DOI:
10.1039/D4RA08825E
(Paper)
RSC Adv., 2025,
15, 10191-10199
A new method for the rapid determination of sodium percarbonate in aqueous samples using a modified HPLC setup†
Received
17th December 2024
, Accepted 27th March 2025
First published on 2nd April 2025
Abstract
Sodium percarbonate (SPC) is a widely used oxidant with applications in environmental remediation, especially within advanced oxidation processes (AOPs). Despite its prevalence, traditional methods for SPC quantification are often limited by complexity, cost, or lack of adaptability, creating a need for rapid, reliable, and scalable analytical approaches. This study presents a novel method for SPC quantification using a modified high-performance liquid chromatography with visible detection (HPLC-VIS) system. The key innovation lies in replacing the conventional separation column with a narrow-diameter loop reactor made of simple PEEK tubing, allowing SPC to react with acidified potassium iodide directly within the system. This modification eliminates the need for separate sample pretreatment, simplifies the analytical workflow, and enables real-time reaction monitoring while using standard HPLC equipment available in most laboratories. The method demonstrated high repeatability, reproducibility, and strong linearity (R2 > 0.99) across a range of pH values and in complex matrices, including highly saline and organic pollutant-containing samples. The method effectively monitored residual SPC levels in AOP-treated tramadol samples, where it confirmed continued SPC activity post-degradation of the target compound, indicating potential for comprehensive degradation of byproducts. Additionally, tests on a commercial SPC-based detergent (Vanish) validated the method's applicability for real-world samples. Overall, this HPLC-based technique provides a streamlined, environmentally friendly, and robust solution for SPC quantification, offering significant advantages for both research and industrial applications involving SPC in various water matrices.
1. Introduction
1.1. Sodium percarbonate
Sodium percarbonate (2Na2CO3·3H2O2) is a crystalline, water-soluble compound often used as a source of hydrogen peroxide.1 It appears as a granular, white powder and acts as a potent oxidizing agent, releasing sodium carbonate and hydrogen peroxide when dissolved in water. Known for its chemical stability, high solubility, and environmentally benign decomposition products,1 sodium percarbonate (SPC) has diverse applications. First introduced in the early 20th century as a stable, solid alternative to liquid hydrogen peroxide, it enabled easier handling and storage, meeting the growing demand for efficient and safe oxidizing agents in the chemical industry.2,3 Its utility spans a variety of sectors, including textiles,4 pulp and paper bleaching,5 and wastewater treatment,6,7 each of which has been key in driving its industrial relevance.
The market is experiencing rapid global expansion, with a projected compound annual growth rate (CAGR) over the coming years. A recent report from Intellectual Market Insights Research estimates that the global SPC market will grow from USD 1281.2 million in 2021 to USD 1816.76 million by 2028, marking an 8.1% CAGR. This growth is largely driven by the increasing use of SPC in household and personal care applications, such as laundry detergents8 as a bleaching agent, in stain removers,9 and in general household cleaners.10 Moreover, a global shift in consumer preference towards oxygen-based bleaches over traditional chlorine-based bleaches and the rise of eco-friendly cleaning formulations, made SPC-based products appealing to environmentally conscious consumers as it degrades into non-toxic byproducts.11,12 Medically, SPC is incorporated into dental care products, such as toothpaste, where it provides whitening and antibacterial benefits13,14 through the release of hydrogen peroxide, aiding in stain removal and bacterial reduction to enhance oral hygiene.15
Recent developments have sparked greater interest in the application of SPC in advanced oxidation processes (AOPs) for wastewater treatment.16–19 Due to its granular coating, SPC has enhanced stability, longer storage life, and reduced risk of explosion, presenting distinct advantages over liquid hydrogen peroxide.20 In addition, SPC is often added to the Fe(II)/persulfate(PS) system to address limitations in activation efficiency and effective pH range.21 This approach has shown promising results; Fu et al. demonstrated benzene removal using Fe2+-catalyzed SPC,22 and Epold et al. reported effective levofloxacin degradation in aqueous solutions using a combined Fenton and PS system.23
The broad and varied applications of SPC across multiple industries highlight the importance of developing fast and precise analytical methods for its determination. Despite its widespread use, rapid and accurate determination methods remain limited; such methods are essential for quality control processes, ensuring product purity and meeting desired SPC concentration levels.
1.2. A review of existing SPC determination method
The only established standard method for determining SPC is ISO 4321-1977 (E), titled Washing Powders – Determination of Active Oxygen Content – Titrimetric Method (ISO 4321-1977).24 This method employs titrimetric analysis, a colorimetric technique that, although accurate, is now considered outdated due to its time-intensive preparation and the substantial amounts of reagents required, which drive up costs (see Table 1S†). Moreover, its high limits of detection (LOD) and quantification (LOQ) make it inadequate for measuring low SPC concentrations. The technique is also susceptible to human error, as slight color variations in the titrant across trials can affect accuracy despite rigorous efforts at consistency.
Alternative methods reported in the literature face similar limitations (Table 1). Many are equally labor-intensive, depend on hazardous reagents, or require specialized equipment that is not widely available, restricting their accessibility in typical laboratory settings. Furthermore, these methods often lack automation capabilities, making them impractical for processing large sample volumes efficiently.
Table 1 Summary of SPC and H2O2 quantification techniques available in the literature, presenting the advantages and drawbacks in comparison to the proposed method
| Analytical technique |
Description |
Application |
Advantages |
Drawbacks |
| Titrimetric24 |
H2O2 is titrated with potassium permanganate (KMNO4) |
ISO number 4321 |
Accuracy |
High reagent requirements, labor-intensive |
| Colorimetric and fluorescent reaction-based probe25 |
Perhydrolysis of resorufin pivalate by H2O2, produced in situ from percarbonate, to restore the optical properties of resorufin dye |
Office-scanner-based sodium percarbonate detection |
Sensitivity (LOD: 0.45 μM), used available apparatus |
High reagent requirements, requires intensive sample preparation, non-automated |
| Attenuated total reflection-Fourier transform infrared (ATR-FTIR)26 |
Partial least squares (PLS) treatment of data obtained by attenuated total reflectance Fourier transform infrared (ATR FT-IR) spectrometry in wavenumber region of 1435–1342 cm−1 |
Determination of percarbonate in commercial washing powder detergent |
Accuracy |
Non-automated |
| Chemiluminescence sensor27 |
Sensor based on oxalic acid esters (bis(2,4,6-trichlorophenyl) oxalate-TCPO and bis(2,4-dinitrophenyl) oxalate-DNPO) |
Determination of percarbonate in commercial washing powder detergent |
Accuracy, LOD: 54 μg L−1 (for TCPO) and 48 μg L−1 (for DNPO) |
Reagents used are expensive and toxic, requires a special apparatus |
| Fluorescence-based Fenton reagent method28 |
Hydroxylation of benzoic acid by Fe(II) and H2O2 (Fenton reaction) forms fluorescent hydroxybenzoic acid, which is enhanced by complexation with Al(III) |
Real-time determination of hydrogen peroxide in aqueous SPC samples |
Sensitivity (LOD: 10−8 M) real-time detection |
Potential interference from SO2 at low pH and requires calibration adjustments for field conditions |
| Ion chromatography (IC) with UV detector29 |
Separation of hydrogen peroxide as hydroperoxyl ion (HO2−) using an anion exchange column (AEC) and quantification via UV absorption at 210 nm |
Detection and quantification of H2O2 in natural waters, wastewater, and environmental samples |
High specificity |
Requires high-pH eluent (>11.6), potential interference from strong oxidants and organics |
| High-performance liquid chromatography with diode array detector (HPLC-DAD)30 |
Uses the vanadium(V)-peroxo complex, HPLC-DAD, and the fluorescence of 7-hydroxycoumarin formed via Fenton reaction for indirect H2O2 detection |
Detection of trace residual H2O2 in processed fishery foods and other food products |
Detection of trace concentrations |
Requires toxic and hazardous reagents (ammonium metavanadate), high cost, includes derivatization reactions, potential interference from matrices and a complex protocol |
| High-performance liquid chromatography with fluorescence detector (HPLC-FLD) |
Uses Fenton reaction, where H2O2 reacts with coumarin to form 7-hydroxycoumarin, a fluorescent product detected at 330 nm (excitation)/460 nm (emission) |
Detection of trace H2O2 levels in food safety applications |
Detection of trace concentrations |
Requires toxic coumarin, and potential interference from other oxidizing agents in food samples |
| Iodometric (I3−) spectrophotometric method31 |
Based on the oxidation of iodide (I−) by H2O2 in the presence of ammonium molybdate, forming triiodide (I3−), which is measured at 351 nm |
Detection of H2O2 in aqueous solutions, environmental and radiation chemistry studies |
Detection of trace concentrations in small sample volumes |
Requires strict pH control, potential interference from side reactions in buffers, ammonium molybdate can be toxic in large quantities, and lacks automation |
| High-performance liquid chromatography with electrochemical detection (HPLC-ED, DC mode)32 |
H2O2 is detected via oxidation at a gold working electrode in a sodium acetate mobile phase at pH 10.5 |
Detection of trace H2O2 levels in forensic samples (post-blast debris), environmental monitoring, and explosive residues |
Highly selective, low detection limit (LOD: 0.02 mg mL−1, 0.6 mM), effective for post-blast forensic analysis |
Requires specialized electrochemical detector, electrode fouling can occur over time |
| High-performance liquid chromatography with fluorescence detection (HPLC-FD)32 |
Uses post-column derivatization, where H2O2 reacts with p-hydroxyphenylacetic acid (POPHA) and hemin to form a fluorescent dimer, detected at 320 nm (excitation)/405 nm (emission) |
Forensic analysis of H2O2 residues (pre- and post-blast), detection in environmental and controlled laboratory samples |
Highly sensitive, LOD: 0.19 mg mL−1 (6 mM), fluorescence detection provides strong selectivity for H2O2 |
Requires toxic hemin reagent, complex post-column derivatization, and precise flow control of multiple reagents |
1.3. Proposed method theory
The proposed approach uses a repurposed HPLC-VIS setup equipped with autosamplers, facilitating rapid determination of oxidants across numerous samples. By making minor, non-damaging modifications, the setup replaces the traditional reverse or normal phase separation column with a PEEK tube. The mobile phase consists of a concentrated acidified potassium iodide (KI) solution (pH = 3–4, [KI] = 40 mM), which swiftly reduces H2O2 in the sample.
In this setup, H2O2 reacts with I− to produce an iodine (I2) suspension in the PEEK tube (eqn (1)). The generated I2 then further reacts with excess I−, forming the triiodide anion (I3−). The PEEK tube functions as a reaction chamber allowing the reaction to reach completion before elution, ensuring accurate measurements. Detection is performed at 352 nm, the maximum absorption wavelength (λmax) of I3−, a wavelength where most common emerging organic pollutants do not absorb. This unique feature, alongside the use of an inorganic yet compatible mobile phase, offers a significant advantage for laboratories focused on advanced oxidation process (AOP) research involving SPC as an oxidant.
This setup allows researchers and industry professionals to process a large number of samples, each requiring minimal volume (<1 mL), and to determine both contaminant and oxidant concentrations using a single instrument. The method is particularly beneficial for laboratories with limited human resources, as it enables high-throughput analysis while reducing the time, labor, and costs associated with traditional techniques.
2. Chemicals
SPC standards were prepared using sodium percarbonate salt (≥99.00% assay) (Na2CO3·1.5H2O2) (Sigma-Aldrich, USA). The HPLC mobile phase was prepared using potassium iodide (KI) (puriss, 99.0–100.5%), sodium bicarbonate (NaHCO3) (Sigma-Aldrich, Germany) in addition to phosphoric acid (H3PO4) (Fisher Chemical, UK). Sodium hydroxide (NaOH) (Fisher Chemical, UK), sodium phosphate monobasic (NaH2PO4) (Sigma-Aldrich, Germany) and phosphoric acid (H3PO4) (Fisher Chemical, UK) were used for the adjustment of the pH. For spectator ions matrix effect experiments, sodium chloride (NaCl) (Fisher Scientific, UK), technical grade humic acid sodium salt, sodium bicarbonate (NaHCO3) (Sigma-Aldrich, Germany) and fumaric acid (Sigma-Aldrich, Switzerland) were used. Tramadol hydrochloride was obtained by dissolving TRAMAL® 50 mg capsules (STADA, Italy) in deionized water and filtering twice using 0.45 μm S-Pak® membrane filters (Merck, Germany) to remove insoluble excipients (microcrystalline cellulose, sodium starch glycolate, colloidal anhydrous silica, and magnesium stearate). Vanish detergent was purchased from a local store. All water used was of Millipore DI grade.
3. Detection setup
3.1. Modified HPLC setup specifications
The setup for our proposed method utilizes an HPLC-VIS system (Agilent 1100 series) equipped with a quaternary pump, a vacuum degasser, and an autosampler compartment maintained at 4 °C. In place of the conventional separation column, a PEEK tube (Restek, length 44 cm, ID 0.508 mm) is connected in series and kept at room temperature (20–25 °C). Detection is carried out using an integrated diode array detector with an embedded flow cell and a visible lamp (see Fig. 1S†).
3.2. Standards and mobile phase preparation
For the HPLC mobile phase, 5 g of sodium bicarbonate (NaHCO3) was dissolved in 1000 mL of deionized (DI) water to remove all dissolved oxygen.33 Next, 6.64 g of potassium iodide (KI) and 5 mL of phosphoric acid were added to a 500 mL portion of this solution, which was stirred until fully dissolved before being recombined with the remaining 500 mL. The prepared mobile phase was stored overnight in an airtight amber bottle to prevent light-catalyzed oxidation of I− to I2 and to ensure complete dissolution. The mobile phase was freshly prepared weekly to maintain solution stability. This preparation protocol was originally developed by our research group for persulfate (PS) and H2O2 determination.33,34
While SPC and H2O2 have similar properties, it is essential to account for the carbonate ions released during SPC dissolution, which buffer the acidity of the mobile phase. Therefore, this study was conducted to establish the optimal conditions for accurate measurements in the presence of these buffering ions.
SPC standards were prepared in 2 mL HPLC vials from a stock solution. Standards were discarded after a maximum of 3 hours, as SPC remains stable and active only for 5–6 hours.35
3.3. Mobile phase optimization
Sodium bicarbonate (NaHCO3) is initially added to the mobile phase to eliminate dissolved oxygen, thereby preventing the oxidation of iodide (I−) to iodine (I2) and enhancing background intensity. Afterward, potassium iodide (KI) is added and fully dissolved, followed by the addition of phosphoric acid. Phosphoric acid is included to promote the Fenton reaction (eqn (2)), which is kinetically favored under acidic conditions, maximizing the generation of I2 as shown in eqn (3). This prepared solution benefits from the use of cost-effective, highly water-soluble, and low-toxicity chemicals, which also avoid any corrosive effects on HPLC components.| | |
Na2CO3·1.5H2O2 → Na2CO3 + 1.5H2O2
| (1) |
| | |
2HO˙ + 2I− → I2 + 2OH−
| (3) |
3.4. Experimental procedures and conditions
To ensure the reliability of the proposed method, we first evaluated its repeatability, reproducibility, limit of detection (LOD), and limit of quantification (LOQ) using SPC standards prepared in deionized (DI) water. The details of these experiments and their results are provided in Section 4.1.
Following this, we assessed potential sample matrix effects by preparing SPC standards in solutions with specific characteristics. These included varying pH levels (pH 2, 7, and 11, adjusted using a 10 mM phosphate buffer) to investigate pH effects, and the presence of various matrix elements: chloride (Cl− at 20![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 mg L−1), bicarbonate (HCO3− at 150 mg L−1), ozonated species (O3− at 150 mg L−1), humic acid (10 mg L−1), and fumaric acid (10 mg L−1). Additionally, samples containing both SPC and tramadol at 10 mg L−1 were tested to simulate a real-life application involving SPC-based AOPs for tramadol degradation.
000 mg L−1), bicarbonate (HCO3− at 150 mg L−1), ozonated species (O3− at 150 mg L−1), humic acid (10 mg L−1), and fumaric acid (10 mg L−1). Additionally, samples containing both SPC and tramadol at 10 mg L−1 were tested to simulate a real-life application involving SPC-based AOPs for tramadol degradation.
SPC standards were also prepared and tested in natural water matrices, including sea and spring water, each at a concentration of 10 mg L−1. The results of these experiments are discussed in detail in Sections 4.2 and 4.3.
4. Results and discussion
4.1. Quality assurance and method validation
The method validation and quality assurance procedures were based on the guidelines provided in Quantitative Chemical Analysis by Harris (2010, Chapter 4 and 5), which was used to assess the SPC quantification procedure under various testing conditions. To ensure specificity, PEEK tubing configurations were accepted only if they produced high-quality peaks. Linearity was confirmed by an R2 value of ≥0.98. Accuracy was evaluated by testing freshly prepared standards during repeatability tests. The methods for determining precision (reproducibility), range, limit of detection (LOD), limit of quantitation (LOQ), and robustness are described in the following subsections. The HPLC pump flow rate was set to 0.1 mL min−1, and various injection volumes (5, 20, 50, and 100 μL) were tested to determine optimal conditions. The highest linearity (R2 = 0.999) was achieved with a flow rate of 0.1 mL min−1 and an injection volume of 100 μL, indicating ideal calibration conditions. The backpressure recorded under these conditions is 4 bars. Furthermore, the PEEK tube had been in use for oxidants determination in our lab for the past 5 years, processing thousands of samples under different methods,34,35 and shows no signs of degradation, and no change in inner diameter as it can be seen in Fig. 5S.†
4.1.1. Repeatability. To confirm the repeatability of the method, three separate PS standards were prepared and tested for each concentration on the same day by a single researcher using a consistent HPLC setup. The average calibration curves obtained are presented in Fig. 1. Error bars were calculated at a 95% confidence interval, using the formula: peak area = mean measurement ± ts/√n, where t is the Student's t-value (t = 4.303 for 2 degrees of freedom at a 95% confidence level), s is the standard deviation from three replicates, and n is the number of calibration standards. The LINEST function in Excel was used to compute the slope, y-intercept, regression coefficient, and other relevant statistical data, including standard deviations for each variable. The method demonstrated strong repeatability, as indicated by the minimal error bar values.
 |
| | Fig. 1 SPC calibration curve for repeatability methods. Vertical bars represent the error on the mean of three injections calculated at 95% confidence interval 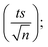 absent bars fall within symbols. absent bars fall within symbols. | |
4.1.2. Reproducibility. Reproducibility tests were carried out over a span of three days, encompassing their respective linear dynamic ranges (LDR). Each day, new mobile phase and standards were prepared, followed by the analysis of three replicates of each standard using the same HPLC instrument. Although it is advised to use a different HPLC instrument each day, as suggested by Harris (2010)36 only one HPLC instrument was accessible. Calibration curves, along with their corresponding error bars, are displayed in Fig. 2. Calibration curves exhibited variation from day to day, while maintaining a very good R2 value greater than 0.99. Therefore, it is strongly recommended to prepare fresh standards and obtain a new calibration curve whenever employing this method to control its low reproducibility.
 |
| | Fig. 2 SPC calibration curve for reproducibility methods. Vertical bars represent the error on the mean of three injections calculated at 95% confidence interval  absent bars fall within symbols. absent bars fall within symbols. | |
4.1.3. Limit of detection and quantification. The Harris method (Method Validation, 5-2) was followed to calculate the limit of detection (LOD) and limit of quantification (LOQ) of SPC.36 Seven tests were conducted to determine the LOD and LOQ using eqn (6) and (7), respectively. The LOD and LOQ for SPC were found to be 1.31 × 10−2 and 4.39 × 10−2 mM, respectively.| |
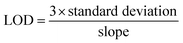 | (6) |
| |
 | (7) |
4.1.4. Method validation. Method validation was conducted following the procedures outlined in Quantitative Chemical Analysis by Harris36 (2010, Chapter 5, Section 5-2: Method Validation) and (2010, Chapter 4, Section 4-3: Comparison of Means with Student's t). The accuracy of the method was validated by having the reference standards' concentration fall within the precision range of the newly developed method as summarized in Table 2. For the t-test, the validation involved both the iodometric titration method, a widely used technique for SPC quantification, and the calibration method to prepare and test the standards. Each standard was measured twice, and the difference in concentrations between the two methods is summarized in Table 3. After data processing, t-test was used to compare the concentrations obtained via the calibration method with those from the iodometric titration approach in Table 4. Having a Student value at a 95% confidence interval greater than the calculated t shows that the new method is accepted.
Table 2 Calibration method results, [SPC] unit: mM
| [SPC]theoretical |
[SPC]1 |
[SPC]2 |
[SPC]avg |
ts/√n |
Range ([SPC]avg ± ts/√n) |
Result |
| 0.2 |
0.151936 |
0.158515 |
0.155225 |
0.059117 |
0.096108 |
0.214342 |
Accepted |
| 0.3 |
0.272179 |
0.267991 |
0.270085 |
0.03762 |
0.232465 |
0.307705 |
Accepted |
| 0.4 |
0.412442 |
0.404309 |
0.418376 |
0.073067 |
0.345308 |
0.491443 |
Accepted |
| 0.5 |
0.513143 |
0.530987 |
0.522065 |
0.160323 |
0.361742 |
0.682388 |
Accepted |
| 0.6 |
0.608389 |
0.606708 |
0.607549 |
0.015105 |
0.592444 |
0.622653 |
Accepted |
| 0.7 |
0.680666 |
0.690507 |
0.685587 |
0.088421 |
0.597166 |
0.774008 |
Accepted |
| 0.8 |
0.764894 |
0.752842 |
0.758868 |
0.108277 |
0.650591 |
0.867145 |
Accepted |
| 0.9 |
0.884192 |
0.900834 |
0.892513 |
0.149518 |
0.742995 |
1.042031 |
Accepted |
| 1 |
0.974672 |
0.988965 |
0.981818 |
0.128417 |
0.853402 |
1.110235 |
Accepted |
Table 3 Calibration method results, [SPC] unit: mM
| [SPC]theoretical |
[SPC]reference method |
[SPC]new method |
D |
| 0.2 |
0.189 |
0.155 |
−0.034 |
| 0.3 |
0.289 |
0.270 |
−0.019 |
| 0.4 |
0.415 |
0.405 |
−0.009 |
| 0.5 |
0.513 |
0.522 |
0.009 |
| 0.6 |
0.594 |
0.608 |
0.014 |
| 0.7 |
0.763 |
0.686 |
−0.077 |
| 0.8 |
0.803 |
0.759 |
−0.044 |
| 0.9 |
0.911 |
0.893 |
−0.018 |
| 1 |
1.102 |
0.982 |
−0.120 |
Table 4 Calibration method results
| Mean (đ) |
−0.033 |
| Std dev. (Sd) |
0.048 |
| t calculated |
1.822 |
| Degrees of freedom (n) |
7 |
| Student value at 95% confidence interval |
2.365 |
| Result |
Accepted |
4.2. pH effect
To evaluate the impact of sample pH on the proposed method, SPC standards were prepared in a 10 mM phosphate buffer (PB) at pH values of 2, 7, and 11, covering acidic, neutral, and basic conditions with substantial buffer strength (Fig. 3). The calibration curves for each buffered matrix demonstrated strong linearity (R2 > 0.99). The calibration curves for the pH 11 solution and the control (non-buffered) sample were nearly identical, suggesting that the method's sensitivity and response remain stable in alkaline environments. This stability may be attributed to the presence of sodium carbonate, a product of SPC dissociation in water. The slight increase in the slope of the calibration curves under acidic conditions (lower pH) likely results from enhanced reactivity, increased sensitivity, improved analyte stability, and reduced buffer interference. Acidic conditions may facilitate more efficient SPC dissolution into hydrogen peroxide and sodium carbonate, contributing to these effects.37
 |
| | Fig. 3 The effect of spectator species on the calibration curve. Vertical bars represent the error on the mean of three injections calculated at 95% confidence interval  absent bars fall within symbols. Experimental conditions: [FA], [HA], [TRA] = 10 mg L−1, [NaCl] = 20000 mg L−1, [HCO3−] = 150 mg L−1. absent bars fall within symbols. Experimental conditions: [FA], [HA], [TRA] = 10 mg L−1, [NaCl] = 20000 mg L−1, [HCO3−] = 150 mg L−1. | |
4.3. Matrix effects
It is essential to consider the effects of common ions and organic and inorganic compounds that may be present in sample matrices. To evaluate potential interference, SPC standards were prepared in aqueous matrices containing high concentrations of these substances, ensuring that naturally occurring ions and compounds—typically present in much lower concentrations—do not interfere with the analysis. The tested additives included chlorides (Cl−), bicarbonates (HCO3−), humic acids (HA), fumaric acid (FA), and tramadol (TRA). Results indicated minimal impact on the linearity and slopes of their respective calibration curves, with variations from the control calibration curve falling within the experimental error range (see Fig. 4). Further details on the selection of tested species and their concentrations are provided in subsequent sections.
 |
| | Fig. 4 The effect of the SPC sample's pH on the calibration curve. Vertical bars represent the error on the mean of three injections calculated at 95% confidence interval  absent bars fall within symbols. absent bars fall within symbols. | |
To assess the effect of salinity, which can enhance H2O2 decomposition,38,39 standards were prepared in a highly saline matrix with NaCl at 20000 mg L−1, classified as highly saline water by the Food and Agriculture Organization of the United Nations (FAO).40
Sample hardness was evaluated by preparing standards with 150 mg L−1 of HCO3−. This concentration was chosen because bicarbonate ions (HCO3−) are commonly present in water due to atmospheric CO2 dissolution and may act as activators of H2O2.41
In addition, natural water matrices often contain organic compounds such as HA and FA, derived from decomposing organic material.42 Therefore, SPC standards with 10 mg L−1 of either HA or FA were prepared to assess their effects. Furthermore, the potential interference of complex organic pollutants, such as micro-contaminants commonly found in AOP-treated water, was examined by preparing standards with tramadol (TRA) at a concentration of 10 mg L−1.
Overall, none of these additives had a significant effect on the method's accuracy, as indicated by Fig. 4.
4.4. Application to natural water matrices
To evaluate the effectiveness of the developed analytical method in natural water matrices, SPC standards were prepared in both sea and spring water. The natural water samples' quality parameters are provided in Table 2S.† As shown in Fig. 5, a slight increase in signal intensity (slope) of 17% was observed in sea water, while no significant effect was noted in spring water. The calibration curves for both natural water matrices displayed strong linearity (R2 > 0.99), indicating reliable performance across different water types.
 |
| | Fig. 5 The effect of natural water matrices on the calibration curve. Vertical bars represent the error on the mean of three injections calculated at 95% confidence interval  absent bars fall within symbols. absent bars fall within symbols. | |
4.5. Application to AOPs research in action
SPC is commonly used as an oxidant in AOPs. However, many studies that focus on optimizing these applications often neglect monitoring the residual oxidant (SPC), a critical step that is labor-intensive and time-consuming with traditional quantification methods. Advanced techniques, meanwhile, are often prohibitively expensive. To address this, we used SPC in a UVC/SPC system for the degradation of tramadol (TRA). Samples collected at various degradation intervals were processed using our method to determine the residual [SPC]. Fig. 4S† shows the decreasing absorbance of TRA during the oxidation control experiment from 220 nm to 300 nm over 6 min of reaction time. The calibration method was applied to quantify the remaining [SPC] and to calculate the percentage reaction stoichiometric efficiency (% RSE) as defined by eqn (8).43 The results (Fig. 6) showed that SPC consumption continued even after the complete degradation of the target compound, indicating that SPC effectively mineralizes the degradation byproducts formed during the process.| |
 | (8) |
 |
| | Fig. 6 Application for SPC quantification in AOPs research. Vertical bars represent the error on the mean of three injections calculated at 95% confidence interval 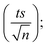 absent bars fall within symbols. Experimental conditions: [TRA] = 10 mg L−1, [SPC] = 0.5 mM. The inset represents the % RSE determined for the experiment. absent bars fall within symbols. Experimental conditions: [TRA] = 10 mg L−1, [SPC] = 0.5 mM. The inset represents the % RSE determined for the experiment. | |
Notably, the developed method facilitated a rapid calculation of % RSE, a crucial factor for evaluating UVC's effect on SPC activation. The relatively acceptable % RSE of 25% suggests a high affinity of SPC toward the degradation of byproducts in the reactive medium. However, this value remains much greater than the % RSE obtained in similar AOP systems using persulfate as oxidant with iron species for persulfate activation.44–46 These findings highlight the importance of further investigating the role of UVC in this context, as well as monitoring TRA degradation byproducts to optimize the applied AOP.
4.6. Application to washing powder detergents
To evaluate the effectiveness of the developed analytical method, we tested it using the washing powder detergent Vanish (Fig. 2S†), which contains between 10 and 20% SPC,47 along with other substances (Table 3S†). Samples with varying detergent concentrations were prepared and analyzed using the proposed method. The resulting “chromatograms” displayed a single peak at the same retention time as SPC (Fig. 3Sa†), confirming the presence of SPC in the samples (Fig. 3Sb†).
The results, shown in Fig. 7, indicate a strong linear relationship between the peak area and the concentration of Vanish. The calibration curve demonstrated high linearity (R2 > 0.99) and consistency, underscoring the method's reliability for quantifying SPC in powder detergents. The small error bars indicate excellent reproducibility and minimal experimental error. Furthermore, the percentage of SPC in Vanish powder was determined using our method to be 10.83%, which is within the range of expected values (10–20%).
 |
| | Fig. 7 Calibration curve showing the area versus the concentration of Vanish. Vertical bars represent the error on the mean of three injections calculated at 95% confidence interval  absent bars fall within symbols. absent bars fall within symbols. | |
5. Conclusions
Sodium percarbonate (SPC), a widely used oxidant with numerous applications, has experienced increased demand in recent years, emphasizing the need for rapid, reliable methods to monitor its concentration in aqueous media. Existing methods for SPC quantification have distinct limitations and specific requirements. Here, we present a novel method using a modified HPLC configuration to quantify SPC across various water matrix conditions. This method offers significant advantages over traditional techniques in terms of simplicity, scalability, automation, and reduced chemical consumption per sample. It has demonstrated excellent repeatability, reproducibility, and adaptability across a wide range of pH and salinity conditions. Furthermore, the method showed minimal interference from organic compounds (OCs) when applied in advanced oxidation process (AOP) research, whether in deionized or natural water matrices. This approach provides an efficient, effective, and environmentally friendly solution for SPC quantification across diverse water matrices.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was funded in part by the University Research Board (award number 103603) of the American University of Beirut and USAID-Lebanon through the National Academy of Sciences under PEER project 5-18 (award number 103262) and the MEPI TLP program (award number 103992). A. Nasreddine would like to thank the FAS Dean's office through the undergraduate research experience award. The authors are thankful to Joan Younes, Simon Al-Ghawi and Boutros Sawaya for their technical assistance and graphic designer Dany Khoury for the design of the graphical abstract. They are also very thankful to the personnel of the K. Shair CRSL for their kind help.
References
- M. Khanmohammadi, N. Mashkuri, M. Rostami and A. B. Garmarudi, J. Anal. Chem., 2012, 67, 330–334 CAS.
- H. Xue, J. Li, G. Zhang, B. Liu, M. Li and H. Wang, Environ. Sci.: Water Res. Technol., 2023, 9, 2171–2187 CAS.
- P. Li, X. Cheng, W. Zhou, C. Luo, F. Tan, Z. Ren, L. Zheng, X. Zhu and D. Wu, J. Cleaner Prod., 2020, 269, 122228 CAS.
- Q. Li, R. Lu, Y. Liang, K. Gao and H. Jiang, Materials, 2022, 15, 5849 CAS.
- E. Pesman, S. Imamoglu, E. Ersoy Kalyoncı and H. Kirci, Bioresources, 2014, 9, 523–536 CAS.
- Y. Huang, L. Bu, Y. Wu, S. Zhu, S. Zhou, Z. Shi and D. D. Dionysiou, Chem. Eng. J., 2022, 442, 135995 CAS.
- X. Ling, J. Deng, C. Ye, A. Cai, S. Ruan, M. Chen and X. Li, Sci. Total Environ., 2021, 799, 149382 Search PubMed.
- G. O. Bianchetti, C. L. Devlin and K. R. Seddon, RSC Adv., 2015, 5, 65365–65384 CAS.
- C. Oldfield, R. M. Morgan, H. F. Miles and J. C. French, Aust. J. Forensic Sci., 2018, 50, 345–354 Search PubMed.
- E. Igos, R. Moeller, E. Benetto, A. Biwer, M. Guiton and P. Dieumegard, Chemosphere, 2014, 100, 160–166 CAS.
- G. L. Liu, Q. L. Cui and C. W. Yu, Adv. Mater. Res., 2011, 183–185, 1423–1427 CAS.
- X. Fu, X. Gu, S. Lu, V. K. Sharma, M. L. Brusseau, Y. Xue, M. Danish, G. Y. Fu, Z. Qiu and Q. Sui, Chem. Eng. J., 2017, 309, 22–29 Search PubMed.
- J. Kaneko, S. Inoue, S. Kawakami and H. Sano, J. Endod., 2000, 26, 25–28 CrossRef CAS PubMed.
- R. Date, J. Yue, A. Barlow, P. Bellamy, M. Prendergast and R. Gerlach, Am. J. Dent., 2003, 16, 3B–8B Search PubMed , https://pubmed.ncbi.nlm.nih.gov/15055980/.
- M. Alam, R. Jagger, R. Vowles and J. Moran, Int. J. Dent. Hyg., 2011, 9, 37–42 CrossRef CAS PubMed.
- K. Fedorov, M. P. Rayaroth, N. S. Shah and G. Boczkaj, Chem. Eng. J., 2023, 456, 141027 CrossRef CAS.
- Y. Li, Y. Zhu, D. Wang, G. Yang, L. Pan, Q. Wang, B.-J. Ni, H. Li, X. Yuan, L. Jiang and W. Tang, Water Res., 2020, 174, 115626 Search PubMed.
- B. Pieczykolan, I. Płonka and K. Barbusiński, Architecture, Civil Engineering, Environment, 2016, 9, 135–140 CrossRef.
- J. Gao, R. F. Nunes, K. O'Shea, G. L. Saylor, L. Bu, Y.-G. Kang, X. Duan, D. D. Dionysiou and S. Luo, Water Res., 2022, 219, 118457 CrossRef CAS PubMed.
- J. Gao, X. Duan, K. O'Shea and D. D. Dionysiou, Water Res., 2020, 171, 115394 CrossRef CAS PubMed.
- J. Mo, T. Lin, X. Zhang, F. Jiang and H. Chen, Desalination, 2023, 547, 116258 CrossRef CAS.
- X. Fu, X. Gu, S. Lu, Z. Miao, M. Xu, X. Zhang, Z. Qiu and Q. Sui, Chem. Eng. J., 2015, 267, 25–33 CrossRef CAS.
- I. Epold, M. Trapido and N. Dulova, Chem. Eng. J., 2015, 279, 452–462 CrossRef CAS.
- ISO 4321:1977 Washing Powders – Determination of Active Oxygen Content – Titrimetric Method, https://www.iso.org/standard/10192.html Search PubMed.
- K. M. Lee, K. Y. Park, N. Y. Kim, J. H. Yoo, M. G. Choi, S. Ahn and S.-K. Chang, Dyes Pigm., 2021, 184, 108804 CrossRef CAS.
- K. K. M. Khanmohammadi, Talanta, 2005, 65, 824–827 Search PubMed.
- E. Omanovic and K. Kalcher, Int. J. Environ. Anal. Chem., 2005, 85, 853–860 CAS.
- J. H. Lee, I. N. Tang, J. B. Weinstein-Lloyd and E. B. Halper, Environ. Sci. Technol., 1994, 28, 1180–1185 CAS.
- M. Song, J. Wang, B. Chen and L. Wang, Anal. Chem., 2017, 89, 11537–11544 CAS.
- J. Park, H. Noh, H. J. Suh, D. Ryu, H. J. Lee and C. Lee, Food Sci. Biotechnol., 2023, 32, 27–37 CAS.
- N. V. Klassen, D. Marchington and H. C. E. McGowan, Anal. Chem., 1994, 66, 2921–2925 CAS.
- M. Tarvin, B. McCord, K. Mount and M. L. Miller, Forensic Sci. Int., 2011, 209, 166–172 CAS.
- O. Tantawi, A. Baalbaki, R. El Asmar and A. Ghauch, Sci. Total Environ., 2019, 654, 107–117 CAS.
- A. Baalbaki, N. Zein Eddine, S. Jaber, M. Amasha and A. Ghauch, Talanta, 2018, 178, 237–245 CAS.
- Household Uses for Sodium Percarbonate, https://www.chemistrystore.com/sodium_percarbonate2.pdf Search PubMed.
- D. C. Harris, Quantitative Chemical Analysis, 8th edn, 2010 Search PubMed.
- A. Rastinfard, B. Dalisson and J. Barralet, Acta Biomater., 2022, 145, 390–402 CrossRef CAS PubMed.
- C.-H. Liao, S.-F. Kang and F.-A. Wu, Chemosphere, 2001, 44, 1193–1200 CrossRef CAS PubMed.
- J. De Laat, G. T. Le and B. Legube, Chemosphere, 2004, 55, 715–723 CAS.
- J. D. Rhoades, A. Kandiah and A. M. Mashali, The use of saline waters for crop production, FAO Irrigation and Drainage paper 48, FAO, Rome, 1992 Search PubMed.
- D. E. Richardson, H. Yao, K. M. Frank and D. A. Bennett, J. Am. Chem. Soc., 2000, 122, 1729–1739 CAS.
- F. J. Stevenson, Humus Chemistry: Genesis, Composition, Reactions, John Wiley & Sons, 1994 Search PubMed.
- A. Ghauch, A. Baalbaki, M. Amasha, R. El Asmar and O. Tantawi, Chem. Eng. J., 2017, 317, 1012–1025 CAS.
- A. Ghauch, G. Ayoub and S. Naim, Chem. Eng. J., 2013, 228, 1168–1181 CAS.
- G. Ayoub and A. Ghauch, Chem. Eng. J., 2014, 256, 280–292 CAS.
- S. Naim and A. Ghauch, Chem. Eng. J., 2016, 288, 276–288 CAS.
- Reckitt Benckiser Group, Safety Data Sheet for Vanish Oxi Action Powder Fabric Stain Remover, 2019 Search PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Sarah Ghazali,
Weam Bou Karroum
,
Sarah Ghazali,
Weam Bou Karroum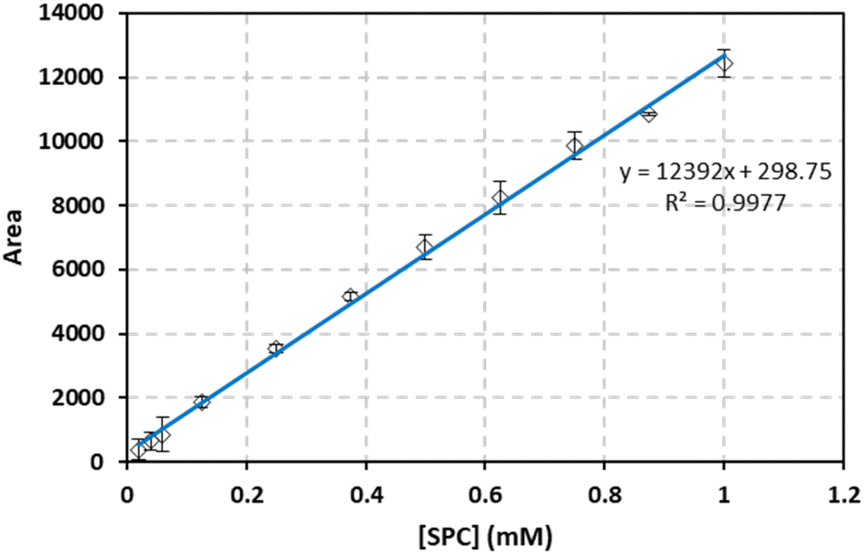
 absent bars fall within symbols.
absent bars fall within symbols.
 absent bars fall within symbols.
absent bars fall within symbols.


 absent bars fall within symbols. Experimental conditions: [FA], [HA], [TRA] = 10 mg L−1, [NaCl] = 20
absent bars fall within symbols. Experimental conditions: [FA], [HA], [TRA] = 10 mg L−1, [NaCl] = 20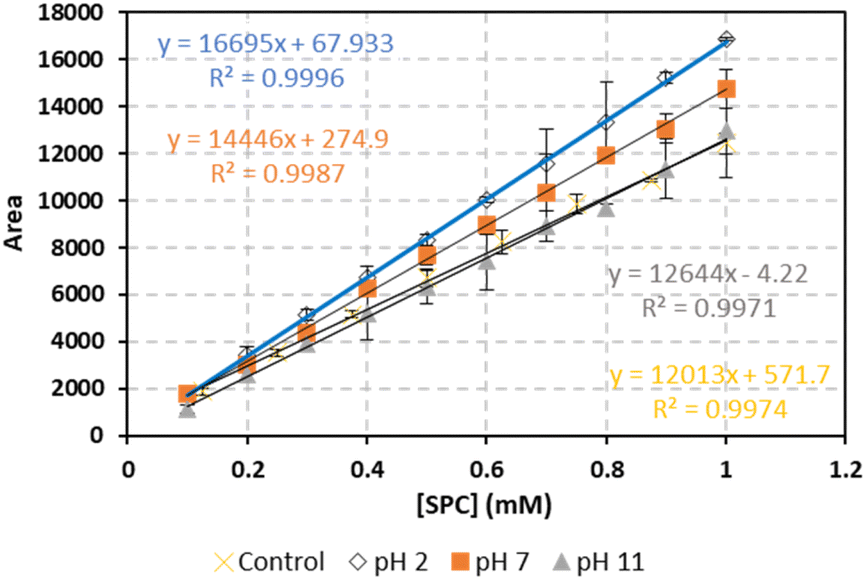
 absent bars fall within symbols.
absent bars fall within symbols.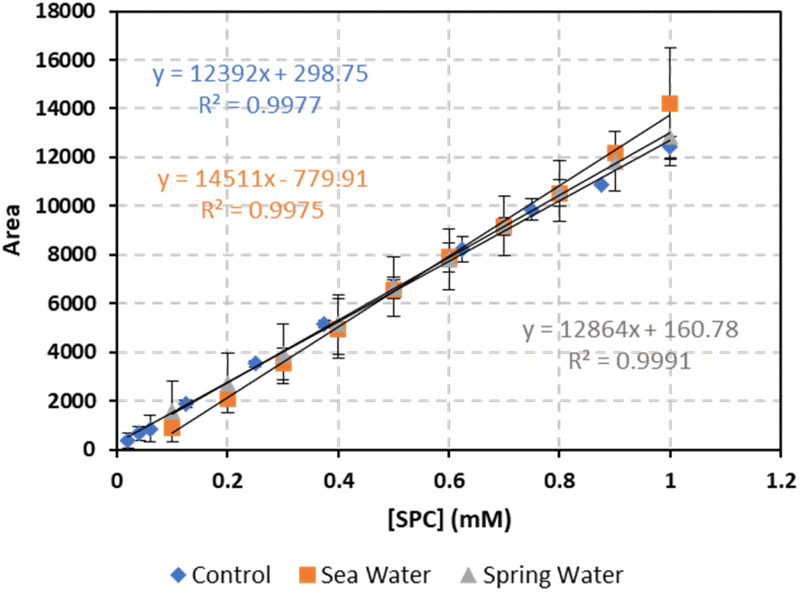
 absent bars fall within symbols.
absent bars fall within symbols.

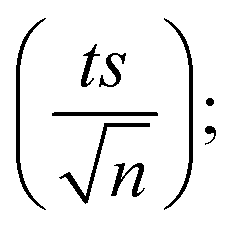 absent bars fall within symbols. Experimental conditions: [TRA] = 10 mg L−1, [SPC] = 0.5 mM. The inset represents the % RSE determined for the experiment.
absent bars fall within symbols. Experimental conditions: [TRA] = 10 mg L−1, [SPC] = 0.5 mM. The inset represents the % RSE determined for the experiment.
 absent bars fall within symbols.
absent bars fall within symbols.