
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceTheoretical evaluation of a bulky ortho-thioalkyl-azobenzene as an alternative to photocontrol structural cytotoxic effects of metal-free and disulfide oxidized hSOD1 in pathogenesis of ALS†
Constanza Galaz-Araya‡
a,
Daniel Zuñiga-Núñez‡b,
Francisca Salas-Sepúlvedaa,
Alejandra Herrera-Morandec,
Alexis Aspée *b,
Horacio Poblete*a and
Ricardo A. Zamora*d
*b,
Horacio Poblete*a and
Ricardo A. Zamora*d
aCentro de Bioinformática, Simulación y Modelado (CBSM), Facultad de Ingeniería, Universidad de Talca, 2 Norte 685, Talca, Chile. E-mail: hopoblete@utalca.cl
bFacultad de Química y Biología, Universidad de Santiago de Chile, Casilla 40, Correo 33 Santiago, Chile
cDepartamento de Física y Química, Facultad de Ingeniería, Universidad Autónoma de Chile, Av. Pedro de Valdivia 425, Providencia 7500000, Chile
dInstituto de Investigación Interdisciplinaria (I3), Vicerrectoría Académica, and Centro de Bioinformática, Simulación y Modelado (CBSM), Facultad de Ingeniería, Universidad de Talca, Campus Lircay, Talca 3460000, Chile. E-mail: ricardo.zamora@utalca.cl
First published on 24th March 2025
Abstract
This study presents a novel photopharmacological strategy to mitigate the cytotoxic effects of apo-hSOD1S–S, a misfolded protein implicated in neurodegenerative diseases. Using quantum chemical calculations and molecular dynamics simulations, we demonstrate that ortho-thio-substituted azobenzene photoswitches (ortho-TABPs) can be employed to precisely modulate the dynamics of the crucial electrostatic loop (EL) in apo-hSOD1S–S. We establish that larger ortho-S-alkyl substituents on the ortho-TABP enhance its redox stability, favouring the cis conformation through the modulation of the position of the n → π* transition. This stability is crucial for operation within the reducing cellular environment. Furthermore, we demonstrate the successful and consistent photomodulation of EL conformational dynamics in apo-hSOD1S–S through covalent tethering of an ortho-TABP. This control is achieved by leveraging the thermodynamically stable trans conformation of the photoswitch, which allosterically influences the EL and consequently, the geometry of the Zn-binding site, a critical determinant of apo-hSOD1S–S cytotoxicity. This work paves the way for developing targeted therapies for neurodegenerative diseases by demonstrating the precise and effective photomodulation of apo-hSOD1S–S via rationally designed ortho-TABPs.
Introduction
Human Superoxide Dismutase-1 (hSOD1) is a soluble copper and zinc-containing enzyme located in the cell cytosol. hSOD1 has been proved to be catalytically active as a homodimer form.1 The function of hSOD1 is to catalyze the dismutation of superoxide to hydrogen peroxide (H2O2) and molecular oxygen (O2).1 Mutations associated with misfolded hSOD1 have been proposed as one of the main causes of amyotrophic lateral sclerosis (ALS), the most common motor neuron degenerative disease.2,3 Some of these ALS-linked hSOD1 mutations cause a gain of pathological functions due mainly to destabilization of the monomer–monomer interface4–6 and also demetallation of their catalytic site.7,8 Interestingly, monomeric and dimeric demetallated hSOD1 forms exhibit prion-like properties such as transmissibility between cells and intercellular propagation.9 Through cell culture experiments, it has been revealed that demetallated hSOD1 located in the cytosol is released into the extracellular medium and can actively spread to adjacent cells.9 Takashima et al. demonstrated that the hSOD1 release to the extracellular medium (hSOD1 wild-type and four ALS-linked hSOD1 mutants) were in the metal-free form and disulfide oxidized form (apo-hSOD1S–S).9 The apo-hSOD1S–S form was found to be the primary species responsible for the prion-like propagation and cytotoxic effects in motor neuron cells.9 It is believed that the internalization of extracellular apo-hSOD1S–S in healthy neuronal cells serves as a conformational template to convert natively well-folded hSOD1 into a misfolded/aggregated species.10–12 The removal of apo-hSOD1S–S from the extracellular medium significantly decreases the cytotoxic effect and the prion-like propagation process. The cytotoxic effect of the extracellular apo-hSOD1S–S in ALS has been reported to be due to its ability to chelate Zn(II) and disrupt intracellular Zn(II) homeostasis.13,14 Recently, Tajiri et al. proposed an apo-hSOD1S–S dependent Zn(II) chelation mechanism, which depends on the redox state of the specific disulfide bond.15 Apo-hSOD1S–S can incorporate more than one Zn(II) ion per monomer by using the Cu metal-binding site to add additional Zn(II). The Zn(II)-chelating mechanism is based on the existence of several abnormal oxidized hSOD1 Zn-metalation states (i.e., [Zn, Zn][Zn, Zn], [E, Zn][Zn, Zn], [E, Zn][Zn, E], [E, Zn][E, E]).15 Conversely, the reduced apo form of hSOD1 (Apo-hSOD1SH) exists solely as a monomer, and it can only bind a maximum of one Zn(II) ion per monomer, even in the presence of a high concentration of Zn(II).15 Under intracellular oxidative stress conditions, the nascent apo-hSOD1SH can undergo oxidation, further increasing the release and extracellular concentration of the apo-hSOD1S–S.15 Apo-hSOD1S–S as well as Zn-bound hSOD1S–S species does not represent a natural substrate for the copper chaperone for SOD1 (CCS chaperone), which causes that both Apo-hSOD1S–S and Zn-bound hSOD1S–S cannot be restored to its native metalated form.15,16 While apo- and Zn-bound hSOD1S–S can exchange Zn(II) for Cu(II) without the assistance of a chaperone, this process results in slow maturation and does not represent a physiologically relevant pathway. The background information indicates that decreasing the affinity of the extracellular apo-hSOD1S–S for Zn(II) would represent a valuable alternative to decrease the cytotoxic effect and the prion-like propagation process of hSOD1 in ALS models during early pre-symptomatic stages. It is well known that almost all ALS-linked hSOD1 mutations alone have a negatively effect on the hSOD1 structure. In particular, thirteen different ALS-linked mutations have been catalogued as responsible for the destabilization of the electrostatic loop (EL).3 EL, in addition to fulfilling a catalytic role during the dismutation of O2·–, has the function of “solvent seal,” isolating the Zn(II) binding site of hSOD1 from water molecules. Mera-Adasme et al. have shown through computational technique that 5 of these mutants affect the interaction of the EL with the hSOD1 structure, propitiating the opening of the EL and the hydration of the Zn binding site.17 Additionally, Souza et al. showed that some of these mutations generates a destabilization of the Zn binding site by opening the EL, which decreases the affinity of hSOD1 for Zn(II).18 In this work, we present a detailed computational study on using azobenzene-based photoswitch (ABP) to modulate the structural cytotoxic conformation of apo-hSOD1S–S. Specifically, we explore the feasibility of use one ortho-thio-ABP (ortho-TABP) covalently coupled at apo-hSOD1S–S structure to generate an allosteric structural modulation of the EL opening/closing process. Based on the premise that the conditions of the extracellular environment in which apo-hSOD1S–S is released are generally oxidizing, and that this redox condition can be modified by physiologically relevant extracellular levels of glutathione (GSH),19,20 in the first part of this work, we focus on characterizing by using quantum calculations based on density functional theory (DFT) and, time-dependent density functional theory (TD-DFT) and free energy calculations, the electronic and mechanical effect of different ortho-S-alkyl substituents on the photochromic and thermodynamic ABP properties. The ortho-S-alkyl substituents were selected because this type of substitution in ABP has improve the resistance of ABPs to the reduction by GSH.21 Ortho-TABPs was compared against their oxygenated analogues (ortho-OABPs) due to the similar chemistry between O and S atoms and that ortho-oxy-alkyl substituents are one of the most widely used modifications in the field of photopharmacology. Subsequently, we select and covalently couple one ortho-TABP to the apo-hSOD1S–S and evaluate the ability of the ortho-TABP to modulate allosterically the positioning of hSOD1 EL, thereby altering the chelating capacity of the enzyme in the apo state. Employing atomistic molecular dynamics simulations and free energy calculations, we investigate the possibility of photocontrolling the EL opening/closing process. By using ABF method, we characterise the free energy profiles of the mechanical isomerization process for one ortho-TABP covalently couple to hSOD1 structure. Taken together, all the computational tools used in this work allowed us to determine in silico that the bulky ortho-S-alkyl substituents in ABP effectively (i) enhance redox stability of the azo group, (ii) allow to separate the position of the n → π* transition in the trans and cis conformations, and (iii) one ortho-TABP covalently coupled to the hSOD1 structure in cis conformation, and its subsequent mechanical isomerization to its trans conformation allows controlling allosterically the opening/closing process of the EL. The computational approaches employed in this work allow us to consider the potential experimental use of ortho-TABPs for targeting apo-hSOD1S–S, aiming to abolish or reduce the cytotoxic effects and prion-like propagation process of hSOD1 with spatiotemporal precision while minimizing unwanted side effects on healthy cells.
to hydrogen peroxide (H2O2) and molecular oxygen (O2).1 Mutations associated with misfolded hSOD1 have been proposed as one of the main causes of amyotrophic lateral sclerosis (ALS), the most common motor neuron degenerative disease.2,3 Some of these ALS-linked hSOD1 mutations cause a gain of pathological functions due mainly to destabilization of the monomer–monomer interface4–6 and also demetallation of their catalytic site.7,8 Interestingly, monomeric and dimeric demetallated hSOD1 forms exhibit prion-like properties such as transmissibility between cells and intercellular propagation.9 Through cell culture experiments, it has been revealed that demetallated hSOD1 located in the cytosol is released into the extracellular medium and can actively spread to adjacent cells.9 Takashima et al. demonstrated that the hSOD1 release to the extracellular medium (hSOD1 wild-type and four ALS-linked hSOD1 mutants) were in the metal-free form and disulfide oxidized form (apo-hSOD1S–S).9 The apo-hSOD1S–S form was found to be the primary species responsible for the prion-like propagation and cytotoxic effects in motor neuron cells.9 It is believed that the internalization of extracellular apo-hSOD1S–S in healthy neuronal cells serves as a conformational template to convert natively well-folded hSOD1 into a misfolded/aggregated species.10–12 The removal of apo-hSOD1S–S from the extracellular medium significantly decreases the cytotoxic effect and the prion-like propagation process. The cytotoxic effect of the extracellular apo-hSOD1S–S in ALS has been reported to be due to its ability to chelate Zn(II) and disrupt intracellular Zn(II) homeostasis.13,14 Recently, Tajiri et al. proposed an apo-hSOD1S–S dependent Zn(II) chelation mechanism, which depends on the redox state of the specific disulfide bond.15 Apo-hSOD1S–S can incorporate more than one Zn(II) ion per monomer by using the Cu metal-binding site to add additional Zn(II). The Zn(II)-chelating mechanism is based on the existence of several abnormal oxidized hSOD1 Zn-metalation states (i.e., [Zn, Zn][Zn, Zn], [E, Zn][Zn, Zn], [E, Zn][Zn, E], [E, Zn][E, E]).15 Conversely, the reduced apo form of hSOD1 (Apo-hSOD1SH) exists solely as a monomer, and it can only bind a maximum of one Zn(II) ion per monomer, even in the presence of a high concentration of Zn(II).15 Under intracellular oxidative stress conditions, the nascent apo-hSOD1SH can undergo oxidation, further increasing the release and extracellular concentration of the apo-hSOD1S–S.15 Apo-hSOD1S–S as well as Zn-bound hSOD1S–S species does not represent a natural substrate for the copper chaperone for SOD1 (CCS chaperone), which causes that both Apo-hSOD1S–S and Zn-bound hSOD1S–S cannot be restored to its native metalated form.15,16 While apo- and Zn-bound hSOD1S–S can exchange Zn(II) for Cu(II) without the assistance of a chaperone, this process results in slow maturation and does not represent a physiologically relevant pathway. The background information indicates that decreasing the affinity of the extracellular apo-hSOD1S–S for Zn(II) would represent a valuable alternative to decrease the cytotoxic effect and the prion-like propagation process of hSOD1 in ALS models during early pre-symptomatic stages. It is well known that almost all ALS-linked hSOD1 mutations alone have a negatively effect on the hSOD1 structure. In particular, thirteen different ALS-linked mutations have been catalogued as responsible for the destabilization of the electrostatic loop (EL).3 EL, in addition to fulfilling a catalytic role during the dismutation of O2·–, has the function of “solvent seal,” isolating the Zn(II) binding site of hSOD1 from water molecules. Mera-Adasme et al. have shown through computational technique that 5 of these mutants affect the interaction of the EL with the hSOD1 structure, propitiating the opening of the EL and the hydration of the Zn binding site.17 Additionally, Souza et al. showed that some of these mutations generates a destabilization of the Zn binding site by opening the EL, which decreases the affinity of hSOD1 for Zn(II).18 In this work, we present a detailed computational study on using azobenzene-based photoswitch (ABP) to modulate the structural cytotoxic conformation of apo-hSOD1S–S. Specifically, we explore the feasibility of use one ortho-thio-ABP (ortho-TABP) covalently coupled at apo-hSOD1S–S structure to generate an allosteric structural modulation of the EL opening/closing process. Based on the premise that the conditions of the extracellular environment in which apo-hSOD1S–S is released are generally oxidizing, and that this redox condition can be modified by physiologically relevant extracellular levels of glutathione (GSH),19,20 in the first part of this work, we focus on characterizing by using quantum calculations based on density functional theory (DFT) and, time-dependent density functional theory (TD-DFT) and free energy calculations, the electronic and mechanical effect of different ortho-S-alkyl substituents on the photochromic and thermodynamic ABP properties. The ortho-S-alkyl substituents were selected because this type of substitution in ABP has improve the resistance of ABPs to the reduction by GSH.21 Ortho-TABPs was compared against their oxygenated analogues (ortho-OABPs) due to the similar chemistry between O and S atoms and that ortho-oxy-alkyl substituents are one of the most widely used modifications in the field of photopharmacology. Subsequently, we select and covalently couple one ortho-TABP to the apo-hSOD1S–S and evaluate the ability of the ortho-TABP to modulate allosterically the positioning of hSOD1 EL, thereby altering the chelating capacity of the enzyme in the apo state. Employing atomistic molecular dynamics simulations and free energy calculations, we investigate the possibility of photocontrolling the EL opening/closing process. By using ABF method, we characterise the free energy profiles of the mechanical isomerization process for one ortho-TABP covalently couple to hSOD1 structure. Taken together, all the computational tools used in this work allowed us to determine in silico that the bulky ortho-S-alkyl substituents in ABP effectively (i) enhance redox stability of the azo group, (ii) allow to separate the position of the n → π* transition in the trans and cis conformations, and (iii) one ortho-TABP covalently coupled to the hSOD1 structure in cis conformation, and its subsequent mechanical isomerization to its trans conformation allows controlling allosterically the opening/closing process of the EL. The computational approaches employed in this work allow us to consider the potential experimental use of ortho-TABPs for targeting apo-hSOD1S–S, aiming to abolish or reduce the cytotoxic effects and prion-like propagation process of hSOD1 with spatiotemporal precision while minimizing unwanted side effects on healthy cells.
Results and discussion
To understand the electronic and geometrical influences of different ortho-S-alkyl and ortho-O-alkyl substitutions on the ABP, we performed quantum calculations to investigate how the variation in the size of the substituents affect the photophysical and redox properties of ABPs. Our primary focus was on the variation of the absorption properties and whether modifying ABPs in the ortho position with these substitutions leads to the modification in the separation of the π → π* and n → π* bands between the trans and cis conformations. Additionally, we evaluate whether the variations in the size of the substituents influence the redox stability of the ABPs (Chart 1). | ||
| Chart 1 Ortho-TABPs and its corresponding hydrazobenzenes. The molecules represent the trans–cis photoisomerization process of the ABPs with ortho-S-alkyl substituents (ortho-TABPs) and their corresponding hydrazobenzenes analogues, respectively. Ortho-ABP substituted with SCHCH3CH3 and SCH3 are abbreviated throughout this work as ortho-TIABP and ortho-TMABP, respectively. For the electronic and structural comparison of these molecules, their oxygenated analogues (ortho-OIABP and ortho-OMABP, respectively) were used, in which the sulphur atom of the original molecules was replaced by an oxygen atom. Hereafter, 1A and 1B represent ortho-TIABP in its trans conformation and its corresponding hydrazobenzene, respectively; 1C and 1D represent ortho-TIABP in its cis conformation and its corresponding hydrazobenzene, respectively; 2A and 2B represent ortho-TMABP in its trans conformation and its corresponding hydrazobenzene, respectively; 2C and 2D represent ortho-TIABP in its cis conformation and its corresponding hydrazobenzene, respectively. 3A and 3B represent ortho-OIABP in its trans conformation and its corresponding hydrazobenzene, respectively; 3C and 3D represent ortho-OIABP in its cis conformation and its corresponding hydrazobenzene, respectively; 4A and 4B represent ortho-OMABP in its trans conformation and its corresponding hydrazobenzene, respectively; 4C and 4D represent ortho-TIABP in its cis conformation and its corresponding hydrazobenzene, respectively. | ||
The functional employed for geometric optimization was selected considering the energy calculated as a parameter. The studies were conducted using different hybrid functionals, with B3LYP being the selected functional (Table S1†). B3LYP was the functional that provided the lowest optimization energy for ortho-S-alkyl azobenzene systems. Subsequently, a functional calibration was performed to select a hybrid functional for the calculations of electronic absorption spectra by TD-DFT (Table S2†). Initially, we determined the n → π* and π → π* absorption bands and molecular orbitals from optimized trans and cis conformations of ortho-TIABP and ortho-TMABP (Chart 1, Table 1, Fig. 1). The Table 1 show the vertical excitation energies related to excitation from HOMO → LUMO (n → π*) and from HOMO−1 → LUMO (π → π*) for the molecules in trans and cis conformations. In relation to the π → π* transition, the theoretical characterization of the ortho-TABP derivatives establish that the substitution of ABPs with ortho-S-isopropyl or ortho-S-methyl substituents does not generate a significant variation in the absorption maxima in the trans conformations (5 nm). For a cis conformation, substitution with an ortho-S-isopropyl substituent generates a large hypsochromic shift of 36 nm in comparison with the ortho-S-methyl substituent. In the case of n → π* transition, the phenomenon is similar. The substitution with ortho-S-isopropyl does not result in a significant variation compared to the use of ortho-S-methyl in the trans conformation (4 nm). However, it does induce a hypsochromic shift of 36 nm relative to the ortho-S-methyl substituent. The results indicate that the use of bulky ortho-S-alkyl substituents would help in the separation of the n → π* bands between the trans and cis conformations, theoretically enabling more efficient photoswitching. The aforementioned is due to that, in addition to irradiating the π–π* band to generate the cis conformer, it would also be possible to irradiate the n → π* band, increasing in this way also the production of the cis conformer.21–23 These results are important in relation to maximizing the photoisomerization process for use in biological systems. TABPs derivatives show that their substitution in the four ortho position allows the photoisomerization process from trans to cis using blue and green light. When comparing ortho-TABPs and ortho-OABPs (Tables 1 and S3†), it becomes apparent that the phenomenon of photoisomerization from trans to cis with two different light is exclusive to ABPs with ortho-S-alkyl substituents. Ortho-OABPs undergoes trans to cis isomerization only with green light.24 Our results show that the use of bulky ortho-S-alkyl substituents would potentiate the trans to cis photoisomerization phenomenon using both blue and green light. The characterization of these new ortho-TABP were aligned with the theoretical-experimental study conducted by Samanta et al.21
| Isomer | Excited state | Transition (CI) | λ (nm)/E (eV) | f | |
|---|---|---|---|---|---|
| 1A | E | n → π* | H → L (0.63) | 529/2.36 | 0.269 |
| π → π* | H−1 → L (0.70) | 444/2.79 | 0.047 | ||
| 1B | E | n → π* | H → L (0.55) | 360/3.44 | 0.456 |
| π → π* | H−1 → L (0.57) | 317/3.91 | 0.172 | ||
| 1C | Z | n → π* | H → L (0.68) | 497/2.49 | 0.128 |
| π → π* | H−1 → L (0.69) | 447/2.78 | 0.027 | ||
| 1D | Z | n → π* | H → L (0.67) | 376/3.29 | 0.116 |
| π → π* | H−1 → L (0.67) | 329/3.75 | 0.085 | ||
| 2A | E | n → π* | H → L (0.64) | 533/2.32 | 0.350 |
| π → π* | H−1 → L (0.70) | 449/2.76 | 0.030 | ||
| 2B | E | n → π* | H → L (0.68) | 356/3.48 | 0.574 |
| π → π* | H−1 → L (0.67) | 317/3.91 | 0.001 | ||
| 2C | Z | n → π* | H → L (0.69) | 533/2.32 | 0.144 |
| π → π* | H−1 → L (0.70) | 483/2.57 | 0.032 | ||
| 2D | Z | n → π* | H → L (0.69) | 380/3.26 | 0.095 |
| π → π* | H−1 → L (0.47) | 333/3.72 | 0.015 |
 | ||
| Fig. 1 Molecular orbital surface for LUMO (L), HOMO (H) and HOMO−1(H−1) of ortho-S-alkyl azobenzenes (ortho-TABPs) and its corresponding hydrazobenzenes calculated with B3LYP/6-31G(d,p). Solvent was simulated with C-PCM model. 1A and 1B represent ortho-TIABP and their corresponding hydrazobenzenes in the trans conformation, while 1C and 1D represent ortho-TIABP and their corresponding hydrazobenzenes in the cis conformation. 2A and 2B represent ortho-TMABP and their corresponding hydrazobenzenes in the trans conformation, while 2C and 2D represent ortho-TMABP and their corresponding hydrazobenzenes in the cis conformation. Representations were made using VMD. | ||
The results presented by Samanta et al. show that ortho-S-alkyl ABPs, in addition to exhibiting a greater redshift in the H → L transition, also has increased electronic stability against azo group reduction. However, Samanta et al. do not specify which conformation of the ABPs with ortho-S-alkyl substitutions is most susceptible to the reduction process or whether a modification in the bulkiness of the S-alkyl substituent would affect the redox process. To address these questions, we carried out the electronic characterization of the LUMO, HOMO, and HOMO−1 orbitals in ortho-TIABP, ortho-TMABP, ortho-OIABP, and ortho-OMABP in both trans and cis conformations, as well as their respective hydrazobenzenes. To TABPs, the electron density in all trans molecules primarily resides in the azo motif (Fig. 1). Upon reduction, the electron density shifts towards the chlorine atoms when the molecules are reduced (Fig. 1). In the case of the cis conformation, for the HOMO and HOMO−1 molecular orbitals, electron density is similarly distributed, mainly centred on the azo motif. For the LUMO orbital, electron density is spread throughout the molecule. Comparing the electron density of the HOMO, HOMO−1, and LUMO orbitals of the ortho-TABPs in relation to the ortho-OABPs (Fig. 1 and S1†) shows that the largest displacement of electron density from the aromatic rings towards the ends of the molecule would be the reason for the reduced propensity to reduce of the ortho-TABPs. The electronic comparison of the energy levels of LUMO, HOMO, and HOMO−1 orbitals of the ortho-TABPs in trans and cis conformation and their corresponding hydrazobenzenes are presented in the Fig. 2A and B. In addition, to quantitatively assess the higher electron hardness of ortho-TABPs compared to ortho-OABPs, we calculated the energy difference between the HOMO–LUMO gap between all ortho-S-alkyl and ortho-O-alkyl azobenzenes under study and their respective hydrazobenzenes. To evaluate the influence of the trans and cis conformations on the electron hardness of ortho-TABPs and ortho-OABPs, we calculated the ΔH–L. In this work, we defined ΔH–L as the energy difference between the HOMO–LUMO gap of the reduced species minus the HOMO–LUMO gap of the oxidized species. This estimation allows us to straightforwardly determine which conformations of ortho-TABPs and ortho-OABPs are more susceptible to reduction.
 | ||
| Fig. 2 Frontier molecular orbital energy levels of ortho-TIABP, ortho-TMABP, and their respective hydrazobenzenes in trans and cis conformations. (A) 1A and 1B represent ortho-TIABP and their corresponding hydrazobenzenes in the trans conformation, while 1C and 1D represent ortho-TIABP and their corresponding hydrazobenzenes in the cis conformation. (B) 2A and 2B represent ortho-TMABP and their corresponding hydrazobenzenes in the trans conformation, while 2C and 2D represent ortho-TMABP and their corresponding hydrazobenzenes in the cis conformation. The light blue, black, and blue lines represent the LUMO, HOMO, and HOMO−1, respectively. (C) ΔH–L gap between ortho-TABP derivatives and their respective hydrazobenzenes in trans (red bars) and cis conformations (blue bars). (D) ΔH–L gap between ortho-OABP derivatives and their respective hydrazobenzenes in trans (red bars) and cis conformations (blue bars). | ||
Fig. 2C and D illustrate that in the ground state, all evaluated trans conformations (ortho-TABPs, ortho-OABPs, and their corresponding hydrazobenzenes) exhibit significant differences in the determined ΔH–L values. This finding suggests that neither the trans conformation itself nor the bulkiness of the S-alkyl or O-alkyl substituents significantly influences the propensity for azo group reduction in these ABPs. Conversely, the cis conformations of both ortho-TABPs and ortho-OABPs demonstrated an increased propensity to reduction compared to their trans counterparts. Notably, among the cis conformations, ortho-TABPs exhibit greater electron hardness towards reduction than ortho-OABPs (Fig. 2C and D). The higher ΔH–L values calculated for ortho-TABPs in comparison to ortho-OABPs quantitatively confirm the lower susceptibility to reduction of the former. Our theoretical results indicate that the use of ortho-S-alkyl substitutions in ABPs would enhance stability against reduction of the azo group reduction under physiological conditions, and the bulkiness of these substituents does not significantly impact the electron hardness of either TABPs or ortho-OABPs. Having established that the cis conformation is responsible for the enhanced electron hardness and reduced susceptibility to reduction of ortho-S-alkyl ABPs, and that the size of the S-alkyl substituent does not drastically alter this effect, we next sought to characterize the thermodynamics properties of isomerization of this class of ABPs. To this end, we employed molecular dynamics simulations and free energy calculations.
Specifically, we aimed to elucidate the impact of different S-alkyl substitutions on the conformational stability of various ortho-TABPs and compare these findings to those obtained for ortho-OABP derivatives. While acknowledging the potential for these ABPs to undergo isomerization through multiple mechanisms, our primary focus was to directly and reliably determine the thermodynamic energy of the cis conformations and assess whether this energy, along with the barrier for trans–cis isomerization, is influenced by the nature of the ortho-alkyl substituent. For this purpose, we employed the Adaptive Biasing Force (ABF) method to calculate the free energy profiles for the mechanical isomerization of substituted ortho-TABPs, ortho-MABPs, and the unsubstituted ABP in an aqueous environment. This approach allowed us to determine the free energy of each cis conformations, as well as the energy barrier separating the trans and cis conformations (Fig. 3). Fig. 3A presents the free energy profiles obtained for the mechanical isomerization of different ABPs evaluated in water. Two key observations emerge from these profiles. First, the energy impact of the ortho-S-isopropyl and ortho-O-isopropyl substituents on the cis conformation is substantially smaller than that observed for the cis conformations of ABPs with ortho-S-methyl and ortho-O-methyl substituents (Fig. 3B). Second, the energy difference between the cis conformation of ABPs with ortho-S-isopropyl and ortho-S-methyl substitutions is negligible (0.74 kcal mol−1) compared to the difference obtained for ABPs with ortho-O-isopropyl and ortho-O-methyl substitutions (2.04 kcal mol−1) (Fig. 3C). Importantly, the free energy results related to the energy barrier of the mechanical isomerization process for ortho-TABPs and ortho-OABPs are consistent with experimental data, which indicate that the faster back isomerization rate for ortho-TABP (minutes) compared to ortho-OABP (days).21,24 Taken together, our quantum and thermodynamic calculations suggest ortho-S-alkyl substitutions effectively enhance the protection of the azo group against reduction. Although our analysis revealed no substantial energetic differences in the isomerization process among the evaluated ortho-S-alkyl substitutions, employing ABPs with bulkier ortho-S-alkyl groups could present a viable alternative to ortho-OABPs. In the context of this study, we are particularly interested in the application of ortho-TIABP within physiological environments. Previous research has shown that the reduction of the azo group under physiological conditions can be catalyzed by the binding of one or two GSH molecules to the ABP's azo group.25 Ortho-TABPs have been reported to exhibit greater stability compared to ortho-OABPs due to the absence of hydrogen bonding.26 Moreover, the increased steric bulk of the ortho-S-isopropyl substituent, relative to ortho-S-methyl or -ethyl substitutions, would likely hinder the interaction of GSH molecules with ortho-TABPs. This steric hindrance could extend the molecule's functional lifespan and enhance its redox stability compared to ABPs possessing ortho-O-alkyl substitutions.
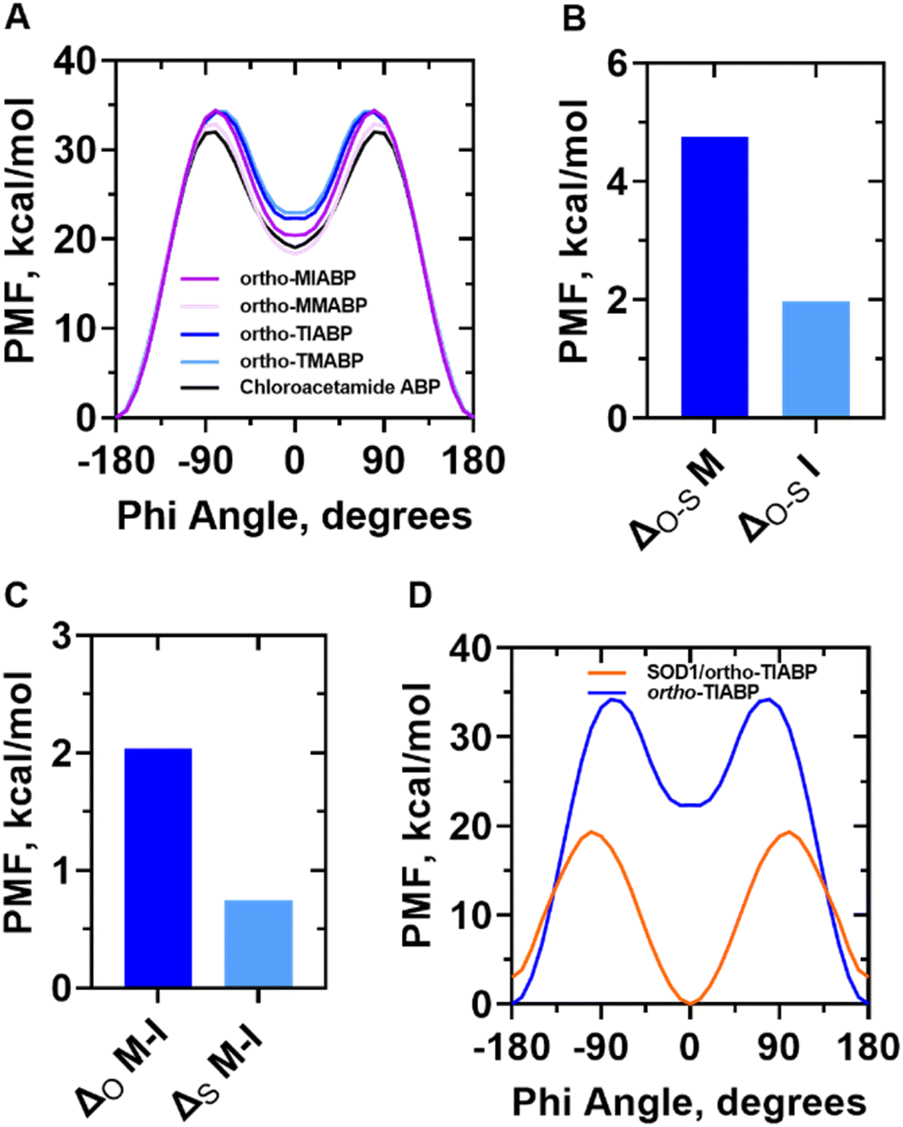 | ||
| Fig. 3 Free energy calculations for the isomerization of ABPs with different types of ortho substituents in water and covalently linked to SOD1. (A) Free-energy (PMF) profiles of the trans–cis isomerization process of ortho-TABPs, ortho-OABPs, and non-substituted ABP at room temperature in water. (B) Difference in free energy between the cis conformations for ortho-TABP and ortho-OABP with different alkyl substitutions. (C) Difference in free energy between the cis conformations for the same type of ABP but with different alkyl substitutions. ‘M’ and ‘I’ represent methyl and isopropyl substitutions, respectively. (D) Free energy (PMF) calculations of the trans–cis isomerization process of one ortho-TIABP in water (blue line) and tethered to the SOD1 dimer in water both at room temperature. | ||
Recognizing that physiologically relevant concentrations of GSH are continuously released into the extracellular milieu, even in the oxidizing environment associated with ALS development, as part of GSH-mediated redox homeostasis, we posit that utilizing substituents capable of preventing or delaying GSH-mediated azo group reduction is crucial, particularly when considering the use of TABP for photomodulation of extracellular processes. Therefore, we selected TIABP to investigate the feasibility of photocontrolling the structural conformation of hSOD1's EL to influence the chelating capacity of apo-hSOD1S–S. To achieve this, we tethered TIABP in the cis conformation to the free cysteine residues (C111) of each monomer within the apo-hSOD1S–S structure. In hSOD1, the C111 residues are in their reduced state, permitting covalent attachment of TIABP exclusively in the cis conformation. The C111 residues have been extensively employed as anchoring sites for molecules in experimental studies investigating hSOD1 structural dynamics.27,28 Each C111 residue is directly connected to the active site of its respective hSOD1 monomer. Because previous reports have indicated that covalent modification of C111 can perturb the position of hSOD1′s EL, we hypothesize that covalently bridging both C111 residues with a single TIABP molecule, followed by its subsequent cis to trans isomerization could enable the allosteric modulation of the EL opening and closing process. Fig. 3D illustrates the free energy profiles for the isomerization of ortho-TIABP both when covalently linked to the apo-hSOD1S–S structure and when free in solution. Comparing the energy profiles obtained for the mechanical isomerization process in both systems allows us to assess how the stability of the ABP's cis and trans configurations changes upon conjugation to apo-hSOD1S–S. Notably, the cis configuration is more stable for TIABP when tethered to apo-SOD1S–S, whereas the trans configuration is more stable for free TIABP in water. This shift in the relative stability of the two configurations aligns with experimental observations, which have demonstrated that the most stable configuration of an ABP tethered to a peptide or protein is dictated by the distance between the cysteine residues used for anchoring.29,30 In our case, the cysteine residues chosen to anchor TIABP to apo-SOD1S–S only permit binding in the cis configuration. While our results indicate that the tethered TIABP in the cis configuration (red line, Fig. 3D) can transition to the trans configuration, this transition requires more energy compared to free TIABP in water (blue line, Fig. 3D). The lower energy barrier for cis-to-trans isomerization of ortho-TIABP in water compared to when tethered to hSOD1S–S suggests that, in a biological context, fewer light cycles could be required to maintain a higher population of the cis isomer, extending the duration of EL modulation. The thermodynamic stability of the cis conformation allows for fewer light cycles to be used and extend the effect on the mechanical modulation of the electrostatic loop.
To investigate whether the cis–trans isomerization of TIABP covalently attached to the apo-hSOD1S–S structure could allosterically influence the enzyme's conformational dynamics, we performed atomistic molecular dynamics simulations. Three distinct states were considered: apo-hSOD1S–S, apo-hSOD1S–Scis (representing apo-hSOD1S–S modified with TIABP in the cis conformation), and apo-hSOD1S–Strans (representing apo-hSOD1S–S modified with TIABP in the trans conformation). Our analysis focused on one specific contact point between EL and the apo-SOD1S–S structure: the interaction between the sidechain of N86 and the backbone of D124. This interaction has been implicated as a critical determinant of EL positioning within hSOD1 structure and in the insertion of Zn(II) into the metal-binding site.17,31
Fig. 4A shows a comparative analysis of the N86-D124 interaction distance in unmodified apo-hSOD1S–S, apo-hSOD1S–Scis, and the apo-hSOD1S–Strans. Notably, the distance comparison between the unmodified apo-SOD1S–S and the one modified with TIABP in the cis conformation reveals that covalent tethering of TIABP in the cis conformation does not significantly alter the EL positioning (Fig. 4A and C). However, upon TIABP isomerization to the trans conformation, a distinct disturbance in the N86-D124 contact point becomes evident (Fig. 4A, B and D). This disturbance leads to a substantial increase in both EL fluctuation and the N86-D124 interaction distance. Molecular dynamics simulations of the apo-hSOD1S–Strans system show no variation of the contact point for the first 250 ns. Subsequently, the EL undergoes repetitive and consistent cycles of opening and closing (Fig. 4B), with our simulations revealing a total of 10 transitions and an average cycle time of 7.94 ns. The events highlighted in the Fig. 4B represent those identified after filtering for pseudo-events that deviated by more than two standard deviations from the average N86-D124 contact distance between 250 ns and 500 ns.
 | ||
| Fig. 4 Molecular dynamics simulations of apo-hSOD1S–S, apo-hSOD1S–Scis, and apo-hSOD1S–Strans. (A) Distances for the interaction between the mass centres of N86 and D124 along the trajectory. (B) Opening-closing events for the last 250 ns of trajectory (250–500 ns) for apo-hSOD1S–S, apo-hSOD1S–Scis, and apo-hSOD1S–Strans. Each event is symbolized by an asterisk and represents an opening–closing cycle of the EL. (C) Structural superposition of apo-hSOD1S–S (green) and apo-hSOD1S–Scis (blue). The dashed line box shows the position of the EL in both models. (D) Structural superposition of apo-hSOD1S–Scis (blue) and apo-hSOD1S–Strans (red). The dashed line box shows the position of the EL in both models. (E) Spatial arrangement of the residues involved in the stabilizing interaction of the EL (green: apo-hSOD1S–S; blue: apo-hSOD1S–Scis). (F) Spatial arrangement of the residues involved in the stabilizing interaction of the EL (blue: apo-hSOD1S–Scis; red: apo-hSOD1S–Strans). | ||
The increased EL fluctuation arising from the disruption of the stabilizing contact between the EL and hSOD1 directly impacts the orientation and geometry of residues involved in the Zn coordination at the binding site (Fig. S2†). The EL displacement promotes the reorganization of the Zn binding site residues and impairs the N86-D124 interaction (Fig. 4E, F, and S2†). Importantly, our results show that the structural transition of TIABP exert a localized effect on the EL region without perturbing the overall structure of apo-SOD1S–S (Fig. 4C and D) or the monomer–monomer interface (Fig. S3†).
The findings presented in Fig. 4 underscore the profound impact of TIABP isomerization on the conformational dynamics of critical EL-stabilizing residues, consequently affecting the geometry of the Zn binding site. Our computational study suggests that TIABP could serve as a valuable tool for the experimental photomodulation apo-SOD1S–S species' cytotoxic effects by inhibiting its chelating capacity. Given the thermodynamic stability of the trans conformation of TIABP covalently linked to hSOD1S–S, this inhibitory effect is expected to be sufficiently persistent to maintain the Zn binding site in a disorganized yet non-aggregating state within apo-hSOD1S–S.
It has been shown that impaired Zn homeostasis is implicated in ALS pathogenesis.32,33 Homma et al. found that, under Zn-deficient conditions, apo-SOD1, by interacting with the ER, can induce the expression of a Zn transporter to normalize intracellular Zn levels.14 Our hypothesis states that the mechanical modulation of the EL by using ortho-TIABP tethered to apo-SOD1S–S could contribute to increasing intracellular Zn levels under Zn-deficient conditions.
Conclusions
In this study, we conducted a systematic assessment of the electronic and mechanical properties of ortho-S-alkyl substituted azobenzenes, focusing on the impact of thio substituent size on azo group reduction and their potential for photocontrolling the cytotoxic effects associated with the chelating capacity of the metal-free and disulfide-oxidized apo-hSOD1S–S. Regarding the allow to separate the position of the n → π* transition in the trans and cis conformations, this separation could enhance the efficiency of photoactivation in either the cis or trans state. By tuning the irradiation wavelength to selectively target the mentioned transition, one could potentially achieve more efficient cis isomer production with minimal off-target effects. This increased efficiency is crucial for applications in biological systems, where minimizing light cycles and maximizing the desired effect are paramount.Besides, our findings reveal that bulky thio substituents exhibit enhanced resistance to azo group reduction compared to smaller substituents, with the cis conformation demonstrating increased electronic stability.
Examining the thermodynamic effects of ortho-S-alkyl substituents in ABPs, our results suggest that thio-substituted ABPs could serve as viable alternative to alkoxy-substituted ABPs in scenarios where rapid cis–trans relaxation is crucial for photocontrolling biological processes within reductive environments.
This comprehensive computational characterization of ortho-TABPs establishes the feasibility of employing ABPs with bulky S-alkyl substituents for the photocontrol of biological processes. The ability to modulate trans-to-cis isomerization using both blue and green light imparts these molecules with a bifunctional character, further broadening their potential applications.
Experimental section
Quantum mechanical calculations for ABPs
All quantum mechanical calculations were performed using Density Functional Theory (DFT) with the Gaussian09 package.34 Geometric optimization for the evaluated molecules was carried out using DFT, employing the B3LYP hybrid density functional35,36 and the 6-311++G(d,p) basis set.37,38 The converged wave function were verified by analytical computation of harmonic vibrational frequencies. Subsequently, electronic absorption spectra calculations were performed using TD-DFT by singlet–singlet vertical transitions Franck–Condon type using 20 excited states, through the B3LYP/6-311++G(d,p) functional as the calculation level. Spectra were calculated in solution phase (DMSO). The modelling of the solvent was performed using conductor-like polarizable continuum model (C-PCM) with standard parameter for DMSO.39ABPs molecular models
All ABP structures were modelled using MolView web application and parametrized with ParamChem server (CHARMM General Force Field version 3.0.1).40 All the systems were solvated with the standard TIP3P water model of CHARMM36 force field41 in a periodic box of approximately 35 × 35 × 35 Å3, according to ABPs size, using VMD 1.9.4a version.42Molecular dynamics parameters
The molecular dynamics simulations were performed in the molecular dynamics software NAMD 2.14,43 including the CHARMM36 force field.40 By the Langevin thermostat and Langevin Piston method, the temperature and pressure were maintained at 300 K and 101.325 kPa (1.0 atm) respectively, and the particle-mesh Ewald algorithm was used to compute the electrostatic interactions with a grid spacing of <1.2 Å.44,45 A smooth 8–9 Å cutoff of van der Waals forces was employed.Free energy calculations
All systems shown in this work followed an independent molecular dynamics protocol that considered a minimization of 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps, followed by an unrestricted equilibration of at least 2 ns. Starting from the equilibration configuration, the adaptive biasing force method (ABF) was applied to estimate the free energy landscape associated with the rotation between ABPs cis to trans conformations in water and covalently coupled to hSOD1. The single transition coordinate used to estimate the Potential Mean Force (PMF), was the dihedral torsional angle formed by: C–N–N–C atoms in azo molecules and was implemented through the Colvars module of NAMD,46 exploring the full range of rotation (from −180° to 180° of dihedral angle) in a single-window with a bin width for ABF samples of 10.0° in 1 us of simulation with a 4 fs time step scheme.
000 steps, followed by an unrestricted equilibration of at least 2 ns. Starting from the equilibration configuration, the adaptive biasing force method (ABF) was applied to estimate the free energy landscape associated with the rotation between ABPs cis to trans conformations in water and covalently coupled to hSOD1. The single transition coordinate used to estimate the Potential Mean Force (PMF), was the dihedral torsional angle formed by: C–N–N–C atoms in azo molecules and was implemented through the Colvars module of NAMD,46 exploring the full range of rotation (from −180° to 180° of dihedral angle) in a single-window with a bin width for ABF samples of 10.0° in 1 us of simulation with a 4 fs time step scheme.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank to Fondecyt grant no. 1240759 (R. Z.), Fondecyt grant no. 1231468 (A. A.) and Fondecyt grant no. 1211143 (H. P.), and the Doctoral Fellowship No. 21200778 (C. G.-A.), and No. 21241753 (F. S.-S.), of the Agencia Nacional de Investigación y Desarrollo of Chile of Chile (ANID). Universidad de Santiago de Chile, Usach, Proyecto Código 021841AL_POSTDOC, and 021841AL, Dirección Científica y Tecnológica, Dicyt (R. Z.).References
- E. C. A. Eleutherio, R. S. Silva Magalhães, A. De Araújo Brasil, J. R. Monteiro Neto and L. De Holanda Paranhos, Arch. Biochem. Biophys., 2021, 697, 108701, DOI:10.1016/j.abb.2020.108701.
- M. Berdyński, P. Miszta, K. Safranow, P. M. Andersen, M. Morita, S. Filipek, C. Żekanowski and M. Kuźma-Kozakiewicz, Sci. Rep., 2022, 12, 103, DOI:10.1038/s41598-021-03891-8.
- K. S. Molnar, N. M. Karabacak, J. L. Johnson, Q. Wang, A. Tiwari, L. J. Hayward, S. J. Coales, Y. Hamuro and J. N. Agar, J. Biol. Chem., 2009, 284, 30965–30973, DOI:10.1074/jbc.M109.023945.
- J. S. Valentine and P. J. Hart, Proc. Natl. Acad. Sci. U. S. A., 2003, 100, 3617–3622, DOI:10.1073/pnas.0730423100.
- M. A. Hough, J. G. Grossmann, S. V. Antonyuk, R. W. Strange, P. A. Doucette, J. A. Rodriguez, L. J. Whitson, P. J. Hart, L. J. Hayward, J. S. Valentine and S. S. Hasnain, Proc. Natl. Acad. Sci. U. S. A., 2004, 101, 5976–5981, DOI:10.1073/pnas.0305143101.
- H. R. Broom, J. A. O. Rumfeldt, K. A. Vassall and E. M. Meiering, Protein Sci., 2015, 24, 2081–2089, DOI:10.1002/pro.2803.
- R. Byström, P. M. Andersen, G. Gröbner and M. Oliveberg, J. Biol. Chem., 2010, 285, 19544–19552, DOI:10.1074/jbc.M109.086074.
- I. Sirangelo and C. Iannuzzi, Molecules, 2017, 22, 1429, DOI:10.3390/molecules22091429.
- C. Takashima, Y. Kosuge, M. Inoue, S.-I. Ono and E. Tokuda, Int. J. Mol. Sci., 2021, 22, 4155, DOI:10.3390/ijms22084155.
- C. Münch, J. O'Brien and A. Bertolotti, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 3548–3553, DOI:10.1073/pnas.1017275108.
- L. I. Grad, W. C. Guest, A. Yanai, E. Pokrishevsky, M. A. O'Neill, E. Gibbs, V. Semenchenko, M. Yousefi, D. S. Wishart, S. S. Plotkin and N. R. Cashman, Proc. Natl. Acad. Sci. U. S. A., 2011, 108, 16398–16403, DOI:10.1073/pnas.1102645108.
- L. I. Grad, J. J. Yerbury, B. J. Turner, W. C. Guest, E. Pokrishevsky, M. A. O'Neill, A. Yanai, J. M. Silverman, R. Zeineddine, L. Corcoran, J. R. Kumita, L. M. Luheshi, M. Yousefi, B. M. Coleman, A. F. Hill, S. S. Plotkin, I. R. Mackenzie and N. R. Cashman, Proc. Natl. Acad. Sci. U. S. A., 2014, 111, 3620–3625, DOI:10.1073/pnas.1312245111.
- A.-S. Johansson, M. Vestling, P. Zetterström, L. Lang, L. Leinartaitė, M. Karlström, J. Danielsson, S. L. Marklund and M. Oliveberg, PLoS One, 2012, 7, e36104, DOI:10.1371/journal.pone.0036104.
- K. Homma, T. Fujisawa, N. Tsuburaya, N. Yamaguchi, H. Kadowaki, K. Takeda, H. Nishitoh, A. Matsuzawa, I. Naguro and H. Ichijo, Mol. Cell, 2013, 52, 75–86, DOI:10.1016/j.molcel.2013.08.038.
- M. Tajiri, H. Aoki, A. Shintani, K. Sue, S. Akashi and Y. Furukawa, Free Radical Biol. Med., 2022, 183, 60–68, DOI:10.1016/j.freeradbiomed.2022.03.014.
- Y. Furukawa, A. S. Torres and T. V. O'Halloran, EMBO J., 2004, 23, 2872–2881, DOI:10.1038/sj.emboj.7600276.
- R. Mera-Adasme, H. Erdmann, T. Bereźniak and C. Ochsenfeld, Metallomics, 2016, 8, 1141–1150, 10.1039/C6MT00085A.
- P. C. T. Souza, S. Thallmair, S. J. Marrink and R. Mera-Adasme, J. Phys. Chem. Lett., 2019, 10, 7740–7744, DOI:10.1021/acs.jpclett.9b02868.
- G. McBean, Antioxidants, 2017, 6, 62, DOI:10.3390/antiox6030062.
- F. Li, S. Li, Y. Shi, F. Lin, L. Rui, J. Shi and K. Sun, Mediators Inflammation, 2024, 2024, 4482642, DOI:10.1155/2024/4482642.
- S. Samanta, T. M. McCormick, S. K. Schmidt, D. S. Seferos and G. A. Woolley, Chem. Commun., 2013, 49, 10314, 10.1039/c3cc46045b.
- D. B. Konrad, G. Savasci, L. Allmendinger, D. Trauner, C. Ochsenfeld and A. M. Ali, J. Am. Chem. Soc., 2020, 142, 6538–6547, DOI:10.1021/jacs.9b10430.
- M. Gao, D. Kwaria, Y. Norikane and Y. Yue, Nat. Sci., 2023, 3, e220020, DOI:10.1002/ntls.20220020.
- A. A. Beharry, O. Sadovski and G. A. Woolley, J. Am. Chem. Soc., 2011, 133, 19684–19687, DOI:10.1021/ja209239m.
- J. E. Sheldon, M. M. Dcona, C. E. Lyons, J. C. Hackett and M. C. T. Hartman, Org. Biomol. Chem., 2016, 14, 40–49, 10.1039/C5OB02005K.
- L. Stricker, M. Böckmann, T. M. Kirse, N. L. Doltsinis and B. J. Ravoo, Chem.–Eur. J., 2018, 24, 8639–8647, DOI:10.1002/chem.201800587.
- C. Solsona, T. B. Kahn, C. L. Badilla, C. Álvarez-Zaldiernas, J. Blasi, J. M. Fernandez and J. Alegre-Cebollada, J. Biol. Chem., 2014, 289, 26722–26732, DOI:10.1074/jbc.M114.565333.
- J. R. Auclair, K. J. Boggio, G. A. Petsko, D. Ringe and J. N. Agar, Proc. Natl. Acad. Sci. U. S. A., 2010, 107, 21394–21399, DOI:10.1073/pnas.1015463107.
- R. J. Mart and R. K. Allemann, Chem. Commun., 2016, 52, 12262–12277, 10.1039/C6CC04004G.
- S. Samanta and G. A. Woolley, ChemBioChem, 2011, 12, 1712–1723, DOI:10.1002/cbic.201100204.
- R. Mera-Adasme, C.-M. Suomivuori, A. Fierro, J. Pesonen and D. Sundholm, JBIC, J. Biol. Inorg. Chem., 2013, 18, 931–938, DOI:10.1007/s00775-013-1039-8.
- R. Sirabella, V. Valsecchi, S. Anzilotti, O. Cuomo, A. Vinciguerra, P. Cepparulo, P. Brancaccio, N. Guida, N. Blondeau, L. M. T. Canzoniero, C. Franco, S. Amoroso, L. Annunziato and G. Pignataro, Front. Neurosci., 2018, 12, 510, DOI:10.3389/fnins.2018.00510.
- W. I. M. Vonk and L. W. J. Klomp, Biochem. Soc. Trans., 2008, 36, 1322–1328, DOI:10.1042/BST0361322.
- D. J. Frisch, M. J. Trucks, G. W. Schlegel, H. B. Scuseria, G. E. Robb, M. A. Cheeseman, J. R. Scalmani, G. Barone, V. Mennucci, B. Petersson, G. A. Nakatsuji, H. Caricato, M. Li, X. Hratchian, H. P. Izmaylov, A. F. Bloino, J. Zheng and G. Sonnenb. “Gaussian 09, Revision and E.01,” Gaussian 09, Revis. E.01, Gaussian 0. Wallingford CT: Gaussian, Inc.; 2009 Search PubMed.
- A. D. Becke, J. Chem. Phys., 1993, 98, 5648–5652, DOI:10.1063/1.464913.
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B, 1988, 37, 785–789, DOI:10.1103/PhysRevB.37.785.
- G. A. Petersson and M. A. Al-Laham, J. Chem. Phys., 1991, 94, 6081–6090, DOI:10.1063/1.460447.
- G. A. Petersson, A. Bennett, T. G. Tensfeldt, M. A. Al-Laham, W. A. Shirley and J. Mantzaris, J. Chem. Phys., 1988, 89, 2193–2218, DOI:10.1063/1.455064.
- M. Cossi, N. Rega, G. Scalmani and V. Barone, J. Comput. Chem., 2003, 24, 669–681, DOI:10.1002/jcc.10189.
- K. Vanommeslaeghe and A. D. MacKerell, J. Chem. Inf. Model., 2012, 52, 3144–3154, DOI:10.1021/ci300363c.
- J. Huang and A. D. MacKerell, J. Comput. Chem., 2013, 34, 2135–2145, DOI:10.1002/jcc.23354.
- W. Humphrey, A. Dalke and K. Schulten, J. Mol. Graphics, 1996, 14, 33–38, DOI:10.1016/0263-7855(96)00018-5.
- J. C. Phillips, D. J. Hardy, J. D. C. Maia, J. E. Stone, J. V. Ribeiro, R. C. Bernardi, R. Buch, G. Fiorin, J. Hénin, W. Jiang, R. McGreevy, M. C. R. Melo, B. K. Radak, R. D. Skeel, A. Singharoy, Y. Wang, B. Roux, A. Aksimentiev, Z. Luthey-Schulten, L. V. Kalé, K. Schulten, C. Chipot and E. Tajkhorshid, J. Chem. Phys., 2020, 153, 044130, DOI:10.1063/5.0014475.
- S. E. Feller, Y. Zhang, R. W. Pastor and B. R. Brooks, J. Chem. Phys., 1995, 103, 4613–4621, DOI:10.1063/1.470648.
- T. Darden, D. York and L. Pedersen, J. Chem. Phys., 1993, 98, 10089–10092, DOI:10.1063/1.464397.
- G. Fiorin, M. L. Klein and J. Hénin, Mol. Phys., 2013, 111, 3345–3362, DOI:10.1080/00268976.2013.813594.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra08972c |
| ‡ These author contributed equally to this work. |
| This journal is © The Royal Society of Chemistry 2025 |