DOI:
10.1039/D4RA08592B
(Paper)
RSC Adv., 2025,
15, 3047-3059
Evaluating the CO2 capture potential of MgO sheets: a DFT study on the effects of vacancy and Ni doping for assessing environmental sustainability
Received
5th December 2024
, Accepted 13th January 2025
First published on 29th January 2025
Abstract
Investigations on two-dimensional materials for efficient carbon dioxide (CO2) capture and storage have recently attracted much attention, especially in the global industrial sector. In this work, the CO2 uptake by three configurations of two-dimensional magnesium oxide was investigated using density functional theory. CO2 capture analysis was performed considering the geometrical, thermophysical, vibrational, electronic and optical properties. Results indicated that CO2 adsorption by magnesium oxide (MgO) sheets is a spontaneous process accompanied by a decrease in Gibbs free energy. Moreover, the CO2 molecular entropy and enthalpy of the CO2 adsorbed sheet were decreased, indicating that the entire process was enthalpy-driven. Among the pristine, vacant and nickel-doped (Ni-doped) MgO sheets, the Ni-doped system was found to have the highest values of Gibbs free energy, enthalpy and entropy in the order of −51.366 kJ mol−1-K, −65.105 kJ mol−1 and 127.606 J mol−1, respectively. It was also found to adsorb CO2 in the ultraviolet and visible (UV-Vis) regions within the range of 100–850 nm. Electronic interactions demonstrated that metallicity was significantly induced on the MgO sheet Ni impurity states, which enhanced the adsorption ability. Notably, hybrid orbitals between py and pz revealed strong physisorption, as confirmed by the partial density of states (PDOS). The findings of this research promote CO2 capture sustainability by encouraging future experimentalists to use two-dimensional MgO as a better surface for CO2 capture.
1. Introduction
Global demands for greener energy are exponentially increasing owing to some confirmed negative effects of fossil fuels, such as atmospheric contamination, global warming and water pollution.1 Similarly, overconsumption of fossil fuels breaks the original balance between carbon and oxygen, leading to a series of serious problems such as rise in sea levels and glacier melting. Statistically, the amount of carbon dioxide (CO2) emission related to energy consumption worldwide peaked at 33.3 Gt in 2019.2 Considering the increased emission from fossil fuel products, amount of CO2 emission was confirmed to increase to 9.54% in 2010 .3 Consequently, CO2 must be used for alternative purposes to mitigate the harmful effects of excessive emissions and so that the activated products can serve as a source for additional chemical raw materials.
Graphene is a two-dimensional conjugated carbon nanomaterial consisting of sp2 hybrid C atoms connected by hexagons, which was first identified and examined in 2004.4 It was discovered and studied by Geim and Novoselov. Subsequently, scientists discovered that graphene exhibits exceptional electrical conductivity properties owing to its high specific surface area.5 Its application prospects in gas adsorption and other fields are favourable because of these properties. Several other two-dimensional (2D) materials have been investigated and were found to be worthy for several optoelectronic applications, such as the adsorption of water vapor, photocatalysis, solar cells, optical glasses, and radiation shielding.6,7 For example, Cao et al. studied the adsorption properties of 2D carbon toward heavy metal ions.8 They found that Cd2+, Hg2+, and Pb2+ were actively adsorbed on the surface of the 2D carbon. In 2019, Francis et al. comprehensively reported the active performance of graphene–MoS2 nanosheets in CO2 capture.9 Their report revealed that the electronic characteristics of graphene and MoS2 were not significantly affected by CO2 physisorption, as demonstrated by the density of state (DOS) plots. In another report, Itas et al. reported remarkable progress in the quantum capacitance of 2D silicon carbide (SiC) by surface modification and band gap engineering.10 According to their work, incorporating boron into defective 2D-SiC significantly improves the CQ of monolayer SiC.
Nowadays, density functional theory is the commonest way to tailor materials (including two-dimensional materials) for future experimental purposes. Chettri et al. reported the tailoring of hexagonal boron nitride materials for hydrogen adsorption using the DFT approach.11 Notably, their results revealed the most favourable adsorption site for hydrogen with a binding energy of 0.21 eV. Using band gap engineering, DFT calculations have revealed that graphene oxide (GO) can be a future material for optoelectronic applications.12 Bulk magnesium oxide (MgO) has been applied very well in waste removal, water purification, dye degradation air emission control, etc., owing to its capacity to stabilize dissolved heavy metal species.13 Additionally, pristine MgO has been studied from both experimental and theoretical perspectives.14 It is highly believed that the optoelectronic performance of MgO can be significantly improved through surface modification and band gap engineering. For example, Jitendra et al. investigated the electronic properties of MgO in terms of surface modification using zinc and iron plantation.15 They found that the implantation of Fe and Zn ions led to a reduction in the coordination geometry of the Mg2+ ions in the host lattice. According to a review report by Ruhaimi et al., magnesium-based adsorbents can actively serve as future materials for CO2 capture and utilization owing to their large surface areas.16 Felix and Christoph also reported the prospect of MgO-based adsorbents for potential CO2 capture at high temperatures.17
However, its 2D form in the pristine, defect and doped states has rarely been investigated both experimentally and theoretically. Out of the few studies, defective and doped MgO nanosheets have been investigated and found to be worthy of optoelectronics and spintronics applications.18 In this work, we report the CO2 adsorption properties of 2D MgO, known as the MgO sheet using the popular density functional theory. Studies were conducted on pristine, vacant and Ni-doped MgO sheet systems. The 2D MgO sheet was considered for this investigation because it is highly reactive and demonstrates good performance in the adsorption process. However, we chose Ni transition metal as a dopant for MgO because previous reports revealed that the introduction of Ni as a dopant led to improved transmittance and absorbance of up to 70% within the visible range.19 Additionally, Ni doping significantly expands the sp2 domain and increases the number of photo-generated charges.20 The innovation related to this research depends on the improved carbonation kinetics and efficiency, which improves 2D MgO's reactivity and stability in terms of CO2 capture.
2. Research methods
2.1 Geometry optimization of the studied structures
In this work, Gaussian 09 and Quantum ESPRESSO codes were used to complete all calculations, which are based on the DFT calculation method.21 We first generated a pristine two-dimensional (2D) MgO material using the VESTA code and ensured that the Mg–O bond lengths were appropriately adjusted according to the experimental values.22 It is commonly known that molecular adsorption is significantly impacted by crystal defects. The surface of the MgO sheet is prone to oxygen or Mg vacancy defects. In this work, the surface model of the vacancy of the MgO sheet was achieved through the elimination of one O atom. We used this information to simultaneously construct the MgO sheet (001) surface with and without vacancies and under Ni-doping. Fig. 1(a)–(c) presents the pristine, vacant and Ni-doped MgO sheets, respectively. A 15 Å vacuum layer was added in the z direction to build the sheet mode and prevent interaction during the adsorption process. The structural appearance of all the three configurations is depicted in Fig. 1.
 |
| | Fig. 1 Geometry of the isolated systems and CO2 exposure of (a) pristine MgO sheet, (b) vacant MgO sheet and (c) Ni-doped MgO sheet. Yellow balls represent Mg atoms, red balls represent O atoms, grey balls represent C atoms, and blue balls represent Ni atoms. | |
2.2 Computational procedure for optimization
For the most part, the geometrical relaxations of each model were used to perform all the computations and ensure that the force on each atom was reduced below 0.05 eV per atom. Using the plane wave basis set and pseudopotential method, the electron wave function was expanded, and the overall exchange–correlation energy of all electrons was handled by the PBE (Perdew–Burke–Ernzerhof) functional of the GGA (generalized gradient approximation).23 To account for the efficiency and accuracy of our calculations, the kinetic energy cutoff value of the plane wave basis group of 100 Ry was chosen. The 3 × 3 × 1 Monkhorst–Pack format k-point grid was used to integrate the Brillouin zone after testing various k-point grid schemes (the k-point setting of 5 × 5 × 2 2D MgO unit cell was optimized). The self-consistent field (SCF) cycle converges to 2.0 × 10−6 eV per atom; the maximum force convergence value of each atom was 0.05 eV Å; the maximum internal stress of the crystal was 0.10 GPa; and the maximum displacement convergence value of the atom was 0.002 Å. These are the convergence criteria for both structure optimization and energy calculation.
2.3 Thermal and adsorption energy calculations
Analysing thermodynamic parameters such as Gibbs free energy, enthalpy, adsorption energy and entropy is crucial for several reasons, such spontaneity of the process, adsorption mechanism, and temperature dependence. To describe the stability of CO2 gas at the selected absorption sites, we determined the formation energy (Ef) and adsorption energy (Eads) of each model in accordance with Raganati et al.24 It is noteworthy that the formation energies of all the systems were determined using the conventional total energy calculation in terms of the total energy of the block systems and their constituent parts:25| | |
Ef(MgO) = Et(MgO) − Et(Mg)/ηMg − Et(O)/ηO,
| (1) |
| | |
Ef(Ni_MgO) = Et(Ni_MgO) − Et(Mg)/ηMg − Et(O)/ηO − Et(Ni)/ηNi,
| (2) |
| | |
Eads = ECO2/sheet − ECO2 − Esheet,
| (3) |
where Et(Mg), Et(O), Et(MgO), and Et(Ni) represent the total energies of isolated Mg, isolated O, isolated MgO, and isolated Ni atoms, respectively. η is the number of atoms in the corresponding Mg, O and Ni elements. The vacancy formation energy is also calculated using eqn (1). However, the total number of oxygen atoms differs owing to vacancy.
Regarding adsorption, ECO2/sheet is the total energy of the system after adsorption, ECO2 is the energy of the adsorbed free CO2 gas molecules, and Esheet is the energy of the MgO sheet before adsorption.
The charge difference density (CDD), which defines the charge transferred during intermolecular interactions, was determined by Alexandros et al.:26
| | |
Δρ = ρCO2/sheet − ρCO2 − ρsheet,
| (4) |
where
ρCO2/sheet,
ρsheet, and
ρCO2 represent the total charge density of the system after adsorption, the charge density of the MgO sheet before adsorption, and the charge density of free CO
2 gas molecules, respectively. Thermodynamic parameters such as Gibbs free energy (Δ
G), enthalpy (Δ
H) and entropy (Δ
S) play significant roles in scaling up the gas adsorption processes. They highlight the estimate of the adsorption mechanism and determine the feasibility of the process. In the current work, we determine the Δ
G and Δ
H of the systems under study using the following equations:
| | |
ΔH (298 K) = ΔHCO2adsorbed sys − ΔHisol system − ΔHisol CO2,
| (5) |
| | |
ΔG (298 K) = ΔGCO2adsorbed sys − ΔGisol system − ΔGisol CO2,
| (6) |
where Δ
HCO2adsorbed sys is the enthalpy of the CO
2 adsorbed MgO sheet, Δ
Hisol system is the enthalpy of the pristine MgO sheet and Δ
Hisol CO2 is the enthalpy of the isolated CO
2 molecule. Similarly, Δ
Gisol system is the enthalpy of the pristine MgO sheet and Δ
Gisol CO2 is the enthalpy of the isolated CO
2 molecule.
2.4 Determination of ultra-violet-visible (UV-Vis) and vibrational properties
In this project, we employed UV-Vis to properly understand the range of wavelengths in which CO2 molecule was adsorbed. The procedure was first introduced by Beer–Lambert, who proposed the following equation:27where μ is the absorption/adsorption coefficient.
3. Results and discussion
3.1 Adsorption of CO2 molecules on optimized MgO systems
To obtain the most favorable adsorption site, we optimized a cubic system of two-dimensional MgO, referred to as an MgO nanosheet or MgO sheet. The material consists of atoms of Mg and O arranged in a cubic lattice with Mg–O bond length and lattice constants of 2.30 Å and 4.20 Å, respectively, according to the reported experimental data.28 Fig. 1(a)–(c) displays the optimized structure of the isolated systems together with their corresponding exposure to CO2 before being adsorbed. The aftermath of adsorbing CO2 molecules is illustrated in Fig. 2(a)–(c). It can be observed that the orientation of all the isolated systems completely changed owing to interactions with CO2 molecules. Weak bonds can be observed attaching adsorbents to the adsorbed, demonstrating van der Waal's interactions. Regarding CO2 adsorption on pristine Mgo sheets, Fig. 2(a) reveals the bending of the CO2 molecule to 130°. Additionally, most of the CO2 molecules were adsorbed on the O site with few participation by mg atoms. CO2 was adsorbed on the O site at 130.80° and then at 155.20° at the Mg site. During adsorption, Mg and O atoms at the bottom side of the sheet completely buckled to 90°, forming a bowl-like structure. Regarding the vacant MgO nanosheet, we found that the C atom was weakly adsorbed on the surface of the Mg and O sites at 126.40° and 24.10°, respectively. Two atoms of Mg on the vacant site were bent towards the CO2 adsorption site at 173°. Generally, we found that CO2 was adsorbed by the nearby Mg and O atoms on the corresponding MgO adsorption surfaces. The results of the formation energies, adsorption energies and binding energies can be found in tables. It has been observed that all the systems demonstrated stability owing to the obtained negative values of energy parameters,29 meaning that energy must be applied to the systems from external sources to change their present states. Further discussions on energy parameters can be found in Subsection 3.3. The calculated results regarding the structural stability and thermodynamic stabilities of the systems are presented in Tables 1 and 2. As shown in Table 1, the formation energy and adsorption energy values all turn out to be negative, meaning that the systems are in a state of high structural stability. For example, the negative value of formation energy for pure MgO (−2.15 eV) indicates that it can undergo a spontaneous adsorption process, requiring an external amount of energy to kick off. Therefore, because all the configurations of the MgO sheets were found to be stable, they can be synthesized experimentally by depositing MgO films at very low temperatures (for example 190 °C) and compact films with small crystallites can be formed.30
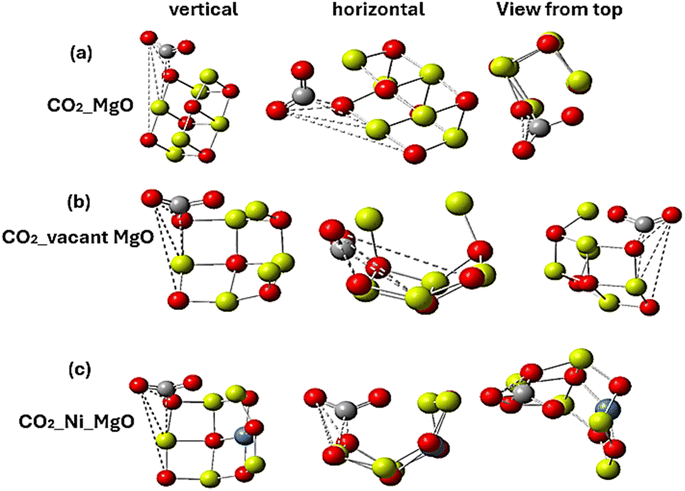 |
| | Fig. 2 CO2 adsorbed on the surface of (a) pristine MgO sheet, (b) vacant MgO sheet and (c) Ni-doped MgO sheet. The broken bonds indicate weak interactions (van der Waals), which favor the adsorption process. | |
Table 1 Stability parameters for CO2 adsorbed on pure, vacant and Ni-doped MgO monolayer
| Material |
Formation energy (eV) |
Adsorption energy (eV) |
Binding energy (eV) |
| Pure MgO |
−2.15 |
— |
— |
| CO2@pure MgO |
— |
−0.52 |
51.21 |
| CO2@vacant MgO |
— |
−0.77 |
51.28 |
| CO2@Ni-doped MgO |
— |
−0.91 |
51.40 |
Table 2 Energy correction values for isolated surfaces and CO2-adsorbed surfaces
| Systems |
DFT energy (hartree) |
ZPE (hartree) |
Etot (hartree) |
Hcorr (hartree) |
Gcorr (hartree) |
| CO2 |
−188.50 |
0.011 |
0.013 |
0.014 |
−0.010 |
| MgO |
−1643.30 |
0.032 |
0.043 |
0.044 |
−0.003 |
| vMgO |
−1577.04 |
0.025 |
0.037 |
0.038 |
−0.012 |
| Ni_MgO |
−2960.23 |
0.027 |
0.042 |
0.043 |
−0.014 |
| CO2_MgO |
−1840.93 |
0.044 |
0.058 |
0.059 |
0.003 |
| CO2_vMgO |
−1765.58 |
0.038 |
0.053 |
0.054 |
−0.003 |
| CO2_Ni_MgO |
−3148.84 |
0.042 |
0.058 |
0.059 |
−0.002 |
3.2 UV-Vis adsorption and vibrational analysis
The ultraviolet-visible (UV-Vis) adsorption analysis provides a very accurate, reliable and sensitive approach to gas sensing analysis.31 It is one of the simplest and most economical methods adopted by researchers to investigate the interactions between adsorbents and adsorbents. Using this method, the only change in adsorbance is measured against the wavelength. In this work, UV-Vis analysis was conducted on the three isolated systems and was then reconducted after adsorbing CO2 molecules. Fig. 3(a)–(c) presents UV-Vis results of isolated pristine, vacant and Ni-doped MgO nanosheets, respectively. As presented in Fig. 3(a), (c) and (e), the long wavelength band with maximums of 1333.10, 492.90 and 2339.81 nm occupied visible and IR adsorption spectra of pristine, vacant, and Ni-doped MgO nanosheets, respectively. These peaks demonstrate the points of potential maximum energy adsorption that correspond to the IR and visible regions. Therefore, the adsorption potentials of these adsorbents are determined by higher electronic excitations, not by thermal decomposition. With respect to the pristine MgO nanosheet, flat peaks below 491.22 nm present zero potential to adsorb in the UV-Vis region, which corresponds to the reported characteristics of covalent compounds.32 As the system adsorbs CO2 (Fig. 3(b)), the intensity of the adsorption was significantly reduced from 1295.74 to 406.21 M−1 cm−1. Additionally, most of the CO2 is not adsorbed from 100 to 300 nm, corresponding to the UV region, and then from 600 to 800 nm, corresponding to the visible region. The adsorption spectrum from 300 to 566.22 nm with a high peak at 403.20 nm corresponds to the small adsorption in the UV-Vis. Fig. 3(d) displays the CO2 adsorption spectra for the vacant MgO nanosheet. This system also demonstrated its capacity to adsorb CO2 in both UV and visible regions with high adsorption from 350 to 653 nm. Except for this range, termination of the spectrum at 800 nm confirmed that the vacant MgO sheet adsorbed CO2 across the UV-Vis range only. A feature distinct from pristine and vacant MgO systems was observed when the Ni-doped MgO nanosheet adsorbed CO2 molecules. As shown in Fig. 3(f), an entirely flat peak was observed up to 780 nm, covering all UV-Vis regions. Therefore, no adsorption was recorded. Additionally, the adsorption peak appeared only in the IR region, starting from 842 nm with its maximum at 1202.24 nm. Based on this, the Ni-doped MgO nanosheet registered no adsorption in the UV-Vis range; it was also observed to adsorb in parts of the far IR and hence regarded as a better adsorbent than the other systems. In other words, this system presented itself with better performance than pristine and vacant MgO nanosheets.
 |
| | Fig. 3 UV-Vis spectra of (a) pristine MgO sheet, (b) CO2_MgO sheet, (c) vacant MgO sheet, (d) CO2_vacant MgO sheet, (e) Ni-doped MgO sheet, and (f) CO2_Ni doped MgO sheet. | |
To achieve a good interpretation of the obtained results, we carried out a frequency-IR vibrational adsorption analysis of the CO2 adsorbed systems, as shown in Fig. 4. With respect to CO2 adsorption on a pristine MgO sheet, Fig. 4(b) illustrates the strongest band 1750 cm−1. Two other stronger bands can be observed at 500 and 750 cm−1, respectively. Below 500 cm−1 is the region of weak bands corresponding to weak adsorption.33 As the vacant system adsorbs CO2 (Fig. 4(b)), several weak bands appear below 1000 cm−1, indicating that CO2 was weakly adsorbed in the region, except for the valley formed at 500 cm−1. Similarly, Fig. 4(c) illustrates continuous harmonic vibrations up to 1000 cm−1, indicating interactions between the UV and visible regions. The presence of several sharp peaks by the pristine MgO sheet indicated irregular adsorption compared to the two other systems. Analysis of vibrational frequencies revealed 34, 36 and 39 vibrations by CO2 adsorbed pristine, vacant and Ni-doped MgO sheets, respectively. Moreover, these vibrations were observed in various directions, demonstrating that the adsorbents are very flexible to CO2 adsorption. Therefore, CO2 is adsorbed at various surface points of the adsorbents according to the intensity of the vibrations. Fig. 5 displays vibrations by the adsorbing systems and various vector displacements of some randomly selected modes 21, 25, 30 and 37, with frequencies. Most of the vibrations were contributed by O (also see Subsection 3.4) atoms because they were more electronegative than Mg.
 |
| | Fig. 4 Vibrational frequencies of (a) CO2_MgO sheet, (b) CO2_vacant MgO sheet and (c) CO2_Ni doped MgO sheet. | |
 |
| | Fig. 5 Vector displacements of some randomly selected vibrations for (a) CO2_MgO sheet, (b) CO2_vacant MgO sheet and (c) CO2_Ni-doped MgO sheet. | |
3.3 Thermophysical analysis
In condensed matter and thermal physics, gaseous adsorption is a process accompanied by the spontaneity of a chemical process. It also depends on the feasibility of the adsorption process. Gas adsorption is a spontaneous process accompanied by a decrease in Gibbs free energy, enthalpy and entropy. As reported by others,34 a negative value of Gibbs free energy (ΔG0) indicates a spontaneous process, as presented in Table 2. Moreover, when free energy is negative, the process shows that the adsorbing system has lost some energy or moved from a higher energy state to a more stable lower energy state; therefore, the process is spontaneous because some amount of energy is released. Table 2 displays the energy correction parameters used to obtain the corrected adsorption parameters presented in Table 3. The parameters were determined for the isolated and adsorbed systems. As shown in Table 3, all energy values presented negative signs, indicating that all the investigated systems were highly stable. To justify the accuracy of this work, the calculated formation energies were evaluated using both the DFT and Hartree–Fock methods. It can be observed from Table 3 that the values obtained from the two approaches agreed well. For example, the DFT and HF values corresponding to CO2_MgO were −1840.89 and −1840.87 hartree, respectively. Among the isolated systems, CO2 presented itself as more stable because of its lower value of free energy (−188.51 H), while Ni-doped MgO was considered less stable owing to its highest value of free energy (−2960.25), which is due to unwanted interactions between the MgO molecule and Ni impurities.
Table 3 Adsorption parameters for isolated and CO2-adsorbed systems
| Systems |
DFT energy (hartree) |
Etot |
H (hartree) |
G (hartree) |
| CO2 |
−188.49 |
−188.49 |
−188.47 |
−188.51 |
| MgO |
−1843.27 |
−1643.26 |
−1843.26 |
−1843.31 |
| vMgO |
−1577.02 |
−1577.01 |
−1577.00 |
−1577.06 |
| Ni_MgO |
−2960.21 |
−2960.19 |
−2960.19 |
−2960.25 |
| CO2_MgO |
−1840.89 |
−1840.87 |
−1840.87 |
−1840.93 |
| CO2_vMgO |
−1765.54 |
−1765.53 |
−1765.53 |
−1765.58 |
| CO2_Ni_MgO |
−3148.80 |
−3148.78 |
−3148.78 |
−3148.84 |
With respect to studies of adsorption properties, enthalpy change (ΔH0) highlights the nature and mechanism of the process. When enthalpy is negative, an exothermic process occurs, while positive enthalpy indicates an endothermic process. Ofomaja et al. confirmed that the positive value of ΔH0 (89.13 kJ mol−1) was the result of the endothermic adsorption of malachite green onto chemically modified rice husk.35 In Table 4, the obtained ΔH0 values for CO2 adsorbed on pristine, vacant and Ni-doped systems are −18.140, −22.13 and −65.11 kJ mol−1, respectively. Therefore, all the systems underwent exothermic adsorption processes. Additionally, the high value of ΔH0 by the Ni-doped system indicates that CO2 is well adsorbed on its surface compared to the two other systems. Other parameters, such as changes in free energy and entropy, can also be observed in Table 4.
Table 4 CO2 adsorption energy parameters for the three adsorption surfaces
| Thermodynamic parameter |
Adsorbent |
Adsorbate |
Value |
Remark |
| Gibbs free energy (ΔG0) (kJ mol−1-kel) |
Pristine MgO nanosheet |
CO2 |
−18.21 |
Spontaneous |
| vMgO nanosheet |
CO2 |
−11.63 |
Spontaneous |
| Ni_MgO nanosheet |
CO2 |
−51.366 |
Spontaneous |
| Enthalpy change (ΔH0) (kJ mol−1) |
Pristine MgO nanosheet |
CO2 |
−18.14 |
Exothermic |
| vMgO nanosheet |
CO2 |
−22.13 |
Exothermic |
| Ni_MgO nanosheet |
CO2 |
−65.11 |
Exothermic |
| Entropy change (ΔP0) (J mol−1) |
Pristine MgO nanosheet |
CO2 |
117.17 |
Physisorption |
| vMgO nanosheet |
CO2 |
121.38 |
Physisorption |
| Ni_MgO nanosheet |
CO2 |
127.61 |
Physisorption |
3.4 Electronic transport analysis
Fig. 6(a) displays an electronic band diagram of the pure MgO sheet. This system revealed significant semiconducting features, demonstrating a band gap value of 2.30 eV. Moreover, the band gap was found to be direct at the gamma point with balance band maxima and conduction band minima at the same momenta.36 In this type of material, energy is produced in the form of light during electron–hole recombination because electrons undergo elastic collisions across the Fermi level. The magnetic properties of these systems were also checked by considering the up and down spins of the total density of states (DOS), as shown in Fig. 6(b). The MgO sheet under investigation was found to be nonmagnetic owing to the symmetry of the up and down spins, which brought the magnitude of the magnetic moment to zero. Fig. 6(b) also depicts that the characteristic features of the MgO sheet in the valence band were mainly due to the high interactions of the p' orbitals of the O atom. Additionally, p' orbitals of Mg were observed with few contributions to the electronic interactions in pristine MgO sheets. The presence of flat energy states around the Fermi level justified the semiconducting features, as observed in Fig. 6(a). The obtained band gap value of this sheet presented it as a better material for several band gap modification applications, such as energy storage, photovoltaic, solar cell, and waste treatment.37 To check the total effects of CO2 adsorptions, we then created a vacancy in the pristine MgO sheet by removing one O atom. Separately, we also doped a pristine MgO sheet with one Ni atom and reported the obtained results, as presented in Fig. 7.
 |
| | Fig. 6 (a) Electronic band diagram of pristine MgO sheet showing valence band maxima and conduction band minima and (b) DOS and PDOS diagrams showing various orbital contributions. | |
 |
| | Fig. 7 PDOS diagrams of (a) CO2 absorbed on pristine MgO sheet, (b) CO2 adsorbed on vacant MgO sheet and (c) CO2 adsorbed on Ni-doped MgO sheet. | |
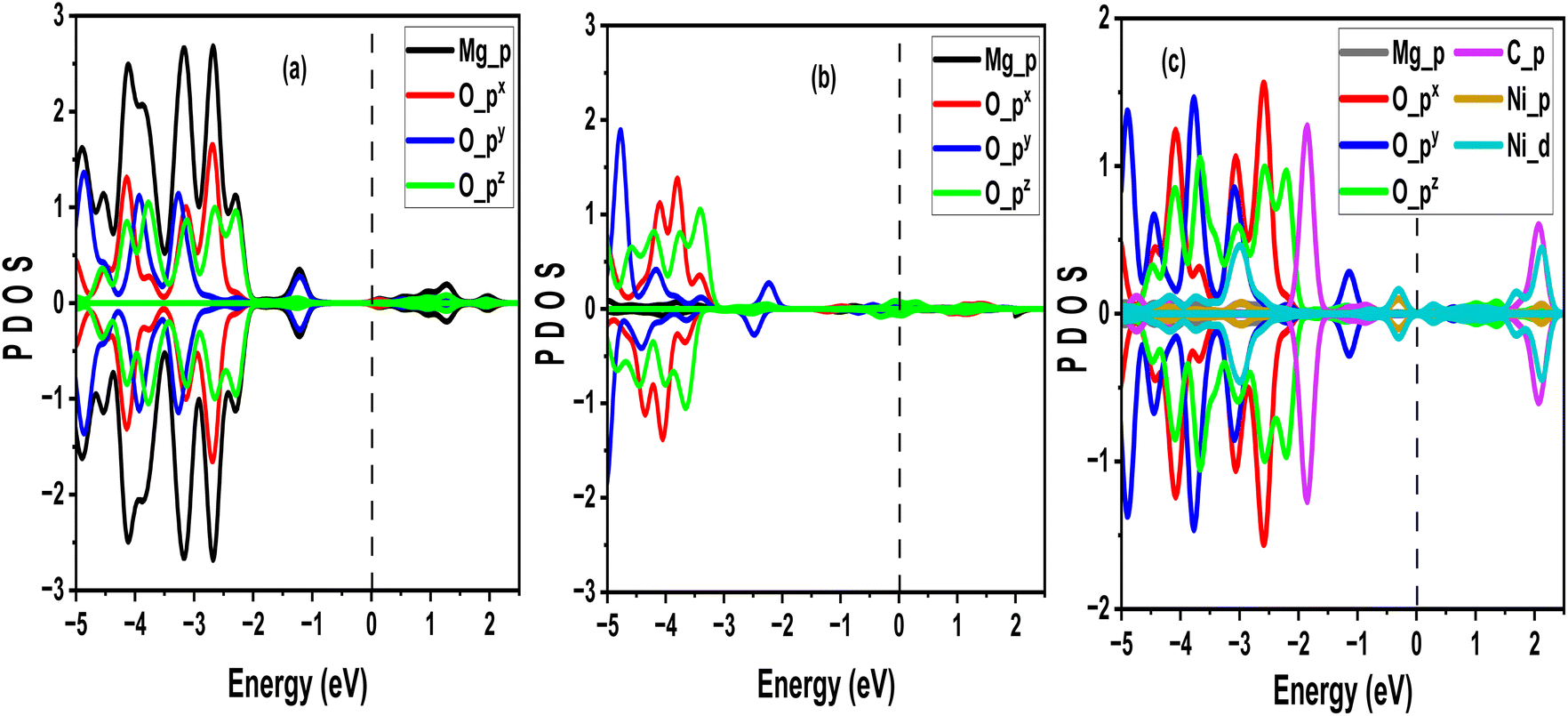
According to previous reports, the partial density of states (PDOS), which offers comprehensive insights into the electronic structure of the adsorbate-substrate system, is essential for explaining the adsorption process.38 Regarding electronic interactions, understanding the interaction between the adsorbate's and substrate's electronic states is made easier through PDOS analysis. Additionally, the precise energy levels at which the adsorbate states are found can be obtained by examining the PDOS spectra, which is essential for understanding whether the process involves physisorption (weak van der Waal's) or chemisorption (strong bonds). Fig. 7(a) describes the PDOS pattern of the CO2 adsorption on a pure MgO sheet. As shown, the PDOS revealed closure of the Fermi level owing to an increase in the interactions of more atoms of O. The majority of the interactions in the vicinity of the Fermi level were mainly due to partial hybridization of py and pz orbitals of O atoms in the CO2 molecules, which is in line with the discussion in Subsection 3.1. An observed symmetry in the up and down spin confirmed that the MgO monolayer is non-magnetic after taking CO2 molecules. To observe a more interesting feature of the results of the interactions between CO2 and MgO sheets, we further reported the effects of CO2 adsorption on the defected (vacant) MgO sheet. Fig. 7(b) illustrates asymmetric PDOS between −2.51 eV and 1.94 eV, indicating that the vacant MgO sheet turned magnetic when it interacted with CO2 on its (001) surface. In this system, the general behaviour of the Fermi level was taken over by the pz orbital of the O atom. These orbitals also dominated the conduction band with few participations by the px orbital of the same O atom. Therefore, we can describe the general adsorption behaviour of vacant MgO sheets with respect to the dominance interaction by p orbitals of O. In Fig. 7(c), when CO2 was adsorbed on the Ni-doped MgO sheet surface, the 3p orbital of the Ni atom broadened in the −11 eV to −2.5 eV energy range. The contributions of the upper and lower spin states of the Ni atom were the same, which is related to zero magnetization. Additionally, the dominant interaction at the Fermi level was by d' orbital of Ni, indicating that CO2 molecules preferred to be adsorbed near the vicinity of the Ni element.
3.5 Analysis of charge density (CD) and charge density difference (CDD)
Solid-state physics and materials science depend heavily on charge density descriptions to accurately understand the electronic interactions and characteristics of materials.39 It helps describe how materials, such as metals, semiconductors, and insulators, are structured electrically. In quantum chemistry, charge density maps are used to show the distribution and occupation of electrons within ions and molecules. The charge density distributions of pure MgO are depicted in Fig. 8(a). The crystal structure of MgO indicates the charge density distributions on the Mg and O sites. As observed, all charges were generally accumulated on the O sites because O is highly electronegative (−2), which also justifies our earlier discussion in Subsection 3.1. As CO2 is adsorbed on pure MgO (Fig. 8(b)), more charges were observed to gather around the CO2 molecule, indicating its readiness to partially interact with MgO. Fig. 8(c) presents the isolated structure of CO2 adsorption on vacant MgO. In this material, CO2 molecules can be observed to accumulate near the vacant site of MgO because, at this time, electronegativity has been introduced in Mg. Regarding Ni-doped MgO, CO2 molecules prefer to be adsorbed near the vicinity of the Ni atom because the nickel atom is one of the strong ferromagnetic materials. In general, the charge density distributions of CO2 (Fig. 8(a)–(c)) adsorbed MgO systems revealed enhanced orbital hybridization between py and pz orbitals of O compared to pure MgO, which was found to be in good agreement with other studies.40
 |
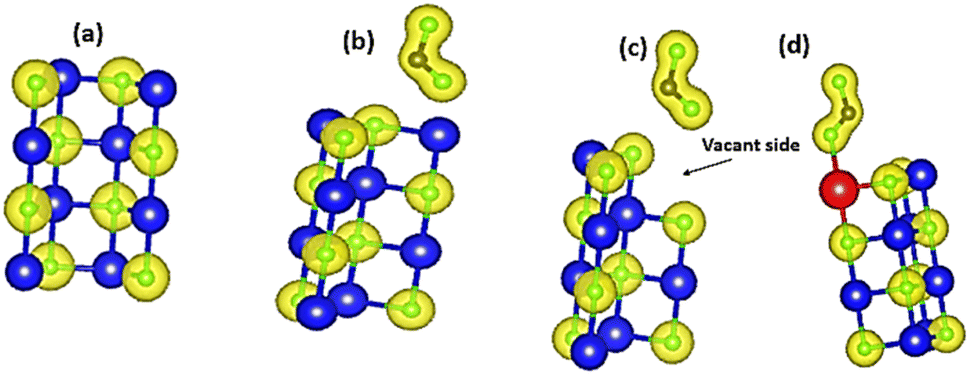
| | Fig. 8 Charge density distributions of (a) pristine MgO sheet, (b) CO2 absorbed on pristine MgO sheet, (c) CO2 adsorbed on vacant MgO sheet, and (d) CO2 adsorbed on Ni-doped MgO sheet. Yellow balls represent the iso-surfaces, blue balls represent Mg atoms; green balls represent oxygen atoms, and grey balls represent C atoms. | |
The fundamental idea of DFT is that the electronic charge density of the ground state, ρ(r), with r being the position vector, determines all the physical properties of a quantum system, including energy. Fig. 9 presents the charge density difference (CDD) of the various configurations of the MgO sheet under investigation. It can be observed that the electron accumulation region of the CO2 was significantly concentrated near the vicinity of the O atoms. However, the charge depletion region was found to be near the Mg atoms. Fig. 9(a)–(c) also depicts that the oxygen atoms near the CO2 adsorption sites greatly took part in most of the absorption processes. Among these three materials, the highest CDD was attributed to Ni-doped MgO (Fig. 9(c)), which is in good agreement with previous studies.41 Generally, Mg atoms in the absorption sites were found to lose some electrons; hence, a large number of charges accumulated between C–O to form chemical bonds.
 |
| | Fig. 9 Charge density difference of (a) CO2 absorbed on pristine MgO sheet, (b) CO2 adsorbed on vacant MgO sheet, and (c) CO2 adsorbed on Ni-doped MgO sheet. | |
3.6 Optical spectra analysis
In solid-state physics and chemistry, adsorption and absorption are also very important optical properties of materials because they play key roles in determining the nature of light interaction with matter.42 When light is incident on a material surface, some part of it is adsorbed, absorbed, reflected, refracted, or transmitted. One of the better examples of a perfect adsorber is a black body, which demonstrates a high ability to adsorb light at all wavelengths. Additionally, when examining energy band structure, impurity levels, excitons, localized defects, lattice vibrations, and specific magnetic excitations in materials, optical properties offer valuable tools. To achieve this, new research methods and a deeper comprehension of quantum materials are needed. In the present study, we further examine the CO2 adsorption behaviors of the optimized MgO sheets using optical spectra analysis. First, we report the optical properties of the pristine MgO sheet in terms of the complex dielectric functions represented by43where ε1(ω) and ε2(ω) are the real and imaginary dielectric constants, respectively. The real part emphasizes the material's refractive index and electronic polarizability, or capacity to store electrical energy, while the imaginary part accounts for the material's energy loss or absorption of light. Fig. 10(a) presents the real spectra of the pristine MgO sheet. The results were computed in both parallel (x) and perpendicular (y) to the surface of the sheet. As can be observed, the static dielectric constants were observed at 4.4 (perpendicular) and 5.2 (parallel). The negative peaks in the parallel direction indicated the potential for plasmon excitations in the MgO sheet.44 Additionally, the high value of a static dielectric constant in the parallel direction revealed that this MgO sheet likely refracted more in that direction. High peaks at 2.4 eV in both directions revealed the ability of the MgO sheet to store electrical energy, which is one of the characteristics of better candidates for energy adsorption, photocatalysis and waste management.45 In Fig. 10(b), the energy loss due to the adsorption of light was reported in both parallel and perpendicular directions. Zero peaks were observed in ranges of 0–1.4 eV and 0–2.2 eV in parallel and perpendicular directions, respectively, confirming good adsorption in the UV-Vis range. These energy points can be termed the edges of adsorptions in the pristine MgO sheet. Moreover, zero peaks in this range demonstrated the optical band gap values of the pristine MgO sheet. They also confirmed that no energy was lost in the visible range, which agrees well with our discussion in Subsection 3.2. The electron energy loss (EELS) spectrum presented in Fig. 10(c) is supported by the information in Fig. 10(b), and it can be observed that no energy was lost due to adsorption between 0 and 3 eV. Therefore, our results show that pristine MgO sheets can be better materials for visible light applications, such as adsorption, absorption, solar cells, and photocatalysis.46
 |
| | Fig. 10 Optical spectra of pure MgO sheet, (a) real dielectric spectrum, (b) imaginary dielectric spectrum and (c) EELS spectrum. | |
Having known the optoelectronic potentials of the MgO sheet in its pure form, we now exposed it to CO2 interactions and then analysed the new changes observed. Results were obtained by calculating the optical adsorptions of the CO2 adsorbed MgO sheet in the pure, vacant and Ni-doped systems. The optical adsorption spectra of the CO2 molecules and MgO sheets are displayed in Fig. 11(a)–(c) The intrinsic exciton adsorption of CO2 is responsible for the adsorption edge of around 1.4 eV, corresponding to the overpotential value of CO2 adsorption. The spectra of CO2 molecules created by O ion implantation are responsible for these clear adsorption peaks. Thus, it is anticipated that there will be an effective energy transfer from the MgO sheet to CO2. In each case, higher electromagnetic interactions were observed in the visible range corresponding to 0–2.1 eV, indicating that CO2 was well adsorbed in the visible range. In the pristine system (Fig. 11(a)), the intensity of the adsorption was generally low in both directions and then diminished towards the UV range. Therefore, CO2 molecules were not adsorbed above the visible range in the MgO sheet material in agreement with Subsection 3.2. Fig. 11(b) illustrates that CO2 was significantly adsorbed (by a factor of 37 a.u.) in the perpendicular direction because of Mg deficiency. With respect to the Ni-doped system, highly precise behaviors were obtained, showing that the Ni-doped system adsorbed CO2 molecules significantly in both the parallel and perpendicular directions. This is because it is very active in methanation and adsorption and limits the production of carbonaceous materials. In contrast to Fig. 11(b), the Ni-doped MgO sheet adsorbed CO2 more in the parallel direction than in the perpendicular direction. In general, CO2 was actively adsorbed in the visible (near IR) region. Additionally, adsorption was inversely related to the total energy of the system.
 |
| | Fig. 11 CO2 adsorption spectra (a) CO2 absorbed on pristine MgO sheet, (b) CO2 adsorbed on vacant MgO sheet and (c) CO2 adsorbed on Ni-doped MgO sheet. | |
To summarize, we presented a summary of the data obtained in this work and compared it with previous works, as shown in Table 5. Generally, spontaneous adsorption processes can be observed with negative values of Gibbs free energy. Electronically, all the adsorbing systems turned metallic after adsorbing their corresponding adsorbents. Additionally, non-spontaneous processes can be identified with negative values of entropy, and most of the adsorptions took place in the UV-Vis range by all the systems. Based on the data obtained, which are well correlated with previous works, we can report that CO2 adsorption on the surface of the MgO sheet is highly sustainable. The process will help downgrade the release of large amounts of CO2 into the atmosphere by large emitters. Therefore, this research recommends two-dimensional MgO as a potential material for mitigating global warming and ocean acidification caused by CO2 emissions from factories and automobiles.
Table 5 Presentation of current results versus previous resultsa
| System |
Band gap (eV) |
UV-Vis |
ΔG |
ΔH |
ΔS |
Thermodynamic process |
Type of adsorption |
Ref |
| NR = not reported. |
| Current work |
| CO2_MgO |
0.00 |
100–800 |
−18.214 |
−18.14 |
117.17 |
Spontaneous |
Physisorption |
|
| CO2_vMgO |
0.00 |
100–800 |
−11.627 |
−22.13 |
121.38 |
Spontaneous |
Physisorption |
| CO2_Ni_MgO |
0.00 |
100–850 and 3135.5 |
−51.366 |
−65.11 |
127.61 |
Spontaneous |
Physisorption |
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) |
| Others |
| Ag/Bi2O3 |
0.00 |
250–760 |
−32.40 |
35.60 |
100.11 |
Spontaneous |
Physisorption |
[47] |
| CO2 on montmorillonite |
NR |
100–810 |
−2.40 |
−12.0 |
8.322 |
Spontaneous |
Physisorption |
[48] |
| CO2 on activated carbon |
0.00 |
NR |
−26.40 |
−15.20 |
−18.00 |
Non-spontaneous |
Chemisorption |
[49] |
| Fluorine-doped boron nitride |
0.00 |
NR |
−33.91 |
−59.43 |
40.00 |
Spontaneous |
Physisorption |
[50] |
4. Conclusion
Conclusively, the structural, electronic, vibrational, thermal and optical properties of MgO sheets were investigated in pristine, vacant and Ni-doped states using DFT. The formation energy was significantly reduced by Ni atom-induced impurities at the Fermi level. This is accompanied by promoting the CO2 adsorption ability. We observe that the optimized MgO sheet underwent several bucklings when it adsorbs the CO2 molecule. For example, during adsorption, Mg and O atoms at the bottom side of the sheet completely buckled to 90°, forming a bowl-like structure. Owing to vacancy, the C atom was weakly adsorbed on the Mg and O sites at 126.4° and 24.1°, respectively. UV-Vis analysis revealed that both pristine and vacant MgO sheets adsorbed only CO2 in the UV-Vis range with significant intensity, while the Ni-doped MgO sheet extended its adsorption to the IR region. 34, 36 and 39 vibrations were produced in various directions by CO2 adsorbed pristine, vacant and Ni-doped MgO sheets, respectively, confirming their ability to adsorb in various directions. The obtained enthalpy values of 18.140, −22.132 and −65.105 kJ mol−1 attributed to pristine, vacant and Ni-doped systems, respectively, confirmed that the adsorption process is exothermic. According to CDD analysis, the electron accumulation region of the CO2 was significantly concentrated near the vicinity of O atoms in MgO sheets. Based on the data obtained, which are well correlated with previous works, we can report that CO2 adsorption on the surface of the MgO sheet is highly sustainable. The process will help downgrade the release of large amounts of CO2 into the atmosphere by large emitters. Sustainably, Ni-doped MgO can actively act as a converter of CO2 into valuable chemicals, such as syngas and methanol. In line with the global mission to achieve carbon neutrality and sustainable development, these advanced materials can actively contribute to reducing greenhouse gas emissions. Therefore, this research recommends two-dimensional MgO as a potential material for mitigating global warming and ocean acidification caused by CO2 emissions from factories and automobiles. However, achieving homogeneous doping requires accurate control of some prevailing synthesis conditions; hence, synthesizing vacant and Ni-doped MgO sheets can be accompanied by synthesis complexity.
Data availability
All data used in this manuscript regarding band structure, DOS and optical spectra analysis were analysed and obtained using the quantum ESPRESSO codes, which are freely available at https://www.quantumespresso.org.
Author contributions
Conceptualization and writing original draft: Y. S. I. Research methodology and software: M. U. K. and Y. S. I. Formal analysis and revision: Y. S. I.; M. U. K. and M. M.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
The authors extend their appreciation to the University Higher Education Fund for funding this research work under Research Support Program for Central Labs at King Khalid University through the project number CL/PRI/B/8.
References
- T. Ghellab, Z. Charifi and H. Baaziz, A DFT insight into optoelectronics and transport phenomena in the monoclinic BiGaIn2S6 compound for applications in renewable energy, Mater. Sci. Semicond. Process., 2024, 172, 108048 CrossRef CAS.
- W. Chengrui, F. Yang and H. Duan, DFT study of CO2 adsorption properties on pristine, vacancy and doped graphenes, Solid State Commun., 2021, 337, 114436 CrossRef.
- R. Masoume and N. Alireza, A DFT study on production of hydrogen from biomass-derived formic acid catalyzed by Pt–TiO2, Int. J. Hydrogen Energy, 2020, 45, 20993–21003 CrossRef.
- A. Imran, B. Al arsh and G. Evgeny, Graphene based adsorbents for remediation of noxious pollutants from wastewater, Environ. Int., 2019, 127, 160–180 CrossRef PubMed.
- K. Qiaoping, X. Shi and M. Weiwei, Strategies to improve the adsorption properties of graphene-based adsorbent towards heavy metal ions and their compound pollutants: a review, J. Hazard. Mater., 2021, 415, 125690 CrossRef PubMed.
- G. Xinyu, Y. Yuan and X. Meng, Recent development on BN-based photocatalysis: a review, Mater. Sci. Semicond. Process., 2020, 120, 105256 CrossRef.
- L. Jian, M. Xu and L. Shiru, Direct-gap semiconducting tri-layer silicene with 29% photovoltaic efficiency, Nano Energy, 2018, 51, 489–495 CrossRef.
- Y. Cao, M. H. Hasanen and W. Zhang, Adsorption properties of two-dimensional carbon material towards the heavy metal ions, J. Mol. Liq., 2021, 342, 117500 CrossRef CAS.
- M. E. Francis, I. E. Collins and W. Michael, A comparative study of mechanisms of the adsorption of CO2 confined within graphene–MoS2 nanosheets: a DFT trend study, Nanoscale Adv., 2019, 1, 1442–1451 RSC.
- I. Yahaya, M. A. Amnah and R. A. Mazen, Envisaging the quantum capacitance in modified monolayer silicon carbide, Appl. Phys. A: Mater. Sci. Process., 2024, 130, 668 CrossRef.
- B. Chettri, P. K. Patra and N. H. Nyugen, Hexagonal boron nitride (h-BN) nanosheet as a potential hydrogen adsorption material: a density functional theory (DFT) study, Surf. Interfaces, 2021, 24, 101043 CrossRef CAS.
- A. Qadoos, R. Muhammad and M. N. Naeem, Bandgap engineering in graphene oxide (GO) via integrating DFT calculations with atmospheric-pressure microplasma (AMP) treatment for optoelectronic applications, Hybrid Advances, 2025, 8, 100353 CrossRef.
- H. C. S. Perera, S. Anoop and G. Das, Magnesium oxide (MgO) nanoadsorbents in wastewater treatment: a comprehensive review, J. Magnesium Alloys, 2024, 12, 1709–1773 CrossRef CAS.
- E. Ola and J. S. Anthony, Adsorption of water on the MgO(001) surface, Surf. Sci., 1999, 437, 239–248 CrossRef.
- P. Jitendra, C. L. Weon and L. Jahyee, Surface and local electronic structure modification of MgO film using Zn and Fe ion implantation, Appl. Surf. Sci., 2018, 432, 132–139 CrossRef.
- A. H. Ruhaimi, M. A. Aziz and A. A. Jalil, Magnesium oxide-based adsorbents for carbon dioxide capture: current progress and future opportunities, J. CO2 Util., 2021, 34, 101357 CrossRef.
- D. Felix and C. Muller, Prospects of MgO-based sorbents for CO2 capture applications at high temperatures, Curr. Opin. Green Sustainable Chem., 2022, 36, 100645 CrossRef.
- V. Vo, V. V. Tuan and D. M. Hoat, Defective and doped MgO monolayer as promising 2D materials for optoelectronic and spintronic applications, Mater. Sci. Semicond. Process., 2022, 149, 106876 CrossRef.
- G. Xin, Q. Zhou and J. Wang, Adsorption of SO2 molecule on Ni-doped and Pd-doped graphene based on first-principle study, Appl. Surf. Sci., 2020, 517, 146180 CrossRef.
- R. Poul, A. Christy and K. Aslam, Significance of Ni doping on structure-morphology-photoluminescence, optical and photocatalytic activity of CBD grown ZnO nanowires for opto-photocatalyst applications, Inorg. Chem. Commun., 2020, 119, 108082 CrossRef.
- S. Y. Aminu, S. Amiruddin and I. Kunya Salisu, Effects of Exchange Correlation Functional (Vwdf3) on the Structural, Elastic, and Electronic Properties of Transition Metal Dichalogenides, J. Niger. Soc. Phys. Sci., 2023, 5, 1094 CrossRef.
- M. Jayapriya, R. Krithikadevi and M. Arulmozhi, Bio fabrication of 2D MgO/Ag nanocomposite for effective environmental utilization in antibacterial, anti-oxidant and catalytic applications, Surf. Interfaces, 2022, 30, 101921 CrossRef.
- A. I. Kamaluddeen, H. N. Awwal and R. Razif, The potentials of boron-doped (nitrogen deficient) and nitrogen-doped (boron deficient) BNNT photocatalysts for decontamination of pollutants from water bodies, RSC Adv., 2023, 13, 23659–23668 RSC.
- F. Raganati, M. Alfe and R. Chirone, Isotherms and thermodynamics of CO2 adsorption on a novel carbon-magnetite composite sorbent, Chem. Eng. Res. Des., 2018, 134, 540–552 CrossRef CAS.
- A. E. Antoine and W. Chris, High-throughput DFT calculations of formation energy, stability and oxygen vacancy formation energy of ABO3 perovskites, Sci. Data, 2017, 4, 170153 CrossRef PubMed.
- E. A. Alexandros, R. T. Maria and W. Lars, Nanolatex architectonics: Influence of cationic charge density and size on their adsorption onto surfaces with a 2D or 3D distribution of anionic groups, J. Colloid Interface Sci., 2023, 634, 610–620 CrossRef PubMed.
- J. Hedi, A. Maha and R. Raja, Activated Carbon as an Adsorbent for CO2 Capture: Adsorption, Kinetics, and RSM Modeling, ACS Omega, 2024, 9, 2080–2087 CrossRef PubMed.
- D. M. Hoat, K. N. Duy and N. Mosayeb, Structural, electronic and optical properties of pristine and functionalized MgO monolayers: a first principles study, RSC Adv., 2020, 10, 40411–40420 RSC.
- B. Daniel, K. Malathe and B. Abderrezak, A DFT study of the adsorption energy and electronic interactions of the SO2 molecule on a CoP hydrotreating catalyst, RSC Adv., 2021, 11, 2947–2957 RSC.
- F. Dieter, Z. Dejan and K. Kathrin, Synthesis of Two Structurally Different MgO Films Containing Dioxygen Species: Dioxygen Embedded at Grain Boundaries, and as Components of a Superfilled Rock Salt Structure, Coatings, 2024, 14, 1563 CrossRef.
- A. Maryam, M. A. Sayed and A. Larimi, UV-Vis light responsive Bi2WO6 nanosheet/TiO2 nanobelt heterojunction photo-catalyst for CO2 reduction, Catal. Commun., 2023, 179, 106681 CrossRef.
- A. I. Alexander, V. K. Andrii and L. Stanislav, UV-Vis absorption spectra and electronic structure of merocyanines in the gas phase, Spectrochim. Acta, Part A, 2018, 190, 332–335 CrossRef PubMed.
- G. Di Lonardo and G. Massiarelli, Infrared absorption cross-sections and integrated absorption intensities of HFC-125 and HFC-143a, J. Quant. Spectrosc. Radiat. Transfer, 2000, 66, 129–142 CrossRef.
- L. Kui and Y. Jao, Entropy and enthalpy changes during adsorption and displacement of shale gas, Energy, 2021, 221, 119854 CrossRef.
- A. E. Ofomaja and Y. S. Ho, Effect of Temperatures and pH on Methyl Violet Biosorption by Mansonia Wood Sawdust, Bioresour. Technol., 2008, 99, 5411–5417 CrossRef CAS PubMed.
- Z. Xiao, L. Yuanyuan and L. Yuzhuo, Indirect-to-direct band gap transition and optical properties of Cs2BiAgX6 with mechanical strains: the density functional theory investigation, J. Mater. Res. Technol., 2022, 17, 425–432 CrossRef.
- L. Zhi, V. C. Cristian and H. Juncheng, Experimental and DFT studies of gold nanoparticles supported on MgO(111) nano-sheets and their catalytic activity, Phys. Chem. Chem. Phys., 2011, 13, 2582–2589 RSC.
- S. Hongbing, C. Junxian and Y. Shengping, DFT study on adsorption of small gas molecules on Pt-capped armchair and zigzag single-walled carbon nanotubes, Mater. Today Commun., 2023, 37, 107200 CrossRef.
- C. Fatah, L.-D. Corinne and T. Hao, DFT studies of the bonding mechanism of 8-hydroxyquinoline and derivatives on the (111) aluminum surface, Phys. Chem. Chem. Phys., 2015, 17, 22243–22258 RSC.
- F. Zhe, Z. Yu and H. Wang, Influence of surface charge density on ligand-metal bonding: a DFT study of NH3 and HCOOH on Mg(0001) surface, Appl. Surf. Sci., 2019, 470, 893–898 CrossRef.
- M. Youcef, Y. Salem and B. Nourdine, Charge Density Study, DFT Calculations, Hirshfield Surface Analysis and Molecular Docking of (Z)-3-N-(Ethyl)-2-N′-(3-methoxyphenyl imino)thiazolidine-4-one, Russ. J. Phys. Chem. A, 2023, 97, 1731–1745 CrossRef.
- L. Qingqing, Y. Shaoqian and S. Xiaoxin, Density functional theory study on the electronic, optical and adsorption properties of Ti-, Fe- and Ni-doped graphene, Diamond Relat. Mater., 2022, 128, 109290 CrossRef.
- A. R. Bahram and H. Jafari, DFT study on electronic and optical properties of halogen-adsorbed hexagonal boron nitride, Comput. Condens. Matter, 2019, 21, e00416 CrossRef.
- A. Tamadhur, M. Nagham and K. Warood, Electronic and optical properties of the BN bilayer as gas sensor for CO2, SO2, and NO2 molecules: a DFT study, Results Chem., 2023, 5, 100978 CrossRef.
- R. Razif, T. Salisu and M. Kolo, Structural, mechanical, electronic and optical properties of N-decorated single-walled silicon carbide nanotube photocatalyst for hydrogen evolution via water splitting: a DFT study, Sci. Technol. Adv. Mater., 2023, 24, 2271912 CrossRef PubMed.
- Z. Shengzhe, Y. Yang and L. Ran, Enhanced selective adsorption and photocatalytic of Ag/Bi2O3 heterostructures modified up-conversion nanoparticles, J. Environ. Chem. Eng., 2022, 10, 107107 CrossRef.
- Z. Shengzhe, Y. Yi and L. Ran, Enhanced selective adsorption and photocatalytic of Ag/Bi2O3 heterostructures modified up-conversion nanoparticles, J. Environ. Chem. Eng., 2022, 10, 107107 CrossRef.
- D. Xidong, G. Wenfeng and C. Yugang, Thermodynamics analysis of the adsorption of CH4 and CO2 on montmorillonite, Appl. Clay Sci., 2020, 192, 105631 CrossRef.
- J. Hedi, M. M. Almoneef and A. Jbara, Experimental study of CO2 and N2 adsorption on activated carbon, Mater. Res. Express, 2023, 10, 075601 CrossRef.
- H. Pengbo, W. Shujuan and Y. Zhuo, CO2 adsorption enhancement over Al/C-doped h-BN: a DFT study, Chemosphere, 2022, 292, 133396 CrossRef PubMed.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *abc,
Mayeen Uddin Khandaker*ade and
Mustafa Mahmoudfg
*abc,
Mayeen Uddin Khandaker*ade and
Mustafa Mahmoudfg