
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Advances in the synthesis of Fe-based bimetallic electrocatalysts for CO2 reduction
Ayesha Zafar†
a,
Adnan Majeed†
 a,
Abdul Ahad
c,
Muhammad Adnan Iqbal
*ab,
Tanveer Hussain Bokhari
c,
Zanira Mushtaq
a and
Shahzaib Ali
a
a,
Abdul Ahad
c,
Muhammad Adnan Iqbal
*ab,
Tanveer Hussain Bokhari
c,
Zanira Mushtaq
a and
Shahzaib Ali
a
aDepartment of Chemistry, University of Agriculture Faisalabad, Faisalabad-38000, Pakistan. E-mail: adnan.iqbal@uaf.edu.pk; aayeshazafar99@gmail.com; 2021ag2578@uaf.edu.pk; zanira14802@gmail.com; zaibsmd@gmail.com
bOrganometallic and Coordination Chemistry Laboratory, University of Agriculture Faisalabad, Faisalabad-38000, Pakistan
cDepartment of Chemistry, Government College University Faisalabad, Faisalabad-38000, Pakistan. E-mail: aa.ahad9998@gmail.com; tanveer.bokhari@yahoo.com
First published on 18th March 2025
Abstract
Achieving carbon neutrality and slowing down global warming requires research into ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) the electrochemical CO2 reduction reaction (CO2RR), which produces useful compounds. Utilizing renewable energy to meet carbon-neutral energy goals produces single-carbon (C1) and multi-carbon (C2+) goods. Efficient and selective electrocatalysts are essential to advancing this revolutionary technology; bimetallic Fe-based catalysts work better than their monometallic counterparts because multiple metals work synergistically to reduce CO2 levels. A thorough summary of recent developments in the synthesis of Fe–X bimetallic catalysts will be provided in this review, with an emphasis on key performance indicators like stability, faradaic efficiency, potential, current density, and primary product production. In addition, this analysis will look at representative instances of Fe bimetallic catalysts that are well-known for their selectivity in generating particular alcohols and hydrocarbons, clarifying the mechanics behind CO2 reduction, pointing out existing difficulties, and examining the potential of electrosynthesis processes in the future.
the electrochemical CO2 reduction reaction (CO2RR), which produces useful compounds. Utilizing renewable energy to meet carbon-neutral energy goals produces single-carbon (C1) and multi-carbon (C2+) goods. Efficient and selective electrocatalysts are essential to advancing this revolutionary technology; bimetallic Fe-based catalysts work better than their monometallic counterparts because multiple metals work synergistically to reduce CO2 levels. A thorough summary of recent developments in the synthesis of Fe–X bimetallic catalysts will be provided in this review, with an emphasis on key performance indicators like stability, faradaic efficiency, potential, current density, and primary product production. In addition, this analysis will look at representative instances of Fe bimetallic catalysts that are well-known for their selectivity in generating particular alcohols and hydrocarbons, clarifying the mechanics behind CO2 reduction, pointing out existing difficulties, and examining the potential of electrosynthesis processes in the future.
 Ayesha Zafar | Miss Ayesha Zafar was born in Punjab-Pakistan in November 1997. She completed her schooling and college education in the city of Jaranwala-Pakistan and did her Graduation in BS Chemistry at Government Postgraduate College Jaranwala Pakistan in October 2020. She then joined the University of Agriculture Faisalabad in September 2021 for an MPhil in Chemistry in organometallics and coordination chemistry under the supervision of Dr Muhammad Adnan Iqbal Associate Professor University of Agriculture Faisalabad and completed her MPhil degree in August 2023. Her research work during MPhil was on the synthesis of metal complexes and their catalytic applications. |
 Adnan Majeed | Mr Adnan Majeed was born in Punjab, Pakistan, in January 1998. He completed his schooling and college education in Sargodha, Pakistan. He earned his Bachelor's degree in Chemistry from The University of Lahore in August 2021, where he was awarded a Gold Medal for outstanding academic performance. In September 2021, he joined the University of Agriculture Faisalabad to pursue an MPhil in Chemistry, specializing in catalysis, organometallics, and coordination chemistry, under the supervision of Dr Muhammad Adnan Iqbal, Associate Professor at the University of Agriculture Faisalabad. He successfully completed his MPhil degree in August 2023. His research interests include organo-photocatalysis, photocatalysis, photooxidation, wastewater treatment, DFT, RSM analysis, and the synthesis of organometallic compounds and their catalytic applications. |
 Abdul Ahad | Mr Abdul Ahad was born in Punjab-Pakistan on 6 June 1999. He completed his schooling and college education in the city of Jaranwala-Pakistan and did his Graduation in BS Chemistry at Punjab Group of College Jaranwala campus affiliated with Government College and University of Faisalabad Pakistan in October 2023. With hands-on experience as an Assistant Lab Chemist at Tariq Corporation and a Cutting Quality Controller at Sadaqat Limited (2020–2023), he has honed his technical and analytical skills. Passionate about applying his expertise in a dynamic setting, he thrives on challenges and innovation. |
 Muhammad Adnan Iqbal | Dr Muhammad Adan Iqbal was born in Punjab-Pakistan in April 1984. He completed his schooling and college education in the city of Faisalabad-Pakistan and his Bachelor's degree in Chemistry at the University of the Punjab-Lahore-Pakistan in August 2007. He completed his Master's (MPhil) in Environmental Sciences at the College of Earth and Environmental Science, University of the Punjab, Lahore in 2010 and in parallel served as Lecturer of Chemistry at Minhaj University Lahore till July 2010. He then joined Universiti Sains Malaysia, Penang-Malaysia in July 2010 for MS leading to PhD study in Dr Rosenani A. Haque's laboratory on a fellowship. He completed his PhD in Organometallic Chemistry in April 2014 and got an opportunity for a postdoctoral fellowship at the same research laboratory. During his PhD studies, Dr Iqbal visited the University of Western Australia, Perth, Australia on a research attachment at Professor Murray Baker's research Laboratory. He finally joined the University of Agriculture Faisalabad in September 2015 as an Assistant Professor. Currently, he has established an organometallic and coordination chemistry laboratory at UAF community college, University of Agriculture Faisalabad-Pakistan with the help of funding from the Higher Education Commission of Pakistan through one SRGP, two NRPU research grants, PSF and PAS. His research interests include the synthesis of metallodrugs. Dr Iqbal has published more than 150 research and review articles in international journals, a book on organometallic chemistry, and three book chapters. He is the managing editor of a reputable research journal, the Journal of Angiotherapy. He has produced 5 PhD and 57 MPhil degree holders in the field of Chemistry. He has organized several workshops, Seminars, and Symposiums. He has national (LUMS, University of the Punjab, Lahore, GC University Faisalabad, etc.) and international (University of Western Australia, Perth, Universiti Sains Malaysia, Malaysia, St John's University, USA) research collaborations. |
 Tanveer Hussain Bokhari | Prof. Dr Tanveer Hussain Bokhari holds a PhD degree in Chemistry from GC University, Lahore after being awarded the Indigenous Scholarship for PhD by the Higher Education Commission Islamabad. He did his post-doctoral studies at BYU, USA. He has published 160 research papers at international and national levels in well-reputed journals and is the author of two books. Currently, he is serving as a Professor of Chemistry at the Department of Chemistry, Government College University Faisalabad, where he was awarded the Research Productivity Award for the years of 2011 and 2012 by the Pakistan Council for Science and Technology, Islamabad-Pakistan. |
 Zanira Mushtaq | Zanira Mushtaq was born in Punjab-Pakistan in March 2000. She completed her Bachelor's degree in Chemistry from Government College University Faisalabad in August 2021 having outstanding academic performance, and her Master's degree in Chemistry from the University of Agriculture Faisalabad, Pakistan in 2023. Her research interests are Computational chemistry, DFT/TD-DFT, Sensitizers, Dyes, Photovoltaics, Solar cell Applications, Dye-sensitized solar cells, the Role of Pi-spacers, and Next-generation cells. |
 Shahzaib Ali | Mr Shahzaib was born in Punjab-Pakistan in March 1997. He completed his schooling and college education at Faisalabad-Pakistan and his Bachelor's degree in Chemistry at Govt College University Faisalabad, Pakistan in January 2021. He then joined the University of Agriculture Faisalabad in September 2021 for an MPhil in Chemistry in organometallics and coordination chemistry under the supervision of Dr Muhammad Adnan Iqbal Associate Professor at the University of Agriculture Faisalabad. He completed his MPhil degree in August 2023. His research interests are Computational Chemistry, Organic Solar Cells, photooxidation, and wastewater treatment. |
1. Introduction
Over the past 170 years, human activity has caused a sharp rise in CO2 emissions, which has resulted in ocean acidification and global climate change.1,2 Terrestrial ecosystems only absorb around 30% of CO2 produced by humans annually, which is insufficient to offset anthropogenic emissions, and the amount of CO2 in the atmosphere has dramatically increased in recent decades, reaching 400 ppm for the first time in human history. Since the late 1950s, the Mauna Loa Observatory in Hawaii has been continually monitoring atmospheric CO2, and as of 2024, CO2 concentrations are approximately 417 ppm. One of the main causes of climate change, this represents a sharp rise from pre-industrial levels of about 280 ppm.3–5 According to predictions, if present emission trends continue, CO2 levels might surpass 450 ppm by 2030. If immediate mitigation measures are not taken, some models forecast significantly higher concentrations.6 Over 450 ppm could dramatically raise the probability of catastrophic climate consequences, including more severe and frequent heatwaves, rising sea levels, and disturbances to ecosystems and food security.7Although CO2 is a very stable molecule that is usually inert, it can undergo electrochemical activation and be transformed into reduced products through the CO2 reduction reaction (CO2RR) with the help of protons in solution and appropriate cathodic reduction potentials.8,9 Several methods have been suggested to change CO2 into value-added products, such as chemical transformation,10,11 reduction by photocatalysis,12–14 reduction by electrocatalysis,15,16 thermal catalysis,17,18 photothermal catalysis,19 as well as biological conversion.20,21 The simplicity, mild reaction conditions, environmental compatibility, and possible integration with energy from renewable sources of electrocatalytic CO2 reduction (eCO2RR) make it stand out among these applications. Since CO2 has a linear, symmetrical structure with a zero-dipole moment, it is stable and challenging to activate in electrocatalytic reduction. Because of the molecule's symmetry, the opposing dipoles of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds cancel each other out, resulting in a low electron density around the molecule's center, especially close to the carbon atom. The molecule's general lack of a dipole moment is a result of the decreased electron density surrounding the carbon center. It is more difficult to activate CO2 due to its low electron density. Effective electrocatalytic reduction requires the catalyst to capture and hold CO2 molecules long enough for the reaction. However, CO2 lacks a strong dipole and other reactive properties, making it more difficult to activate for the reduction process. This demands extremely effective catalysts that can change CO2's electron density or aid in breaking its strong CO bonds.22,23 A significant overpotential is needed for CO2 activation due to the dissociation energy needed to break the CO bond, which is more than 750 kJ mol−1, leading to poor energy efficiency and high operational costs.24 The shale gas revolution of the last twenty years, especially the large amounts of ethane (3–12% fraction) from shale gas, has changed the dynamics of the global energy market and produced an excess of ethane despite its relatively low market price, which is especially noticeable in the United States.25,26 Studies on CO2 reduction for CO and carbon-based energy sources27–29 have gained increasing attention owing to rising atmospheric CO2 levels and expanding energy demands, with a focus on finding inexpensive, efficient, and selective catalysts.30–32 These catalysts include homo-bimetallic sites (Fe–Fe, Co–Co, Ni–Ni, Cu–Cu) that indicate enhanced reactivity in comparison to monometallic counterparts, while hetero-bimetallic catalysts remain relatively overlooked.33–41
O bonds cancel each other out, resulting in a low electron density around the molecule's center, especially close to the carbon atom. The molecule's general lack of a dipole moment is a result of the decreased electron density surrounding the carbon center. It is more difficult to activate CO2 due to its low electron density. Effective electrocatalytic reduction requires the catalyst to capture and hold CO2 molecules long enough for the reaction. However, CO2 lacks a strong dipole and other reactive properties, making it more difficult to activate for the reduction process. This demands extremely effective catalysts that can change CO2's electron density or aid in breaking its strong CO bonds.22,23 A significant overpotential is needed for CO2 activation due to the dissociation energy needed to break the CO bond, which is more than 750 kJ mol−1, leading to poor energy efficiency and high operational costs.24 The shale gas revolution of the last twenty years, especially the large amounts of ethane (3–12% fraction) from shale gas, has changed the dynamics of the global energy market and produced an excess of ethane despite its relatively low market price, which is especially noticeable in the United States.25,26 Studies on CO2 reduction for CO and carbon-based energy sources27–29 have gained increasing attention owing to rising atmospheric CO2 levels and expanding energy demands, with a focus on finding inexpensive, efficient, and selective catalysts.30–32 These catalysts include homo-bimetallic sites (Fe–Fe, Co–Co, Ni–Ni, Cu–Cu) that indicate enhanced reactivity in comparison to monometallic counterparts, while hetero-bimetallic catalysts remain relatively overlooked.33–41
Efficient CO2 reduction catalyst design is hard due to stability, huge potentials,29,42 restricted solubility, competing HER, and slow kinetics.43–46 Reduced activation barriers are critical for improving electrocatalyst efficiency and selectivity. Despite the beneficial features of iron-group metallic alloys and compounds, their efficacy as catalysts in CO2 reduction remains insufficient due to low activity and stability.47,48 Because Fe is so readily available, it is essential to build highly efficient Fe–N–C catalysts. With the abundance of iron, there is a need to produce these catalysts. Fe-porphyrins treated with phenolic groups showed remarkable CO faradaic yields exceeding 90% without degradation, emphasizing Fe–N4 sites in macrocycles as active centers.49 The incorporation of Fe atomically into nitrogen-doped carbon substrates, such as Fe–N–C catalysts, has demonstrated remarkable catalytic reactivity towards CO2 reduction to CO, with Fe–N4 sites largely studied as active sites in several investigations.50–53 Fe species provide dynamic surface manipulation, which is critical for understanding structure dynamics and rational catalyst designing in CO2 electro-reduction reaction (CO2ERR).54–56 Recent studies reveal that heteroatom inclusion in carbon support alters the electrical environment, allowing tailored Fe sites to lower the energy of activation limitations in electrocatalysis.57 Fe or Cu-based metals/alloys are widely used as catalysts in CO2 reduction; Fe has significant catalytic activity and a minimal energy barrier, whereas Cu has excellent CO2 adsorption traits and resistance to coking.58–61 In addition to heteroatom inclusion, the use of a second metal atom in Fe-based materials also termed dual-atomic catalysts (DACs) improves catalytic activity synergistically. CO2 reduction relies on DACs, while Ru, Fe, Mn-based homogeneous, and Cu-based heterogeneous catalysis provide viable alternatives.62–72 The adsorbate–metal surface interaction in Fe–N–C single-atom catalysts (also known as SACs) is influenced by the shift of the d-band center.73 Determining the intensity and kind of these interactions between molecules is largely dependent on this change. Consequently, it has a major effect on the catalytic activity. Integrating heteronuclear metal atoms such as Ni, Co, and Zn permits electronic structure adjustment, which facilitates adsorbate absorption as well as desorption on the surface of the catalyst.74–76 Recently, multiple reviews have investigated the CO2ERR, spanning diverse catalysts like as copper–palladium nanoalloys,9 Cu-based nanocrystals,77 bimetallic chalcogenides,78 bimetallic catalysts with atomic sites,79 Bi-based,80 Ni-based,81 Sn-based,82 and carbide-based bimetallic catalysts.83 Fe-based bimetallic electrocatalysts are superior to other metals because they are more affordable, widely available, and have better selectivity for CO2 reduction products. When it comes to stability, efficiency, and scalability, they can perform better than single-metal catalysts like Cu. There is currently no review that provided in-depth analysis of the research on Fe-based bimetallic electrocatalysts for CO2 reduction. Given the growing importance of electrolytic CO2 reduction, the performance of Fe-based bimetallic catalysts merits a thorough examination. This research focuses on the production, implementation, and mechanistic understanding of these catalysts in CO2 electrocatalysis, covering a wide spectrum of product forms. Furthermore, the study highlights the problems and opportunities in developing and comprehending Fe-based bimetallic electrocatalysts, which offer useful insights for future research paths in this sector.
2. CO2 reduction pathways over Fe-based bimetallic electrocatalysts
Fe/Ni–N–C materials were used as electrocatalytic reduction (ECR) catalysts by Huiying Tian and coworkers. These substances were utilized to speed up the electrochemical processes that produce CO.84 Fig. 1A illustrates the CO2 reduction into CO over the Fe/Ni–N–C catalyst. For the catalytic reduction of CO2 into CO, the Fe/Ni–N–C catalyst provides sites for CO2 to bind, and Fe/Ni acts as active centers for the reduction. For binding, CO2 accepts electrons and protons from the electrolyte solution and converts them into intermediate CO precursors such as CHOOH. This reaction is typically completed in two steps CO2 → COOH → CO* as mentioned in Fig. 1. The Fe/Ni–N–C electrocatalyst achieved an impressive (faradaic efficiency of CO) FECO of 92.9% at −0.677 V vs. (reversible hydrogen electrode) RHE, indicating great efficiency. The system retained an elevated current density and faradaic efficiency for the generation of CO (FECO) when applied in a continuous flow cell at scale, holding onto over 89% shortly after 40 hours of electrolysis. Because of the binary metals combined effect, charge transfer rates were increased, resulting in favorable kinetics and long-term, effective electrochemical performance.57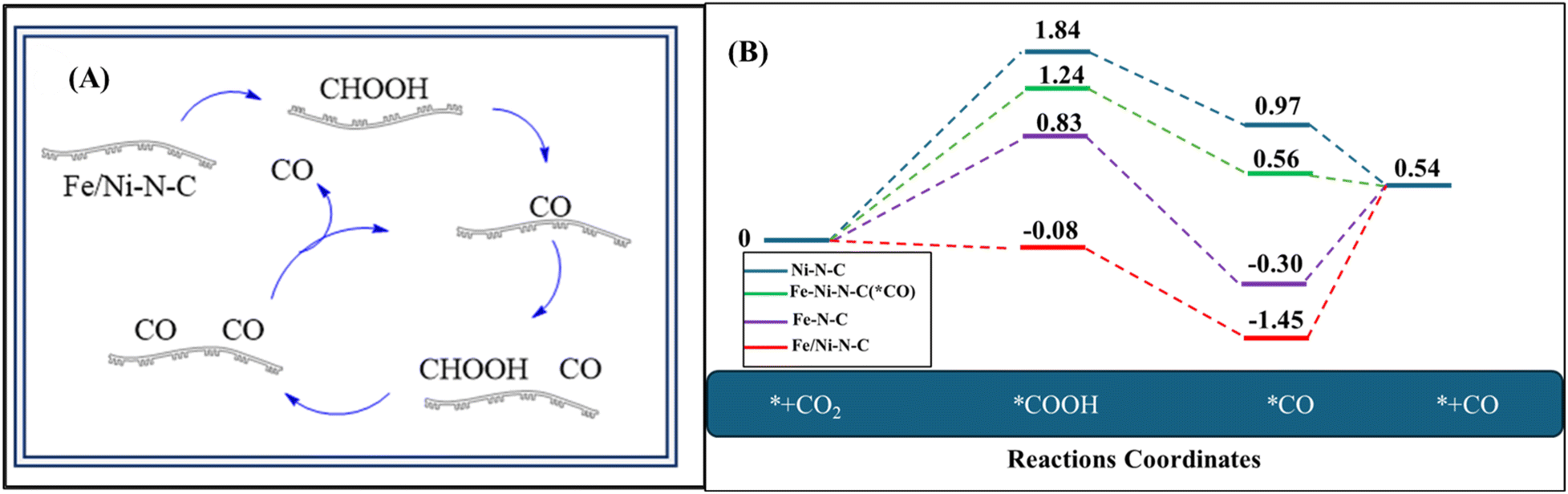 | ||
| Fig. 1 (A) Proposed paths for reduction of CO2 into CO over Fe/Ni–N–C. (B) Using DFT, the electrochemical reduction of CO2 to CO on Fe–N–C, Ni–N–C, and Fe/Ni–N–C with and without adsorbed *CO was represented by a free energy diagram. | ||
To thoroughly investigate the combined effect of Ni–N–C, Fe–N–C, and Fe/Ni–N–C on ECR, DFT studies were performed utilizing the computational hydrogen electrode technique.85 In electrocatalytic CO2 reduction, the Fe atom's electrical properties and catalytic action are largely determined by its spin orientation.86,87 Fe can exist in both high and low spin states in bimetallic Fe-based catalysts, which affects the electron distribution in the d-orbitals and changes the CO2 and intermediate adsorption strength.88 While a low-spin level may produce more stable, less reactive configurations, a high-spin state can increase the activation of CO2 by offering more accessible electron states.89 The impact of these spin arrangements on the CO2 reduction reaction mechanism and efficiency is clarified using DFT simulations. The major catalytic sites, Me–N4 motifs, were used as single-site models based on prior investigations.51,90 The electrocatalytic reduction (ECR) process involved typical two-electron and two-proton transfer reactions, culminating in the creation of *COOH and *CO intermediates. The symbol asterisk (*) represents the active site. Ni–N–C, Fe–N–C, and Fe/Ni–N–C optimized geometries served as computational models for the investigation.91,92 On Fe–N–C sites, the rate-determining step was *CO → CO(g), while on Ni–N–C sites, it was CO2(g) → *COOH. Fe/Ni–N–C generated *COOH intermediates easily, while *CO desorption was difficult. Fe/Ni–N–C adsorbed with *CO intermediates had a much smaller free energy shift for the rate-determining phase *CO → CO(g), indicating easier desorption. This shows that Fe/Ni–N–C provides more active sites by efficiently combining the benefits of Ni–N–C and Fe–N–C sites, increasing CO generation catalytic activity. CO2 adsorption on Fe–Ni bimetallic sites, electron and proton transfer pathways to form *COOH within *CO intermediate, and subsequent CO(g) desorption to regenerate Fe–Ni–N–C (*CO) are the suggested ECR reaction routes on Fe/Ni–N–C. This highlights the increased catalytic activity seen in the studies. The Fe, Ni bimetallic nitrogen-doped carbon successfully lowered the energy barriers of *COOH intermediate production and *CO-to-CO, improving favorable kinetics and increasing ECR activity, verified by the DFT calculations. According to the findings of the calculations above, the suggested ECR reaction pathways of CO2 to CO on Fe/Ni–N–C and their energy diagram using DFT calculation are shown in Fig. 1.84
3. Bimetallic graphene catalysts: mechanistic pathways for CO2 reduction and CH4 production
Previous studies have shown that single-atom-doped graphene is exceptionally efficient for catalyzing CO2RR.93–96 Researchers have studied diverse doping techniques for adding transition metals to the graphene, demonstrating that bimetal single-atom-doped catalysts have greater catalytic performance than standard single-atom-doped catalysts.97–100 Run Zhang et al., employed DFT calculations for examining CO2RR on three bimetal-doped graphene catalysts, Cu–Ni/DG, Cu–Fe/DG, and Fe–Ni/DG. Different reduction pathways yield various products such as CH4, CH3OH, HCOOH, and CO. In the initial stages of CO2RR on doped graphene, CO2 adsorption occurs, characterized by analysis of Eads, electron density difference, density of state (DOS), as well as Integrated Crystal Orbital Hamiltonian Population (ICOHP). Compared to Cu–Fe/DG, CO2 reacts more strongly with iron-based Fe–Ni/DG. The catalytic performance of the material is improved by this greater contact. Electron density difference, DOS, and ICOHP studies reveal more robust interactions between certain dopants (Fe and Ni) and CO2.84,101 Table 1 summarizes the adsorption energies of process intermediates on bimetal-doped Fe catalysts. When CO2 is first protonated, it produces *COOH or *OCHO, which can then be hydrogenated again to generate CO or HCOOH. These changes proceed in many ways:| COOH → *CO + H2O → * + CO, |
| OCHO → *HCOOH → * + HCOOH |
| Reaction intermediates | Potential energy (eV) | |
|---|---|---|
| Cu–Fe/DG | Fe–Ni/DG | |
| *CO | −1.7 | −2.81 |
| *HCOOH | 0.78 | −0.27 |
| *CH2O | −1.46 | −2.05 |
| *CH3OH | 0.97 | −0.44 |
| *CH4 | 0.70 | −0.76 |
On Cu–Fe/DG, Cu–Ni/DG, and Fe–Ni/DG catalysts, the high adsorption of CO and HCOOH encourages continued reduction as intermediates. Nevertheless, significant free energy barriers prevent CO and HCOOH from being desorbed from the catalyst's surface, which presents problems for product release. In the CO2 reduction reaction, CH3OH is a potential product. Four pathways for CO2 → CH3OH involve *CO or *HCOOH as intermediates. *CO undergoes the following reactions:
| *CO → *CHO → *CH2O (exothermic) |
| *CHOH → CH3OH |
*CHO is exothermically converted to *CH2O on Cu–Ni/DG and Cu–Fe/DG. On the other hand, *CHO into *CHOH conversions on Fe–Ni/DG are endothermic.
| *CHO → *CHOH (endothermic) |
This distinction draws attention to the different energetics of different catalysts for these reactions.
The thermodynamic favorability of *CH2O formation is highlighted by its reduced variance in free energy, which is why it is preferred over *CHOH formation. This preference highlights the role that energetics play in identifying the paths of reactions. Six routes for CO2 → CH4 reduction were investigated, using *CO or *HCOOH as intermediaries. *CO undergoes the following conversions:
| *CO → *CHO |
| *CO → *COH |
It resulted in *CHO or *COH and finally CH4 by additional hydrogenation. *CO prefers *CHO production because of the lower free energy fluctuation. On Cu–Ni/DG and Cu–Fe/DG,
| *CHO → *CH2O (exothermic) |
| *CHO → *CHOH (endothermic) |
While, Fe–Ni/DG prefers,
| *CHO → *CH2O |
Path 3 is the best route from Path 1 to Path 5, demonstrating its effectiveness and fit for the intended change. Nevertheless, route 6 (*OCHO → *HCOOH) doesn't work as the best route for Fe–Ni/DG due to significant free energy fluctuation. The optimized pathway is Path 6, which is exothermic on Cu–Fe/DG and Cu–Ni/DG.101
| *CHO → *CH2OH → *OCH3 → *CH4 (exothermic) |
Because CO2 interacts with Fe or Ni atoms more strongly than its interaction with Cu, Fe–Ni/DG is more stable than Cu–Ni/DG and Cu–Fe/DG. The catalytic potential of Fe–Ni/DG for CO2 conversion reactions is highlighted by its improved stability. Fe–Ni/DG becomes more prominent in bimetal-doped graphene systems because of its increased stability, which implies that it can support effective and long-lasting catalytic activity. Graphene doped with Cu, Fe, and Ni shows significant selectivity for CO2 reduction over hydrogen evolution (HER), suggesting that these materials are effective catalysts for CO2 conversion processes, with various product outcomes seen for the initial protonation step of CO2 on these catalysts.
4. FeCo-Pc catalysts for multi-carbon (C2) product formation
FeCo-Pc catalyst with dual metal–nitrogen active sites for efficient CO2RR. FeCo-Pc overcomes the challenge of C–C coupling seen in single-atom catalysts,102,103 enabling the production of C2 products, as shown in Table 2. These C2 products include C2H4, CH2OHCH2OH, C2H5OH, and CH3COOH with enhanced selectivity, due to the cumulative effects of Fe and Co dual active sites anchored within phthalocyanine (FeCo-Pc).104 The computations are performed using the Gaussian 09 program, PBE exchange–correlation functional, and 6-31G* basis sets.105,106 C–C coupling processes in CO2 reduction are critical for comprehending multi-carbon product generation.107,108 Water plays an important role in the reduction of CO2 because it influences intermediate hydration, provides protons for product production, and functions as a solvent for ion transport. Additionally, through the oxygen evolution reaction (OER), it competes with CO2 reduction and affects catalyst behavior.109,110 CO was identified as a crucial intermediate, discovered by in situ spectroscopy.111,112 PCETs generate C1 intermediates like CHO* and COH*, with thermodynamics and kinetics assessed on FeCo-Pc surfaces. CO to CHO* (formyl group) is produced by further reducing *CO and adding an extra proton and electron. Usually, this process produces more valuable chemicals such as alcohols and aldehydes. In the case of COH* protonation step is required for the formation of COH*, but not the complete reduction required to generate *CHO. In the synthesis of other C1 products, such as ethanol or methane, this is frequently a transitional stage. CO dimerization, proposed as the initial step to C2+ products, and carbene coupling with CO* to form
coupling with CO* to form 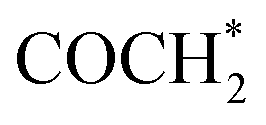 or CH2CO*, are investigated.42,113 However, the generation of “dead-end” intermediates, such as COCO* and other C–C linked intermediates, tightly bound to FeCo-Pc, incurs high energy consumption, creating kinetic obstacles and surface contamination. Electrolytes C–C coupling pathways are influenced by pH,114 with high pH favoring CO* to COH* conversion and CO* coupling with CHO* to generate COCHO*, boosting C2 synthesis.115,116 COCHO* formation has a lower activation barrier compared to CHO*–CO* precursors, demonstrating its thermodynamic favorability and potential to provide more C2 products in CO2RR on FeCo-Pc surfaces.117 In an aqueous solution, the relative stability of COOH* as well as OCHO* in CO2RR against H* in HER affects the competing processes of the H2 evolution reaction (HER).118 FeCo-Pc prefers CO2RR over HER due to larger free energy changes for COOH* and OCHO* production against adsorbed H*. COOH* has a smaller overpotential than OCHO*, indicating FeCo-Pc prefers CO2RR.119,120 The electro-conversion of CO2 to C2H4 is important for the C2H4 industry, however, it faces challenges with an elevated overpotential and multi-electron transfer processes.121,122
or CH2CO*, are investigated.42,113 However, the generation of “dead-end” intermediates, such as COCO* and other C–C linked intermediates, tightly bound to FeCo-Pc, incurs high energy consumption, creating kinetic obstacles and surface contamination. Electrolytes C–C coupling pathways are influenced by pH,114 with high pH favoring CO* to COH* conversion and CO* coupling with CHO* to generate COCHO*, boosting C2 synthesis.115,116 COCHO* formation has a lower activation barrier compared to CHO*–CO* precursors, demonstrating its thermodynamic favorability and potential to provide more C2 products in CO2RR on FeCo-Pc surfaces.117 In an aqueous solution, the relative stability of COOH* as well as OCHO* in CO2RR against H* in HER affects the competing processes of the H2 evolution reaction (HER).118 FeCo-Pc prefers CO2RR over HER due to larger free energy changes for COOH* and OCHO* production against adsorbed H*. COOH* has a smaller overpotential than OCHO*, indicating FeCo-Pc prefers CO2RR.119,120 The electro-conversion of CO2 to C2H4 is important for the C2H4 industry, however, it faces challenges with an elevated overpotential and multi-electron transfer processes.121,122
| Compound | Reaction intermediate | Potential energy (eV) |
|---|---|---|
| C2H4 | COOH* | 0.13 |
| CO* | 0.23 | |
| CHO* | 0.68 | |
| COCHO* | 0.70 | |
| CHOCHO* | 0.38 | |
| CHOHCHO* | 0.55 | |
| CHCHO* | 0.27 | |
| CH2CHO* | 0.17 | |
| CH2CHOH* | 0.06 | |
| CH2CH* | 0.51 | |
| C2H5OH | CHOHCH2O* | 0.41 |
| CHCH2O* | 0.21 | |
| CH2CH2O* | 0.40 | |
| C2H2OH* | 0.71 | |
| CH3COOH | COCHOH* | 0.57 |
| COCH2O* | 1.59 | |
| COHCH2O* | 0.26 | |
| CH2COOH* | 0.60 | |
| CH2OHCH2OH | CHOHCH2OH* | 0.16 |
| CH2OHCH2OH* | 0.20 |
FeCo-Pc catalysts increase CO2RR by favoring COOH* over H*, resulting in C2H4 generation. During CO reduction, CHO* formation takes precedence over COH* formation. The rate-limiting step for C2H4 generation is coupling CHO* with CO* to generate COCHO*.22 FeCo-Pc surfaces aid in producing C2H4 by reducing COCHO* to glycolaldehyde and hydrogenating further. FeCo-Pc catalysts provide a viable avenue for the electrochemical process to transform CO2 into ethanol (C2H5OH), a critical commodity chemical, via C–C coupling reactions. The procedure is optimized by hydrogenating typical intermediates with ethylene (C2H4). CHOHCHO* is found as a selectivity-determining molecule. Thermodynamically, C2H5OH is produced through the optimum process of CHOHCHO* hydrogenation to CHOHCH2O*.117,123,124
The rate-limiting step (RLS) for C2H5OH creation involves the hydrogenation process of CH2CH2O* to C2H4OH*, which has a greater barrier than the formation of C2H4. The preference for CHCHO* or CHOHCH2O* production during CHOHCHO* reduction determines the selectivity of C2H4 and C2H5OH. Increasing potential increases the feasibility of producing C2H4 and C2H5OH on FeCo-Pc, with all fundamental stages downward energetically at −0.66 V-RHE. Previous investigations have discovered ethylene glycol to be a negligible product in CO2RR utilizing catalysts such as Au, Ru, and Cu.125–127 Calvinho et al., recently proved that CO2RR may be converted to ethylene glycol (CH2OHCH2OH) using a transition-metal phosphide catalyst. CHOHCHO*, like C2H4 and C2H5OH, determines the selectivity of CH2OHCH2OH production. Protonation of CHOHCH2O* results in the formation of CHOHCH2OH*, which is preferred over CHCH2O*. This is then transformed into ethylene glycol. Geometry optimization demonstrates that CH2OH is not chemisorbed, indicating that it prefers the formation pathway over C2H5OH. Both CH2OHCH2OH and C2H5OH have an identical kinetic barrier for CHOHCH2O* production, resulting in C2H4 selectivity.104,128–130 Possible electrochemical reduction pathways for CO2 into C2 products are shown in Scheme 1.
 | ||
| Scheme 1 Possible electrochemical reaction pathways of CO2 over Fe-based bimetallic catalyst into C2+ products. | ||
5. Fe-based bimetallic electrocatalysts: advanced pathways for CO2 reduction
As previously reported, nitrogen-doped carbon nanotubes with Fe/Fe3N nanoparticles improve the catalytic performance of the oxygen reduction process (ORR) by exposing active areas and enabling electron transport.131–134 Before pyrolysis, Fe-doped zinc-imidazole frameworks (ZIF-8) were changed with phosphomolybdic acid hydrate (PMo), resulting in the formation of Fe nanoparticles contained within molybdenum and nitrogen-co-doped carbon scaffolds (Fe-NP/MNCF). In CO2 electrolysis powered by a Zn–air battery (ZAB), Fe-NP/MNCF served as a dual-functional catalyst during ORR and CO2RR. To synthesize, Fe(NO3)3·9H2O, Zn(NO3)2·6H2O, and PMo were dispersed in 2.5 mL of water that was deionized by ultrasound. The precursor was calcined at 900 °C in an argon environment for 2 hours to produce Fe-NP/MNCF, as illustrated in Fig. 2A.135,136 | ||
| Fig. 2 (A) Synthesis of Fe-NP/MNCF (B) synthesis of FeNi@N-CNTs and (C) synthesis of Fe3Ni7–N–C. | ||
Nitrogen-doped carbon was obtained by annealing ZIF-8 at 950 °C in an argon environment. The final product, called ZIF-NC, has a three-dimensional porous structure.137 FeNi@N-CNTs catalysts were developed by wet impregnation and thermal processing. The heat was applied at 1100 °C to produce FeNi@N-CNTs-X, where X is the temperature at which they were annealed (Fig. 2B). By encasing the FeNi alloy in N-CNTs, this synthesis method enhances the catalytic properties for potential CO2 reduction uses. Better CO2ER activity and stability were demonstrated by FeNi@N-CNTs-1100, which showed over 90% CO faradaic efficiency spanning a wide potential range (−0.47 to −0.97 V vs. RHE). Optimized *COOH adsorption and *CO desorption were credited with improving catalytic activity and CO selectivity.138,139
Xiao Han et al. employed a solution approach to create FeNi precursors, which were subsequently transformed into a variety of FeNi-NC catalysts via one-step pyrolysis. Dissolved Ni(NO3)2·6H2O, Zn(NO3)2·6H2O, and Fe(NO3)3·9H2O in 80 mL of methanol and stirred thoroughly. Separately, another 80 mL of methanol was used to dissolve 2-methylimidazole and added to the metal nitrate solution. The resultant mixture was agitated constantly for 8 hours to produce the catalyst precipitate. The resulting precipitate washed away with the solvent methanol centrifuged, evaporated at 60 °C, and powdered to produce Fe3Ni7-ZIF samples with different Fe/Ni ratios, as illustrated in Fig. 2C. These catalysts attain about 100% overall Faraday efficiency by promoting CO2 electroreduction into CO and H2. Furthermore, we discovered that altering the applied potential across a large range throughout the procedure makes it simple to change the syngas ratio from 1:1 to 6:1 (CO/H2). Because of its versatility, syngas can be utilized to manufacture fuels and raw materials for chemicals.140
The unique features of doped Cu in Fe–N–C catalysts, including its numerous oxidation states, which facilitate fast electron transfer,87,141 ability to particularly manufacture C2+ products,142–144 and improved interaction with CO2 to limit hydrogen development, have sparked great interest in CO2RR. The Fe/Cu–N–C catalyst, which was produced by adding a copper promoter to a mixture of iron and carbon sources and then pyrolyzing it, has outstanding CO2 reduction efficiency with more than 90% CO faradaic productivity (FECO) in a broad potential range (−0.5 to −0.7 V) and remarkable stability, with FECO maintained after 10 hours of electrolysis. To make the Fe/Cu–N–C catalyst, Shulin Zhao et al., mixed tris(2,4-pentanedionato)iron(III), Cu-acetylacetonate, along with meso-tetra(4-methoxyphenyl) porphin in CHCl3 and stirred it at 60 °C for the period of 3 h. Rotational evaporation was used to extract the solvent from the mixture after 30 minutes of sonication following the addition of zinc oxide. After the powder was produced, it was heated to 900 °C in an argon atmosphere for two hours, then it was leached for six hours at 80 °C in 0.5 M H2SO4 and allowed to dry overnight.145 Table 3 shows the comparative analysis of heteronuclear Fe-based catalysts for CO2 electroreduction.
| Catalyst | Synthesis method | Key features | CO2 reduction efficiency | Stability | Reference |
|---|---|---|---|---|---|
| Fe-NP/MNCF | Fe-doped ZIF-8 modified with PMo, pyrolysis at 900 °C | Molybdenum and nitrogen co-doped carbon scaffold | The dual-functional catalyst for ORR and CO2RR | Used in Zn–air battery-powered CO2 electrolysis | 135 and 136 |
| ZIF-NC | Annealing ZIF-8 at 950 °C | Three-dimensional porous structure | — | — | 137 |
| FeNi@N-CNTs-X | Wet impregnation and thermal processing at 1100 °C | Encapsulated FeNi alloy in N-CNTs, enhanced CO2 reduction | >90% CO faradaic efficiency (−0.47 to −0.97 V vs. RHE) | High stability | 138 and 139 |
| Fe3Ni7-ZIF | Solution approach, pyrolysis | Various Fe/Ni ratios to adjust performance | ∼100% faraday efficiency, tunable syngas ratio (1:1 to 6:1 CO/H2) |
High stability, broad potential range | 140 |
| Fe/Cu–N–C | Cu promoter added to Fe/carbon mixture, pyrolysis at 900 °C, acid leaching | Enhanced electron transfer, improved CO2 adsorption | >90% CO faradaic efficiency (−0.5 to −0.7 V) | Stable after 10 hours of electrolysis | 145 |
| C–Fe–Co-ZIF | Impregnation of ZIF-8 with Fe and Co, pyrolysis | Bimetallic Co–Fe catalyst for CO2 electroreduction | +10% CO faradaic efficiency vs. pure Co-ZIF | H2/CO ratios tunable (0.8 to 4.2), 93% FECO + H2 over 10 hours | 146 |
| Fe/Mn–N–C | Potassium citrate calcination, Fe and Mn doping, pyrolysis at 800 °C | Atomic dispersion of Fe and Mn for CO selectivity | 94% CO faradaic efficiency at −0.5 V (RHE) | >80% FECO after 12 hours | 146 and 147 |
The production of Fe/Mn–N–C, a unique bimetallic catalyst consisting of iron and manganese atomic dispersion, involved the elevated temperatures calcination of an organic carbon-based porous precursor. The solution of potassium citrate monohydrate was initially calcined for an hour at 800 °C in a nitrogen atmosphere to create porous black carbon compounds. The resulting solid was dried in the oven for 12 hours at 80 °C after being rinsed with deionized water and a 1 M H2SO4 solution until it attained a neutral pH. A mixture consisting of carbon material, Fe(NO3)3·9H2O, and MnCl2·4H2O in deionized H2O was ultrasonically treated for an hour, centrifuged, and dried afterward. The resultant solid was combined with melamine in a particular mass ratio and then calcined at 800 °C, over a nitrogen environment for two hours to generate the Fe/Mn–N–C catalyst,146 Fig. 3A. At a −0.5 V overpotential (RHE), the Fe/Mn–N–C catalyst produced a 94% Faraday efficiency (FE) for CO in the 0.1 M KHCO3 electrolyte. This shows that, in these electrochemical circumstances, the catalyst has a high selectivity for CO synthesis. The catalyst's performance is notable when compared to previously published iron-based and manganese-based electrocatalysts, which include FeMn–N–C (FECO 80% at −0.5 V RHE), NFe-CNT/CNS (FECO 69% at −0.6 V RHE), and Mn–N–C (FECO 70% at −0.6 V RHE).53,148,149 Following just 12 hours of uninterrupted catalysis, the FECO was above 80%, suggesting good stability. Density functional theory (DFT) calculations show that the interaction of neighboring Fe–Mn centers lowers the potential for COOH* production and CO desorption.146
 | ||
| Fig. 3 Methodology for the synthesis of (A) Fe/Mn–N–C and (B) C–Fe–Co-ZIF catalysts. | ||
6. Development of atomically distributed Co–Fe catalysts for CO2 reduction
Bimetallic Co–Fe catalysts that are atomically distributed were developed in two steps. Using this method, the catalysts were synthesized with accurate atomic-level dispersion of iron and cobalt through a series of synthesis steps. To ensure a successful yield without interference in the crystallization of Co-ZIF, Fe–Co-ZIF precursors were generated by an impregnation process that modified ZIF-8 into Co-ZIF and absorbed Fe source. Pyrolysis was then used to manufacture the final catalysts (C–Fe–Co-ZIF) for CO2 electro-reduction,150 shown in Fig. 3B. The bimetallic catalysts produced more CO, with an additional 10% in CO Faradaic efficiency (FE) when compared to pure C–Co-ZIF. Adjustable H2/CO ratios (0.8 to 4.2) reached across a wide potential range, with a high overall FE CO + H2 of 93% over 10 hours, showing the catalyst's capacity for efficient syngas production from CO2.1477. Graphene oxide-based catalysts for CO2 reduction
Graphene oxide (GO) was produced with graphite using the modified Hummers' method.151 GO suspension (2 mg mL−1) was made by sonicating it in deionized water for 5 hours. Iron and nickel nitrates were introduced to the GO solution, which was sonicated for three more hours. The resulting slurry was heated to 180 °C in an autoclave lined with Teflon for 12 h before being freeze-dried to generate a columnar product. Subsequently, a chemical vapor deposition (CVD) process at 1000 °C with Ar and NH3 was used to synthesize the H–NiFe/NG composite, followed by annealing with hot steam. A novel method involving steam-assisted chemical vapor deposition introduces surface oxygen vacancies (VO) into Ni–Fe BM NPs, creating electron-rich centers that activate CO2 molecules.152,153 This method reduces the energy barrier for creating COOH* intermediates, increasing the reduction of carbon dioxide to CO while maintaining a faradaic efficiency of as high as 94% at −0.80 V (vs. RHE) along with excellent stability. Surface VO-modified atoms of nickel have a vital role in increasing the electrocatalytic efficacy of reduction of CO2 to CO, according to density functional theory simulations.1548. Molecular catalyst-based heterostructures for CO2 reduction
The design and synthesis of a molecular catalyst-based heterostructure for the reduction of CO2 is still a serious issue. Molecular catalysts with transition-metal elements (Co, Ru, Fe, Ni, Cu) and ligands made of organic compounds (phthalocyanine, polypyridine, porphyrin) provide precise active sites and structural tunability for researching CO2ER processes.155–159 These catalysts facilitate detailed investigations into CO2 reduction catalysis. A crystalline bimetallic phthalocyanine heterostructure electrocatalyst (CoPc/FePc HS) was developed for CO2 reduction, achieving a remarkable CO2 to CO conversion efficiency of 99% at the potential of −0.87 vs. RHE and demonstrating outstanding stability over 10 h of electrocatalysis. Different Co/Fe molar ratios (3:1, 1:1, 1:3)160 of CoPc/FePc heterostructures, along with CoPc and FePc controls were synthesized by dispersing a mixture of CoPc and FePc in DMF and subjecting it to solvothermal treatment at 180 °C for 24 hours. Precipitates in the shape of purple microrods were gathered and cleaned with ethanol. They were then calcined for three hours at 450 °C in an Ar environment. CoPc/FePc heterostructures were formed as a consequence of this technique. This method provides a controlled approach to tailor the composition of bimetallic phthalocyanine heterostructures for CO2 reduction applications.161
9. Cu–Fe–N6–C: a high-performance diatomic site catalyst for CO2 reduction
Metal–nitrogen–carbon (M–N–C) catalysts have great potential for CO2 electrocatalytic reduction because of their abundance of active sites and low-cost raw ingredients.52,162–165 Cu–Fe–N6–C, a new diatomic site catalyst coordinated with nitrogen and embedded into a carbon matrix, was developed, and synthesized. Cu–Fe–N6–C was synthesized in two primary stages. First, PcCu-Fe-ZIF-8 is created by combining PcCu, zinc nitrate, iron nitrate, and 2-Me–imidazole, resulting in a blue precipitate that indicates uniform dispersion of Cu and Fe species inside the framework. PcCu-Fe-ZIF-8 becomes Cu–Fe bimetallic sites distributed on a nitrogen-doped carbon framework upon annealing at 1000 °C under Ar. The necessity for extra acid leaching treatment is eliminated by this technique. For a variety of processes, the resulting catalyst structure improves catalytic performance (Fig. 4). This catalyst outperformed individual Cu–N–C and Fe–N–C catalysts thanks to synergistic effects at bimetallic sites. Cu–Fe–N6–C demonstrated outstanding CO selectivity, with an exceptional faradaic efficiency of 98% at −0.7 V, and maintained selectivity after 10 hours of electrolysis. Experimental and theoretical investigations revealed that the combined catalysis of several metallic sites increased CO2 adsorption enthalpy, and lowered activation energy, resulting in enhanced selectivity, activity, and stability, as well as decreased impedance in CO2 hydrogenation.166 | ||
| Fig. 4 Synthesis of Cu–Fe–N6–C. | ||
For CO2 conversion, some Na-promoted Co–Fe bimetallic catalysts ranging in proximity and compositions were investigated. These catalysts are designed to use the strong selectivity of iron for olefins during CO2 hydrogenation, along with the high activity and reducibility of cobalt. The goal of this combination is to improve CO2 conversion operations' overall efficiency and selectivity.167–170 Co-precipitation was used to produce Co–Fe bimetallic catalysts, which were then hydrothermally treated. The manufacture of uniform catalysts with regulated compositions and architectures is made easier by this technique.171,172 Co(NO3)2·6H2O and Fe(NO3)3·9H2O were dissolved sequentially in deionized water to achieve a [Co]2+ + [Fe]3+ concentration of 0.09 M, followed by the addition of 5 mol L−1 NaOH solution until pH 11 was reached. The resultant hydroxide precipitates were hydrothermally treated at 150 °C for 24 hours before being centrifuged, washed, and dried at 80 °C. The dried products were calcined at 400 °C for 3 hours to produce Co–Fe catalysts with various Co/Fe molar ratios (1/4, 1/2, 1/1, 2/1, and 4/1), designated as Co1Fe4, Co1Fe2, Co1Fe1, Co2Fe1, and Co4Fe1, respectively. The Co1Fe2 catalyst, having a Co/Fe molar proportion of 1/2 and proximity, permitted the quick reduction of CoFe2O4 to CoxFey alloy and subsequently carbonization to χ-(CoxFe1−x)5C2 alloy carbide. It demonstrated improved stability and performance in olefin production without deactivation over 500 h on-stream.173
10. Fe/Ni–N–C catalysts with 3D carbon-based structures for CO2 reduction
A 3D carbon-based material was produced, featuring bimetallic centers174 that include NiNC and FeNC, which demonstrated synergistic effects advantageous to the CO2RR. The synthesis procedure involved numerous steps to produce various catalyst materials. Tripotassium citrate monohydrate was cooked at 800 °C under nitrogen, and then treated with sulfuric acid and water to create a porous carbon material. Next, a mixture containing carbon, nickel nitrate, iron nitrate, and glucose in water was processed using ultrasound and then combined with melamine. This mixture was heated at 800 °C under nitrogen to produce the NiNC/FeNC catalyst.175 Further, specific catalysts like FePc@NiNC and NiPc@FeNC were prepared by treating NiNC or FeNC with N,N-dimethylformamide and adding iron phthalocyanine (FePc) or nickel phthalocyanine (NiPc), respectively (Fig. 5). Each stage required precise chemical reactions and thermal treatments to generate the correct catalyst compositions. DFT models and observations show176 Fe atoms are reactive and adsorption sites for CO2RR, while substantial CO* adsorption reduces stability. By adding Ni atoms, CO* adsorption on Fe is decreased, changing the energy barriers and improving stability. The Fe–N4 and Ni–N4 sites work in concert to facilitate the rate-limiting processes (CO2(g) → COOH*, +0.95 eV) in FePc@NiNC. Flexible syngas composition is made possible by this synergy while high catalytic activity is maintained.177 | ||
| Fig. 5 Methodology for the synthesis of FePc@NiNC catalyst. | ||
11. Ni/Fe–N–C: diatomic metal–nitrogen catalysts for CO2 reduction
A ZIF-8 was used to create a catalyst consisting of isolated diatomic metal–nitrogen species. Initially, Fe-doped ZIF-8 was made by combining zinc nitrate, iron nitrate, and 2-methylimidazole, maintaining that Fe ions were chemically bound to the organic ligand rather than being physically absorbed.178 Fe-doped ZIF-8 was dissolved in n-hexane, and nickel nitrate methanol solution was added gradually. Nickel was contained within ZIF-8's tiny cavities using this method. Nickel was well incorporated into the framework owing to the steady infusion.90,179 After thermal treatment at 1000 °C, the resulting catalyst, Ni/Fe–N–C, containing nitrogen-coordinated diatomic Ni–Fe species, was obtained (Fig. 6). For comparison, crystalline Ni–N–C and Fe–N–C catalysts were synthesized similarly. After 30 hours, the Ni/Fe–N–C catalyst retains 99% selectivity and over 90% CO faradaic efficiency from −0.5 to −0.9 V, which ended at 98% at −0.7 V. Synergistic Ni–Fe interactions lower CO2 reduction reaction barriers and cause structural changes upon CO2 adsorption, according to DFT research, improving the catalyst's performance.180 | ||
| Fig. 6 Methodology for the synthesis of Ni/Fe–N–C catalyst. | ||
12. Micelle-encapsulated Fe-based nanoparticles for CO2 reduction
Inverse micelle encapsulation was used to produce size-selected nanoparticles (NPs) of Fe, FeCu, FeAg, Ag, and Cu. The poly(styrene)-block-poly(2-vinylpyridine) (PS-P2VP) diblock copolymer, which was obtained from Polymer Source Inc., was used in this procedure. Metallic salts (FeCl2, AgNO3, CuCl2, FeCl3) and the copolymer were dissolved in toluene. By taking advantage of the micelles encapsulating attributes, this technique made controlled nanoparticle manufacturing easier.181,182 Specifically, isotopically enriched 57FeCl2 salt was employed for NRIXS measurements, prepared from iron foil with 95% 57Fe isotopic enrichment using adapted literature procedures.183 Following NP synthesis, the samples were soaked with carbon black powder and then treated using N2-plasma to eliminate the polymer, resulting in clean NP surfaces. The NPs were subsequently distributed into an ethanol/Nafion solution enabling electrode deposition, accompanied by further N2-plasma treatment to remove any remaining polymer before electrochemical evaluation. Fig. 7 shows that the production of 57Fe NPs involves mixing PS-P2VP in toluene to create reverse micelles, which were then added to 57FeCl2 salt and stirred for 72 h. Similar methods were utilized to create 57FeCu and 57FeAg NPs by changing the ratios of 57FeCl2 to CuCl2 or AgNO3 in the micellar solution. FeAg NPs had 36% CO faradaic selectivity at −1.1 V vs. RHE in 0.1 M KHCO3, similar to pure Ag NPs, but FeCu NPs prefer H2 evolution, similar to pure Fe NPs.184 Table 4 summary of recently reported Fe-based bimetallic electrocatalysts for CO2 reduction. | ||
| Fig. 7 Synthesis of 57FeCu and 57FeAg NPs for CO2ERR. | ||
| Catalyst | Electrolyte | Major product | FE (%) | Potential (V) | Current density (mA cm−2) | Stability |
|---|---|---|---|---|---|---|
| Fe-NP/MNCF | 0.5 M KHCO3 | CO, H2 | 87.50% | −0.7 | 10 | 36 h |
| FeNi@N-CNTs | 0.5 M KHCO3 | CO | 90% | −0.47 to −0.97 | 20.18 | 35 h |
| Fe3Ni7-ZIF | 0.5 M KHCO3 | CO, H2 | 81.30% | −0.9 | −22.5 | Good |
| Fe/Cu–N–C | 0.5 M KHCO3 | CO | 97% | −0.6 | 74 | 10 h |
| Fe/Mn–N–C | 0.1 M KHCO3 | CO | 94% | −0.5 | −83.5 | 12 h |
| Fe/Ni-ZIF-8 | 0.5 M KHCO3 | CO | 89% | −0.677 | 26.92 | 40 h |
| FePc@NiNC | 0.5 M KHCO3 | CO, H2 | 100% | −0.8 | 260 | 18 h |
| Ni/Fe–N–C | 0.5 M KHCO3 | CO | 98% | −0.7 | 7.4 | 30 h |
| H–NiFe/NG | 0.1 M KHCO3 | CO | 94% | −0.8 | 18.2 | 20 h |
| PcCu-Fe-ZIF-8 | 0.1 M KHCO3 | CO | 98% | −0.7 | 7 | 10 h |
| 57FeAg NPs | 0.1 M KHCO3 | CO | 36% | −1.1 | 0.35 | 2.5 h |
| Fe–Co-ZIF | 0.5 M KHCO3 | CO, H2 | 93% | −0.55 | 8 | 10 h |
| CuFe/OG | — | CH4 | — | 0.97 | — | — |
| FeNi/DG | — | CH4 | — | −0.44 | — | — |
| FeCo-Pc | — | C2+ | — | −0.66 | — | — |
13. Summary and outlook
Possibilities for the advancement of sustainable energy technology look promising for future studies on electrochemical CO2 reduction with bimetallic catalysts. Optimizing catalyst compositions and structures to increase selectivity and efficiency in the production of CO, syngas, and other multi-carbon products is a crucial field of research. Fe–Ni, Fe–Ag, Fe–Mo, Fe–Co, Cu–Fe, and Fe/Mn–N are examples of novel metal combinations that present the potential for enhanced catalytic performance. The main goals of the research will be to comprehend the fundamental structure–activity correlations and stability of these bimetallic catalysts in practical working environments. The scalability of bimetallic catalysts for large-scale commercial applications is limited by their typical synthesis, which involves intricate deposition, pyrolysis, and reduction methods. The development of more affordable, optimized synthesis techniques with improved loading capacities is necessary to meet this challenge and permit Fe-based bimetallic catalysts to be widely used in renewable energy systems. Moreover, a major challenge presented by the chemical instability of these catalysts is the reduction of active sites and changed performance caused by corrosion of the carbon substrate. Under practical circumstances, Fe-based bimetallic electrocatalysts for CO2 reduction encounter difficulties such as low catalytic activity, poor selectivity for target products, and restricted stability. Controlling the chemical intermediates, improving the electrical and geometric properties, and interpreting the synergistic effects between metals are still major challenges. Precise control of Fe-based bimetallic catalysts' shape, structure, and atomic coordination is required to strike a compromise between stability and catalytic activity. The aim is to design specialized bimetallic catalysts with enhanced stability features and active sites outperforming existing catalysts. This will facilitate the development of scalable CO2 conversion technologies for use in sustainable energy applications, assisting in the shift to a world without carbon emissions. The development of effective CO2 electroreduction catalysts will be speed up by collaborative, multidisciplinary research that combines theoretical and experimental methods. Future research should concentrate on investigating novel bimetallic combinations that improve performance and customizing catalyst structures by nano-structuring. Enhancing these catalysts' scalability for industrial applications is also essential. Developments in reaction mechanism research, computational modeling, and in situ characterization methods will improve catalyst design and propel more effective CO2 conversion systems.Data availability
No primary research results, software, or code have been included, and no new data were generated or analyzed as part of this review.Author contributions
Ayesha Zafar: writing – original draft. Adnan Majeed: writing – review & editing and software. Abdul Ahad: formal analysis. Muhammad Adnan Iqbal: conceptualization, resources, supervision. Tanveer Hussain Bokhari: validation. Zanira Mushtaq: data curation, validation. Shahzaib Ali: visualization.Conflicts of interest
The authors declare no conflict of interest.Acknowledgements
The authors are thankful to the Pakistan Science Foundation (PSF) for awarding the research grant PSF/CRP/Consr-676.References
- S. A. Rekker, K. R. O'Brien, J. E. Humphrey and A. C. Pascale, Nat. Clim. Change, 2018, 8, 489–492 CrossRef.
- J. Rogelj, P. M. Forster, E. Kriegler, C. J. Smith and R. Séférian, Nature, 2019, 571, 335–342 CrossRef CAS.
- Z. Chen, G. Zhang, H. Chen, J. Prakash, Y. Zheng and S. Sun, Renewable Sustainable Energy Rev., 2022, 155, 111922 CrossRef CAS.
- K. Heine, in The Quaternary in the Tropics: A Reconstruction of the Palaeoclimate, Springer, 2024, pp. 11–84 Search PubMed.
- S. Munassar, Development of pre-operational mesoscale inverse modelling system to quantify CO2 sources and sinks over Europe, PhD dissertation, Friedrich-Schiller-Universität Jena, Jena 2024.
- V. P. N. Bharathi, K. Muthuswamy, B. Natarajan, B. Vasudevan, S. Appavu, R. Marimuthu, K. Shanmugam and D. Rajaram, Fresenius Environ. Bull., 2024, 1581 CAS.
- A. Lucas, Clim. Risk Manag., 2021, 31, 100257 CrossRef.
- A. Vasileff, C. Xu, Y. Jiao, Y. Zheng and S.-Z. Qiao, Chem, 2018, 4, 1809–1831 CAS.
- F. Zhou, J. Zhang, Y. Zhang, Y. Wu, Y. Wang and W. Luo, Coord. Chem. Rev., 2024, 509, 215802 CrossRef CAS.
- F. Juliá-Hernández, T. Moragas, J. Cornella and R. Martin, Nature, 2017, 545, 84–88 CrossRef.
- K. Sekine and T. Yamada, Chem. Soc. Rev., 2016, 45, 4524–4532 RSC.
- H. Rao, L. C. Schmidt, J. Bonin and M. Robert, Nature, 2017, 548, 74–77 CrossRef CAS.
- W. Kim, B. A. McClure, E. Edri and H. Frei, Chem. Soc. Rev., 2016, 45, 3221–3243 RSC.
- S. Fang, M. Rahaman, J. Bharti, E. Reisner, M. Robert, G. A. Ozin and Y. H. Hu, Nat. Rev. Methods Primers, 2023, 3, 61 CrossRef CAS.
- F. Li, A. Thevenon, A. Rosas-Hernández, Z. Wang, Y. Li, C. M. Gabardo, A. Ozden, C. T. Dinh, J. Li and Y. Wang, Nature, 2020, 577, 509–513 CrossRef CAS PubMed.
- Y. Chen, X.-Y. Li, Z. Chen, A. Ozden, J. E. Huang, P. Ou, J. Dong, J. Zhang, C. Tian and B.-H. Lee, Nat. Nanotechnol., 2024, 19, 311–318 CrossRef CAS PubMed.
- D. M. Koshy, S. S. Nathan, A. S. Asundi, A. M. Abdellah, S. M. Dull, D. A. Cullen, D. Higgins, Z. Bao, S. F. Bent and T. F. Jaramillo, Angew. Chem., 2021, 133, 17613–17621 CrossRef.
- Q. Wang, H. Wang, X. Ren, R. Pang, X. Zhao, L. Zhang and S. Li, J. Phys. Chem. Lett., 2023, 14, 8421–8427 CrossRef CAS.
- Y. Dong, R. Song, Z. Zhang, X. Han, B. Wang, S. Tao, J. Zhao, A. N. Alodhayb, Z. Chen, X. Yi and N. Zhang, Cell Rep. Phys. Sci., 2024, 5(10), 102227 CrossRef CAS.
- S. Gleizer, R. Ben-Nissan, Y. M. Bar-On, N. Antonovsky, E. Noor, Y. Zohar, G. Jona, E. Krieger, M. Shamshoum and A. Bar-Even, Cell, 2019, 179, 1255–1263 CrossRef CAS.
- T. E. Miller, T. Beneyton, T. Schwander, C. Diehl, M. Girault, R. McLean, T. Chotel, P. Claus, N. S. Cortina and J.-C. Baret, Science, 2020, 368, 649 CrossRef CAS PubMed.
- Y. Y. Birdja, E. Pérez-Gallent, M. C. Figueiredo, A. J. Göttle, F. Calle-Vallejo and M. T. Koper, Nat. Energy, 2019, 4, 732–745 CrossRef CAS.
- L. Zhang, Z.-J. Zhao, T. Wang and J. Gong, Chem. Soc. Rev., 2018, 47, 5423–5443 RSC.
- S. Lu, H. Hu, H. Sun, F. Yang, H. Zhu, M. Du, Y. Jin and W. Zhang, Green Chem., 2024, 26(10), 5744–5769 RSC.
- B. M. Tackett, E. Gomez and J. G. Chen, Nat. Catal., 2019, 2, 381–386 CrossRef CAS.
- Z. Xie, L. R. Winter and J. G. Chen, Matter, 2021, 4, 408–440 CrossRef CAS.
- C. S. Diercks, Y. Liu, K. E. Cordova and O. M. Yaghi, Nat. Mater., 2018, 17, 301–307 CrossRef CAS.
- J. Qiao, Y. Liu, F. Hong and J. Zhang, Chem. Soc. Rev., 2014, 43, 631–675 RSC.
- E. E. Benson, C. P. Kubiak, A. J. Sathrum and J. M. Smieja, Chem. Soc. Rev., 2009, 38, 89–99 RSC.
- H. Takeda, C. Cometto, O. Ishitani and M. Robert, ACS Catal., 2017, 7, 70–88 CrossRef CAS.
- S. Zhang, Q. Fan, R. Xia and T. J. Meyer, Acc. Chem. Res., 2020, 53, 255–264 CrossRef CAS PubMed.
- H. Yang, Q. Lin, C. Zhang, X. Yu, Z. Cheng, G. Li, Q. Hu, X. Ren, Q. Zhang and J. Liu, Nat. Commun., 2020, 11, 593 CrossRef CAS.
- M. Cheng, Y. Yu, X. Zhou, Y. Luo and M. Wang, ACS Catal., 2018, 9, 768–774 CrossRef.
- D. E. Polyansky, D. C. Grills, M. Z. Ertem, K. T. Ngo and E. Fujita, ACS Catal., 2022, 12, 1706–1717 CrossRef CAS.
- T. Ouyang, H. H. Huang, J. W. Wang, D. C. Zhong and T. B. Lu, Angew. Chem., 2017, 129, 756–761 CrossRef.
- C. Zhang, P. Gotico, R. Guillot, D. Dragoe, W. Leibl, Z. Halime and A. Aukauloo, Angew. Chem., Int. Ed., 2023, 62, e202214665 CrossRef CAS.
- M. E. Ahmed, S. Adam, D. Saha, J. Fize, V. Artero, A. Dey and C. Duboc, ACS Energy Lett., 2020, 5, 3837–3842 CrossRef CAS.
- J. A. Intrator, D. A. Velazquez, S. Fan, E. Mastrobattista, C. Yu and S. C. Marinescu, Chem. Sci., 2024, 15(17), 6385–6396 RSC.
- X. Jiang, X. Li, Y. Kong, C. Deng, X. Li, Q. Hu, H. Yang and C. He, J. Energy Chem., 2023, 76, 462–469 CrossRef CAS.
- Y. Yao, J.-H. Wu, G. Liu, R. Zhang, Z.-S. Yang, S. Gao, T.-C. Lau and J.-L. Zhang, ChemCatChem, 2024, 16(10), e202301705 CrossRef CAS.
- Y. Yao, J. H. Wu, G. Liu, R. Zhang, Z. S. Yang, S. Gao, T. C. Lau and J. L. Zhang, ChemCatChem, 2024, e202301705 CrossRef CAS.
- R. Kortlever, J. Shen, K. J. P. Schouten, F. Calle-Vallejo and M. T. Koper, J. Phys. Chem. Lett., 2015, 6, 4073–4082 CrossRef CAS PubMed.
- W. Zhang, Q. Qin, L. Dai, R. Qin, X. Zhao, X. Chen, D. Ou, J. Chen, T. T. Chuong and B. Wu, Angew. Chem., Int. Ed., 2018, 57, 9475–9479 CrossRef CAS PubMed.
- J. Zou, C. Y. Lee and G. G. Wallace, Advanced Science, 2021, 8, 2004521 CrossRef CAS.
- J. E. Huang, F. Li, A. Ozden, A. Sedighian Rasouli, F. P. García de Arquer, S. Liu, S. Zhang, M. Luo, X. Wang and Y. Lum, Science, 2021, 372, 1074–1078 CrossRef CAS.
- Y. Jia, F. Li, K. Fan and L. Sun, Adv. Powder Mater., 2022, 1, 100012 CrossRef.
- S. Gao, Z. Sun, W. Liu, X. Jiao, X. Zu, Q. Hu, Y. Sun, T. Yao, W. Zhang and S. Wei, Nat. Commun., 2017, 8, 14503 CrossRef CAS PubMed.
- T. Li, G. Luo, K. Liu, X. Li, D. Sun, L. Xu, Y. Li and Y. Tang, Adv. Funct. Mater., 2018, 28, 1805828 CrossRef.
- H.-J. Yang, J. Dong, Y.-H. Hong, W.-F. Lin, Z.-Y. Zhou and S.-G. Sun, Electrochem. Commun., 2018, 97, 82–86 CrossRef CAS.
- Q. Cheng, K. Mao, L. Ma, L. Yang, L. Zou, Z. Zou, Z. Hu and H. Yang, ACS Energy Lett., 2018, 3, 1205–1211 CrossRef CAS.
- A. S. Varela, W. Ju and P. Strasser, Adv. Energy Mater., 2018, 8, 1703614 CrossRef.
- X. Wang, Y. Pan, H. Ning, H. Wang, D. Guo, W. Wang, Z. Yang, Q. Zhao, B. Zhang and L. Zheng, Appl. Catal., B, 2020, 266, 118630 CrossRef CAS.
- A. S. Varela, N. Ranjbar Sahraie, J. Steinberg, W. Ju, H. S. Oh and P. Strasser, Angew. Chem., Int. Ed., 2015, 54, 10758–10762 CrossRef CAS PubMed.
- Z.-F. Huang, J. Song, Y. Du, S. Xi, S. Dou, J. M. V. Nsanzimana, C. Wang, Z. J. Xu and X. Wang, Nat. Energy, 2019, 4, 329–338 CrossRef CAS.
- J. Wang, L. Gan, W. Zhang, Y. Peng, H. Yu, Q. Yan, X. Xia and X. Wang, Sci. Adv., 2018, 4, eaap7970 CrossRef.
- X. Liang, J. Xiao, W. Weng and W. Xiao, Angew. Chem., 2021, 133, 2148–2152 CrossRef.
- F. Pan, B. Li, E. Sarnello, S. Hwang, Y. Gang, X. Feng, X. Xiang, N. M. Adli, T. Li and D. Su, Nano Energy, 2020, 68, 104384 CrossRef CAS.
- H. Lv, L. Lin, X. Zhang, D. Gao, Y. Song, Y. Zhou, Q. Liu, G. Wang and X. Bao, J. Mater. Chem. A, 2019, 7, 11967–11975 RSC.
- Y. Li, B. Hu, C. Xia, W. Q. Xu, J. P. Lemmon and F. Chen, J. Mater. Chem. A, 2017, 5, 20833–20842 RSC.
- J. Lu, C. Zhu, C. Pan, W. Lin, J. P. Lemmon, F. Chen, C. Li and K. Xie, Sci. Adv., 2018, 4, eaar5100 CrossRef PubMed.
- J. Ko, B.-K. Kim and J. W. Han, J. Mater. Chem. C, 2016, 120, 3438–3447 CAS.
- M. Behrens, F. Studt, I. Kasatkin, S. Kühl, M. Hävecker, F. Abild-Pedersen, S. Zander, F. Girgsdies, P. Kurr and B.-L. Kniep, Science, 2012, 336, 893–897 CrossRef CAS PubMed.
- Z. Chen, P. Kang, M.-T. Zhang, B. R. Stoner and T. J. Meyer, Energy Environ. Sci., 2013, 6, 813–817 RSC.
- S. C. Mandal, K. S. Rawat, S. Nandi and B. Pathak, Catal. Sci. Technol., 2019, 9, 1867–1878 RSC.
- H. Peng, M. T. Tang, X. Liu, P. S. Lamoureux, M. Bajdich and F. Abild-Pedersen, Energy Environ. Sci., 2021, 14, 473–482 RSC.
- Y. Hori, H. Wakebe, T. Tsukamoto and O. Koga, Surf. Sci., 1995, 335, 258–263 CrossRef CAS.
- S. C. Mandal and B. Pathak, ACS Appl. Nano Mater., 2021, 4, 11907–11919 CrossRef CAS.
- S. Kar, A. Goeppert and G. S. Prakash, J. Am. Chem. Soc., 2019, 141, 12518–12521 CrossRef CAS PubMed.
- S. C. Mandal, A. Das, D. Roy, S. Das, A. S. Nair and B. Pathak, Coord. Chem. Rev., 2022, 471, 214737 CrossRef CAS.
- A. Zafar, T. H. Mawat, E. M. Atiyah, M. Adnan Iqbal, A. Majeed, G. Iram, R. Qureshi and S. Khalid, ChemistrySelect, 2024, 9, e202304566 CrossRef CAS.
- A. Zafar, C. Takeda, A. Manzoor, D. Tanaka, M. Kobayashi, Y. Wadayama, D. Nakane, A. Majeed, M. A. Iqbal and T. Akitsu, Molecules, 2024, 29, 398 CrossRef CAS.
- H. A. Petersen, T. H. Myren and O. R. Luca, Inorganics, 2020, 8, 62 CrossRef CAS.
- B. Hammer and J. K. Norskov, Nature, 1995, 376, 238–240 CrossRef CAS.
- H. X. Zhong, J. Wang, Q. Zhang, F. Meng, D. Bao, T. Liu, X. Y. Yang, Z. W. Chang, J. M. Yan and X. B. Zhang, Adv. Sustainable Syst., 2017, 1, 1700020 CrossRef.
- C. Zhu, H. Li, S. Fu, D. Du and Y. Lin, Chem. Soc. Rev., 2016, 45, 517–531 RSC.
- Y. Yan, H. Cheng, Z. Qu, R. Yu, F. Liu, Q. Ma, S. Zhao, H. Hu, Y. Cheng and C. Yang, J. Mater. Chem. A, 2021, 9, 19489–19507 RSC.
- B. Talukdar, S. Mendiratta, M. H. Huang and C. H. Kuo, Chem. - Asian J., 2021, 16, 2168–2184 CrossRef CAS.
- Q. Li, Y.-C. Wang, J. Zeng, X. Zhao, C. Chen, Q.-M. Wu, L.-M. Chen, Z.-Y. Chen and Y.-P. Lei, Rare Met., 2021, 40, 3442–3453 Search PubMed.
- J. Fu, K. Liu, H. Li, J. Hu and M. Liu, Environ. Chem. Lett., 2021, 1–20 Search PubMed.
- W. Chen, Y. Wang, Y. Li and C. Li, CCS Chem., 2023, 5, 544–567 CrossRef CAS.
- X.-H. Liu, X.-L. Jia, Y.-L. Zhao, R.-X. Zheng, Q.-L. Meng, C.-P. Liu, W. Xing and M.-L. Xiao, Advanced Sensor and Energy Materials, 2023, 100073 CrossRef.
- F. Cheng, X. Zhang, K. Mu, X. Ma, M. Jiao, Z. Wang, P. Limpachanangkul, B. Chalermsinsuwan, Y. Gao and Y. Li, Energy Technol., 2021, 9, 2000799 CrossRef CAS.
- X. Bao, T. Wang and Y. Yang, Mater. Chem. Front., 2024, 8(3), 627–651 RSC.
- H. Tian, Z. Shui, M. A. Raza, L. Zhu and X. Chen, J. Alloys Compd., 2023, 958, 170544 CrossRef CAS.
- J. K. Nørskov, J. Rossmeisl, A. Logadottir, L. Lindqvist, J. R. Kitchin, T. Bligaard and H. Jonsson, J. Phys. Chem. B, 2004, 108, 17886–17892 CrossRef PubMed.
- Y. Li, D. Wang, Y. Guan, H. Liu, Y. Bao, N. Wu, X. Zhao, C. Sun, Z. Li and L. Lei, Nano Energy, 2024, 132, 110370 CrossRef.
- Y. Zhang, Q. Wu, J. Z. Y. Seow, Y. Jia, X. Ren and Z. J. Xu, Chem. Soc. Rev., 2024, 53, 8123–8136 RSC.
- A. Mahsud, M. Arif, W. U. Khan, T. Zhang, S. Hussain, M. Azam and Z. Lu, Mol. Catal., 2023, 550, 113526 CrossRef CAS.
- A. Sen and G. Rajaraman, Inorg. Chem., 2023, 62, 2342–2358 CrossRef CAS PubMed.
- C. Zhao, X. Dai, T. Yao, W. Chen, X. Wang, J. Wang, J. Yang, S. Wei, Y. Wu and Y. Li, J. Am. Chem. Soc., 2017, 139, 8078–8081 CrossRef CAS.
- X. Wang, S. Ding, T. Yue, Y. Zhu, M. Fang, X. Li, G. Xiao and L. Dai, Nano Energy, 2021, 82, 105689 Search PubMed.
- M. Duarte, N. Daems, J. Hereijgers, D. Arenas-Esteban, S. Bals and T. Breugelmans, J. CO2 Util., 2021, 50, 101583 CrossRef.
- C. Zhang, S. Yang, J. Wu, M. Liu, S. Yazdi, M. Ren, J. Sha, J. Zhong, K. Nie and A. S. Jalilov, Adv. Energy Mater., 2018, 8, 1703487 CrossRef.
- Y. Cheng, S. Zhao, H. Li, S. He, J.-P. Veder, B. Johannessen, J. Xiao, S. Lu, J. Pan and M. F. Chisholm, Appl. Catal., B, 2019, 243, 294–303 CrossRef CAS.
- L. R. L. Ting and B. S. Yeo, Curr. Opin. Electrochem., 2018, 8, 126–134 CrossRef CAS.
- N. Li, X. Chen, W.-J. Ong, D. R. MacFarlane, X. Zhao, A. K. Cheetham and C. Sun, ACS Nano, 2017, 11, 10825–10833 CrossRef CAS PubMed.
- H. Zhang, J. Li, S. Xi, Y. Du, X. Hai, J. Wang, H. Xu, G. Wu, J. Zhang and J. Lu, Angew. Chem., 2019, 131, 15013–15018 CrossRef.
- L. J. Arachchige, Y. Xu, Z. Dai, X. L. Zhang, F. Wang and C. Sun, J. Mater. Sci. Technol., 2021, 77, 244–251 CrossRef CAS.
- L. Jasin Arachchige, Y. Xu, Z. Dai, X. Zhang, F. Wang and C. Sun, J. Mater. Chem. C, 2020, 124, 15295–15301 Search PubMed.
- S. Dongare, N. Singh and H. Bhunia, Appl. Surf. Sci., 2021, 556, 149790 CrossRef CAS.
- R. Zhang, Y. Zhang, L. Liu, X. Li, Y. Tang, Y. Ni, C. Sun and H. Zhang, Appl. Surf. Sci., 2022, 582, 152472 CrossRef CAS.
- X. Cui, W. An, X. Liu, H. Wang, Y. Men and J. Wang, Nanoscale, 2018, 10, 15262–15272 RSC.
- W. Bi, C. Wu and Y. Xie, ACS Energy Lett., 2018, 3, 624–633 CrossRef CAS.
- L. Guo and S. Guo, Int. J. Hydrogen Energy, 2024, 51, 1532–1544 CrossRef CAS.
- M. J. E. A. Frisch, gaussian 09, Revision d. 01, Gaussian. Inc, Wallingford CT 201, 2009. Search PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865 Search PubMed.
- G. Rostamikia, A. J. Mendoza, M. A. Hickner and M. J. Janik, J. Power Sources, 2011, 196, 9228–9237 CrossRef CAS.
- Y. Zhao, X. Chang, A. S. Malkani, X. Yang, L. Thompson, F. Jiao and B. Xu, J. Am. Chem. Soc., 2020, 142, 9735–9743 CAS.
- X. Nie, M. R. Esopi, M. J. Janik and A. Asthagiri, Angew. Chem., Int. Ed., 2013, 52(9), 2611 CrossRef CAS.
- Q. Zhao and E. A. Carter, J. Chem. Theory Comput., 2020, 16, 6528–6538 Search PubMed.
- A. D. Handoko, F. Wei, Jenndy, B. S. Yeo and Z. W. Seh, Nat. Catal., 2018, 1(12), 922–934 CrossRef CAS.
- R. Kas, O. Ayemoba, N. J. Firet, J. Middelkoop, W. A. Smith and A. Cuesta, ChemPhysChem, 2019, 20, 2904–2925 CrossRef CAS.
- Y. Zheng, A. Vasileff, X. Zhou, Y. Jiao, M. Jaroniec and S.-Z. Qiao, J. Am. Chem. Soc., 2019, 141, 7646–7659 CrossRef CAS PubMed.
- T. K. Todorova, M. W. Schreiber and M. Fontecave, ACS Catal., 2019, 10, 1754–1768 CrossRef.
- H. Xiao, T. Cheng, W. A. Goddard III and R. Sundararaman, J. Am. Chem. Soc., 2016, 138, 483–486 CrossRef CAS PubMed.
- H. Xiao, T. Cheng and W. A. Goddard III, J. Am. Chem. Soc., 2017, 139, 130–136 CrossRef CAS.
- A. J. Garza, A. T. Bell and M. Head-Gordon, ACS Catal., 2018, 8, 1490–1499 CrossRef CAS.
- J. Zhao, J. Zhao, F. Li and Z. Chen, J. Mater. Chem. C, 2018, 122, 19712–19721 CAS.
- Z. Zhao and G. Lu, ACS Catal., 2018, 8, 3885–3894 CrossRef CAS.
- Y. Feng, W. An, Z. Wang, Y. Wang, Y. Men and Y. Du, ACS Sustainable Chem. Eng., 2019, 8, 210–222 Search PubMed.
- H. Mei, H. Cheng, Z. Wang and J. Li, Chem. Eng. Sci., 2017, 164, 81–89 CrossRef CAS.
- X.-F. Qiu, H.-L. Zhu, J.-R. Huang, P.-Q. Liao and X.-M. Chen, J. Am. Chem. Soc., 2021, 143, 7242–7246 CrossRef CAS.
- T. Cheng, H. Xiao and W. A. Goddard III, Proc. Natl. Acad. Sci. U. S. A., 2017, 114, 1795–1800 CrossRef CAS.
- K. P. Kuhl, E. R. Cave, D. N. Abram and T. F. Jaramillo, Energy Environ. Sci., 2012, 5, 7050–7059 RSC.
- J. Tamura, A. Ono, Y. Sugano, C. Huang, H. Nishizawa and S. Mikoshiba, Phys. Chem. Chem. Phys., 2015, 17, 26072–26078 RSC.
- K.-i. Tominaga, Y. Sasaki, T. Watanabe and M. Saito, J. Chem. Soc. Chem. Commun., 1995, 1489–1490 RSC.
- K. Schouten, Y. Kwon, C. Van Der Ham, Z. Qin and M. Koper, Chem. Sci., 2011, 2, 1902–1909 Search PubMed.
- K. U. Calvinho, A. W. Alherz, K. M. Yap, A. B. Laursen, S. Hwang, Z. J. Bare, Z. Clifford, C. B. Musgrave and G. C. Dismukes, J. Am. Chem. Soc., 2021, 143, 21275–21285 CrossRef CAS PubMed.
- X. Xu, Z. Xia, X. Zhang, H. Li, S. Wang and G. Sun, Nanoscale, 2020, 12, 3418–3423 RSC.
- M. Qiao, Y. Wang, Q. Wang, G. Hu, X. Mamat, S. Zhang and S. Wang, Angew. Chem., Int. Ed., 2020, 59, 2688–2694 CrossRef CAS PubMed.
- Y. Zheng, F. He, J. Wu, D. Ma, H. Fan, S. Zhu, X. Li, Y. Lu, Q. Liu and X. Hu, ACS Appl. Nano Mater., 2019, 2, 3538–3547 CrossRef CAS.
- H. Tan, J. Tang, J. Henzie, Y. Li, X. Xu, T. Chen, Z. Wang, J. Wang, Y. Ide and Y. Bando, ACS Nano, 2018, 12, 5674–5683 CrossRef CAS PubMed.
- M. Lefèvre, E. Proietti, F. Jaouen and J.-P. Dodelet, science, 2009, 324, 71–74 Search PubMed.
- B.-C. Hu, Z.-Y. Wu, S.-Q. Chu, H.-W. Zhu, H.-W. Liang, J. Zhang and S.-H. Yu, Energy Environ. Sci., 2018, 11, 2208–2215 RSC.
- T. Wang, Q. Zhang, K. Lian, G. Qi, Q. Liu, L. Feng, G. Hu, J. Luo and X. Liu, J. Colloid Interface Sci., 2024, 655, 176–186 CrossRef CAS PubMed.
- G. Li, X. Wan, Q. Zheng, M. Yang, Y. Xia, X. Qi, T. Wang and Z. Wu, Colloids Surf., A, 2024, 699, 134713 CrossRef CAS.
- E. Luo, H. Zhang, X. Wang, L. Gao, L. Gong, T. Zhao, Z. Jin, J. Ge, Z. Jiang and C. Liu, Angew. Chem., 2019, 131, 12599–12605 CrossRef.
- L. Zhang, B. Geng, Y. Gao, H. Kang, P. Wang, C. Liu, H. Xiao, M. Zhao, J. Jia and H. Wu, Chem. Eng. J., 2024, 481, 148086 CrossRef CAS.
- G. Li, X. Qi, G. Zhang, S. Wang, K. Li, J. Wu, X. Wan, Y. Liu and Q. Li, Microchem. J., 2022, 179, 107515 Search PubMed.
- X. Han, Y. Chang, T. Yue, M. Jia and J. Jia, J. Alloys Compd., 2024, 971, 172772 Search PubMed.
- W. Fan, Z. Li, C. You, X. Zong, X. Tian, S. Miao, T. Shu, C. Li and S. Liao, Nano energy, 2017, 37, 187–194 Search PubMed.
- Z.-Z. Wu, X.-L. Zhang, Z.-Z. Niu, F.-Y. Gao, P.-P. Yang, L.-P. Chi, L. Shi, W.-S. Wei, R. Liu and Z. Chen, J. Am. Chem. Soc., 2021, 144, 259–269 CrossRef PubMed.
- S. Zhang, S. Zhao, D. Qu, X. Liu, Y. Wu, Y. Chen and W. Huang, Small, 2021, 17, 2102293 CrossRef CAS PubMed.
- C. E. Creissen and M. Fontecave, Nat. Commun., 2022, 13, 2280 CrossRef CAS PubMed.
- S. Zhao, Y. Sun, K. Lu, J. Wang, M. Qiao, Y. Wang, Y. Huang, Y. Wu and Y. Chen, J. CO2 Util., 2023, 70, 102420 CrossRef CAS.
- C. Ma, H. Zhang, W. Kong, B. Shen and H. Lyu, Environ. Funct. Mater., 2022, 1, 284–297 Search PubMed.
- Z. Chen, G. Zhang, Y. Wen, N. Chen, W. Chen, T. Regier, J. Dynes, Y. Zheng and S. Sun, Nano-Micro Lett., 2022, 14, 25 CrossRef CAS.
- F. Pan, H. Zhao, W. Deng, X. Feng and Y. Li, Electrochim. Acta, 2018, 273, 154–161 CrossRef CAS.
- F. Pan, W. Deng, C. Justiniano and Y. Li, Appl. Catal., B, 2018, 226, 463–472 CrossRef CAS.
- X. Wang, Z. Chen, X. Zhao, T. Yao, W. Chen, R. You, C. Zhao, G. Wu, J. Wang and W. Huang, Angew. Chem., 2018, 130, 1962–1966 CrossRef.
- D. C. Marcano, D. V. Kosynkin, J. M. Berlin, A. Sinitskii, Z. Sun, A. Slesarev, L. B. Alemany, W. Lu and J. M. Tour, ACS Nano, 2010, 4, 4806–4814 CrossRef CAS PubMed.
- Z. Geng, X. Kong, W. Chen, H. Su, Y. Liu, F. Cai, G. Wang and J. Zeng, Angew. Chem., 2018, 130, 6162–6167 CrossRef.
- S. Huygh, A. Bogaerts and E. C. Neyts, J. Mater. Chem. C, 2016, 120, 21659–21669 CAS.
- Q. He, Y. Zhang, H. Li, Y. Yang, S. Chen, W. Yan, J. Dong, X. M. Zhang and X. Fan, Small, 2022, 18, 2108034 CrossRef CAS.
- C. Costentin, S. Drouet, M. Robert and J.-M. Savéant, Science, 2012, 338, 90–94 CrossRef CAS PubMed.
- S. Lin, C. S. Diercks, Y.-B. Zhang, N. Kornienko, E. M. Nichols, Y. Zhao, A. R. Paris, D. Kim, P. Yang and O. M. Yaghi, Science, 2015, 349, 1208–1213 CrossRef CAS PubMed.
- C. Yang, S. Li, Z. Zhang, H. Wang, H. Liu, F. Jiao, Z. Guo, X. Zhang and W. Hu, Small, 2020, 16, 2001847 CrossRef CAS.
- H.-x. Zhong, Q. Zhang, J. Wang, X.-b. Zhang, X.-l. Wei, Z.-j. Wu, K. Li, F.-l. Meng, D. Bao and J.-m. Yan, ACS Catal., 2018, 8, 3965–3970 CrossRef CAS.
- K.-H. Liu, H.-X. Zhong, S.-J. Li, Y.-X. Duan, M.-M. Shi, X.-B. Zhang, J.-M. Yan and Q. Jiang, Prog. Mater. Sci., 2018, 92, 64–111 CrossRef CAS.
- Y. Ma, J. Li, X. Liao, W. Luo, W. Huang, J. Meng, Q. Chen, S. Xi, R. Yu and Y. Zhao, Adv. Funct. Mater., 2020, 30, 2005000 CrossRef CAS.
- C. Yang, Z. Gao, D. Wang, S. Li, J. Li, Y. Zhu, H. Wang, W. Yang, X. J. Gao and Z. Zhang, Sci. China Mater., 2022, 65, 155–162 CrossRef CAS.
- J. Wang, X. Huang, S. Xi, H. Xu and X. Wang, Angew. Chem., Int. Ed., 2020, 59, 19162–19167 CrossRef CAS PubMed.
- Q. Wang, Y. Lei, D. Wang and Y. Li, Energy Environ. Sci., 2019, 12, 1730–1750 RSC.
- J. Li, Y. Kuang, Y. Meng, X. Tian, W.-H. Hung, X. Zhang, A. Li, M. Xu, W. Zhou and C.-S. Ku, J. Am. Chem. Soc., 2020, 142, 7276–7282 CrossRef CAS PubMed.
- L. Chen, C. He, R. Wang, Q. Li, J. Zeng, W. Liu, Y. Wang, Q. Wang, T. Ye and Y. Tang, Chin. Chem. Lett., 2021, 32, 53–56 CrossRef CAS.
- R. Yun, F. Zhan, X. Wang, B. Zhang, T. Sheng, Z. Xin, J. Mao, S. Liu and B. Zheng, Small, 2021, 17, 2006951 CrossRef CAS PubMed.
- H. Yang, Y. Dang, X. Cui, X. Bu, J. Li, S. Li, Y. Sun and P. Gao, Appl. Catal., B, 2023, 321, 122050 CrossRef CAS.
- F. Song, X. Yong, X. Wu, W. Zhang, Q. Ma, T. Zhao, M. Tan, Z. Guo, H. Zhao and G. Yang, Appl. Catal., B, 2022, 300, 120713 CrossRef CAS.
- A. S. Ismail, M. Casavola, B. Liu, A. Gloter, T. W. van Deelen, M. Versluijs, J. D. Meeldijk, O. Stéphan, K. P. de Jong and F. M. de Groot, ACS Catal., 2019, 9, 7998–8011 CrossRef CAS.
- R. Satthawong, N. Koizumi, C. Song and P. Prasassarakich, Catal. Today, 2015, 251, 34–40 Search PubMed.
- G. Wu, J. Wang, W. Ding, Y. Nie, L. Li, X. Qi, S. Chen and Z. Wei, Angew. Chem., Int. Ed., 2016, 55, 1340–1344 CrossRef CAS.
- M. Ding, Asian J. Chem., 2014, 26, 1808 CrossRef CAS.
- N. Liu, J. Wei, J. Xu, Y. Yu, J. Yu, Y. Han, K. Wang, J. I. Orege, Q. Ge and J. Sun, Appl. Catal., B, 2023, 328, 122476 CrossRef CAS.
- H. Zhong, M. Ghorbani-Asl, K. H. Ly, J. Zhang, J. Ge, M. Wang, Z. Liao, D. Makarov, E. Zschech and E. Brunner, Nat. Commun., 2020, 11, 1409 CrossRef CAS PubMed.
- W. Cheng, P. Yuan, Z. Lv, Y. Guo, Y. Qiao, X. Xue, X. Liu, W. Bai, K. Wang and Q. Xu, Appl. Catal., B, 2020, 260, 118198 CrossRef CAS.
- W. Ju, A. Bagger, G.-P. Hao, A. S. Varela, I. Sinev, V. Bon, B. Roldan Cuenya, S. Kaskel, J. Rossmeisl and P. Strasser, Nat. Commun., 2017, 8, 1–9 CrossRef CAS.
- X. Yang, T. Tat, A. Libanori, J. Cheng, X. Xuan, N. Liu, X. Yang, J. Zhou, A. Nashalian and J. Chen, Mater. Today, 2021, 45, 54–61 Search PubMed.
- F. Pan, H. Zhang, K. Liu, D. Cullen, K. More, M. Wang, Z. Feng, G. Wang, G. Wu and Y. Li, ACS Catal., 2018, 8, 3116–3122 Search PubMed.
- X. C. Huang, Y. Y. Lin, J. P. Zhang and X. M. Chen, Angew. Chem., Int. Ed., 2006, 45, 1557–1559 CrossRef CAS.
- W. Ren, X. Tan, W. Yang, C. Jia, S. Xu, K. Wang, S. C. Smith and C. Zhao, Angew. Chem., Int. Ed., 2019, 58, 6972–6976 CrossRef CAS PubMed.
- J. R. Croy, S. Mostafa, J. Liu, Y.-h. Sohn and B. Roldan Cuenya, Catal. Lett., 2007, 118, 1–7 CrossRef CAS.
- B. Roldan Cuenya, J. R. Croy, S. Mostafa, F. Behafarid, L. Li, Z. Zhang, J. C. Yang, Q. Wang and A. I. Frenkel, J. Am. Chem. Soc., 2010, 132, 8747–8756 CrossRef CAS.
- G. Winter, D. Thompson and J. Loehe, Inorganic syntheses, 1973, 14, 99–104 CrossRef.
- S. Kunze, P. Grosse, M. Bernal Lopez, I. Sinev, I. Zegkinoglou, H. Mistry, J. Timoshenko, M. Y. Hu, J. Zhao and E. E. Alp, Angew. Chem., 2020, 132, 22856–22863 CrossRef.
Footnote |
| † Both are the first authors. |
| This journal is © The Royal Society of Chemistry 2025 |