DOI:
10.1039/D5RA00016E
(Review Article)
RSC Adv., 2025,
15, 14001-14029
The power of DNA-encoded chemical libraries in the battle against drug-resistant bacteria
Received
1st January 2025
, Accepted 8th April 2025
First published on 30th April 2025
Abstract
Drug-resistant bacteria are increasingly posing an imminent existential threat, as many bacteria have developed resistance mechanisms that render most antibiotics ineffective. In the meantime, the number of newly approved antibiotics or new clinical antibacterial drug candidates is sharply declining. A key challenge is finding effective pharmacophores that can penetrate and accumulate inside bacterial cells. DNA-encoded chemical libraries (DECLs) play vital roles in accelerating hit identification and screening against various bacterial protein targets. In this review, we highlight the pivotal role of DECLs in accelerating the identification of new pharmacophores and hit compounds against drug-resistant bacteria. This review focuses on the protein targets, where DECLs have directly contributed to the rapid identification of new inhibitors. In addition, this review explores the methods used to screen DECLs against various bacterial targets and discusses the current outlook and perspectives on the role of DECLs in tackling antimicrobial resistance.
1. Introduction
In late September 1928, the Nobel laureate Alexander Fleming discovered the world's first antibiotic, penicillin. Although penicillin was a game-changer against bacteria in the last century, nowadays, bacteria have developed resistance mechanisms to penicillin and to the vast majority of approved antibiotics. Consequently, there is an ongoing bacterial threat as the discovery of new antibiotics or new druggable bacterial targets is currently in sharp decline.1 The World Health Organisation (WHO) estimated that bacterial drug resistance was directly responsible for 1.27 million global deaths in 2019 and contributed to 4.95 million deaths.2 WHO has also estimated that antimicrobial resistance (AMR) will be responsible for 10 million deaths globally per year by 2050.3 The number of approved antibacterial drugs has steadily declined since 1980.4 In June 2021, 76 antibacterial candidates were being evaluated in clinical trials. Of those clinical candidates, 54% targeted the WHO priority pathogens, 25% were new pharmacophores, and 4 candidates possessed new modes of action.5 These numbers indicate that there are still few clinical candidates encompassing new pharmacophores or new modes of action targeting the WHO priority pathogens. One of the reasons for this modest response to this existential threat is the lack of a sustainable and suitable economical model to support the discovery of new antibacterial agents. The long time and high costs associated with traditional research and development models hindered several biotechnology and pharmaceutical companies to invest in developing new antibacterial agents. Identifying new pharmacophores or hit compounds with physicochemically compatible properties against the resistant bacteria is one of the key challenges in this battle.6 The traditional hit identification toolbox includes public-knowledge-based rational design and various screening platforms, such as high-throughput, virtual, and fragment screens, along with traditional synthetic library screening technologies. These methods are resource and time-intensive, especially high-throughput screening (HTS) and often requires dedicated automated infrastructures with extensive liquid dispensing systems, plate handling robots, and storage capabilities. HTS requires large quantities of consumables like individual library members, targets of interest, buffers, and other reagents. In addition, it involves analysing individual members one at a time, which can take days to weeks depending on the library size and the number of targets co-screened. The overall experimental time is further extended by the need to validate a functional assay for each new target type. These constraints limit the number of molecules that can be screened, resulting in a limited chemical space.7 The enormous power of DNA encoded chemical libraries (DECLs) is playing an increasingly important role in this battle by providing wide and diverse chemical space to accelerate the identification of new hit compounds.8 DECLs have become more prominent as a powerful tool for hit identification in drug discovery.9 The DECL technique involves the synthesis of DNA-tagged molecules in which each DNA sequence uniquely codes for the attached molecule's chemical structure. They can be screened as mixtures via affinity selection against an immobilised protein target. The binding molecules are identified by amplification and sequencing of the DNA tag. Libraries comprising millions of compounds can be rapidly synthesised and screened on a microscale.
In this review, we shed light on the pivotal role of DECLs in accelerating the identification of new pharmacophores and hit compounds against drug-resistant bacteria. The review analyses all hit compounds identified through DECL technology, focusing on the targets and bacteria where DECLs has directly contributed to the accelerated identification of new antibacterial agents. In addition, the review explores the methods used to screen DECLs against various bacterial targets and discusses the current outlook and perspectives on DECLs' role in tackling antimicrobial resistance.
2. DNA encoded chemical library technology
Before delving into role of DECLs in identifying new antibacterial agents, it is important to cover its key principles and practices. The DECL concept was pioneered by Brenner and Lerner in 1992.10 The technology involves encoding chemical libraries with sequenced DNA tags. The DNA tags act as identification key for each chemical compound in the library. The process of using DECLs in drug discovery involves two main stages. The first stage is DECL synthesis, which depends on split and pool combinatorial synthesis. In this stage, DNA oligonucleotide is covalently linked through a chemical linker to bifunctional small chemical building blocks. This DNA conjugate undergoes two-three cycles of split and pool combinatorial chemical synthesis and DNA barcode elongation. The outcome of this process is a small molecule, ideally with druglike properties, covalently linked to unique DNA barcode (Fig. 1). The split and pool combinatorial technique leads to exponential library growth. Libraries up to 109 could be easily accessed, covering novel chemical space.11
 |
| | Fig. 1 DNA-encoded chemical library (DECL) synthesis: each chemical building block is covalently attached to a DNA tag. These tagged building blocks are then pooled and split into separate reaction wells. In each well, different building blocks react with the DNA conjugates, which are subsequently elongated with unique DNA tags corresponding to the reacted building blocks. This cycle can be repeated two to three times before pooling the final DNA conjugates to form the DNA-encoded chemical library. Created in https://BioRender.com. | |
The second stage is the selection process. The ultimate advantage of DECL technology is the ability to simultaneously screen billions of compounds against immobilised targets using affinity selection. High throughput screening is limited by the robotic facilities and the available protein quantities. In DECLs, 30–300 μg of tagged protein is required for the selection process. Tagging proteins is a selection requirement to enable immobilisation to biocompatible resin or magnetic beads. Several tags could be installed in any protein of interest (e.g. biotin, or expression with a poly-histidine tag, GST fusion, or FLAG tag). The immobilised protein of interest is incubated with DECLs. The DECL member with strong affinity for the protein remains bound, while the non-binders are washed away. After washing, the retained binders are eluted from the protein and amplified by qPCR. The amplified products are analyzed by next-generation sequencing (NGS) (Fig. 2). The obtained sequences are used to decode the chemical structure of the binders (hits). As a validation step, the confirmed hits were resynthesized off-DNA, and their binding was verified using biochemical or biophysical assays.
 |
| | Fig. 2 DNA-encoded chemical library selection: an affinity-based method is used to identify compounds that bind to an immobilised target protein. (1) Incubation: the DECL is incubated with the immobilised target protein. (2) Binding and washing: DECL members that bind to the target protein form complexes, then the unbound ones are washed away. (3) Elution: bound DECL members are eluted from the protein. (4) Selection process is repeated again with the eluted DECL members from the first round. (5) DECL members are amplified and submitted for next generation sequencing (NGS) and data analysis. Created in https://biorender.com/. | |
De-Luca-Flaherty and coworkers pioneered the first attempt to screen encoded combinatorial libraries to identify novel antimicrobials.12 This combinatorial library was not DNA encoded, although it represented the first attempt to screen pooled encoded combinatorial library to find new antibacterial compounds. The authors built a 46K triazine based combinatorial library. Each member of the library was tethered to a bead bearing a unique chemical tag (Fig. 3). The tag is a secondary amine chemical-based tag that could be decoded by appropriate mass spectrometry techniques. The bead based 46K combinatorial library was evaluated for their antibacterial activity through an agar-based whole-cell format. This assay shows that the lawn-based approach can be effectively utilised for large libraries, marking an initial step in applying encoded combinatorial chemistry to discover antimicrobial hits. Several triazine based hits were identified which provided new antibacterial pharmacophores. For example, compound 1 shows a minimum inhibitory concentration (MIC) value of 4 μg mL−1 against Staphylococcus aureus and Bacillus subtilis (Fig. 4).
 |
| | Fig. 3 Split and pool synthesis of combinatorial bead-based library tagged with peptide-based chemical structure. Created in https://biorender.com/. | |
 |
| | Fig. 4 Triazine-based hit identified from the bead-based library tagged with a peptide-based chemical structure. | |
3. Screening platform
One of the comprehensive and important attempts to discover new antibacterial pharmacophores was conducted by GSK scientists.4 The company's scientists designed parallel selection platform to assess the ligandability of multiple bacterial protein targets. The main concept is to screen a wide collection of the bacterial targets against a pool of billions of different DECLs. Hypothetically, if the target is ligandable, it will bind to one of the molecules present in this multibillion pool of DECLs. This platform allows the user to prioritise targets based on their ligandability before investing time and money to find a ligand to an unligandable target. Although the concept of ligandability is different from druggability, discovering new ligands to proteins helps understand the biology behind the targeted protein. The authors discovered new ligands for six different target proteins in S. aureus and Acinetobacter baumannii. This strategy was extended to find ligands to several protein targets in Mycobacterium tuberculosis. The selection process started by immobilising the affinity-tagged proteins onto affinity matrix. Then, the individual proteins were exposed to the DECL containing billions of DNA tagged compounds. The non-binding DECL members were washed away and the bound molecules were eluted from the proteins by heat. The process was automated, enabling the parallel evaluation of hundreds of proteins. This was repeated multiple times to enrich the bound molecules against the background. The protein target ligandability was assessed by the number of enriched chemical series (Fig. 5).
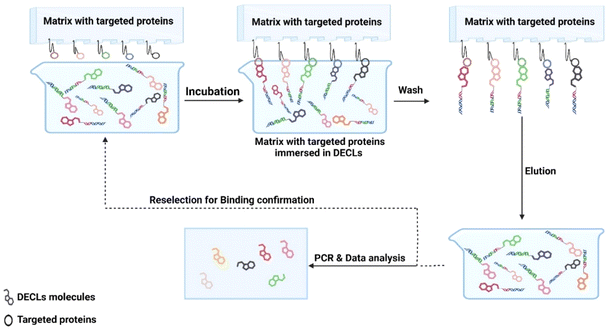 |
| | Fig. 5 Prioritising protein targets based on DECL selection. The process involves immobilising the affinity-tagged proteins, individually, onto an affinity matrix. The proteins were incubated individually in the multibillion pool of DECLs. The unbound DECL members were washed away. The bound DECL members were eluted from the protein targets. The eluted DECL members were submitted for another round of selection before being amplified and submitted for NGS and data analysis. Created in https://biorender.com/. | |
The first bacterial targets screened in this platform were S. aureus proteins. S. aureus is a Gram-positive bacterium that primarily colonises human and animal mucous membranes and regions of the skin.13 It also consistently colonises about 20–30% of the nose in humans.14,15 The bacteria's quorum sensing system controls several virulence factors that bacteria produce, such as leukocidins, enterotoxins, proteases, hemolysins, exfoliative toxins and immune-modulatory factors.13,16 Methicillin-resistant Staphylococcus aureus (MRSA) is a significant strain of S. aureus that induces nosocomial infections and presents a serious problem.16,17 In S. aureus, the authors evaluated 39 potential enzymes targets. These targets were selected based on their role as potential antibacterial drug targets and the availability of biochemical assays for the follow up assessment. Five antibacterial drug targets were prioritised in this evaluation: methionyl-tRNA synthetase (MRS), isoleucyl-tRNA synthetase (IRS), methionine aminopeptidase (MetAP), undecaprenyl pyrophosphate synthase (UppS) and acetyl-CoA carboxylase (ACC), which are involved in critical processes such as protein synthesis,18 aminoacylate tRNAIle with isoleucine and correcting any defect in this process through a post-transfer editing function,19 protein maturation and survival,20 maintaining cell wall integrity via carriage of the oligosaccharide in peptidoglycan synthesis,21 and lipid metabolism,22 respectively. The authors disclosed three chemical series active against three targets in S. aureus. The phenyl benzimidazole chemical series is an inhibitor of MRS (benzimidazole 2) with an IC50 of 0.00083 μM in the biochemical assay and exhibits a moderate level of antibacterial activity with a minimum inhibitory concentration (MIC) value of 0.5 μg mL−1. In parallel, (t-butylphenyl-piperazine amide 3) shows binding affinity to IRS, with an IC50 of 0.75 μM and a MIC of 4 μg mL−1. For MetAP, benzimidazole-triazole 4 shows an IC50 of 0.35 μM and a very weak MIC (>128 μg mL−1). In a separate research paper,23 the authors reported the pyrazole chemical series represented by compound 5 as an inhibitor of undecaprenyl pyrophosphate synthase (UppS) in S. aureus with an IC50 of 190 nM and a MIC of 8 μg mL−1 (Scheme 1).
 |
| | Scheme 1 The active chemical structures against four targets in S. aureus. | |
Based on the results from S. aureus screening, the authors decided to expand the screening platform to include targets from A. Baumannii. A. Baumannii is an aerobic, Gram-negative opportunistic pathogen that is responsible for various healthcare-associated infections, including urinary tract infections, ventilator-associated pneumonia, bloodstream infections, secondary meningitis, surgical site infections, as well as burn and soft tissue infections.24 It presents a worldwide health issue and poses a treatment challenge due to the emergence and continuous escalation of resistance to antibacterial drugs.25 The authors chose up to 80 proteins in A. Baumannii. After purification and quantification, 70 proteins were screened; 52 of 70 gave positive DECL selection signals, and 18 were prioritised for off-DNA synthesis. Unlike the biochemical assays used in off-DNA hit validation in S. aureus screening, the authors directly tested the off-DNA synthesised hit compounds in cellular assays for their antibacterial activity against a panel of Gram-negative bacteria. This tactic allowed them to simplify and accelerate the screening validation and prioritise the chemical series that have cell penetration. Three targets and their corresponding chemical series were disclosed: LpxA, UppS, and loLA, which are responsible for lipid biosynthesis,26 membrane integrity,21 and lipoprotein transport,27 respectively. In LPxA, two chemical series were identified. Compound 6, which is a representative structure of one of the series, showed an MIC of <64 μg mL−1 with a confirmed target mode of action. In UppS, three chemical series were identified with confirmed modes of action. Compound 7 gave an IC50 of 0.043 μM, and MIC of <128 μg mL−1 and is a representative of one of the series. In loLA, one chemical series with a confirmed mode of action was identified. Tetrahydropyrido-pyrimidine 8 showed a MIC of <128 μg mL−1 (Scheme 2).
 |
| | Scheme 2 The active chemical structures against three targets in A. Baumannii. | |
The authors expanded the platform screening to include tuberculosis (TB), which remains a significant global health challenge. Although treatments for TB are available, the rise of multidrug-resistant TB (MDR-TB) and extensively drug-resistant TB (XDR-TB) has exacerbated the problem. The causative agent, M. tuberculosis, has become more widespread and resistant to key frontline therapies, such as rifampicin and isoniazid. In a similar manner to S. aureus and A. Baumannii, 42 proteins targets in M. tuberculosis were assessed for their ligandability. The authors opened the platform for academic collaborators to provide purified and his- or biotinylated-tagged proteins. Of the 42 proteins, 27 showed enriched DECL signals. The authors prioritised three chemical series for off-DNA validation. The biochemical assay against dfrA (also known as DHFR) confirmed the potency of three chemical series. Compounds 9–11 showed potencies and MICs between 0.492–4.9 μM and 2.5–80 μg mL−1, respectively (Scheme 3).
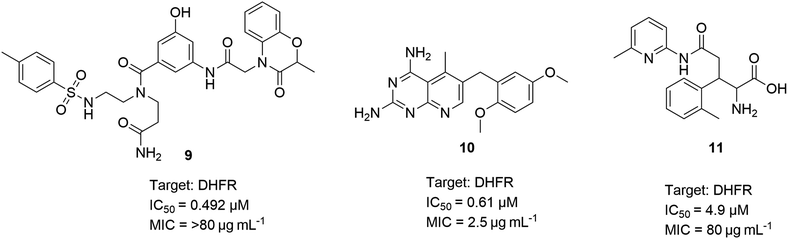 |
| | Scheme 3 The active chemical structures against DHFR targets in M. tuberculosis. | |
To the best of our knowledge, this is the most comprehensive screening platform, with 151 bacterial protein targets for their ligandability. The data generated from this platform will serve the drug discovery community to prioritise bacterial targets based on their ligandability. Interestingly, the authors discovered that the DECL platform has a success rate comparable to high-throughput screening (HTS) platforms within GSK. Moreover, the DECL platform identified hits for all targets during the screening of 29 targets in S. aureus, whereas HTS was only successful in 21 targets. In addition, DECLs predicted the same outcomes as HTS. Ultimately, the key advantage of DECL is the ability to get similar and better outcomes than HTS in a fraction of the cost, effort, and time.
In the upcoming sections, we will delve into the specifics of bacterial targets, the modes of resistance they employ, and the various screening approaches utilised. We will also highlight the contributions of DECLs in identifying potential hits through these screening methods.
4. Mycobacterium tuberculosis
As briefly mentioned above, tuberculosis (TB) is a remarkable health challenge worldwide. It was the main cause of death from infectious diseases, accounting for about 1.3 million deaths in 2022. It is ranked among the top ten causes of death worldwide.28 Treatment of standard TB usually lasts for six months, while that of the drug-resistant TB can take up to 9–20 months with a high relapse rate. Despite the availability of TB treatments, the emergence of both multidrug-resistant TB (MDR-TB) and extensively drug-resistant TB (XDR-TB) has increased the severity of this disease.29 M. tuberculosis, the causative agent of TB, became more prevalent and showed resistance to the frontline therapies: rifampicin and isoniazid. A recent study shows that 7.5% of the tested infected patients are resistant to the first-line drug rifampicin.30,31 In addition, the WHO reported that there was a 37% increase in diagnosed drug resistant cases in 2021 compared to 2020. There is an urgent need to develop new drugs to tackle these multidrug resistance strains. In the following section, we shed light on the role of DECLs in identifying new inhibitors to two well-known M. tuberculosis targets.
4.1. Enoyl–acyl-carrier protein (ACP) reductase, InhA
Isoniazid is the first line therapy for the TB infections32 and it inhibits the essential NADH dependent enoyl–acyl-carrier protein (ACP) reductase, InhA of M. tuberculosis. InhA is crucial for the bacteria's growth and survival. It catalyses a critical step in the fatty acid elongation process essential for synthesising mycolic acids, which are key components of the cell wall of M. tuberculosis.33 This step catalyses the NADH-dependent reduction of enoyl-ACP 15, converting it to acyl-ACP 16 (Scheme 4) to complete a round of elongation. This enzyme is seen as a desirable target for TB drugs, such as isoniazid, due to its involvement in the fatty acid synthesis II (FAS-II) pathway, which is not present in humans.34 This difference confers a high degree of selective targeting of bacterial cells without affecting human cells. Since isoniazid is a prodrug, it is activated with KatG (antioxidant enzyme).35 Activated isoniazid reacts with NADH to form isoniazid–NADH adduct. This adducts binds tightly to InhA, competes with NADH in InhA, and hinders InhA's normal function in fatty acid synthesis.36 Many M. tuberculosis strains are becoming resistant to multiple drugs (MDR-TB). Most of this resistance is due to mutations in the katG gene or the area that controls it. These mutations prevent it from being activated, making it ineffective.37
 |
| | Scheme 4 Fatty acid synthesis II (FAS II), demonstrating the role of InhA. | |
InhA inhibitors do not require activation and would effectively target isoniazid-resistant strains of M. tuberculosis and prevent cross-resistance development. However, discovering InhA inhibitors that are active in cells has been difficult. The lack of compounds with cellular activity has hindered the development of InhA-targeting compounds with suitable characteristics for in vivo studies. Using DECLs has enabled the identification of new molecules that act on various targets, including InhA.38 Soutter and coworkers39 presented previous InhA inhibitors that exhibit different binding preferences for InhA (Table 1 and Scheme 5). Building upon this knowledge of InhA inhibition, the authors sought to discover new inhibitors having cellular activity using DECLs. The data from these previously reported InhA inhibitors were used as benchmarks for evaluating the potency of their newly discovered inhibitors.
Table 1 Previously reported InhA inhibitors. Compound 17: isoniazid adduct.40 Compound 18: PT70.41 Compound 19: pyridomycin.42 Compound 20: methyl thiazole.43 Compound 21: pyrazole ELT hit.44 Compound 22: pyridine dione.45 α: Ki measurement. β: cLogP calculated for prodrug isoniazid rather than active drug INH–NAD adduct. γ: K1 measurement. δ: Kd measurement. Ω: IC50. ††: MIC90. ◆: a close analog, NITD-529, showed binding only to the InhA: NADH. cLogP: calculated log10 of water–octanol partition coefficients. LLE: logarithmic ligand efficiency (log10 of reported affinity minus cLogP). ND: not determined
| Compound |
Reported affinity (nM) |
Cellular activity: MIC50 (μM) (H37Rv) |
cLogP |
LLE |
Predominant InhA binding |
| 17 |
0.79α |
0.158 |
−0.7β |
9.8 |
Apo |
| 18 |
0.02γ |
10.00 |
7.0 |
3.7 |
NAD+ |
| 19 |
6309.57δ |
0.398 |
3.1 |
2.1 |
Apo |
| 20 |
3.16δ |
0.199 |
2.3 |
6.2 |
NADH |
| 21 |
3.98Ω |
0.501†† |
1.4 |
7.0 |
ND |
| 22 |
630.96Ω |
0.079 |
5.3 |
0.9 |
NADH◆ |
 |
| | Scheme 5 Structures of previously reported InhA inhibitors, demonstrating cellular activity in M. tuberculosis. | |
The authors exploited 66-billion-member DECLs supplied from X-Chem, a major provider of DECLs. The InhA target protein was expressed with an N-terminal 6xHis tag. This His tag consists of six consecutive histidine residues and exhibits a high affinity for metal ions such as nickel (Ni2+). Different selection conditions were tested in parallel to identify compounds with diverse binding preferences and potential mechanisms of action. Their analysis revealed four distinct enrichment profiles: (1) compounds enriched only with apo (no cofactor) InhA, (2) compounds enriched only with InhA:NAD+ complex, (3) compounds enriched only with InhA:NADH complex but not when a tight-binding inhibitor was present, and (4) compounds enriched with both InhA:NAD+ and InhA:NADH, but not when a tight-binding inhibitor was present. Notably, 70% of the enriched compound families (groups of structurally related compounds with the same profile) exhibit a preference for the InhA:NADH complex (third profile). The authors selected approximately 50 representative compounds across all enrichment profiles for off-DNA synthesis using the following selection criteria: (a) potential mechanism of action, (b) enrichment level, and (c) physicochemical properties. Two in vitro assay formats were used to test the 50 off-DNA compounds. The first one was protein incubated with only NADH, and the second was protein incubated with a mixture of NADH and excess NAD+. Compounds with IC50 values below 20 μM in the previous two in vitro assays were further tested in a tuberculosis (TB) panel. The TB panel included various assays: (1) minimum inhibitory concentration (MIC), (2) IC50 and IC90 against M. tuberculosis H37Rv (aerobic and anaerobic conditions), (3) minimum bactericidal concentration (MBC) for M. tuberculosis H37Rv, (4) intracellular activity assay (IC50 and IC90) in M. tuberculosis-infected human cells, and (5) MIC against five drug-resistant M. tuberculosis strains. The authors identified 10 series (series 1–10) compounds from enrichment results of different DECLs (Scheme 6 and Table 2). The lead compound 10a was discovered from a family of DECLs containing members that showed specific enrichment when screened against InhA:NAD+, with the compounds sharing a 4-phenylpiperidine-4-carboxylate core structure. The potency of the lead compound 10a against InhA:NAD+ complex exhibited 5-times improvement than that against the InhA:NADH complex. This improvement confirmed a preferential binding towards the InhA: NAD+ complex. To validate these results, SPR experiments were run. Results demonstrated that the compound 10a exhibited a Kd value for the InhA:NAD+ complex, which was 19 times better than that for the InhA:NADH complex. Compound 10a exhibits a MIC value of 12 μM under aerobic conditions (Table 2). These results suggest that it would be more helpful to concentrate on the discovery of inhibitors that specifically bind to InhA bound with NAD+, rather than discovering inhibitors that solely target InhA bound with NADH.
 |
| | Scheme 6 Main structures of the series (1–10). | |
Table 2 Assay results of the most potent compound in each series. Mtb = M. tuberculosis, (N/T) – Not tested
| Series |
Series 1a |
Series 2a |
Series 1c |
Series 4a |
Series 4b |
Series 6a |
Series 7 |
Series 8a |
Series 8a |
| Structure |
 |
 |
 |
 |
 |
 |
 |
 |
 |
| InhA NADH assay IC50, μM |
0.065 |
0.057 |
0.026 |
0.297 |
0.026 |
5.917 |
0.577 |
0.130 |
0.198 |
| Mtb H37Rv aerobic assay MIC, μM |
13 |
N/T |
18.5 |
25.0 |
7.9 |
50.0 |
45.5 |
12 |
12 |
| Mtb H37Rv aerobic assay MBC, μM |
13 |
N/T |
20 |
N/T |
5.1 |
N/T |
N/T |
N/T |
N/T |
| Mtb H37Rv aerobic assay IC50, μM |
10.5 |
N/T |
9.65 |
N/T |
5.1 |
N/T |
N/T |
N/T |
N/T |
| Mtb H37Rv aerobic assay IC90, μM |
13.5 |
N/T |
19.5 |
N/T |
8.6 |
N/T |
N/T |
N/T |
N/T |
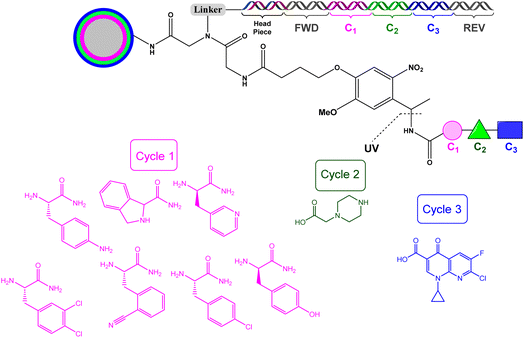
In a subsequent effort, Evindar and coworkers utilised the DECLs to target InhA.44 The DECL was built in three cycles; the first cycle included 22 orthogonally protected diamines, while the second and third cycles included carboxylic acids, aldehydes, sulfonyl chlorides, and isocyanates used as a capping building block for the deprotected diamines (Scheme 7). The DECL composed of 16.1 million compounds and was screened against InhA in three selection conditions (protein alone, protein with NAD+, and protein with NADH).
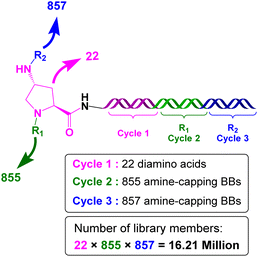 |
| | Scheme 7 DECLs of 3 cycles. The first cycle included 22 orthogonally protected diamines, while the second and third cycles included carboxylic acids, aldehydes, sulfonyl chlorides, and isocyanates used as capping building blocks for the deprotected diamines; cycle 1 in pink, cycle 2 in green, and cycle 3 in blue. | |
The enrichment results revealed distinct pharmacophores under various selection conditions. The aminoproline building block in cycle 1 emerges as a significant hit when the selection conditions include protein with NADH. Off-DNA synthesis of the aminoproline derivatives was conducted to confirm the activity. The key amoniproline core was decorated with different building blocks and screened against InhA assay. The structure activity relationship (SAR) divided the chemical space around the aminoproline core to three attachment points (P1, P2, and P3) (Scheme 8). Pyrazole at the P2 position in derivative 24 perfectly occupies the cleft which is normally occupied by the substrate/product. In addition, the pyrazole nitrogen forms hydrogen bonding interactions with the 2′hydroxyl group of NADH. Compound 24 is potent against the InhA assay and shows a favourable ADMET profile; however, it has modest antibacterial activity against M. tuberculosis. The authors explored novel modification in the three points of the attachments to expand the chemical space beyond building blocks used in DECLs and to increase the inhibition potency against InhA and antibacterial activity. They concluded that compound 25 shows the best balance between InhA inhibition, antibacterial activity and ADMET properties. They progressed compound 25 for in vivo studies against a murine TB acute infection model; however, it turned out to be inactive.
 |
| | Scheme 8 Chemical space around the aminoproline core divided into three attachment points (P1, P2, and P3). Compound 24 shows the best balance between InhA inhibition, antibacterial activity and ADMET properties. | |
4.2. Polyketide synthase 13 (Pks13)
Polyketide synthase 13 (Pks13-TE) is the thioesterase domain of M. tuberculosis and was chosen as the target for DECLs screening.46 Mycobacteria, including M. tuberculosis, depend on specific lipids known as mycolic acids to make an extra layer of protection in their cell wall.47 The mycolic acid biosynthesis pathway is an important target for making drugs against M. tuberculosis (Scheme 4). Pks13 is a multifunctional enzyme that catalyses the last step of mycolic acid synthesis by combining a long-chain fatty acid and a very long-chain meromycolic acid through a condensation reaction.48 Many genetic in vitro and in vivo experiments have proven that Pks13 is essential for the viability of M. tuberculosis.49,50 The disruption of the formation of mycolic acid through the inhibition of Pks13 will compromise the M. tuberculosis cell wall.
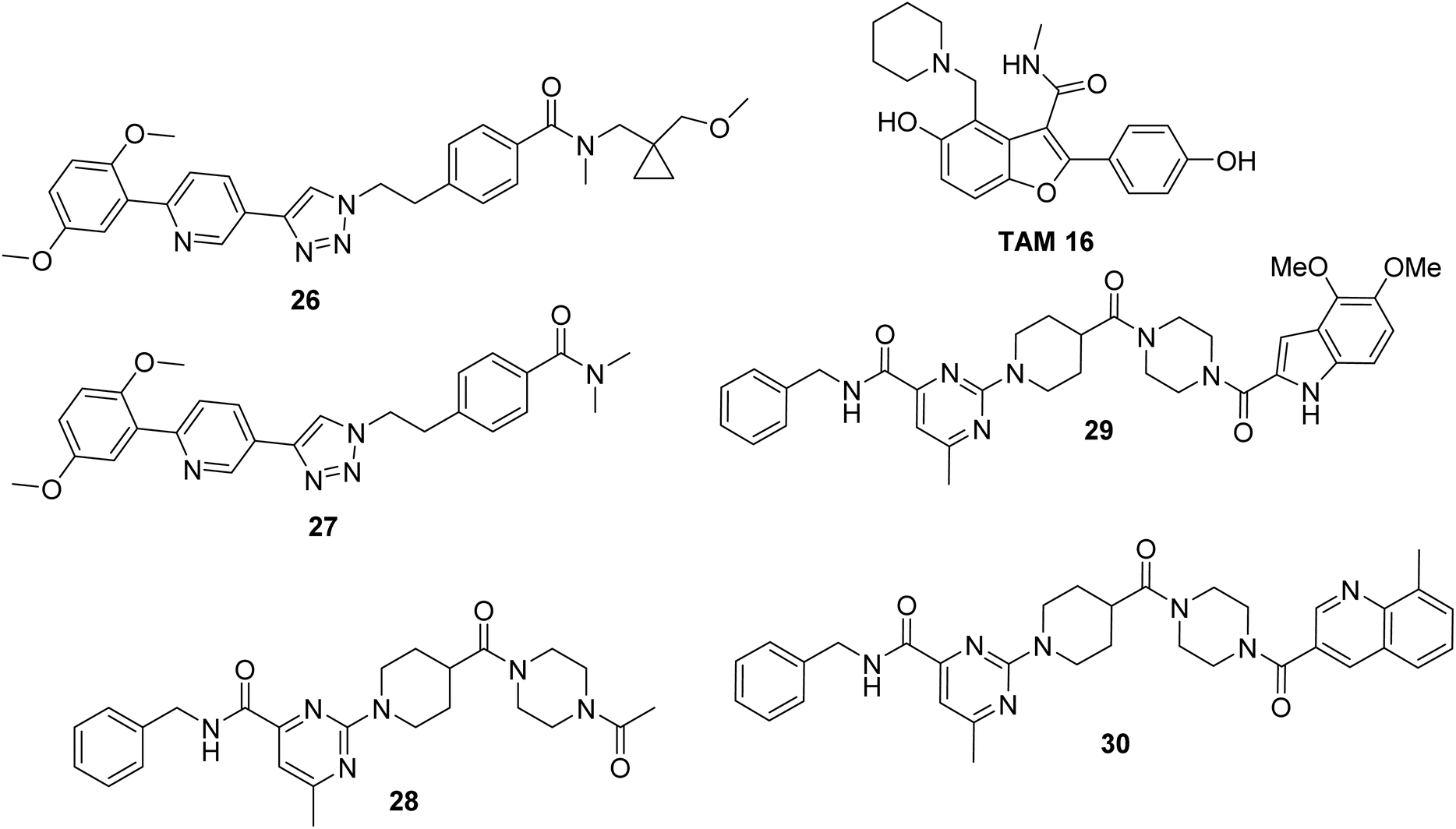
TAM 16 is a lead inhibitor for Pks13-TE with a remarkable efficacy in the TB mouse model compared with that of isoniazid, the first-line drug used for TB treatment (Scheme 9). However, further development of TAM 16 was hindered by hERG toxicity.50 Therefore, the authors decided to exploit 59 DECLs, representing 125 billion compounds for screening against Pks13-TE at 0.5, 2, and 10 μM. The screening conditions included the target protein Pks13-TE alone, Pks13-TE combined with the inhibitor TAM 16, and a no-target control. The selection outputs were PCR-amplified and sequenced into chemical structures. Eighty compounds showed a remarkable enrichment among the three concentrations of Pks13-TE and were prioritised for synthesis off-DNA based on their physicochemical properties and clarity of the SAR. To test these compounds for their enzyme inhibition ability, TAMRA biochemical assay was used. This assay is composed of TAMRA fluorophore linked to a C10 hydrocarbon linker followed by a catalytic serine specific reactive group called fluorophosphonate which was found to bind to the active site of Pks13-TE.51,52 Only 30 of the 80 synthesised off-DNA hits were active, with 18 hits showing IC50 values of less than 20 μM (threshold was set at <50 μM). After extensive hit-to-lead optimisations, compounds 26–30 were progressed for further in vitro studies (Scheme 9). The compounds were tested for inhibiting M. tuberculosis growth in culture medium using wild-type (WT) M. tuberculosis H37Rv and pks13-TetON (it overexpresses Pks13 of the wild-type level when cultivated with anhydrotetracycline [ATc] to ∼400% and under expresses Pks13 when cultivated without ATc to ∼20% of the WT level). Table 3 shows the IC50, MIC results, and fold shifts for each inhibitor.
 |
| | Scheme 9 Structures of DECL hits from series A–B. | |
Table 3 IC50, MIC results and fold shifts for hit compounds
| Hit ID |
IC50 (μM) |
WT H37Rv MIC (μM) |
Hypermorph MIC fold shift (+ATc/WT) |
Hypomorph MIC fold shift (WT/ATc) |
| 26 |
0.5 |
0.05 |
2.3 |
0.88 |
| 27 |
0.43 |
0.25 |
2.3 |
2.9 |
| 28 |
15.1 |
390 |
n.d. |
1.5 |
| 29 |
0.48 |
0.044 |
2.5 |
1.8 |
| 30 |
0.85 |
0.21 |
2.6 |
3.5 |
The four inhibitors (26, 27, 29 and 30) were tested for in vivo efficacy and pharmacokinetic parameters using a female mouse. Inhibitor 26 achieved the best results with higher concentrations in the blood plasma compared to the other three inhibitors. Therefore, this inhibitor was used for further efficacy study on a mouse model with acute infection.
5. Pseudomonas aeruginosa
Pseudomonas aeruginosa is one of the top-listed Gram-negative nosocomial bacteria causing hospital acquired infections.53 It can grow in moist surfaces (e.g., ventilations) in a wide range of temperatures with minimum nutritional requirements, which allows it to spread through contaminated hands, surfaces, and equipment.54 P. aeruginosa is a multi-drug resistance (MDR) opportunistic pathogen. It may be associated with a variety of life-threatening infections, especially in immunocompromised individuals with cystic fibrosis, hospital-acquired pneumonia, urinary tract infections, chronic obstructive pulmonary disease (COPD) and infections requiring ventilation, such as COVID-19.53,55 The discovery of new pharmacophores to combat this multidrug-resistant bacteria is still not at the forefront of addressing this imminent threat. In the following section, we highlight the role of DECLs in identifying potential hits for two targets in P. aeruginosa.
5.1. PqsE thioesterase
PqsE is an enzyme in P. aeruginosa crucial for the bacteria's biofilm formation and virulence.56 This enzyme complexes with RhlR to regulate the bacteria's quorum sensing system and controls several virulence factors, such as pyocyanin, elastase, and rhamnolipids.57 PqsE is seen as a desirable therapeutic target because of its multi-target impact, as it regulates several virulence factors essential for the bacterium's pathogenicity. This enzyme has esterase activity, but the substrate or product resulting from this esterase activity remains unidentified.58 Bross and coworkers59 developed a plasmid by linking the psqE gene with an N-terminal 6xHis tag and stimulating protein production by introducing this plasmid into Escherichia coli BL21.60 After purifying 6xHis-PqsE with nickel, the researchers used cobalt resin for their DECL screen and to eliminate unwanted proteins that co-purified with 6xHis-PqsE on nickel during initial purification. To ensure that the DNA does not interfere with the reaction between small molecules and PqsE, the researchers constructed negative and positive controls. The negative one was only 120 bp of DNA barcode. The positive one was Hartman's61 inhibitor 31, 2-(pyridin-3′-yl) benzoic acid, modified to 32, linked to eight bp DNA via an azide linkage by click chemistry, and ligated to 120 bp DNA to give the positive control 33 (Scheme 10). The researchers screened the two controls against PqsE prior to the large library screening to validate their DECL selection protocol's selectivity and efficiency, ensuring its reliability for the subsequent 550 million compound screen. The screening started with the 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :100 ratio of positive to negative, and ended with recovering a 50:1 ratio of positive to negative after three rounds, which represents a 5000-fold enrichment (from 1:100 to 50:1) of positive control. This dramatic enrichment reveals the power of DECLs to identify specific binders to the target.
:100 ratio of positive to negative, and ended with recovering a 50:1 ratio of positive to negative after three rounds, which represents a 5000-fold enrichment (from 1:100 to 50:1) of positive control. This dramatic enrichment reveals the power of DECLs to identify specific binders to the target.
 |
| | Scheme 10 Positive control synthesis from the known inhibitor. | |
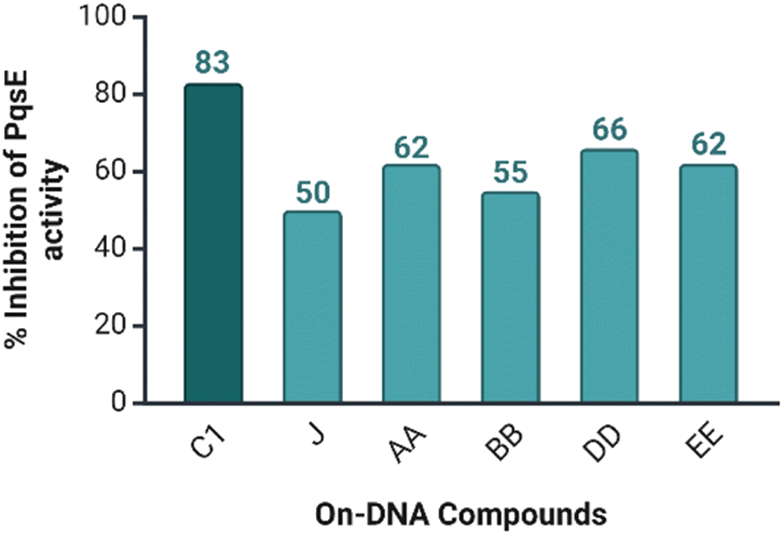
Five libraries were constructed containing 550 million small molecules. The screening results showed a central p-aminobenzoic acid with pendant nitrogen heterocycles (5-, 6-, or 7-membered) as the most enriched central building blocks. Based on this observation, 31 compounds were chosen for resynthesis using (on-DNA resynthesis) to evaluate their ability to inhibit the catalytic activity of PqsE. Meta-bromo-thiolactone (mBTL) was selected as the artificial substrate of PqsE since the natural substrate of PqsE is unknown. The Ellman assay was chosen to evaluate the ability of the 31 DNA conjugated compounds to inhibit the catalytic PqsE activity. Among the 31 compounds, the five most potent DNA conjugates are J, AA, BB, DD, and EE, and they reduced the enzyme activity by 50% at least (Fig. 6).
 |
| | Fig. 6 Bar chart of the five most potent DNA conjugated compounds among the 31 tested ones. C1 was measured at 100 μM, while the remaining DNA conjugated compounds were measured at 10 μM. Compound C1 has the highest activity in the bar chart. Created in https://biorender.com/. | |
Although compound BB was not the most efficient of the five compounds, it was chosen for the off-DNA synthesis because of its structural similarity to the known inhibitor 31. Four analogues (34–37) were synthesised (Table 4). The four analogues (34–37) were tested in different biophysical and biochemical assays, leading to the confirmation of binding and activity of compounds 34 and 36. Furthermore, the researchers synthesised twelve additional analogues. The most potent compounds (IC50 < 20 μM) were tested in vitro to assess their impact on P. aeruginosa's production of pyocyanin, which is dependent on PqsE. During the monitoring of pyocyanin production, there was no reduction in its production during the test of the most potent compounds. This led to further investigation about the fates of 34 and 36 in the in vitro assay. Mass spectrometry was used to quantify the level of the two compounds over time after the administration of mBTL as a positive control to P. aeruginosa. As expected, the mBTL concentration increased in the cell lysate and decreased in the medium surrounding the bacteria. In the meantime, compounds 34 and 36 initially remained in the same concentration in the medium surrounding the bacteria, but they were not detected after hours inside the cell lysate or in the medium surrounding it. This indicates that they were quickly effluxed from the bacteria, quickly metabolised or did not enter at all. This explained why pyocyanin production was not reduced in the in vivo assay. Finally, a study on mechanism of action was carried out, and the results showed that compounds 34 and 36 behaved as non-competitive inhibitors, but it was unknown whether the compounds were allosteric or orthosteric.
Table 4 Structure and IC50 values of the compounds (34–37) in PqsE enzyme inhibition activity assay
| Compound |
Structure |
IC50 (μM) |
Compound |
Structure |
IC50 (μM) |
| 34 |
 |
18.1 |
36 |
 |
19.1 |
| 35 |
 |
>100 |
37 |
 |
>100 |
5.2. UDP-N-acetylglucosamine acyltransferase (LpxA)
UDP-N-acetylglucosamine acyltransferase (LpxA) is a vital enzyme that contributes to the biosynthetic pathway of the outer lipopolysaccharide membrane of P. aeruginosa and other Gram-negative bacteria.62 It is essential for the viability of P. aeruginosa and other Gram-negative bacteria. In contrast, Gram-positive bacteria are missing this enzyme. Therefore, inhibition of this enzyme will lead to inhibition of P. aeruginosa and other Gram-negative bacteria. There is no known inhibitor targeting LpxA. Therefore, targeting this enzyme will develop a new class of antibiotics with a novel mode of action. LpxA is functional trimer with active site between the monomer units. It is part of the Raetz pathway for the biosynthesis of lipopolysaccharide in Gram-negative bacteria.63 The enzyme catalyses the reversible transfer of (R)-3-hydroxydecanoate from the acyl carrier protein (ACP) onto the 3-OH position of UDP-N-acetylglucosamine (UDP-GlcNAc). The carbon chain length of the fatty acid linked to ACP varies by species (Scheme 11).
 |
| | Scheme 11 Biosynthesis of lipopolysaccharide through the enzymatic reaction of LpxA, which is part of the Raetz pathway in Gram-negative bacteria. | |
Ryan and coworkers exploited DECL technology to discover novel inhibitors targeting LpxA.64 Eleven diverse DECLs with a total of 66 billion DNA tagged compounds (provided from X-Chem) were incubated with histidine- and FLAG-tagged P. aeruginosa LpxA (FLAG-P. aeruginosa LpxA-His) in the presence and absence of LpxA substrate (UDP N-acetyl-α-D-glucosamine or the inhibitory peptide 920) or its product UDP-3-O-((R)-3-hydroxydecanoyl)-N-acetylglucosamine. The same incubation conditions were replicated to histidine- and FLAG-tagged E. coli LpxA (His-E. coli LpxA-FLAG) with or without its substrate (UDP N-acetyl-α-D-glucosamine or the inhibitory peptide 920) or its product UDP-3-O-((R)-3-hydroxymyristoyl)-N-acetylglucosamine. The enrichment results of these selections showed a cluster of structurally related building blocks that can bind to P. aeruginosa LpxA but not to E. coli LpxA. No reduction in enrichment was observed when peptide 920 or the substrate of P. aeruginosa LpxA were present; however, a significant reduction in the enrichment was observed when the product of P. aeruginosa LpxA was present. This suggested that the enriched compounds bind to LpxA in a competitive way with its product. The resynthesis (off-DNA) of the most enriched compound 38 and testing it against P. aeruginosa LpxA gave IC50 = 400 nM. However, the same compound shows no inhibition against E. coli LpxA (IC50 > 50 μM). Although compound 38 shows promising potency against P. aeruginosa LpxA, the compound shows no cellular activity in a bacterial assay against P. aeruginosa: MIC > 128 μg mL−1 (Scheme 12).
 |
| | Scheme 12 Most enriched compound from the selection process against P. aeruginosa LpxA and E. coli LpxA. Lead optimisation campaign gave compound 39. | |
Extensive hit-to-lead optimisation of hit compound 38 aimed at enhancing target inhibition, cellular activity, and metabolic stability led to the discovery of a highly potent compound 39 against P. aeruginosa LpxA. Compound 39 demonstrated an IC50 of 2 nM and an MIC of 8 μg mL−1 against P. aeruginosa. The authors concluded that further efforts should be directed towards increasing the accumulation of the active compounds into the bacterial cells. They spotted one of the key reasons of the low compound accumulations inside bacterial cells to be the efflux pumps, which expel the active compounds outside the bacteria.
6. Mechanisms of antimicrobial resistance
Resistance mechanisms to antimicrobials can be classified into two primary categories: intrinsic and acquired resistance.65 Intrinsic resistance occurs when bacteria inherently possess specific characteristics or structures that render them impervious to the effects of an antibiotic. In contrast, acquired resistance arises when bacteria develop resistance mechanisms through either vertical evolution, involving mutations, horizontal evolution, or the acquisition of resistance genes from other organisms.66 The mechanisms of acquired resistance include modifications to the target protein to which the drug binds, reduction in the accumulation or uptake of the antimicrobial agent, overexpression of enzymes that inactivate the drug, and overproduction of the antimicrobial target.67 In the following section we will shed light on the role of β-lactamase in resisting antibiotics.
6.1. β-Lactamase enzymes
β-Lactam antibiotics such as penicillins, cephalosporins, and carbapenems are the most widely used antibiotics. However, the ability of β-lactamase to hydrolysis these antibiotics became a major source of antibiotic resistance. β-Lactamases are composed of main four groups (A–D) based on their amino acid sequence homology. Classes A, C, and D are serine-based hydrolases,68–70 while class B is based on zinc metallo-enzymes. KPC-2 (class A), NDM, VIM, and IMP enzymes (class B), and OXA-48 (class D) β-lactamases are among the most common carbapenemases. Inhibition of β-lactamase will lead to increasing the treatment options of multidrug resistance pathogens. Several β-lactamase inhibitors were developed to restore the β-lactams antibiotics activity;71 however, bacterial resistance is still in progress. One of the important reasons of the inefficacy of the β-lactamases inhibitors in Gram-negative bacteria is the lack of accumulation of inhibitors in the periplasm, where the β-lactamases are expressed.72–74 Therefore, discovering new pharmacophores that able to penetrate the bacteria outer membrane and accumulate in the periplasm is vital to restore β-lactam antibiotic efficacy.
Palzkill and coworkers built a focused DECL exploiting the highly conserved carboxylate-binding pocket, which is located next to the catalytic residues of the active site of β-lactamases. The authors designed the DECL to specifically target the carboxylate-binding pocket by building three different libraries (L1, L2, and L3). These two cycle libraries were built by combining different chemical building blocks (BB1 and BB2). Each compound in the library ends up with a carboxylate group or tetrazole group designed to interact with residues of the carboxylate binding pocket (Scheme 13). The combined focused libraries were screened against the OXA-48 and NDM-1 β-lactamases, leading to the identification of several hits.
 |
| | Scheme 13 Synthetic scheme of the three focused DECLs including COOH/tetrazole as a binding motif. | |
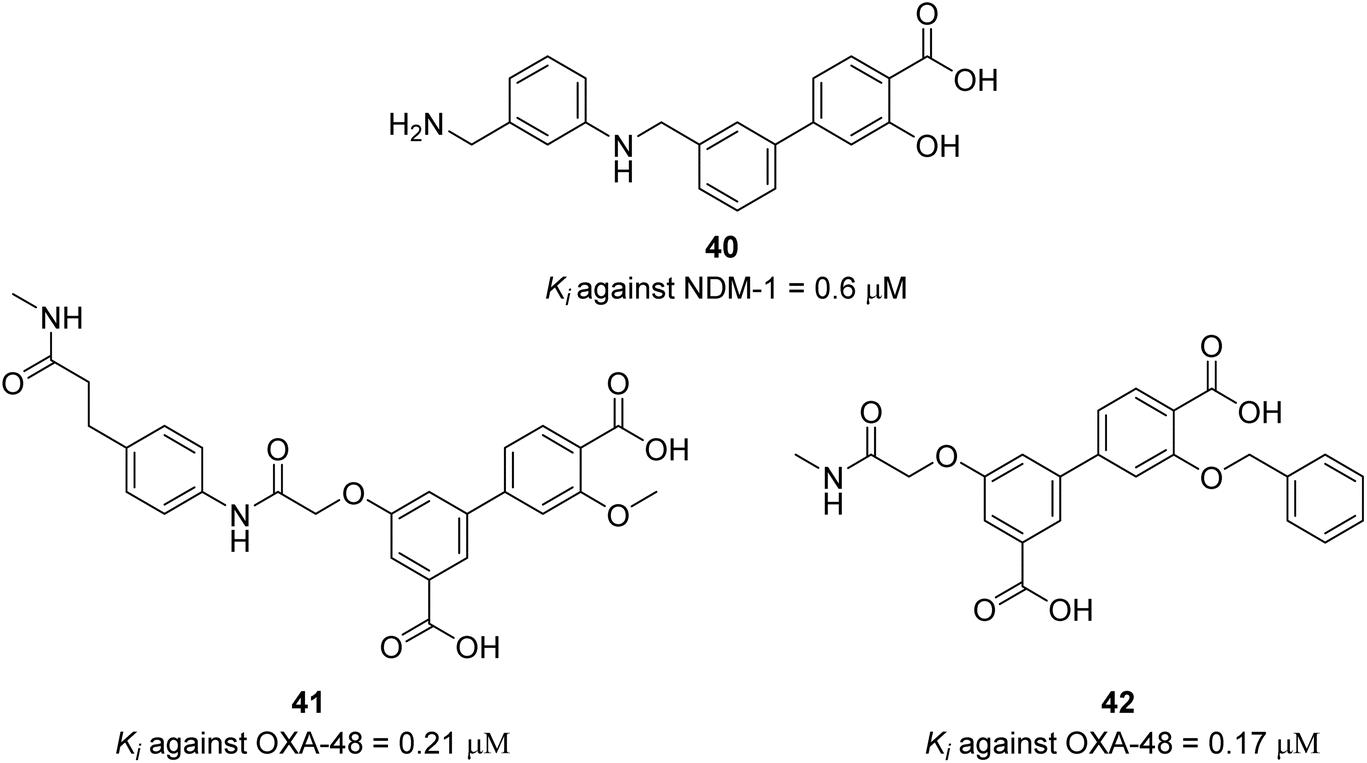
The authors screened the focused DECLs L1, L2, L3 and another 13 different DECLs against NDM-1 (class B metallo-β-lactamase). The only library which gave potential binders is DECL L1 which have carboxylate-binding pocket-targeting motifs (carboxylic acid/tetrazole). The approach of focused DECL to exploit the available biochemical information about the target enzyme to direct the DECL synthesis is facilitating the library design and is very efficient to produce highly selective compounds. Several binders were identified containing salicylic acid derivatives. Salicylic acids are known to chelate metal ions, which is an indication that the identified binders have strong affinity with zinc in the active site. The three most enriched compounds were prioritised for off-DNA synthesis and tested in enzymatic inhibition assay against NDM-1 with the carbapenem antibiotic imipenem as the reporter substrate and zinc ions, which are essential for NDM-1 activity. After extensive SAR optimisation, compound 40 was identified as the best compound with submicromolar enzymatic inhibition potency (Scheme 14). The compound's ability to inhibit NDM-1 in the MIC assay in the presence of imipenem against E. coli expressing NDM-1 enzyme was examined. Compound 40 had a two-fold reduction in imipenem's MIC at 8 μg mL−1, suggesting that it had a weak synergistic activity with imipenem. One of the potential reasons of this weak synergistic activity is the ability of compound 40 to accumulate in the periplasm of the bacteria. The authors confirmed that compound 40 had a comparable accumulation level to tetracycline, which is known for its high accumulation levels. They concluded that the modest in vitro inhibition potency (Ki) of compound 40 could be the reason behind the weak synergistic activity with imipenem. The focused DECL approach leverages biochemical data about the target enzyme, along with topological and pharmacophoric information to guide the synthesis of the DECLs. This strategy enhances library design and efficiently yields highly selective compounds.
 |
| | Scheme 14 Two best OXA-48 inhibitors and NDM-1 hit compound from DECL screening and SAR optimisation. | |
In a similar vein, OXA-48 (class D) β-lactamase is among the most common carbapenemases.75 It is the most resistant β-lactamase of all clinically available β-lactamase inhibitors except avibactam.76–78 The majority of β-lactamase inhibitors are directed to class A β-lactamases; however, they are not active against OXA-48 which have low sequence homology with class A β-lactamases.79 Avibactam is a non-β-lactam based inhibitor that can inactivate OXA-48 β-lactamase with significant efficacy. However, the ability of other β-lactamases to hydrolyse avibactam, such as KPC-2, raises concerns about the potential development of resistance mechanisms that could inactivate non-β-lactam-based inhibitors like avibactam.
Similar to the screening against NDM-1, the screening of the combined focused libraries (L1, L2, and L3) against the his-tagged OXA-48 (class D β lactamase) gave four potential binders, which were tested in an enzyme inhibition assay. Two compounds were confirmed as potential hits and show promising enzymatic inhibition activity. Further structure activity relationship (SAR) revealed compounds 40 and 41 as the potent inhibitors for OXA-48 (Scheme 14).
In another study, the authors exploited triazine focused DECLs to find non-β-lactam inhibitors for OXA-48 β-lactamases.73 A triazine DECL was synthesised generating 162 million unique compounds (Scheme 15). The triazine DECL synthesis started by attaching a DNA headpiece to building block 1 (BB1) though a linker to give DNA conjugate 45, which completed cycle 1 of split and pool combinatorial synthesis. The further two cycles led to the installation of building blocks 2 and 3 (BB2 and BB3) into the triazine core (Scheme 15). The final DECL 47 was pooled and submitted for selection.
 |
| | Scheme 15 Triazine DECL synthesis through 3 cycles of split and pool combinatorial chemistry. | |
The selection process of the triazine DECL led to the identification of 5 hits (48–52) (Scheme 16). Each of these compounds were resynthesised off-DNA and tested for their inhibition against OXA-48. Compound 48 exhibited micromolar activity against OXA-48 with Ki = 0.9 ± 0.1 μM. Compounds 49 and 50 showed minimal activity with Ki values >50 μM revealing that building block 1 (fluoro-N-methylazetidine-3-carboxamide group) did not contribute to OXA-48 inhibition. Compound 51 was the most potent against OXA-48 with a Ki of 0.53 μM. It has a carboxylate group that can bind the carboxylate binding pocket of OXA-48 making it the lead compound for further investigations. The alkylated form of compound 51 gave compound 52 which showed loss of activity against OXA-48 with Ki values >50 μM. The potent activity of compound 51 was confirmed from the crystal structure of compound 51 with OXA-48, which showed that 51 binds in the active site of OXA-48 through several interactions.73
 |
| | Scheme 16 Hit compounds identified from DECL selection against OXA-48. Orange is building block 1, green is building block 2, and purple is building block 3. | |
Compound 51 was also tested for its inhibition ability against other OXA enzymes that are prevalent in different bacterial families (Table 5). This compound inhibits OXA-48 with higher potency than any of the other OXA enzymes.
Table 5 Inhibition constants (Ki) of compound 51 with different OXA β-lactamases
| Enzyme |
Sequence identity (%) |
51 |
| Ki (μM) |
| OXA-48 |
100.0 |
0.53 |
| OXA-10 |
49.4 |
61 |
| OXA-24 |
36.1 |
14 |
| OXA-58 |
36.2 |
45 |
| OXA-163 |
97.9 |
0.44 |
The minimum inhibitory concentrations (MIC) studies were performed against E. coli MG1655OXA-48 strain expressing OXA-48. The two β-lactam antibiotics (ampicillin and imipenem) were used separately with increased concentrations of compound 51 and avibactam as a positive control (MIC values are found in Table 6). Compound 51 showed poor activity compared to avibactam in the MIC assays which was due to poor accumulation and its susceptibility to efflux pumps by bacteria that drive the compound out of the cell. Modifications to compound 51 for optimised accumulation in the bacteria and enhanced efficacy in vivo was achieved by the addition of one or more primary amines and by decreasing its flexibility.
Table 6 Minimum inhibitory concentrations (MICs) of ampicillin (AMP) and imipenem (IMP) in the presence of compound 51 and avibactam
| [Compound 51] (μg mL−1) |
MIC (μg mL−1) |
[Avibactam] (μg mL−1) |
MIC (μg mL−1) |
| AMP |
IMP |
AMP |
IMP |
| 0 |
512 |
0.625 |
0 |
512 |
0.625 |
| 4 |
1024 |
0.625 |
0.125 |
256 |
0.3125 |
| 8 |
512 |
0.3125 |
0.25 |
128 |
0.3125 |
| 16 |
512 |
0.3125 |
0.5 |
64 |
0.1563 |
| 32 |
512 |
0.3125 |
1 |
32 |
0.1563 |
| 64 |
512 |
0.3125 |
2 |
16 |
0.1563 |
| 128 |
512 |
0.3125 |
4 |
4 |
0.781 |
| 256 |
512 |
0.625 |
7. Phenotypic DNA-encoded chemical library screening
In a significant advancement in the DECL screening approach, Paegel and his coworkers80 synthesised DECL beads that can be directly tested using a bead diffusion assay81,82 to evaluate their ability to inhibit bacterial growth. This approach circumvents the need to identify and isolate purified protein targets and their binders and subsequently resynthesise these binders with or without their DNA tags for further screening and optimisation of analogues for cellular activity. This means that compounds that are unable to penetrate cells or inactive in cellular environments are excluded from the drug discovery process early on. It is also an attempt to resolve the paradox that screening against purified isolated proteins requires the compound to be hydrophobic, but screening against bacterial cells requires the compound to be hydrophilic to accumulate in bacterial cells.83,84 The DECL bead library comprised 7488 members and was synthesised in three cycles using the split-and-pool combinatorial synthesis. Each bead contained a DNA tag and a single library member which covalently attached to the bead via a photocleavable linker. This linker is designed to break when exposed to light, releasing the compound from the bead (Scheme 17). The whole cell bead diffusion assay was used to screen the library against Gram negative (E. coli) and Gram positive (B. subtilis) bacteria. Ciprofloxacin (PC-cipro) was used as a positive control. The library contained 85000 beads for screening, 45000 of which were screened against E. coli, and the remaining 40000 against B. subtilis. Twenty-four beads formed a growth inhibition zone (GIZ) against E. coli and 73 against B. subtilis, and the total hit ratio to the total number screened was 0.114%. The DNA tags of these 97 hits were amplified by PCR, sequenced and decoded. The observations indicated that the identity of the building block at position 1 seemed to have a minimal effect on the activity of these structurally related compounds. The broth microdilution assay was used to determine the MIC of hits. The off DNA resynthesised hits were tested against the same bacterial strains used in the initial screening against E. coli and B. subtilis. Despite hits 53 and 54 exhibiting MIC values of approximately 32 μg mL−1 for E.coli and 1.5 μg mL−1 for B. subtilis, they were not selected for further investigation because they did not demonstrate a clear structure activity relationship (SAR). The most potent hit was compound 60, which exhibited a MIC value of 0.016 μg mL−1 against E.coli and 0.063 μg mL−1 against B. subtilis.
 |
| | Scheme 17 One bead structure; (C) letter denotes cycle. | |
The optimisation pathway for the hits is illustrated in Fig. 7 and the structure activity relationship is in Scheme 18. The researchers selected the most potent compound 60 and the positive control (PC-cipro) to test their activity against E. coli DNA gyrase and also determine the IC50. The IC50 of PC-cipro was 0.28 μM, which outperformed compound 60 by five times (IC50 of 1.43 μM). At the same time, compound 60 succeeded in inhibiting the biochemical activity of E.coli DNA gyrase. This indicates that compound 60 worked similarly to (PC-cipro) but exhibited less potency. Compounds 57 and 60 were selected for testing their potential as broad-spectrum antibiotic activity using antibacterial disk diffusion assays against three panels. The three panels included (a) standard ATCC (American Type Culture Collection) organisms, (b) clinical isolates (bacteria isolated from patients), and (c) carbapenem-resistant (MDR) Enterobacter strains (Table 7). The results indicate that compound 60 shows promise as a broad-spectrum antibiotic that is effective against both Gram-positive and Gram-negative bacteria. Compound 60 specifically forms growth inhibition zones for two out of four clinical tested isolates and was active just against one of eleven multi-drug resistant (MDR) strains.
 |
| | Fig. 7 Flow chart highlighting the progression and structural optimisation of fluoronaphthyridone-based compounds. MIC values in μg mL−1. Created in https://biorender.com/. | |
 |
| | Scheme 18 Nine compounds and the positive control (ciprofloxacin) with their MIC values (μg mL−1), where red indicates activity against E. coli and blue indicates activity against B. subtilis. | |
Table 7 Antibacterial activity of compound 60 compared with ciprofloxacin and cefiderocol against ATCC control strains and clinical isolates. Values represent zone of inhibition diameters (mm) in antibacterial disk diffusion assays. N/D indicates no detectable inhibition zone. CRE denotes carbapenem-resistant Enterobacterales
| Organism |
Compound 60 (5 μg) |
Ciprofloxacin (5 μg) |
Cefiderocol (30 μg) |
| AATCC control strains |
| E. coli ATCC 25922 |
31 |
33 |
25 |
| B. subtilis ATCC 23857 |
28 |
31 |
15 |
| P. aeruginosa ATCC 27853 |
24 |
27 |
25 |
| K. pneumoniae ATCC 700603 |
20 |
21 |
23 |
| K. pneumoniae BAA 1705 (CRE) |
N/D |
N/D |
19 |
| S. aureus ATCC 25923 |
20 |
23 |
9 |
|
| Clinical strains |
| E. coli |
N/D |
N/D |
25 |
| K. pneumoniae |
23 |
23 |
26 |
| K. pneumoniae |
N/D |
N/D |
25 |
| S. marcescens |
20 |
18 |
29 |
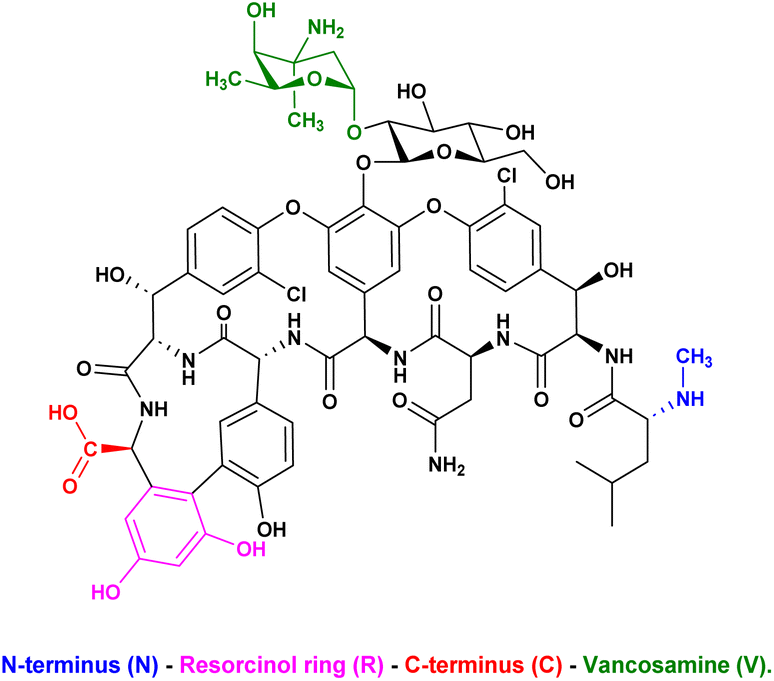
To combat antimicrobial resistance, it is necessary to consistently enhance the effectiveness of antibiotics such as vancomycin.85 In another phenotypic screening, Huang and his coworkers86 developed a novel DECL termed a vancomycin-templated DEL, which possesses 14100 derivatives of vancomycin that was screened against vancomycin-intermediate resistant Staphylococcus aureus (VISA) and a vancomycin-resistant Enterococcus faecium (VRE) strain through a binding affinity assay. The standardised antibiotic assay, which means the expected death of bacteria, is unsuitable for DECL screening. The affinity-based assay is facilitated by the binding of vancomycin to the Lys–DAla–DAla arrangement found in peptidoglycan precursor peptides, which are integral components of the bacterial cell wall.87 In parallel, the potential bactericidal activity remains significant during the binding affinity assay, which complicates the screening of the library. To easily observe the influence of the activity of the vancomycin-templated DEL against VISA and VRE, vancomycin was tested in different concentrations against the two strains. Both the VISA-Mu50 and VRE-0649 strains survived at concentrations of vancomycin up to 0.1 and 3 mg mL−1, respectively. Based on these results, measuring bacterial binding with concentrations of vancomycin below 0.1 mg mL−1 is possible. Despite the variety of modifications that can be performed on vancomycin, such as alterations on the core cyclopeptide88 or cationic moieties,89 peptide conjugates,90 lipid tails91 and saccharides92 at peripheral positions, the authors focused on modifications at peripheral positions (Scheme 19).
 |
| | Scheme 19 Chemical structure of vancomycin, highlighting the N-terminus (blue), resorcinol ring (magenta), C-terminus (red), and vancosamine (green). | |
The modification on the N-terminal will have a minimal impact on antimicrobial effect.93 The modifications on the C-terminal and resorcinol positions are less common than modifications on the vancosamine position. The authors installed dibenzoazacyclooctyne (DBCO) onto vancomycin, which is termed Van-DBCO (Scheme 20). Following this, the DNA headpiece was attached onto the N-terminus through copper-free strain-promoted azide–alkyne cycloaddition (SPAAC). Then, a Cu-catalysed click reaction installed 150 azides in the resorcinol peripheral position. Afterwards, the vancosamine DNA conjugates reacted through the C-terminus amine group with another 150 aldehydes, under reductive amination conditions. The final Van-templated DEL library encompassed 14100 members (Scheme 21).
 |
| | Scheme 20 Synthesis pathway of Van-DBCO. Step 1: Norvancomycin is modified with an alkyne group using formaldehyde and N-methylpropargylamine. Step 2: The modified compound is attached to a PEG chain (PEGylated) and coupled with a DBCO group using NHS, EDCI, and NH2–PEG4–COOH. | |
 |
| | Scheme 21 Synthesis of Van-derived DNA encoded library. | |
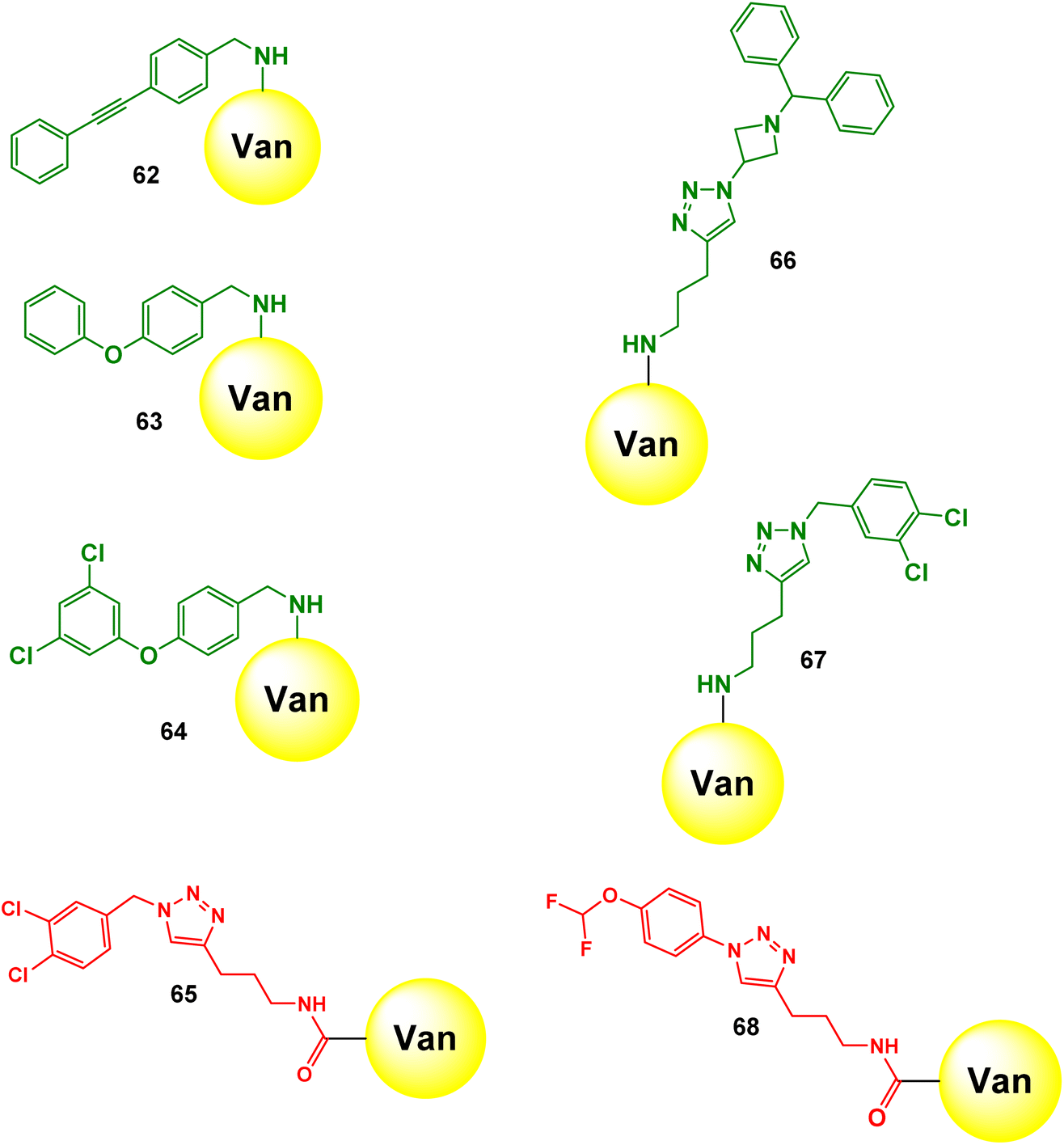
The authors screened the Van-focused DEL through affinity binding assays against both targets: VISA and VRE. The high binding compounds were identified after quantitative PCR (qPCR), NGS, and decoding. During analysing the results, the authors measured copy number (how many copies of a specific compound are present in a sample) before and after screening. This means that the compound that successfully binds has a significant higher copy number and is likely to be a potential effective antibacterial agent. They also measured select-E (the percentage of the copy number of a specific compound after the screening divided by the percentage of the copy number of that compound before the screening). Select-E means how well and copy number means how many. After analysing the screening result, the authors prioritised several derivatives that showed higher bacterial binding affinity than all the other building blocks to synthesise and to perform the antibacterial test to determine the minimum inhibitory concentration (MIC) values. The selection of the resulting derivatives was not dependent on copy number and select-E because their calculation was separately based on each individual compound. Therefore, they preferred to use the average select-E because it provided a more comprehensive perspective by showing how different compounds work together. It enables authors to assess the overall effect of a group of compounds and facilitates comparison between different sets of compounds. The authors assumed that the R1 and R2 groups in the Van-templated DEL may not synergise well, despite their significant individual effects. In addition, they noticed that the decrease of activity in the antibacterial assay is due to unsuitable additional modifications that may affect the vancomycin pharmacophore. Thus, they adopted the building block rational to select only single modification during the resynthesis process. The synthesised compounds (62–68) were tested against clinically isolated strains of S. aureus, Staphylococcus epidermidis, Enterococcus faecalis, Enterococcus faecium, and S. pneumoniae (Scheme 22). The results showed remarkable activity with MIC values as low as 0.015–0.03 μg mL−1. These compounds outperformed vancomycin 64–128 times against drug-resistant strains of S. aureus and S. epidermidis and up to 128 times against E. faecalis and E. faecium. They outperformed levofloxacin by up to 1024 times against drug-resistant strains of S. aureus and S. epidermidis, and even 1024–4096 times in some cases (Table 8).
 |
| | Scheme 22 Chemical structures of 7 novel vancomycin-derived compounds (62–68). | |
Table 8 Minimum inhibitory concentration (MIC) values (μg mL−1) of vancomycin, levofloxacin, and 7 novel compounds (62–68) against various bacterial strains including Staphylococcus, Enterococcus, and Streptococcus species. Color-coded MIC values indicate compound efficacy, with blue representing the lowest (most effective) concentrations and red representing the highest (least effective) concentrations. (Van) – vancomycin, (Lev) – levofloxacin, (N/T) – not tested
8. Targeting membrane synthesis enzymes
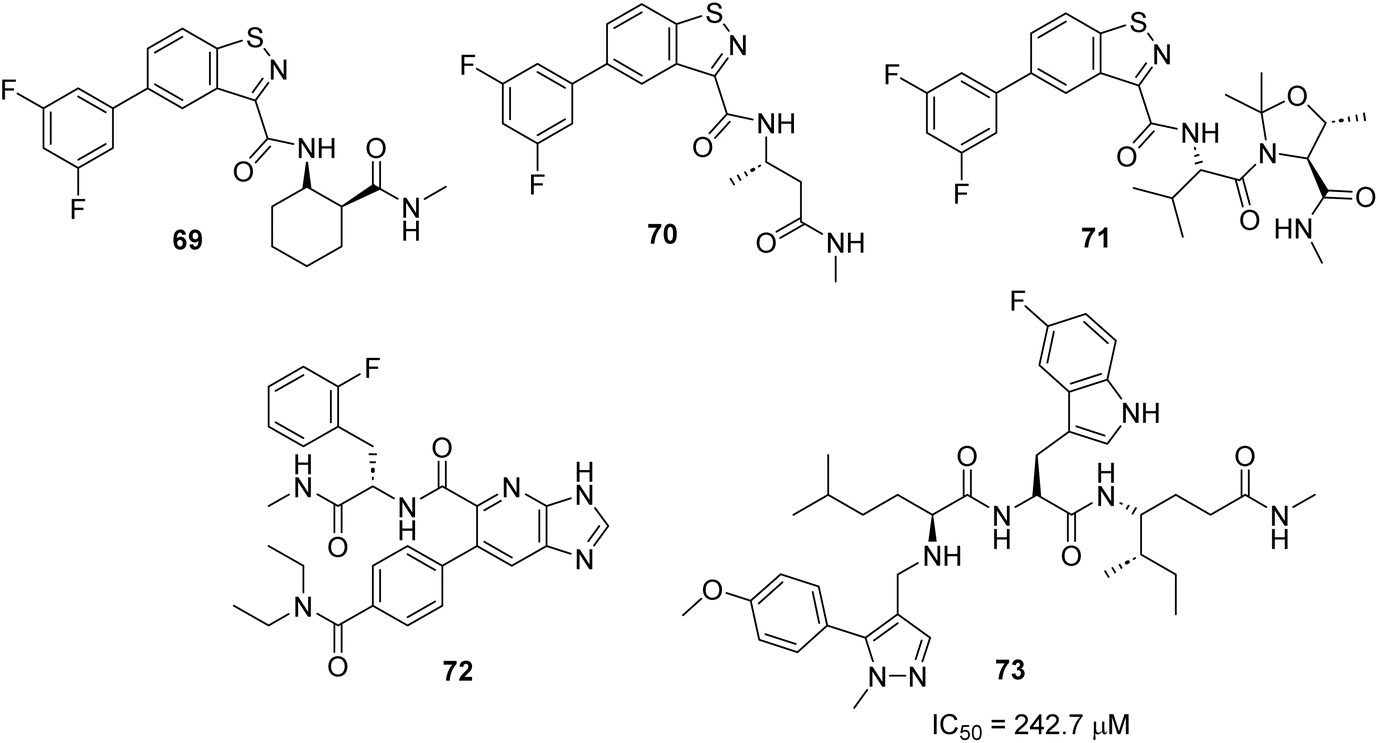
The bacterial cell wall is essentially composed of peptidoglycan as a building block. The two intracellular enzymes: UDP-N-acetylglucosamine-enolpyruvyl transferase (MurA) and D-alanine:D-alanine ligase (Ddl) are mainly involved in the biosynthesis of peptidoglycan, so the inhibition of these two enzymes has been successfully targeted.94,95 Fosfomycin is a broad-spectrum antibiotic used to treat lower urinary tract infections that targets MurA enzyme inhibition through covalent reaction with the catalytic cysteine.96 Several resistance bacteria such as M. tuberculosis, Borrelia burgdorferi, and Chlamydia trachomati do not have the Cys residues in the active site of the MurA enzyme, making them resistant to fosfomycin. There are other reasons reported to be cause resistance to fosfomycin (e.g., enzymatic inactivation of the drug itself, changes in the active site of the antibiotic and decreased permeability).96,97 D-Cycloserine inhibits Ddl and is used to treat multidrug-resistant tuberculosis. However, it causes severe side effects, including peripheral neuropathy and seizures.98 Gobec and coworkers used a DELopen library99 that includes 4.4 billion compounds that have been pooled into a single mixture from 30 different subsets to find inhibitors for DdlB and MurA enzymes.100 The enzymes were His-tagged and immobilised on magnetic Ni-NTA beads for the affinity-based DECL screening. In the first DECL screening against Ddlb, five compounds (69–73) bound DdlB and were prioritised for off-DNA resynthesis to confirm their ability to inhibit the enzyme. Compounds (69–72) did not inhibit the enzyme, whereas compound 73 showed an inhibitory effect with an IC50 value of 242.7 μM (Scheme 23).
 |
| | Scheme 23 Hits from screening against DdlB. | |
In parallel, DECL screening against MurA revealed two compounds (74, 75). The two compounds exhibited an inhibitory activity with IC50 values of 14.1 μM and 15.5 μM, respectively (Scheme 24).
 |
| | Scheme 24 Hits from screening against MurA. | |
9. Conclusions and future perspectives
The full potential of DNA encoded chemical libraries (DECLs) to identify new hits tackling antimicrobial resistance has yet to be fully achieved. These libraries offer a unique opportunity to explore chemical space and identify the best binders for proteins of interest. However, their success in addressing antimicrobial resistance has been limited. The pioneering work by GSK scientists in exploring the ligandability of numerous bacterial proteins was groundbreaking. Although their 66 billion compound libraries did not yield hits for all tested targets, it highlighted the importance of DECL selection conditions in hit-protein binding. Some proteins may require cofactors or complexes that are not accessible under standard DECL selection conditions. It is hard to believe that such mega-libraries did not produce suitable hits. Therefore, addressing the selection processes to go beyond immobilisation to resins and allowing screening in proteins' native environments could overcome the hit identification hurdles for certain protein classes. For instance, DECL selection in cell lysates can provide new opportunities to identify new bacterial targets. In addition, screening DECL in cell lysates provides a very similar native environments for proteins of interest.101 Target specificity can be achieved by spiking-in or overexpression of targets of interest. Phenotypic screening of DECLs against membrane proteins and bacterial efflux pumps can provide a synergistic solution to the issue of antibiotic accumulation within bacterial cells.
Exploring a vast chemical space is crucial for identifying the best hits. However, the accumulation of hit compounds within bacterial cells is key to a successful drug discovery program in the antimicrobial field. The physicochemical properties of the screened compounds play a vital role in overcoming this challenge. Throughout this review, several potent hits were identified from diverse and extensive DECLs, but none have reached clinical trials or succeeded in in vivo tests, to the best of our knowledge. Designing libraries that adhere to the physicochemical properties of the “antimicrobial properties chemical space” will help identify more potent hits that can accumulate and act within bacterial cells. The accumulation of compounds inside bacterial cells requires effective permeation through the bacterial outer membrane. This raises questions about the effectiveness of screening compounds through biochemical assays without ensuring their permeability. This is where phenotypic screening becomes valuable, as it can bypass the permeability checkpoint and directly assess the compound's ability to act within the bacterial environment. In this review, only two limited success stories described the screening of DECLs through a phenotypic screening approach.80,86 The key properties102 that ensure positive penetration of compounds through the bacterial cell walls include molecular weight, cLog D, and polar surface area. In addition, incorporating a zwitterionic design, using fewer rotatable bonds and sterically accessible primary amines, has been proven to enhance cell penetration.84,103–106 We recommend several strategies to enhance the effectiveness of DECLs in antimicrobial drug discovery research. First, it is crucial to design more focused DECLs that align with antimicrobial physicochemical properties guidelines while also covering a broad chemical space. Secondly, leveraging existing medicinal chemistry knowledge related to the chemical structures of successful antibacterial agents is essential. This approach follows the well-known drug discovery wisdom “starting with a known target or compound is often more effective than beginning with a new one”. This is based on the understanding that bacteria do not create new proteins but rather modify existing ones to render antibacterial drugs ineffective. Similarly, we can modify known drugs to restore their activity. Thirdly, it is important to develop new screening methods that allow DECLs to be tested against proteins of interest in their native environments. Finally, it is highly recommended to bypass traditional biochemical assays through phenotypic screening or other screening methods that demonstrate the ability of hit compounds to penetrate bacterial cell walls and accumulate inside cells. We believe that DECLs still have promising potential to be fully realised.
Data availability
No new data were created or analysed in this study. All the references used in this study are mentioned in the bibliography section of the manuscript. Data sharing is not applicable to this article.
Author contributions
HH, RES, ZEMF, HHQ: investigation, visualization, writing original draft, review & editing. HH: methodology, supervision & review. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
There are no conflicts to declare.
Notes and references
- V. H. Krishnaprasad and S. Kumar, Antimicrobial Resistance: An Ultimate Challenge for 21st Century Scientists, Healthcare Professionals, and Policymakers to Save Future Generations, J. Med. Chem., 2024, 67, 15927–15930 CrossRef CAS PubMed.
- C. J. L. Murray, K. S. Ikuta, F. Sharara, L. Swetschinski, G. Robles Aguilar, A. Gray, C. Han, C. Bisignano, P. Rao, E. Wool, S. C. Johnson, A. J. Browne, M. G. Chipeta, F. Fell, S. Hackett, G. Haines-Woodhouse, B. H. Kashef Hamadani, E. A. P. Kumaran, B. McManigal, S. Achalapong, R. Agarwal, S. Akech, S. Albertson, J. Amuasi, J. Andrews, A. Aravkin, E. Ashley, F.-X. Babin, F. Bailey, S. Baker, B. Basnyat, A. Bekker, R. Bender, J. A. Berkley, A. Bethou, J. Bielicki, S. Boonkasidecha, J. Bukosia, C. Carvalheiro, C. Castañeda-Orjuela, V. Chansamouth, S. Chaurasia, S. Chiurchiù, F. Chowdhury, R. Clotaire Donatien, A. J. Cook, B. Cooper, T. R. Cressey, E. Criollo-Mora, M. Cunningham, S. Darboe, N. P. J. Day, M. De Luca, K. Dokova, A. Dramowski, S. J. Dunachie, T. Duong Bich, T. Eckmanns, D. Eibach, A. Emami, N. Feasey, N. Fisher-Pearson, K. Forrest, C. Garcia, D. Garrett, P. Gastmeier, A. Z. Giref, R. C. Greer, V. Gupta, S. Haller, A. Haselbeck, S. I. Hay, M. Holm, S. Hopkins, Y. Hsia, K. C. Iregbu, J. Jacobs, D. Jarovsky, F. Javanmardi, A. W. J. Jenney, M. Khorana, S. Khusuwan, N. Kissoon, E. Kobeissi, T. Kostyanev, F. Krapp, R. Krumkamp, A. Kumar, H. H. Kyu, C. Lim, K. Lim, D. Limmathurotsakul, M. J. Loftus, M. Lunn, J. Ma, A. Manoharan, F. Marks, J. May, M. Mayxay, N. Mturi, T. Munera-Huertas, P. Musicha, L. A. Musila, M. M. Mussi-Pinhata, R. N. Naidu, T. Nakamura, R. Nanavati, S. Nangia, P. Newton, C. Ngoun, A. Novotney, D. Nwakanma, C. W. Obiero, T. J. Ochoa, A. Olivas-Martinez, P. Olliaro, E. Ooko, E. Ortiz-Brizuela, P. Ounchanum, G. D. Pak, J. L. Paredes, A. Y. Peleg, C. Perrone, T. Phe, K. Phommasone, N. Plakkal, A. Ponce-de-Leon, M. Raad, T. Ramdin, S. Rattanavong, A. Riddell, T. Roberts, J. V. Robotham, A. Roca, V. D. Rosenthal, K. E. Rudd, N. Russell, H. S. Sader, W. Saengchan, J. Schnall, J. A. G. Scott, S. Seekaew, M. Sharland, M. Shivamallappa, J. Sifuentes-Osornio, A. J. Simpson, N. Steenkeste, A. J. Stewardson, T. Stoeva, N. Tasak, A. Thaiprakong, G. Thwaites, C. Tigoi, C. Turner, P. Turner, H. R. van Doorn, S. Velaphi, A. Vongpradith, M. Vongsouvath, H. Vu, T. Walsh, J. L. Walson, S. Waner, T. Wangrangsimakul, P. Wannapinij, T. Wozniak, T. E. M. W. Young Sharma, K. C. Yu, P. Zheng, B. Sartorius, A. D. Lopez, A. Stergachis, C. Moore, C. Dolecek and M. Naghavi, Global burden of bacterial antimicrobial resistance in 2019: a systematic analysis, Lancet, 2022, 399, 629–655 CrossRef CAS PubMed.
- No Time to Wait: Securing the Future from Drug-Resistant Infections Report to the Secretary-General of the United Nations, 2019.
- C. A. Machutta, C. S. Kollmann, K. E. Lind, X. Bai, P. F. Chan, J. Huang, L. Ballell, S. Belyanskaya, G. S. Besra, D. Barros-Aguirre, R. H. Bates, P. A. Centrella, S. S. Chang, J. Chai, A. E. Choudhry, A. Coffin, C. P. Davie, H. Deng, J. Deng, Y. Ding, J. W. Dodson, D. T. Fosbenner, E. N. Gao, T. L. Graham, T. L. Graybill, K. Ingraham, W. P. Johnson, B. W. King, C. R. Kwiatkowski, J. Lelièvre, Y. Li, X. Liu, Q. Lu, R. Lehr, A. Mendoza-Losana, J. Martin, L. McCloskey, P. McCormick, H. P. O'Keefe, T. O'Keeffe, C. Pao, C. B. Phelps, H. Qi, K. Rafferty, G. S. Scavello, M. S. Steiginga, F. S. Sundersingh, S. M. Sweitzer, L. M. Szewczuk, A. Taylor, M. F. Toh, J. Wang, M. Wang, D. J. Wilkins, B. Xia, G. Yao, J. Zhang, J. Zhou, C. P. Donahue, J. A. Messer, D. Holmes, C. C. Arico-Muendel, A. J. Pope, J. W. Gross and G. Evindar, Prioritizing multiple therapeutic targets in parallel using automated DNA-encoded library screening, Nat. Commun., 2017, 8, 16081 CrossRef CAS PubMed.
- M. S. Butler, G. Valeria, S. Hatim, P. Sarah, L. Al-Sulaiman, J. H. Rex, F. Prabhavathi, C. A. Arias, P. Mical, G. E. Thwaites, C. Lloyd, R. A. Alm, L. Christian, S. Melvin, L. L. Silver, N. Ohmagari, R. Kozlov, H. Stephan and B. Peter, Analysis of the Clinical Pipeline of Treatments for Drug-Resistant Bacterial Infections: Despite Progress, More Action Is Needed, Antimicrob. Agents Chemother., 2022, 66, e01991 CAS.
- S. Zhao, J. W. Adamiak, V. Bonifay, J. Mehla, H. I. Zgurskaya and D. S. Tan, Defining new chemical space for drug penetration into Gram-negative bacteria, Nat. Chem. Biol., 2020, 16, 1293–1302 CAS.
- Y. K. Sunkari, V. K. Siripuram, T. L. Nguyen and M. Flajolet, High-power screening (HPS) empowered by DNA-encoded libraries, Trends Pharmacol. Sci., 2022, 43, 4–15 CrossRef CAS PubMed.
- A. A. Peterson and D. R. Liu, Small-molecule discovery through DNA-encoded libraries, Nat. Rev. Drug Discovery, 2023, 22, 699–722 CrossRef CAS PubMed.
- A. Mullard, DNA-encoded drug libraries come of age, Nat. Biotechnol., 2016, 34, 450–451 CrossRef PubMed.
- S. Brenner and R. A. Lerner, Encoded combinatorial chemistry, Proc. Natl. Acad. Sci. U. S. A., 1992, 89, 5381–5383 CrossRef CAS PubMed.
- A. L. Satz, A. Brunschweiger, M. E. Flanagan, A. Gloger, N. J. V. Hansen, L. Kuai, V. B. K. Kunig, X. Lu, D. Madsen, L. A. Marcaurelle, C. Mulrooney, G. O'Donovan, S. Sakata and J. Scheuermann, DNA-encoded chemical libraries, Nat. Rev. Methods Primers, 2022, 2, 3 CrossRef CAS.
- J. L. Silen, A. T. Lu, D. W. Solas, M. A. Gore, D. Maclean, N. H. Shah, J. M. Coffin, N. S. Bhinderwala, Y. Wang, K. T. Tsutsui, G. C. Look, D. A. Campbell, R. L. Hale, M. Navre and C. R. DeLuca-Flaherty, Screening for Novel Antimicrobials from Encoded Combinatorial Libraries by Using a Two-Dimensional Agar Format, Antimicrob. Agents Chemother., 1998, 42, 1447–1453 CrossRef CAS PubMed.
- W. Fayisa and N. Tuli, Review on Staphylococcus aureus, International Journal of Nursing Care and Research, 2023, 1, 1–8 Search PubMed.
- H. F. L. Wertheim, D. C. Melles, M. C. Vos, W. van Leeuwen, A. van Belkum, H. A. Verbrugh and J. L. Nouwen, The role of nasal carriage in Staphylococcus aureus infections, Lancet Infect. Dis., 2005, 5, 751–762 CrossRef PubMed.
- B. P. Howden, S. G. Giulieri, T. Wong Fok Lung, S. L. Baines, L. K. Sharkey, J. Y. H. Lee, A. Hachani, I. R. Monk and T. P. Stinear, Staphylococcus aureus host interactions and adaptation, Nat. Rev. Microbiol., 2023, 21, 380–395 CrossRef CAS PubMed.
- Y. Oogai, M. Matsuo, M. Hashimoto, F. Kato, M. Sugai and H. Komatsuzawa, Expression of virulence factors by Staphylococcus aureus grown in serum, Appl. Environ. Microbiol., 2011, 77, 8097–8105 CrossRef CAS PubMed.
- R. H. Deurenberg, C. Vink, S. Kalenic, A. W. Friedrich, C. A. Bruggeman and E. E. Stobberingh, The molecular evolution of methicillin-resistant Staphylococcus aureus, Clin. Microbiol. Infect., 2007, 13, 222–235 CrossRef CAS PubMed.
- M. A. Deniziak and J. Barciszewski, Methionyl-tRNA synthetase, Acta Biochim. Pol., 2001, 48, 337–350 CrossRef CAS PubMed.
- E. Kermgard, Z. Yang, A.-M. Michel, R. Simari, J. Wong, M. Ibba and B. A. Lazazzera, Quality Control by Isoleucyl-tRNA Synthetase of Bacillus subtilis Is Required for Efficient Sporulation, Sci. Rep., 2017, 7, 41763 CrossRef CAS PubMed.
- S. C. Chai, W.-L. Wang, D.-R. Ding and Q.-Z. Ye, Growth inhibition of Escherichia coli and methicillin-resistant Staphylococcus aureus by targeting cellular methionine aminopeptidase, Eur. J. Med. Chem., 2011, 46, 3537–3540 CrossRef CAS PubMed.
- T. P. Ko, C. H. Huang, S. J. Lai and Y. Chen, Structure of undecaprenyl pyrophosphate synthase from Acinetobacter baumannii, Acta Crystallogr., Sect. F: Struct. Biol. Commun., 2018, 74, 765–769 CrossRef CAS PubMed.
- S. W. Polyak, A. D. Abell, M. C. J. Wilce, L. Zhang and G. W. Booker, Structure, function and selective inhibition of bacterial acetyl-coa carboxylase, Appl. Microbiol. Biotechnol., 2012, 93, 983–992 CrossRef CAS PubMed.
- N. Concha, J. Huang, X. Bai, A. Benowitz, P. Brady, L. C. Grady, L. H. Kryn, D. Holmes, K. Ingraham, Q. Jin, L. Pothier Kaushansky, L. McCloskey, J. A. Messer, H. O'Keefe, A. Patel, A. L. Satz, R. H. Sinnamon, J. Schneck, S. R. Skinner, J. Summerfield, A. Taylor, J. D. Taylor, G. Evindar and R. A. Stavenger, Discovery and Characterization of a Class of Pyrazole Inhibitors of Bacterial Undecaprenyl Pyrophosphate Synthase, J. Med. Chem., 2016, 59, 7299–7304 CrossRef CAS PubMed.
- O. O. Adeniji, E. A. E. Elsheikh and A. I. Okoh, Prevalence of classes 1 and 2 integrons in multidrug-resistant Acinetobacter baumanni isolates recovered from some aquatic environment in South Africa, Sci. Rep., 2022, 12, 20319 CrossRef CAS PubMed.
- I. Kyriakidis, E. Vasileiou, Z. D. Pana and A. Tragiannidis, Acinetobacter baumannii antibiotic resistance mechanisms, Pathogens, 2021, 10, 373 CrossRef CAS PubMed.
- W. Zhang, B. Aurosree, B. Gopalakrishnan, J.-M. Balada-Llasat, V. Pancholi and P. Pancholi, The role of LpxA/C/D and pmrA/B gene systems in colistin-resistant clinical strains of Acinetobacter baumannii, Frontiers in Laboratory Medicine, 2017, 1, 86–91 CrossRef.
- S.-i. Narita and H. Tokuda, Biogenesis and Membrane Targeting of Lipoproteins, EcoSal Plus, 2010, 4 DOI:10.1128/ecosalplus.4.3.7.
- E. H. Tobin and D. Tristram, Tuberculosis Overview, in StatPearls, Treasure Island, FL, 2025 Search PubMed.
- K. J. Seung, S. Keshavjee and M. L. Rich, Multidrug-resistant tuberculosis and extensively drug-resistant tuberculosis, Cold Spring Harbor Perspect. Med., 2015, 5, a017863 CrossRef PubMed.
- J. C. Palomino and A. Martin, Drug resistance mechanisms in Mycobacterium tuberculosis, Antibiotics, 2014, 3, 317–340 CrossRef PubMed.
- A. S. Dean, O. Tosas Auguet, P. Glaziou, M. Zignol, N. Ismail, T. Kasaeva and K. Floyd, 25 years of surveillance of drug-resistant tuberculosis: achievements, challenges, and way forward, Lancet Infect. Dis., 2022, 22, e191–e196 CrossRef PubMed.
- C. O'Connor, P. Patel and M. F. Brady, Isoniazid, in StatPearls, Treasure Island, FL, 2025 Search PubMed.
- C. E. Barry, R. E. Lee, K. Mdluli, A. E. Sampson, B. G. Schroeder, R. A. Slayden and Y. Yuan, Mycolic acids: structure, biosynthesis and physiological functions, Prog. Lipid Res., 1998, 37, 143–179 CrossRef CAS PubMed.
- L. Bibens, J. P. Becker, A. Dassonville-Klimpt and P. Sonnet, A Review of Fatty Acid Biosynthesis Enzyme Inhibitors as Promising Antimicrobial Drugs, Pharmaceuticals, 2023, 16 CrossRef CAS PubMed.
- A. Dessen, A. Quemard, J. S. Blanchard, W. R. Jacobs Jr and J. C. Sacchettini, Crystal structure and function of the isoniazid target of Mycobacterium tuberculosis, Science, 1995, 267, 1638–1641 CrossRef CAS PubMed.
- A. Chollet, L. Mourey, C. Lherbet, A. Delbot, S. Julien, M. Baltas, J. Bernadou, G. Pratviel, L. Maveyraud and V. Bernardes-Génisson, Crystal structure of the enoyl-ACP reductase of Mycobacterium tuberculosis (InhA) in the apo-form and in complex with the active metabolite of isoniazid pre-formed by a biomimetic approach, J. Struct. Biol., 2015, 190, 328–337 CrossRef CAS PubMed.
- H. M. Hernando, B. Michael, B. d. V. Miriam, C. Magali, G. M. Inírida, V.-B. Mandira, B.-J. Helen, L. Caroline, F. Janet, G.-G. Lourdes, L. C. Inés, B. Mridula, C. Fernando, M. Megan, K. D. Eisenach, S.-O. José, C. M. Donald, P. d. L. Alfredo and A. David, Population Genetics Study of Isoniazid Resistance Mutations and Evolution of Multidrug-Resistant Mycobacterium tuberculosis, Antimicrob. Agents Chemother., 2006, 50, 2640–2649 CrossRef PubMed.
- H. Yang, P. F. Medeiros, K. Raha, P. Elkins, K. E. Lind, R. Lehr, N. D. Adams, J. L. Burgess, S. J. Schmidt, S. D. Knight, K. R. Auger, M. D. Schaber, G. J. Franklin, Y. Ding, J. L. DeLorey, P. A. Centrella, S. Mataruse, S. R. Skinner, M. A. Clark, J. W. Cuozzo and G. Evindar, Discovery of a Potent Class of PI3Kα Inhibitors with Unique Binding Mode via Encoded Library Technology (ELT), ACS Med. Chem. Lett., 2015, 6, 531–536 CrossRef CAS PubMed.
- H. H. Soutter, P. Centrella, M. A. Clark, J. W. Cuozzo, C. E. Dumelin, M.-A. Guie, S. Habeshian, A. D. Keefe, K. M. Kennedy, E. A. Sigel, D. M. Troast, Y. Zhang, A. D. Ferguson, G. Davies, E. R. Stead, J. Breed, P. Madhavapeddi and J. A. Read, Discovery of cofactor-specific, bactericidal Mycobacterium tuberculosis InhA inhibitors using DNA-encoded library technology, Proc. Natl. Acad. Sci. U. S. A., 2016, 113, E7880–E7889 CrossRef CAS PubMed.
- R. Rawat, A. Whitty and P. J. Tonge, The isoniazid-NAD adduct is a slow, tight-binding inhibitor of InhA, the Mycobacterium tuberculosis enoyl reductase: adduct affinity and drug resistance, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 13881–13886 CrossRef CAS PubMed.
- S. R. Luckner, N. Liu, C. W. am Ende, P. J. Tonge and C. Kisker, A slow, tight binding inhibitor of InhA, the enoyl-acyl carrier protein reductase from Mycobacterium tuberculosis, J. Biol. Chem., 2010, 285, 14330–14337 CrossRef CAS PubMed.
- R. C. Hartkoorn, F. Pojer, J. A. Read, H. Gingell, J. Neres, O. P. Horlacher, K.-H. Altmann and S. T. Cole, Pyridomycin bridges the NADH- and substrate-binding pockets of the enoyl reductase InhA, Nat. Chem. Biol., 2014, 10, 96–98 CrossRef CAS PubMed.
- P. S. Shirude, P. Madhavapeddi, M. Naik, K. Murugan, V. Shinde, R. Nandishaiah, J. Bhat, A. Kumar, S. Hameed, G. Holdgate, G. Davies, H. McMiken, N. Hegde, A. Ambady, J. Venkatraman, M. Panda, B. Bandodkar, V. K. Sambandamurthy and J. A. Read, Methyl-Thiazoles: A Novel Mode of Inhibition with the Potential to Develop Novel Inhibitors Targeting InhA in Mycobacterium tuberculosis, J. Med. Chem., 2013, 56, 8533–8542 CrossRef CAS PubMed.
- L. Encinas, H. O'Keefe, M. Neu, M. J. Remuiñán, A. M. Patel, A. Guardia, C. P. Davie, N. Pérez-Macías, H. Yang, M. A. Convery, J. A. Messer, E. Pérez-Herrán, P. A. Centrella, D. Álvarez-Gómez, M. A. Clark, S. Huss, G. K. O'Donovan, F. Ortega-Muro, W. McDowell, P. Castañeda, C. C. Arico-Muendel, S. Pajk, J. Rullás, I. Angulo-Barturen, E. Álvarez-Ruíz, A. Mendoza-Losana, L. Ballell Pages, J. Castro-Pichel and G. Evindar, Encoded Library Technology as a Source of Hits for the Discovery and Lead Optimization of a Potent and Selective Class of Bactericidal Direct Inhibitors of Mycobacterium tuberculosis InhA, J. Med. Chem., 2014, 57, 1276–1288 CrossRef CAS PubMed.
- U. H. Manjunatha, S. P. S. Rao, R. R. Kondreddi, C. G. Noble, L. R. Camacho, B. H. Tan, S. H. Ng, P. S. Ng, N. L. Ma, S. B. Lakshminarayana, M. Herve, S. W. Barnes, W. Yu, K. Kuhen, F. Blasco, D. Beer, J. R. Walker, P. J. Tonge, R. Glynne, P. W. Smith and T. T. Diagana, Direct inhibitors of InhA are active against Mycobacterium tuberculosis, Sci. Transl. Med., 2015, 7, 269ra3 Search PubMed.
- I. V Krieger, S. Yalamanchili, P. Dickson, C. A. Engelhart, M. D. Zimmerman, J. Wood, E. Clary, J. Nguyen, N. Thornton, P. A. Centrella, B. Chan, J. W. Cuozzo, M. Gengenbacher, M.-A. Guié, J. P. Guilinger, C. Bienstock, H. Hartl, C. D. Hupp, R. Jetson, T. Satoh, J. T. S. Yeoman, Y. Zhang, V. Dartois, D. Schnappinger, A. D. Keefe and J. C. Sacchettini, Inhibitors of the Thioesterase Activity of Mycobacterium tuberculosis Pks13 Discovered Using DNA-Encoded Chemical Library Screening, ACS Infect. Dis., 2024, 10, 1561–1575 CrossRef PubMed.
- S. M. Batt, D. E. Minnikin and G. S. Besra, The thick waxy coat of mycobacteria, a protective layer against antibiotics and the host's immune system, Biochem. J., 2020, 477, 1983–2006 CrossRef PubMed.
- S. K. Kim, M. S. Dickinson, J. Finer-Moore, Z. Guan, R. M. Kaake, I. Echeverria, J. Chen, E. H. Pulido, A. Sali, N. J. Krogan, O. S. Rosenberg and R. M. Stroud, Structure and dynamics of the essential endogenous mycobacterial polyketide synthase Pks13, Nat. Struct. Mol. Biol., 2023, 30, 296–308 CrossRef CAS PubMed.
- R. K. Meade, J. E. Long, A. Jinich, K. Y. Rhee, D. G. Ashbrook, R. W. Williams, C. M. Sassetti and C. M. Smith, Genome-wide screen identifies host loci that modulate Mycobacterium tuberculosis fitness in immunodivergent mice, G3: Genes, Genomes, Genet., 2023, 13, jkad147 CrossRef CAS PubMed.
- C. Wilson, P. Ray, F. Zuccotto, J. Hernandez, A. Aggarwal, C. Mackenzie, N. Caldwell, M. Taylor, M. Huggett, M. Mathieson, D. Murugesan, A. Smith, S. Davis, M. Cocco, M. K. Parai, A. Acharya, F. Tamaki, P. Scullion, O. Epemolu, J. Riley, L. Stojanovski, E. M. Lopez-Román, P. A. Torres-Gómez, A. M. Toledo, L. Guijarro-Lopez, I. Camino, C. A. Engelhart, D. Schnappinger, L. M. Massoudi, A. Lenaerts, G. T. Robertson, C. Walpole, D. Matthews, D. Floyd, J. C. Sacchettini, K. D. Read, L. Encinas, R. H. Bates, S. R. Green and P. G. Wyatt, Optimization of TAM16, a Benzofuran That Inhibits the Thioesterase Activity of Pks13; Evaluation toward a Preclinical Candidate for a Novel Antituberculosis Clinical Target, J. Med. Chem., 2022, 65, 409–423 CrossRef CAS PubMed.
- M. D. Hall, A. Yasgar, T. Peryea, J. C. Braisted, A. Jadhav, A. Simeonov and N. P. Coussens, Fluorescence polarization assays in high-throughput screening and drug discovery: a review, Methods Appl. Fluoresc., 2016, 4, 022001 Search PubMed.
- D. A. Bachovchin, S. J. Brown, H. Rosen and B. F. Cravatt, Identification of selective inhibitors of uncharacterized enzymes by high-throughput screening with fluorescent activity-based probes, Nat. Biotechnol., 2009, 27, 387–394 CrossRef CAS PubMed.
- M. F. Moradali, S. Ghods and B. H. Rehm, Pseudomonas aeruginosa Lifestyle: A Paradigm for Adaptation, Survival, and Persistence, Front. Cell. Infect. Microbiol., 2017, 7, 39 Search PubMed.
- S. Qin, W. Xiao, C. Zhou, Q. Pu, X. Deng, L. Lan, H. Liang, X. Song and M. Wu, Pseudomonas aeruginosa: pathogenesis, virulence factors, antibiotic resistance, interaction with host, technology advances and emerging therapeutics, Signal Transduction Targeted Ther., 2022, 7, 199 CrossRef CAS PubMed.
- N. Sathe, P. Beech, L. Croft, C. Suphioglu, A. Kapat and E. Athan, Pseudomonas aeruginosa: infections and novel approaches to treatment “Knowing the enemy” the threat of Pseudomonas aeruginosa and exploring novel approaches to treatment, Infectious Medicine, 2023, 2, 178–194 CrossRef PubMed.
- D. G. Davies, M. R. Parsek, J. P. Pearson, B. H. Iglewski, J. W. Costerton and E. P. Greenberg, The Involvement of Cell-to-Cell Signals in the Development of a Bacterial Biofilm, Science, 1998, 280, 295–298 CrossRef CAS PubMed.
- S. Mukherjee, D. Moustafa, C. D. Smith, J. B. Goldberg and B. L. Bassler, The RhlR quorum-sensing receptor controls Pseudomonas aeruginosa pathogenesis and biofilm development independently of its canonical homoserine lactone autoinducer, PLoS Pathog., 2017, 13, e1006504 CrossRef PubMed.
- I. R. Taylor, P. D. Jeffrey, D. A. Moustafa, J. B. Goldberg and B. L. Bassler, The PqsE Active Site as a Target for Small Molecule Antimicrobial Agents against Pseudomonas aeruginosa, Biochemistry, 2022, 61, 1894–1903 CrossRef CAS PubMed.
- J. S. Valastyan, M. R. Tota, I. R. Taylor, V. Stergioula, G. A. B. Hone, C. D. Smith, B. R. Henke, K. G. Carson and B. L. Bassler, Discovery of PqsE Thioesterase Inhibitors for Pseudomonas aeruginosa Using DNA-Encoded Small Molecule Library Screening, ACS Chem. Biol., 2020, 15, 446–456 CrossRef CAS PubMed.
- S. Yu, V. Jensen, J. Seeliger, I. Feldmann, S. Weber, E. Schleicher, S. Häussler and W. Blankenfeldt, Structure Elucidation and Preliminary Assessment of Hydrolase Activity of PqsE, the Pseudomonas Quinolone Signal (PQS) Response Protein, Biochemistry, 2009, 48, 10298–10307 CrossRef CAS PubMed.
- M. Zender, F. Witzgall, S. L. Drees, E. Weidel, C. K. Maurer, S. Fetzner, W. Blankenfeldt, M. Empting and R. W. Hartmann, Dissecting the Multiple Roles of PqsE in Pseudomonas aeruginosa Virulence by Discovery of Small Tool Compounds, ACS Chem. Biol., 2016, 11, 1755–1763 CrossRef CAS PubMed.
- P. Zhou and J. Zhao, Structure, inhibition, and regulation of essential lipid A enzymes, Biochim. Biophys. Acta, Mol. Cell Biol. Lipids, 2017, 1862, 1424–1438 CrossRef CAS PubMed.
- T. J. O. Wyckoff and C. R. H. Raetz, The Active Site of Escherichia coli UDP-N-acetylglucosamine Acyltransferase: Chemical Modification and Site-Directed Mutagenesis, J. Biol. Chem., 1999, 274, 27047–27055 CrossRef CAS PubMed.
- M. D. Ryan, A. L. Parkes, D. Corbett, A. P. Dickie, M. Southey, O. A. Andersen, D. B. Stein, O. R. Barbeau, A. Sanzone, P. Thommes, J. Barker, R. Cain, C. Compper, M. Dejob, A. Dorali, D. Etheridge, S. Evans, A. Faulkner, E. Gadouleau, T. Gorman, D. Haase, M. Holbrow-Wilshaw, T. Krulle, X. Li, C. Lumley, B. Mertins, S. Napier, R. Odedra, K. Papadopoulos, V. Roumpelakis, K. Spear, E. Trimby, J. Williams, M. Zahn, A. D. Keefe, Y. Zhang, H. T. Soutter, P. A. Centrella, M. A. Clark, J. W. Cuozzo, C. E. Dumelin, B. Deng, A. Hunt, E. A. Sigel, D. M. Troast and B. L. M. DeJonge, Discovery of novel UDP-N-acetylglucosamine acyltransferase (LpxA) inhibitors with activity against Pseudomonas aeruginosa, J. Med. Chem., 2021, 64(19), 14377–14425 CrossRef CAS PubMed.
- C. A. Hart and S. Kariuki, Antimicrobial resistance in developing countries, BMJ, 1998, 317, 647–650 CrossRef CAS PubMed.
- F. C. Tenover, Mechanisms of antimicrobial resistance in bacteria, Am. J. Infect. Control, 2006, 34, S3–S10 CrossRef PubMed ; discussion S64–S73..
- M. A. Abushaheen, Muzaheed, A. J. Fatani, M. Alosaimi, W. Mansy, M. George, S. Acharya, S. Rathod, D. D. Divakar, C. Jhugroo, S. Vellappally, A. A. Khan, J. Shaik and P. Jhugroo, Antimicrobial resistance, mechanisms and its clinical significance, Disease-a-Month, 2020, 66, 100971 CrossRef PubMed.
- B. Jaurin and T. Grundström, ampC cephalosporinase of Escherichia coli K-12 has a different evolutionary origin from that of beta-lactamases of the penicillinase type, Proc. Natl. Acad. Sci. U. S. A., 1981, 78, 4897–4901 CrossRef CAS PubMed.
- M. Ouellette, L. Bissonnette and P. H. Roy, Precise insertion of antibiotic resistance determinants into Tn21-like transposons: nucleotide sequence of the OXA-1 beta-lactamase gene, Proc. Natl. Acad. Sci. U. S. A., 1987, 84, 7378–7382 CrossRef CAS PubMed.
- R. P. Ambler, J. Baddiley and E. P. Abraham, The structure of β-lactamases, Philos. Trans. R. Soc., B, 1997, 289, 321–331 Search PubMed.
- S. Park, J. Fan, S. Chamakuri, M. Palaniappan, K. Sharma, X. Qin, J. Wang, Z. Tan, A. Judge, L. Hu, B. Sankaran, F. Li, B. V. V. Prasad, M. M. Matzuk and T. Palzkill, Exploiting the Carboxylate-Binding Pocket of β-Lactamase Enzymes Using a Focused DNA-Encoded Chemical Library, J. Med. Chem., 2024, 67, 620–642 CrossRef CAS PubMed.
- B. A. Lund, T. Christopeit, Y. Guttormsen, A. Bayer and H.-K. S. Leiros, Screening and Design of Inhibitor Scaffolds for the Antibiotic Resistance Oxacillinase-48 (OXA-48) through Surface Plasmon Resonance Screening, J. Med. Chem., 2016, 59, 5542–5554 CrossRef CAS PubMed.
- D. M. Taylor, J. Anglin, S. Park, M. N. Ucisik, J. C. Faver, N. Simmons, Z. Jin, M. Palaniappan, P. Nyshadham, F. Li, J. Campbell, L. Hu, B. Sankaran, B. V. V. Prasad, H. Huang, M. M. Matzuk and T. Palzkill, Identifying Oxacillinase-48 Carbapenemase Inhibitors Using DNA-Encoded Chemical Libraries, ACS Infect. Dis., 2020, 6, 1214–1227 CrossRef CAS PubMed.
- K. Palica, M. Vorácová, S. Skagseth, A. Andersson Rasmussen, L. Allander, M. Hubert, L. Sandegren, H.-K. Schrøder Leiros, H. Andersson and M. Erdélyi, Metallo-β-Lactamase Inhibitor Phosphonamidate Monoesters, ACS Omega, 2022, 7, 4550–4562 CrossRef CAS PubMed.
- L. K. Logan and R. A. Weinstein, The Epidemiology of Carbapenem-Resistant Enterobacteriaceae: The Impact and Evolution of a Global Menace, J. Infect. Dis., 2017, 215, S28–S36 CrossRef CAS PubMed.
- Z. Aktaş, C. Kayacan and O. Oncul, In vitro activity of avibactam (NXL104) in combination with β-lactams against Gram-negative bacteria, including OXA-48 β-lactamase-producing Klebsiella pneumoniae, Int. J. Antimicrob. Agents, 2012, 39, 86–89 CrossRef PubMed.
- D. E. Ehmann, H. Jahić, P. L. Ross, R.-F. Gu, J. Hu, T. F. Durand-Réville, S. Lahiri, J. Thresher, S. Livchak, N. Gao, T. Palmer, G. K. Walkup and S. L. Fisher, Kinetics of Avibactam Inhibition against Class A, C, and D β-Lactamases, J. Biol. Chem., 2013, 288, 27960–27971 CrossRef CAS PubMed.
- D. E. Ehmann, H. Jahić, P. L. Ross, R.-F. Gu, J. Hu, G. Kern, G. K. Walkup and S. L. Fisher, Avibactam is a covalent, reversible, non-β-lactam β-lactamase inhibitor, Proc. Natl. Acad. Sci. U. S. A., 2012, 109, 11663–11668 CrossRef CAS PubMed.
- B. A. Evans and S. G. B. Amyes, OXA β-Lactamases, Clin. Microbiol. Rev., 2014, 27, 241–263 CrossRef PubMed.
- W. G. Cochrane, P. R. Fitzgerald and B. M. Paegel, Antibacterial Discovery via Phenotypic DNA-Encoded Library Screening, ACS Chem. Biol., 2021, 16, 2752–2756 CrossRef CAS PubMed.
- V. S. Fluxà, N. Maillard, M. G. P. Page and J.-L. Reymond, Bead diffusion assay for discovering antimicrobial cyclic peptides, Chem. Commun., 2011, 47, 1434–1436 RSC.
- K. J. Fisher, J. A. Turkett, A. E. Corson and K. L. Bicker, Peptoid Library Agar Diffusion (PLAD) Assay for the High-Throughput Identification of Antimicrobial Peptoids, ACS Comb. Sci., 2016, 18, 287–291 CrossRef CAS PubMed.
- E. D. Brown and G. D. Wright, Antibacterial drug discovery in the resistance era, Nature, 2016, 529, 336–343 CrossRef CAS PubMed.
- M. F. Richter, B. S. Drown, A. P. Riley, A. Garcia, T. Shirai, R. L. Svec and P. J. Hergenrother, Predictive compound accumulation rules yield a broad-spectrum antibiotic, Nature, 2017, 545, 299–304 CrossRef CAS PubMed.
- G. Dhanda, P. Sarkar, S. Samaddar and J. Haldar, Battle against Vancomycin-Resistant Bacteria: Recent Developments in Chemical Strategies, J. Med. Chem., 2019, 62, 3184–3205 CrossRef CAS PubMed.
- D. Guan, J. Liu, F. Chen, J. Li, X. Wang, W. Lu, Y. Suo, F. Tang, L. Lan, X. Lu and W. Huang, A Vancomycin-Templated DNA-Encoded Library for Combating Drug-Resistant Bacteria, J. Med. Chem., 2024, 67, 3778–3794 CrossRef CAS PubMed.
- Y. Nitanai, T. Kikuchi, K. Kakoi, S. Hanamaki, I. Fujisawa and K. Aoki, Crystal Structures of the Complexes between Vancomycin and Cell-Wall Precursor Analogs, J. Mol. Biol., 2009, 385, 1422–1432 CrossRef CAS PubMed.
- A. Okano, N. A. Isley and D. L. Boger, Peripheral modifications of [Ψ[CH2NH]Tpg4]vancomycin with added synergistic mechanisms of action provide durable and potent antibiotics, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, E5052–E5061 CrossRef CAS PubMed.
- D. Guan, F. Chen, Y. Qiu, B. Jiang, L. Gong, L. Lan and W. Huang, Sulfonium, an Underestimated Moiety for Structural Modification, Alters the Antibacterial Profile of Vancomycin against Multidrug-Resistant Bacteria, Angew. Chem., Int. Ed., 2019, 58, 6678–6682 CrossRef CAS PubMed.
- F. Umstätter, C. Domhan, T. Hertlein, K. Ohlsen, E. Mühlberg, C. Kleist, S. Zimmermann, B. Beijer, K. D. Klika, U. Haberkorn, W. Mier and P. Uhl, Vancomycin Resistance Is Overcome by Conjugation of Polycationic Peptides, Angew. Chem., Int. Ed., 2020, 59, 8823–8827 CrossRef PubMed.
- M. S. Butler, K. A. Hansford, M. A. T. Blaskovich, R. Halai and M. A. Cooper, Glycopeptide antibiotics: back to the future, J. Antibiot., 2014, 67, 631–644 CrossRef CAS PubMed.
- D. Guan, F. Chen, L. Xiong, F. Tang, Faridoon, Y. Qiu, N. Zhang, L. Gong, J. Li, L. Lan and W. Huang, Extra Sugar on Vancomycin: New Analogues for Combating Multidrug-Resistant Staphylococcus aureus and Vancomycin-Resistant Enterococci, J. Med. Chem., 2018, 61, 286–304 CrossRef CAS PubMed.
- D. Guan, F. Chen, W. Shi, L. Lan and W. Huang, Single Modification at the N-Terminus of Norvancomycin to Combat Drug-Resistant Gram-Positive Bacteria, ChemMedChem, 2023, 18, e202200708 CrossRef CAS PubMed.
- W. Vollmer, D. Blanot and M. A. De Pedro, Peptidoglycan structure and architecture, FEMS Microbiol. Rev., 2008, 32, 149–167 CrossRef CAS PubMed.
- H. Barreteau, A. Kovač, A. Boniface, M. Sova, S. Gobec and D. Blanot, Cytoplasmic steps of peptidoglycan biosynthesis, FEMS Microbiol.
Rev., 2008, 32, 168–207 CrossRef CAS PubMed.
- M. Hrast, I. Sosič, R. Šink and S. Gobec, Inhibitors of the peptidoglycan biosynthesis enzymes MurA-F, Bioorg. Chem., 2014, 55, 2–15 CAS.
- M. Jukič, S. Gobec and M. Sova, Reaching toward underexplored targets in antibacterial drug design, Drug Dev. Res., 2019, 80, 6–10 Search PubMed.
- T. Fujihira, S. Kanematsu, A. Umino, N. Yamamoto and T. Nishikawa, Selective increase in the extracellular d-serine contents by d-cycloserine in the rat medial frontal cortex, Neurochem. Int., 2007, 51, 233–236 Search PubMed.
- https://delopen.org.
- M. Proj, K. Bozovičar, M. Hrast, R. Frlan and S. Gobec, DNA-encoded library screening on two validated enzymes of the peptidoglycan biosynthetic pathway, Bioorg. Med. Chem. Lett., 2022, 73, 128915 CAS.
- Y. Huang, Y. Li and X. Li, Strategies for developing DNA-encoded libraries beyond binding assays, Nat. Chem., 2022, 14, 129–140 CrossRef CAS PubMed.
- G. Krishnamoorthy, I. V. Leus, J. W. Weeks, D. Woloscheck, V. V. Rybenkov and H. I. Zgurskaya, Synergy between Active Efflux and Outer Membrane Diffusion Defines Rules of Antibiotic Permeation into Gram-Negative Bacteria, mBio, 2017, 8 DOI:10.1128/mbio.01172-17.
- D. G. Brown, T. L. May-Dracka, M. M. Gagnon and R. Tommasi, Trends and Exceptions of Physical Properties on Antibacterial Activity for Gram-Positive and Gram-Negative Pathogens, J. Med. Chem., 2014, 57, 10144–10161 Search PubMed.
- R. O'Shea and H. E. Moser, Physicochemical Properties of Antibacterial Compounds: Implications for Drug Discovery, J. Med. Chem., 2008, 51, 2871–2878 Search PubMed.
- F. Ruggiu, S. Yang, R. L. Simmons, A. Casarez, A. K. Jones, C. Li, J. M. Jansen, H. E. Moser, C. R. Dean, F. Reck and M. Lindvall, Size Matters and How You Measure It: A Gram-Negative Antibacterial Example Exceeding Typical Molecular Weight Limits, ACS Infect. Dis., 2019, 5, 1688–1692 CrossRef CAS PubMed.
- S. Acosta-Gutiérrez, L. Ferrara, M. Pathania, M. Masi, J. Wang, I. Bodrenko, M. Zahn, M. Winterhalter, R. A. Stavenger, J.-M. Pagès, J. H. Naismith, B. van den Berg, M. G. P. Page and M. Ceccarelli, Getting Drugs into Gram-Negative Bacteria: Rational Rules for Permeation through General Porins, ACS Infect. Dis., 2018, 4, 1487–1498 CrossRef PubMed.
Footnote |
| † These authors contributed equally to this work. |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence a,
Zainab E. M. Farahat†
a,
Zainab E. M. Farahat†