
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNi–Co bimetallic phosphide catalyst toward electrocatalytic ammonia synthesis under ambient conditions†
Dongnan Zhao‡
ab,
Zhixian Mao‡bc,
Shengbo Zhang *bc,
Min Liub,
Kui Hub,
Daopeng Libc,
Zhengguo Qub,
Li Zhoubc and
Tongfei Shi*bc
*bc,
Min Liub,
Kui Hub,
Daopeng Libc,
Zhengguo Qub,
Li Zhoubc and
Tongfei Shi*bc
aSchool of Energy Materials and Chemical Engineering, Hefei University, Hefei 230601, China
bKey Laboratory of Materials Physics, Centre for Environmental and Energy Nanomaterials, Anhui Key Laboratory of Nanomaterials and Nanotechnology, CAS Center for Excellence in Nanoscience Institute of Solid State Physics, HFIPS, Chinese Academy of Sciences, Hefei 230031, China. E-mail: tfshi@issp.ac.cn; shbzhang@issp.ac.cn
cUniversity of Science and Technology of China, Hefei 230026, China
First published on 3rd April 2025
Abstract
Electrocatalytic nitrate reduction reaction (NitRR) under ambient conditions is a promising sustainable and eco-friendly method for ammonia (NH3) synthesis, which currently highly relies on the energy-consuming Haber–Bosch process with enormous CO2 emissions. In this work, we report the synthesis of a Ni–Co bimetallic phosphide catalyst (NiCoP) using the traditional hydrothermal combined high-temperature phosphorization method. Compared with monometallic phosphides such as Ni2P and CoP, the as-synthesized NiCoP catalyst with synergistic effects exhibits remarkable NitRR performance with the highest faradaic efficiency (FE) of 91.3 ± 4.4% at −1.2 V (vs. RHE) with the maximum NH3 yield rate of 5553.4 ± 400.8 μg h−1 cm−2 at −1.4 V (vs. RHE). Further in situ different electrochemical mass spectrometry (DEMS) analysis is employed to identify the intermediate produced during the electrocatalytic NitRR process, confirming NiCoP as a promising electrocatalyst for NH3 synthesis.
Ammonia (NH3) has a wide range of applications in various industries, including dyes, synthetic fibers, and nitric acid production.1 The Haber–Bosch process is the conventional approach employed in the industrial synthesis of NH3.2,3 This approach consumes substantial amounts of energy while generating significant quantities of CO2, which hinders the achievement of “dual carbon” goals encompassing peak carbon emissions followed by carbon neutrality.4 Therefore, there is an urgent need to discover a new efficient clean synthesis method for NH3 synthesis at this stage.5 Currently, there are several emerging research directions in the field of NH3 synthesis.6,7 The primary areas of focus include electrochemical nitrogen/nitrate reduction reaction (NitRR/NRR).8 Among these, NRR benefits from abundant nitrogen sources. However, the formidable challenge lies in breaking the strong triple bond of N
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) N due to its high dissociation energy, compounded by N2's limited solubility in water.9 These constraints significantly impede both production yield and faradaic efficiency (FE) of NH3 synthesis. On the other hand, NitRR overcomes the limitation posed by low solubility of N2 in aqueous environments. Furthermore, compared to the dissociation energy (941 kJ mol−1) required for breaking the triple bond in NN, nitrate reduction benefits from a lower dissociation energy (204 kJ mol−1) for breaking the double bond in N
N due to its high dissociation energy, compounded by N2's limited solubility in water.9 These constraints significantly impede both production yield and faradaic efficiency (FE) of NH3 synthesis. On the other hand, NitRR overcomes the limitation posed by low solubility of N2 in aqueous environments. Furthermore, compared to the dissociation energy (941 kJ mol−1) required for breaking the triple bond in NN, nitrate reduction benefits from a lower dissociation energy (204 kJ mol−1) for breaking the double bond in N![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O during catalytic reactions facilitated by electrochemical processes.10,11
O during catalytic reactions facilitated by electrochemical processes.10,11
Transition-metal phosphides (TMPs) have recently emerged as one type of earth-abundant electrocatalyst with impressive activity.12,13 Considerable attention has been paid to these types of catalysts due to their excellent electronic conductivity, hybrid orbitals, and low cost.14 Additionally, in the process of forming transition metal phosphides, with the relatively large radius of phosphorus atoms, stable interstitial compounds are challenging to form. Therefore, phosphorus atoms occupy internal structural units in metals through radius filling, exposing more coordination unsaturated surface atoms.15 An increased number of active sites leads to enhanced catalytic activity. For instance, Ni2P with (111) crystal plane grown in situ on nickel foam (NFP) serves as a high-performance catalyst for NitRR,16 exhibiting a NH3 yield rate (RNH3) of 0.056 mmol h−1 mg−1 and a FE reaching up to 99.23%.17 Besides, Sun and co-workers reported a Co1-P/NPG catalyst and demonstrated the Co atoms serve as the main NO3− activation sites,18 while the P atoms can regulate the distribution of water at the catalyst's surface and simultaneously promote water dissociation, accelerating the proton-coupled electron transfer (PCET) kinetics of NitRR to achieve a RNH3 of 8.6 mg h−1 mg−1 with a FE of 93.8%.19 These findings hold significant implications for electrocatalysis research focused on studying TMPs.
Inspired by the aforementioned work, we successfully synthesized a Ni–Co bimetallic phosphide catalyst (NiCoP) for the electrocatalytic reduction of nitrate to produce NH3. Compared with monometallic phosphides such as Ni2P and CoP, NiCoP exhibited enhanced performance in NH3 synthesis. As a result, the as-synthesized NiCoP affords an excellent RNH3 of 5553.4 ± 400.8 μg h−1 cm−2 at −1.4 V (vs. RHE) and a maximum FE of 91.3 ± 4.4 at −1.2 V (vs. RHE), with outstanding stability during 10 h NitRR. The 15N isotope labeling experiments confirmed that the produced NH3 indeed originated from the nitrate reduction reaction catalyzed by NiCoP.
Fig. 1a illustrates the synthetic process of the NiCoP catalyst using the traditional hydrothermal combined high-temperature phosphorization method. For meaningful comparison, Ni2P and CoP were also fabricated using similar synthetic method (Fig. S1, ESI†). The X-ray diffraction (XRD) pattern of the synthesized NiCoP sample is presented in Fig. 1b. The observed peaks at 2θ = 40.9°, 44.9°, 47.6° and 54.4° correspond to the crystal planes (111), (201), (210) and (300) of NiCoP (JCPDS Card No. 04-001-6153),20 respectively. The high-resolution transmission electron microscopy (HR-TEM) image shown in Fig. 1c reveals a lattice spacing approximately equal to 0.21 nm corresponding to the crystal plane (111) of NiCoP as consistent with XRD results. The high-angle annular dark-field scanning transmission electron microscopy (HAADF-STEM) along with elemental mapping images (Fig. 1d and e), further corroborate the homogeneous distribution of P, O, Ni and Co elements over the entire sample, further confirming the formation of NiCoP.
 | ||
| Fig. 1 (a) Schematic diagram illustrating the synthetic procedure of NiCoP. (b) XRD pattern and (c) TEM image (inset is the HR-TEM image) of NiCoP. (d) HAADF-STEM image and (e) corresponding elemental mapping images of NiCoP. | ||
The as-synthesized NiCoP sample exhibits favorable mesoporous and microporous structures (Fig. S2, ESI†), which is beneficial for exposuring of more active sites and facilitates charge transfer during electrocatalytic processes.21 Surface composition and oxidation states of NiCoP were investigated using X-ray photoelectron spectroscopy (XPS) (Table S1, ESI†). Fig. 2a displays the XPS spectrum obtained from the surface measurement, indicating the presence of Ni, Co, O, and P elements. Fig. 2b shows the high-resolution XPS spectra of Ni 2p, where Ni 2p3/2 peaks at 853.3 eV and 856.8 eV correspond to Ni2+ and Ni3+, respectively;22 while peaks at 870.6 eV and 874.7 eV belong to Ni2+ and Ni3+ in the case of Ni 2p1/2;23 the satellite peaks are observed at binding energies around 880.1 eV and 863.5 eV in combination with 859.5 eV.24 The high-resolution XPS spectra of Co 2p is depicted in Fig. 2c, wherein the Co 2p3/2 peak encompasses both Co3+ (778.8 eV) and Co2+ (782.0 eV), while the peaks at 793.8 eV for Co3+ and 797.9 eV for Co2+ correspond to the Co 2p1/2 state. The peaks observed at 785.3 eV and 802.8 eV are identified as satellite peaks. In Fig. 2d, a P–O peak is discernible at a binding energy of 134.1 eV, which likely originates from phosphate compounds,25 whereas the peak at 129.6 eV corresponds to P 2p3/2.26 The above XPS results further confirm the formation of NiCoP after phosphorization treatment. Furthermore, we characterized Ni2P and CoP using XPS spectroscopy (Fig. S3, ESI†). In comparison with CoP, NiCoP exhibits Co3+ species, which offer additional empty d orbitals. This characteristic enhances the affinity for Hads, thereby promoting NitRR.
 | ||
| Fig. 2 (a) Surface survey XPS spectrum of NiCoP. High-resolution (b) Ni 2p, (c) Co 2p, and (d) P 2p spectra of NiCoP. | ||
The electrochemical NitRR performance of the as-synthesized NiCoP was investigated under ambient temperature and pressure in a standard three-electrode H-type cell. The linear sweep voltammetry (LSV) curves in Ar-saturated 0.1 M Na2SO4 electrolyte with or without NO3− were first measured to examine the catalytic responsiveness of NO3−. As depicted in Fig. 3a, when NO3− was present in the electrolyte solution, the increase of current density is due to the excellent correspondence of the NiCoP catalyst to NO3− at the reduction potential. Constant potential electrolysis ranging from −0.8 V to −1.4 V (vs. RHE) were selected to further quantitatively assess NH3 electrosynthesis activity. The time dependent current density curves recorded for 2 h at various potentials illustrate the superior stability of NiCoP (Fig. S4, ESI†). In this study, the main product NH3 and by-product NO2− and NH3 were quantified by UV-vis spectrophotometry with calibration curves (Fig. S5 and S6, ESI†). As shown in Fig. 3b, the catalytic NiCoP catalyst result in remarkable NH3 yield rate (RNH3) of 5553.4 ± 400.8 μg h−1 cm−2 at −1.4 V (vs. RHE), and the superior faradaic efficiency (FE) of 91.3 ± 4.4% at −1.2 V (vs. RHE), superior to the most reported NitRR electrocatalysts (Table S2 and Fig. S7, ESI†). Both the RNH3 and the FE were times higher than those of Ni2P (RNH3: 1807.2 μg h−1 cm−2; FE: 54.6%) and CoP (RNH3: 3600.1 μg h−1 cm−2; FE: 90.3%) (Fig. S8, ESI†), which is attributed to synergistic effect of Ni–Co bimetallic catalyst.27
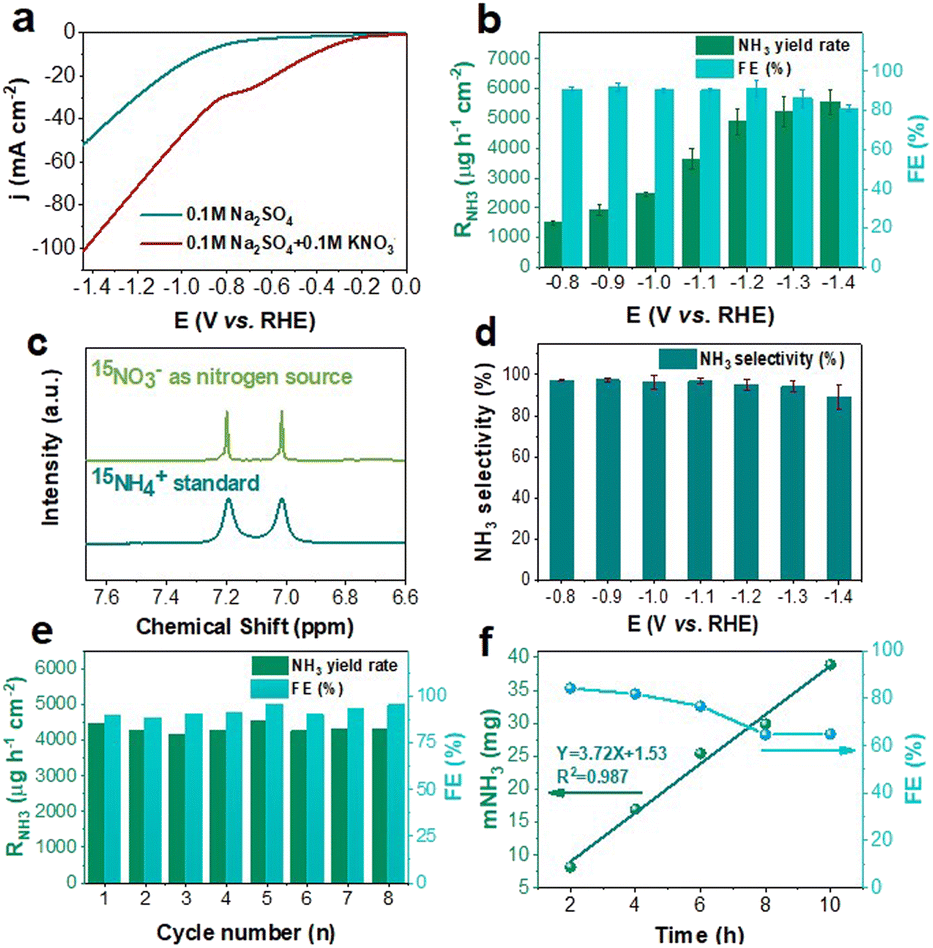 | ||
| Fig. 3 (a) LSV curves of NiCoP catalyst in 0.1 M Na2SO4 and 0.1 M Na2SO4 + 0.1 M KNO3 solution. (b) RNH3 and FE of NiCoP catalyst obtained at different potentials for 2 h NitRR measurement. (c) 1H NMR spectra of NiCoP catalyst using 15NO3− as nitrogen source for NitRR and standards (15NH4)2SO4. (d) SNH3 of NiCoP catalyst obtained at different potentials. (e) Recycling tests for NiCoP catalyst during NitRR at −1.2 V (vs. RHE). (f) The FE and NH3 yield were monitored over a 10 h NitRR conducted at −1.2 V (vs. RHE). | ||
In addition, LSV tests were also conducted on Ni2P and CoP (Fig. S9, ESI†). At the same potential, the current densities of Ni2P and CoP were both lower than that of NiCoP. These experimental results further substantiate the aforementioned conclusion. To verify the source of ammonium, 15N isotope labeling experiments are conducted.28 As shown in Fig. 3c, the 1H nuclear magnetic resonance (NMR) spectra of electrolyte adopting 15NO3− as reactants show typical double peaks of 15NH4+. In addition, we also conducted other representative control experiments, including: (1) open circuit potential (OCP), (2) 0.1 M Na2SO4 electrolyte without NO3− and (3) blank carbon paper (CP) without NiCoP catalyst (Fig. S10, ESI†). The control experiments can effectively diagnose possible errors caused by environmental influences including raw material, electrolyte, catalyst decomposition, and other factors. Obviously, the extremely low absorbance obtained by the indophenol blue method confirms the validity of our experimental process and results (Fig. S11, ESI†).
Moreover, due to the low levels of NO2− detected (<0.056 mM) after NitRR at all applied potentials (Fig. S5, ESI†), the selectivity of NH3 (SNH3) of NiCoP at different applied potentials was calculated as displayed in Fig. 3d. The SNH3 of NiCoP can reach the maximum FE of 97.4 ± 0.8% at −0.9 V (vs. RHE) and excellent FE of NH3 over 95% under a broad range from −0.8 to −1.2 V (vs. RHE) (Fig. 3d), indicating the high NitRR-to-NH3 selectivity of NiCoP. Notably, when the applied potential exceeds −1.2 V (vs. RHE), the RNO2− increases, while both the FE and SNH3 of NiCoP decrease significantly, primarily due to the occurrence of competitive hydrogen evolution reaction (HER) on NiCoP.29,30 Besides superior activity, the durability of the catalyst is another crucial parameter for practical applications. As shown in Fig. 3e, both NH3 yields and FEs have no obvious fluctuation during 8 recycling tests at −1.2 V (vs. RHE), confirming the high stability of NiCoP toward electrochemical NitRR. The corresponding UV-vis absorption spectra and current density curves are displayed in Fig. S12 (ESI†). Additionally, the superior durability of NiCoP for 10 h at −1.2 V (vs. RHE) was confirmed by chronoamperometry measurements (Fig. S13, ESI†). The corresponding NH3 yield and time show a good linear relationship, with a slight decrease in FE (Fig. 3f). The LSV test was conducted on the reacted NiCoP (Fig. S14, ESI†), and its LSV curve basically coincides with that of the pre-reaction sample, further demonstrating the excellent stability of NiCoP. To determine the morphological and structural stability, careful examination of XRD, XPS and SEM characterizations on NiCoP after NitRR test (Fig. S15–S17, ESI†) confirms the high structural stability of NiCoP.
To further investigate the influence mechanism of the chemical microenvironment on catalytic activity, electrochemical impedance spectroscopy (EIS) and double-layer capacitance (DLC) tests were also performed (Fig. S18 and S19, ESI†). EIS measurements at various potentials revealed that NiCoP exhibits a lower charge transfer impedance at higher potentials, facilitating the efficient conversion of nitrate to ammonia under these conditions. The DLC test results for CoP, Ni2P, and NiCoP reveal that the electrochemical surface area (ECSA) of NiCoP lies between those of Ni2P and CoP. Despite having a lower ECSA compared to CoP, NiCoP exhibits superior NitRR performance. This demonstrates that the Ni–Co synergistic effect promotes the transformation of nitrate to ammonia. Furthermore, the in situ differential electrochemical mass spectrometry (DEMS) is employed for in situ detection of molecular intermediate and product over NiCoP.31 The gaseous molecules can be identified based on m/z values, and the relative quantities of intermediates and products can be estimated in real-time based on signal intensity.32 As shown in Fig. 4, the strongest m/z signals of 17 are recorded, which confirms the high NH3 selectivity of NiCoP. The signal intensity of the intermediate is significantly lower than that of NH3, indicating that NH3 is the main product of NiCoP catalyst. The in situ DEMS results are inconsistent with the UV-vis results. This result suggests that the main reaction pathway of NitRR on NiCoP catalyst may be NO3− → *NO3 → *NO2 → *NO → *N → *NH → *NH2 → *NH3 → NH3 (g).
 | ||
| Fig. 4 In situ DEMS measurements of NitRR over NiCoP. | ||
In conclusion, the Ni–Co bimetallic phosphide catalyst is experimentally proven as a superb NitRR electrocatalyst for ambient NH3 production with a large yield of 5553.4 ± 400.8 μg h−1 cm−2 at −1.4 V (vs. RHE) and a maximum FE of 91.3 ± 4.4 at −1.2 V (vs. RHE), respectively. The in situ DEMS analysis is employed to identify the intermediate produced during the electrocatalytic NitRR process, confirmed NiCoP as a promising electrocatalyst for NH3 synthesis. The results indicate that traditional hydrothermal combined high-temperature phosphorization can be considered as a promising method for the preparation of NitRR electrocatalysts. Meanwhile, this discovery promotes the application of transition-metal phosphides catalysts in room-temperature electrochemical reactions.
Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Dongnan Zhao: conceptualization, investigation, visualization, writing – original draft. Shengbo Zhang: data curation, resources, supervision, writing – review & editing. Kui Hu: investigation, resources, formal analysis. Daopeng Li: investigation, resources, formal analysis. Zhixian Mao: investigation, resources, formal analysis. Min Liu: software. Zhengguo Qu: software. Li Zhou: software. Tongfei Shi: funding acquisition, supervision, resources, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the Natural Science Foundation of Anhui Provincial Natural Science Foundation (Grant No. 2408085MB021).Notes and references
- H. Zhang, H. Wang, X. Cao, M. Chen, Y. Liu, Y. Zhou, M. Huang, L. Xia, Y. Wang, T. Li, D. Zheng, Y. Luo, S. Sun, X. Zhao and X. Sun, Adv. Mater., 2024, 36, e2312746 Search PubMed.
- J. Cao, Y. Hu, Y. Zheng, W. Zhang and B. Yu, Front. Energy, 2024, 18, 128–140 CrossRef.
- H. Liu, L. Bai, A. Bergmann, B. R. Cuenya and J. Luo, Chem, 2024, 10, 2963–2986 CAS.
- Y. Wang, C. Wang, M. Li, Y. Yu and B. Zhang, Chem. Soc. Rev., 2021, 50, 6720–6733 RSC.
- J. Martínez, A. Ortiz and I. Ortiz, Appl. Catal., B, 2017, 207, 42–59 Search PubMed.
- D. Liu, L. Qiao, S. Peng, H. Bai, C. Liu, W. F. Ip, K. H. Lo, H. Liu, K. W. Ng and S. Wang, Adv. Funct. Mater., 2023, 33, 2303480 Search PubMed.
- J. Theerthagiri, J. Park, H. T. Das, N. Rahamathulla, E. S. F. Cardoso, A. P. Murthy, G. Maia, D. V. N. Vo and M. Y. Choi, Environ. Chem. Lett., 2022, 20, 2929–2949 Search PubMed.
- O. Q. Carvalho, R. Marks, H. K. Nguyen, M. E. Vitale-Sullivan, S. C. Martinez, L. Árnadóttir and K. A. Stoerzinger, J. Am. Chem. Soc., 2022, 144, 14809–14818 Search PubMed.
- Z. Wu, Y. Song, Y. Liu, W. Luo, W. Li and J. Yang, Chem Catal., 2023, 3, 100786 Search PubMed.
- M. T. de Groot and M. T. M. Koper, J. Electroanal. Chem., 2004, 562, 81–94 Search PubMed.
- Y. Zhao, Y. Liu, Z. Zhang, Z. Mo, C. Wang and S. Gao, Nano Energy, 2022, 97, 107124 CrossRef CAS.
- W. Wen, S. Fang, Y. Zhou, Y. Zhao, P. Li and X. Y. Yu, Angew. Chem., Int. Ed., 2024, 63, e202408382 CAS.
- W. Zheng, L. Zhu, Z. Yan, Z. Lin, Z. Lei, Y. Zhang, H. Xu, Z. Dang, C. Wei and C. Feng, Environ. Sci. Technol., 2021, 55, 13231–13243 CAS.
- J. Zhou, S. Gao and G. Hu, Energy Fuels, 2024, 38, 6701–6722 CAS.
- Q. Yao, J. Chen, S. Xiao, Y. Zhang and X. Zhou, ACS Appl. Mater. Interfaces, 2021, 13, 30458–30467 Search PubMed.
- R. Zhang, Y. Guo, S. Zhang, D. Chen, Y. Zhao, Z. Huang, L. Ma, P. Li, Q. Yang and G. Liang, Adv. Energy Mater., 2022, 12, 2103872 CAS.
- Q. Yao, J. Chen, S. Xiao, Y. Zhang and X. Zhou, ACS Appl. Mater. Interfaces, 2021, 13, 30458–30467 CAS.
- J. Ni, J. Yan, F. Li, H. Qi, Q. Xu, C. Su, L. Sun, H. Sun, J. Ding and B. Liu, Adv. Energy Mater., 2024, 14, 2400065 CAS.
- J. Wu, J.-H. Li and Y.-X. Yu, J. Phys. Chem. Lett., 2021, 12, 3968–3975 CrossRef CAS.
- Y. Lin, Y. Pan, S. Liu, K. Sun, Y. Cheng, M. Liu, Z. Wang, X. Li and J. Zhang, Appl. Catal., B, 2019, 259, 118039 CrossRef CAS.
- H. Xie, D. Jiang, H. Chen, X. Ma, X. Liu, Q. Qi and Y. Wang, Nanoscale, 2023, 15, 7430–7437 RSC.
- Z. Zhou, L. Zeng, G. Xiong, L. Yang, H. Yuan, J. Yu, S. Xu, D. Wang, X. Zhang and H. Liu, Chem. Eng. J., 2021, 426, 129214 CrossRef CAS.
- T. Liu, A. Li, C. Wang, W. Zhou, S. Liu and L. Guo, Adv. Mater., 2018, 30, 1803590 CrossRef.
- S. Mahmood, M. S. Riaz, M. Ammar, Z. Wang, M. J. Iqbal, G. A. Ashraf, N. Afshan, N. Hassan, A. Bahadur and S. Iqbal, Int. J. Hydrogen Energy, 2024, 70, 315–324 CrossRef CAS.
- J. Liang, W.-F. Hu, B. Song, T. Mou, L. Zhang, Y. Luo, Q. Liu, A. A. Alshehri, M. S. Hamdy, L.-M. Yang and X. Sun, Inorg. Chem. Front., 2022, 9, 1366–1372 RSC.
- Y. Li, H. Zhang, M. Jiang, Y. Kuang, X. Sun and X. Duan, Nano Res., 2016, 9, 2251–2259 CAS.
- J. Wang, J. Feng, T.-O. Soyol-Erdene, Z. Wei and W. Tang, Sep. Purif. Technol., 2023, 320, 124155 CrossRef CAS.
- Y. Zhao, R. Shi, X. Bian, C. Zhou, Y. Zhao, S. Zhang, F. Wu, G. I. N. Waterhouse, L.-Z. Wu, C.-H. Tung and T. Zhang, Adv. Sci., 2019, 6, 1802109 CrossRef PubMed.
- Y. Zhang, X. Chen, W. Wang, L. Yin and J. C. Crittenden, Appl. Catal., B, 2022, 310, 121346 CrossRef CAS.
- Y. Wang, W. Zhou, R. Jia, Y. Yu and B. Zhang, Angew. Chem., Int. Ed., 2020, 59, 5350–5354 CrossRef CAS.
- W.-J. Sun, L.-X. Li, H.-Y. Zhang, J.-H. He and J.-M. Lu, ACS Sustain. Chem. Eng., 2022, 10, 5958–5965 CrossRef CAS.
- Y. Wang, W. Yu, X. Li, J. Yu and W. Zhou, Chem. Eng. J., 2023, 469, 143889 CrossRef CAS.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra00391a |
| ‡ These authors contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |