
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSimultaneous H2 production and water purification with surface-modified nanostructured TiO2 photoelectrodes†
Enrica Maria Malannata ,
Alessandro Auditore*,
Roberto Fiorenza,
Maria Teresa Armeli Iapichino,
Francesca Lo Presti,
Nunzio Tuccitto and
Antonino Licciardello
,
Alessandro Auditore*,
Roberto Fiorenza,
Maria Teresa Armeli Iapichino,
Francesca Lo Presti,
Nunzio Tuccitto and
Antonino Licciardello
Dipartimento di Scienze Chimiche, Università di Catania, Viale A. Doria 6, 95125 Catania, Italy. E-mail: alessandro.auditore@unict.it
First published on 2nd May 2025
Abstract
The removal of emerging contaminants from water and production of green energy are some of the pressing needs of today's world. The use of water pollutants for the production of H2 can be a powerful strategy for solving both these problems. Several approaches have been proposed for this purpose, such as photocatalysis, electrocatalysis and photoelectrocatalysis. In this context, TiO2 is the most commonly used material, but it has several performance limitations. However, they can be improved with appropriate surface modifications. In this work, the inner surfaces of nanostructured TiO2-based films were modified to improve their photoelectrocatalytic and photocatalytic performances, with an aim to simultaneously remove a water pollutant (rhodamine B dye) and generate H2 in a custom-designed dual-chamber reactor. For this purpose, the TiO2 nanostructure, which can be used as a photoanode in photocatalytic and photoelectrocatalytic experiments, was functionalized by introducing a zirconium phosphate monolayer (ZP modification). The excellent performances of the ZP-modified photoanodes in simultaneously achieving photoelectrocatalytic dye removal and H2 evolution indicate that they are interesting candidates for attaining sustainable and circular solutions for environmental protection.
Introduction
The demand for better environmental quality and carbon-neutral economy have become key issues in our society. In this context, water pollution is an increasingly serious and urgent problem that must be solved. The presence of several contaminants in water, such as pesticides, pharmaceuticals, microplastics, and organic dyes (emerging pollutants, EPs) can cause negative effects on the environment and human health, and hence, their removal is crucial.1At the same time, to reduce dependence on fossil fuels and to reconcile the ever-increasing demand for energy with environmental concerns, the development of clean energy sources is imperative. Energy production needs to follow green processes by avoiding the use of fossil fuels and using sustainable energy carriers such as, molecular hydrogen. H2 is considered a promising energy vector that can drive the transition from a fossil fuel-based economy to near zero anthropogenic carbon emission.2 In particular, green hydrogen (i.e., H2 produced with renewable energy) represents a versatile solution for both environmental and energy problems. A potential solution that matches water pollution remediation and production of green hydrogen is the use of EPs as feedstock for H2 generation. Indeed, with new and efficient processes, such as photocatalytic (PC) and photoelectrocatalytic (PEC) reactions, it is possible to use organic substances to enhance H2 evolution.3,4
A photocatalytic process5 involves the use of a semiconductor, which, under light stimulus of appropriate wavelengths, leads to the formation of electrons in the conduction band (CB) and holes in the valence band (VB). These species, when interacting with water or air, promote the formation of various radicals (hydroxide OH*, superoxide O2−*, and hydroperoxide OOH*), which allow the rapid degradation of a wide range of pollutants.5 Since the pioneering work of Fujishima and Honda about 50 years ago,6 the photocatalytic water splitting reaction for hydrogen production has been largely investigated. In such a reaction, the photoexcited electrons in the semiconductor's CB can be used to promote the reduction of protons arising from the oxidation of water (made by the holes in the semiconductor's VB). The addition of a small amount of an organic hole-scavenger to the reaction mixture limits electron/hole recombination (a common reason for deactivation of photocatalysts), favouring the H2 evolution.7,8
The photoelectrocatalytic approach improves the photocatalytic process using an external bias to further reduce the recombination of holes and electrons.9 In both the PC and PEC approaches, which have been studied in a variety of cell configurations,10 the oxygen evolution reaction (OER) is the rate-determining step of the overall processes.11 The addition of an organic substrate to the reaction mixture or the presence of pollutants in water can be exploited because these compounds can react with photogenerated holes12 acting as sacrificial agents. This approach allows contextual water purification13 and further enhancement of H2 production14 by replacing the OER with photo-oxidation of the target water contaminant.15
In the literature, only a few examples of simultaneous H2 formation and water pollutant removal have been reported,16 whereas the two reactions are usually studied separately.
Compared to single PC or PEC reactions, the simultaneous approach exhibits differences in the reaction mechanisms and performances, depending on the experimental setup, the used materials and the target pollutants involved in the oxidation reaction.15,17
In this paper, we investigate such a simultaneous approach in PEC experiments performed in a custom-designed twin-chamber reactor where the degradation of a model pollutant occurs in the photoanode chamber at a TiO2-based photoanode, while hydrogen evolution is monitored in the cathodic chamber. Titanium dioxide (TiO2) is one of the most studied materials in PC and PEC applications.18 Nevertheless, it has several disadvantages, including a wide bandgap that limits light absorption to the UV-A region of the solar spectrum, low quantum efficiency and relatively fast charge-carrier recombination.19 To improve the photocatalytic activity of TiO2, several strategies have been reported, such as doping,20 composite formation21 and surface modification.22
In particular, surface engineering approaches enabled improving the light-harvesting, interaction with the target substrates and generation of active species responsible for the photo(electro) activity, to finally enhance the performance of the material compared to the unmodified samples.23–27
This study exploits the properties of TiO2 films that have undergone surface modification via the formation of a zirconium phosphate monolayer. In a previous study28 we investigated, by time-of-flight secondary ion mass spectrometry (ToF-SIMS), the photodegradation of Rhodamine B (RhB) directly at the surface of TiO2 and surface-modified TiO2 films. The presence of various intermediate degradation products was detected, including by-products originating from the loss of ethyl chains from the initial RhB dye. This prompted us to investigate the possibility of exploiting the degradation of RhB on TiO2-based photoanodes to facilitate hydrogen production in a PEC arrangement.
The twin-chamber PEC approach is expected to require a lower external bias than the electrocatalytic approach29,30 and is susceptible to extension to other water pollutants and possible scale-up.31
Experimental
Samples preparation
Nanostructured TiO2 films were prepared using the doctor blade technique32 by depositing a layer (about 50 μm) of a commercial titania paste for screen-printing (anatase TiO2 nanoparticle paste, average size: 20 nm, Solaronix Nanoxide S/P) on conductive substrates (fluorinated tin oxide, FTO, Solaronix). The FTO substrate was previously cleaned to remove any organic impurities by sonication in a 50![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :50 (v/v) mixture of 2-propanol (99.9%, VWR Chemicals) and acetone (99,8%, Carlo Erba) and subsequent O2 plasma treatment (75 W, 60 s, 0.4 torr) performed in a March Instrument Plasmod plasma asher. After deposition, the samples were annealed at 500 °C (heating ramp 5 °C min−1) for 1 h to obtain a 5 μm thick nanostructured TiO2 layer. The samples were then treated in O2 plasma (75 W, 300 s, 0.4 Torr) to remove any residual organic contaminant and then immersed in ultrapure water (Milli-Q quality) to promote surface hydroxylation. Finally, they were dried under N2 steam at room temperature. Samples prepared as described above were coded as TiO2.
:50 (v/v) mixture of 2-propanol (99.9%, VWR Chemicals) and acetone (99,8%, Carlo Erba) and subsequent O2 plasma treatment (75 W, 60 s, 0.4 torr) performed in a March Instrument Plasmod plasma asher. After deposition, the samples were annealed at 500 °C (heating ramp 5 °C min−1) for 1 h to obtain a 5 μm thick nanostructured TiO2 layer. The samples were then treated in O2 plasma (75 W, 300 s, 0.4 Torr) to remove any residual organic contaminant and then immersed in ultrapure water (Milli-Q quality) to promote surface hydroxylation. Finally, they were dried under N2 steam at room temperature. Samples prepared as described above were coded as TiO2.
The surface of some of the TiO2 films was modified by the ‘ZP process’. This type of surface modification has been widely investigated previously by our research group for various applications, including as a photoanode material for dye-sensitized cells.28,33–35 Absolute ethanol (99.8%, Aldrich), phosphorus(V) oxychloride (POCl3) (99%, Aldrich) and hydrated zirconyl chloride (ZrOCl2·xH2O) (99,99%, Aldrich) were used as received. The water used in all experiments was of Milli-Q quality (Millipore Corp.).
Two sets of TiO2 samples with different surface treatments were prepared. The first set of samples, coded as TiO2-P, was modified (after cleaning and surface hydroxylation) by 1 h immersion in pure POCl3, followed by thorough rinsing in ultrapure water and final drying in N2 steam at room temperature.
The second set of TiO2 samples, coded as TiO2-PZr, underwent an additional modification step after treatment with POCl3 (as described for TiO2-P), involving sample immersion in a solution of ZrOCl2 (10−3 M) in water for 1 hour, rinsing in ultrapure water and drying in N2 steam at room temperature.
Samples characterization
XRD analysis was carried out using a SmartLab Rigaku diffractometer in Bragg–Brentano mode with a rotating anode of Cu Kα radiation operating at 45 kV and 200 mA. The average size of the crystalline grains was determined using the Scherrer formula.36The UV-Vis Diffuse Reflectance spectra (UV-Vis DRS) were obtained using a Jasco V-750 spectrometer and an integrating sphere using barium sulphate as a reference. The optical band gap of the materials was estimated using the Kubelka–Munk function.37
ToF-SIMS measurements were performed in a TOFSIMS IV (IONTOF GmbH, Münster, Germany) equipped with a Cs ion source for sputtering and a Bi/Mn liquid metal ion source for analysis. In the experimental conditions used in this study, the mass resolution was M/ΔM ≥ 7000 at m/z 27. Depth profiles were acquired in the so-called “non-interlaced” dual beam mode, where sputtering cycles (Cs+ ions, 10 keV, 20 nA, 80 × 80 μm2 rastered area) alternated with low current analysis cycles (Bi3+, 25 keV, <1 pA) rastered over an area of 30 × 30 μm2 concentric to the sputter crater. The depth scale was calibrated by measuring the depth of the TiO2 layer thickness with a KLA Tencor P-7 stylus profilometer while assuming a constant sputtering rate through the entire TiO2-based film.
X-ray photoelectron spectra (XPS) were acquired in a PHI 5000 Versa Probe II system (ULVAC-PHI, INC.) with an analyzer pass energy of 23.50 eV using monochromatized Al Kα X-ray radiation. The XPS peak intensities were obtained after subtracting the Shirley background.
The morphologies of the samples were examined using a field emission scanning electron microscope (FE-SEM, ZEISS SUPRA 55 VP).
Photocatalytic and photoelectrocatalytic experiments
The photocatalytic performance of the nanostructured films was evaluated in the solar photocatalytic oxidation of RhB.The TiO2-based films (area 1 cm2) were placed in a custom-made Pyrex jacked photoreactor [reported in Fig. S1†] containing a 10−6 M solution of RhB (≥95%, Merck). This concentration implies an absorbance that falls within the validity of the Lambert–Beer law. The photoreactor was maintained at 25 °C with constant stirring. After the adsorption–desorption process in the dark (120 min), the sample was irradiated with a solar lamp (Osram Ultra Vitalux 300 W, irradiance of 10.7 mW cm−2, 300–2000 nm).
The degradation of RhB was monitored by measuring the UV-vis absorbance at 554 nm. Small aliquots of RhB solution were sampled at regular time intervals during a total irradiation time of 4 hours (for the photocatalytic tests) and 2 hours (for the photoelectrocatalytic tests), and the concentration was calculated using a standard Lambert–Beer calibration curve.
The kinetic constant k was obtained using the Langmuir–Hinshelwood model and the pseudo-first-order rate equation.38 According to the literature,38,39 k values were calculated by plotting, as a function of irradiation time, the ratio C/C0 between the concentration C at time t and the initial concentration in the dark C0 and fitting the data with the equation:
| C/C0 = exp[−kt] |
For comparison purposes, photolysis tests (i.e., tests performed by irradiating the dye solution in the absence of the titania catalyst) were performed using the same irradiation times for both the photocatalytic and PEC tests.
The simultaneous photocatalytic oxidation of RhB target pollutants and hydrogen production were monitored in photoelectrocatalytic experiments conducted in a two-electrode configuration in a custom-designed two-chamber reactor. The chambers were separated by a Nafion 117 membrane (Redox.me, Sweden), which allows the exchange of H+ ions only. The membrane was activated according to the standard procedure reported in the literature.40 Each chamber of the twin reactor has a volume of 240 mL and is equipped with a flat quartz window to allow uniform irradiation of the photocatalytic film. The TiO2-based films, as described in the previous section, were used as the working electrodes in the photoanode chamber. A Pt wire (Redox.me, Sweden) was used as the counter electrode, and 0.1 M Na2SO4 (≥99%, Carlo Erba Reagents) was used as the supporting electrolyte. The cathode chamber was fluxed with Ar flow directly connected to a gas chromatograph (Agilent 6890, equipped with a packed column, Carboxen 1000, and a TCD detector) to quantify the amount of H2 produced. The target pollutant was added only to the photoanode chamber, and its abatement was evaluated by UV-Vis measurements, as described before for the photocatalytic tests. A Keithley 2611B System Source Meter was used to apply an external bias during the photoelectrocatalytic tests in the two electrode configuration. The volume of solution used in each reactor chamber was 180 mL.
To investigate the mechanism underlying RhB degradation, radical scavenger tests were performed. The degradation of RhB was carried out using the procedures described above, but adding isopropanol (OH* scavenger, ≥99.5%, ACS reagent from Merck) or p-benzoquinone (O2−* scavenger, reagent grade, ≥98% from Meck), both 10−3 M, during the photocatalytic and photoelectrocatalytic tests.41
The photocatalytic and PEC measurements were repeated three times, resulting in an experimental error of less than 1%.
Results and discussion
Photocatalytic remediation of rhodamine B: catalytic screening
In the photocatalytic tests for the photo-oxidation of RhB, the activity of the different samples was evaluated to identify the most promising material to be used as a photoanode for photoelectrocatalytic tests in the twin-chamber reactor.As shown in Fig. 1a, all samples produce complete degradation of the target dye during the examined irradiation time. In particular, the bare TiO2 and modified TiO2-PZr samples exhibited similar performances, with the TiO2-P sample showing a slightly lower activity. Notably, in the photolysis test (i.e., in irradiation runs without the presence of the catalyst), only 5% of the RhB dye was degraded under the same irradiation conditions. The kinetic constants reported in Fig. 1b, calculated considering a pseudo-first order reaction,38 confirm a lower performance of the TiO2-P sample compared to TiO2 and TiO2-PZr, which display similar k values. Consequently, the best performing photocatalysts (namely TiO2 and TiO2-PZr) were used as photoanode materials for investigating the simultaneous RhB degradation and H2 formation.
 | ||
| Fig. 1 (a) Photocatalytic degradation of RhB dye (error bars fall within the symbol size) and (b) calculated kinetics constants. | ||
Photoelectrocatalytic tests for simultaneous rhodamine B abatement and hydrogen production
The experiments were carried out by irradiating the photoanode with a solar lamp (the same used in the photocatalytic screening) placed 3 cm away from the flat quartz window of the photoanode chamber of the twin-chamber reactor and maintaining a voltage of +1 V between the two electrodes. The 1 V bias value was established based on preliminary screening (data not shown). Under our experimental conditions, this value was the most suitable for the simultaneous approach adopted in this study. Lower applied voltages were not sufficient to promote H2 formation (within the experimental detection limits), whereas higher voltages did not provide further improvement in terms of hydrogen evolution.Fig. 2a shows that the bare TiO2 mesoporous electrode exhibited approximately linear hydrogen production up to about 60 minutes with a contextual RhB abatement of 10% after 60 minutes and about 30% after 120 minutes. The TiO2-PZr (Fig. 2b), while showing similar hydrogen production, exhibits a rather larger efficiency in the removal of RhB (about 60% of RhB abatement after 120 minutes).
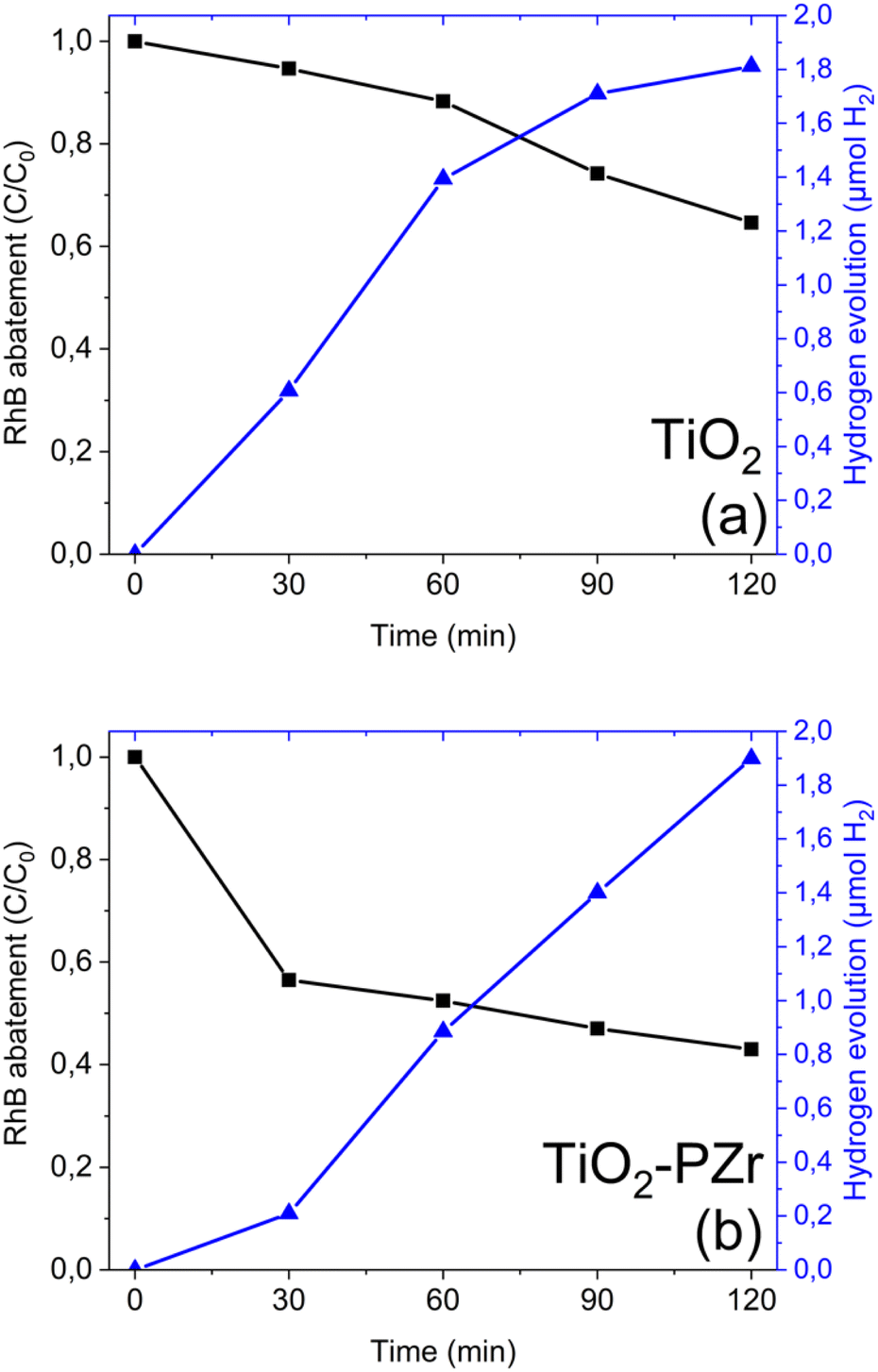 | ||
| Fig. 2 Photoelectrocatalytic trends of simultaneous reactions for (a) TiO2 and (b) TiO2-PZr. Error bars fall within the symbol size. | ||
Remarkably, the performance of the TiO2-PZr film remain stable after 5 consecutive runs of the simultaneous reactions, as shown in Fig. S3 (ESI†). In these tests, the TiO2-PZr film was irradiated in each run for 120 minutes. After each cycle, the film was removed from the dual-chamber reactor, washed in pure water, allowed to dry at room temperature, and subsequently re-tested using a fresh dye solution.
To correlate the measured photoelectrocatalytic performance with the change in the physico-chemical properties of TiO2 due to the surface functionalization, structural, optical, morphological and surface characterizations were performed.
Structural, optical and surface characterization
The XRD patterns of the examined samples are shown in Fig. 3. Each sample exhibited distinctive peaks corresponding to the polycrystalline anatase TiO2 structure. The peaks at 2θ = 25.30°, 37.93°, 48.09°, 54.08°, 55.16°, and 62.75° correspond to the typical reflections 101, 004, 200, 105, 211, and 213 of the anatase phase (ICDD no. 00-084-1286), whose intensities match those reported in the database.42,43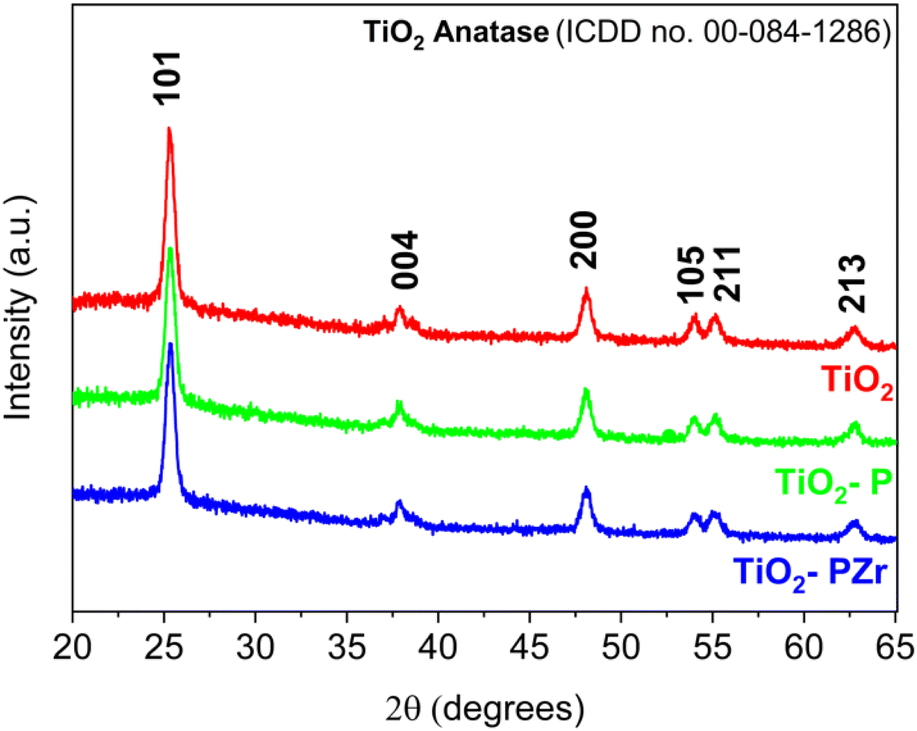 | ||
| Fig. 3 XRD patterns of TiO2, TiO2-P, and TiO2-PZr samples. | ||
The crystallite sizes of the examined samples were estimated using the Scherrer equation33 D = Kλ/βcosθ where K is the shape factor, λ is the X-ray wavelength, and β is the full width at half maximum (FWHM) of the most intense peak. The selected peaks were at 2θ = 25.30° for TiO2, 2θ = 25.29° for TiO2-P, and 2θ = 25.34° for TiO2-PZr. The calculated crystallite sizes were 15.8 nm, 14.9 nm, and 15.1 nm for TiO2, TiO2-P, and TiO2-PZr, respectively. Moreover, no substantial shift in the XRD patterns was observed due to surface functionalization (i.e., in the TiO2-P and TiO2-PZr patterns). Additional information on the particle shape after functionalization was obtained from the SEM images (Fig. S4a and b†), which show the presence of regular nanoparticles in both shape and size with a quasi-ellipsoidal morphology.
Fig. 4 displays the UV-vis diffuse reflectance spectra (UV-DRS) of the examined samples. It is possible to note a blue shift of the TiO2-PZr corresponding to a slight increase in the optical band gap, as evaluated using the Kubelka–Munk function37 (inset in Fig. 4). Actually, the calculated values of the optical band gap are very similar for all the samples (3.2 eV for TiO2 and TiO2-P; 3.5 eV for TiO2-PZr). These results are consistent with the reported optical band gap of TiO2 in the anatase crystalline phase.44
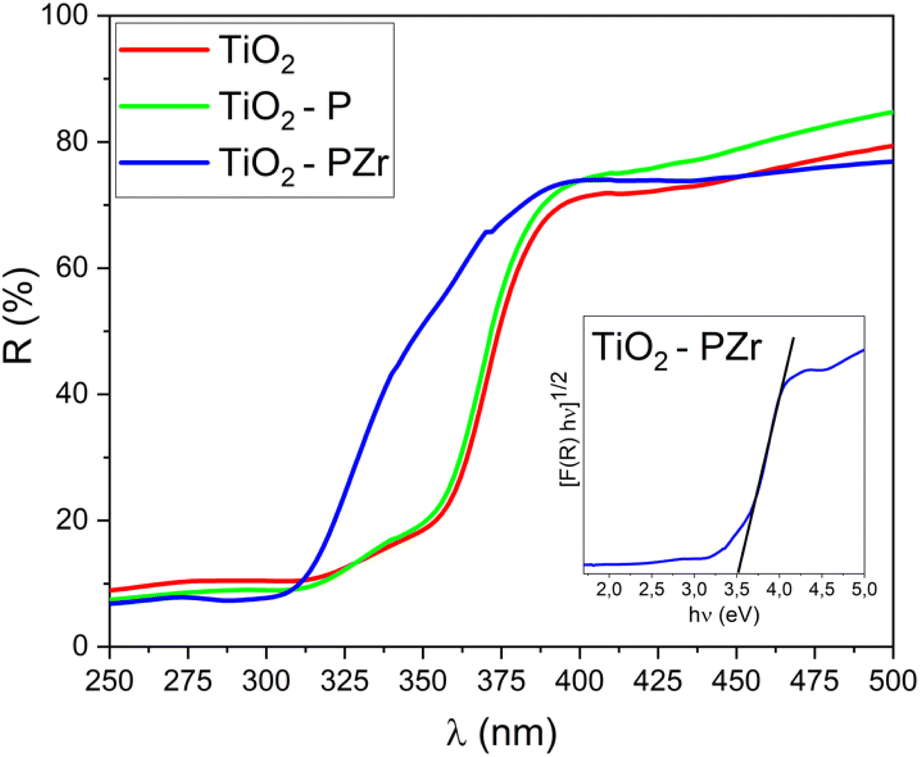 | ||
| Fig. 4 UV-vis diffuse reflectance spectra (UV-DRS) of TiO2, TiO2-P, and TiO2-PZr samples. Inset: estimation of optical band gap using the Kubelka–Munk function (TiO2-PZr is shown as a representative sample). | ||
To check the success of the surface modification strategy, samples were investigated by time-of-flight secondary ion mass spectrometry (ToF-SIMS) and X-ray photoelectron spectroscopy (XPS). In particular, ToF-SIMS was used to obtain information on the in-depth homogeneity of the modification.
Fig. 5 reports the ToF-SIMS depth profile of a TiO2-PZr sample, where the intensity of some characteristic mass peaks is reported as a function of depth. In particular, TiO−, SnO2− and Si− signals identify, respectively, the titania layer, FTO layer and glass substrate, while the PO−, ZrO−, and Cl signals are the signature of the ZP treatment and they are not present in the depth profiles of the unmodified titania layers (Fig. S5†). ZP-related peaks are present along the entire nanostructured film, demonstrating that the functionalization occurred along the entire film thickness.
 | ||
| Fig. 5 ToF-SIMS depth profile of TiO2-PZr film. | ||
Further insight into the effect of ZP modification on the surface composition of nanostructured titania films was provided by XPS, and the main results are reported in Fig. 6 and 7. In particular, Fig. 6a and b show the Ti 2p and O 1s regions of the XPS spectra of bare TiO2, whereas Fig. 7a–d report the same spectral regions for the TiO2-PZr sample, with the addition of the P 2p (Fig. 7c) and Zr 3d (Fig. 7d) regions (the latter two peaks are not present in the spectrum of unmodified titania). In bare TiO2 (Fig. 6a), the position of the Ti 2p band (Ti 2p3/2 at 458.5 eV and Ti 2p1/2 at 464.3 eV) is consistent with that reported in the literature for Ti(IV) in anatase.45,46 In the TiO2-PZr sample (Fig. 7a), the peaks were slightly shifted to higher binding energies (458.8 and 464.6, respectively), consistent with the attachment of phosphate groups at the titania surface.47,48 The binding energies of P 2p (133.6) and Zr 3d5/2 (182.8) (Fig. 7c and d) in the TiO2-PZr sample are compatible with the formation of a ZP monolayer,49 at the oxide surface. The O 1s region of TiO2 (Fig. 6b) shows a major component at 529.8 ± 0.1 eV, which is assigned to the lattice oxygen in the oxide, and a smaller component at 531.4 ± 0.1 eV which was assigned to the surface hydroxyl species.50 In the TiO2-PZr sample it is possible to observe (Fig. 7b), the same components (529.9 ± 0.1 eV and 531.3 ± 0.1 eV, respectively). However, the higher BE content was more intense and broad than that in bare TiO2, consistent with an increased concentration of hydroxyl species at the surface and the presence of slightly different chemical environments (including the presence of phosphate groups).47
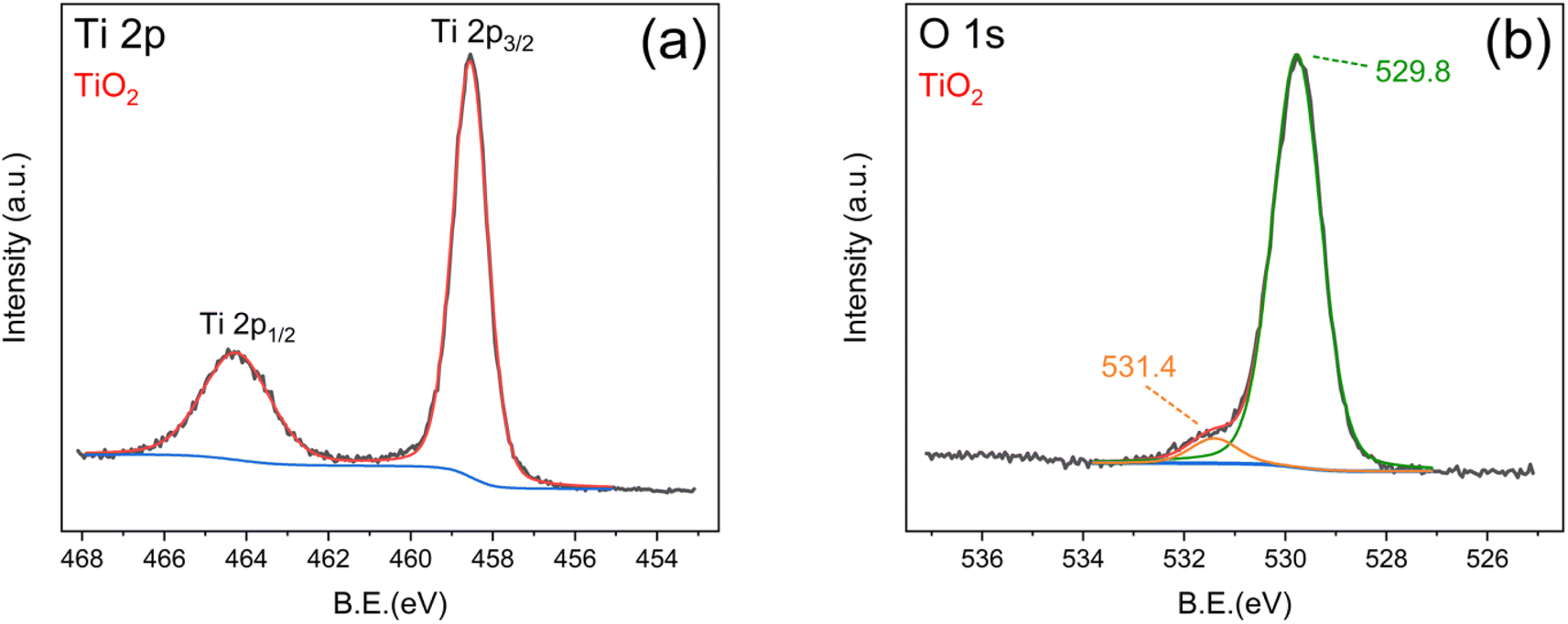 | ||
| Fig. 6 XPS spectrum of TiO2 film: (a) Ti 2p region and (b) O 1s region. | ||
 | ||
| Fig. 7 XPS spectrum of TiO2-PZr film: (a) Ti 2p region (b) O 1s region (c) P 2p region and (d) Zr 3d region. | ||
In summary, the above data confirm the successful ZP functionalization, which occurred along the whole depth of the nanostructured titania film, as demonstrated by ToF-SIMS depth profiles, and caused an increase in the concentration of hydroxyl moieties, as shown by the XPS results, which also confirm the presence of Zr(IV) and phosphate groups. Moreover, the ZP treatment did not appreciably affect the integrity of the anatase phase, the average size of the crystallites (XRD results), or the surface morphology (SEM images), and had little or no influence on the band gap (UV-DRS data).
However, as discussed above (Fig. 4), the ZP-treated samples showed increased activity in the PEC tests compared with the bare TiO2 film. This suggests that the presence of zirconium phosphate and the increased concentration of surface hydroxyl groups has a favourable effect on improving the photo-oxidation of RhB in the twin reactor photoelectrocatalytic tests, where the degradation of the dye was approximately twice (60%) compared with that of unmodified TiO2 (30%). Furthermore, it can be hypothesized that the presence of Zr(IV) favours the surface charge mobility,51–53 contributing to the synchronous reactions of dye degradation and hydrogen production.54
In contrast to the pronounced effect of surface functionalization of titania in PEC experiments, in the simple PC ones, the activities of bare TiO2 and TiO2-PZr films in RhB abatement appear to be rather similar. Such behaviour could be ascribed to differences in the reaction mechanisms involved in the two approaches, i.e., the single reactor photocatalytic oxidation of RhB and the twin reactor simultaneous photoelectrocatalytic RhB oxidation and H2 production. In PC, the photoexcitation of TiO2 leads to the formation of several radical species (mainly ˙OH, ˙O2−, ˙OOH). All of them can contribute to the oxidative degradation of the dye according to a series of reaction pathways (a–e) proposed in the literature:55,56
(a) TiO2 activation:
| TiO2 + hv → TiO2(eCB− + hVB+) |
(b) Formation of hydroxyl radicals:
| hVB+ + H2O→ ˙OH + Haq+ |
(c) Formation of superoxide radicals:
| eCB− + O2 → ˙O2− |
(d) Formation of hydroperoxide radical:
| ˙O2− + H+ → ˙HO2 |
(e) Photo-oxidation of RhB:
| RhB + ˙HO2 + ˙OH → H2O + intermediate products + CO2 |
According to the above scheme, the formation of radicals that initiate degradation pathways is a consequence of the charge separation produced by light absorption. In particular, the formation of the superoxide radical ion ˙O2−, which is expected to contribute to some dye degradation pathways, is due to the photogenerated electrons in the conduction band of titania.57 Since TiO2 and TiO2-PZr films exhibit similar photocatalytic activity, this mechanism, if present, should occur in the same way in both cases.
In the case of the photoelectrocatalytic experiments with a dual-chamber reactor, where modified titania photoanodes (TiO2-PZr) perform better than the unmodified ones, electrons photogenerated at the anode move directly to the cathode, where they are used for the H2 evolution reaction,17 according to the scheme:
| 2eCB− + 2Haq+ → H2 |
Thus, it is expected that in the photoelectrocatalytic approach, the formation of superoxide radicals (reaction c) will be reduced because the photogenerated eCB− will move directly to the cathodic chamber at a variance of the PC case, where they will contribute to the generation of superoxide radicals. Consequently, in the PEC case, ˙OH species are expected to be the main radicals involved in RhB degradation, whereas in the PC approach, ˙O2− should play a major role.58
To verify the above hypotheses and, in particular, the contribution of radicals to dye degradation, additional PC and PEC experiments were carried out in the presence of radical scavengers, which were added to the reaction mixtures both in the batch PC reactor and in the photoanode chamber of the PEC experiment. In separate tests, we used isopropanol (IPA) to impair the formation of hydroxyl radicals and benzoquinone (BQ) to remove the superoxide radicals.41
The results of these tests are summarized in Fig. 8. We note that in the PC tests (Fig. 8a), the activity significantly decreased for both TiO2 and TiO2-PZr films upon the addition of BQ, which is known to quench ˙O2− radicals, confirming that they are the main species involved in the RhB degradation path. The addition of BQ in the PEC case (Fig. 8b) affected RhB degradation to a limited extent. By contrast, in the PEC approach, the addition of IPA, associated with the removal of ˙OH, greatly hinders RhB degradation compared with the PC case, where the effect of IPA, although present, is much less pronounced. Moreover, in the PEC tests, the influence of IPA on RhB abatement was less pronounced in the case of TiO2-PZr. We hypothesize that the latter effect is related to the higher concentration of surface OH species in the ZP-functionalized sample, as shown by the XPS results, although at the present stage, this remains to be clarified.
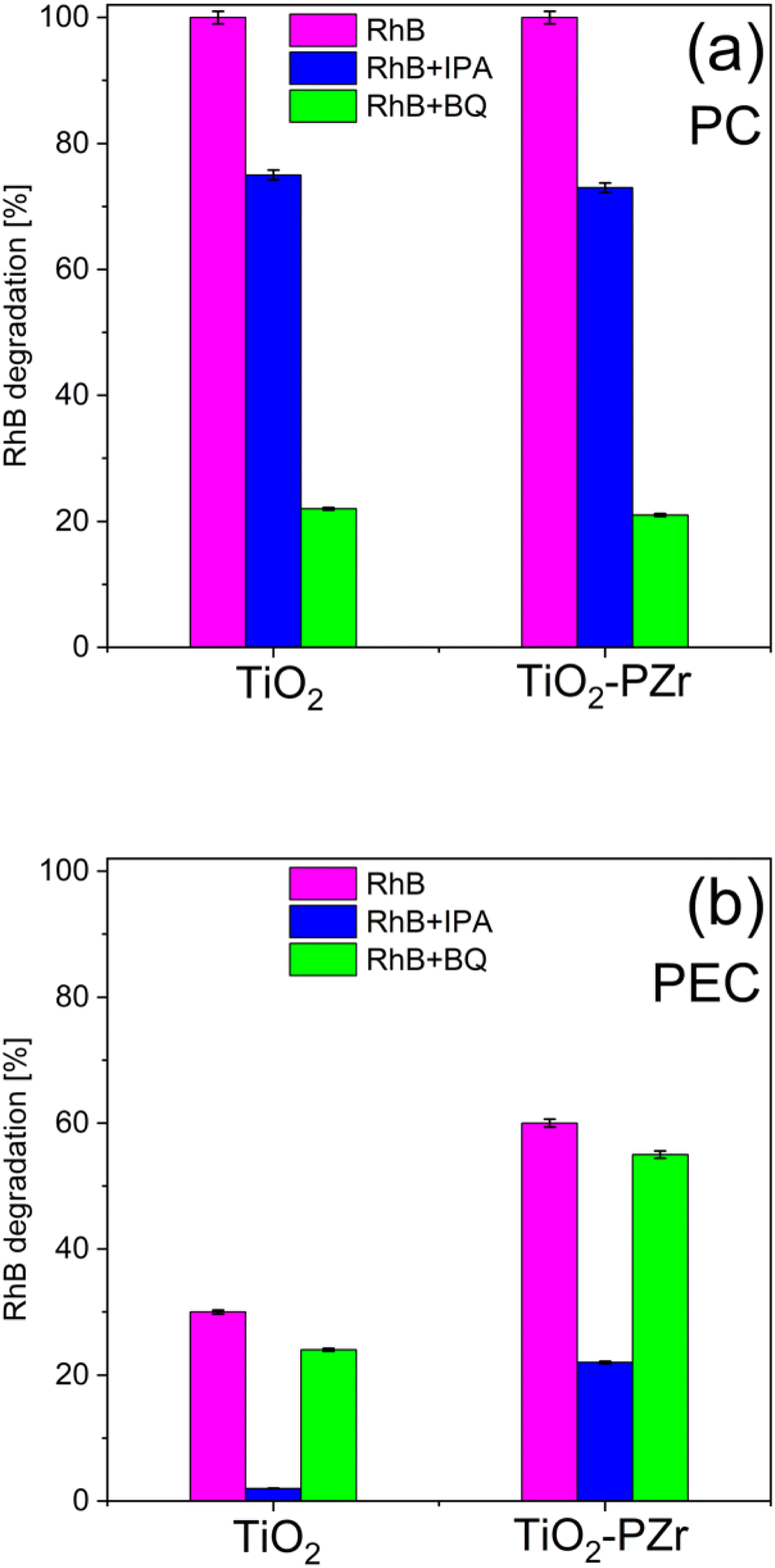 | ||
| Fig. 8 (a) Photocatalytic degradation of RhB dye by adding radical scavengers after 240 min of simulated solar irradiation. (b) Photoelectrocatalytic degradation of RhB dye by adding radical scavengers after 120 min of simulated solar irradiation. | ||
Conclusions
In this study, different nanostructured TiO2 films were tested for the photoelectrocatalytic removal of rhodamine B with simultaneous hydrogen production in a custom-built dual-chamber reactor.Nanostructured TiO2 films (anatase phase) were surface-modified with zirconium-phosphate functionalization (ZP process) that, while introducing new surface functionalities, does not affect the anatase phase and the morphology of the film. Compared with unmodified titania, the use of modified films as photoanodes in a dual-chamber photoelectrocatalytic arrangement leads to an increase in dye degradation, with comparable contextual H2 production.
The approach of simultaneous pollutant abatement and hydrogen production in a dual chamber PEC arrangement combined with the use of ZP-modified photoanodes appears to be a promising approach for new solutions for both environmental remediation and green hydrogen formation. Our results obtained on a model pollutant are proof of concept, which is worth exploring in the application to real-world situations, such as the use of wastewater as a matrix with the goal of water purification and high-added hydrogen production.
Data availability
The data used and analysed during this study are available in the ESI† accompanying this document.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was funded by the European Union (NextGeneration EU) through the MUR-PNRR project SAMOTHRACE (No. ECS00000022). The authors thank Prof. G. Malandrino and Prof. G. G. Condorelli (University of Catania) for their help in XRD and XPS measurements, respectively. The Bio-nanotech Research and Innovation Tower (BRIT) laboratory of the University of Catania is thanked for providing the SmartLab diffractometer and the PHI Versa Probe XPS facilities.References
- V. Geissen, H. Mol, E. Klumpp, G. Umlauf, M. Nadal, M. van der Ploeg, S. E. A. T. M. van de Zee and C. J. Ritsema, Int. Soil Water Conserv. Res., 2015, 3, 57–65 CrossRef.
- A. Kovač, M. Paranos and D. Marciuš, Int. J. Hydrogen Energy, 2021, 46, 10016–10035 Search PubMed.
- S. A. Balsamo, R. Fiorenza, M. T. A. Iapichino, F. J. Lopez-Tenllado, F. J. Urbano and S. Sciré, Mol. Catal., 2023, 547, 113346 CrossRef CAS.
- M. Wang, S. Shen, L. Li, Z. Tang and J. Yang, J. Mater. Sci., 2017, 52, 5155–5164 CrossRef CAS.
- M. N. Chong, B. Jin, C. W. K. Chow and C. Saint, Water Res., 2010, 44, 2997–3027 CrossRef CAS PubMed.
- A. Fujishima and K. Honda, Nature, 1972, 238, 37–38 CrossRef CAS PubMed.
- P. Hota, A. Das and D. K. Maiti, Int. J. Hydrogen Energy, 2023, 48, 523–541 CrossRef CAS.
- Y. Wu, T. Sakurai, T. Adachi and Q. Wang, Nanoscale, 2023, 15, 6521–6535 RSC.
- K. Poonia, P. Singh, A. Singh, S. Thakur, Q. Van Le, T. Ahamad, P. Raizada, C. Wang, L. H. Nguyen and V.-H. Nguyen, Environ. Chem. Lett., 2023, 21, 265–283 Search PubMed.
- S. McMichael, P. Fernández-Ibáñez and J. A. Byrne, Water, 2021, 13, 1198 CrossRef CAS.
- N. Yuan, Q. Jiang, J. Li and J. Tang, Arabian J. Chem., 2020, 13, 4294–4309 CrossRef CAS.
- R. Kaushik, S. Gandhi and A. Halder, ACS Appl. Nano Mater., 2023, 6, 4297–4308 CrossRef CAS.
- X. M. C. Ta, R. Daiyan, T. K. A. Nguyen, R. Amal, T. Tran-Phu and A. Tricoli, Adv. Energy Mater., 2022, 12, 2201358 CrossRef CAS.
- E. Doukas, P. Balta, D. Raptis, G. Avgouropoulos and P. Lianos, Materials, 2018, 11, 1269 CrossRef PubMed.
- M. A. U. Olea, J. de J. P. Bueno and A. X. M. Pérez, J. Environ. Chem. Eng., 2021, 9, 106480 CrossRef CAS.
- S. Kampouri and K. C. Stylianou, ACS Catal., 2019, 9, 4247–4270 CrossRef CAS.
- P. Fernandez-Ibanez, S. McMichael, A. Rioja Cabanillas, S. Alkharabsheh, A. Tolosana Moranchel and J. A. Byrne, Curr. Opin. Chem. Eng., 2021, 34, 100725 CrossRef.
- A. Fujishima, T. N. Rao and D. A. Tryk, J. Photochem. Photobiol., C, 2000, 1, 1–21 CrossRef CAS.
- S.-Y. Lee and S.-J. Park, J. Ind. Eng. Chem., 2013, 19, 1761–1769 CrossRef CAS.
- X. Jaramillo-Fierro and R. León, Nanomaterials, 2023, 13, 1068 CrossRef CAS PubMed.
- R. Fiorenza, C. Contarino, V. Spanò, M. T. A. Iapichino and S. A. Balsamo, Catal. Today, 2023, 423, 114251 CrossRef CAS.
- H. Park, Y. Park, W. Kim and W. Choi, J. Photochem. Photobiol., C, 2013, 15, 1–20 CrossRef CAS.
- P. Innocenzi and L. Malfatti, J. Photochem. Photobiol., C, 2024, 58, 100646 CrossRef CAS.
- L. Yang, F. Li and Q. Xiang, Mater. Horiz., 2024, 11, 1638–1657 RSC.
- H. Kaneko, T. Minegishi and K. Domen, Chem. - Eur. J., 2018, 24, 5697–5706 CrossRef CAS PubMed.
- L. Jing, J. Zhou, J. R. Durrant, J. Tang, D. Liu and H. Fu, Energy Environ. Sci., 2012, 5, 6552 RSC.
- S. Challagulla, R. Nagarjuna, R. Ganesan and S. Roy, Nano-Struct. Nano-Objects, 2017, 12, 147–156 CrossRef CAS.
- E. M. Malannata, A. Auditore and A. Licciardello, J. Vac. Sci. Technol. B, 2023, 41, 044008 CrossRef CAS.
- C. M. Pecoraro, M. Bellardita, V. Loddo, D. Virtù, F. Di Franco and M. Santamaria, Appl. Catal., A, 2023, 650, 118987 CrossRef CAS.
- A. Bachmeier, V. C. C. Wang, T. W. Woolerton, S. Bell, J. C. Fontecilla-Camps, M. Can, S. W. Ragsdale, Y. S. Chaudhary and F. A. Armstrong, J. Am. Chem. Soc., 2013, 135, 15026–15032 CrossRef CAS PubMed.
- M. Rumayor, J. Fernández-González, A. Domínguez-Ramos and A. Irabien, Cleaner Prod. Lett., 2023, 4, 100041 CrossRef CAS.
- R. Ramos-Hernández, E. Pérez-Gutiérrez, F. D. Calvo, M. F. Beristain, M. Cerón and M. J. Percino, Coatings, 2023, 13, 425 CrossRef.
- S. Vitale, G. Zappalà, N. Tuccitto, A. Torrisi, E. Napolitani and A. Licciardello, J. Vac. Sci. Technol., B:Nanotechnol. Microelectron.:Mater., Process., Meas., Phenom., 2016, 34, 03H110 Search PubMed.
- S. Vitale, B. Laramée-Milette, A. Valenti, M. E. Amato, G. S. Hanan, N. Tuccitto and A. Licciardello, J. Vac. Sci. Technol., B:Nanotechnol. Microelectron.:Mater., Process., Meas., Phenom., 2020, 38, 032802 CAS.
- V. Spampinato, S. Vitale, S. Quici, A. Torrisi, V. Stepanenko, F. Würthner and A. Licciardello, Surf. Interface Anal., 2013, 45, 206–210 CrossRef CAS.
- A. L. Patterson, Phys. Rev., 1939, 56, 978–982 CrossRef CAS.
- E. S. Welter, S. Garg, R. Gläser and M. Goepel, ChemPhotoChem, 2023, 7, e202300001 CrossRef CAS.
- S. Ghasemi, A. Esfandiar, S. Rahman Setayesh, A. Habibi-Yangjeh, A. Iraji zad and M. R. Gholami, Appl. Catal., A, 2013, 462–463, 82–90 CrossRef CAS.
- M. Ahmad, M. T. Qureshi, W. Rehman, N. H. Alotaibi, A. Gul, R. S. Abdel Hameed, M. Al Elaimi, M. F. H. Abd el-kader, M. Nawaz and R. Ullah, J. Alloys Compd., 2022, 895, 162636 CrossRef CAS.
- S. Pujiastuti and H. Onggo, AIP Conf. Proc., 2016, 1711, 060006 CrossRef.
- S. Chen, Y. Hu, S. Meng and X. Fu, Appl. Catal., B, 2014, 150–151, 564–573 Search PubMed.
- Y. Wang, L. Li, X. Huang, Q. Li and G. Li, RSC Adv., 2015, 5, 34302–34313 RSC.
- D. A. H. Hanaor and C. C. Sorrell, J. Mater. Sci., 2011, 46, 855–874 CrossRef CAS.
- A. Fujishima, X. Zhang and D. Tryk, Surf. Sci. Rep., 2008, 63, 515–582 CrossRef CAS.
- Z. Bian, J. Zhu, S. Wang, Y. Cao, X. Qian and H. Li, J. Phys. Chem. C, 2008, 112, 6258–6262 CrossRef CAS.
- R. Fiorenza, M. Bellardita, T. Barakat, S. Scirè and L. Palmisano, J. Photochem. Photobiol., A, 2018, 352, 25–34 CrossRef CAS.
- M. C. O. Monteiro, G. Cha, P. Schmuki and M. S. Killian, ACS Appl. Mater. Interfaces, 2018, 10, 6661–6672 CrossRef CAS PubMed.
- M. Testa, V. La Parola, F. Mesrar, F. Ouanji, M. Kacimi, M. Ziyad and L. Liotta, Catalysts, 2019, 9, 148 CrossRef.
- V. Spampinato, N. Tuccitto, S. Quici, V. Calabrese, G. Marletta, A. Torrisi and A. Licciardello, Langmuir, 2010, 26, 8400–8406 CrossRef CAS PubMed.
- J. Zhu, J. Yang, Z.-F. Bian, J. Ren, Y.-M. Liu, Y. Cao, H.-X. Li, H.-Y. He and K.-N. Fan, Appl. Catal., B, 2007, 76, 82–91 CrossRef CAS.
- F. Lei, S. Chen, H. Sun, H. Han, J. Yang, J. Huang, D. Li and D. Sun, Mater. Chem. Phys., 2021, 263, 124377 CrossRef CAS.
- A. Bartkowiak, O. Korolevych, G. L. Chiarello, M. Makowska-Janusik and M. Zalas, Materials, 2021, 14, 2955 Search PubMed.
- J. J. Carey and K. P. McKenna, J. Phys. Chem. C, 2019, 123, 22358–22367 Search PubMed.
- K. R. Davies, M. G. Allan, S. Nagarajan, R. Townsend, V. Asokan, T. Watson, A. R. Godfrey, M. M. Maroto-Valer, M. F. Kuehnel and S. Pitchaimuthu, Ind. Eng. Chem. Res., 2023, 62, 19084–19094 Search PubMed.
- G. Palmisano, M. Addamo, V. Augugliaro, T. Caronna, A. Di Paola, E. G. López, V. Loddo, G. Marcì, L. Palmisano and M. Schiavello, Catal. Today, 2007, 122, 118–127 Search PubMed.
- Y. Nosaka, Catalysts, 2022, 12, 1557 Search PubMed.
- L. Kőrösi, B. Bognár, S. Bouderias, A. Castelli, A. Scarpellini, L. Pasquale and M. Prato, Appl. Surf. Sci., 2019, 493, 719–728 Search PubMed.
- J. Liu, S. Zhang, W. Wang and H. Zhang, J. Energy Chem., 2023, 86, 84–117 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra00251f |
| This journal is © The Royal Society of Chemistry 2025 |