
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceInvestigation of the anticancer activity of modified 4-hydroxyquinolone analogues: in vitro and in silico studies†
Yousra Ouafa Bouonea,
Abdeslem Bouzina *a,
Abdelhak Djemelbc,
Sanaa K. Bardaweeld,
Malika Ibrahim-Oualie,
Boulanouar Bakchichef,
Farouk Benaceurc and
Nour-Eddine Aoufa
*a,
Abdelhak Djemelbc,
Sanaa K. Bardaweeld,
Malika Ibrahim-Oualie,
Boulanouar Bakchichef,
Farouk Benaceurc and
Nour-Eddine Aoufa
aLaboratory of Applied Organic Chemistry, Bioorganic Chemistry Group, Department of Chemistry, Sciences Faculty, Badji-Mokhtar - Annaba University, Box 12, 23000 Annaba, Algeria. E-mail: abdeslem.bouzina@univ-annaba.dz; bouzinaabdeslem@yahoo.fr
bLaboratory of Phytochemistry and Pharmacology, Department of Chemistry, Faculty of Exact Sciences and Informatics, University of Jijel, BP 98, 18000, Jijel, Algeria
cResearch Unit of Medicinal Plants, RUMP, Attached to Biotechnology Research Center, CRBt, 03000 Laghouat 25000, Constantine, Algeria
dDepartment of Pharmaceutical Sciences, School of Pharmacy, University of Jordan, Amman 11942, Jordan
eEnsemble TPR, 52 Av. Escadrille Normandie Niémen, Marseille, 13013, France
fLaboratory of Biological and Agricultural Sciences (LBAS), Amar Telidji University, Laghouat 03000, Algeria
First published on 5th February 2025
Abstract
A set of nitrogen-based heterocycles derived from the quinoline ring as well as cyclohexanedione and dimedone cores were subjected to in vitro anticancer activity evaluation against four different cancerous cell lines namely; HCT116, A549, PC3, and MCF-7 respectively to colon, lung, prostate, and breast cancers. Compound 3g presented promising results exhibiting the best IC50 values among the investigated compounds for the four tested cell lines. In vitro results were supported with in silico studies including molecular docking simulation in order to learn more about the binding mode of the studied derivatives with relevant drug targets in cancer treatment, namely; anaplastic lymphoma kinase and cyclin-dependent kinase 2. Compound 3g showing the best in vitro results exhibited the most promising docking scores among the studied compounds. Moreover, molecular dynamics simulation was performed to the best ligand studying its stability inside the selected enzymes. Furthermore, a DFT study was performed to investigate the structural composition, electron density, and reactivity of tested compounds to identify the most important parts of the derivatives and elaborate a structure–activity relationship.
Introduction
In the recent years, cancer and related pathologies emerged among the deadliest diseases worldwide. According to the latest GLOBACAN report updated in 2022 by the International Agency for Research on Cancer, 20 million new cases attained from cancer were reported along with 9.7 million deaths worldwide with lung cancer being the most frequent in both attainability and mortality representing 12.4% of all cancer cases and 18.7% of all deaths due to cancer. Besides lung cancer, other types like breast, colorectum, and prostate cancers were classified among the most commonly diagnosed representing 11.6%, 9.6%, and 7.3% of all global cases.1 Alongside with harming the physical and mental health of millions of patients and families, it is recognized that carcinogenic pathologies require very long treatment incurring fees not affordable by all patients.2 Facing cancerous diseases is nowadays a primordial challenge in view of the high mortality caused by cancer and complications related to cancer treatment. Oncology has seen huge advancements since the appearance of what is called targeted therapy with small molecules inhibitors. The latter is the treatment that directly target the main causes of cancer development that could include enzymes and proteins with dysregulation.3 Among the most known drug targets classified as carcinogenic; the protein kinase family responsible of the phosphorylation reaction crucial in different physiological processes. Overexpression, dysregulation, mutation, and translocation of these proteins were the origin of several diseases' development especially cancerous diseases. Furthermore, in the past few years, 80 small molecules inhibiting various kinases have secured the approval of the FDA making the proteins belonging to the kinase family the most targeted proteins in cancer research.4Heterocycles are a large class of compounds constituted of heteroatom-containing rings of different sizes that originate from natural sources as well as synthetic processes. These compounds are known for their high efficiency in many important fields including specifically the medicinal area.5 Among the most pharmacologically beneficial heterocycles, are those that contain one or more nitrogen atoms.6 These latter represented, in 2020, 75% of the commercialized drugs as well as the entities approved by the FDA exhibiting various biological effects against a high number of diseases like cancer,7,8 HIV,9 tuberculosis,10 alzheimer's,11 and diabetes12 as well as presenting excellent antimicrobial activity.13 Quinolines and derivatives represent an integral part of the nitrogen-based interesting heterocycles presenting a remarkable number of pharmacological activities.14 Compounds inclosing a quinoline core have been widely used in the clinical treatment of several pathologies.
As an example, chloroquine 1 was first commercialized and used as an antimalarial agent for a long time,15 besides, it was associated later to Covid-19 treatment along with hydroxychloroquine 2 (Fig. 1).16 Many other compounds featuring the quinoline core were employed as active substances against several health issues, such as ciproflaxin 3 and pitavastatin 4 as antibiotic and cholesterol lowering agents respectively (Fig. 1).13,15,17
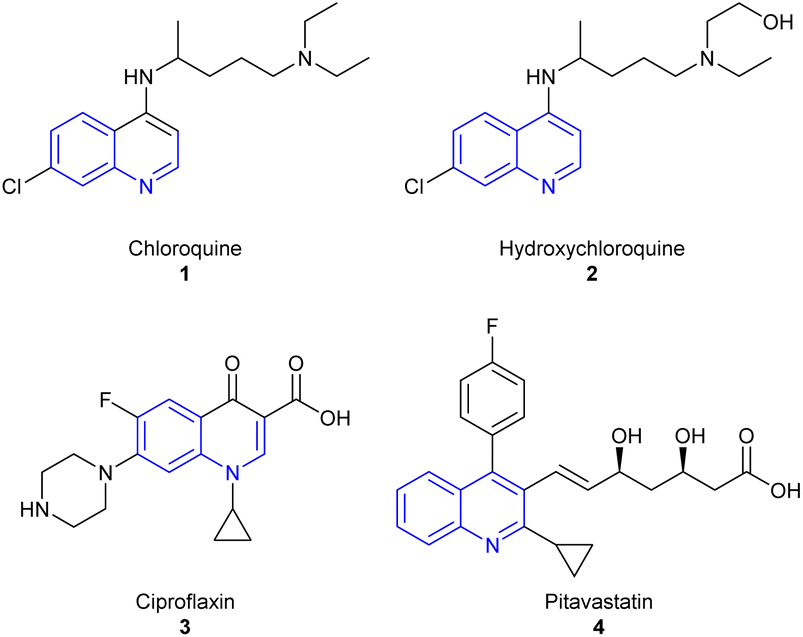 | ||
| Fig. 1 Structure of commercialized active quinoline derivatives. | ||
On the other hand, six-membered heterocycles fused to non-aromatic rings such as cyclohexanedione and dimedone moieties have attracted much interest in the domain of drug design and development of novel drug candidates.18 Numerous scientific papers were dedicated to synthesize and evaluate the biological activities of such compounds, and many of the synthesized products were found to be efficient including xanthenone 5, chromenone 6, hydroquinolines 7, and acridinedione 8 displaying respectively; antimicrobial,19 anticancer,18 calcium channels inhibition,20 and anti-inflammatory21 activities (Fig. 2).
 | ||
| Fig. 2 Structures of dimedone-based fused heterocycles. | ||
The present work is dedicated to the evaluation of the anticancer activity of modified quinoline rings consisting of 6-membered nitrogen-based heterocycles fused to cyclohexane-1,3-dione or dimedone cores. Ten derivatives of the stated family of compounds were tested against four different cancer cell lines involving HCT116 (colon cancer), A549 (lung cancer), PC3 (prostate cancer), and MCF-7 (breast cancer). The in vitro results were reinforced through in silico studies including molecular docking in which the activity of investigated compounds was predicted against two proteins belonging to the kinase family citing anaplastic lymphoma kinase and cyclin-dependent kinase 2. Furthermore, molecular dynamics simulation was performed to study the stability of the best ligand inside the cavities of both above-mentioned proteins. Additionally, a DFT complementary study was carried out to enrich our knowledge about the electronic behavior and the reactivity of the studied 4-hydroxyquinolone analogues (Scheme 1).
 | ||
| Scheme 1 Schematic representation of the design strategy employed in this work. | ||
Experimental
Chemistry
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :30, v/v) with 0.1% of formic acid to H2O/CH3CN (10:90, v/v) with 0.1% of formic acid at a flow rate of 0.5 mL min−1, with UV monitoring at the wavelength of 254 nm with a run time of 30 min. Chemical shifts are reported in δ units (ppm) relative to CDCl3 or DMSO (δ 77.0 and 39.0–40.0). Microanalysis spectra were performed by Elemental Analyser (Euro E.A. 3000-V3.0-single-2007), and the determined values were within the acceptable limits of the calculated values. Melting points were recorded on a Büchi B-545 apparatus in open capillary tubes.
:30, v/v) with 0.1% of formic acid to H2O/CH3CN (10:90, v/v) with 0.1% of formic acid at a flow rate of 0.5 mL min−1, with UV monitoring at the wavelength of 254 nm with a run time of 30 min. Chemical shifts are reported in δ units (ppm) relative to CDCl3 or DMSO (δ 77.0 and 39.0–40.0). Microanalysis spectra were performed by Elemental Analyser (Euro E.A. 3000-V3.0-single-2007), and the determined values were within the acceptable limits of the calculated values. Melting points were recorded on a Büchi B-545 apparatus in open capillary tubes.The crystallographic data and experimental details for structural analysis are summarized in Table 1. The reported structure was solved with the SHELXT-2014/5 (ref. 22) solution program by Intrinsic Phasing with Olex2 (ref. 23) as the graphical interface. The model was refined with SHELXL-2018/3 (ref. 24) using full matrix least-squares minimization on F2. All absorption corrections were performed with the CrysAlisPro 1.171.42.51a25 using spherical harmonics implemented in SCALE3 ABSPACK scaling algorithm. Crystal structure visualization and construction of crystal packing diagrams were performed using Mercury 3.8 software.26
| Moiety formula | 2(C17H17NO3) | Density (calculated) (g cm−3) | 1.293 |
| Sum formula | C34H34N2O6 | Absorption coefficient (mm−1) | 0.722 |
| Formula weight (g mol−1) | 566.63 | F (000) | 1200.0 |
| Crystal habit, color | Prism | Crystal size (mm) | 0.22 × 0.16 × 0.12 |
| Crystal system | Yellow | 2θ range for data collection (°) | 7.83 to 152.696 |
| Space group | P21/n | Reflections collected | 25203 |
| a (Å) | 11.3460(7) | Independent reflections | 6077 |
| b (Å) | 16.9005(8) | Rint | 0.0278 |
| c (Å) | 15.8105(10) | Number of parameters | 386 |
| α (°) | 90 | Goodness-of-fit on F2 | 1.043 |
| β (°) | 106.296(7) | Final R indexes [I ≥ 2σ(I)] | R1 = 0.0395, wR2 = 0.1062 |
| γ (°) | 90 | R Indexes [all data] | R1 = 0.0462, wR2 = 0.1121 |
| Volume (Å3) | 2909.9(3) | Largest difference peak and hole (e Å−3) | 0.21/−0.17 |
| Z, Z′ | 4, 0 | CCDC deposition no. | 2259912 |
CCDC 2259912, contains the supplementary crystallographic data for compound 3f. These data can be obtained free of charge from The Cambridge Crystallographic Data Centre via https://www.ccdc.cam.ac.uk/data_request/cif.
:1 mixture of ethyl acetate and petroleum ether. Pure layers were then concentrated under vacuum.
4-Hydroxy-1-phenyl-7,8-dihydroquinoline-2,5(1H,6H)-dione (Scheme 2, entry 3a). Brown powder; 55% yield; mp = 162–164 °C; Rf = 0.35 (AcOEt/petroleum ether, 60
:40). IR (KBr, cm−1): 3261.97, 3063.51, 2943.69, 2890.57, 1710.86, 1592.00, 1574.67, 1530.95, 1492.38; 1H-NMR (400 MHz, DMSO-d6): δ = 1.91 (p, 2H, J = 6.4 Hz, CH2), 2.43 (t, 2H, J = 6.2 Hz, CH2–C), 2.54 (t, 2H, J = 5.6 Hz, 2H, CH2–CO), 5.63 (s, 1H, CH), 7.23–7.42 (m, 2H, Ar-H), 7.42–7.64 (m, 3H, Ar-H), 12.71 (s, 1H, OH); 13C NMR (101 MHz, DMSO-d6): δ = 19.98, 28.86, 35.82, 95.86 (CH), 104.70, 128.14, 128.93, 129.48, 137.35, 162.12, 162.62 (N–C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), 167.24 (C–OH), 202.53 (CO); MS: (m/z) = 256 (M + 1); anal. calc. for C15H13NO3 C, 70.58; H, 5.13; N, 5.49; found: C, 70.62; H, 5.10; N, 5.44.
O), 167.24 (C–OH), 202.53 (CO); MS: (m/z) = 256 (M + 1); anal. calc. for C15H13NO3 C, 70.58; H, 5.13; N, 5.49; found: C, 70.62; H, 5.10; N, 5.44.
4-Hydroxy-1-(p-tolyl)-7,8-dihydroquinoline-2,5(1H,6H)-dione (Scheme 2, entry 3b). Crystal; 65% yield; mp = 222–224 °C; Rf = 0.45 (AcOEt/petroleum ether, 60
:40). IR (KBr, cm−1): 3391.87, 2957.15, 1655.76, 1607.02, 1511.88, 1441.96; 1H-NMR (400 MHz, CDCl3): δ = 1.97–2.03 (m, 2H, CH2), 2.42 (s, 3H, CH3), 2.47 (t, 2H, J = 6.2 Hz, CH2–C), 2.57 (t, 2H, J = 6.0 Hz, CH2–CO), 5.87 (s, 1H, CH), 7.05 (d, 2H, J = 8.2 Hz, Ar-H), 7.32 (d, 2H, J = 8.0 Hz, Ar-H), 12.43 (s, 1H, OH); 13C NMR (101 MHz, CDCl3): δ = 20.81, 21.36, 29.41, 36.59, 98.01 (CH), 105.98, 127.63, 130.82, 134.77, 139.70, 160.40, 164.05 (N–CO), 167.78 (C–OH), 201.59 (CO); MS: (m/z) = 270 (M + 1); anal. calc. for C16H15NO3 C, 71.36; H, 5.61; N, 5.20; C, 71.31; H, 5.64; N, 5.23.
1-(4-Chlorophenyl)-4-hydroxy-7,8-dihydroquinoline-2,5(1H,6H)-dione (Scheme 2, entry 3c). Crystal; 60% yield; mp = 240–242 °C; Rf = 0.62 (AcOEt/petroleum ether, 60
:40). IR (KBr, cm−1): 3258.20, 2924.99, 1673.74, 1532.55, 1489.79, 1403.58; 1H-NMR (400 MHz, DMSO-d6): δ = 1.92 (p, 2H, J = 6.3 Hz, CH2), 2.44 (t, 2H, J = 6.2 Hz, CH2–C), 2.54 (t, 2H, J = 6.0 Hz, CH2–CO), 5.64 (s, 1H, CH), 7.32–7.40 (m, 2H, Ar-H), 7.58–7.66 (m, 2H, Ar-H), 12.70 (s, 1H, OH); 13C NMR (101 MHz, D MSO-d6): δ = 19.96, 28.86, 35.82, 95.81 (CH), 104.82, 129.53, 130.21, 133.64, 136.18, 162.10, 162.52 (N–CO), 167.33 (C–OH), 202.53 (CO); MS: (m/z) = 290 (M + 1); anal. calc. for C15H12ClNO3 C, 62.19; H, 4.18; N, 4.83; found: C, 62.15; H, 4.14; N, 4.80.
1-(4-Fluorophenyl)-4-hydroxy-7,8-dihydroquinoline-2,5(1H,6H)-dione (Scheme 2, entry 3d). Crystal; 60% yield; mp = 226–228 °C; Rf = 0.49 (AcOEt/petroleum ether, 60
:40). IR (KBr, cm−1): 3398.70, 2921.08, 1728.10, 1661.78, 1605.05, 1584.15, 1559.12, 1401.03; 1H-NMR (400 MHz, CDCl3): δ = 1.97–2.08 (m, 2H, CH2), 2.48 (t, 2H, J = 6.2 Hz, CH2–C), 2.58 (t, 2H, J = 6.0 Hz, CH2–CO), 5.86 (s, 1H, CH), 7.13–7.19 (m, 2H, Ar-H), 7.19–7.25 (m, 2H, Ar-H), 12.43 (s, 1H, OH); 13C NMR (101 MHz, CDCl3): δ = 20.78, 29.45, 36.54, 98.02 (CH), 106.16, 117.17, 117.40, 129.82, 129.91, 133.19, 133.22, 160.13, 163.89 (N–CO), 167.89 (C–OH), 201.58 (CO); MS: (m/z) = 274 (M + 1); anal. calc. for C15H12FNO3 C, 65.93; H, 4.43; N, 5.13; found: C, 65.99; H, 4.47; N, 5.10.
4-Hydroxy-1-(2-methoxyphenyl)-7,8-dihydroquinoline-2,5(1H,6H)-dione (Scheme 2, entry 3e). Crystal; 65% yield; mp = 171–173 °C; Rf = 0.29 (AcOEt/petroleum ether, 60
:40). IR (KBr, cm−1): 3401.19, 2952.72, 1682.96, 1650.30, 1528.11, 1503.17, 1410.66; 1H-NMR (400 MHz, DMSO-d6): δ = 1.66–2.05 (m, 2H, CH2), 2.22–2.35 (m, 2H, CH2–C), 2.42–2.68 (m, 2H, CH2–CO), 3.76 (s, 3H, CH3), 5.61 (s, 1H, CH), 7.10 (td, 1H, J = 1.2, 7.6 Hz, Ar-H), 7.23 (dd, 2H, J = 1.7, 7.7 Hz, Ar-H), 7.49 (ddd, 1H, J = 1.7, 7.4, 8.3 Hz, Ar-H), 12.68 (s, 1H, OH); 13C NMR (101 MHz, DMSO-d6): δ = 19.97, 27.77, 35.82, 55.86, 95.83 (CH), 104.65, 112.57, 120.93, 125.47, 129.27, 130.72, 154.09, 162.10, 162.36 (N–CO), 167.19 (C–OH), 202.42 (CO); MS: (m/z) = 286 (M + 1); anal. calc. for C16H15NO4 C, 67.36; H, 5.30; N, 4.91; found: C, 67.31; H, 5.25; N, 4.87.
4-Hydroxy-7,7-dimethyl-1-phenyl-7,8-dihydroquinoline-2,5(1H,6H)-dione (Scheme 2, entry 3f). Crystal; 68% yield; mp = 210–212 °C; Rf = 0.47 (AcOEt/petroleum ether, 60
:40). IR (KBr, cm−1): 3429.88, 2963.91, 1676.72, 1592.40, 1536.24, 1455.51, 1405.54; 1H-NMR (400 MHz, CDCl3): δ = 1.03 (s, 6H, 2CH3), 2.32 (s, 2H, CH2–C), 2.44 (s, 2H, CH2–CO), 5.87 (s, 1H, CH), 7.13 (d, 2H, J = 7.2 Hz, Ar-H), 7.11–7.18 (m, 2H, Ar-H), 7.46–7.59 (m, 3H, Ar-H), 12.39 (s, 1H, OH); 13C NMR (101 MHz, CDCl3): δ = 28.14, 32.63, 42.81, 50.16, 97.91 (CH), 104.99, 128.03, 129.58, 130.26, 137.45, 158.73, 164.13 (N–CO), 167.62 (C–OH), 201.28 (CO); MS: (m/z) = 284 (M + 1); anal. calc. for C17H17NO3 C, 72.07; H, 6.05; N, 4.94; found: C, 72.10; H, 6.08; N, 4.99.
1-Benzyl-4-hydroxy-7,7-dimethyl-7,8-dihydroquinoline-2,5(1H,6H)-dione (Scheme 2, entry 3g). Crystal; 69% yield; mp = 168–170 °C; Rf = 0.64 (AcOEt/petroleum ether, 60
:40). IR (KBr, cm−1): 3350.20, 3031.30, 2954.97, 1659.17, 1632.99, 1586.34, 1443.85; 1H-NMR (400 MHz, DMSO-d6): δ = 0.92 (s, 6H, 2CH3), 2.48 (s, 2H, CH2–C), 2.84 (s, 2H, CH2–CO), 5.37 (s, 2H, N–CH2), 5.68 (s, 1H, CH), 7.13 (d, 2H, J = 7.2 Hz, Ar-H), 7.25–7.30 (m, 1H, Ar-H), 7.36 (dd, 2H, J = 4.6, 10.1 Hz, Ar-H), 12.65 (s, 1H, OH); 13C NMR (101 MHz, DMSO-d6): δ = 27.33, 31.85, 45.70, 48.78, 95.58 (CH), 104.08, 125.91, 127.21, 128.69, 136.27, 160.56, 162.70 (N–CO), 166.71 (C–OH), 202.00 (CO); MS: (m/z) = 298 (M + 1); anal. calc. for C18H19NO3 C, 72.71; H, 6.44; N, 4.71; found: C, 72.74; H, 6.40; N, 4.65.
1-(4-Fluorophenyl)-4-hydroxy-7,7-dimethyl-7,8-dihydroquinoline-2,5(1H,6H)-dione (Scheme 2, entry 3h). Yellow powder; 70% yield; mp = 193–195 °C; Rf = 0.66 (AcOEt/petroleum ether, 60
:40). IR (KBr, cm−1): 3373.70, 2956.29, 1665.73, 1625.38, 1526.83, 1508.41; 1H-NMR (400 MHz, CDCl3): δ = 1.04 (s, 6H, 2CH3), 2.31 (s, 2H, CH2–C), 2.44 (s, 2H, CH2–CO), 5.85 (s, 1H, CH), 7.11–7.14 (m, 2H, Ar-H), 7.20–7.25 (m, 2H, Ar-H), 12.37 (s, 1H, OH); 13C NMR (101 MHz, CDCl3): δ = 28.15, 32.63, 42.89, 50.07, 97.84 (CH), 105.11, 117.27, 117.50, 129.92, 133.20, 158.68, 161.63, 164.11 (N–CO), 167.67 (C–OH), 201.27 (CO); MS: (m/z) = 302 (M + 1); anal. calc. for C17H16FNO3 C, 67.76; H, 5.35; N, 4.65; found: C, 67.71; H, 5.34; N, 4.61.
4-Hydroxy-1-(4-methoxyphenyl)-7,7-dimethyl-7,8-dihydroquinoline-2,5(1H,6H)-dione (Scheme 2, entry 3i). Crystal; 71% yield; mp = 186–188 °C; Rf = 0.44 (AcOEt/petroleum ether, 60
:40). IR (KBr, cm−1): 3431.97, 2960.15, 1676.28, 1609.50, 1534.74, 1510.66, 1457.69, 1403.88; 1H-NMR (400 MHz, CDCl3): δ = 1.03 (s, 6H, 2CH3), 2.34 (s, 2H, CH2–C), 2.43 (s, 2H, CH2–CO), 3.86 (s, 3H, CH3), 5.86 (s, 1H, CH), 7.04 (s, 4H, Ar-H), 12.37 (s, 1H, OH); 13C NMR (101 MHz, CDCl3): δ = 28.15, 32.58, 42.87, 50.13, 55.68, 97.79 (CH), 104.98, 115.48, 128.98, 129.85, 159.26, 160.16, 164.41 (N–CO), 167.55 (C–OH), 201.28 (CO); MS: (m/z) = 314 (M + 1); anal. calc. for C18H19NO4 C, 69.00; H, 6.11; N, 4.47; found: C, 69.04; H, 6.13; N, 4.49.
4-Hydroxy-1-(2-methoxyphenyl)-7,7-dimethyl-7,8-dihydroquinoline-2,5(1H,6H)-dione (Scheme 2, entry 3j). Yellow powder; 61% yield; mp = 176–178 °C; Rf = 0.52 (AcOEt/petroleum ether, 60
:40). IR (KBr, cm−1): 3236.20, 2928.01, 1738.11, 1668.33, 1532.10, 1496.44, 1455.64; 1H-NMR (400 MHz, CDCl3): δ = 1.03 (d, 6H, J = 2.2 Hz, 2CH3), 2.23 (d, 1H, J = 17.6 Hz, CH–C), 2.36 (d, 2H, J = 17.6 Hz, CH–C), 2.43 (s, 2H, CH2–CO), 3.80 (s, 3H, CH3), 5.86 (s, 1H, CH), 7.04–7.17 (m, 3H, Ar-H), 7.47 (ddd, 1H, J = 3.5, 5.8, 8.3 Hz, Ar-H), 12.38 (s, 1H, OH); 13C NMR (101 MHz, CDCl3): δ = 27.74, 28.61, 32.47, 41.82, 50.19, 55.99, 97.76 (CH), 104.94, 112.55, 121.67, 125.90, 129.31, 131.17, 154.41, 159.75, 163.85 (N–CO), 167.63 (C–OH), 201.31 (CO); MS: (m/z) = 314 (M + 1); anal. calc. for C18H19NO4 C, 69.00; H, 6.11; N, 4.47; found: C, 69.05; H, 6.17; N, 4.50.
Anticancer activity evaluation
The human cancer cell lines HCT116 (colon carcinoma), A549 (lung carcinoma), PC3 (prostate carcinoma), MCF-7 (breast carcinoma), and normal fibroblasts were maintained in DMEM medium, supplemented with 10% fetal bovine serum (FBS), 1% penicillin–streptomycin, and 1% L-glutamine, as previously described.29 Cells were cultured at 37 °C in a 5% CO2 incubator. The test compounds were dissolved in dimethyl sulfoxide (DMSO) to prepare a 10 mM stock solution. The stock was further diluted in culture medium to achieve final concentrations ranging from 0.1 μM to 500 μM, ensuring the solvent concentration did not exceed 0.1%. Cells were seeded in 96-well plates at 10000 cells per well and incubated for 24 hours. After attachment, cells were treated with varying concentrations of the test compound and incubated for 48 hours. After treatment, 20 μL of MTT (5 mg mL−1) was added to each well, and cells were incubated for 4 hours. The formazan crystals formed were dissolved in 150 μL DMSO, and absorbance was measured at 570 nm using a microplate reader.
The cytotoxicity of the compound was assessed by calculating the IC50 (half-maximal inhibitory concentration) using GraphPad Prism software (version 8.0).30 Negative control cells were treated with DMSO, and positive controls were treated with cisplatin or doxorubicin. Data were presented as mean ± SD from three independent experiments (n = 9).
Computational methods
084 water molecules/35131 atoms for 3g-CDK2. To neutralize the system, sodium and chloride ions were added as counterions. Following solvation, energy minimization was performed with a convergence threshold of 1 kcal mol−1 Å−1, succeeded by pre-equilibration through the six-step relaxation protocol of Desmond. The first two steps involved energy minimization; initially restraining the solute, then removing these restraints followed by three short MD simulations lasting 12 ps, 12 ps, and 24 ps, respectively, under NPT ensemble conditions at temperatures of 10 K and 300 K. A subsequent 300 ns production run was conducted under periodic boundary conditions and isothermal–isobaric conditions with a relaxation time of 0.2 ps. Visualization of the protein–ligand complexes and analysis of the MD trajectory were carried out using Maestro. In-depth analyses were conducted using Simulation Interactions Diagram, offering a comprehensive exploration of the simulation data.37Results and discussion
Chemistry
The synthesis of interesting products using microwave irradiation41 in addition to their biological evaluation42,43 against multiple drug targets together with our interest grabbed by the remarkable versatility of β-enaminones as precursors to medicinally effective molecules and reactive chemical intermediates44,45 constituted the main axis of our previous works. In this study, we aim to extend this research by exploring the anticancer activity of prepared β-enaminone-derived N-heterocycles similar to 4-hydroxyquinolone ring.The general method for synthesizing these 4-hydroxyquinolone analogues 3(a–j) is evocated in Scheme 2. This protocol that is advantaged with its rapidity and respect for the environment is based on the condensation of simple and available reagents; namely previously prepared β-enaminones 1(a–j) and commercialized diethyl malonate 2 in the presence of ethanol. Reaction mixture was then subjected to microwave irradiation as an activation tool under the catalysis of bismuth chloride (BiCl3).28
 | ||
| Scheme 2 Synthesis of modified 4-hydroxyquinolone analogues. | ||
X-ray diffraction analysis
Suitable crystals of compound 3f were subjected to a total structural elucidation using a single crystal X-ray diffraction method.The resulting ORTEP diagram was drawn at 50% probability and is depicted in Fig. 3. Results showed that compound 3f 4-hydroxy-7,7-dimethyl-1-phenyl-7,8-dihydroquinoline-2,5(1H,6H)-dione crystallized in the monoclinic crystal system with P21/n space group as two molecules constituting the asymmetrical unit. The presence of the hydroxyl group allowed the formation of an intramolecular hydrogen bond with the ketone group of the adjacent cycle in both molecules of the asymmetrical unit forming two six-membered pseudocycles with S(6) graph sets. Distances of the stated hydrogen bonds were found to be in the range of 1.79 and 1.81 Å (Table 2). These observations are consistent with our previously reported crystal structure of an analogue of the same set.28
 | ||
| Fig. 3 ORTEP diagram for compound 3f. | ||
| D–H⋯A | d(D–H) | d(H⋯A) | d(D–A) | D–H–A | Symmetry |
|---|---|---|---|---|---|
| O2–H2⋯O3 | 0.820 | 1.798 | 2.538(2) | 149.25 | x, y, z |
| O5–H5A⋯O6 | 0.820 | 1.805 | 2.542(2) | 148.8 | x, y, z |
| C6–H6⋯O2 | 0.930 | 2.452 | 3.356(2) | 163.80 | x, y, z; −x, 1 − y, 2 − z |
| C2–H2A⋯O4 | 0.930 | 2.411 | 3.283(2) | 156.17 | x, y, z; −1 + x, y, z |
| C17–H17B⋯O6 | 0.960 | 2.685 | 3.644(2) | 177.3 | x, y, z; −x, 1 − y, 1 − z |
| C29–H29B⋯O1 | 0.970 | 2.598 | 3.234(2) | 123.26 | x, y, z; −1/2 + x, 1.5 − y, 1/2 + z |
| C19–H19⋯O1 | 0.930 | 2.699 | 3.480(2) | 142.13 | x, y, z; −1/2 + x, 1.5 − y, 1/2 + z |
| C31–H31A⋯O4 | 0.970 | 2.519 | 3.436(2) | 157.6 | x, y, z; −1/2 + x, 1.5 − y, −1/2 + z |
Several intermolecular hydrogen bonds were noticed in the crystal structure of compound 3f as shown in the crystal packing diagram (Fig. 4), with distances ranging between 2.41 and 2.70 Å (Table 2). The observed contacts permitted the reinforcement of the crystal components keeping the cohesion of the crystal.
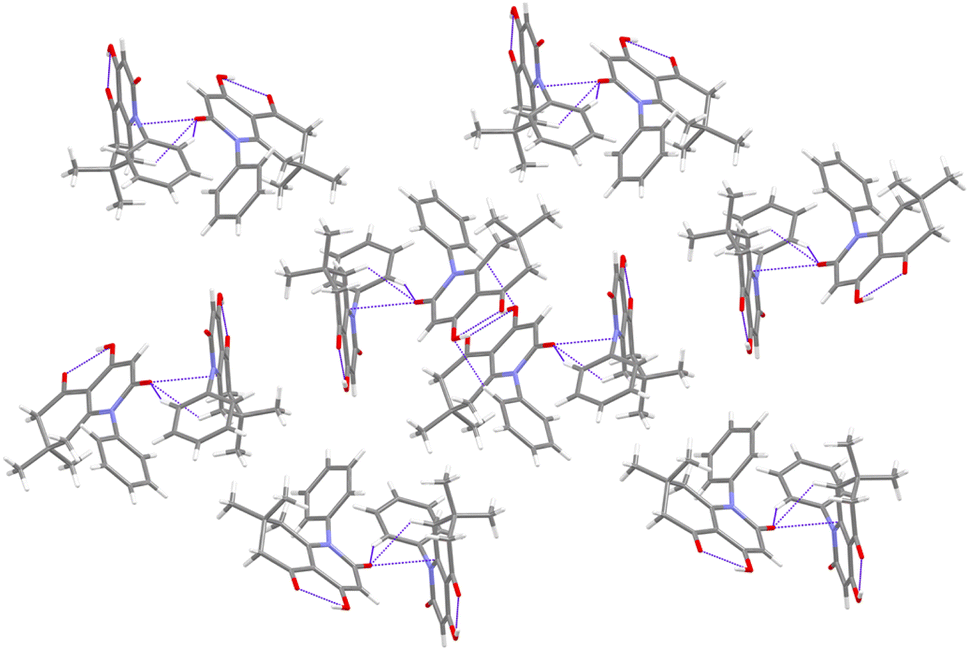 | ||
| Fig. 4 Packing diagram viewed along (a) axis. | ||
Anticancer evaluation
The anticancer activity of the modified 4-hydroxyquinolone analogues was assessed in vitro against four human cancer cell lines: HCT116 (colon carcinoma), A549 (lung carcinoma), PC3 (prostate carcinoma), and MCF-7 (breast carcinoma). The IC50 values, which represent the concentration of the compound required to inhibit 50% of cell viability, were determined for each compound across these cell lines (Table 3).| Entry | HCT116 | A549 | PC3 | MCF-7 | Fibroblasts |
|---|---|---|---|---|---|
| a SD never exceeded 5%. Data was collected for triplicates in three independent experiments, n = 9. | |||||
| 3a | 148.3 | 189.0 | 151.0 | 189.0 | >500 |
| 3b | 162.0 | 159.0 | 239.4 | 213.1 | >500 |
| 3c | 139.4 | 257.0 | 250.6 | 260.9 | >500 |
| 3d | 46.5 | 98.5 | 65.9 | 34.2 | >500 |
| 3e | 29.5 | 67.0 | 45.0 | 62.6 | >500 |
| 3f | 118.0 | 127.0 | 95.5 | 70.6 | >500 |
| 3g | 18.4 | 32.4 | 29.1 | 26.9 | >500 |
| 3h | 28.9 | 87.9 | 29.3 | 32.4 | >500 |
| 3i | 33.4 | 73.0 | 34.1 | 50.1 | >500 |
| 3j | 38.2 | 78.8 | 43.6 | 81.4 | >500 |
| Doxorubicin | 5.8 | — | — | 2.3 | — |
| Cisplatin | — | 17.0 | 23.5 | — | — |
Compound 3a showed relatively high IC50 values, ranging from 148.3 μM in HCT116 to 189 μM in MCF-7, indicating moderate activity. Similarly, compound 3b exhibited IC50 values of 162.0 μM (HCT116) to 239.4 μM (PC3), suggesting selective but moderate efficacy.
Compound 3d exhibited a potent anticancer activity, with IC50 values as low as 34.2 μM in MCF-7 and 46.5 μM in HCT116, suggesting strong inhibition of cell proliferation across all tested lines. Compound 3e also showed significant activity, with IC50 values ranging from 29.5 μM in HCT116 to 67.0 μM in A549, indicating its potential as an effective anticancer agent. Compound 3h exhibited IC50 values of 87.9 μM in A549 and 29.3 μM in PC3, while compound 3i showed promising effects with IC50 values ranging from 33.4 μM in HCT116 to 73.0 μM in A549. Compound 3j demonstrated moderate activity, with IC50 values ranging from 38.2 μM in HCT116 to 81.4 μM in MCF-7.
Finally, compound 3g exhibited the most potent anticancer activities across the cell lines, with IC50 values significantly lower than those of the other analogs, making it a promising candidate for further development in anticancer therapies. Importantly, the IC50 values of compound 3g are comparable to those of the clinically approved standard drugs, doxorubicin and cisplatin, as shown in Table 3.
All compounds (3a–3j) were also tested against normal fibroblast cells to evaluate their potential toxicity. At the examined concentrations, no significant cytotoxicity was observed in the normal fibroblast cells, indicating that the compounds did not exhibit harmful effects on non-cancerous cells within the tested concentration range.
In silico study
To this end, we selected two known drug targets highly linked to tumor growth; firstly, the anaplastic lymphoma kinase; an insulin receptor protein belonging to the superfamily of tyrosine kinase46 that participates in the pathogenesis of different diseases especially the NSCLC that refers to the non-small cell lung cancer presenting nearly 90% of lung cancers.47 Secondly, the cyclin-dependent kinase 2 that is widely partitioned in the human organism plays crucial roles in cell cycle transition and is a part of many cancers development due to abnormalities such as breast, lung, colorectal, prostate, and cervical carcinomas.48
A molecular docking study was guaranteed employing PDB files 5FTO and 6GUE corresponding to ALK in complex with Entrectinib and CDK2 in complex with AZD5438 respectively.
After completion of the molecular docking process, we were able to collect the docking scores of different investigated products inside the cavities of ALK and CDK2 (Table 4). Being reliable numerical indicators of ligand–protein stability, docking score values allowed the identification of the best ligands among the studied modified 4-hydoxyquinolones.
| Entry | ALK (5FTO) | CDK2 (6GUE) | ||||
|---|---|---|---|---|---|---|
| Docking scores (kcal mol−1) | H-bonds | Hydrophobic interactions | Docking scores (kcal mol−1) | H-bonds | Hydrophobic interactions | |
| 3a | −7.706 | Met1199, Glu1197 | Leu1122, Leu1256, Val1130, Phe1127, Ala1148, Val1180, Leu1196, Leu1198, Met1199 | −7.897 | Leu83 | Ala144, Ala31, Val18, Tyr15, Ileu10, Val64, Phe80, Phe82, Leu83, Leu134 |
| 3b | −7.697 | Met1199, Glu1197 | Leu1122, Leu1256, Val1130, Phe1127, Ala1148, Val1180, Leu1196, Leu1198, Met1199 | −7.612 | Leu83 | Ala144, Ala31, Val18, Tyr15, Ileu10, Val64, Phe80, Phe82, Leu83, Leu134 |
| 3c | −7.731 | Met1199 | Leu1122, Leu1256, Val1130, Phe1127, Ala1148, Val1180, Leu1196, Leu1198, Met1199 | −7.705 | Leu83 | Ala144, Ala31, Val18, Tyr15, Ileu10, Val64, Phe80, Phe82, Leu83, Leu134 |
| 3d | −7.192 | Met1199 | Leu1122, Leu1256, Val1130, Phe1127, Ala1148, Leu1196, Leu1198, Met1199 | −7.993 | Leu83 | Ala144, Ala31, Val18, Tyr15, Ileu10, Val64, Phe80, Phe82, Leu83, Leu134 |
| 3e | −7.471 | — | Leu1122, Phe1127, Val1130, Leu1256, Ala1200, Met1199, Leu1198, Leu1196, Ala1148 | −7.770 | Leu83 | Ala144, Ala31, Val18, Tyr15, Ileu10, Val64, Phe80, Phe82, Leu83, Leu134 |
| 3f | −7.935 | Arg1253 | Leu1122, Leu1256, Val1130, Phe1127, Ala1148, Leu1196, Met1199 | −7.982 | Lys33 | Ala144, Ala31, Val18, Tyr15, Ileu10, Val64, Phe80, Phe82, Leu83, Leu134 |
| 3g | −9.070 | Met1199, Glu1197 | Leu1122, Leu1256, Val1130, Phe1127, Ala1148, Val1180, Leu1196, Leu1198, Met1199, Cys1255 | −8.233 | Lys33, Leu83, Asp145 | Ala144, Ala31, Val18, Tyr15, Ileu10, Val64, Phe80, Phe82, Leu83, Leu134 |
| 3h | −8.266 | Arg1253 | Leu1122, Leu1256, Val1130, Phe1127, Ala1148, Leu1196, Leu1198, Met1199 | −8.028 | Lys33 | Ala144, Ala31, Val18, Tyr15, Ileu10, Val64, Phe80, Phe82, Leu83, Leu134 |
| 3i | −7.451 | — | Leu1122, Leu1256, Val1130, Phe1127, Ala1148, Ala1200, Leu1196, Leu1198, Met1199 | −7.889 | Lys33 | Ala144, Ala31, Val18, Tyr15, Ileu10, Val64, Phe80, Phe82, Leu83, Leu134 |
| 3j | −7.920 | Arg1253 | Leu1122, Leu1256, Val1130, Phe1127, Ala1148, Leu1196, Leu1198, Met1199 | −7.126 | Lys33 | Ala144, Ala31, Val18, Tyr15, Ileu10, Val64, Phe80, Phe82, Leu83, Leu134, Ileu135 |
| Entrectinib | −11.966 | Met1199, Glu1197 | Leu1122, Leu1256, Val1130, Phe1207, Ala1148, Ala1200, Leu1196, Leu1198, Met1199, Cys1255, Pro1260 | — | — | — |
| AZD5438 | — | — | — | −10.580 | Lys33, Leu83, Asp86 | Ala144, Ala31, Val18, Tyr15, Ileu10, Val64, Phe80, Phe82, Leu83 |
Docking scores of simulated ligand–protein complexes corresponding to the ALK protein ranged between −7.19 and −9.07 kcal mol−1 indicating an acceptable to good stability of our products inside the active site of ALK compared to the reference re-docked ligand, especially for the benzyl-containing compound 3g having the best score (−9.07 kcal mol−1). Most 4-hydroxyquinolone analogues derived from dimedone-based β-enaminones exhibited better affinities according to their docking score values which suggests a significant importance of the two methyl groups contained in their structures in enhancing the stability. In addition, several interactions including H-bonds were observed between the studied compounds and the residues of the ALK pocket contributing to the enhancement of their stability. Compounds 3c and 3d containing chlorine and fluorine respectively were implicated in one H-bond with Met1199. Further, compounds 3a, 3b, and 3g exhibiting the best docking score were involved in two H-bonds formation with both Met1199 and Glu1197 (Table 5) which are considered key residues of the active site since the co-crystalized ligand Entrectinib exhibited H-bonds with the same two residues. Further, the stability of the studied compounds inside the cavity of ALK was enhanced by significant hydrophobic interactions with several key residues including Leu1122, Leu1256, Val1130, Phe1127, Ala1148, Ala1200, Leu1196, Leu1198, Met1199, and Cys1255. Compounds 3f, 3h, and 3j engaged in Pi–Pi stacking with the side chain of Phe1127 (Tables 4 and S1 ESI†).





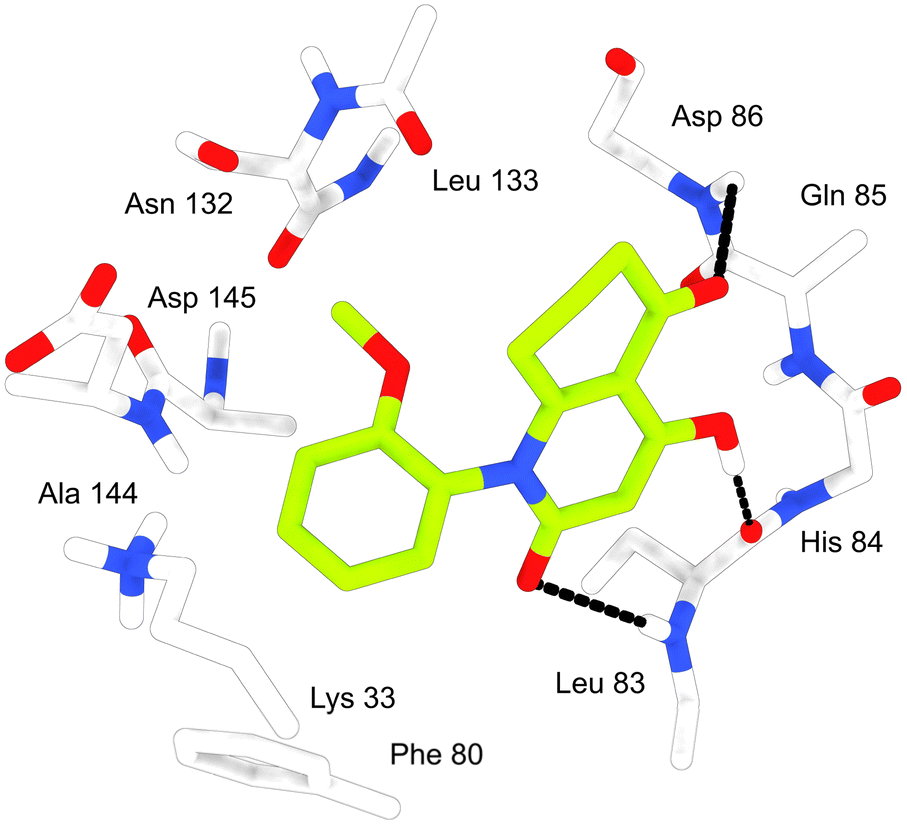




In the case of CDK2, results were comparable to the ALK results with docking scores lying from −7.12 to −8.24 kcal mol−1 which corresponds to acceptable values of affinities. As noticed in the docking study using ALK as the target, compound 3g exhibited the best results in the docking study involving CDK2 as well. Compounds 3a, 3b, 3c, 3d, and 3e displayed an H-bond with Leu83 residue, besides, derivatives 3f, 3h, 3i, and 3j were able to contribute to H-bond formation with Lys33. Among the studied compounds only derivative 3g presenting the best docking score succeeded in the formation of 3 H-bonds with Leu83, Lys33, and Asp145. It is important to point out that the co-crystalized ligand AZD5438 also exhibited 3 H-bonds with Leu83, Lys33, and Asp86 making compound 3g the most analogous to the reference ligand in terms of demonstrated interactions. Besides, all compounds were involved in important hydrophobic interactions with key residues of the CDK2 active site (Ala144, Ala31, Val18, Tyr15, Ileu10, Val64, Phe80, Phe82, Leu83, Leu134, and Ileu135) contributing in the stability of the formed ligand-protein complexes (Tables 4 and S1 ESI data†).
According to the promising results and to optimize the studied derivatives as effective drug candidates with enhanced activity, we propose modifying the substitutions on the nitrogen atom. Notably, the 3g derivative exhibits the highest activity in both in vitro and in silico studies. This increased activity is likely attributed to the structural difference in 3g, where a methylene group separates the nitrogen atom from the aromatic nucleus, unlike the other derivatives.
This separation reduces steric hindrance, allowing the 3g molecule to position itself more effectively within the active site pockets of CDK2 and ALK. Consequently, we are exploring the use of aliphatic or aromatic amines linked via side chains to further improve activity.
The RMSD plot for the 3g-ALK complex demonstrated remarkable stability, with minimal fluctuations observed in the protein structure. These small variations suggest that the protein likely stabilized around a consistent thermal average during the second half of the simulation. This behavior indicates that the protein reached equilibrium, a critical prerequisite for the meaningful interpretation of binding interactions and conformational analyses.
Notably, the RMSD range for the system did not exceed 0.2 Å. Similarly, the RMSD plot for the ligand exhibited a highly stable trajectory, indicating that the ligand remained securely bound to the protein's active site without significant displacement. This observed stability strongly supports the high affinity of the 3g ligand and its proper fit within the binding pocket of the ALK enzyme (Fig. 5).
 | ||
| Fig. 5 RMSD (3g-ALK: left, 3g-CDK2: right). | ||
For the second system, 3g-CDK2, the ligand demonstrated significant stability within the active pocket throughout the simulation. From 0 to 250 ns, the alignment between the C-alpha atoms and the ligand-fitted protein was highly stable, with the RMSD remaining close to 0 Å. After 250 ns, a slight deviation in the ligand was observed, with the RMSD increasing to 2.5 Å. Despite this deviation, the RMSD consistently stayed below 3 Å, indicating that the ligand maintained stable interactions within the binding site across the entire simulation.
These findings underscore the strong affinity and stability of ligand 3g for CDK2 enzyme. The minimal fluctuations and sustained alignment suggest that forms strong interactions with the active site without disrupting the structural integrity of the protein. This consistent stability highlights the ligand's potential for targeted therapeutic applications, as it maintains its binding efficiency and supports the functional conformation of protein throughout the simulation period (Fig. 5).
In the analysis of the obtained results for the two studied systems, 3g-ALK and 3g-CDK2, no significant fluctuations were observed in the curve of the atomic displacement, indicating consistency between simulated and experimental flexibility metrics.
These findings confirm the success of the simulation, as the RMSF and B-factor curves remained parallel throughout most of the 300 ns trajectory. The overall RMSF patterns for both ALK and CDK2 systems demonstrated structural stability during the simulation. Notably, residues critical for ligand binding exhibited low RMSF values in both systems, indicating that the ligand 3g effectively stabilized the binding pockets of the enzymes.
Additionally, both systems showed an RMSF range within acceptable limits for stable proteins, with significant flexibility restricted to loop regions and terminal residues. This localized flexibility likely facilitates the functional dynamics of the proteins while preserving their overall structural integrity (Fig. 6).
 | ||
| Fig. 6 RMSF (3g-ALK: left, 3g-CDK2: right). | ||
The radius of gyration (rGyr) reflects the spatial distribution of a molecule's atoms around its center of mass. For ligands, rGyr provides insights into compactness and conformational changes during a simulation. A lower rGyr indicates that the ligand adopts a more compact and stable conformation, which is generally favorable for binding as compact ligands tend to fit more snugly into binding pockets. Variations in rGyr over the simulation reflect flexibility. While moderate flexibility can enable the ligand to adapt to the binding pocket of the protein, excessive flexibility might indicate instability. The rGyr results for ligand 3g in the active pockets of the two studied enzymes, ALK and CDK2, demonstrated significant stability during the 300 ns simulations. In both systems, the rGyr remained consistently within the range of 3.5–3.8 Å, with a very small deviation of approximately 0.3 Å. This limited fluctuation suggests that the ligand 3g maintained a stable and compact conformation throughout the simulation (Fig. 7).
 | ||
| Fig. 7 Radius of gyration (3g-ALK: left, 3g-CDK2: right). | ||
All contacts identified through molecular docking were preserved during molecular dynamics simulations for the two studied complexes, 3g-ALK and 3g-CDK2, indicating the reliability of the molecular docking results. For the ALK enzyme, ligand 3g demonstrated the formation of three hydrogen bonds observed in the docking study, along with an additional hydrogen bond involving Lys 1150 and the carbonyl oxygen, with a persistence rate of 42%. This residue also participated in a Pi–cation interaction with the benzylic ring at 21%. Additionally, all hydrophobic interactions identified during docking were maintained throughout the simulation.
For the CDK2 target, ligand 3g retained the three hydrogen bonds observed in the molecular docking study and formed an additional hydrogen bond with Glu 81. Furthermore, ligand 3g exhibited strong hydrophobic interactions with key residues, including Leu 134, Tyr 15, Ala 131, and other residues within the active pocket of CDK2. These interactions highlight the stability of ligand 3g within the binding sites of CDK2, further validating its potential as a therapeutic candidate (Fig. 8).
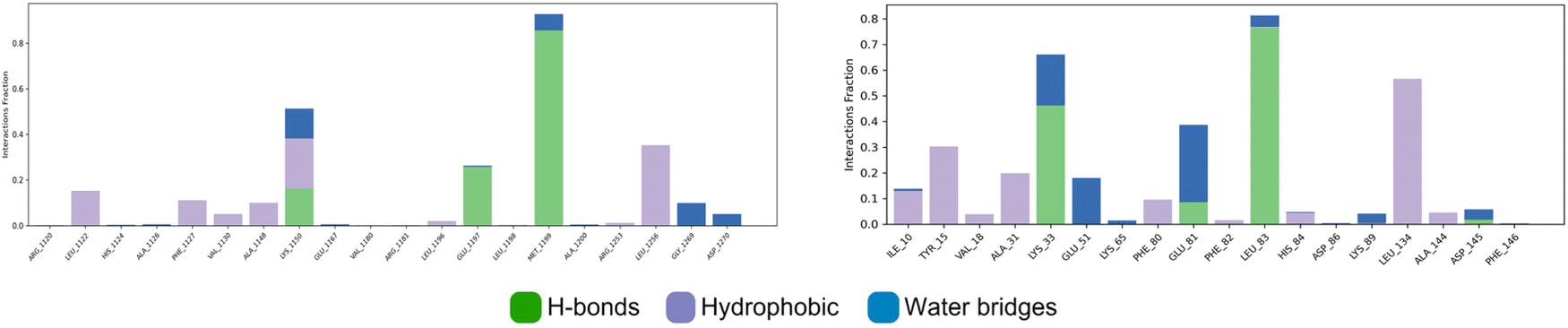 | ||
| Fig. 8 P–L contacts (3g-ALK: left, 3g-CDK2: right). | ||
| Entry | μ (D) | EHOMO (eV) | ELUMO (eV) | ΔEgap | (η) | (S) | (μ) | (χ) | (ω) |
|---|---|---|---|---|---|---|---|---|---|
| 3a | 4.0139 | −5.9601 | −1.2267 | 4.7334 | 2.3667 | 0.4225 | −3.5934 | 3.5934 | 2.7279 |
| 3b | 4.4347 | −5.9171 | −1.1823 | 4.7348 | 2.3674 | 0.4224 | −3.5497 | 3.5497 | 2.6613 |
| 3c | 2.5369 | −6.1011 | −1.3818 | 4.7193 | 2.3596 | 0.4238 | −3.7414 | 3.7414 | 2.9662 |
| 3d | 2.8784 | −6.0475 | −1.3124 | 4.7351 | 2.3675 | 0.4224 | −3.6799 | 3.6799 | 2.8599 |
| 3e | 4.9604 | −5.8115 | −1.0754 | 4.7361 | 2.3681 | 0.4223 | −3.4435 | 3.4435 | 2.5036 |
| 3f | 4.0406 | −5.9416 | −1.2104 | 4.7312 | 2.3656 | 0.4227 | −3.5760 | 3.5760 | 2.7028 |
| 3g | 3.4627 | −5.9977 | −1.2139 | 4.7838 | 2.3919 | 0.4181 | −3.6058 | 3.6058 | 2.7179 |
| 3h | 2.9442 | −6.0279 | −1.2961 | 4.7318 | 2.3659 | 0.4227 | −3.6620 | 3.6620 | 2.8340 |
| 3i | 7.3766 | −5.8115 | −1.5742 | 4.2374 | 2.1187 | 0.4720 | −3.6929 | 3.6929 | 3.2183 |
| 3j | 4.7610 | −8.9558 | −5.0513 | 3.9046 | 1.9523 | 0.5122 | −7.0035 | 7.0035 | 12.5621 |
Dipolar moments of investigated compounds ranged between 2.5369 and 7.3766 D starting from the chlorine-substituted derivative 3c to the methoxy-substituted derivative 3i as the less and most polar compounds respectively. Optimized structures and FMOs graphical representations of all synthesized 4-hydroxyquinolone analogues are summarized in Table 7.
| Code | Optimized structure | HOMO | LUMO |
|---|---|---|---|
| 3a |  |
 |
 |
| 3b |  |
 |
 |
| 3c |  |
 |
 |
| 3d |  |
 |
 |
| 3e | 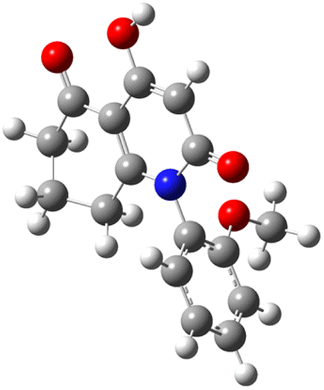 |
 |
 |
| 3f |  |
 |
 |
| 3g |  |
 |
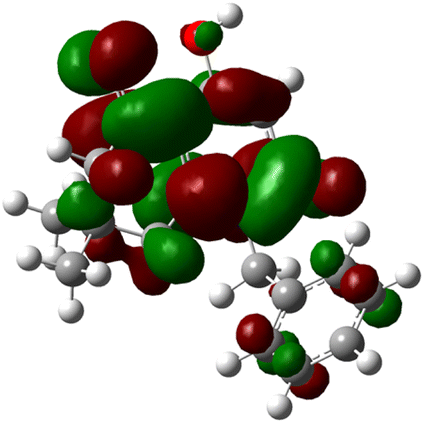 |
| 3h |  |
 |
 |
| 3i | 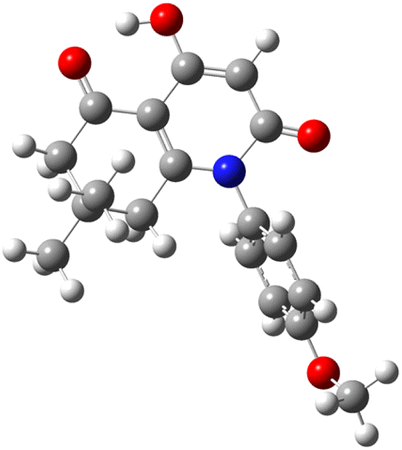 |
 |
 |
| 3j |  |
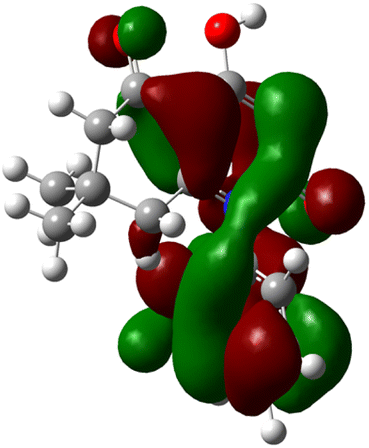 |
 |
The energy gap values between HOMO and LUMO for studied compounds were located in the interval of [3.9046–4.7838 eV], in which compound 3g is the most stable among studied derivatives.
Additionally, HOMO and LUMO orbitals as presented in Table 7 are situated in the key functional groups of the heterocyclic core of the investigated compounds including the amide and the enolic part as well as the carbonyl of the cyclohexene ring suggesting a high ability of these parts to donate and accept electrons permitting their interactions with molecular targets through electron transfer.
MEP of investigated compounds were represented as maps with color codes lying between the deepest red showing the most nucleophilic parts and the deepest blue showing the most electrophilic ones (Fig. 9).Generally, red color was situated in the carbonyl groups corresponding to ketone and amide moieties. It can be assumed that these groups have a high potency to form interactions by acting as proton acceptors in H-bonds formation. This observation is reliable with the molecular docking results since carbonyl groups were involved in H-bond formation. Moreover, red color corresponding to electron-rich parts of the molecules was also located on the oxygen atom of the hydroxyl group. In addition, blue color that indicates electron-deficient regions was strongly located in hydrogen atoms of the hydroxyl groups suggesting a high electronegativity gap between the two mentioned atoms, thus a significant lability of corresponding hydrogens and the ability of these latter to act as H-bond donors, which was also noticed in molecular docking results in which the hydroxyl group acted as proton donor in H-bond formation with several residues of the corresponding active sites.
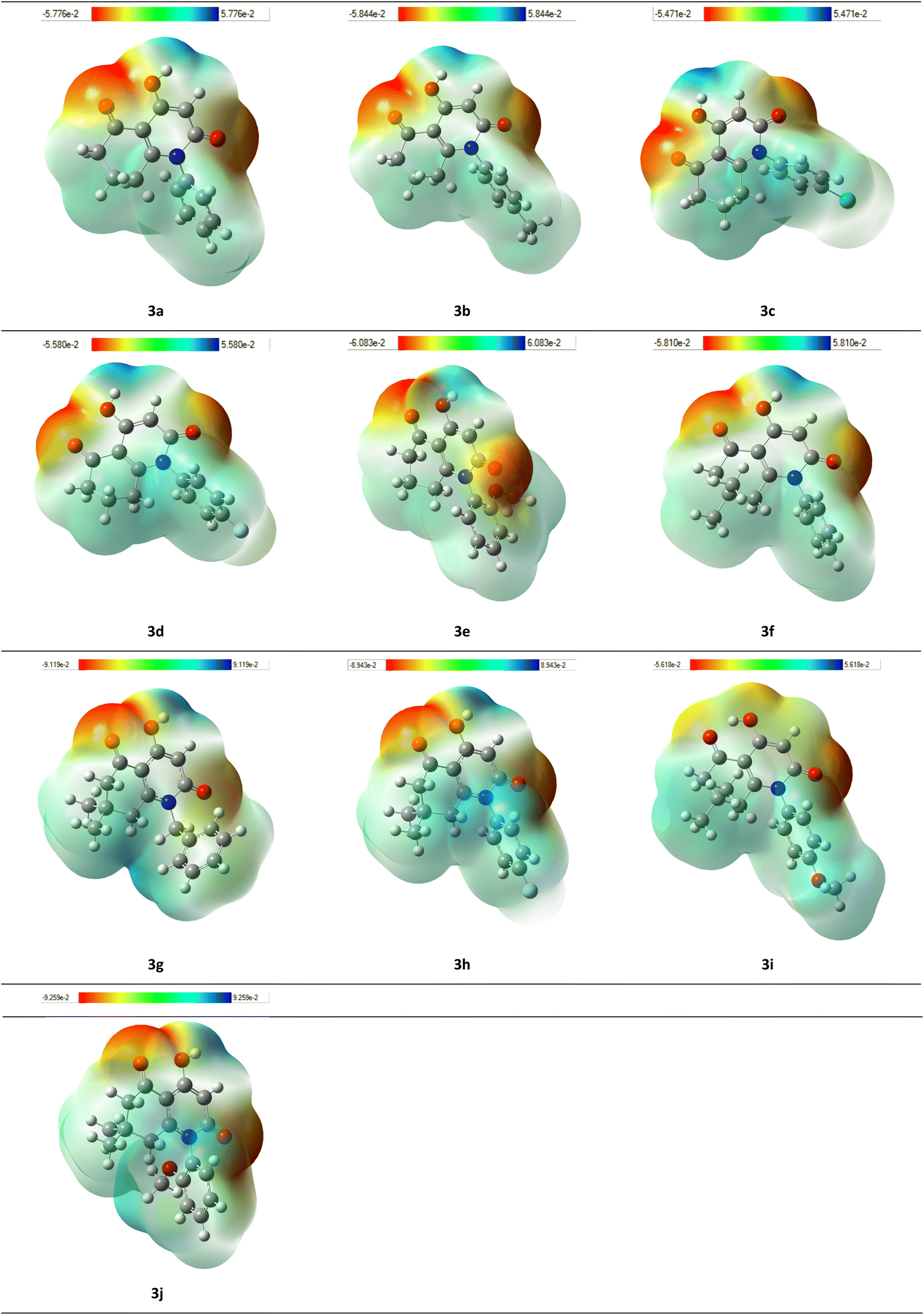 | ||
| Fig. 9 MEP maps of synthesized 4-hydroxyquinolone analogues. | ||
Compound 3g presents a balance between electronic stability and reactivity characterized by a relatively high HOMO–LUMO energy gap of around 4.78 eV. The electronic stability is likely provided by the resonance of the benzyl group as well as the presence of the hydroxyl group that can enhance stability through H-bond formation. On the other hand, the hydroxyl group and the two methyl groups in 3g contributes in donating electron density to the dihydroquinolone core while the amide and carbonyl functional groups tend to withdraw electron density, these features are of a great interest in terms of reactivity since it enables the compound to engage in both nucleophilic and electrophilic reactions, in that way, resulting in interactions with specific residues of significant molecular targets.
In view of the DFT results including energy gap as well as the HOMO, LUMO, and MEP representations correlated to the observed interactions in the molecular docking assessment, the main parts of the studied molecules are clearly identified as the functional groups of the nitrogen-based heterocycle especially the hydroxyl group which played the role of an excellent H-bond donor and acceptor. In addition to hydroxyl, the amide and carbonyl groups were also identified as key parts of the molecule participating in the interactions, thereby, contributing to the activity.
Conclusions
Anticancer activity evaluation was performed on synthesized derivatives analogous to 4-hydroxyquinolone core. The synthesis of these products was carried out using a previously reported procedure described by our group involving simple synthons; diethyl malonate and easily prepared β-enaminones. The in vitro anticancer activity was evaluated on prepared 4-hydroxyquinolone analogues employing four cancerous cell lines; HCT116, A549, PC3, and MCF-7. Results showed that several derivatives of the studied set of compounds were able to inhibit the growth of the aforementioned cell lines especially compound 3g containing the benzyl group. The molecular docking study that was performed on synthesized products involving known protein kinases; ALK and CDK2 showed promising results with acceptable to good docking scores in which the 3g derivative was observed as the most potent. The latter exhibiting good docking scores was able to form significant interactions with key residues of the active sites of both ALK and CDK2 owing to the presence of carbonyl and hydroxyl groups within its structure. It is worth-mentioning that the MEP maps generated using DFT identified the carbonyl and hydroxyl groups respectively as the most reactive nucleophilic and electrophilic parts of the molecules. These results displayed a clear consistency between the in vitro and in silico investigations. This study enabled us to prove once more the importance of β-enaminones as precursors to interesting compounds besides to the high interest of N-heterocycles in drug design. In view of the observed results, it is important to further explore the core of tested 4-hydroxyquinolone analogues in cancer treatment through bringing structural modifications especially on the 3g derivative, which could enhance the activity while leaning on different in silico predictable tools as well as past bibliographic observations.Data availability
All data generated or analysed during this study are included in this published article and its ESI.† Crystallographic data for 3f has been deposited at the Cambridge Crystallographic Data Centre CCDC under CCDC 2259912 number and can be obtained from [https://doi.org/10.5517/ccdc.csd.cc2fvmdb].Author contributions
Y. O. B. synthesis and characterization of compounds, DFT study, manuscript writing, A. B. molecular docking, molecular dynamics simulation, manuscript writing and reviewing, A. D. in vitro tests, S. K. B. determining and discussing the anticancer activity, writing and revising the manuscript. M. I. O. analyzed the crystallography data, B. B. contributing to the review of the paper, F. B. contributing to the review of the paper, N. E. A. contributing to the review of the paper.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was supported financially by The General Directorate for Scientific Research and Technological Development (DG-RSDT), Algerian Ministry of Scientific Research, Applied Organic Chemistry Laboratory (FNR 2000). PRFU Project: B00L01UN230120230002.Notes and references
- F. Bray, M. Laversanne, H. Sung, J. Ferlay, R. L. Siegel, I. Soerjomataram and A. Jemal, Ca-Cancer J. Clin., 2024, 74, 229 CrossRef PubMed.
- M. Siddiqui and S. V. Rajkumar, Mayo Clin. Proc., 2012, 87, 935 CrossRef PubMed.
- L. Zhong, Y. Li, L. Xiong, W. Wang, M. Wu, T. Yuan, W. Yang, C. Tian, Z. Miao, T. Wang and S. Yang, Signal Transduction Targeted Ther., 2021, 6, 1 CrossRef PubMed.
- R. Roskoski Jr, Pharmacol. Res., 2024, 107059 CrossRef PubMed.
- (a) L. Maram and F. Tanaka, Org. Lett., 2020, 22, 2751 CrossRef CAS PubMed; (b) L. Maram and F. Tanaka, Org. Lett., 2019, 21, 1165 CrossRef CAS PubMed.
- L. Maram, J. M. Michael, H. Politte, V. S. Srirama, A. Hadji, M. Habibi, M. O. Kelly, R. T. Brookheart, B. N. Finck, L. Hegazy, K. S. McCommis and B. Elgendy, Eur. J. Med. Chem., 2025, 283, 117150 CrossRef CAS PubMed.
- J. Akhtar, A. A. Khan, Z. Ali, R. Haider and M. S. Yar, Eur. J. Med. Chem., 2017, 125, 143 CrossRef CAS PubMed.
- P. Martins, J. Jesus, S. Santos, L. R. Raposo, C. R. Rodrigues, P. V. Baptista and A. R. Fernandes, Molecules, 2015, 20, 16852 CrossRef CAS PubMed.
- R. V. Patel, Y. S. Keum and S. W. Park, Eur. J. Med. Chem., 2015, 97, 649 CrossRef CAS PubMed.
- K. Chaudhari, S. Surana, P. Jain and H. M. Patel, Eur. J. Med. Chem., 2016, 124, 160 CrossRef CAS PubMed.
- B. Sameem, M. Saeedi, M. Mahdavi and A. Shafiee, Eur. J. Med. Chem., 2017, 128, 332 CrossRef CAS PubMed.
- R. Kaur, L. Dahiya and M. Kumar, Eur. J. Med. Chem., 2017, 141, 473 CrossRef CAS PubMed.
- N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, Molecules, 2020, 25, 1909 CrossRef CAS PubMed.
- X. M. Chu, C. Wang, W. Liu, L. L. Liang, K. K. Gong, C. Y. Zhao and K. L. Sun, Eur. J. Med. Chem., 2019, 161, 101 CrossRef CAS PubMed.
- S. Jain, V. Chandra, P. K. Jain, K. Pathak, D. Pathak and A. Vaidya, Arabian J. Chem., 2019, 12, 4920 CrossRef CAS.
- S. A. Meo, D. C. Klonoff and J. Akram, Eur. Rev. Med. Pharmacol. Sci., 2020, 24, 4539 CAS.
- J. Zhang, S. Wang, Y. Ba and Z. Xu, Eur. J. Med. Chem., 2019, 174, 1 CrossRef CAS PubMed.
- D. Sharma, M. Kumar and P. Das, Synth. Commun., 2021, 51, 2553 CrossRef CAS.
- M. Kaya, E. Demir and H. Bekci, J. Enzyme Inhib. Med. Chem., 2013, 28, 885 CrossRef CAS PubMed.
- E. K. Aslan, K. Lam, C. Dengiz, K. Denzinger, I. Y. D. Erdamar, S. Huang, G. W. Zamponi, G. Wolber and M. G. Gündüz, J. Mol. Struct., 2024, 1307, 137983 CrossRef.
- G. Periyasami, P. Antonisamy, K. Perumal, A. Stalin, M. Rahaman and A. A. Alothman, Bioorg. Chem., 2019, 90, 103047 CrossRef CAS PubMed.
- G. M. Sheldrick, Acta Crystallogr., Sect. A:Found. Adv., 2015, 71, 3 CrossRef PubMed.
- O. V. Dolomanov, L. J. Bourhis, R. J. Gildea, J. A. K. Howard and H. Puschmann, J. Appl. Crystallogr., 2009, 42, 339 CrossRef CAS.
- G. M. Sheldrick, Acta Crystallogr., Sect. C:Struct. Chem., 2015, 71, 3 Search PubMed.
- Rigaku Oxford Diffraction, CrysAlisPro Software System, 2022.
- C. F. Macrae, I. Sovago, S. J. Cottrell, P. T. A. Galek, P. McCabe, E. Pidcock, M. Platings, G. P. Shields, J. S. Stevens, M. Towler and P. A. Wood, J. Appl. Crystallogr., 2020, 53, 226 CrossRef CAS PubMed.
- R. Redjemia, A. Bouzina, Y. O. Bouone, A. Mansouri, R. Bahadi and M. Berredjem, Res. Chem. Intermed., 2022, 48, 4947 CrossRef CAS.
- Y. O. Bouone, A. Bouzina, R. Sayad, A. Djemel, F. Benaceur, A. Zoukel, M. Ibrahim-Ouali, N.-E. Aouf and F. Bouchareb, RSC Adv., 2023, 13, 28030 RSC.
- D. A. Sabbah, S. E. Hasan, R. Abu Khalaf, S. K. Bardaweel, R. Hajjo, K. M. Alqaisi, K. A. Sweidan and A. M. Al-Zuheiri, Molecules, 2020, 25, 5348 CrossRef CAS PubMed.
- (a) N. Teggar, B. Bakchiche, M. E. S. Abdel-Aziz, S. K. Bardaweel and M. A. Ghareeb, Jordan J. Pharm. Sci., 2023, 16, 184 CrossRef; (b) L. Alsous and S. Bardaweel, Adv. Anticancer Agents Med. Chem., 2022, 22, 1826 CrossRef CAS PubMed.
- M. Menichincheri, E. Ardini, P. Magnaghi, N. Avanzi, P. Banfi, R. Bossi, L. Buffa, G. Canevari, L. Ceriani, M. Colombo, L. Corti, D. Donati, M. Fasolini, E. Felder, C. Fiorelli, F. Fiorentini, A. Galvani, A. Isacchi, A. L. Borgia, C. Marchionni, M. Nesi, C. Orrenius, A. Panzeri, E. Pesenti, L. Rusconi, M. B. Saccardo, E. Vanotti, E. Perrone and P. Orsini, J. Med. Chem., 2016, 59, 3392 CrossRef CAS PubMed.
- D. J. Wood, S. Korolchuk, N. J. Tatum, L. Z. Wang, J. A. Endicott, M. E. M. Noble and M. P. Martin, Cell Chem. Biol., 2019, 26, 121 CrossRef CAS PubMed.
- Release S, 2 LigPrep, version 3.4. Schrödinger, LLC, New York, NY, 2015, 26400175.
- R. A. Friesner, J. L. Banks, R. B. Murphy, T. A. Halgren, J. J. Klicic, D. T. Mainz, M. P. Repasky, E. H. Knoll, D. E. Shaw, M. Shelley, J. K. Perry, P. Francis and P. S. Shenkin, J. Med. Chem., 2004, 47, 1739 CrossRef CAS PubMed.
- (a) E. C. Meng, T. D. Goddard, E. F. Pettersen, G. S. Couch, Z. J. Pearson, J. H. Morris and T. E. Ferrin, Protein Sci., 2023, 32, 4792 CrossRef PubMed; (b) E. F. Pettersen, T. D. Goddard, C. C. Huang, E. C. Meng, G. S. Couch, T. I. Croll, J. H. Morris and T. E. Ferrin, Protein Sci., 2021, 30, 70 CrossRef CAS PubMed.
- (a) K. J. Bowers, E. Chow, H. Xu, R. O. Dror, M. P. Eastwood, B. A. Gregersen, J. L. Klepeis, I. Kolossvary, M. A. Moraes, F. D. Sacerdoti, J. K. Salmon, Y. Shan and D. E. Shaw, Proceedings of the ACM/IEEE Conference on Supercomputing (SC06), Tampa, Florida, 2006, November 11-17 Search PubMed; (b) Schrödinger Release 2024-3: Desmond Molecular Dynamics System, D. E. Shaw Research, New York, NY, 2024.
- Schrödinger Release 2023-4: Maestro, Schrödinger, LLC, New York, NY, 2024.
- (a) A. D. Becke, J. Chem. Phys., 1993, 98, 5648 CrossRef CAS; (b) M. M. Francl, W. J. Pietro, W. J. Hehre, J. S. Binkley, M. S. Gordon, D. J. DeFrees and J. A. Pople, J. Chem. Phys., 1982, 77, 3654 CrossRef CAS.
- M. J. Frisch, Gaussian-09 Revision A.02, Gaussian Inc, Wallingford CT, 2009 Search PubMed.
- G. Zhang and C. B. Musgrave, J. Phys. Chem. A, 2007, 111, 1554 CrossRef CAS PubMed.
- A. Bouzina, M. Berredjem, B. Belhani, S. Bouacida, C. Marminon, M. Le Borgne, Z. Bouaziz and M. Aissaoui, Res. Chem. Intermed., 2021, 47, 1359 CrossRef CAS.
- A. Bouzina, Y. O. Bouone, O. Sekiou, M. Aissaoui, T. S. Ouk, A. Djemel, R. Mansouri, M. Ibrahim-Ouali, Z. Bouslama and N. E. Aouf, RSC Adv., 2023, 13, 19567 RSC.
- A. Bouzina, A. Djemel, O. Sekiou, I. Kadi, Y. O. Bouone, R. Mansouri, Z. Aouf, M. Ibrahim-Ouali and N.-E. Aouf, J. Mol. Struct., 2023, 1285, 135527 CrossRef CAS.
- I. J. Amaye, R. D. Haywood, E. M. Mandzo, J. J. Wirick and P. L. Jackson-Ayotunde, Tetrahedron, 2021, 83, 131984 CrossRef CAS.
- Y. O. Bouone, A. Bouzina, N.-E. Aouf and M. Ibrahim-Ouali, Res. Chem. Intermed., 2023, 49, 1349 CrossRef CAS.
- R. Roskoski Jr, Pharmacol. Res., 2017, 117, 343 CrossRef PubMed.
- R. Roskoski Jr, Pharmacol. Res., 2013, 68, 68 CrossRef PubMed.
- S. Ghafouri-Fard, T. Khoshbakht, B. M. Hussen, P. Dong, N. Gassler, M. Taheri, A. Baniahmad and N. A. Dilmaghani, Cancer Cell Int., 2022, 22, 325 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. CCDC 2259912. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5ra00252d |
| This journal is © The Royal Society of Chemistry 2025 |