
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceStudy on gas adsorption and separation performance of alkyl functionalized MOF materials under wet conditions†
Yuqiu Jiaoa,
Zishen Duab,
Qingying Xiea,
Xinai Wena,
Wansong Zhanga,
Jianwei Zhua,
Guiwu Lu a,
Guanggang Zhou*a,
Zhenqing Yang*a and
Yuhong Xia*a
a,
Guanggang Zhou*a,
Zhenqing Yang*a and
Yuhong Xia*a
aCollege of Science, China University of Petroleum, Beijing 102249, P. R. China. E-mail: bjpeuzgg@163.com; yangzhq@cup.edu.cn; xiayuhong1999@163.com
bBeijing Gas Group Company Limited, P. R. China
First published on 27th January 2025
Abstract
Metal–organic framework materials exhibit significant potential for diverse applications in gas adsorption and separation. We have studied the performance changes of Cu-BTC, Cu-MBTC and Cu-EBTC under different water-containing conditions. GCMC studies shows that, compared with Cu-BTC, the water absorption properties of Cu-MBTC and Cu-EBTC have a certain degree of decline, which is consistent with the experimental results. The incorporation of alkyl groups led to a decrease in the heat of adsorption for both Cu-MBTC and Cu-EBTC when exposed to water. The gas adsorption performance of Cu-BTC, Cu-MBTC and Cu-EBTC exhibits inconsistent variations in gas adsorption performance under water-containing conditions. The addition of coordinated water molecules enhances the ability of Cu-BTC to adsorb CO2. The capacity of Cu-MBTC with 4 wt% water to adsorb CO2 increased, but the capacity of Cu-MBTC with 8 wt% water to adsorb CO2 decreased. The presence of coordinated water molecules reduces the CO2 adsorption performance of Cu-EBTC. The study of the heat of adsorption explains this phenomenon from the mechanism. At the same time, we found an interesting phenomenon. The existence of trace water molecules can greatly improve the adsorption selectivity of Cu-EBTC to CO2/N2, while the pressure and gas ratio have little effect. It can provide more possibilities for the widespread use of MOF in the fields of gas adsorption and separation.
1 Introduction
Metal–organic frameworks (MOFs) are crystalline materials formed by the coordination of metal ions and organic ligands,1 characterized by their periodic structures and diverse applications in gas storage, separation, and catalysis.2–5 Among these, HKUST-1 (Cu-BTC), composed of benzene-1,3,5-tricarboxylic acid and Cu2+ ions, is a widely studied MOF due to its large surface area, high pore volume, and open metal sites, making it highly promising for gas adsorption and separation.6–8 However, the hydrothermal stability of MOFs, such as Cu-BTC, is a critical concern as water molecules can interact with metal sites fiercely, causing hydrolysis and structural degradation, which limits their practical applications.9–12Michela et al. identified three stages of hydrolysis in Cu-BTC, ultimately leading to crystal collapse, while Calero et al. used Monte Carlo simulations to model the water adsorption, and achieve results that align closely with the experimental data.13–15 Similarly, Sholl et al.16 developed a novel force field based on dispersion-corrected density functional theory (DFT-D2) to improve the adsorption predictions.15,17–19 Despite these advances, challenges remain in accurately simulating the strong water–copper interactions in Cu-BTC.20,21
Density functional theory (DFT) has been widely used to explore MOF-water interactions at a molecular level. Studies have investigated the adsorption of various small molecules, such as water, to Cu-BTC and similar MOFs.16 For example, Rubeš et al. evaluated the accuracy of different density functionals for MOFs containing transition metals,22,23 and Heine et al. studied vibrational characteristics of the adsorbed species.24 Additionally, ab initio molecular dynamics simulations have provided insights into the hydrolysis and decomposition mechanisms of Cu–Cu nodes in HKUST-1.25,26
Recent research has also explored modifications to the organic linkers in MOFs to enhance their performance.27,28 For example, the introduction of alkyl groups, typically considered hydrophobic, can alter water adsorption behavior and improve gas selectivity. Notably, the presence of trace water has been reported to influence the adsorption selectivity of MOFs for CO2 and N2.29 Despite extensive studies on Cu-BTC, limited attention has been given to methyl-modified Cu-BTC (Cu-MBTC) and ethyl-modified Cu-BTC (Cu-EBTC), particularly under humid conditions.27
This study uses computational methods to explore the water adsorption behavior of Cu-MBTC and Cu-EBTC. By calculating water adsorption isotherms and heats of adsorption, the effects of alkyl functionalization in water adsorption are elucidated. Furthermore, the adsorption isotherms and selectivity of these materials for CO2 and N2 in the presence of trace water are evaluated. The findings reveal that alkyl groups can cause reduction of water adsorption and adsorption heat while they have a influence on CO2 adsorption and selectivity. Specifically, coordinated water enhances CO2 adsorption in Cu-BTC but reduces it in Cu-EBTC. These insights highlight the potential applications of functionalized MOFs in CO2 adsorption and separation.
2 Calculation method
2.1 First-principles calculation
The cluster models representing the chemical characteristics of the skeleton are extracted from the unit cell of the MOF materials, as shown in Fig. 1. Generally, when MOFs (Cu-BTC, Cu-MBTC and Cu-EBTC) containing unsaturated metal sites adsorb water molecules, they will preferentially adsorb near the metal sites. Previous research results show that when there are three or more water molecules in the local environment surrounding the Cu–Cu node in the cluster model, the structure of the cluster model will be completely separated and disintegrated.25 Therefore, we built a model by adding 1–2 water molecules near the Cu atom in the cluster model, and subsequently calculated the most stable water adsorption structure and specific configuration of Cu-BTC, Cu-MBTC, and Cu-EBTC under the given local water concentration. Additionally, we computed the adsorption energy and atomic charge distribution. The specific calculation method is to use the 6-31+g(d) basis set to optimize the geometric structure at the B3LYP functional theory level, with the inclusion of D3 dispersion corrections. Subsequently, the def2-TZVP basis set is employed to calculate the energy with greater accuracy. The Chelpg method is used to fit the electrostatic potential to obtain the atomic charge distribution. All DFT calculations are done using Gaussian 16 software.30 First-principles calculations are used in two aspects: (1) calculate the adsorption heat and adsorption position of Cu-BTC, Cu-MBTC and Cu-EBTC for water molecules; (2) calculate the atomic distribution charge of Cu-BTC, Cu-MBTC and Cu-EBTC for GCMC calculation. | ||
| Fig. 1 First-principles calculations (a) Cu-BTC, (b) Cu-MBTC and (c) Cu-EBTC initial cluster model. | ||
2.2 GCMC calculation
The GCMC method was used to calculate the gas adsorption isotherms of Cu-BTC, Cu-MBTC and Cu-EBTC. We used the Sorption Module in Materials Studio 8.0. The initial model used the crystal structure data reported by Williams6 and Yang Cai31 after removing impurities. As shown in Fig. 2, for the Cu-BTC, Cu-MBTC and Cu-EBTC models under water conditions, based on the initial model, we have added water molecules at a certain distance in the vicinity of the Cu atom, based on the initial model and according to the DFT calculation results. We have established several supercell models. Periodic boundary conditions are used, and the cutoff radius is 15.5 Å. The universal force field is used in the GCMC simulations, and the water molecules are represented as SPC models, and we used 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000000 steps for the equilibration and 5000000 steps for the production for each GCMC simulation. The N2 molecule is represented as a three-point model, in which two points are located on two N atoms and the third point is located on its center of mass (COM). Each N2 molecule has a negative charge on each N atom (q = −0.482e) and a positive charge at the COM site (q = 0.964e). CO2 is also modeled as a three-point model, with positive charges (q = 0.70e) assigned to the C atom and negative charges (q = −0.35e) assigned to oxygen atoms. These models and calculation parameters have been extensively utilized in previous studies.27,32
000000 steps for the equilibration and 5000000 steps for the production for each GCMC simulation. The N2 molecule is represented as a three-point model, in which two points are located on two N atoms and the third point is located on its center of mass (COM). Each N2 molecule has a negative charge on each N atom (q = −0.482e) and a positive charge at the COM site (q = 0.964e). CO2 is also modeled as a three-point model, with positive charges (q = 0.70e) assigned to the C atom and negative charges (q = −0.35e) assigned to oxygen atoms. These models and calculation parameters have been extensively utilized in previous studies.27,32
 | ||
| Fig. 2 (a) Cu-BTC unit cell. (b) Cu-BTC unit cell with coordinated water molecules (4 wt%). (c) Front view of Cu-BTC first-principles calculation model for coordinated water molecules (4 wt%). (d) The top view of the Cu-BTC first-principles calculation model with coordinated water molecules (4 wt%). Copper atoms are orange, oxygen is red, carbon is gray, hydrogen is white. | ||
The adsorption isotherms were fitted by the Dual Site Langmuir–Freundlich equation (DSLF), and the adsorption selectivity of Cu-BTC, Cu-MBTC and Cu-EBTC for binary gas mixtures of CO2 and N2 was calculated by combining the obtained parameters with the IAST theory. The specific method can be found in the literature.33
3 Results and discussion
3.1 Adsorption of water by alkyl functionalized Cu-BTC materials
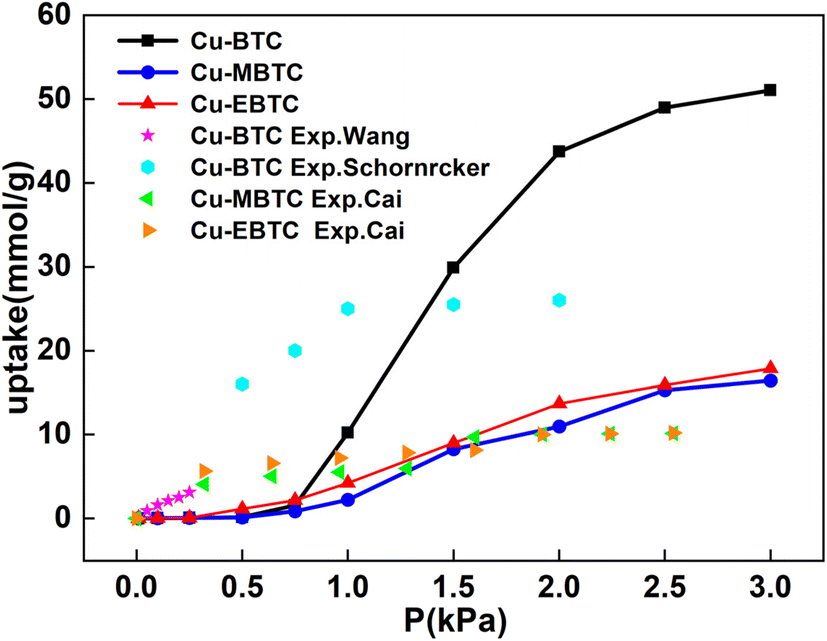 | ||
| Fig. 3 H2O adsorption isotherms of Cu-BTC, Cu-MBTC and Cu-EBTC at 298 K.34 | ||
The adsorption heat of Cu-BTC, Cu-MBTC and Cu-EBTC calculated by GCMC can be seen in Fig. S1 of the ESI.† At a lower adsorption capacity, the calculated adsorption heat is lower due to underestimation of the adsorption effect of water molecules and metal sites. At a higher adsorption capacity, the interaction between water molecules will increase rapidly, resulting in a rapid increase in the calculated adsorption heat. Similar to previous literature reports,16 because the general molecular force field is difficult to describe the strong interaction between water molecules and metal sites, the calculation results of the adsorption heat are not accurate, and it is difficult to further study the adsorption mechanism. So we use first-principles calculations to further study the adsorption behavior of H2O on Cu-BTC, Cu-MBTC and Cu-EBTC.
 | ||
| Fig. 4 DFT optimized structure of Cu node sites of Cu-BTC, Cu-MBTC and Cu-EBTC that absorb water molecules. (a) Cu-BTC containing one water molecule (b) Cu-MBTC containing one water molecule (c) Cu-EBTC containing one water molecule (d) Cu-BTC containing two water molecules (e) Cu-MBTC containing two water molecules (f) Cu-EBTC containing two water molecules. Copper atoms are orange, oxygen is red, carbon is gray, hydrogen is white. | ||
The heat of adsorption calculated by DFT is shown in Table 1. For the three materials Cu-BTC, Cu-MBTC and Cu-EBTC, the heat of adsorption for two water molecules is about 2 kJ mol−1 lower than that for one water molecule. At the same time, the adsorption heat of Cu-MBTC and Cu-EBTC to water molecules is slightly lower than that of Cu-BTC to water molecules, which indicates that the addition of alkyl groups may reduce the strength of the material's adsorption of water. In previous studies, Henning et al. obtained through experiments that the heat of adsorption of Cu-BTC for water molecules is about 50.7 kJ mol−1 at 313 K.35 Nachtigall et al. calculated by DFT/CC the periodic model Cu-BTC adsorption heat of water molecules is about 49 kJ mol−1,19 and the adsorption heat obtained by our DFT calculation is relatively close to it.36
| Cu-BTC | Cu-MBTC | Cu-EBTC | |
|---|---|---|---|
| a (1) Water molecules adsorbed onto the one-side Cu site and the other Cu site empty (4 wt%). (2) Water molecules adsorbed on both Cu sites (8 wt%). | |||
| (1) | 58.7 kJ mol−1 | 57.3 kJ mol−1 | 57.1 kJ mol−1 |
| (2) | 55.7 kJ mol−1 | 54.7 kJ mol−1 | 55.2 kJ mol−1 |
3.2 Adsorption of gas by alkyl functionalized MOF under wet conditions
Typically, the air contains a small amount of water molecules, and Cu-BTC will preferentially adsorb water molecules in the air. Therefore, in practical industrial applications process of Cu-BTC material, in addition to considering its water absorption performance, it is also necessary to consider its adsorption capacity for other gases under the condition of containing a small amount of water. Simultaneously, prior studies have reported that the modification of Cu-BTC may change its gas adsorption capacity under trace water conditions.29 In order to study the effect of alkyl functionalization on the gas adsorption performance of materials under humid conditions, we constructed models of Cu-BTC, Cu-MBTC and Cu-EBTC containing different numbers of water molecules, and calculated their atomic distribution charges. The adsorption capacity and mechanism for gases of Cu-BTC, Cu-MBTC and Cu-EBTC under different water-containing conditions were studied.The result of the atomic distribution charge calculation is used as the parameter input of the GCMC calculation. The specific details are presented in Fig. S2–S44 of the ESI.† In addition, Tables S1–S3 of the ESI† the specific values of the partition charge for each atom. Table S4† shows the pore structure characterization parameters of Cu-BTC, Cu-MBTC, and Cu-EBTC under aqueous and anhydrous conditions. The incorporation of water molecules changes the atomic distribution charges of Cu-BTC, Cu-MBTC and Cu-EBTC. The charges of the copper atoms near the water molecule increase obviously, which have effects on the gas adsorption and separation capabilities of the material.
The adsorption isotherms of Cu-BTC, Cu-MBTC and Cu-EBTC for CO2 under different water conditions at 298 K are shown in Fig. 5. As the incorporation of water molecules reduces the pore volume of the material, the saturated adsorption of CO2 by the three materials has decreased. Previous studies usually focus on the influence of water molecules on the CO2 adsorption performance of materials under low pressure conditions. Our calculation results show that in the pressure range of 0–500 kPa, the presence of coordinated water molecules enhances the CO2 adsorption performance of Cu-BTC, which is similar to that previously reported in the literature.27 However, Cu-MBTC and Cu-EBTC show different performance under the same conditions. The capacity of Cu-MBTC with 4 wt% water to adsorb CO2 increased, but the capacity of Cu-MBTC with 8 wt% water to adsorb CO2 decreased. In contrast, for Cu-EBTC, the presence of coordinated water molecules reduces its CO2 adsorption performance.
 | ||
| Fig. 5 The adsorption isotherms of Cu-BTC, Cu-MBTC and Cu-EBTC on CO2 under different water conditions at 298 K and the partial enlarged view under low pressure (a) Cu-BTC (b) Cu-MBTC (c) Cu-EBTC (d) Cu-BTC partial enlargement (e) Cu-MBTC partial enlargement (f) Cu-EBTC partial enlargement. | ||
The adsorption isotherms of N2 by Cu-BTC, Cu-MBTC and Cu-EBTC under different water conditions at 298 K are shown in Fig. 6. Compared with CO2, the presence of coordinated water molecules has little effect on the material's ability to adsorb N2. The adsorption capacity of Cu-BTC for N2 remained basically unchanged, but the adsorption capacity of Cu-MBTC and Cu-EBTC for N2 decreased slightly. This may be due to the different pore volume of the three materials. The pore volume of Cu-BTC (0.78 cm3 g−1) is greater than that of Cu-MBTC (0.64 cm3 g−1), and the pore volume of Cu-MBTC is greater than that of Cu-EBTC (0.50 cm3 g−1). The presence of water molecules will further reduce the pore volume of the material. Therefore, under the same water load condition, the material with a smaller pore volume is more likely to decrease the adsorption capacity due to the reduction in pore volume.
 | ||
| Fig. 6 The adsorption isotherms of Cu-BTC, Cu-MBTC and Cu-EBTC for N2 under different water conditions at 298 K and the partial enlarged view under low pressure (a) Cu-BTC (b) Cu-MBTC (c) Cu-EBTC (d) Cu-BTC partial enlargement (e) Cu-MBTC partial enlargement (f) Cu-EBTC partial enlargement. | ||
In order to understand the effect of coordinated water molecules on material gas adsorption, we studied the interaction energy in the GCMC simulation. Fig. 7 shows the interaction energy of CO2 and N2 with different numbers of coordinated water molecules Cu-BTC, Cu-MBTC and Cu-EBTC. The interaction energy can be divided into Coulomb energy and non-Coulomb (LJ) energy. Similar to previous literature reports,27,29 for the interaction energy between Cu-BTC and CO2 (Fig. 7a), the non-Coulomb (LJ) energy increases by about 10% at a lower adsorption capacity, which corresponds to conditions under lower pressure. However, the Coulomb energy can increase by more than double. This indicates that the increase in the Coulomb interaction between the material and carbon dioxide is the primary factor contributing to the enhanced adsorption of carbon dioxide. The electrostatic interaction is caused by the interaction between the quadrupole moment of CO2 and the electric field gradient associated with the adsorbent. When water occupy the open metal sites on the copper surface, the electric field gradient of the adsorbent will increase.
 | ||
| Fig. 7 The interaction energy between CO2 and N2 and Cu-BTC, Cu-MBTC and Cu-EBTC with different numbers of coordinated water molecules obtained by GCMC simulation at 298 K (a) CO2 and Cu-BTC (b) CO2 and Cu-MBTC (c) CO2 and Cu-EBTC (d) N2 and Cu-BTC (e) N2 and Cu-MBTC (f) N2 and Cu-EBTC. | ||
At the same time, for the interaction energy between Cu-MBTC and CO2 (Fig. 7b), the non-Coulomb (LJ) energy also has no significant change, but the Coulomb energy changes with the number of water molecules in a different trend than Cu-BTC. The Coulomb energy of Cu-MBTC and CO2 with 4 wt% coordinated water molecules is significantly higher than the Coulomb energy of Cu-MBTC and CO2. However, there is no significant increase in the Coulomb energy of Cu-MBTC and CO2 with 8 wt% coordinated water molecules has no significant increase compared with the Coulomb energy of Cu-MBTC and CO2 with 4 wt% coordinated water molecules. This may be the reason why the adsorption capacity of Cu-MBTC loaded with 4 wt% water, as observed in the adsorption isotherm (Fig. 4b and d) has increased, but the capacity of Cu-MBTC loaded with 8 wt% water has decreased. The interaction energy between Cu-EBTC and CO2 (Fig. 7c) is similar to the interaction energy between Cu-BTC and CO2. However, we observed that the Coulomb energy of Cu-EBTC and CO2 is greater than that of Cu-BTC, and Cu-MBTC and CO2, indicating a stronger interaction between Cu-EBTC and CO2. For N2, the increase of water molecules did not significantly change the interaction energy. This may be attributed to the quadrupole moment of N2 is much smaller than the quadrupole moment of CO2, resulting in a correspondingly smaller Coulomb energy between N2 and the adsorbent, in contrast to the larger Coulomb energy observed between CO2 and the adsorbent.
3.3 Adsorption selectivity
Adsorption selectivity is a crucial parameter that characterizes the degree of separation of each component in the mixed gas. We used the Dual Site Langmuir–Freundlich equation (DSLF) to fit the adsorption isotherm, and the fitting parameters are presented in Table 2. Where R is the goodness of fit, it is evident that the correlation coefficients of the fit are all above 99%, indicating that these coefficients are credible.| Adsorption system | Fitting parameters and regression coefficients | ||||||
|---|---|---|---|---|---|---|---|
| A | b | c | q | k | t | R | |
| Cu-BTC + CO2 | 14.7 | 0.001246 | 1.033 | 0.07649 | 0.002106 | 0.1742 | 0.9997 |
| Cu-BTC + 4 wt% H2O + CO2 | 13.76 | 0.002965 | 0.9293 | 0.07948 | 0.01905 | 0.3313 | 0.9996 |
| Cu-BTC + 8 wt% H2O + CO2 | 10.91 | 0.002125 | 1.004 | 1.514 | 0.01396 | 1.124 | 0.9997 |
| Cu-BTC + N2 | 10.39 | 0.0001004 | 1.099 | 0.6864 | 0.01341 | 0.6813 | 0.9996 |
| Cu-BTC + 4 wt% H2O + N2 | 14.12 | 0.0004599 | 0.8709 | 0.002796 | 0.003334 | 0.8714 | 0.9999 |
| Cu-BTC + 8 wt% H2O + N2 | 11.81 | 0.0004742 | 0.8934 | 0.7946 | 0.02921 | 0.1633 | 0.9999 |
| Cu-MBTC + CO2 | 11.01 | 0.001385 | 1.039 | 0.001 | 0.8678 | 0.6121 | 0.9995 |
| Cu-MBTC + 4 wt% H2O + CO2 | 10.13 | 0.003067 | 0.9456 | 0.000017 | 0.02022 | 0.8366 | 0.9996 |
| Cu-MBTC + 8 wt% H2O + CO2 | 8.924 | 0.003826 | 0.8889 | 0.0001732 | 0.006874 | 0.7464 | 0.9995 |
| Cu-MBTC + N2 | 9.908 | 0.0002275 | 0.9833 | 0.245 | 0.09168 | 0.07603 | 0.9996 |
| Cu-MBTC + 4 wt% H2O + N2 | 7.303 | 0.0002719 | 0.9749 | 2.595 | 0.0003074 | 0.8316 | 0.9999 |
| Cu-MBTC + 8 wt% H2O + N2 | 7.498 | 0.0002915 | 0.956 | 0.000622 | 0.08228 | 0.4698 | 0.9999 |
| Cu-EBTC + CO2 | 9.418 | 0.00456 | 0.9295 | 0.001007 | 0.1685 | 0.2361 | 0.9982 |
| Cu-EBTC + 4 wt% H2O + CO2 | 7.665 | 0.00557 | 0.9132 | 0.0004559 | 0.05167 | 0.9233 | 0.9973 |
| Cu-EBTC + 8 wt% H2O + CO2 | 6.475 | 0.01018 | 0.8314 | 0.0005068 | 0.0002514 | 0.9333 | 0.9989 |
| Cu-EBTC + N2 | 7.817 | 0.0002544 | 1.016 | 0.3733 | 0.06098 | 0.2238 | 0.9996 |
| Cu-EBTC + 4 wt% H2O + N2 | 7.228 | 0.0003219 | 0.9632 | 0.008587 | 0.0000770 | 0.001225 | 0.9999 |
| Cu-EBTC + 8 wt% H2O + N2 | 6.274 | 0.000301 | 0.9579 | 0.5651 | 0.0004379 | 0.01004 | 0.9998 |
The adsorption selectivity of Cu-BTC, Cu-MBTC and Cu-EBTC towards different gas mixtures of CO2/N2 at 298 K containing different numbers of coordinated water molecules is shown in Fig. 8. For the three materials, within the calculation range, the presence of water molecules significantly increases their adsorption selectivity. And the adsorption selectivity of the material with 8 wt% of coordinated water molecules is greater than that of the material with 4 wt% of coordinated water molecules. For Cu-BTC and Cu-MBTC, when the coordinated water molecules reach 8 wt%, the adsorption selectivity for CO2/N2 reaches about 14. At the same time, we also found an interesting phenomenon. When the coordinated water molecules reach 8 wt%, the adsorption selectivity of Cu-EBTC for CO2/N2 exceeds 25. The adsorption selectivity exhibits a significant increase, and it is much higher than the adsorption selectivity of Cu-BTC and Cu-MBTC for CO2. This has potential application prospects in the fields of CO2 adsorption and separation. Compared with coordinated water molecules, other influencing factors (pressure, CO2/N2 ratio) have less influence on the material's adsorption selectivity. Compared with coordinated water molecules, other influencing factors (pressure, CO2/N2 ratio) have a smaller impact on the adsorption selectivity of the three materials. This means that the method of enhancing the material's selective adsorption ability by adding water molecules will not be affected by changes in pressure and mixed gas concentration ratio. This has potential application prospects in CO2 adsorption and separation, and also provides new ideas for further designing MOF materials with high adsorption selectivity.
 | ||
| Fig. 8 The adsorption selectivity of Cu-BTC, Cu-MBTC and Cu-EBTC with different amounts of coordinated water molecules at 298 K for different gas ratios of CO2/N2 (a) Cu-BTC CO2/N2 = 1:4 (b) Cu-BTC CO2/N2 = 1:1 (c) Cu-BTC CO2/N2 = 4:1 (d) Cu-MBTC CO2/N2 = 1:4 (e) Cu-MBTC CO2/N2 = 1:1 (f) Cu-MBTC CO2/N2 = 4:1 (g) Cu-EBTC CO2/N2 = 1:4 (h) Cu-EBTC CO2/N2 = 1:1 (i) Cu-EBTC CO2/N2 = 4:1. | ||
4 Conclusion
In this paper, GCMC and DFT methods are used to simulate the influence of organic linkers on the water absorption performance of MOFs and the gas adsorption and separation performance under water-containing conditions. Studies have shown that the addition of alkyl groups reduces the water absorption performance of the material and reduces the heat of adsorption of the material to water. GCMC research shows that the addition of coordinated water molecules enhances the CO2 adsorption capacity of Cu-BTC. The capacity of Cu-MBTC with 4 wt% water to adsorb CO2 increased, but the capacity of Cu-MBTC with 8 wt% water to adsorb CO2 decreased. At the same time, the presence of coordinated water molecules reduces the CO2 adsorption performance of Cu-EBTC. However, the coordinated water molecules have little effect on the ability of the material to adsorb nitrogen. This may be attributed to the fact that the quadrupole moment of N2 is much smaller than that of CO2. Studies on the heat of adsorption have shown that when water occupies the open metal sites on copper, it will enhance the interaction between CO2 and the adsorbent. The study of adsorption selectivity shows that, compared with Cu-BTC and Cu-MBTC, the adsorption selectivity of Cu-EBTC at 8 wt% water loading has a very significant improvement, and the pressure and the ratio of CO2/N2 have little effect on the material adsorption selectivity. This provides new possibilities for the application of MOF materials in the fields of adsorption and separation of CO2.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.References
- T. Düren, Y.-S. Bae and R. Q. Snurr, Chem. Soc. Rev., 2009, 38, 1237–1247 RSC.
- G. A. González-Martínez, J. A. Zárate, A. Martínez, E. Sánchez-González, J. R. Álvarez, E. Lima, E. González-Zamora and I. A. Ibarra, RSC Adv., 2017, 7, 24833–24840 RSC.
- J. Ethiraj, F. Bonino, C. Lamberti and S. Bordiga, Microporous Mesoporous Mater., 2015, 207, 90–94 CrossRef CAS.
- D. Zhang, X. Jing, D. S. Sholl and S. B. Sinnott, J. Phys. Chem. C, 2018, 122, 18456–18467 CrossRef CAS.
- H. Huang, W. Zhang, D. Liu and C. Zhong, Ind. Eng. Chem. Res., 2012, 51, 10031–10038 CrossRef CAS.
- S. S.-Y. Chui, S. M.-F. Lo, J. P. H. Charmant, A. G. Orpen and I. D. Williams, Science, 1999, 283, 1148–1150 CrossRef CAS PubMed.
- M.-L. Han, G.-X. Wen, W.-W. Dong, Z.-H. Zhou, Y.-P. Wu, J. Zhao, D.-S. Li, L.-F. Ma and X. Bu, J. Mater. Chem. C, 2017, 5, 8469–8474 RSC.
- A. Hou, Y. Du, Y. Su, Z. Pang, S. liu, S. Xian, X. Zhao, L. Ma, B. Liu, H. Wu and Z. Zhou, ACS Appl. Nano Mater., 2024, 7, 10998–11007 CrossRef CAS.
- D. Hu, Z. Sun, Y. Han, H. Meng and X. Zhang, Dalton Trans., 2023, 52, 11441–11450 RSC.
- F. Wang, W. Hu, S. Zhang, C. Zhu, Y. Fan and Q. Wang, Microporous Mesoporous Mater., 2023, 351, 112480 CrossRef CAS.
- T. Yan, Y. Huo and W.-G. Pan, ACS Appl. Nano Mater., 2023, 6, 10903–10924 CrossRef CAS.
- M. Hemdan, A. H. Ragab, N. F. Gumaah and M. F. Mubarak, Int. J. Biol. Macromol., 2024, 274, 133498 CrossRef CAS PubMed.
- Q. Min Wang, D. Shen, M. Bülow, M. Ling Lau, S. Deng, F. R. Fitch, N. O. Lemcoff and J. Semanscin, Microporous Mesoporous Mater., 2002, 55, 217–230 CrossRef.
- M. Todaro, G. Buscarino, L. Sciortino, A. Alessi, F. Messina, M. Taddei, M. Ranocchiari, M. Cannas and F. M. Gelardi, J. Phys. Chem. C, 2016, 120, 12879–12889 CrossRef CAS.
- S. Calero and P. Gómez-Álvarez, J. Phys. Chem. C, 2015, 119, 467–472 CrossRef CAS.
- J. Zang, S. Nair and D. S. Sholl, J. Phys. Chem. C, 2013, 117, 7519–7525 CrossRef CAS.
- T. R. C. Van Assche, T. Duerinck, J. J. Gutiérrez Sevillano, S. Calero, G. V. Baron and J. F. M. Denayer, J. Phys. Chem. C, 2013, 117, 18100–18111 CrossRef CAS.
- J. J. Gutiérrez-Sevillano, J. M. Vicent-Luna, D. Dubbeldam and S. Calero, J. Phys. Chem. C, 2013, 117, 11357–11366 CrossRef.
- J. M. Castillo, T. J. H. Vlugt and S. Calero, J. Phys. Chem. C, 2008, 112, 15934–15939 CrossRef CAS.
- H. Li, P. Jing, J. Lu, L. Xi, Q. Wang, L. Ding, W.-M. Wang and Z. Song, Dalton Trans., 2021, 50, 2854–2863 RSC.
- Y. Zhao, J. Liu, M.-L. Han, G.-P. Yang, L.-F. Ma and Y.-Y. Wang, Rare Met., 2021, 40, 499–504 CrossRef CAS.
- L. Grajciar, P. Nachtigall, O. Bludský and M. Rubeš, J. Chem. Theory Comput., 2015, 11, 230–238 CrossRef CAS PubMed.
- M. Rubeš, L. Grajciar, O. Bludský, A. D. Wiersum, P. L. Llewellyn and P. Nachtigall, ChemPhysChem, 2012, 13, 488–495 CrossRef PubMed.
- B. Supronowicz, A. Mavrandonakis and T. Heine, J. Phys. Chem. C, 2013, 117, 14570–14578 CrossRef CAS.
- W. Xue, Z. Zhang, H. Huang, C. Zhong and D. Mei, J. Phys. Chem. C, 2020, 124, 1991–2001 CrossRef CAS.
- L. Grajciar, O. Bludský and P. Nachtigall, J. Phys. Chem. Lett., 2010, 1, 3354–3359 CrossRef CAS.
- A. Ö. Yazaydın, A. I. Benin, S. A. Faheem, P. Jakubczak, J. J. Low, R. R. Willis and R. Q. Snurr, Chem. Mater., 2009, 21, 1425–1430 CrossRef.
- D. V. Gonçalves, R. Q. Snurr and S. M. P. Lucena, Adsorption, 2019, 25, 1633–1642 CrossRef.
- J. Yu, Y. Wu and P. B. Balbuena, ACS Sustainable Chem. Eng., 2016, 4, 2387–2394 CrossRef CAS.
- M. J. Frisch, et al., Gaussian 16, Gaussian, Inc., Wallingford, CT, 2016 Search PubMed.
- Y. Cai, Y. Zhang, Y. Huang, S. R. Marder and K. S. Walton, Cryst. Growth Des., 2012, 12, 3709–3713 CrossRef CAS.
- Q. Yang, C. Xue, C. Zhong and J.-F. Chen, AIChE J., 2007, 53, 2832–2840 CrossRef CAS.
- G. Zhou, Z. Du, Y. Ma, Y. Zhang, H. Wu, X. Sun, W. Song, X. Zhang, Y. Jiao and G. Lu, Comput. Mater. Sci., 2020, 181, 109755 CrossRef CAS.
- Y. Cai, Y. Zhang, Y. Huang, S. R. Marder and K. S. Walton, Cryst. Growth Des., 2012, 12, 3709–3713 CrossRef CAS.
- S. K. Henninger, F. P. Schmidt and H.-M. Henning, Appl. Therm. Eng., 2010, 30, 1692–1702 CrossRef CAS.
- W. You, Y. Liu, J. D. Howe, D. Tang and D. S. Sholl, J. Phys. Chem. C, 2018, 122, 27486–27494 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra08720h |
| This journal is © The Royal Society of Chemistry 2025 |