
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSize-dependent metal–support interactions in Co/CeO2–Y2O3 catalysts for enhanced methane dry reforming†
Yanhui Longab,
Liboting Gaoa,
Yilin Zhanga,
Wee-Liat Ong b,
Hao Zhang*acd and
Yan Jianhuaacd
b,
Hao Zhang*acd and
Yan Jianhuaacd
aState Key Laboratory of Clean Energy Utilization, Zhejiang University, Hangzhou 310027, China. E-mail: zhang_hao@zju.edu.cn
bCollege of Energy Engineering, ZJU-UIUC, Zhejiang University, Hangzhou 310027, China
cNingbo Innovation Center, Zhejiang University, Ningbo 315100, China
dInner Mongolia Daqingshan Laboratory, Hohhot 017000, China
First published on 7th April 2025
Abstract
Metal–support interactions (MSI) significantly influence the effectiveness of heterogeneous catalysts. Specific sites at the metal support interface can exhibit high reactivity, prompting increasing interest in optimizing not only the properties of metal particles but also the metal–support interface. Nevertheless, a precise modulation of MSI strength for optimal metal dispersion and size remains a significant challenge. Here, we demonstrate that tuning the particle size of the support by varying calcination temperature can effectively modulate the interaction between Co and CeO2–Y2O3(CY), thereby greatly enhancing the dry reforming of methane (DRM). A combination of X-ray diffraction, H2-TPR, X-ray photoelectron spectroscopy, and transmission electron microscopy reveals that cobalt nanoparticles, stabilized on CY supports with an intermediate particle size of ∼75 nm, exhibit superior strength and enhanced DRM activity due to improved MSI after reduction at 800 °C. For catalysts with optimal MSI, we demonstrate that facile formation of oxygen vacancies is pivotal for the high DRM activity.
Introduction
The rational design of supported metal catalysts is pivotal in heterogeneous catalysis.1 The interaction between metal species and their supports plays a crucial role in catalyst performance, particularly on reducible oxide supports such as TiO2, Nb2O5, and CeO2, where metal–support interactions (MSI) originate from multiple mechanisms.2 A notable example is the encapsulation of dispersed metal nanoparticles by an oxide support layer, induced by reduction, oxidation, or reactant adsorption, known as strong metal–support interaction (SMSI).3 Additionally, charge transfer, support-induced restructuring, and the formation of specific metal–support interfaces contribute to MSI.4 Although SMSI may negatively impact catalytic activity by reducing the active metal surface area, it can enhance the reaction performance through other mechanisms. Control over MSI typically involves manipulating the reduction temperature to adjust the extent of decoration or encapsulation of metallic nanoparticles by a thin layer of reducible support.5 Furthermore, a series of reduction–oxidation–reduction pretreatments can induce structural changes in metal particles on reducible supports like TiO2 and Nb2O5, impacting adsorptive properties and catalytic performance.6,7Ceria and its derivatives are unique among reducible oxide supports due to their ability to modulate MSI through both electronic effects and structural decoration or alloying.8–12 This distinctive property makes ceria-based catalysts highly effective for catalysis, particularly in applications like dry reforming of methane (DRM), a promising pathway for the simultaneous conversion of methane and carbon dioxide into syngas (H2 and CO), a vital precursor for alternative fuels and chemicals from non-petroleum sources.13
Ceria-supported late transition metals such as Co, Ni, and Ru have proven effective in DRM.14–16 Studies have highlighted the critical role of the metal–support interface.17,18 Most recent researches, utilizing both experimental methods and density functional theory calculations, demonstrated that a robust metal–support interaction reduced the energy barrier for methane activation and prevented the carbon deposits, thereby facilitating its dissociation.19,20 Especially, Liu and colleagues conducted a comprehensive investigation into the mechanism of methane dry reforming over Ni–CeO2 catalysts, revealing that mesoporous structures (MSI) significantly facilitate the cleavage of C–H bonds.21 Notably, well-dispersed cobalt species on ceria, prepared via co-precipitation or sol–gel methods, exhibit atypical reduction behavior, characterized by multiple reduction stages across a wide temperature range. This behavior has been linked to varying interaction strengths between Co and Ce, alongside contributions from both surface and bulk Ce4+ to Ce3+ reduction, and the reduction of Co–Ce–O solid solution.8,9,18 However, a precise modulation of MSI strength to achieve optimal cobalt dispersion and stability remains challenging, particularly under high-temperature reactions such as DRM.
In this study, we introduce a simple strategy to optimize MSI strength in Co/CeO2–Y2O3 (Co/CY) for DRM by adjusting the particle size of the support. We found that cobalt nanoparticles on CY supports with an intermediate particle size of ∼20 nm exhibits higher dispersion of metallic cobalt compared to both larger and smaller sizes. This result can be attributed to an optimal strength of the interaction between cobalt and CY, which stabilizes cobalt nanoparticles during reduction. Consequently, these catalysts demonstrate high ceria reduction levels at low temperatures and markedly improved catalytic activity in C–H bond dissociation.
Methods
All chemicals were obtained from Sigma-Aldrich: cobalt(II) acetate tetrahydrate, ammonia solution (30% NH3 in H2O), cerium(III) nitrate hexahydrate and yttrium nitrate.Preparation of CY supports
CY supports were prepared by co-precipitation. An aqueous solution of metal precursors was prepared from Y(NO3)3·6H2O and Ce(NO3)3·6H2O. Afterwards, it was precipitated by adding an aqueous solution of ammonia (30%) dropwise at room temperature. The pH of the solution was adjusted to 9. The resulting precipitate was aged for 24 h, followed by filtration, washing and drying in a convection oven at 110 °C overnight. To prepare CY supports with different particle sizes, dried precipitate was calcined in an air flow at temperatures between 700 °C and 900 °C for 4 h. The obtained samples are denoted as CY700, CY800 and CY900, corresponding to the calcination temperature.Preparation of Co/CY catalysts
A series of CY-supported cobalt catalysts with different supports was prepared using a wet impregnation method. For this purpose, the desired amount of cobalt acetate was dissolved in 30 mL of ammonia solution (30 wt%). The suspension obtained by adding 2 g of CY to the solution was stirred for 2 h, after which water was removed by evaporation. The catalysts were dried in air at 110 °C overnight and then calcined at 350 °C for 4 h. Prior to the catalytic activity measurements, the catalysts were reduced at 500 °C in H2 for 4 h.Characterization
The crystalline structure of the catalysts was determined by recording X-ray diffraction patterns with a Bruker D2 Phaser diffractometer using Cu Kα radiation. The particle size was estimated with the Scherrer equation. Reduced samples were transferred to the sample holder via a glovebox. The samples were placed in an X-ray diffraction sample holder and covered with Kapton tape. Hydrogen TPR (H2-TPR) experiments were performed with a Micromeritics Autochem II 2920 instrument. Typically, 100 mg of catalyst was loaded in a tubular quartz reactor. The sample was reduced in 4 vol.% H2 in N2 at a flow rate of 50 mL min−1, while heating from room temperature up to 600 °C at a rate of 10 °C min−1. N2 physisorption was performed at −196 °C on a Micromeritics TriStar II 3020 to determine the specific surface area (SSA) of the CZ supports. The samples were degassed at 160 °C for at least 12 h prior to N2 physisorption measurements. Surface areas were calculated using the Brunauer–Emmett–Teller (BET) method. TEM images were acquired with a Tecnai 20 transmission electron microscope (FEI) equipped with a LaB6 filament and operated at an acceleration voltage of 200 kV. Calcined catalysts were prepared by dropping a suspension of finely ground material in analytical grade absolute ethanol onto Quantifoil R 1.2/1.3 holey carbon films supported on a copper grid. Reduced catalysts were transferred to an argon-filled glovebox and dispersed in dry n-hexane; then a few droplets were placed on Cu TEM grids. The grid was transported in a GATAN vacuum transfer holder (model number CHVT3007). Raman spectra were obtained on Horiba Jobin Yvon iHR 550 imaging spectrometer, with laser wavelength of 532 nm. Raman spectra of 1000 h stability test sample were obtained on Horiba LabRAM HR Evolution Confocal RAMAN.Catalytic activity
A schematic of the experimental system is provided in Fig. S1.† Catalytic activity measurements were performed in a ten-tube parallel microflow reactor. The samples were pressed, sieved and crushed, and the fraction between 20 and 40 mesh was used. Each quartz reactor was filled with 200 mg of sample diluted with 800 mg of SiC of the same sieve fraction. The obtained mixture was enclosed between two quartz wool plugs. The reaction was performed at atmospheric pressure. Prior to reaction, the catalysts were reduced in situ in a flow consisting of 10 vol.% H2 in He (total flow rate 100 mL min−1 at STP per reactor tube), while heating from room temperature to 500 °C at a rate of 10 °C min−1, followed by an isothermal dwell of 4 h. After cooling to the reaction temperature in the same flow, the pretreatment gas was replaced by a feed consisting of 10 vol.% CO2 and 10 vol.% CH4 in N2 (total flow rate 200 mL min−1 at STP). The temperature was increased by steps of 25 °C at a rate of 5 °C min−1. When the target temperature was reached, a period of 20 min was allowed for stabilization. Then, the effluent gas was analyzed by NDIR gas analyzer (GASBOARD-3100, Wuhan cubic optoelectronic Co., Ltd).Results and discussion
The particle size of the CY support was adjusted between 25 and 300 nm by varying the calcination temperature of the initial precipitate from 700 °C to 900 °C (Fig. 1a): (i) CY700 ∼25 nm, (ii) CY800 ∼75 nm, and (iii) CY900 ∼120 nm. These CY materials were then used to create Co/CY catalysts (2.5 wt% cobalt) via the wet impregnation method, labeled as Co/CYx (x = 700, 800, 900). The space velocity and gas composition for the DRM reaction were optimized to 200 mL min−1 and 10%CH4/10%CO2/80%N2, respectively, at 650 °C. As seen in Fig. 1b, both CH4 and CO2 conversions exhibit a volcanic trend with increasing calcination temperature. Co/CY700 exhibits the lowest catalytic performance with CH4 and CO2 conversions of 32% and 38%, respectively, and those for Co/CY900 reach 44% and 53%, respectively. Notably, the Co/CY800 catalyst achieves the highest conversions, with CH4 at 58% and CO2 at 70%, as well as CO and H2 productivities of ∼133 mmol gcat−1 h−1 and ∼108 mmol gcat−1 h−1, respectively(Fig. 1c). In addition, the intrinsic turnover frequency (TOF) of CH4 was evaluated at low conversions (below 10%, Fig. 1d), consistently revealing the following trend: Co/CY800 (4.7 molCH4 molCo−1s−1) > Co/CY900 (3.3 molCH4 molCo−1 s−1) > Co/CY700 (2.1 molCH4 molCo−1 s−1). As for the optimal Co/CY800 sample, the CH4 conversions reached an exceptional level at 650 °C (Table 1).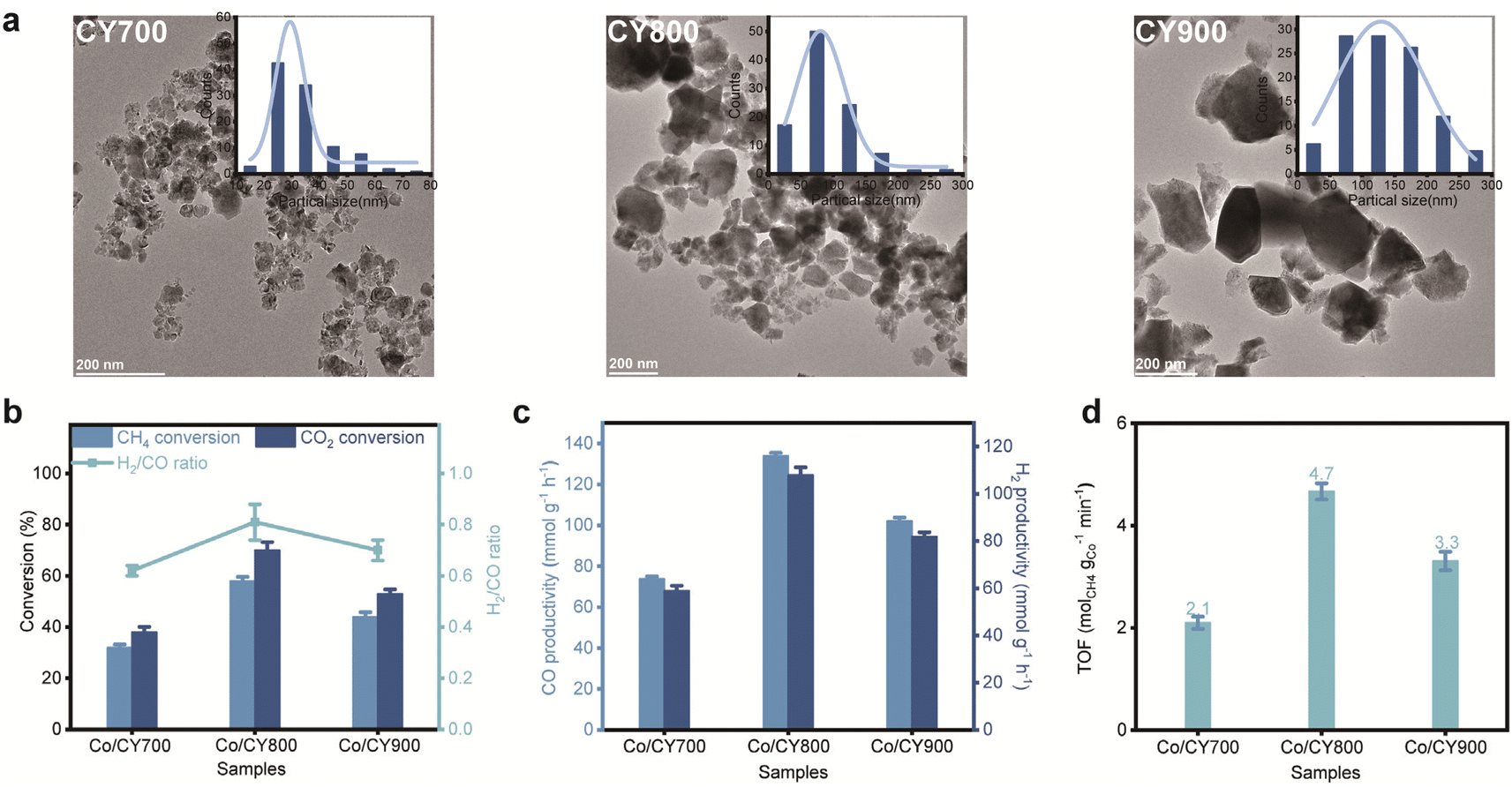 | ||
| Fig. 1 Effect of support calcination pretreatment on catalytic activity. (a) Transmission electron microscopy (TEM) images of as-prepared CY700, CY800 and CY900. (b and c) CH4 and CO2 conversion over various catalysts at 650 °C. (d) Intrinsic TOF over various catalysts within the catalytic dynamic range. | ||
The X-ray diffraction (XRD) patterns for various Co/CYx are depicted in Fig. 2a. The diffraction peaks for all samples align with the fluorite phase of CeO2 (PDF# [43-1002]), with no distinct peaks for Co species, likely due to either a low Ce content or the high dispersion of Co species on the support. As presented in Fig. 2b, Co/CY800 featured the highest specific surface area (48 m2 g−1) among the various Co/CYx catalysts. The volcanic trend in activity observed for Co/CYx catalysts concerning CY particle size are likely linked to MSI. To further explore this effect, we examined the reducibility of Co/CYx catalysts using temperature-programmed reduction (TPR) analysis (Fig. 2c). The TPR profile for Co/CY700 shows two typical reduction features associated with the stepwise reduction of Co3O4 to CoO(β) and then to Co(γ). In contrast, the TPR profile of Co/CZ800 presents additional features, with a low-temperature reduction peak (α) likely due to the partial surface reduction of ceria. Complete reduction of the samples occurs at a much higher temperature, characteristic of cobalt catalysts with SMSI.27 Additionally, the high-temperature reduction region splits into two peaks, attributed to the reduction of cobalt species weakly (γ) and strongly (δ) interacting with the support.9,27 The extra high-temperature features could also stem from the reduction of the cobalt–support interface Co–O–Ce species or from hydrogen spillover, which leads to bulk reduction of the support.28 A notable excess of H2 consumption (Fig. 2d) observed for catalysts with enhanced MSI indicates deeper reduction of the support (Ce4+ → Ce3+). Stabilization of the intermediate CoO phase over a wider temperature range suggests that the γ feature in the TPR profile relates to support reduction rather than CoO reduction. Indeed, the CoO → Co0 transformation occurred at a higher temperature for Co/CY800, typical for systems with strong MSI. The TPR results reveal a correlation between catalytic activity and the extent of MSI—the most active catalysts exhibit complex reduction behavior with high-temperature features associated with both cobalt and cerium oxides.
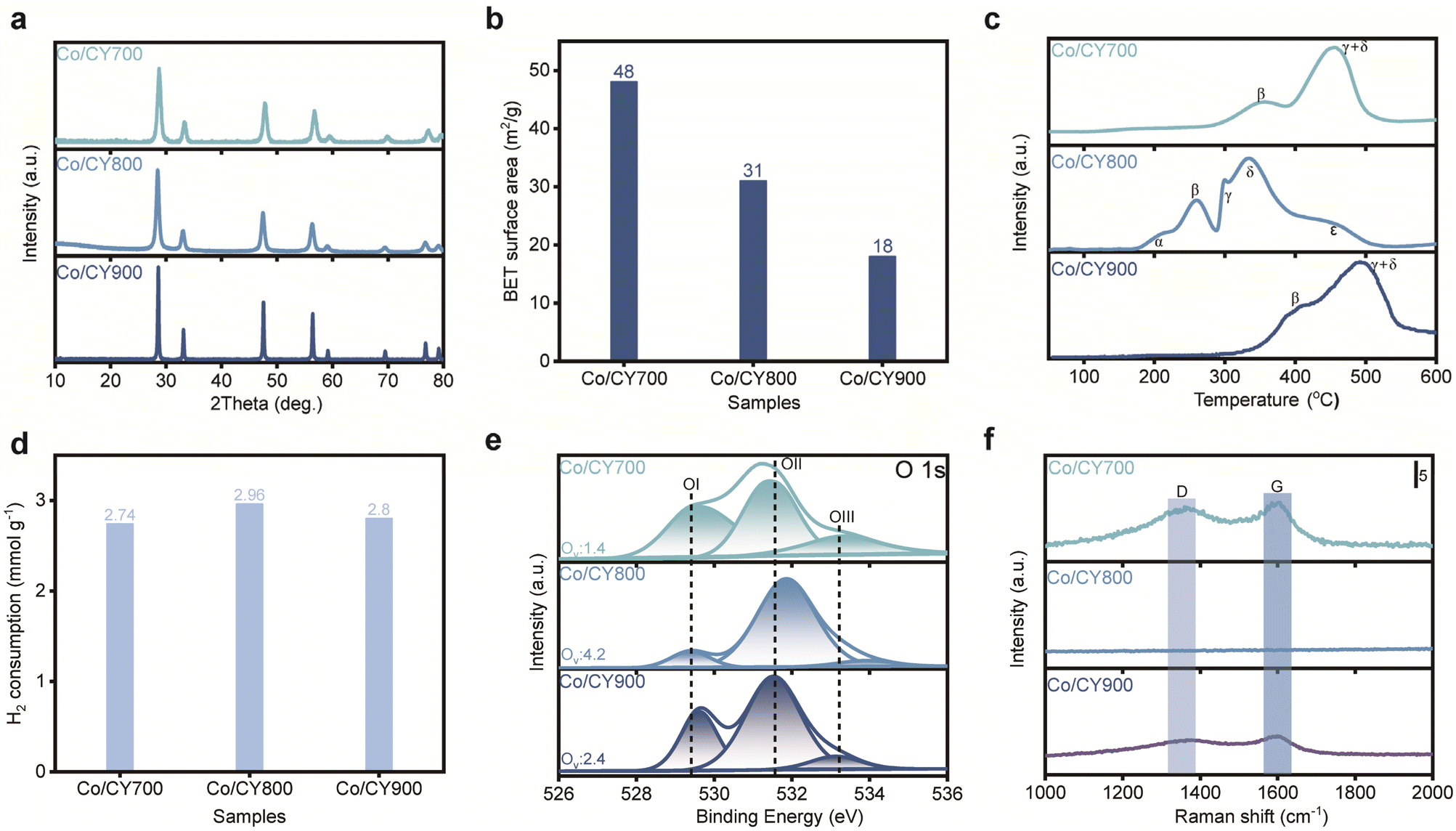 | ||
| Fig. 2 Characterization of Co/CYx catalysts. (a) XRD patterns of fresh Co/CYx. (b) BET surface area of Co/CYx. (c) H2-TPR profiles of fresh Co/CYx and (d) quantification of hydrogen consumption during H2-TPR analysis. (e) XPS analysis of the O 1s peaks of reduced Co/CYx and (f) Raman spectra of the spent catalysts. | ||
MSI in ceria-based materials commonly correlate with the generation of surface oxygen vacancies. X-ray photoelectron spectroscopy (XPS) analysis was performed to quantify vacancies concentration in Co/CYx. By deconvoluting the O 1s spectra (Fig. 2e), OI (∼529.5 eV) corresponds to lattice oxygen (Olatt) and the highly oxidative oxygen species (O22− or O−) associate with the surface oxygen vacancies (Oads), respectively, while OII (∼530.5 eV) and OIII (∼531.5 eV) can be assigned to hydroxyl (OH−) and/or carbonate (CO32−) species. Following established methods, the concentration of surface oxygen vacancies can be estimated from the ratio (Ov = Oads/Olatt) of surface oxygen species (O22− or O−) to lattice oxygen species, as derived from XPS data.3,29,30 Obviously, Co/CY800 exhibits a higher Ov ratio compared to Co/CY700 and Co/CY900, suggesting a higher concentration of oxygen vacancies. Furthermore, as reported previously, lowering the lattice oxygen content on the surface leads to higher oxygen vacancy concentration, further supporting that Co/CY800 also possesses the richest surface oxygen vacancies, testifying the enhancement of oxygen vacancy concentration due to CeO2 morphology. Deconvolution of the Co 2p3/2 spectrum shows Co0 and Co2+ on all catalysts, implying that the variations in catalytic activity are independent of Co valence state (see Fig. S2†). Raman spectra of spent Co/CYx (Fig. 2f) show slight D- and G-band carbon peaks on the spent Co/CY700 and Co/CY900, but none on Co/CY800, attributable to the abundant oxygen vacancies present in Co/CY800. Together, H2-TPR and XPS data underscore the correlation between catalytic activity and the extent of the MSI-the most active catalysts display complex reduction behaviour with high-temperature features related to both cobalt and cerium oxides.
A key feature of Co/CYx catalysts is the stabilization of cobalt nanoparticles by the support under high temperature reducing conditions. Transmission electron microscopy (TEM) and scanning transmission electron microscopy–energy-dispersive X-ray spectroscopy (STEM-EDX) analysis of calcined Co/CYx catalysts reveals that the initial dispersion of cobalt oxide is consistent, with particle sizes ranging from 7 to 20 nm (Fig. S3†). However, after reduction in H2 at 500 °C, significant differences emerge in cobalt metal particle size (Fig. 3). Upon reduction, the cobalt phase undergoes more pronounced sintering in Co/CY700 (Fig. 3a). STEM-EDX and size distribution images also show the formation of significantly larger cobalt particles (about 50 nm). This pronounced sintering in Co/CY700, indicated by the lack of high-temperature TPR features, can be attributed to its weak MSI. Similarly, Co/CY900 exhibits large cobalt agglomerates, attributed to its lower surface area. In contrast, Co/CY800 showed much better resistance to sintering during high-temperature reduction, maintaining consistent particle size distributions before and after reduction without larger aggregates. In summary, intermediate-sized carriers ensure the stabilization of cobalt (Co) species' dimensions, which in turn fosters a consistent dispersion of Co particles while inhibiting their aggregation. This arrangement maximizes the interaction between the Co particles and the carrier, thereby cultivating a higher concentration of Co–CY interfaces and fortifying the interfacial interaction between the metal and the substrate. Consequently, the catalytic efficacy is notably improved in comparison to particles of either smaller or larger size. This analysis highlights that optimal strategy for preparing efficient Co/CYx catalysts is to use supports with sufficiently large particle sizes to ensure strong interfacial MSI without compromising the surface area.
 | ||
| Fig. 3 TEM images of reduced Co/CYx (a) Co/CY700, (b) Co/CY800 and (c) Co/CY900); insets show particle size distribution and STEM-EDX images of Co/CYx after reduction at 500 °C. | ||
Conclusions
We introduce a novel strategy for tuning the mesoporous structure intensity in the Co/CYx catalyst. During the high-temperature reduction process crucial for catalyst activation, cobalt nanoparticles anchored on smaller CY particles frequently succumb to sintering. Notably, cobalt nanoparticles deposited on CY particles with an intermediate diameter of 75 nm demonstrate improved stability. Mesoporous structure enhancement has been demonstrated to optimize the dispersion of the reduced cobalt, thereby maximizing the dehydrogenation-methane (DRM) activity. Detailed analyses utilizing X-ray diffraction (XRD), hydrogen temperature-programmed reduction (H2-TPR), and X-ray photoelectron spectroscopy (XPS) validated the robust mesoporous structure at the Co–CY interface. The distinctive chemical attributes of the optimized Co–CY interface facilitate the stabilization of metal nanoparticles under high-temperature reduction conditions and augment the activation rate of CH4. While the precise nature of the intrinsic active sites remains unclear, this study holds significant potential for the application of methane catalytic combustion, water-gas shift (reverse), Fischer–Tropsch synthesis, and associated catalytic processes.Data availability
Data will be made available on reasonable request.Author contributions
All authors contributed to the investigation, supervise, writing original draft, review, data analysis & editing.Conflicts of interest
The authors have no conflicts of interest to declare with respect to the research, authorship and publication of this article.Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 52276214), Inner Mongolia Autonomous Region “Science and Technology Breakthrough” Project (No. 2024KJTW0013) and Inner Mongolia Daqingshan Laboratory Scientific and Technological Support Program (2023KYPT0033).References
- C. Vogt and B. M. Weckhuysen, Nat. Rev. Chem, 2022, 6, 89–111 Search PubMed.
- T. W. Van Deelen, C. Hernández Mejía and K. P. De Jong, Nat. Catal., 2019, 2, 955–970 CAS.
- Y. Long, K. Yang, Z. Gu, S. Lin, D. Li, X. Zhu, H. Wang and K. Li, Appl. Catal., B, 2022, 301, 120778 CAS.
- J. Liu, L. Chen and X. Liu, ACS Catal., 2024, 14, 1987–2002 CAS.
- H. Wang, G. Cui, H. Lu, Z. Li, L. Wang, H. Meng, J. Li, H. Yan, Y. Yang and M. Wei, Nat. Commun., 2024, 15, 3765 CAS.
- J. Zhou, Z. Gao, G. Xiang, T. Zhai, Z. Liu, W. Zhao, X. Liang and L. Wang, Nat. Commun., 2022, 13, 327 CAS.
- C. Hernández Mejía, T. W. Van Deelen and K. P. De Jong, Nat. Commun., 2018, 9, 4459 Search PubMed.
- A. Parastaev, V. Muravev, E. H. Osta, T. F. Kimpel, J. F. M. Simons, A. J. F. Van Hoof, E. Uslamin, L. Zhang, J. J. C. Struijs, D. B. Burueva, E. V. Pokochueva, K. V. Kovtunov, I. V. Koptyug, I. J. Villar-Garcia, C. Escudero, T. Altantzis, P. Liu, A. Béché, S. Bals, N. Kosinov and E. J. M. Hensen, Nat. Catal., 2022, 5, 1051–1060 CAS.
- A. Parastaev, V. Muravev, E. Huertas Osta, A. J. F. Van Hoof, T. F. Kimpel, N. Kosinov and E. J. M. Hensen, Nat. Catal., 2020, 3, 526–533 CAS.
- C. Yang, X. Yu, S. Heißler, A. Nefedov, S. Colussi, J. Llorca, A. Trovarelli, Y. Wang and C. Wöll, Angew. Chem., Int. Ed., 2017, 56, 375–379 CAS.
- Y. Lykhach, S. M. Kozlov, T. Skála, A. Tovt, V. Stetsovych, N. Tsud, F. Dvořák, V. Johánek, A. Neitzel, J. Mysliveček, S. Fabris, V. Matolín, K. M. Neyman and J. Libuda, Nat. Mater., 2016, 15, 284–288 CAS.
- J. Graciani, K. Mudiyanselage, F. Xu, A. E. Baber, J. Evans, S. D. Senanayake, D. J. Stacchiola, P. Liu, J. Hrbek, J. F. Sanz and J. A. Rodriguez, Science, 2014, 345, 546–550 CAS.
- A. T. Ashcroft, A. K. Cheetham, M. L. H. Green and P. D. F. Vernon, Nature, 1991, 352, 225–226 CAS.
- J. Yu, T. Le, D. Jing, E. Stavitski, N. Hunter, K. Lalit, D. Leshchev, D. E. Resasco, E. H. Sargent, B. Wang and W. Huang, Nat. Commun., 2023, 14, 7514 CAS.
- C. Palmer, D. C. Upham, S. Smart, M. J. Gordon, H. Metiu and E. W. McFarland, Nat. Catal., 2020, 3, 83–89 CrossRef CAS.
- Q. Zhu, H. Zhou, L. Wang, L. Wang, C. Wang, H. Wang, W. Fang, M. He, Q. Wu and F.-S. Xiao, Nat. Catal., 2022, 5, 1030–1037 CrossRef CAS.
- P. Kumar Yadav and S. Sharma, Fuel, 2024, 358, 130163 CrossRef CAS.
- F. Zhang, Z. Liu, S. Zhang, N. Akter, R. M. Palomino, D. Vovchok, I. Orozco, D. Salazar, J. A. Rodriguez, J. Llorca, J. Lee, D. Kim, W. Xu, A. I. Frenkel, Y. Li, T. Kim and S. D. Senanayake, ACS Catal., 2018, 8, 3550–3560 CrossRef CAS.
- B. O. Yusuf, M. Umar, E. Kotob, A. Abdulhakam, O. A. Taialla, M. M. Awad, I. Hussain, K. R. Alhooshani and S. A. Ganiyu, Chem.–Asian J., 2024, 19, e202300641 CrossRef CAS PubMed.
- A. Tanimu, B. O. Yusuf, S. Lateef, G. Tanimu, A. M. Alhassan, M. O. Azeez, K. Alhooshani and S. A. Ganiyu, J. Environ. Chem. Eng., 2024, 12, 113873 CrossRef CAS.
- Z. Liu, D. C. Grinter, P. G. Lustemberg, T. Nguyen-Phan, Y. Zhou, S. Luo, I. Waluyo, E. J. Crumlin, D. J. Stacchiola, J. Zhou, J. Carrasco, H. F. Busnengo, M. V. Ganduglia-Pirovano, S. D. Senanayake and J. A. Rodriguez, Angew. Chem., Int. Ed., 2016, 55, 7455–7459 CrossRef CAS PubMed.
- Z. Xie, B. Yan, S. Kattel, J. H. Lee, S. Yao, Q. Wu, N. Rui, E. Gomez, Z. Liu, W. Xu, L. Zhang and J. G. Chen, Appl. Catal., B, 2018, 236, 280–293 CrossRef CAS.
- D. Liang, Y. Wang, M. Chen, X. Xie, C. Li, J. Wang and L. Yuan, Appl. Catal., B, 2023, 322, 122088 CrossRef CAS.
- J. Yu, T. Le, D. Jing, E. Stavitski, N. Hunter, K. Lalit, D. Leshchev, D. E. Resasco, E. H. Sargent, B. Wang and W. Huang, Nat. Commun., 2023, 14, 7514 CrossRef CAS PubMed.
- T.-Y. Liang, D. Senthil Raja, K. C. Chin, C.-L. Huang, S. A. Sethupathi, L. K. Leong, D.-H. Tsai and S.-Y. Lu, ACS Appl. Mater. Interfaces, 2020, 12, 15183–15193 CrossRef CAS.
- J.-H. Park, S. Yeo and T.-S. Chang, J. CO2 Util., 2018, 26, 465–475 CrossRef CAS.
- F. Zhang, Z. Liu, S. Zhang, N. Akter, R. M. Palomino, D. Vovchok, I. Orozco, D. Salazar, J. A. Rodriguez, J. Llorca, J. Lee, D. Kim, W. Xu, A. I. Frenkel, Y. Li, T. Kim and S. D. Senanayake, ACS Catal., 2018, 8, 3550–3560 CrossRef CAS.
- M. Li, H. Amari and A. C. Van Veen, Appl. Catal., B, 2018, 239, 27–35 Search PubMed.
- Y. Long, X. Wang, H. Zhang, K. Wang, W.-L. Ong, A. Bogaerts, K. Li, C. Lu, X. Li, J. Yan, X. Tu and H. Zhang, JACS Au, 2024 DOI:10.1021/jacsau.4c00153.
- Y. Zheng, K. Li, H. Wang, D. Tian, Y. Wang, X. Zhu, Y. Wei, M. Zheng and Y. Luo, Appl. Catal., B, 2017, 202, 51–63 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d5ra00007f |
| This journal is © The Royal Society of Chemistry 2025 |