclusters of both GEM–CPT ADDCs and DOX–HCPT PNDs, hereafter referred to as the large and small drug nanoclusters, were observed in this aqueous setting. The binding energy was examined at three critical sites: the outer leaflet, the centre of the bilayer, and the inner leaflet. Initially, the drug nanocluster contacts the outer leaflet (A1) and begins to internalize into the membrane (A2). As it advances to the bilayer centre (A3), it interacts robustly with the hydrophobic core created by the lipid tails, showing considerable binding energy due to favourable hydrophobic interactions. Finally, the nanocluster penetrates the inner leaflet (A4), moving further into the cell. The distance for each interaction phase was maintained at 2 nm, with the membrane initially set at a distance of zero relative to the centre of the simulation. In the analysis of binding energy, distances situated above the membrane's centre were designated as negative, while those located below were assigned positive values. These positional arrangements facilitated the evaluation of the binding energy relative to the distance throughout this study. | ||
| Fig. 4 Pathway of drug nanocluster penetration through the cell membrane. The 4 nm thick membrane comprises the outer leaflet, bilayer centre, and inner leaflet. The nanocluster transitions from floating near the surface (A1) to interacting with the outer layer (A2), embedding in the hydrophobic core (A3), and penetrating the inner layer (A4), entering the cell. | ||
Experimental findings indicate that drug–drug conjugates have an average size of approximately 50 nm.9,63 However, molecular simulations reduce this size to 1.5–4 nm to optimise computational efficiency.6,9,63–67 Despite this reduction, simulations provide valuable insights into drug behaviour and development, as evidenced by studies on carrier-free self-assembled nanorods, even at reduced doses.68 Fig. 5(a) and (b) highlights the critical role of nanocluster size in membrane permeability and cellular uptake. For larger GEM–CPT ADDC nanoclusters, the binding energy increases sharply as the cluster approaches −3.1 nm from the membrane centre, peaking at high positive values before declining after crossing this point. The minimum binding energy occurs at 2.7 nm for cancer membranes and 0.8 nm for normal membranes, followed by fluctuations as the nanocluster exits the membrane. MD simulations also reveal the extraction of cholesterol from the membrane, which adheres to the nanocluster and induces temporary deformation with slight curvature (Fig. 5(c) and (d)). This process, driven by hydrophobic interactions and steric effects, underscores the importance of sterically induced self-assembly. In normal membranes, higher concentrations of phosphatidylethanolamine (PE) in the inner leaflet increase rigidity, which presents a substantial barrier to drug penetration.69 This rigidity correlates with a greater binding energy difference from the global minimum until the nanocluster exits, emphasising the interplay between membrane composition and nanocluster dynamics.
 | ||
| Fig. 5 Binding energies of GEM–CPT ADDC nanoclusters interacting with (a) cancer and (b) normal membranes. (c) and (d) present snapshots of the nanoclusters at local energy maxima or minima during their translocation through the cancer and normal membrane models, respectively. Cross-sectional views highlight the nanocluster's position, excluding Na+, Cl−, and water molecules for clarity. | ||
The transport of the small GEM–CPT ADDC nanocluster through cancer and normal membranes revealed distinct differences in drug–membrane interactions. In cancer membranes, the binding energy increased steadily as the nanocluster penetrated the membrane, reflecting a stable and efficient transport process (Fig. S4(a)†). In contrast, normal membranes exhibited significant fluctuations in binding energy after reaching the global minimum, indicative of structural disruptions during penetration (Fig. S4(b)†). The cancer membrane remained relatively stable throughout nanocluster transport, resulting in a consistent increase in binding energy with minimal structural changes (Fig. S4(c)†). However, in normal membranes, cholesterol and lipid tails disrupted the nanocluster structure, causing a disordered lipid arrangement that impeded drug release and led to pronounced variations in binding energy (Fig. S4(d)†). These observations underscore the critical role of sterically controlled self-assembly in drug delivery, where disulfide bonds in the GEM–CPT nanocluster contribute to membrane disruption, particularly in normal membranes, posing challenges for efficient release and highlighting the need for optimised nanocluster design.
The penetration of both large and small DOX–HCPT PND nanoclusters resulted in less membrane disruption due to their π–π stacking structure, which is more sophisticated than the GEM–CPT ADDC nanocluster. For the larger nanocluster, the binding energy exhibited significant variations as it traversed the cancer membrane. In contrast, the normal membrane showed notable fluctuations at 4.0 nm beyond the global minimum, as illustrated in Fig. S5(a) and (b).† The minimal disruption occurred in the outer leaflet of the cancer membrane by the drug nanocluster, while the orientation of their π–π stacking resulted in considerable disruption of the inner leaflet of the normal membrane by increasing the contact surface area and binding energy, as demonstrated in Fig. S5(c) and (d).† For the smaller nanocluster, the binding energy decreased significantly in the cancer membrane but increased with fluctuations within a 4 to 6 nm range in the normal membrane, as seen in Fig. S6(a) and (b).† This reduction in the cancer membrane was attributed to van der Waals and electrostatic interactions with the lipid head groups of the inner leaflet, which allowed the membrane to restore its original structure, shown in Fig. S6(c).† In the normal membrane, cholesterol molecules were expelled from the membrane, as shown in Fig. S6(d).† These findings emphasise the role of drug orientation in membrane interaction and disruption, due to the π–π stacking structure of the drug nanocluster, providing information for the development of selective and effective drug delivery systems across various types of membranes.
3.3 Interactions between nanoclusters and lipid bilayer of normal and cancer cells
To gain insights into the interlization mechanisms of the nanoclusters, we examined interactions with lipid head groups (HG) and tail groups (TG). Notable differences in binding energy were observed for large GEM–CPT and DOX–HCPT nanoclusters that interact with HG and TG in the inner and outer layers of the membrane, as shown in Fig. 6. For HG interactions, the binding energy decreased at approximately −2.9 nm and then increased to a global maximum, which differs from the global minimum observed in the overall membrane model (Fig. 6(a)). The global maximum appeared at roughly 3.2 nm for GEM–CPT and around 1.1 nm for DOX–HCPT, indicating varying HG disruption due to drug penetration. After global maximum, the binding energy showed pronounced fluctuations until drug release, while both nanoclusters preserved their original structures during penetration (Fig. 6(b) and (c)). In contrast, in TG interactions, the binding energy showed an inverse pattern, slowly declining to a global minimum at approximately 2.0 nm, with minor increases and negligible fluctuations thereafter, as shown in Fig. 6(d). This suggests weaker nanocluster–TG interactions. The GEM–CPT nanocluster altered its structure due to the function of the disulfide bond as a foldable joint, while the DOX–HCPT nanocluster preserved its structure (Fig. 6(e) and (f)). To provide an atomistic view of the large drug nanoclusters interacting with the cancer membrane, rescaled MD snapshots are available in Fig. S7(a)–(d),† relating to Fig. 6(b), (c), (e) and (f). These results emphasize the unique interaction behaviours of large GEM–CPT and DOX–HCPT nanoclusters with HG and TG in the cancer membrane, highlighting how structural integrity and lipid perturbation impact binding energy patterns. | ||
| Fig. 6 Binding energies of large GEM–CPT (black line) and DOX–HCPT (red line) nanoclusters interacting with (a) HG cancer membranes and (d) TG cancer membranes. Snapshots (b and c) show GEM–CPT and DOX–HCPT nanoclusters at the surface of the HG cancer membrane, while (e and f) depict their positions at the surface of the TG cancer membrane, respectively. | ||
Moreover, the interactions between large drug nanoclusters and the HG/TG of a normal membrane displayed distinct variations in the binding energy patterns and drug conformations compared to those of a cancer membrane. Initially, the binding energy decreased around −3.1 nm and increased sharply at ≈3.9 nm, with only minor fluctuations and no steady rise observed between −2.4 and 3.4 nm. This behaviour contrasts with that of the cancer membrane, as shown in Fig. S8(a).† Molecular dynamics snapshots of the GEM–CPT and DOX–HCPT models that interact with HG are presented in Fig. S8(b) and (c).† In interactions with TG, the binding energy formed a well-shaped curve with variable depths, with the GEM–CPT model achieving a more pronounced minimum binding energy than DOX–HCPT, indicating a preference for membrane penetration by GEM–CPT, as depicted in Fig. S8(d).† However, the GEM–CPT nanocluster showed significant structural loss, likely due to its flexible disulfide bond, while the DOX–HCPT nanocluster largely preserved its structure, as evidenced in Fig. S8(e) and (f).† These results suggest that the GEM–CPT model displays stronger interactions with TG and enhanced structural flexibility, whereas the DOX–HCPT model maintains superior structural stability, indicating different interaction mechanics and membrane penetration efficiencies.
For smaller GEM–CPT and DOX–HCPT nanoclusters, interactions with the HG portion of the cancer cell membrane lead to different trends in binding energy. In contrast, both drug nanoclusters exhibit similar binding energy patterns when interacting with TG. As shown in Fig. S9(a),† the binding energy of GEM–CPT decreases at approximately −2.9 nm and increases at around 3.7 nm. Meanwhile, the DOX–HCPT nanocluster displays a wave-like pattern caused by electrostatic interactions between its aromatic rings and negatively charged HG lipids (PS and PE). These favourable interactions enable the small DOX–HCPT nanocluster to embed into the membrane's outer leaflet. Similar behaviour is seen when the larger DOX–HCPT nanocluster crosses the cancer membrane. Interestingly, as shown in Fig. S9 (b) and (c),† DOX–HCPT is surrounded by a higher concentration of PS (pink) and PE lipids (orange) compared to GEM–CPT. For TG interactions, the binding energy demonstrates a well-shaped profile, with the GEM–CPT/TG binding energy having a shallower profile than DOX–HCPT, suggesting DOX–HCPT's superior membrane accessibility, as shown in Fig. S9(d).† In particular, as depicted in Fig. S9(e) and (f),† the DOX–HCPT nanocluster undergoes more structural alteration, including π–π stacking deformation, while GEM–CPT maintains its structural integrity. These observations imply that the strong affinity of DOX–HCPT for PS and PE lipids, combined with its structural flexibility, enhances its binding strength and membrane accessibility compared to GEM–CPT.
The interactions of small drug nanoclusters with both HG and TG of the normal membrane mirrored those seen with larger nanoclusters, with binding energy decreasing around −3.0 nm and peaking at approximately 3.0 nm, accompanied by significant fluctuations as the drug nanoclusters engaged with HG. Between −2.6 and 2.5 nm, there were minor fluctuations without a steady increase, as illustrated in Fig. S10(a).† The GEM–CPT and DOX–HCPT drugs interacted with HG by aligning their oxygen-containing groups toward the membrane, as shown in Fig. S10(b) and (c).† Regarding TG interactions, the binding energy showed a well-defined valley for both drug models, although GEM–CPT achieved lower energy values beyond 4 nm, as shown in Fig. S10(d),† suggesting similar membrane access for both models. Importantly, both drugs sustained significant structural damage, which is shown in Fig. S10(e) and (f).† These results suggest that despite the fact that both small drug models have comparable TG interaction patterns and membrane access, their structural degradation points to potential stability issues during membrane entry.
The cellular uptake of drugs is significantly influenced by the interactions between drug nanoclusters and HG lipids of cancer cell membranes as well as the structural characteristics of the nanoclusters. Analysis of hydrogen bonds revealed that for larger nanoclusters, the DOX–HCPT model formed a higher average number of hydrogen bonds with PE lipids compared to GEM–CPT, while no substantial differences were observed with PS lipids. In particular, the highest number of hydrogen bonds in the DOX–HCPT nanocluster occurred at approximately −3 nm, corresponding to the global minimum binding energy, as depicted in Fig. S11.† For smaller nanoclusters, the DOX–HCPT model similarly demonstrated hydrogen bonds with PE lipids that were stronger than those with GEM–CPT. Furthermore, hydrogen bonds involving PS lipids were observed exclusively in the DOX–HCPT model and were absent in the GEM–CPT model, as shown in Fig. S12.† These findings highlight that the enhanced π–π stacking structure of DOX–HCPT nanoclusters facilitates more efficient drug uptake by cells compared to the steric self-assembly of GEM–CPT, emphasising the critical role of structural design in optimising drug delivery systems.
3.4 Impact of co-delivery on membrane deformation
The physicochemical and structural characteristics of a membrane result in significant variations in its deformation and drug-delivery attributes. Initially, the mass density profile of the cancer membrane and the large GEM–CPT nanocluster exhibited a balanced distribution of lipids, cholesterol, and water, with no water molecules detected inside the membrane at the beginning of the simulation. By 16 ns, notable shifts in lipid density peaks were observed with the GEM–CPT nanocluster, indicating that it disrupts the membrane structure and facilitates lipid rearrangement. In addition, the presence of water molecules within the membrane was observed, as illustrated in Fig. 7(a). MD simulations showed that the hydrophilicity of the GEM–CPT nanocluster attracted water molecules, drawing them in as the nanocluster moved through the cancer membrane, as shown in Fig. 7(b). In contrast, the DOX–HCPT nanocluster did not cause water infiltration during its penetration, as shown in Fig. 7(c). The hydrophobic nature of DOX–HCPT repelled nearby water molecules, discouraging their entry into the membrane and resulting in only slight membrane disturbance, indicated by stable lipid density and relatively shallow insertion, as reflected in Fig. 7(d). | ||
| Fig. 7 Mass density profiles of main lipid groups for (a) large GEM–CPT and (c) large DOX–HCPT nanoclusters interacting with the cancer membrane at different simulation times. Snapshots of solvent molecules during the translocation of (b) GEM–CPT and (d) DOX–HCPT nanoclusters across the cancer membrane. | ||
In the examination of large drug models that invade the normal membrane, no water molecules were identified within the membrane, as demonstrated by the MD snapshots in Fig. S13(a) and (b).† Both large nanoclusters caused minimal disturbance to the membrane, the mass density profile indicating a lack of water, and slight alterations in the lipid profiles after drug translocation (occurring at 18 ns for GEM–CPT and 17 ns for DOX–HCPT), as illustrated in Fig. S13(c) and (d).† Likewise, during the passage of small drug models through the cancer membrane, the hydrophobic core remained devoid of water, as shown in Fig. S14(a) and (b).† However, water was located near the entry points, aligning with membrane restructuring and notable interference. After drug translocation, an asymmetry in the lipid profile was observed, depicted in Fig. S14(c) and (d).† In contrast, the small GEM–CPT nanocluster enabled water infiltration into the membrane's interior, while the small DOX–HCPT nanocluster resulted in water concentration at the penetration site, as shown in Fig. S15(a) and (b).† These findings were supported by the mass density profile, which revealed water in the centre of the membrane for both small nanoclusters, as shown in Fig. S15(c) and (d).† Consequently, drug attributes such as size, charge, and amphiphilicity are crucial in defining the mechanisms of translocation and membrane permeability related to water infiltration.
Cholesterol molecules, crucial for maintaining membrane stability, were observed outside the membrane during drug administration. For larger drug models that traverse the cancer cell membrane, CHL molecules remained consistently external to the membrane. However, in normal membranes, the GEM–CPT nanocluster effectively extracted cholesterol molecules, a behaviour not observed with the DOX–HCPT nanocluster, as shown in Fig. S16.† In the case of smaller drug nanoclusters, cholesterol extraction occurred in both cancerous and normal membranes. This phenomenon is attributed to the high penetration potential of small nanoclusters and structural disturbances induced by TG interactions, which promote cholesterol removal, as shown in Fig. S17.† These findings highlight the structural adaptability of GEM–CPT nanoclusters, enabling stronger cholesterol extraction, while DOX–HCPT nanoclusters maintain their structural integrity with minimal cholesterol interaction.
4 Conclusions
This study provides a comprehensive molecular understanding of drug penetration through lipid membranes by examining amphiphilic drug–drug conjugates (GEM–CPT) and pure nanodrugs (DOX–HCPT) using molecular dynamics simulations. GEM–CPT demonstrated significant membrane penetration capabilities, effectively disrupting cancer cell membranes by extracting cholesterol and inducing structural alterations, particularly in the inner leaflet, although its flexible disulfide bond led to notable structural instability. In contrast, DOX–HCPT maintained structural integrity through stable π–π stacking interactions, allowing selective binding to HG regions and minimal interaction with cholesterol, particularly in normal membranes. Binding energy analysis revealed distinct interaction patterns: GEM–CPT exhibited stronger interactions with TG lipids due to its hydrophilic nature, whereas DOX–HCPT showed enhanced interactions with HG regions, driven by aromatic stacking and electrostatic complementarity. The size of the nanocluster also played a critical role, with larger clusters exhibiting steric-driven assembly and higher binding fluctuations in cancer membranes, while smaller clusters showed a higher penetration potential but greater structural deformation. Hydration dynamics further distinguished the two systems; GEM–CPT attracted water molecules during penetration, enhancing hydrophilic accessibility, while DOX–HCPT repelled water due to its hydrophobicity. These findings underscore the distinct interaction mechanisms of hydrophilic GEM–CPT and hydrophobic DOX–HCPT with lipid membranes, emphasising their respective advantages in disrupting cancerous membranes or maintaining stability in normal membranes. This study offers valuable insights for the design of nanocarriers to improve co-delivery efficiency, target specificity, and therapeutic efficacy.Data availability
Because of the extensive amount of data associated with the calculation results in this paper, it is not feasible to upload all the data to a public network. However, the relevant calculation data supporting the findings of this study are available within the article and its ESI.† Raw data supporting the findings of this study are available from the corresponding author upon reasonable request.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could influence the integrity and objectivity of the findings presented in this article.Acknowledgements
This work was supported by the Research of Khon Kaen University (RP68-3-002). CC would also like to express our gratitude to the Science Achievement Scholarship of Thailand for financial support.Notes and references
- F. Jing, Q. Guo, W. Xu, H. Qu and Z. Sui, Bioorg. Med. Chem. Lett., 2018, 28, 826–830 CrossRef PubMed.
- V. T. Devita Jr, R. C. Young and G. P. Canellos, Cancer, 1975, 35, 98–110 CrossRef.
- C. He, Z. Tang, H. Tian and X. Chen, Adv. Drug Delivery Rev., 2016, 98, 64–76 CrossRef PubMed.
- S.-Y. Qin, A.-Q. Zhang, S.-X. Cheng, L. Rong and X.-Z. Zhang, Biomater. Sci, 2017, 112, 234–247 CrossRef PubMed.
- Q. Song, X. Wang, Y. Wang, Y. Liang, Y. Zhou, X. Song, B. He, H. Zhang, W. Dai and X. Wang, et al., Mol. Pharmaceutics, 2016, 13, 190–201 CrossRef PubMed.
- C. Gao, P. Bhattarai, M. Chen, N. Zhang, S. Hameed, X. Yue and Z. Dai, Bioconjugate Chem., 2018, 29, 3967–3981 CrossRef PubMed.
- P. Huang, D. Wang, Y. Su, W. Huang, Y. Zhou, D. Cui, X. Zhu and D. Yan, J. Am. Chem. Soc., 2014, 136, 11748–11756 CrossRef PubMed.
- M. Sun, Q. Qian, L. Shi, L. Xu, Q. Liu, L. Zhou, X. Zhu, J.-M. Yue and D. Yan, Sci. China:Chem., 2020, 63, 35–41 CrossRef CAS.
- M. Hou, P. Xue, Y.-E. Gao, X. Ma, S. Bai, Y. Kang and Z. Xu, Biomater. Sci., 2017, 5, 1889–1897 RSC.
- S. Fu, G. Li, W. Zang, X. Zhou, K. Shi and Y. Zhai, Acta Pharm. Sin. B, 2022, 12, 92–106 CrossRef CAS.
- M. N. Holme, I. A. Fedotenko, D. Abegg, J. Althaus, L. Babel, F. Favarger, R. Reiter, R. Tanasescu, P.-L. Zaffalon and A. Ziegler, et al., Nat. Nanotechnol., 2012, 7, 536–543 CrossRef CAS PubMed.
- C.-M. J. Hu and L. Zhang, Biochem. Pharmacol., 2012, 83, 1104–1111 CrossRef CAS PubMed.
- L. Feng, M. Gao, D. Tao, Q. Chen, H. Wang, Z. Dong, M. Chen and Z. Liu, Adv. Funct. Mater., 2016, 26, 2207–2217 CrossRef CAS.
- Q. Liu, J. Zhang, W. Sun, Q. R. Xie, W. Xia and H. Gu, Int. J. Nanomed., 2012, 999–1013 CAS.
- S. Shirazi-Fard, A. R. Zolghadr and A. Klein, New J. Chem., 2023, 47, 22063–22077 RSC.
- M. Ibrahim, W. H. Abuwatfa, N. S. Awad, R. Sabouni and G. A. Husseini, Pharm., 2022, 14, 254 CAS.
- F. Chen, Y. Zhao, Y. Pan, X. Xue, X. Zhang, A. Kumar and X.-J. Liang, Mol. Pharm., 2015, 12, 2237–2244 CrossRef CAS PubMed.
- R. Zhang, X. Qin, F. Kong, P. Chen and G. Pan, Drug Delivery, 2019, 26, 328–342 CrossRef CAS PubMed.
- A. Preetha, R. Banerjee and N. Huilgol, et al., J. Cancer Res. Ther., 2005, 1, 180–186 CrossRef CAS PubMed.
- P. P. Hsu and D. M. Sabatini, Cell, 2008, 134, 703–707 CrossRef CAS PubMed.
- C. R. Santos and A. Schulze, FEBS J., 2012, 279, 2610–2623 CrossRef CAS.
- J. V. Swinnen, F. Vanderhoydonc, A. A. Elgamal, M. Eelen, I. Vercaeren, S. Joniau, H. Van Poppel, L. Baert, K. Goossens and W. Heyns, et al., Int. J. Cancer, 2000, 88, 176–179 CrossRef CAS.
- F. P. Kuhajda, Cancer Res., 2006, 66, 5977–5980 CrossRef CAS PubMed.
- N. Azordegan, V. Fraser, K. Le, L. M. Hillyer, D. W. Ma, G. Fischer and M. H. Moghadasian, Mol. Cell. Biochem., 2013, 374, 223–232 CrossRef CAS PubMed.
- T. E. Merchant, J. N. Kasimos, T. Vroom, E. de Bree, J. L. Iwata, P. W. de Graaf and T. Glonek, Cancer Lett., 2002, 176, 159–167 CrossRef CAS PubMed.
- P. V. Escriba, A. V. Ferrer-Montiel, J. A. Ferragut and J. M. Gonzalez-Ros, Biochem., 1990, 29, 7275–7282 CrossRef CAS.
- G. van Meer, Cold Spring Harbor Perspect. Biol., 2011, 3, a004671 Search PubMed.
- G. Van Meer, D. R. Voelker and G. W. Feigenson, Nat. Rev. Mol. Cell Biol., 2008, 9, 112–124 CrossRef CAS PubMed.
- T. Rivel, C. Ramseyer and S. Yesylevskyy, Sci. Rep., 2019, 9, 1–14 CrossRef PubMed.
- R. Hirano, T. Kagamiya, Y. Matsumoto, T. Furuta and M. Sakurai, Biochem. Biophys. Rep., 2021, 25, 100913 Search PubMed.
- E. Lindahl, M. Abraham, B. Hess and D. Van der Spoel, GROMACS Development Team: Stockholm, Sweden, 2020 Search PubMed.
- D. A. Case, T. E. Cheatham III, T. Darden, H. Gohlke, R. Luo, K. M. Merz Jr., A. Onufriev, C. Simmerling, B. Wang and R. J. Woods, J. Comput. Chem., 2005, 26, 1668–1688 CrossRef PubMed.
- R. Salomon-Ferrer, D. A. Case and R. C. Walker, Wiley Interdiscip. Rev.: Comput. Mol. Sci., 2013, 3, 198–210 Search PubMed.
- W. Phanchai, J. Thonghlueng and T. Puangmali, ACS Appl. Nano Mater., 2024, 7, 5554–5563 CrossRef.
- P. Toomjeen, W. Phanchai, C. Choodet, A. Chompoosor, R. Thanan, C. Sakonsinsiri and T. Puangmali, J. Phys. Chem. B, 2019, 123, 1129–1138 CrossRef PubMed.
- K. L. Rhinehardt, G. Srinivas and R. V. Mohan, J. Phys. Chem. B, 2015, 119, 6571–6583 CrossRef CAS PubMed.
- M. Rezaeisadat, A.-K. Bordbar and R. Omidyan, J. Mol. Liq., 2021, 332, 115862 CrossRef CAS.
- P. Stipa, S. Marano, R. Galeazzi, C. Minnelli, G. Mobbili and E. Laudadio, Eur. Polym. J., 2021, 147, 110292 CrossRef CAS.
- A. Kabedev, S. Hossain, M. Hubert, P. Larsson and C. A. Bergström, J. Pharm. Sci., 2021, 110, 176–185 CrossRef CAS PubMed.
- U. Srikulwong, W. Phanchai, P. Srepusharawoot, C. Sakonsinsiri and T. Puangmali, J. Phys. Chem. B, 2021, 125, 6697–6708 CrossRef CAS PubMed.
- W. Phanchai, U. Srikulwong, A. Chuaephon, S. Koowattanasuchat, J. Assawakhajornsak, R. Thanan, C. Sakonsinsiri and T. Puangmali, ACS Appl. Nano Mater., 2022, 5, 9042–9052 CrossRef CAS.
- W. L. Jorgensen, J. Chandrasekhar, J. D. Madura, R. W. Impey and M. L. Klein, J. Chem. Phys., 1983, 79, 926–935 CrossRef CAS.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38 CrossRef CAS PubMed.
- J. Wang, W. Wang, P. A. Kollman and D. A. Case, J. Am. Chem. Soc., 2001, 222, 2001 Search PubMed.
- S. Jo, T. Kim, V. G. Iyer and W. Im, J. Comput. Chem., 2008, 29, 1859–1865 CrossRef CAS PubMed.
- J. Lee, D. S. Patel, J. Ståhle, S.-J. Park, N. R. Kern, S. Kim, J. Lee, X. Cheng, M. A. Valvano and O. Holst, et al., J. Chem. Theory Comput., 2018, 15, 775–786 CrossRef PubMed.
- A.-R. Allouche, J. Comput. Chem., 2011, 32, 174–182 CrossRef CAS PubMed.
- D. L. Daleke, Curr. Opin. Hematol., 2008, 15, 191–195 CrossRef CAS PubMed.
- A. C. Alves and D. Ribeiro and Nunes, Biochim. Biophys. Acta, Biomembr., 2016, 1858, 2231–2244 CrossRef CAS PubMed.
- T. J. Desai, J. E. Toombs, J. D. Minna, R. A. Brekken and D. G. Udugamasooriya, Oncotarget, 2016, 7, 30678 CrossRef PubMed.
- B. Park, Biophys. J., 2024, 123, 372a CrossRef.
- F. Włodek, W. Kulig and A. Stachowicz-Kuśnierz, Biochim. Biophys. Acta, 2024, 1866, 184327 CrossRef.
- C. A. Lochbaum, A. K. Chew, X. Zhang, V. Rotello, R. C. Van Lehn and J. A. Pedersen, ACS Nano, 2021, 15, 6562–6572 CrossRef CAS PubMed.
- T. Lunnoo, J. Assawakhajornsak and T. Puangmali, J. Phys. Chem. C, 2019, 123, 3801–3810 CrossRef.
- P. A. Kollman, I. Massova, C. Reyes, B. Kuhn, S. Huo, L. Chong, M. Lee, T. Lee, Y. Duan and W. Wang, et al., Acc. Chem. Res., 2000, 33, 889–897 CrossRef PubMed.
- D. Greene, R. Qi, R. Nguyen, T. Qiu and R. Luo, J. Chem. Inf. Model., 2019, 59, 3041–3056 CrossRef PubMed.
- R. Kumari, R. Kumar, O. S. D. D. Consortium and A. Lynn, J. Chem. Inf. Model., 2014, 54, 1951–1962 CrossRef PubMed.
- K. Sztandera, M. Gorzkiewicz, E. A. Zizzi, N. Dybczak, L. Poltorak, M. A. Deriu and B. Klajnert-Maculewicz, Bioelectrochemistry, 2023, 152, 108449 CrossRef PubMed.
- A. Kumar, B. Mishra, A. D. Konar, E. Mylonakis and A. Basu, J. Phys. Chem. B, 2024, 128, 6049–6058 CrossRef PubMed.
- Z. Cao, L. Liu, G. Hu, Y. Bian, H. Li, J. Wang and Y. Zhou, Biochim. Biophys. Acta, 2020, 1862, 183402 CrossRef PubMed.
- E. Wang, H. Sun, J. Wang, Z. Wang, H. Liu, J. Z. Zhang and T. Hou, Chem. Rev., 2019, 119, 9478–9508 CrossRef PubMed.
- N. Homeyer and H. Gohlke, Mol. Inf., 2012, 31, 114–122 CrossRef PubMed.
- Y. Xu, Y. Huang, X. Zhang, W. Lu, J. Yu and S. Liu, Int. J. Pharm., 2018, 550, 45–56 CrossRef PubMed.
- R. Menichetti, K. H. Kanekal and T. Bereau, ACS Cent. Sci., 2019, 5, 290–298 CrossRef PubMed.
- P. K. Tang, K. Chakraborty, W. Hu, M. Kang and S. M. Loverde, J. Chem. Theory Comput., 2020, 16, 3373–3384 CrossRef PubMed.
- C. H. Tse, J. Comer, Y. Wang and C. Chipot, J. Chem. Theory Comput., 2018, 14, 2895–2909 CrossRef PubMed.
- P. Siani, E. Donadoni, L. Ferraro, F. Re and C. Di Valentin, Biochim. Biophys. Acta, 2022, 1864, 183763 CrossRef PubMed.
- Y. Zhao, Y. Zhao, Q. Ma, B. Sun, Q. Wang, Z. Ding, H. Zhang, X. Chu, M. Liu and Z. Wang, et al., Int. J. Nanomed., 2019, 8665–8683 CrossRef PubMed.
- R. Dawaliby, C. Trubbia, C. Delporte, C. Noyon, J.-M. Ruysschaert, P. Van Antwerpen and C. Govaerts, J. Biol. Chem., 2016, 291, 3658–3667 CrossRef PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra00480b |
| This journal is © The Royal Society of Chemistry 2025 |