
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceThe reverse water gas shift reaction (RWGS) mechanism study on the γ-MoC(100) surface†
Xiaoshu Yao,
Zhihong Wei *,
Jingyuan Mei,
Xianhui Guo and
Xinxin Tian*
*,
Jingyuan Mei,
Xianhui Guo and
Xinxin Tian*
Institute of Molecular Science, Key Laboratory of Chemical Biology and Molecular Engineering of Ministry of Education, Shanxi University, Taiyuan 030006, China. E-mail: weizhihong@sxu.edu.cn; tianxx@sxu.edu.cn
First published on 3rd January 2025
Abstract
CO2 conversion and reuse technology are crucial for alleviating environmental stress and promoting carbon cycling. Reverse water gas shift (RWGS) reaction can transform inert CO2 into active CO. Molybdenum carbide (MoC) has shown good performance in the RWGS reaction, and different crystalline phases exhibit distinct catalytic behaviors. Here, we performed a systematic study on the RWGS reaction mechanism on the hexagonal-phase γ-MoC(100) surface by using density functional theory (DFT). It is found that the redox mechanism, i.e. the direct dissociation of CO2, is the dominant pathway. CO2 firstly adsorbs on the surface with an adsorption energy of −2.14 eV, and then dissociates into CO* and O* with a barrier of 0.83 eV. Surface O* hydrogenating into OH* has a high barrier of 2.15 eV. OH* further hydrogenating into H2O* has a barrier of 1.48 eV, and the disproportionation of OH* considerably lowers this value to 0.06 eV. However, the desorption of product CO is particularly challenging due to the large energy demand of 3.06 eV. This characteristic, in turn, provides feasibility and opportunity for CO2 to serve as a potential alternative carbon source for CO on the γ-MoC(100) surface. In contrast, other Mo-based catalysts such as hexagonal MoP and cubic α-MoC have better RWGS catalytic efficiency.
1 Introduction
Currently, the prevalent view is that greenhouse gases emitted by human activities are the primary cause of global climate change,1,2 with CO2 being the most significant component among these gases.3,4 Consequently, reducing CO2 emissions and achieving its conversion and reuse are essential methods for addressing the environmental threats posed by its excessive atmospheric concentration.5–9 As an abundant, renewable, and inexpensive C1 resource, converting inert CO2 into reactive CO is a key process for reuse after its capture. The CO or syngas produced from this process can be coupled with Fischer–Tropsch synthesis or carbonylation reactions to convert CO2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 10,11 into hydrocarbon liquid fuels or high-value chemicals such as aldehydes, alcohols, carboxylic acids and esters,2,9,12,13 thereby realizing the carbon cycling. Therefore, developing key technologies for CO2 conversion has attracted extensive research.
10,11 into hydrocarbon liquid fuels or high-value chemicals such as aldehydes, alcohols, carboxylic acids and esters,2,9,12,13 thereby realizing the carbon cycling. Therefore, developing key technologies for CO2 conversion has attracted extensive research.
The reverse water gas shift (RWGS) reaction is an effective means of converting CO2 into CO (CO2 + H2 → CO + H2O, ΔH298K = 41.1 kJ mol−1).14,15 This reaction skillfully circumvents the complex technical challenges and high energy costs associated with the direct conversion of CO2 into liquid hydrocarbons, demonstrating significant potential in the field of carbon recycling. Advances in the RWGS process have focused on improving catalyst efficiency, reducing energy consumption, especially the exploration of new catalytic materials and reaction conditions that enhance CO selectivity and conversion rates.16,17
Transition metal carbides (TMCs) are a class of materials with unique electronic structures and noble-metal-like properties.18–20 They are inexpensive and abundant, and have been widely studied as promising alternative catalysts for noble metal catalysts or supports.21,22 Molybdenum carbide (MoC), as a typical representative of TMC catalysts, has shown good performance in RWGS reaction, including high activity, selectivity towards CO, and resistance to coking.23–29 Especially, MoC catalysts can operate effectively at lower temperatures than traditional metal-based catalysts, while maintaining good stability.24 The inherent properties of MoC, such as its ability to activate CO2 and H2, make it a promising candidate for RWGS applications.
MoC exists in various crystalline structures,30 including hexagonal and cubic forms, each exhibiting distinct catalytic behaviors in the RWGS reaction. The face-centered cubic α-MoC catalyst (also known as δ-MoC in some literature) exhibits excellent water dissociation capabilities, producing numerous hydroxyl groups on its surface during the reaction. Metals loaded on the α-MoC support can be highly dispersed or form stable single-atom catalysts through strong interactions with the support, creating new active sites at the interface. Consequently, high reaction activity for the WGS/RWGS reaction can be observed even at low temperatures.23–26 For hexagonal phase, Wang et al.31 calculated the adsorption of hydrogen on different surfaces of hexagonal phase Mo2C under various coverages using density functional theory (DFT) and found that the dissociative adsorption of H2 on the Mo termination of (001) and (100) surfaces, as well as Mo/C mixed termination of (101) and (201) surfaces, is kinetically and thermodynamically favorable. The results show that MoC has excellent ability to activate H2. Zhang et al.32 synthesized Cu/β-Mo2C catalysts by using β-Mo2C as support, which significantly promoted the uniform dispersion of Cu on the surface of β-Mo2C and avoided the deactivation of the catalysts due to sintering at high temperatures, thus exhibiting high RWGS catalytic activity and excellent stability. Galallah et al.33 prepared Mo2C@CN using carbon–nitride (CN) as the support, achieving a CO2 conversion rate of approximately 76% at 700 °C, with high CO selectivity (87%) and very low CH4 selectivity (2%). The addition of potassium as a promoter further improved the selectivity for CO to 99%. This outcome exemplifies the remarkable RWGS reaction activity.
γ-MoC, similar to β-Mo2C, is a hexagonal-phase MoC34 and its crystal surfaces exhibit typical metallic properties, resulting in high electrocatalytic performance. It shows outstanding hydrogen evolution reaction (HER) and oxygen evolution reaction (OER)35–39 activity. Compared to the extensive research on its electrochemical activity, there is little research on its RWGS activity or other thermal catalytic reaction, partly due to the poor thermal stability of this phase at high temperatures. In 2014, γ-MoC was successfully prepared as a stable pure nanomaterial for the first time.36 Further exploration of the RWGS reaction activity on this γ-MoC therefore has become valuable. This work aims to study the RWGS reaction mechanism on γ-MoC through DFT calculations, and the Mo-terminated γ-MoC(100) surface was used as the model. Based on the DFT results, the RWGS activity of γ-MoC is further compared with α-MoC, MoP and metallic Mo.
2 Computational methods and models
2.1 Calculation methods
All calculations in this work were performed using the DS-PAW package which is based on the projector augmented wave (PAW) pseudopotential.40 The Perdew–Burke–Ernzerhof (PBE) functional with the generalized gradient approximation (GGA) was adopted.41 The plane-wave cutoff energy was set to 400 eV. For structure relaxation, the force tolerance and energy difference were respectively set to lower than 0.02 eV Å−1 and 10−4 eV. All the energies in this work included zero-point energy (ZPE) correction. Transition state calculations were done using the NEB method within DS-PAW.The adsorption energy Eads of species on the catalyst surface is defined as:
| Eads = E(x/slab) − E(x) − E(slab) |
2.2 Model
A WC-type γ-MoC was chosen and the bulk parameters after optimized were found to be a = b = 2.93 Å and c = 2.84 Å, which were in good agreement with experimental results.42,43 The Mo-terminated (100) surface that has only Mo atoms exposed was selected. A p(3 × 3) supercell with eight atomic layers was used as the model (Fig. 1), and the bottom four layers of the model are fixed, and the top four layers are allowed to relax. In order to avoid strong interactions between the layers, the vacuum layer was set to 15 Å. The Brillouin zone was sampled by Monkhorst–Pack scheme using 3 × 3 × 1 k-points grids. | ||
| Fig. 1 Top and side views of the γ-MoC(100) surface (blue and gray balls represent Mo and C atoms, respectively). | ||
3 Results and discussion
3.1 Adsorption of RWGS intermediates on γ-MoC(100) surface
The RWGS reaction may commonly follow four primary mechanisms.28,29 Among them, the carbonate mechanism is generally more favorable on oxide catalyst surfaces, particularly on basic oxides and transition metal oxides.28,29,44–48 Due to the nature of MoC catalyst surface, surface O can only be obtained from dissociation of CO2, and the combination of CO2 and O is endothermic by at least 1.61 eV (Fig. S1†). The energy is considerably high. Therefore, detailed carbonate mechanism was not considered in this work, and only the other three mechanisms were discussed in the following. As shown in Table 1, the first one is the redox mechanism, also known as the direct dissociation mechanism, where CO2* directly dissociates into CO* and O* species (R2), H2 does not directly participate in CO2 reduction, just reduces the surface O* atom to form H2O* (R10 and R11). The second one is carboxy mechanism, which dissociated H* reacts firstly with CO2* to form COOH* intermediate (R3), and then COOH* undergoes one or two step decomposition reactions to generate CO* (R4–R6). The third one is formate mechanism, which dissociated H* reacts firstly with CO2* to form HCOO* intermediate (R7), and then HCOO* undergoes two step decomposition reactions to generate CO* (R8 and R9).| Mechanism | Label | Elementary reaction |
|---|---|---|
| Redox mechanism | R0 | CO2* + * → CO2* |
| R1 | H2 + 2* → H* + H* | |
| R2 | CO2* + * → CO* + O* | |
| R10 | O* + H* → OH* + * | |
| R11 | OH* + H* → H2O* + * | |
| Carboxy mechanism | R0 | CO2 + * → CO2* |
| R1 | H2 + 2* → H* + H* | |
| R3 | CO2* + H* → COOH* + * | |
| R4 | COOH* + * → CO* + OH* | |
| R5 | COOH* + * → COH* + O* | |
| R6 | COH* + * → CO* + H* | |
| R10 | O* + H* → OH* + * | |
| R11 | OH* + H* → H2O* + * | |
| Formate mechanism | R0 | CO2 + * → CO2* |
| R1 | H2 + 2* → H* + H* | |
| R7 | CO2* + H* → HCOO* + * | |
| R8 | HCOO* + * → CHO* + O* | |
| R9 | CHO* + * → CO* + H* | |
| R10 | O* + H* → OH* + * | |
| R11 | OH* + H* → H2O* + * |
As shown in Table 1, 10 main intermediates, including CO2, H2, CO, H2O, C, H, O, OH, COOH and HCOO are involved in these three mechanisms. We first studied the adsorption of these intermediates on the γ-MoC(100) surface. The configurations of these intermediates at the most stable adsorption site are shown in Fig. 2. Table 2 lists the adsorption energy and the corresponding adsorption site of each intermediate.
 | ||
| Fig. 2 The most stable adsorption configurations of intermediates on the γ-MoC(100) surface (blue, red, white and gray balls represent Mo, O, H and bulk-C atoms, respectively. To distinguish the C atoms in the adsorbate, they are represented in black balls). | ||
| Species | γ-MoC(100) | |
|---|---|---|
| Site | Eads | |
| C | Hollow | −10.33 |
| H | Bridge | −0.74 |
| O | Hollow | −4.50 |
| CO2 | Hollow | −2.14 |
| H2 | Top | −0.51 |
| CO | Hollow | −3.06 |
| H2O | Bridge | −0.84 |
| OH | Hollow | −5.05 |
| COOH | Hollow | −4.29 |
| HCOO | Hollow | −5.47 |
As shown in Fig. 2, on the γ-MoC(100) surface, CO2 and CO preferentially adsorb at the hollow sites, interacting with the surrounding Mo atoms through their C and O atoms, and in the chemisorbed state, CO2 is transformed from a linear to a bent structure. H2 molecule stably adsorbs parallel to the surface at the Mo top site, while H2O stably adsorbs at the Mo–Mo bridge site. The most stable adsorption sites for C and O are the hollow sites, whereas H species locates at the bridge site. OH adsorbs vertically at the hollow site, with O interacting with the 4 surrounding surface Mo atoms. Both COOH and HCOO adsorb at the hollow sites and interact with the surface through both C and O atoms. For the convenience of comparison with future experimental infrared spectral data, the calculated vibration frequencies of different intermediates are afforded (Table S1†).
3.2 RWGS reaction mechanism on γ-MoC(100) surface
The reaction begins with the adsorption of CO2 and H2 on the catalyst surface. CO2 adsorbs at the hollow site formed by four surface Mo atoms with Eads = −2.14 eV, while H2 adsorbs at the Mo top site with Eads = −0.51 eV. Subsequently, H2* undergoes dissociation (R1), with the dissociation energy barrier of only 0.08 eV (Table 3) and the process is exothermic by 0.87 eV. Therefore, we deduce that the H2 molecule is very easy to dissociate and surface H* is abundant. Our previous studies of RWGS on Mo and MoP catalysts49,50 suggested redox mechanism is more preferable, therefore, we calculated mainly redox mechanism on the γ-MoC(100) surface while considering the possibility of CO2 direct hydrogenation into COOH* or HCOO*, i.e. the initial steps of the carboxy and formate mechanisms. The potential energy surface based on the calculation results is shown in Fig. 3. The initial states (IS), transition states (TS) and final states (FS) of the RWGS elementary steps on the γ-MoC(100) surface are shown in Fig. 4.| Label | Elementary reaction | γ-MoC(100) | ||
|---|---|---|---|---|
| Ea | Er | d | ||
| R1 | H2* → H* + H* | 0.08 | −0.87 | 1.173 |
| R2 | CO2* → CO* + O* | 0.83 | −2.04 | 1.866 |
| R3 | CO2* + H* → COOH* | 1.66 | 1.19 | 1.352 |
| R7 | CO2* + H* → HCOO* | 0.62 | 0.27 | 1.673 |
| R8 | HCOO* → CHO* + O* | 1.13 | −1.59 | 1.919 |
| R10 | O* + H* → OH* | 2.15 | 1.52 | 1.239 |
| R11 | OH* + H* → H2O* | 1.48 | 1.33 | 1.244 |
| R12 | OH* + OH* → H2O* + O* | 0.06 | 0.15 | 1.257/1.168 |
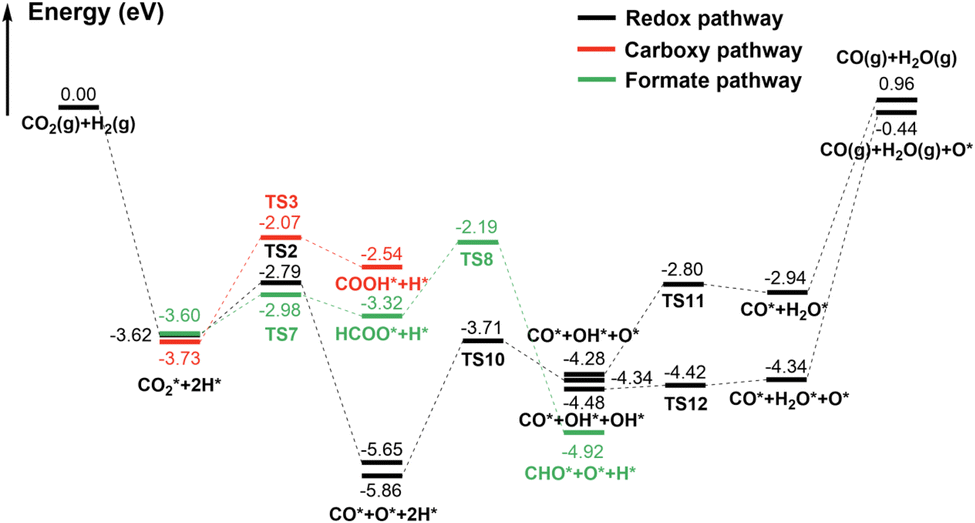 | ||
| Fig. 3 The potential energy surface of RWGS reaction on the γ-MoC(100) surface. | ||
 | ||
| Fig. 4 The initial states (IS), transition states (TS) and final states (FS) of the RWGS elementary steps on the γ-MoC(100) surface. | ||
In the redox pathway, CO2 initially adsorbs at the hollow site as the IS2, as shown in Fig. 4. After passing through the transition state TS2, one of the C–O bonds is broken, forming co-adsorbed CO* and O* on the surface with the dissociation energy barrier of 0.83 eV. Subsequently, the hydrogenation process of O* to form OH* has a high barrier of 2.15 eV, and is endothermic by 1.52 eV, indicating that the formation of OH* is relatively difficult. The reaction energy barrier for the further hydrogenation of OH* to form H2O* is 1.48 eV, and the reaction is endothermic by 1.33 eV. Notably, H2O* can also be obtained by the disproportionation of OH* (2OH* → H2O* + O*), with the barrier significantly reduced to 0.06 eV. The results prove that H2O* formation is more likely through the disproportionation pathway.
In contrast, through the carboxy mechanism pathway, the energy barrier of CO2* hydrogenation to form COOH* is 1.66 eV, and the reaction is endothermic with Er = 1.19 eV. The barrier is considerably higher than that of redox pathway. However, through the formate mechanism pathway, the H* interacts with the C atom of CO2* to form HCOO* species, with a barrier of only 0.62 eV, which is still lower than that of CO2* direct dissociation (0.83 eV). Although this step is kinetically advantageous, it is endothermic by 0.27 eV, which is thermodynamically unfavored compared to the strongly exothermic process of CO2* direct dissociation. Furthermore, the further dissociation of HCOO* needs to overcome a barrier of 1.13 eV, which makes the formate mechanism more difficult. Interestingly, Bader charge analysis showed that, the charge of C and O atom in CO2* is +0.49e and −1.05e, respectively. The charge of surface H is −0.46e. As the configuration of CO2 is transformed from a linear to a bent structure in the chemisorbed state, its HUMO and LUMO orbitals also changed correspondingly. As shown in Fig. S2,† the HUMO orbital is mainly contributed by O atoms while the LUMO orbital is mainly contributed by C atom. Therefore, the negatively charged surface H can easily interact with the LUMO orbital of bent CO2, that is, H attacks C to undergo nucleophilic reactions and generate HCOO*. On the contrary, it is very difficult for surface H to attack O, which is also negatively charged, and interact with the LUMO orbital of bent CO2. This also explains why CO2* hydrogenation into COOH* has much higher barrier than into the HCOO* species. Just as shown in Fig. 3, the redox pathway is more favorable than the other two pathways, and may be the mechanism with the greatest contribution of RWGS reaction on γ-MoC(100) surface. Considering the whole reaction process, it is found that the CO desorption from the surface requires a substantial energy of 3.06 eV. The huge energy gap made this desorption very difficult, and it is rational to deduce that the further dissociation or hydrogenation of CO* is much possible. We suppose that with the RWGS reaction, CO2 can be directly used as a substitute carbon source for CO on the γ-MoC(100) surface for further reaction, without firstly generating CO and then cascade other reactions.
3.3 Comparison of γ-MoC and other Mo-based catalysts
We have previously investigated the RWGS reaction mechanism on the Mo(100) and MoP(100) surfaces.49,50 Comparing the results on these three surfaces (Fig. 5), it can be seen that the adsorption energies of CO2 on the γ-MoC(100) and Mo(100) surfaces are higher than that on the MoP(100) surface. The order of adsorption energy is: MoP(100) (−1.20 eV) < γ-MoC(100) (−2.14 eV) < Mo(100) (−2.54 eV). The dissociation energy barriers of CO2* on these three surfaces follow the order: MoP(100) (0.47 eV) < Mo(100) (0.67 eV) < γ-MoC(100) (0.83 eV). The barriers of OH* formation on the Mo(100) and MoP(100) surfaces were also listed for comparison due to the high value of that on the γ-MoC(100) surface. It is found that the OH* formation barrier is very close on the Mo(100) and MoP(100) surfaces (1.23 vs. 1.18 eV), and much lower than that on the γ-MoC(100) surface (2.15 eV). Interestingly, the desorption of CO on the Mo(100) surface is also very high (3.22 eV) just like on the γ-MoC(100) surface (3.06 eV), while it is lower on the MoP(100) surface (2.07 eV). In contrast, H2O desorption is much easier on all three surfaces. The desorption energies are 0.91, 0.58 and 0.84 eV, respectively. | ||
| Fig. 5 The adsorption energies (Eads, eV) of CO2, energy barriers (Ea, eV) of CO2* dissociation, energy barriers of OH* formation and the desorption energies (Eads, eV) of CO2 and CO on the (100) surfaces of different catalysts. | ||
It is noted that MoP and γ-MoC are both WC-type and have very similar crystal structure, and they have the same Mo:P/Mo:C stoichiometric ratio (1:1). Bader charge analysis showed that, the charge transfer from Mo to P in bulk MoP is 0.74e, while from Mo to C in bulk γ-MoC is 1.25e. We suppose that the charge transfer leads to the weaker adsorption of CO2 on MoP(100) and γ-MoC(100) surfaces when compared to the Mo(100). And the significant differences of CO2 adsorption energy on MoP(100) and γ-MoC(100) surfaces may be due to their different adsorption sites. Unlike the 4-fold hollow site adsorption on the γ-MoC(100) surface, CO2 is adsorbed at a bridge site on the MoP(100) surface, interacting only with two surface Mo atoms.
Overall, MoP(100) surface has moderate CO2, CO and H2O adsorption energies, as well as the lowest CO2* dissociation barrier and OH* formation barrier. From the perspective of RWGS reaction efficiency, MoP(100) should be the best among the three surfaces.
Lin et al.23 investigated the WGS reaction mechanism on Pt/α-MoC catalysts under low-temperature conditions. They modeled the Mo termination of α-MoC(111) surface based on experimental characterization results. Using their data, we deduced the RWGS reaction pathway (the energy difference caused by calculation method was ignored). For the convenience of comparison, the potential energy surfaces of the redox mechanism of RWGS reaction on the γ-MoC(100) and α-MoC(111) surfaces are shown in Fig. 6. It can be found that, the adsorption energy of CO2 on the α-MoC(111) surface (−1.44 eV) is lower than that on the γ-MoC(100) surface (−2.14 eV). According to the projected density of states (PDOS) of the two surfaces (Fig. S3†), near the Fermi level, the total electron density and Mo orbital density of the γ-MoC(100) surface are higher than those of α-MoC(111) surface, indicating the stronger interaction with CO2. Furthermore, CO2* has lower dissociation barrier on the α-MoC(111) surface (0.45 eV) than that on the γ-MoC(100) surface (0.83 eV). The moderate adsorption energy of CO2 and lower energy barrier of CO2 dissociation result in more effective redox of CO2 on the α-MoC(111) surface. In addition, the CO desorption is also easier on the α-MoC(111) surface (2.28 eV or 1.62 eV when considering 4H co-adsorption). However, the H2O* formation barrier on the α-MoC(111) surface is much higher than that on the γ-MoC(100) surface (2.01 vs. 1.48/0.06 eV). We speculate that the disproportionation of OH* may also lower this energy barrier, but we do not have corresponding data. If that's the case, the whole RWGS efficiency on α-MoC(111) is also higher than that on γ-MoC(100), especially due to the easy desorption of CO (1.62/2.28 vs. 3.06 eV).
 | ||
| Fig. 6 The potential energy surfaces of RWGS reaction on the γ-MoC(100) and α-MoC(111) surfaces. | ||
4 Conclusion
The RWGS reaction mechanism on the γ-MoC(100) surface has been investigated using DFT. CO2 has a strong interaction with the γ-MoC(100) surface, and the adsorption energy is 2.14 eV. The redox mechanism is found to be the most preferred pathway. CO2* directly dissociated into CO* and O*, with a moderate barrier (0.83 eV), and the formation of OH* has high barrier of 2.15 eV. OH* further hydrogenates into H2O* has a barrier of 1.48 eV. However, the disproportionation of OH* considerably lower this value to 0.06 eV. Interestingly, the most difficult step is the desorption of CO due to the large energy demand of 3.06 eV. CO* may tend to undergo further hydrogenation or direct dissociation on the surface instead. Although the RWGS performance of γ-MoC(100) surface is not as good as other Mo-based catalysts, it has a relative low energy barrier for activating CO2, and the strong binding energy of CO to the surface is actually beneficial for further carbon conversion reactions. This offers a unique opportunity for CO2 to serve as an alternative carbon source for CO, bypassing the traditional need to first convert CO2 into CO before proceeding with subsequent reactions.Data availability
Structure files, supported figures and tables used in this article have been included as part of the ESI.†Author contributions
Xiaoshu Yao: conceptualization, investigation, data curation, software, visualization, writing – original draft. Jingyuan Mei and Xianhui Guo: investigation, formal analysis. Zhihong Wei: conceptualization, formal analysis, funding acquisition, supervision. Xinxin Tian: conceptualization, methodology, funding acquisition, supervision, resources, writing – review & editing.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (no. 21903049, 22202123) and Fundamental Research Program of Shanxi Province (no. 202403021211195). We gratefully acknowledge HZWTECH for providing computation facilities.References
- Y. Ou, C. Roney, J. Alsalam, K. Calvin, J. Creason, J. Edmonds, A. A. Fawcett, P. Kyle, K. Narayan, P. O'Rourke, P. Patel, S. Ragnauth, S. J. Smith and H. McJeon, Nat. Commun., 2021, 12, 6245–6253 CrossRef CAS PubMed.
- Z. Zhang, S. Y. Pan, H. Li, J. C. Cai, A. G. Olabi, E. J. Anthony and V. Manovic, Renewable Sustainable Energy Rev., 2020, 125, 109799–109815 CrossRef CAS.
- S. Davoodi, M. Al-Shargabi, D. A. Wood, V. S. Rukavishnikov and K. M. Minaev, J. Nat. Gas Sci. Eng., 2023, 117, 205070–205097 CrossRef CAS.
- J. Yao, H. Han, Y. Yang, Y. Song and G. Li, Appl. Sci., 2023, 13, 1169–1195 CrossRef CAS.
- S. Yuan, D. Ma, J. Li, T. Zhou, Z. Ji and H. Han, Adv. Pet. Explor. Dev., 2022, 49, 955–962 CrossRef.
- K. Jiang, P. Ashworth, S. Zhang, X. Liang, Y. Sun and D. Angus, Renewable Sustainable Energy Rev., 2020, 119, 109601–109615 CrossRef.
- J. F. D. Tapia, J. Y. Lee, R. E. H. Ooi, D. C. Y. Foo and R. R. Tan, Sustain. Prod. Consum., 2018, 13, 1–15 CrossRef.
- B. Dziejarski, R. Krzyżyńska and K. Andersson, Fuel, 2023, 342, 127776–127813 CrossRef CAS.
- T. A. Saleh, RSC Adv., 2022, 12, 23869–23888 RSC.
- J. Ren, J. P. Cao, X. Y. Zhao, F. L. Yang and X. Y. Wei, Renewable Sustainable Energy Rev., 2019, 116, 109426–109450 CrossRef CAS.
- J. Ren, Y. L. Liu, X. Y. Zhao and J. P. Cao, J. Energy Inst., 2020, 93, 1083–1098 CrossRef CAS.
- T. A. Saleh, Bull. Mater. Sci., 2022, 45, 101413–101434 Search PubMed.
- Y. He, F. H. Müller, R. Palkovits, F. Zeng and C. Mebrahtu, Appl. Catal., B, 2024, 345, 123663–123693 CrossRef CAS.
- A. M. Bahmanpour, M. Signorile and O. Kröcher, Appl. Catal., B, 2021, 295, 120319–120329 CrossRef CAS.
- Y. A. Daza and J. N. Kuhn, RSC Adv., 2016, 6, 49675–49691 RSC.
- X. Zhang, Y. Liu, M. Zhang, T. Yu, B. Chen, Y. Xu, M. Crocker, X. Zhu, Y. Zhu, R. Wang, D. Xiao, M. Bi, D. Ma and C. Shi, Chem, 2020, 6, 3312–3328 CAS.
- A. Saravanan, P. S. Kumar, D. V. N. Vo, S. Jeevanantham, V. Bhuvaneswari, V. A. Narayanan, P. R. Yaashikaa, S. Swetha and B. Reshma, Chem. Eng. Sci., 2021, 236, 116515–116530 CrossRef CAS.
- P. Chen, J. Ye, H. Wang, L. Ouyang and M. Zhu, J. Alloys Compd., 2021, 883, 160833–160853 CrossRef CAS.
- Q. Gao, W. Zhang, Z. Shi, L. Yang and Y. Tang, Adv. Mater., 2018, 31, 1802880 CrossRef PubMed.
- D. Tian, S. R. Denny, K. Li, H. Wang, S. Kattel and J. G. Chen, Chem. Soc. Rev., 2021, 50, 12338–12376 RSC.
- H. Prats and M. Stamatakis, J. Phys. Chem. Lett., 2024, 15, 3450–3460 CrossRef CAS PubMed.
- H. Prats and M. Stamatakis, J. Mater. Chem. A, 2022, 10, 1522–1534 RSC.
- L. Lin, W. Zhou, R. Gao, S. Yao, X. Zhang, W. Xu, S. Zheng, Z. Jiang, Q. Yu, Y. W. Li, C. Shi, X. D. Wen and D. Ma, Nature, 2017, 544, 80–83 CrossRef CAS PubMed.
- S. Y. Yao, X. Zhang, W. Zhou, R. Gao, W. Q. Xu, Y. F. Ye, L. L. Lin, X. D. Wen, P. Liu, B. B. Chen, E. Crumlin, J. H. Guo, Z. J. Zuo, W. Z. Li, J. L. Xie, L. Lu, C. J. Kiely, L. Gu, C. Shi, J. A. Rodriguez and D. Ma, Science, 2017, 357, 389–393 CrossRef CAS PubMed.
- L. Sun, J. Xu, X. Liu, B. Qiao, L. Li, Y. Ren, Q. Wan, J. Lin, S. Lin, X. Wang, H. Guo and T. Zhang, ACS Catal., 2021, 11, 5942–5950 CrossRef CAS.
- J. Li, L. Sun, Q. Wan, J. Lin, S. Lin and X. Wang, J. Phys. Chem. Lett., 2021, 12, 11415–11421 CrossRef CAS PubMed.
- Q. Zhang, L. Pastor-Pérez, S. Gu and T. Ramirez Reina, Catalysts, 2020, 10, 955–975 CrossRef CAS.
- X. Su, X. Yang, B. Zhao and Y. Huang, J. Energy Chem., 2017, 26, 854–867 CrossRef.
- M. González-Castaño, B. Dorneanu and H. Arellano-García, React. Chem. Eng., 2021, 6, 954–976 RSC.
- C. Chu, C. Li, X. Liu, H. Zhao, C. Wu, J. Li, K. Liu, Q. Li and D. Cao, Catal. Sci. Technol., 2022, 12, 1130–1143 RSC.
- T. Wang, Y. W. Li, J. Wang, M. Beller and H. J. Jiao, J. Phys. Chem. C, 2014, 118, 8079–8089 CrossRef CAS.
- X. Zhang, X. Zhu, L. Lin, S. Yao, M. Zhang, X. Liu, X. Wang, Y. W. Li, C. Shi and D. Ma, ACS Catal., 2016, 7, 912–918 CrossRef.
- A. G. Galallah, M. K. Albolkany, A. E. Rashed, W. Sadik, A. G. El-Demerdash and A. A. El-Moneim, J. Environ. Chem. Eng., 2024, 12, 113380–113391 CrossRef CAS.
- J. Lu, H. Hugosson, O. Eriksson, L. NordstroÈm and U. Jansson, Thin Solid Films, 2000, 370, 203–212 CrossRef CAS.
- X. Zhang, L. Huang, Y. Han, M. Xu and S. Dong, Nanoscale, 2017, 9, 5583–5588 RSC.
- C. Wan, Y. N. Regmi and B. M. Leonard, Angew. Chem., Int. Ed., 2014, 53, 6407–6410 CrossRef CAS PubMed.
- Y. Cheng, J. Gong, B. Cao, X. Xu, P. Jing, B. Liu, R. Gao and J. Zhang, ACS Catal., 2021, 11, 3958–3974 CrossRef CAS.
- T. T. Yang and W. A. Saidi, J. Phys. Chem. Lett., 2020, 11, 2759–2764 CrossRef CAS PubMed.
- G. Q. Yu, W. J. Yin and X. B. Li, Int. J. Hydrogen Energy, 2022, 47, 13664–13673 CrossRef CAS.
- P. E. Blöchl, Phys. Rev. B:Condens. Matter Mater. Phys., 1994, 50, 17953–17979 CrossRef PubMed.
- J. P. Perdew, K. Burke and M. Ernzerhof, Phys. Rev. Lett., 1996, 77, 3865–3868 CrossRef CAS PubMed.
- A. Chrysanthou and P. Grieveson, J. Mater. Sci. Lett., 1991, 10, 145–146 CrossRef CAS.
- H. W. Hugosson, O. Eriksson, L. Nordström, U. Jansson, L. Fast, A. Delin, J. M. Wills and B. Johansson, J. Appl. Phys., 1999, 86, 3758–3767 CrossRef CAS.
- D. Ferri, T. Bürgi and A. Baiker, Phys. Chem. Chem. Phys., 2002, 4, 2667–2672 RSC.
- A. Gogue, F. C. Meunier, D. Tibiletti, J. P. Breen and R. Burch, J. Phys. Chem. B, 2004, 108, 20240–20246 CrossRef.
- P. Panagiotopoulou, D. I. Kondarides and X. E. Verykios, J. Phys. Chem. C, 2011, 115, 1220–1230 CrossRef CAS.
- L. L. Zhou, S. Q. Li, C. Ma, X. P. Fu, Y. S. Xu, W. W. Wang, H. Dong, C. J. Jia, F. R. Wang and C. H. Yan, J. Am. Chem. Soc., 2023, 145, 2252–2263 CrossRef CAS PubMed.
- M. A. Khoshooei, X. Wang, G. Vitale, F. Formalik, K. O. Kirlikovali, R. Q. Snurr, P. Pereira-Almao and O. K. Farha1, Science, 2024, 384, 540–546 CrossRef PubMed.
- Q. S. Lei, DFT Study on the Mechanism of Reverse Water Gas Shift Reaction on Metallic Mo Surface, Undergraduate thesis, Shanxi University, 2022 Search PubMed.
- X. S. Yao, The Mechanism Study of Reverse Water Gas Shift Reaction on Mo-Based Catalyst, Master Thesis, Shanxi University, 2024 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra08671f |
| This journal is © The Royal Society of Chemistry 2025 |