
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceFormation mechanism of anionic isotactic polystyrene initiated by Li carbanions in cyclohexane in the presence of Na-tosylate II: a DFT calculation study†
Tianwen Bai *a,
Jun Lingbc and
Thieo E. Hogen-Esch*b
*a,
Jun Lingbc and
Thieo E. Hogen-Esch*b
aCollege of Biological, Chemical Sciences and Engineering, Jiaxing University, Jiaxing 314001, China. E-mail: baitw@zjxu.edu.cn
bDepartment of Chemistry and Loker Hydrocarbon Research Institute, University of Southern California, Los Angeles, CA 90089-1661, USA. E-mail: hogenesc@gmail.com
cMOE Key Laboratory of Macromolecular Synthesis and Functionalization, Department of Polymer Science and Engineering, Zhejiang University, Hangzhou 310027, China
First published on 7th May 2025
Abstract
The stereochemistry of t-BuLi initiated styrene polymerization in cyclohexane yields isotactic-rich polystyrene in the presence of sodium 4-methylbenzenesulfonate (SMBS). Herein, we report the results of DFT calculations in cyclohexane, both in the presence and absence of SMBS, using the B3PW91 hybrid functional and 6-311++G(d,p) basis set with DFT-D3 correction, allowing for the detailed evaluation of the transition state structures confirmed by benchmark analysis. We focus on the addition of a single styrene to a 1-lithio-1,3(S)-diphenyl-pentane (LDPP) model dimer anion, producing the expected four stereoisomers of 1-lithio-1,3,5-triphenylheptane (LTPH). The addition proceeds through four styrene stereoisomeric complexes, consisting of two pseudo-enantiomeric sets with the corresponding transition states (TSs), which exhibit similar Gibbs free energy (≤0.1 kcal mol−1), in agreement with previously reported results. The geometries of the complexes and transition states are consistent with the major rotational (rather than lateral) motions of the aryl and vinyl groups as the complexes evolve into transition states and trimer anions. In the presence of SMBS, similar but higher energy monomer complexes are formed that surprisingly have nearly the same free energy. However, the TS free energy barriers increase in the following order: m-pro-m <r-pro-m ≅ m-pro-r <r-pro-r. For instance, the TS energies for monomer addition differ by as much as 1.4 kcal mol−1 for the r-pro-r compared to the m-pro-m transition states. However, due to the 1.9 kcal mol−1 lower free energy of the pro-r compared to the pro-m dimer anions, the corresponding activation energies differ by as much as 3.3 kcal mol−1. This would tend to favor the formation for mm triads over rr dyads by a factor of about 1.9 × 102, which is consistent with the prevailing isotactic stereochemistry.
Introduction
Stereochemistry plays an important role in determining the properties of many vinyl polymers, including polystyrene (PS), as highly isotactic and syndiotactic polystyrene (PS) forms exhibit high crystallinity, high melting points and outstanding mechanical properties.1–4 The stereochemistry of Group-3 and Group-4 single-site mechanisms has been reviewed.5Anionic styrene isotactic polymerization involving alkali metal complexes has been reported as early as 1960.6 However, the origin of this process was later attributed to the presence of impurities that are often present.7,8 Worsfold and Bywater reported that the presence of water significantly increased the isotactic content in PS, suggesting that the results were plausibly due to the presence of LiOH formed by adventitious water. Accordingly, Makino et al. demonstrated the synthesis of highly isotactic PS (with triad and pentad contents of 95% and 90%, respectively) at −60 °C, uncontaminated by significant quantities of stereoirregular PS. This was achieved by initiating the polymerization with 3,3-dimethyl-1,1-diphenyl-1-lithiobutane (DMPBL) in the presence of equimolar LiOH, generated in situ in hexane, at temperatures between −30 and −60 °C.9 In the absence of LiOH, only low contents of mm triads (11%) were observed.
Cazzaniga et al. also prepared fractions of isotactic polystyrene (iPS), along with stereoirregular PS at −30 °C using n-BuLi/lithium tert-butoxide (t-BuOLi) complexes.10 They also synthesized semicrystalline ABA type isotactic PS-polybutadiene triblock copolymers.11 The ratio of isotactic polystyrene to polybutadiene (iPS wt% ≥66%) in these copolymers resulted in various microphase-separated morphologies at a scale that is typical of diblock copolymers. However, the synthesis of isotactic PS block copolymers still requires fractionation of the isotactic polystyrene. More recent developments have shown that alkyllithium-alkoxide (RLi/ROMt) with (Mt![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) Li, Na, and K) and R2MgOR/KOR-initiated styrene polymerizations in methylcyclohexane at −40 °C gave isotactic-rich PS (mm contents as high as 85%).12
Li, Na, and K) and R2MgOR/KOR-initiated styrene polymerizations in methylcyclohexane at −40 °C gave isotactic-rich PS (mm contents as high as 85%).12
We have recently reported a polymerization of styrene initiated by t-BuLi in hexane or cyclohexane, in the presence of one or less equivalent of sodium dodecylbenzenesulfonate (SDBS), which produces isotactic-rich PS (mm triads and mmmm pentads at 77% and 51%, respectively) at ambient temperature or above with relatively narrow distributions (Mw/Mn = 1.1–1.4).13 Lower temperatures gave inferior results. The advantage of these anionic polymerizations is the formation of isotactic-rich PS at ambient temperatures and the potential to produce the corresponding isotactic PS block copolymers.
Previous DFT calculations using a B3LYP hybrid functional and 6-31G(d) basis set were carried out on 1-lithio-1,3(S)-diphenyl butane (LDPB) in vacuum or cyclohexane as a model for PSLi, and their complexes with sodium 4-methylbenzenesulfonate (SMBS) as a model for SDBS.13 The results suggested that in cyclohexane and similar solvents at ambient conditions, LDPB adequately models the stereochemical properties of PSLi. This includes the predominant formation of unreactive LDPB and PSLi dimers that are in equilibrium with a reactive monomeric LDPB (PSLi) in cyclohexane and similar hydrocarbons, in agreement with calculations by Yakimansky et al.13,14 Furthermore, the absence of spontaneous epimerization of the pro-chiral LDPB ion pair configuration (1-pro-m or 1-pro-r) (Scheme 1), or the formation and dissociation of the dimer has been shown to be consistent with these calculations.13,14
 | ||
| Scheme 1 Stereochemistry of the conversions of 1 into 4. | ||
The simulated reactions of 3(S)-LDPB with styrene also indicated a strongly preferred Li side (syn) monomer attack through coordination of the Li ion with styrene to give styrene complexes prior to monomer addition.13 Hence, the relative thermodynamic stabilities of the various pro-chiral Li ion pairs complexes are of interest. In the absence of SMBS, the formation of either 1-pro-S or 1-pro-r LDPB-styrene complexes were found, which burdens the Li ion coordinating either the pro-si or pro-re pro-chiral monomer, with the remaining Li ion coordinated to the penultimate 3-phenyl of LDPB.14 The relative free energies of four stereoisomeric LDPB-styrene complexes were consistent with the demonstrated slight preference for heterotactic and syndiotactic PS triads in the absence of SMBS.13,14 Thus, upon monomer addition, the relative ΔG values of formation of 2-m-pro-m, 2-m-pro-r, 2-r-pro-r and2-r-pro-m LDPB-styrene complexes were calculated as 0, -0.53, −1.23 and −0.85 kcal mol−1, respectively, consistent with the experimental data showing the preferred syndiotactic (rr) and heterotactic (mr) triads. In the presence of SMBS, however, these computations showed a strongly favored (ΔG ≈ −30 kcal mol−1) 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1 LDBP-SMBS complex by reaction of the LDPB dimer anion with SMBS.13 The simulated LDPB–SMBS styrene complexes show structures with the Li and Na ions both being close to the benzyl carbanion, with the Na ions being syn with respect to Li and close to the center of the 1-phenyl group. Dimerization of these complexes were shown to be absent. However, the calculations of the transition states corresponding to the monomer addition were inaccessible at the time;15 thus, any conclusions were considered to be tentative.
:1 LDBP-SMBS complex by reaction of the LDPB dimer anion with SMBS.13 The simulated LDPB–SMBS styrene complexes show structures with the Li and Na ions both being close to the benzyl carbanion, with the Na ions being syn with respect to Li and close to the center of the 1-phenyl group. Dimerization of these complexes were shown to be absent. However, the calculations of the transition states corresponding to the monomer addition were inaccessible at the time;15 thus, any conclusions were considered to be tentative.
Here, we report more advanced calculations (B3PW91/6-311++G(d,p)) methods to model the complexes of styrene with a slightly modified 1-lithio-1,3(S)-diphenylpentane (LDPP). The results of the calculations of the detailed structures of LDPP, LDPP-monomer complexes 2, transition 3, and trimer anions 4 in the absence or presence of SMBS are reported. In the absence of SMBS, the free energies of the four isomeric transition states were found to be quite similar, which is consistent with the formation of atactic stereochemistry. However, in the presence of SMBS, the transition state free energies for styrene addition to LDPP are consistent with a preference of isotactic-like polymerizations.
Results and discussion
LDPP
We start with a brief discussion of the model addition of the styrene monomer to the LDPBB precursor (rather than LDPB) that likewise had an arbitrary (3S) configuration (Scheme 1).The pro-si or pro-re styrene addition to the two pro-m or pro-r diastereomers of LDPP should give four m-pro-m, m-pro-r, r-pro-r and r-pro-m stereoisomers of 1-lithio-1,3,5-triphenylheptane 4 (LTPH) via the corresponding four stereoisomeric styrene complexes 2 and transition states 3 (Schemes 1–3). First, the results both with respect to atomic positions and dihedral angles are analyzed. We then report on the analogous reactions in the presence of sodium-4-methyl-benzenesulfonate (SMBS).
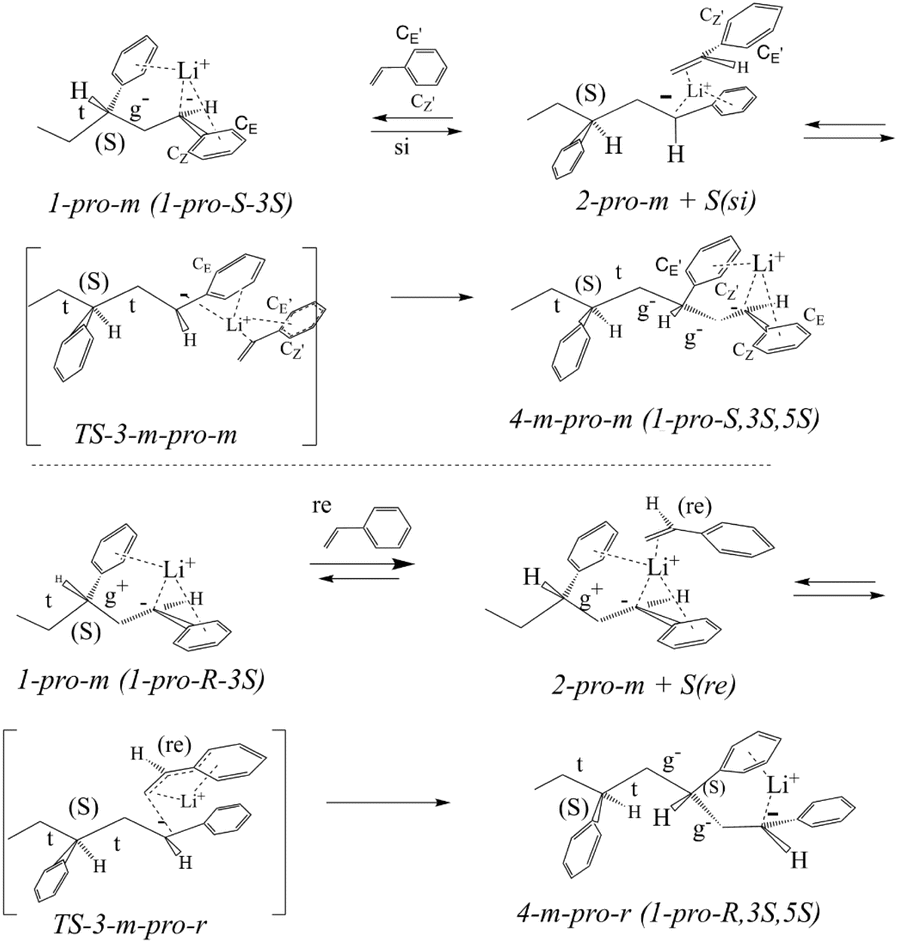 | ||
| Scheme 2 Syn addition of styrene to 1-pro-m LDPP at its pro-si or pro-re faces, giving the 4-m-pro-m or 4-m-pro-r trimer anion via monomer complexes 2, and transition states 3. | ||
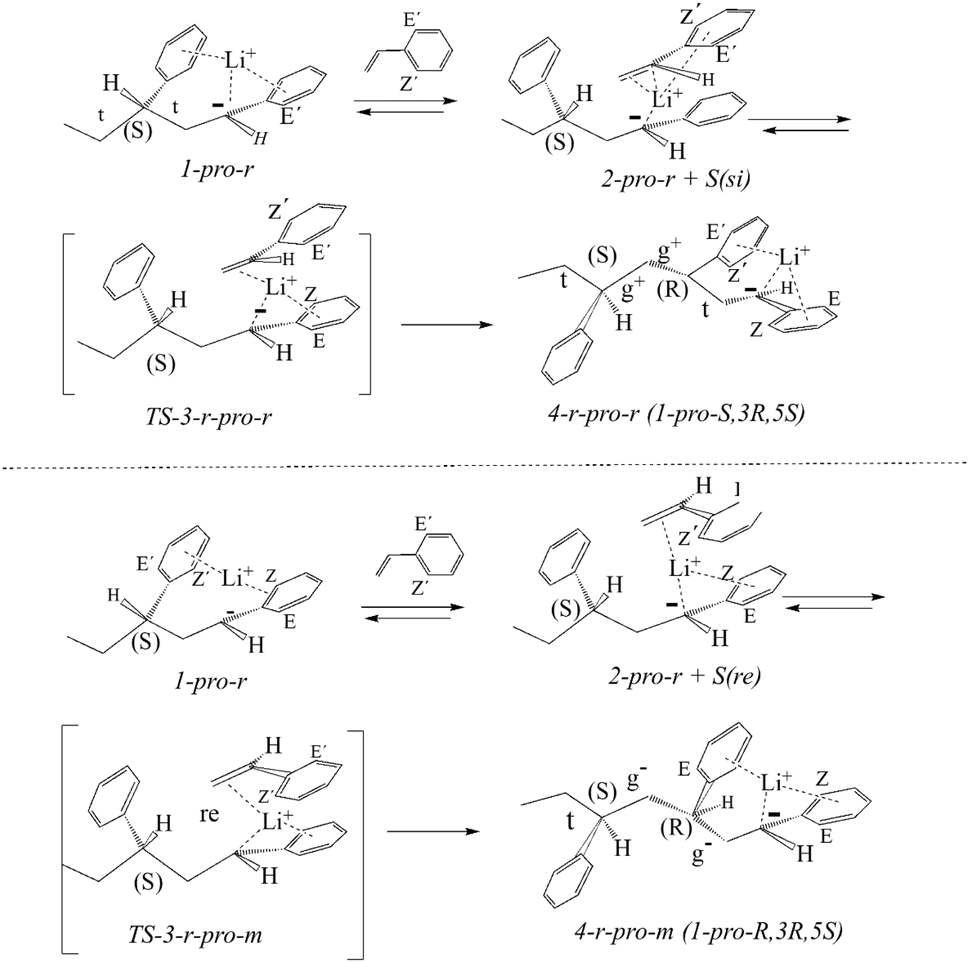 | ||
| Scheme 3 Syn addition of styrene to 1-pro-r LDPP at its pro-si or pro-re faces, giving the 4-r-pro-r [t g−g−t] or 4-r-pro-m trimer [t g−g−t] anions. | ||
Intermediates
The Cahn–Ingold–Prelog rules are modified with the following priorities: phenyl > Li > carbanion > chain, allowing for a convenient link between pro-chirality and configuration with the si and re monomer presentations leading to pro-S and pro-r carbanion configurations, respectively (Scheme 1).13 Along with the quaternary CQ carbon, the ortho (CZ and CE) carbons of LDPP, defined as being S-cis (Z) or S-trans (E) to the chain, remain the same as the corresponding carbons in the coordinating phenyl group (CQ′, CZ′ and CE′) in the styrene complexes 2, transition states 3 and products 4, as also indicated in Schemes 1–3. Thus, the CZ′ and CE′ carbons of the monomer in the Li-coordinated LDPP-monomer complexes result in the CZ and CE carbons in the transition states and LTPH products (Schemes 2 and 3).Dimer precursor model
The relative configurations of carbons 1 and 3 in LDPP can represent either a pro-meso pro-1S, 3S (1-pro-m) or a pro-racemic 1-pro-1R, 3S (1-pro-r) epimer.14 Upon conversion of the four isomeric transition states 3 into the corresponding trimer anions 4, the CQ′, CZ′ and CE′ aryl carbons of 2 and 3 are transformed into CQ, CZ and CE aryl carbons in carbanions 4-m-pro-m and 4-m-pro-r, respectively (Scheme 2 and 3 and Tables S1–S4†). In addition, the CZ and CE aryl carbons of 1-pro-m are transformed into the coordinating CZ′ and CE′ carbons of the 4-m-pro-m or 4-m-pro-r trimer anion, respectively (Scheme 2). Thus, these carbons maintain their stereochemical identities throughout the reaction from complexes to transition state and trimer anions.The surprisingly similar Li distances to the 1-phenyl carbanion and the 3-phenyl coordinating group of the dimer precursor 1 are of special interest in being very similar (Table S1†). For both the 1- pro-m and 1- pro-r isomers (Table S1†), the Li ion is located much closer to CZ (0.22–0.23 nm, 2.2–2.3 Å) than CE (0.30 nm, 3.0 Å). However, its coordination to the 3-phenyl group depends on stereochemistry with the Li ion of 1-pro-m being more strongly coordinated to the CZ′ carbon (0.23 nm, 2.3 Å) than to the CE′ carbon, while the opposite is the case for the 1-pro-r isomer with distances of 0.27 nm (2.7 Å) and 0.24 nm (2.4 Å), respectively. Although the 1-phenyl torsions are nearly the same but with opposite signs, the torsions of the 3-phenyl groups for 1-pro-m (31.3°) and 1-pro-r (17.2°) are not (Table S1†). As seen above, this is consistent with 1-pro-m and 1-pro-r being epimers. The (C2–C3) gauche dihedral angle in 1-pro-m and the larger 3-phenyl torsion account for its greater conformational strain (1.4 kcal mol−1) compared to the 1-pro-r isomer (Fig. 2). The unusual values of the C1–C2 dihedral angle may be seen as distorted trans-like conformations (t) due to the strong intramolecular Li-3-phenyl interactions (see below).14
Trimer anions
The LDPP chain-end (C1–C3) portions of the LTPH trimer anions may be seen to exist as two “sets” of enantiomer-like conformations of carbons 1 and 3: 4-m-pro-m (1-pro-S, 3S) or 4-r-pro-m (1-pro-r, 3R), and 4-r-pro-r (1-pro-S, 3R) or 4-r-pro-m (1-pro-r, 3S). This is supported by the nearly equal magnitudes and opposite signs of the 1- and 3-phenyl groups, which is consistent with a lack of influence of the 5S carbon (Table S2†).A comparison of Tables S1 and S4† show that the distances with respect to the Li ion of the aryl carbons of 1 and 3 (pro-m or pro-r) are nearly identical, as are the magnitudes and torsions of the dihedral angles (CH2–C1–CQ–CZ). For instance, the four isomers of 4 show nearly the same Li–CZ and Li–CE distances in the 1-phenyl carbanion that is nearly independent of stereochemistry, with the Li–CQ (0.21 nm, 2.1 Å) and Li–CZ (0.22–0.23 nm, 2.2–2.3 Å) distances being significantly smaller than that of Li–CE (0.30 nm, 3.0 Å) (Tables S1 and S2†).
Monomer complexes
Four monomer complexes may be formed depending on the two pro-chiral carbanion faces (1-pro-m or 1-pro-r) of LDPP and the reacting monomer at its si or re faces through coordinative displacement of the Li-coordinating 3-phenyl group.This gives four diastereomeric monomer complexes 2, which differ significantly from the two LDPP precursors (Schemes 2 and 3) formed through reaction of 1-pro-m on the si or re face of the monomer. This results in the displacement of the 3-phenyl group, accompanied by rotation of the (C2–C3) bonds, giving all-trans (t t) 2-pro-m-pro-si or 2-pro-m-pro-re complexes with smaller 1-phenyl torsions of −21° and −14° (Tables S1 and S3†). The alternative 2-pro-r-pro-si and 2-pro-r-pro-re monomer complexes are formed similarly, but generate (g− t) complexes with smaller 1-phenyl torsions of 13° and 17° (Table S3†). It is of some interest that the 2-pro-m-pro-si complexes have higher torsions compared with all others, as this may be a factor (but not the only one), considering its much higher free energy compared with the other three, as shown in Fig. 1.
 | ||
| Fig. 1 The whole Gibbs free energy plot of complexes 1 through 4. | ||
The 2-pro-m-pro-si and 2-pro-r-pro-re complexes have much smaller Li–CZ (0.23 nm, 2.3 Å) than Li–CE (0.31 nm, 3.1 Å) distances. Meanwhile, for the 2-pro-m-pro-re and the 2-pro-r-pro-si complexes, this is almost exactly the reverse, with the Li–CE distances being smallest (0.23 nm, 2.3 Å) compared to the coordinating aryl carbons (Table S3†). Interestingly, the Li distances to the coordinating aryl carbons are much smaller for the 2-pro-m-pro-re or 2-pro-r-pro-si complexes. The aryl carbons all have lower values (0.05–0.07 nm, 0.5–0.7 Å) compared with the other two isomers (Fig. 1), indicating that this may correlate with their lower Gibbs free energies (see below).
Transition states
As seen for the trimer anions, there appear to be two “sets” of transition states: 3-pro-m-pro-si/3-pro-r-pro-re and 3-pro-m-pro-r/3-pro-r-pro-r, with each set (unlike the complexes) being quite close to enantiomeric. Thus, for set-1, the Li–CZ carbon distances are 0.23 nm (2.3 Å) and about 0.30 nm (3.0 Å) for Li–CE. Meanwhile, for set-2, the Li–CE and –CZ distances are 0.23 nm and about 0.26–0.27 nm (2.6–2.7 Å, CZ), respectively (Table S4†). The Li aryl carbon distances for the coordinating styrene are virtually the same (Table S4†).Conformations, LDPP and monomer complexes
In this case, the dihedral angles for the LDPP chain carbons are listed conventionally with the carbanion carbon as C1 (Table S5†). For the realistic description of the conformations of LDPP-styrene complexes, transition states and trimer anions, it is necessary to describe their mutual orientations of LDPP and styrene as well. For instance, for the LDPP-styrene complexes, the styrene CH and CH2 vinyl carbons are given as C1′ and C2′, respectively. As indicated above, upon formation of complexes 2, the LDPP (1-pro-m (g t) and 1-pro-r (t t) dimer precursors) give (t t) and (g−t) conformations, respectively, a striking “reversal” of the main conformations (Tables S5 and S6†).The displacement by styrene of the Li-coordinating 3-phenyl group leads to the formation of 2-pro-m or 2-pro-r LDPP monomer complexes, in which the 1-phenyl group is nearly coplanar with the LDPP chain, with the 1-phenyl torsions being much smaller than LDPP with values of about −21°/17° and −14°/13° for 2-pro-m-pro-si/2-pro-r-pro-re and 2-pro-m-pro-re/2-pro-r-pro-si, respectively, and the 3-phenyl groups being sufficiently far away from the carbanions (Table S6†).
Monomer orientations relative to the LDPP chain are represented by intermolecular dihedral angles (C1′–C2′), (C2′–C1) and (C1–C2), where the primes refer to the monomer CH and CH2 vinyl carbons, respectively (Table S6†). It seems plausible that these dihedral angles are consistent with both 2-pro-m-pro-si/2-pro-r-pro-re and 2-pro-m-pro-re/2-pro-r-pro-si being enantiomeric-like. For the 2-pro-m-pro-re//2-pro-r-pro-si pair, the C1′–C2′ and C1–C2 dihedrals are quite close, while these are a little larger (∼6°) for the (C1–C2) dihedrals. However, in all cases, the dihedral angles remain opposite in signs, indicating at least a reasonable argument for pairs of enantiomer-like isomers, given that dihedral angles are far more sensitive measures than the Li aryl carbon distances. In that context, it is interesting that the largest deviation in dihedral angles (16°) is seen for the (C1–C2) dihedral of the 2-pro-m-pro-si/2-pro-r-pro-re pair of complexes, given the extraordinarily large free energy value (9.4 kcal mol−1) for the 2-pro-m-pro-si complex (Fig. 1).
With respect to the LDPP parts of the 2-pro-m-pro-si and 2-pro-m-pro-re (2-pro-m) complexes, this leads to all-trans (t t) conformations with (C1–C2), (C2–C3) and (C3–C4) dihedral angles of about 170°–175° (Table S6†). The two 2-pro-r complexes show (t g t) conformations of LDPP, where the carbanion is likewise fully exposed, but has a single LDPP (C2–C3) gauche conformation. This indicates again that the conformations are much more revealing in the distinction of stereoisomers. As seen from the values of the intermolecular (C2′–C1) dihedrals (Table S6†), the “steepness” of the monomer approach with respect to the chain is greater for 2-pro-m-pro-si/2-pro-r-pro-re and 2-pro-m-pro-re/2-pro-r-pro-si. This is also seen for the dihedrals of the long axes of the monomer and benzyl carbanion that give values of 19° and −6° for set 2-pro-m-pro-si/2-pro-r-pro-re, and −67° and 66° for set 2-pro-m-pro-re/2-pro-r-pro-si (Table S6†).
Transition states
As seen in Table S7,† the near-enantiomeric character of the two sets of transition states (3-pro-m-pro-si/3-pro-r- pro-re, 3-pro-m-pro-re/3-pro-r-pro-si) is closer. Thus, the (C1′–C2′), (C2′–C1) and (C1–C2) monomer dihedrals for the 3-pro-m-pro-si and 3-pro-r-pro-re transition states are about 91°, 50°, 72° and −91°, −52°, −78°, respectively, with the reversals of signs being clear (Table S7†). Likewise, the 3-pro-m-pro-re and 3-pro-r-pro-si transition states (TSs) have quite similar (C1′–C2′) and (C1–C2) dihedrals values, but with very different (C2′–C1) dihedrals, along with opposite signs within each set. This is consistent with the enantiomeric character of the first three carbons of the TS (Table S7†). The virtually identical values (C1′–C2′) dihedrals of both sets at nearly 90° may correlate with the similar free energies of the transition states. In addition, the signs of the dihedral angles in the complexes and transition states remain the same with no exceptions (Tables S6 and S7†).Trimer anions
With the formation of the monomer carbanion bond, the transition states 3 convert into trimer anion 4, with the (C1′–C2′), (C2′–C1), and (C1–C2) intermolecular dihedrals of complexes 3 being converted into the (C1–C2) (C2–C3) and (C3–C4) dihedrals of LTPH (4), respectively, while the LDPP (C2–C3) and (C3–C4) dihedral angles of 3 are virtually unchanged and relabeled as (C4–C5) and (C5–C6) dihedral angles of 4, respectively (Tables S7 and S8†). The nearly constant TS (C1–C2) intermolecular dihedrals of the LDPP chain (∼127°–130°) are consistent with the interaction of the Li ion with the monomer and the LDPP carbanion (Tables S2 and S8†), and resemble the stereochemistry of the carbons 1–3 of LDPP (Table S2†). In going from transition states 3 to trimer anions 4, there are increases in the intermolecular (C1′–C2′), (C2′–C1) and (C1–C2) dihedral angles of about 38°–40°, 14°–17° and small increases or decreases (3°–7°), respectively, as these are converted into the (C1–C2), (C2–C3) and (C3–C4) dihedral. Meanwhile, (C2–C3) and (C3–C4) convert into the (C4–C5) and (C5–C6) dihedrals of 4, with minor changes (≤4.0°, Tables S7 and S8†). The dihedral signs remain unchanged, which i consistent with the increases or decreases in magnitudes (Tables S7 and S8†).The very similar magnitudes of the TS dihedrals in each set are consistent with the very small (≤4.0°) transition state free energy differences. This also holds for 4-m-pro-r and 4-r-pro-r (3S-1-pro-r and 3R-1-pro-S), indicating that the 5-phenyl group in trimer anion 4 has little or no effect on stereochemistry, as concluded earlier, and supports both the LDPP and LTPH anions being appropriate models for the anionic styrene polymerization stereochemistry.13
The results of the computations shown in Table 1 list the free energies of 1-pro-m and 1-pro-r, the energies of the four styrene complexes (2-pro-m + S(si), 2-pro-m + S(re), 2-pro-r + S(si), 2-pro-r + S(re)), the corresponding transition states 3, and the resulting 4-m-pro-r, 4-m-pro-m, 4-m-pro-r and 4-m-pro-m trimer anions that are only between 1.1, 1.7, 0.1 and 0.6 kcal mol−1 more stable than 1-pro-m, respectively.
| Structures | ΔG of 1 (kcal mol−1) | ΔG of 2 (kcal mol−1) | ΔG± of 3 (kcal mol−1)b | ΔG of 4 (kcal mol−1) |
|---|---|---|---|---|
| a Free energies of 1-pro-m and 1-pro-r, corresponding complexes: 2-pro-m + (si), 2-pro-m + (re), 2-pro-r + (si), 2-pro-r + (re).b Activation free energies relative to 1-pro-m and 1-pro-r. | ||||
| 1-m-pro-si | 0 | 9.4 | 17.1 | −1.1 |
| 1-m-pro-re | 0 | 7.9 | 17.1 | −1.7 |
| 1-r–pro-si | −1.4 | 8.8 | 18.9 | −0.1 |
| 1-r-pro-re | −1.4 | 8.1 | 18.8 | −0.6 |
Our calculations shown in Fig. 2 appear to show a slight preference for isotactic-like additions, but this may be offset by a greater tendency for the isotactic chain ends to associate into unreactive dimers. This is fully consistent with a nearly complete absence of stereoregularity when initiated by carefully purified alkyllithium or reactive lithium carbanions occurring in hexane, cyclohexane or similar solvents, which is consistent with our computations.7,9,12
 | ||
| Fig. 2 (A) The whole Gibbs free energy plot of complexes from 5 to 8 under B3PW91/6-311++G(d,p). (B) Benchmark of the whole reaction route under B3PW91/6-311++G(d,p) (orange), B3LYP/6-311++G(d,p) (red) and M06-2X/6-311++G(d,p) (blue) with 3D structures. | ||
LDPP-SMBS complexes
The 5-pro-m and 5-pro-r isomers show (C2–C3) and (C3–C4) dihedral angles with (t g t) and (t t) conformations for the C1–C4 chain segments of the 1-pro-m and 1-pro-r isomers, respectively, with the torsion signs remaining the same in all cases (Tables S2 and S10†).
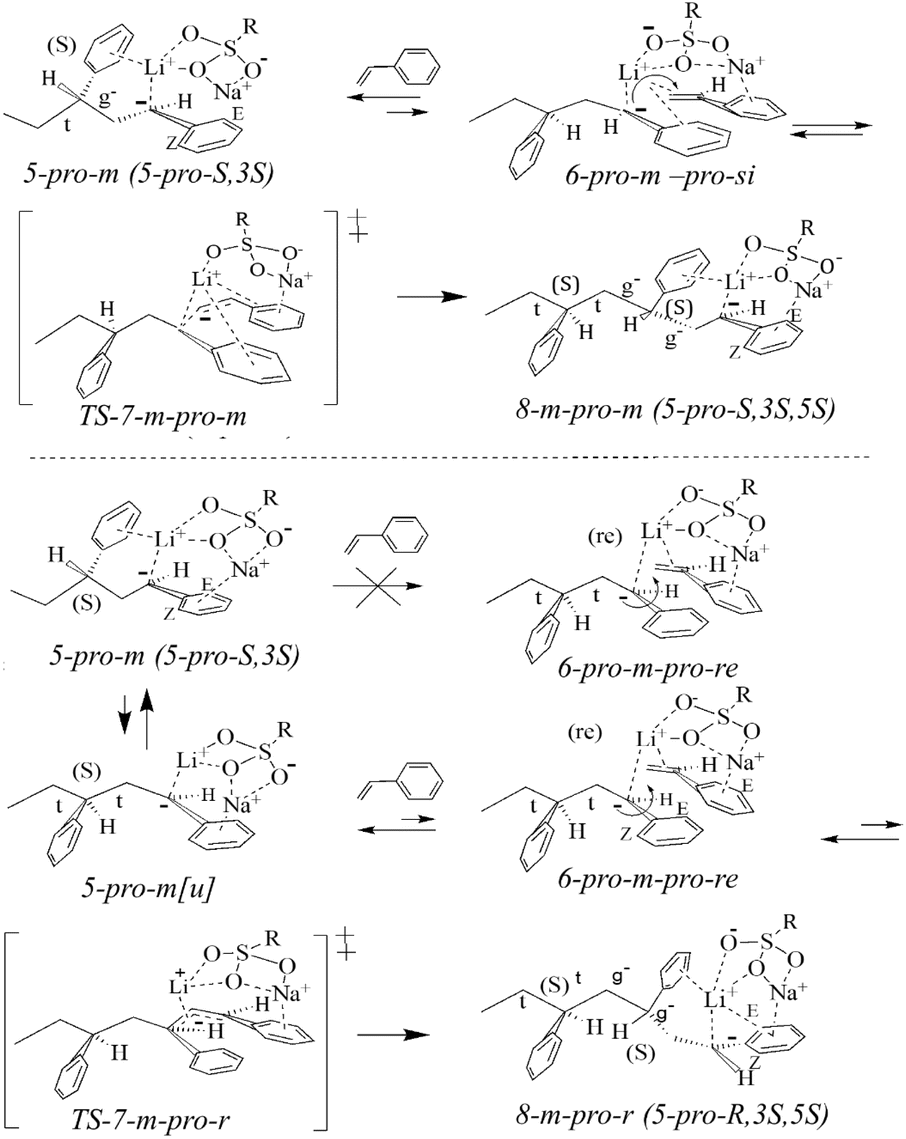 | ||
| Scheme 4 Monomer addition to the 5-pro-m dimer anions. Formation of LTPH (trimer anions) 8-m-pro-m and 8-m-pro-r. | ||
The changes in the formation of the complexes are major and remarkable (Scheme 4). Thus, the 3-phenyl group is displaced from the Li ion by the styrene monomer, and the Na ion is now only coordinated with the monomer phenyl group being 0.28 nm (2.8 Å), which is nearly equidistant to the phenyl carbons. It remains located above the methylene and methine vinyl carbons at 0.24 (2.4 Å) and 0.27 nm (2.7 Å, Table S11†).
Unlike the complexes of 2, the free energies of the four monomer-SMBS complexes 6 are nearly equal in free energy (<0.7 kcal mol−1, Fig. 2). However, one isomer 6-pro-r + Ssi stands out in having dual coordination of the Li ion by sulfonate (Scheme 5), and the 1-phenyl torsion dihedral angle is lower (5°–9°) than the other three cases, suggesting correlation of the Li charge density and 1-phenyl torsion (Table S11†).
 | ||
| Scheme 5 Stereochemistry of the monomer addition to the 6-pro-r dimer anions. Formation of the 8-r-pro-r and 8-r-pro-m isomers. | ||
As indicated above, the formation of the monomer complexes 6 involves large changes in the deployment of Li and Na ions. That includes a reversal of 5-pro-m and 5-pro-r having t g t and t t t conformations, respectively, into two 6-pro-m complexes and one 6-pro-r-pro-re complex having t t t and t g t LDPP backbones.
However, the 6-pro-r-pro-si complex has a LDPP gauche-like (g)g− t conformation with an unusual (C1–C2) dihedral angle (∼88.5°). The (C2–C3) and (C3–C4) LDPP dihedrals of 6, not being involved directly in the reaction, are relabeled as (C4–C5) and (C5–C6) dihedrals upon formation of 8 with virtually unchanged values (Table S16†).
As seen before, in the presence of SMBS, the values and signs of the dihedral angles of the complexes, transition states and trimer anions, though consistent with symmetry properties, differ far more than that seen in the absence of SMBS (Tables S6–S8†). For example, although the 6-pro-m-pro-re and 6-pro-r-pro-si complexes have the expected opposite signs, their (C1′–C2′), (C2′–C1) and (C1–C2) dihedral angle values vary by as much as 73° (Table S14†). Thus, symmetry considerations are not suited in interpreting the free energies that are remarkably close (Fig. 2). The value of the LDPP (C1–C2) dihedral angle of the 6-pro-r-pro-si isomer complex has an unusual value of about 88° that evolves into an equally unusual value of 145° for the corresponding 7-pro-r-pro-si TS, suggesting significant steric strain. However, as shown in Fig. 2, the Gibbs free energies of the monomer complexes are almost identical.
Given the lower (−1.7 kcal mol−1) energies of the chain-end r-compared to m-dyads, this leads to increased differences in activation energies with values of 30.2 kcal mol−1 for 7-pro-r-pro-si compared to the lowest value (26.9 kcal mol−1 for 7-pro-m-pro-si, indicating a difference in the free energy of activation of about 3.3 kcal mol−1 (Table 2). This would suggest that the rates of formation of the mm triads would exceed that of the rr triad formation. Of course, that would be misleading, as the activation free energies of formation of mr and rm dyads are quite a bit lower (Table 2, Fig. 2). Given the multiple and plausibly overlapping effects, the above is likely to play a significant role in explaining the virtual absence of rr triads in LTHP. The benchmark results under B3LYP/6-311++G(d,p) and M06-2X/6-311++G(d,p) agreed well with these conclusions, and all key conclusions (e.g., stereoselectivity trends, rate-limiting steps) remained robust across all tested functionals. M06-2X indeed provided improved thermodynamic data for reaction pathways and lower energy barriers (22.8, 23.5, 26.2, 23.9 kcal mol−1).
| Isomers | 5 | 6 | 7 | (ΔG≠) b | ΔΔ(G≠)c | kmm/kp d |
|---|---|---|---|---|---|---|
| a In cyclohexane.b Differences in the activation Gibbs free energy in kcal mol−1 mol−1 (ΔG≠) between 7 and 5.c Differences in the activation free energies compared to the free energy of activation of pro-m-pro-si.d Propagation rate constant kmm for isotactic triad formation at 300 K relative to: kmr, krr and krm from top to bottom. | ||||||
| pro-m-pro-si | 0 | 22.8 | 26.9 | 26.9 | 0 | 1.0 |
| pro-m-pro-re | 0 | 22.9 | 27.3 | 27.3 | 0.4 | 1.8 |
| pro-r-pro-si | −1.9 | 22.8 | 28.3 | 30.2 | 3.3 | 194 |
| pro-r-pro-re | −1.9 | 22.2 | 28.0 | 29.9 | 3.0 | 119 |
Discussions
Nonpolar media
This study supports the occurrence of four stereoisomeric LDPP-styrene complexes as precursors for monomer addition. The assembly of the complexes is exo-entropic (negative ΔS) and endothermic (positive ΔH), which is consistent with their relatively high free energies: 4.7 to 9.0 kcal mol−1. Especially, the high value of 9.0 kcal mol−1 for the 2-m-pro-m complex is unusual compared to the three other complexes, given that the four transition states have very similar free energies. There may be several reasons, as shown from Table S6.† The (C2′–C1) dihedral angle is quite high (81.6°) compared to its 2-r-pro-re quasi-enantiomer (−65.2°), and to the other two (2-m-pro-re and 2-r-pro-si) complexes that have nearly identical (C2′–C1) dihedral angle values (118.2° and −117.7°), respectively (Table S6†). The corresponding intermolecular (C2′–C1) dihedral angles of the transition states of 3 are now nearly identical (≤3.1°), but have opposite signs (Table S7†). From the data shown in Fig. 2, the complexes 2 should very rapidly convert to the transition states 3 and into LTPH, so that the detection of their small steady state concentrations (for instance, by UV-visible or IR) would be difficult. However, the transition states have nearly the same free energy of about 15.8 ± 0.4 kcal mol−1 given their small (<1 kcal mol−1) free energy differences (Fig. 1), indicating that the rates of monomer addition should be nearly the same for all four stereoisomers. Closer inspection of their structures suggests the presence of two sets of pseudo-enantiomeric transition states (Table S7†). This is even the case for the transition state conformations, and to a lesser degree for the corresponding complexes (Table S7†).Provided that LDPP is a reasonable model for the polymer, the low isotactic content (in the absence of SMBS) can be ascribed to the lower degree of association of r-LDPP into dimers (3.3 kcal mol−1), hence favoring the formation of r dyads at the chain ends, and thus slightly favoring the formation syndiotactic chains (Table 3). However, this may be compensated for by the preferred association of the greater steady state concentrations of the pro-r species, thus giving atactic PS without significant fractions of mm triads and or mmm tetrads. The faster rates of the polymerization in the presence of SMBS, the higher free energy and transition states notwithstanding, are due the larger concentrations of the SMBS complexes (5-pro-m, 5-pro-r), as these do not seem to dimerize to any significant extent. This simple model would also be consistent with the kinetics of this polymerization being reported by Yakimanski et al.14 The formation of long meso (m) sequences of isotactic PS for the case of SMBS may be kinetically favored by the lower TS, as discussed above. In addition, there is the possible participation of an uncoordinated 5-pro-m[u] LDPP intermediate that should rapidly form both 6-m-pro-Ssi and 6-m-pro-Sre monomer complexes. Further simulations with other coordinating agents could point to additives with even more stereo-selective properties. This approach could be of interest in applications for block copolymers.
| No. | ΔG (kcal mol−1) at 298.2 Kb | |
|---|---|---|
| a The pro-meso and pro-racemic structures are indicated as m- and r-, respectively.b Calculation details: B3PW91/6-31+G(d,p) in vacuum. Previous data are shown in parentheses.13 | ||
| 1 | 2 m-LDPB ⇒ (m-LDPB)2 | −19.0 (−21.7) |
| 2 | 2 r-LDPB ⇒ (r-LDPB)2 | −15.7 (−19.5) |
| 3 | m-LDPB + r-LDPB ⇒ (m-LDPB-r-LDPB) | −17.5 (−18.0) |
| 4 | m-LDPB + SMBS ⇒ (m-LDPB-SMBS) | −27.1 (−29.6) |
| 5 | r-LDPB + SMBS ⇒ (r-LDPB-SMBS) | −26.8 (−30.8) |
| 6 | 2 SMBS ⇒ (SMBS)2 | −34.5 (−34.7) |
| 7 | 0.5 (m-LDPB)2 + 0.5 (SMBS)2 ⇒ (m-LDPB-SMBS) | −0.40 (−1.4) |
| 8 | (m-LDPB)2 + 0.5 (SMBS)2 ⇒ (m-LDPB-SMBS-mLDPB) | −4.50 (−7.2) |
As illustrated in Tables S6–S8,† the changes in the dihedral angles of the monomer and LDPP chain-end provide more details into the nature of the molecular changes that mediate the conversions of complexes into transition states, and of transition states into trimer anions. The magnitudes of the changes are typically 30° or less, and the dihedral signs (positive or negative) remain the same as the intermediates interconvert (Tables S6–S8†). This would be consistent with the relatively rapid (∼10−9 to 10−12 s−1) and large rotations of the phenyl or vinyl groups of the monomer, and the phenyl groups of polymer chain ends compared to translational motions.
A comparison of the dimer and trimer anions ion show that the conformations of the first two asymmetric carbons are nearly identical, as shown by the first two dihedral angles of the chain (Tables S2 and S8†), which is consistent with the lack of any influence of the asymmetric carbon 5(S) in LTPH or 3-C in LDPP. In addition, the intramolecular coordination of the lithium ion by the 3-phenyl groups is quite strong, as suggested by the nearly constant CH2–C1–CZ–CQ dihedral angles (1-phenyl torsions) of the two isomers of LDPP and all four isomers of LTPH being nearly identical (±26°–29°). Meanwhile, the (C1–C2) dihedral angles of the LDPP and LTPH are both between 127° and 130° degrees (Tables S2 and S8†), so that the dimer and trimer anions are equally suitable as models.
Polar media, effects of coordinating benzylsulfonates
The polymerization of styrene and nonpolar media in the presence of sodium dodecylbenzene sulfonates has indicated a marked increase in the isotactic PS content. Thus, in the presence of one molar equivalent of SMBS with respect to the initiator, the isotactic content of the polystyrene initiated by t-butyllithium increased markedly with the isotactic triad and pentad contents being close to about 50% and 20%, respectively, indicating clear evidence for a crucial role of the Li-coordinating sulfonate groups.As shown in Fig. 2, the transition state free energies decrease by 2.7 kcal mol−1 as follows: 7-r-pro-r > 7-m-pro-r > 7-r-pro-m > 7-m-pro-m. As the rates of formation of 7-m-pro-si and 7-r-pro-re would represent the corresponding formation of mm and rr dyads, this would entail differences in the free energy of activation (below). The corresponding differences in activation free energies decrease by even more (3.3 kcal mol−1), given the lower free energies of the racemic (r,r) dyads. The formation of the mr and rm triads are lower, but these still should have a significant effect. This trend is qualitatively consistent with the considerable non-bonded interactions in the relative free energies of the TS's (Table 2). There are several reasons: (a) the relatively high values value of the intermolecular (C2′–C1) and (C1–C2) dihedral angles of 7-r-pro-si, (∼73° and 76°, respectively) compared to the other three isomers (Table S15†); (b) the 145° intramolecular (C1–C2) dihedral angle of 7-r-pro-r that indicates a severe conformational strain, which should exceed the free energy of a single gauche conformation; (c) the low value (169°) of the intramolecular (C3–C4) dihedral angle of 7-r-pro-r compared to that of the other three isomers; and (d) the higher values of the 1-phenyl torsions (∼27°) of 7-m-pro-re and 7-r-pro-si compared to the other two (19°–20°, Table S15†). Although these are listed separately, there may well be considerable overlap between these factors.
Thus far, interactions of the SMBS phenyl group in any of the intermediates have not been discussed. In general, the benzene sulfonate phenyl group is not in close proximity to any of the aromatic groups of any of the monomer complexes and transition states.
However, there is an exception in that the 7-pro-r-pro-si transition state, there is a proximity of the ortho-carbon or hydrogen atom of the ortho-carbon of the benzene sulfonate phenyl group to the meta carbon (hydrogen) of the 1-phenyl carbanion, as measured by the distance of the 1-phenyl CZ carbon to the ortho-carbon of the SMBS, being on the order of 3.8 Å. The hydrogens are even closer at about 3.4 Å. Even though these interactions may be small and may not appreciably increase the energy of this transition state, this raises the possibility that this proximity effect may be enhanced by the introduction of a methyl or larger group at the ortho-carbon(s) of the benzenesulfonate. Thus, future explorations in this directions would be interesting.
The free energies of activation for the monomer addition for r-pro-r and r-pro-m are 3.3 and 3.0 kcal mol−1 higher, respectively, than that of the competing m-pro-m and m-pro-r additions. Hence, the direct m-pro-m addition in the presence of the styrene monomer is favored to repeat, as the competing m-pro-r process may be “handicapped” by the absence of a directly accessible TS state, and may depend on the occurrence of a spontaneous cleavage of the Li-3-phenyl coordination (Table S5†).
In addition, the monomer vinyl-phenyl dihedral angles for transition states 7-m-pro-re and 7-r-pro-si are unusually low (∼10°) compared with that (∼27°) of the corresponding isomers of the trimer anion 8 (Table S16†), while the other two (7-m-pro-si and 7-r-pro-re) TS isomers both have torsions (∼19°) that are well matched with regard to the 1-phenyl torsions (∼20°), as the complexes are converted into transition states (Tables 2 and S15†). This would result in an additional negative entropy penalty for the conversions of 6-m-pro-re and 6-r-pro-si into 7-m-pro-re and 7-r-pro-si. Thus, the dependence on stereochemistry with respect to the transition state (TS) free energies appear to indicate mainly non-bonded interactions associated with conformational effects.
As shown in Table 3, the formation constants of 2-pro-m or 2-pro-r 1:1 LDPP-SMBS complexes are very high and nearly identical. In contrast, the LDPP-SMBS complex also dimerizes but with relatively low formation constants (Table 3). Such dimers have been demonstrated for similar carbanions in hydrocarbons, but have been shown to be unreactive.13,14 The same is plausible in the present case, given the data in Table 3. Thus, the computations carried out are based on 1:1 LDPP-SMBS complexes with a stoichiometry that is independent of the LDPP stereoisomer, as both pro-m and pro-r LDPP have nearly the same formation constant with SMBS, which are very large and nearly the same order of magnitude (Table 3).
Conclusions
The anionic polymerization of styrene in cyclohexane initiated by alkyllithium gives atactic polystyrene. However, in the presence of one molar equivalent of SMBS, isotactic-rich polystyrene was obtained. The effects of SMBS was studied through advanced DFT molecular modeling studies on the reaction of a single styrene monomer to the LDPP dimer in the presence and absence of SMBS existing as pro-meso pro-1S, 3S (1-pro-m) or pro-racemic 1-pro-1R, 3S (1-pro-r) epimers to give four 4-m-pro-m, 4-m-pro-r, 4-r-pro-r, 4-r-pro-m, isomeric trimer anions.In conclusion: (A) The polymerization mechanism has been shown to undergo a process through four stereoisomeric 1:1 styrene complexes 2, the corresponding transition states 3 and 1-lithio-1,3,5-triphenylheptane trimer anions 4; (B) LDPP-styrene complexes 2 formed by coordination of Li ion of LDPP to styrene at its pro-si or pro-re faces give 2-pro-m-pro-si/2-pro-m-pro-re or 2-pro-r-pro-si/2-pro-r-pro-re, which are short-lived but stable complexes that equilibrate with the 1-pro-m and 1-pro-r LDPP, and that vary in free energy between 9.0 and 4.7 kcal mol−1, in which the Li-3-phenyl coordination has been disrupted; (C) The four transition states differ from the complexes in that their free energies are nearly equal (within 1.0 kcal mol−1), which is consistent with the formation of atactic polystyrene; (D) The transition state structures closely resemble that of the complexes, but differ primarily through different dihedral angles of the vinyl carbons (C1′, C2′) and the first two carbons of LDPP (C1, C2). Hence, the transition states involve primarily bond rotations of the monomer, as the complexes are transformed into transition states and then into trimer anions. (E) In the resting state, the 1-pro-m and 1-pro-r epimers and the four stereoisomers of 4 have (C1–C2), dihedral angles of about 128°–130°, characterized as distorted trans conformations due to a strong intramolecular coordination of Li to the 3-phenyl group (Tables S2 and S8†). The (C2–C3) and (C3–C4) dihedral angles are either gauche (g ∼67°–73°) or trans (t ∼173°–176°).
SMBS effects.
(F) In the presence of SMBS, the interaction of the Li ion with the 3-phenyl group of LDPP is weakened, but strengthened by Na ion interactions with the monomer phenyl group. This gives 5-pro-m and a 5-pro-r dimer anion with t g t and t t t conformations, respectively. However, the (C1–C2) dihedral angles are nearly 179° and 178°, respectively, rather than the distorted value of about 130° (Table S2†). The free energies of the 5-pro-m are slightly higher (1.7 kcal mol−1) than the 5 pro-r epimers, which is plausibly due to the all-trans conformation of 5-pro-r (Fig. 2). (G) The formation of monomer complexes here changes the LDPP conformation to an all-trans conformation for 6-pro-m, regardless of the monomer presentation (si or re). However, for the case of the pro-r dimer anions, the si monomer complexes show an unusual (C1–C2) conformation of 88.5° denoted as a distorted gauche conformation, while the re monomer leads to a normal t g t conformation. The complexes in this case have much higher, but nearly the same free energies (21.0–21.7 kcal mol−1). (H) The corresponding transition states 7, however, show very different free energies of 24.9, 26.6, 27.6 and 26.4 kcal mol−1 for the formation of the 7-m-pro-m, 7-m-pro-r, 7-r-pro-si and 7-r-pro-re transition states, respectively. As the rates of formation of 7-m-pro-si and 7-r-pro-re would represent the corresponding formation of mm and rr dyads, this would favor the former by a factor of about 1.5 × 103. (I) The structure of the 7-r-pro-r transition state is the only one of the four transition states where the phenyl group of the phenolate is relatively close to the 1-phenyl carbanion with aromatic carbon–carbon distances as small as 3.8 Å. It would seem then that this could be used to raise the corresponding TS even further by introduction of isopropyl or similar groups at the ortho or meta positions of a sodium phenolate derivative. This above approach should be applicable to the stereochemical analysis of anionic and related polymerizations of styrene and similar vinyl hydrocarbon monomers in the presence of alkali cations, and various added cation electrophiles and coordinating nucleophilic bases.Calculation details
All geometries of intermediates and transition states (TSs) were optimized under tight criteria using the B3PW91/6-311++G(d,p) method with DFT-D3 correction. Benchmarks were carried out under B3PW91/6-311++G(d,p), B3LYP/6-311++G(d,p) and M06-2X/6-311++G(d,p), all with DFT-D3 correction. Diffuse functions were considered to describe the ionic force,16 and polarization functions were employed to achieve accurate results.17 Frequency calculations confirmed that the intermediates and TSs had zero and one imaginary frequency, respectively. The reaction pathway for each TS was verified using the intrinsic reaction coordinate (IRC) method. Energies were evaluated with the same basis set. Thermal correction to Gibbs free energies were obtained at 298.2 K and 1.013 × 105 Pa. All calculations were performed using the Gaussian 16 program.18Data availability
The data supporting this article are included in the ESI.†Author contributions
This manuscript was written through the contributions of all authors. All authors have approved the final version of the manuscript.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
Financial support for this research was provided by the Loker Hydrocarbon Research Institute at the University of Southern California, the National Natural Science Foundation of China (No. 22201105) and the Youth Foundation of Jiaxing Municipal (2024AY40028).References
- K. Ziegler, E. Holzkamp, H. Breil and H. Martin, Das mülheimer normaldruck-polyäthylen-verfahren, Angew. Chem., 1955, 67, 541–547 CrossRef CAS.
- G. Natta, Une Nouvelle Classe de Polymeres d′ α-Olefines ayant une Régularité de Structure Exceptionnelle, J. Polym. Sci., Part A: Polym. Chem., 1996, 34, 321–332 CrossRef.
- K. Beckerle, C. Capacchione, H. Ebeling, R. Manivannan, R. Mülhaupt, A. Proto, T. P. Spaniol and J. Okuda, Stereospecific post-metallocene polymerization catalysts: the example of isospecific styrene polymerization, J. Organomet. Chem., 2004, 689, 4636–4641 CrossRef CAS.
- A. S. Rodrigues, E. Kirillov, T. Roisnel, A. Razavi, B. Vuillemin and J. F. Carpentier, Highly isospecific styrene polymerization catalyzed by single-component bridged bis (indenyl) allyl yttrium and neodymium complexes, Angew. Chem., 2007, 119, 7378–7381 Search PubMed.
- A. S. Rodrigues, E. Kirillov and J. F. Carpentier, Group 3 and 4 single-site catalysts for stereospecific polymerization of styrene, Coord. Chem. Rev., 2008, 252, 2115–2136 CrossRef CAS.
- R. Kern, Homogeneous synthesis of isotactic polystyrene using n-butyllithium initiator, Nature, 1960, 187, 410 CrossRef CAS.
- S. Brownstein, S. Bywater and D. Worsfold, Proton resonance spectra and tacticity of polystyrene and deuteriopolystyrenes1, 2, J. Phys. Chem., 1962, 66, 2067–2068 CrossRef CAS.
- H. Hsieh and R. P. Quirk, Anionic Polymerization: Principles and Practical Applications, Marcel Dekker, CRC Press, 1996 Search PubMed.
- T. Makino and T. E. Hogen-Esch, Anionic synthesis of highly isotactic polystyrene in hexane in the presence of lithium hydroxides, Macromolecules, 1999, 32, 5712–5714 CrossRef CAS.
- L. Cazzaniga and R. Cohen, Anionic synthesis of isotactic polystyrene, Macromolecules, 1989, 22, 4125–4128 CrossRef CAS.
- L. Cazzaniga and R. E. Cohen, Synthesis and characterization of isotactic polystyrene/polybutadiene block copolymers, Macromolecules, 1991, 24, 5817–5822 CrossRef CAS.
- J. M. Marechal, S. Carlotti, L. Shcheglova and A. Deffieux, Stereoregulation in the anionic polymerization of styrene initiated by superbases, Polymer, 2003, 44, 7601–7607 CrossRef CAS.
- F. Shi, J. Ling, J. Lu, B. Han, L. Liu and T. E. Hogen-Esch, Synthesis of isotactic polystyrene in hydrocarbons by initiation with t-BuLi in the presence of sodium dodecylbenzenesulfonate, Polymer, 2012, 53, 94–105 CrossRef CAS.
- A. Yakimansky, G. Wang, K. Janssens and M. Van Beylen, Influence of π-complexing agents on the anionic polymerization of styrene with lithium as counterion in cyclohexane. 2. Quantum-chemical density functional theory calculations, Polymer, 2003, 44, 6457–6463 CrossRef CAS.
- A. H. E. Müller and T. E. Hogen-Esch, Stereochemistry of anionic vinyl polymerization. Effects of ion pair prochirality on chain statistics, Macromolecules, 1988, 21, 2336–2339 CrossRef.
- T. A. Manz and D. S. Sholl, A dimensionless reaction coordinate for quantifying the lateness of transition states, J. Comput. Chem., 2010, 31, 1528–1541 CrossRef CAS PubMed.
- T. Li, D. Wang, Y. Heng, G. Hou, G. Zi, W. Ding, L. Maron and M. D. Walter, Experimental and Computational Studies on Uranium Diazomethanediide Complexes, Angew. Chem., Int. Ed., 2023, 62, e202313010 CrossRef CAS PubMed.
- M. Frisch, G. Trucks, H. B. Schlegel, G. Scuseria, M. Robb, J. Cheeseman, G. Scalmani, V. Barone, G. Petersson and H. Nakatsuji, Gaussian 16, Gaussian, Inc.Wallingford, CT, 2016 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: 3D structures and geometry data of the critical transitions states. See DOI: https://doi.org/10.1039/d5ra00332f |
| This journal is © The Royal Society of Chemistry 2025 |