
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceComprehensive assessment of 3-benzyloxyflavones as β-glucosidase inhibitors: in vitro, in vivo, kinetic, SAR and computational studies†
Nafeesa Naeem and
Ehsan Ullah Mughal*
and
Ehsan Ullah Mughal*
Department of Chemistry, University of Gujrat, Gujrat-50700, Pakistan. E-mail: ehsan.ullah@uog.edu.pk
First published on 4th April 2025
Abstract
In this study, a series of 3-benzyloxyflavone derivatives (1–10) was designed and, for the first time, evaluated for both in vitro and in vivo inhibitory activity against the β-glucosidase enzyme. The enzyme inhibitory potential of these derivatives was further assessed in an antihyperglycemic context using in vivo mechanism-based assays on p-nitrophenyl-β-D-glucopyranoside (PGLT) induced diabetic models. Additionally, structure–activity relationship (SAR) was employed to identify structural features crucial for activity. Molecular docking analyses revealed that both the potent compounds and co-crystallized ligands shared similar binding orientations within the active sites of β-glucosidase (PDB IDs: 3AJ7; 66K1). Molecular dynamics (MD) simulations validated the stability of the inhibitor–enzyme complexes under physiological conditions, while density functional theory (DFT) calculations helped elucidate electronic properties critical for activity. Drug-likeness analysis was also conducted to assess the pharmacokinetic potential of the derivatives. The results highlighted several derivatives with significant inhibitory activity, desirable pharmacokinetic profiles, and promising drug-like properties, making them potential candidates for therapeutic development. The target derivatives (1–10) demonstrated strong potential as lead compounds for developing new anti-diabetic agents with effective anti-hyperglycemic properties.
1. Introduction
β-Glucosidase is an essential enzyme involved in the hydrolysis of β-glycosidic bonds in β-D-glucosides, oligosaccharides, and other complex carbohydrates, leading to the release of glucose and other monosaccharides.1 This enzyme plays a pivotal role in biological processes like cellulose degradation, where it aids in breaking down cellobiose into glucose, which is essential for energy production in microorganisms.2 Additionally, β-glucosidase is crucial in the metabolism of glycosides in plants and mammals, contributing to defense mechanisms and lysosomal degradation, respectively.3 The deficiency of β-glucosidase in humans is linked to lysosomal storage disorders such as Gaucher's disease, highlighting its clinical importance.4In microorganisms, especially fungi, and bacteria, β-glucosidase is an important component of the cellulolytic enzyme system, facilitating the complete saccharification of cellulose by converting cellobiose into glucose.5,6 In plants, the enzyme contributes to defense mechanisms, as it helps in the release of bioactive compounds from glycosides in response to external stress or pathogen attack.7–9 In mammals, β-glucosidase plays a role in the lysosomal degradation of glycosphingolipids, with its deficiency leading to disorders such as Gaucher's disease.4,10
In industrial biotechnology, β-glucosidase has attained significant attention due to its role in biofuel production, particularly in the enzymatic breakdown of lignocellulosic biomass.11 Its ability to enhance the saccharification process by hydrolyzing cellobiose into glucose has made it a key target in optimizing bioethanol production.6,12 Additionally, β-glucosidase plays a critical role in the food and beverage industry, where it helps release flavor-enhancing compounds from glycosides in fruits and wines.13,14
Flavonoids, as naturally occurring polyphenolic structures, have drawn extensive interest in scientific research due to their wide-ranging biological activities, including antioxidant, anti-inflammatory, anticancer, and enzyme-inhibitory properties.15–22 3-O-Benzylflavonol, commonly known as 3-benzyloxyflavone (Fig. 1), represents a prominent subclass within the broader category of flavonoid compounds, characterized by the substitution of a benzyloxy group at the third position of the flavonol core. These are one of the pharmacologically active synthetic derivatives of flavonols.23,24 The structural modification in 3-benzyloxyflavone not only enhances its hydrophobic nature but also confers unique molecular interactions that have been linked to increased biological efficacy.25–27 This modification impacts its pharmacokinetic profile, remarkably improving cellular permeability and enzyme-binding affinity.28,29 Recent research highlights the compound's enhanced interaction with hydrophobic enzyme active sites, particularly glycosidases like β-glucosidase, making it a promising candidate for enzyme inhibition.30,31 The compound's structural flexibility and synthetic accessibility further support its potential as a lead molecule for designing analogs with optimized selectivity and biological efficacy.32,33
 | ||
| Fig. 1 Representative structure of 3-O-benzylflavonol or 3-benzyloxyflavone. | ||
Research into β-glucosidase inhibition has gained importance, particularly in the context of discovering new therapeutic agents for managing diseases such as type-2 diabetes and Gaucher's disease.34,35 Traditional inhibitors like acarbose have been effective in controlling glucose metabolism by inhibiting glucosidases, but the search for more potent and selective inhibitors continues.36–38 Recent studies have shown that flavonoids, including flavones and their derivatives, exhibit significant inhibitory activity against β-glucosidase.39,40 These compounds act by interacting with the active site of the enzyme, preventing the hydrolysis of glucosidic substrates.41,42
Among flavonoids, 3-benzyloxyflavones have emerged as promising candidates due to their unique structure–activity relationship (SAR) that enhances their binding affinity to β-glucosidase. Their ability to inhibit the enzyme has been attributed to the presence of the benzyloxy group, which enhances lipophilicity and facilitates interactions with hydrophobic regions of the enzyme's active site.43,44 These flavones are also being studied for their ability to modulate other biological targets, expanding their therapeutic potential beyond enzyme inhibition.45,46
Additionally, to the best of our knowledge, for the first time, this motif has been evaluated against β-glucosidase. Recent advancements in computational docking studies, alongside in vitro, and in vivo enzyme assays, have provided valuable insights into the molecular interactions between 3-benzyloxyflavones and β-glucosidase. These studies have not only confirmed their inhibitory potential but also provided a framework for optimizing their structure for enhanced activity and selectivity. This growing body of evidence suggests that 3-benzyloxyflavones could serve as promising leads for developing novel glucosidase inhibitors with applications in both therapeutic and industrial applications.
2. Experimental section
2.1. General procedure for the synthesis of 3-benzyloxyflavones (1–10)
The 3-benzyloxyflavones (1–10) were synthesized according to the procedure outlined in our previously published literature.232.2. Enzyme inhibitory assay
 | (1) |
2.3. Kinetic assay
A kinetic study was carried out to evaluate the inhibition mechanism of the most potent compounds 8 and 10 against β-glucosidase using earlier methodology.48,51 For compound 8, 20 μL of enzyme solution (1 U mL−1) was incubated at 37 °C with varying concentrations of the inhibitor (0, 55, 75, and 95 μM). The reaction was initiated by adding p-nitrophenyl-β-D-glucopyranoside (PGLT) (1–4 mM) as the substrate, and absorbance changes were monitored at 405 nm for 20 minutes. Lineweaver–Burk plots were used to identify the mode of inhibition, and the Michaelis–Menten constant (Km) and inhibition constant (Ki) were calculated. Additionally, compound 10 was examined under the same conditions at inhibitor concentrations of 0, 30, 50, and 75 μM, with absorbance recorded over 20 minutes using a Gen5 PowerWave XS2 spectrophotometer (BioTek, USA).2.4. Computational studies
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 000 steps under the NVT ensemble and 100000 steps under the NPT ensemble at 310 K.56 The Berendsen thermostat and Parrinello–Rahman barostat were employed to maintain a constant temperature (310 K) and pressure (1 atm). Relaxation of the system was achieved with time constants (τP) of 2.0 ps for pressure and (τT) of 0.1 ps for temperature. The LINCS algorithm was used to constrain hydrogen bond lengths to their equilibrium positions,57 while the Verlet algorithm calculated non-bonded interactions.58 Long-range electrostatic interactions were computed using the Particle Mesh Ewald (PME) method.59 Periodic boundary conditions were applied in all three dimensions (x, y, z), and a production MD run was executed. Trajectories were saved every 10 ps and analyzed using the R BIO3D package and GROMACS commands60 The CHARMM36 force field and GROMACS simulation software were utilized to perform the MD simulation.61P, and the counts of hydrogen bond donors and acceptors, were calculated using cheminformatics software i.e., ChemAxon. The compounds were then screened against Lipinski's rule of five to ensure they met the thresholds for molecular weight (<500 Da), logP (<5), hydrogen bond donors (≤5), and acceptors (≤10). Furthermore, compounds were assessed for synthetic accessibility and a bioavailability score to predict their oral bioavailability.
000 steps under the NVT ensemble and 100000 steps under the NPT ensemble at 310 K.56 The Berendsen thermostat and Parrinello–Rahman barostat were employed to maintain a constant temperature (310 K) and pressure (1 atm). Relaxation of the system was achieved with time constants (τP) of 2.0 ps for pressure and (τT) of 0.1 ps for temperature. The LINCS algorithm was used to constrain hydrogen bond lengths to their equilibrium positions,57 while the Verlet algorithm calculated non-bonded interactions.58 Long-range electrostatic interactions were computed using the Particle Mesh Ewald (PME) method.59 Periodic boundary conditions were applied in all three dimensions (x, y, z), and a production MD run was executed. Trajectories were saved every 10 ps and analyzed using the R BIO3D package and GROMACS commands60 The CHARMM36 force field and GROMACS simulation software were utilized to perform the MD simulation.61P, and the counts of hydrogen bond donors and acceptors, were calculated using cheminformatics software i.e., ChemAxon. The compounds were then screened against Lipinski's rule of five to ensure they met the thresholds for molecular weight (<500 Da), logP (<5), hydrogen bond donors (≤5), and acceptors (≤10). Furthermore, compounds were assessed for synthetic accessibility and a bioavailability score to predict their oral bioavailability.3. Results and discussion
3.1. Chemistry
The synthesis of 3-O-benzylated flavonols/3-benzyloxyflavones (1–10) (Scheme 1), was achieved using the established Algar–Flynn–Oyamada (AFO) reaction. The spectroscopic data for all synthesized 3-benzyloxyflavones (1–10) can be found in our previous publication.23 | ||
| Scheme 1 Synthesis of 3-benzyloxyflavone derivatives (1–10). | ||
3.2. β-Glucosidase inhibitory activity
The 3-benzyloxyflavone derivatives (1–10) were evaluated for their in vitro inhibitory potential against the β-glucosidase enzyme, with IC50 values measured to assess their activity (Table 1). Among the derivatives, compound 8 demonstrated the most potent inhibitory effect with an IC50 = 0.17 μM, indicating a strong affinity for β-glucosidase. This was followed by compound 3, which also showed significant activity with an IC50 = 0.22 μM. In contrast, compound 1 displayed the lowest inhibitory activity among the series, with an IC50 = 1.02 μM, though still showing effectiveness.





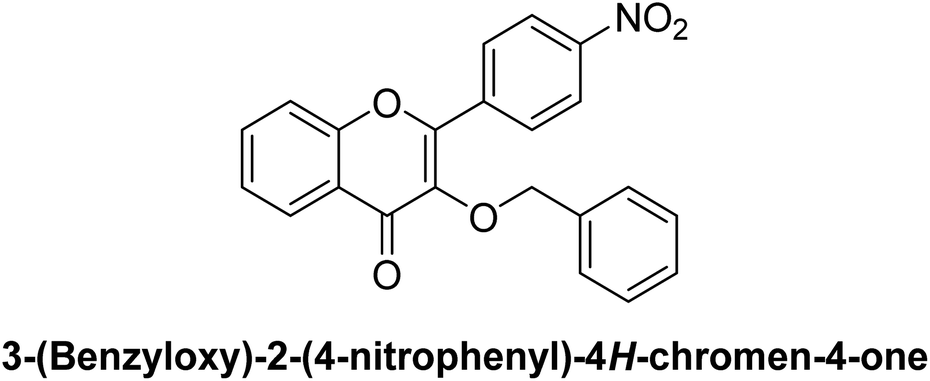

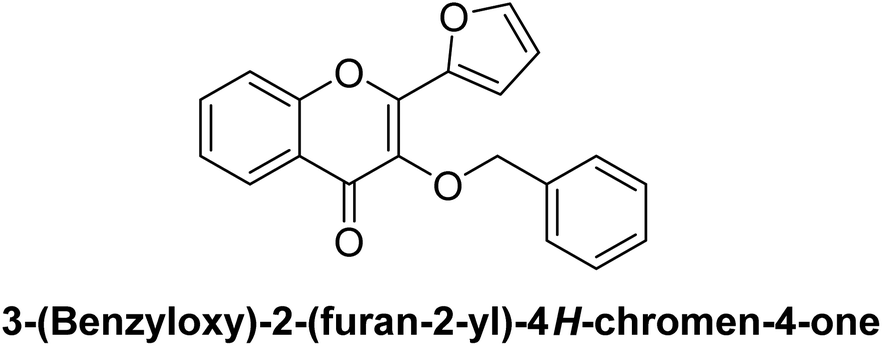

Furthermore, other derivatives exhibited moderate inhibitory activity, with IC50 values ranging from 0.25 μM (compound 10) to 0.51 μM (compound 2). All target compounds showed greater potency compared to the standard reference, acarbose (IC50 = 34.09 μM). These results indicate that the benzyloxyflavone framework is effective in generating potent β-glucosidase inhibitors, with certain structural features likely enhancing binding affinity and enzyme interaction. The SAR for these derivatives suggests that specific substitutions within the benzyloxyflavone core may contribute to variations in potency. These findings indicate that derivatives like 3, 5, 6, 7, 8, and 10 have potential as lead candidates for developing new β-glucosidase inhibitors for antihyperglycemic therapy.
3.3. In vivo study
| Blood glucose level in mg per dL per day | ||||||
|---|---|---|---|---|---|---|
| Groups | 1st | 7th | 14th | 21st | 28th | |
| a All values are presented as mean ± SEM, with n = 8. Statistical significance was evaluated using one-way ANOVA followed by Dunnett's post hoc multiple comparison test. Significance levels were denoted as follows: !!!P < 0.001 for diabetic control versus normal control; *P < 0.05, **P < 0.01, and ***P < 0.001 for comparisons between diabetic control and both test samples and p-nitrophenyl-β-D-glucopyranoside-treated groups. | ||||||
| Normal control | 107.81 ± 5.12 | 110.32 ± 4.81 | 106.70 ± 4.91 | 108.51 ± 4.59 | 103.21 ± 4.41 | |
| Diabetic control | 397.61 ± 5.04!!! | 412.37 ± 4.88!!! | 409.57 ± 4.98!!! | 401.45 ± 5.03!!! | 396.21 ± 4.79!!! | |
| 3 | 5 mg | 398.51 ± 4.91 | 332.97 ± 4.89 | 218.68 ± 4.90* | 172.59 ± 4.80** | 142.84 ± 4.90** |
| 7.5 mg | 400.63 ± 5.12 | 329.73 ± 3.96 | 278.39 ± 4.98* | 189.47 ± 5.00** | 149.62 ± 3.89** | |
| 5 | 5 mg | 398.40 ± 4.68 | 334.91 ± 4.81** | 209.93 ± 4.77** | 175.86 ± 4.79** | 135.81 ± 4.51*** |
| 7.5 mg | 440.70 ± 4.97 | 340.47 ± 4.25** | 235.85 ± 4.97** | 190.56 ± 5.32** | 139.86 ± 4.64*** | |
| 6 | 5 mg | 393.37 ± 4.71 | 341.56 ± 4.71* | 235.71 ± 4.68** | 185.67 ± 4.91** | 153.19 ± 4.73** |
| 7.5 mg | 400.52 ± 4.89 | 335.72 ± 4.48* | 240.93 ± 4.90** | 182.58 ± 4.63** | 148.25 ± 3.98** | |
| 7 | 5 mg | 401.06 ± 5.02 | 339.11 ± 4.90* | 243.12 ± 4.79* | 188.70 ± 4.65* | 165.39 ± 4.88* |
| 7.5 mg | 407.17 ± 4.99 | 328.42 ± 4.54* | 287.53 ± 5.38* | 198.87 ± 4.80* | 161.16 ± 4.41* | |
| 8 | 5 mg | 401.69 ± 5.31 | 306.17 ± 4.58** | 194.85 ± 4.97*** | 148.70 ± 4.83*** | 126.13 ± 4.91*** |
| 7.5 mg | 395.48 ± 4.24 | 310.19 ± 5.10** | 192.77 ± 4.46*** | 158.92 ± 4.14*** | 139.28 ± 5.61*** | |
| 10 | 5 mg | 407.17 ± 4.12 | 331.39 ± 4.73* | 208.09 ± 4.88* | 169.60 ± 4.63** | 137.70 ± 4.89** |
| 7.5 mg | 467.53 ± 5.48 | 325.44 ± 4.89* | 210.16 ± 4.56* | 164.57 ± 4.42** | 140.81 ± 4.90** | |
| p-Nitrophenyl-β-D-glucopyranoside (PGLT) | 10 mg | 402.22 ± 4.97 | 229.56 ± 4.80*** | 154.06 ± 4.59*** | 119.70 ± 4.87*** | 102.27 ± 4.31*** |
Among the benzyloxyflavone derivatives, compound 8 exhibited notable antidiabetic effects. At 5 mg kg−1, glucose levels decreased from 401.69 ± 5.31 mg dL−1 on day 1 to 126.13 ± 4.91 mg dL−1 on day 28, representing the most substantial reduction across the derivatives tested. At 7.5 mg kg−1, compound 8 showed a similar trend, reducing glucose levels from 395.48 ± 4.24 mg dL−1 on day 1 to 139.28 ± 5.61 mg dL−1 by day 28. Other derivatives, such as compounds 3, 6, 7, and 5, also demonstrated dose-dependent decreases in blood glucose, although to a lesser degree than compound 8.
Compound 3, administered at 5 mg kg−1, decreased glucose levels from 398.51 ± 4.91 mg dL−1 initially to 142.84 ± 4.90 mg dL−1 by day 28. At 7.5 mg kg−1, it further reduced levels to 149.62 ± 3.89 mg dL−1. Compound 6 also showed progressive reductions, with glucose levels at 153.19 ± 4.73 mg dL−1 at 5 mg kg−1 and 148.25 ± 3.98 mg dL−1 at 7.5 mg kg−1 by the end of the study period. Compounds 7, 5, and 10 demonstrated a similar gradual decline, with compound 5 at 5 mg kg−1 reducing glucose from 398.40 ± 4.68 mg dL−1 to 135.81 ± 4.51 mg dL−1 by day 28, marking it as one of the more effective derivatives in the lower dose range.
For comparison, the positive control, p-nitrophenyl-β-D-glucopyranoside (PGLT), at 10 mg kg−1, significantly lowered glucose levels from 402.22 ± 4.97 mg dL−1 on day 1 to near-normal levels of 102.27 ± 4.31 mg dL−1 on day 28. This outcome underscores the potency of PGLT as a standard antidiabetic agent.
Overall, these results indicate that benzyloxyflavone derivatives possess significant antihyperglycemic potential, with compound 8 emerging as the most promising candidate due to its substantial impact on glucose reduction. The lower dose (5 mg kg−1) demonstrated effective glucose control across several derivatives, suggesting that further exploration of benzyloxyflavone structures may yield potent candidates for antidiabetic therapy.
| Groups/dose mg kg−1 | 1st day | 7th day | 14th day | 21st day | 28th day | |
|---|---|---|---|---|---|---|
| a All values are presented as means ± SEM, with n = 8. Statistical analysis was conducted using one-way ANOVA followed by Dunnett's post hoc test. Significant differences are indicated as follows: !!!P < 0.001 for comparisons between diabetic and normal control groups, and *P < 0.05, **P < 0.01, ***P < 0.001 for comparisons between diabetic control and test groups or PGLT groups. | ||||||
| Normal control | 20.34 ± 2.18 | 21.71 ± 2.01 | 21.41 ± 1.89 | 20.61 ± 2.18 | 20.98 ± 1.94 | |
| Diabetic control | 21.31 ± 1.52 | 20.56 ± 1.90 | 18.41 ± 1.48 | 17.30 ± 1.70 | 15.17 ± 1.39 | |
| 3 | 5 mg | 22.07 ± 1.81 | 22.11 ± 1.67 | 20.50 ± 1.69 | 20.11 ± 1.81 | 19.41 ± 1.61 |
| 7.5 mg | 22.51 ± 1.73 | 21.98 ± 1.74 | 20.87 ± 1.66 | 20.24 ± 1.78 | 19.75 ± 1.68 | |
| 5 | 5 mg | 20.70 ± 1.73 | 20.66 ± 1.90 | 19.48 ± 1.53 | 19.34 ± 1.71 | 18.96 ± 1.81 |
| 7.5 mg | 21.04 ± 1.79 | 20.83 ± 1.69 | 19.92 ± 1.60 | 19.58 ± 1.74 | 19.03 ± 1.77 | |
| 6 | 5 mg | 19.97 ± 1.81 | 18.91 ± 1.58 | 18.65 ± 1.79 | 18.03 ± 1.48 | 18.16 ± 1.77 |
| 7.5 mg | 20.28 ± 1.80 | 19.32 ± 1.54 | 18.79 ± 1.64 | 18.27 ± 1.56 | 18.34 ± 1.65 | |
| 7 | 5 mg | 21.41 ± 1.73 | 21.16 ± 1.81 | 20.64 ± 1.87 | 19.80 ± 1.70 | 17.91 ± 1.80 |
| 7.5 mg | 21.92 ± 1.76 | 21.37 ± 1.68 | 20.82 ± 1.75 | 19.93 ± 1.77 | 18.21 ± 1.75 | |
| 8 | 5 mg | 22.61 ± 1.98 | 21.12 ± 1.91 | 20.77 ± 1.88 | 20.69 ± 1.69 | 19.66 ± 1.72 |
| 7.5 mg | 22.88 ± 1.91 | 21.36 ± 1.76 | 21.03 ± 1.71 | 20.81 ± 1.74 | 20.13 ± 1.63 | |
| 10 | 5 mg | 19.84 ± 1.70 | 19.67 ± 1.66 | 19.34 ± 1.48 | 18.90 ± 1.60 | 18.75 ± 1.58 |
| 7.5 mg | 20.11 ± 1.72 | 19.85 ± 1.69 | 19.56 ± 1.51 | 19.12 ± 1.54 | 18.89 ± 1.60 | |
| PGLT | 10 mg | 20.16 ± 1.71 | 19.88 ± 1.81 | 20.01 ± 1.61 | 19.70 ± 1.69 | 19.66 ± 1.51 |
Treatment with the test compounds at both 5 mg kg−1 and 7.5 mg kg−1 doses led to a moderation in weight loss compared to the diabetic control. For example, compound 3 at 7.5 mg kg−1 prevented significant weight loss, maintaining a relatively stable weight profile, similar to the control. Other compounds, including 5, 6, and 8 at both doses, also demonstrated protective effects on body weight, although to varying extents. The PGLT (10 mg) treated group exhibited weight maintenance comparable to that of the normal control, validating its protective efficacy. However, treatment with the selected compounds at doses of 5 and 7.5 mg per kg b.w. effectively reversed the PGLT-induced weight loss. Remarkably, these findings suggest that several benzyloxyflavone derivatives at appropriate dosages can positively influence body weight maintenance in diabetic models, potentially reflecting broader antidiabetic properties.
| Groups/dose mg kg−1 | TG (mg dL−1) | Total CH (mg dL−1) | LDL (mg dL−1) | HDL (mg dL−1) | |
|---|---|---|---|---|---|
| a All values are presented as mean ± SEM, with n = 8 per group. Statistical significance was determined using one-way ANOVA followed by Dunnett's post hoc test. Significance levels are indicated as follows: !!!P < 0.001 for diabetic control versus normal control, and *P < 0.05, **P < 0.01, ***P < 0.001 for diabetic control compared with test samples and the PGLT-treated group. | |||||
| Normal control | 91.72 ± 3.72 | 102.44 ± 4.87 | 85.61 ± 2.02 | 57.91 ± 2.03 | |
| Diabetic control | 176.80 ± 3.13!!! | 197.81 ± 5.03!!! | 189.70 ± 3.81!!! | 26.37 ± 1.80!!! | |
| 3 | 5 mg | 129.55 ± 3.76** | 149.11 ± 4.80* | 128.77 ± 3.41** | 42.81 ± 1.90** |
| 7.5 mg | 120.30 ± 3.59** | 138.30 ± 4.72* | 117.44 ± 3.29** | 44.10 ± 1.84** | |
| 5 | 5 mg | 136.30 ± 3.61** | 145.33 ± 4.79** | 124.79 ± 3.91** | 45.34 ± 2.14* |
| 7.5 mg | 130.00 ± 3.58** | 140.20 ± 4.76** | 118.44 ± 3.85** | 46.10 ± 2.11* | |
| 6 | 5 mg | 147.12 ± 4.03* | 152.30 ± 4.80* | 127.64 ± 3.39** | 43.66 ± 1.98* |
| 7.5 mg | 142.40 ± 4.01* | 148.50 ± 4.77* | 122.21 ± 3.31** | 45.00 ± 1.94* | |
| 7 | 5 mg | 145.34 ± 4.40* | 160.04 ± 4.91* | 140.39 ± 4.14** | 39.90 ± 1.92** |
| 7.5 mg | 138.20 ± 4.35* | 153.80 ± 4.85* | 135.02 ± 4.09** | 41.02 ± 1.89** | |
| 8 | 5 mg | 113.09 ± 4.41*** | 125.17 ± 4.92*** | 102.49 ± 3.19*** | 51.11 ± 2.01*** |
| 7.5 mg | 108.01 ± 4.28*** | 119.80 ± 4.89*** | 97.90 ± 3.10*** | 52.70 ± 2.08*** | |
| 10 | 5 mg | 130.09 ± 4.29** | 135.56 ± 4.70** | 113.60 ± 3.40*** | 46.89 ± 1.97* |
| 7.5 mg | 125.20 ± 4.24** | 130.21 ± 4.66** | 110.08 ± 3.31*** | 48.02 ± 1.99* | |
| PGLT | 10 mg | 96.36 ± 4.51*** | 104.73 ± 4.83*** | 87.58 ± 2.96*** | 55.72 ± 2.01*** |
Triglycerides (TG): compounds 8 and 10 at both 5 mg kg−1 and 7.5 mg kg−1 showed substantial reductions in TG levels, approaching those of the standard PGLT-treated group. Remarkably, compound 5 at 7.5 mg kg−1 brought TG levels close to baseline values, demonstrating a remarkable lipid-lowering effect.
Total cholesterol (CH): treatment with compounds 8 and 10, especially at the higher dose, resulted in significant reductions in total cholesterol. Compound 8 at 7.5 mg kg−1 showed a reduction close to that of the PGLT standard group, highlighting its potency in cholesterol management.
Low-density lipoprotein (LDL): most compounds effectively lowered LDL levels, with compounds 8 and 10 at 7.5 mg kg−1 producing the most pronounced effects. These results emphasize their ability to reduce atherogenic lipids, thus contributing to cardiovascular protection.
High-density lipoprotein (HDL): increased HDL levels were observed in animals treated with the derivatives, with compound 8 at 7.5 mg kg−1 showing the highest increase. Elevated HDL levels are beneficial, as they play a crucial role in reverse cholesterol transport.
Henceforth, these findings suggest that benzyloxyflavone derivatives, particularly compounds 8 and 10, hold promising lead candidates for antihyperlipidemic treatment. Their lipid-regulating effects are comparable to standard treatments, and further investigations into their mechanisms could elucidate their potential therapeutic roles.
| Groups/dose mg kg−1 | (SGOT) IU | (SGPT) IU | (ALP) IU | |
|---|---|---|---|---|
| a Data are presented as mean ± SEM, with n = 8 per group. Statistical analysis was performed using one-way ANOVA followed by Dunnett's post hoc test. Significance levels are indicated as !!!P < 0.001 for comparisons between the diabetic and normal control groups, and *P < 0.05, **P < 0.01, ***P < 0.001 for comparisons between the diabetic control and test groups, as well as the PGLT-treated group. | ||||
| Normal control | 21.15 ± 1.98 | 19.34 ± 1.67 | 169.83 ± 4.10 | |
| Diabetic control | 47.53 ± 2.12!!! | 60.21 ± 2.44!!! | 288.67 ± 4.56!!! | |
| 3 | 5 mg | 26.96 ± 2.34** | 38.21 ± 2.61** | 198.77 ± 4.11* |
| 7.5 mg | 24.55 ± 2.02** | 33.78 ± 2.43* | 192.11 ± 4.05** | |
| 5 | 5 mg | 24.61 ± 2.06*** | 31.70 ± 2.11*** | 184.32 ± 4.09*** |
| 7.5 mg | 22.40 ± 2.10*** | 29.32 ± 2.21*** | 181.73 ± 4.08*** | |
| 6 | 5 mg | 31.41 ± 2.12** | 44.56 ± 2.98* | 210.70 ± 4.82** |
| 7.5 mg | 28.69 ± 2.08** | 40.91 ± 2.96* | 205.56 ± 4.72** | |
| 7 | 5 mg | 33.09 ± 2.31* | 45.98 ± 3.04* | 217.89 ± 4.90** |
| 7.5 mg | 29.98 ± 2.15* | 42.83 ± 3.00* | 212.49 ± 4.80** | |
| 8 | 5 mg | 32.44 ± 2.65** | 38.69 ± 2.61** | 197.65 ± 4.71* |
| 7.5 mg | 28.62 ± 2.55** | 33.40 ± 2.50** | 193.43 ± 4.69* | |
| 10 | 5 mg | 29.75 ± 2.40** | 36.29 ± 2.40*** | 196.70 ± 4.11** |
| 7.5 mg | 26.51 ± 2.31** | 31.12 ± 2.35*** | 192.53 ± 4.03** | |
| PGLT | 10 mg | 21.39 ± 1.79*** | 21.17 ± 1.88*** | 171.04 ± 3.91*** |
 | ||
| Fig. 2 Effects of compounds on serum profiles: (A) serum glutamic-oxaloacetic transaminase (SGOT), (B) serum glutamic pyruvic transaminase (SGPT), and (C) alkaline phosphatase (ALP). All values are presented as means ± SEM (n = 8). Statistical significance was assessed using one-way ANOVA followed by Dunnett's post hoc test. A comparison between the diabetic control group and the normal control group revealed significant differences (!!!P < 0.001). Comparisons of the diabetic control group with test samples and PGLT-treated groups showed *P < 0.05, **P < 0.01, and ***P < 0.001. | ||
The liver enzyme profile of diabetic rats treated with benzyloxyflavone derivatives (compounds 3, 5, 6, 7, 8, and 10) showed a significant improvement in serum levels of SGOT, SGPT, and ALP compared to the untreated diabetic control group. Diabetic control rats displayed notably elevated levels of SGOT (47.53 ± 2.12 IU), SGPT (60.21 ± 2.44 IU), and ALP (288.67 ± 4.56 IU), indicating marked liver stress and potential hepatic dysfunction induced by the diabetic condition.
Among the tested compounds, compound 5 (7.5 mg) produced the most pronounced effects, reducing SGOT to 22.40 ± 2.10 IU, SGPT to 29.32 ± 2.21 IU, and ALP to 181.73 ± 4.08 IU, comparable to the levels observed in the PGLT-treated group, which is a standard for comparison. This suggests that compound 5 may have a protective effect on liver function, effectively lowering liver enzyme levels to near-normal values. Additionally, compounds 8 and 10 at 7.5 mg doses also exhibited substantial improvements, reducing SGOT, SGPT, and ALP levels significantly (P < 0.05 to P < 0.001) when compared to the diabetic control, demonstrating their potential for liver protection in diabetic states.
3.4. Kinetic study
Kinetic analysis of compound 8, the most potent β-glucosidase inhibitor among the synthesized derivatives, revealed its competitive inhibition mode. This was determined through Lineweaver–Burk plotting, showing no change in Vmax and an increase in Km, with a Ki value of 90 μM (Fig. S1 in ESI file†). Additionally, the kinetic study of compound 10 also indicated competitive inhibition, as evidenced by the Lineweaver–Burk plot showing a constant Vmax and an increased Km. The Ki value for compound 10 was found to be 75 μM, based on the secondary replot derived from the Lineweaver–Burk plot (Fig. S2 in ESI file†).3.5. Structure–activity relationship (SAR) based on the IC50 values against β-glucosidase
The SAR analysis of the synthesized 3-(benzyloxy)-2-aryl-4H-chromen-4-one derivatives reveals the influence of various substituents on β-glucosidase inhibitory activity (Fig. 3). Each compound's IC50 value indicates the effect of different aryl substitutions at the 2-position on the chromenone ring, highlighting both electron-donating and electron-withdrawing effects. | ||
| Fig. 3 SAR for β-glucosidase enzyme activity of 3-benzyloxyflavones (1–10). | ||
Compounds with electron-donating groups exhibit enhanced inhibitory activity. For example, compound 8, containing a 4-methoxyphenyl group, shows the lowest IC50 value (0.17 μM), suggesting that electron-donating substituents contribute to increased activity by stabilizing interactions within the enzyme's active site. Similarly, compound 3, with a p-tolyl group (an electron-donating methyl group on the phenyl ring), also demonstrates potent inhibition (IC50 = 0.22 μM), reinforcing the role of electron-donating effects in enzyme binding affinity.
On the other hand, the presence of electron-withdrawing groups generally results in moderate β-glucosidase inhibition. For instance, compound 7, featuring a 4-nitrophenyl group, shows an IC50 of 0.37 μM, while compound 6, with a 3-nitrophenyl group, yields an IC50 of 0.30 μM. The nitro group, known for its electron-withdrawing character, likely decreases the electron density on the aryl ring, leading to slightly lower binding efficacy in the enzyme's active site.
Furthermore, compound 5, substituted with a 4-chlorophenyl group (electron-withdrawing chlorine), demonstrates moderate inhibition with an IC50 of 0.36 μM. Interestingly, heterocyclic substitutions like those in compounds 4 (thiophen-2-yl) and 9 (furan-2-yl) result in IC50 values of 0.42 μM and 0.39 μM, respectively. These heterocycles likely provide different steric and electronic properties, subtly influencing their inhibitory profiles.
Additionally, the diphenylamino substitution in compound 2 (IC50 = 0.51 μM) and the dimethylamino substitution in compound 10 (IC50 = 0.25 μM) highlight that more extensive π-conjugation and electron-donating nitrogen substituents can variably enhance binding, with smaller amino groups demonstrating stronger activity. Remarkably, all synthesized compounds show substantially higher activity compared to the standard, acarbose (IC50 = 34.09 μM), indicating a promising scaffold for β-glucosidase inhibition.
Henceforth, electron-donating substituents such as methoxy and methyl enhance β-glucosidase inhibition in the chromenone derivatives, while electron-withdrawing groups like nitro and chloro result in moderate activity, with slight differences arising from the distinct electronic and steric demands of each substituent. This SAR study provides insight into the optimized design of chromenone-based β-glucosidase inhibitors.
3.6. Computational studies
| Sr. no. | PDB ID | Resolution | Year | Ligand | Organism | Reference |
|---|---|---|---|---|---|---|
| 1 | 3AJ7 | 1.30 Å | 2010 | Calcium ion | Saccharomyces cerevisiae | 64 |
| 2 | 6KK1 | 2.80 Å | 2019 | N-{4-[(R)-(3,3-dimethylcyclobutyl)({6-[4-(trifluoromethyl)-1H-imidazol-1-yl]pyridin-3-yl}amino)methyl]benzene-1-carbonyl}-beta-alanine, C26H28F3N5O3, MYZIDYJMNWEJMC-QHCPKHFHSA-N | Homo sapiens | 65 |
Compounds 8 and 10, identified as lead candidates in both in vitro and in vivo studies, display robust interactions with their respective target proteins, as shown in Fig. S3 and S4 in the ESI file,† outperforming the reference ligand, acarbose. The protein–ligand interaction diagrams illustrate that both compounds form hydrophobic contacts and stable hydrogen bonds with active site residues. The 2D interaction diagrams emphasize key hydrogen bond donors and acceptors that enhance binding efficiency, while the hydrophobic maps highlight the non-polar regions that contribute to ligand stability. Compound 8 demonstrates significant stability with 3AJ7 in Fig. S3 in the ESI file,† whereas compound 10 exhibits superior hydrophobic compatibility and binding energy with 6KK1 in Fig. S4 in the ESI file.† These results indicate that compounds 8 and 10 are potent inhibitors of both targets, with their strong hydrophobic interactions, high binding affinities, and substantial hydrogen bonding reinforcing their potential as effective antidiabetic agents.
 | ||
| Fig. 4 MD trajectory analysis highlighting various stability parameters. (a) RMSD of the protein backbone atoms over the simulation. (b) RMSF plot depicting the flexibility of protein residues in the presence of the ligand. (c) Rg analysis reflects the structural compactness of the protein. (d) Hydrogen bonding profile showing the number of hydrogen bonds formed between the protein and the ligand during the simulation. | ||
To further investigate protein flexibility, the Root Mean Square Fluctuation (RMSF) was calculated and represented in a plot (Fig. 4b). High RMSF values correspond to flexible residues, while lower values indicate rigid regions. Significant fluctuations were observed in residues 75–95, 245–255, and 325–345, which correspond to loop regions. The remaining residues exhibited minimal fluctuations, suggesting that the protein retained its rigidity and did not undergo notable flexibility during the simulation.
The compactness of the protein structure was examined by calculating the radius of gyration (Rg) over the simulation period (Fig. 4c). Elevated Rg values indicate protein unfolding events. The Rg plot revealed that the protein maintained structural stability throughout the simulation, with values starting around ∼2.05 nm and remaining consistent between ∼2.06 and ∼2.07 nm.
Additionally, hydrogen bonding between the protein and ligand was analyzed to evaluate the stability of the complex (Fig. 4d). A greater number of hydrogen bonds generally correlates with enhanced stability. At the beginning of the simulation, up to four hydrogen bonds were observed, which gradually decreased to three towards the end. The average number of hydrogen bonds throughout the simulation was 2.5, indicating strong binding and a stable protein–ligand complex.
3.6.7.1. Dipole moment. It is a measurement of molecule polarity that affects solubility and reactivity.72 The dipole moment of compound 1 (3.288292), compound 2 (4.502051), compound 8 (2.893935), and compound 9 (3.785607) was substantially lower than that of acarbose (9.201717 debye). But out of all the derivatives, compound 10 had the largest dipole moment (6.387456 debye), suggesting a polarity closest to acarbose. This implies compound 10 might resemble the reference chemical regarding solubility and intermolecular interactions.
3.6.7.2. Frontier molecular orbitals (HOMO–LUMO) analysis. In quantum chemistry, the Frontier Molecular Orbitals (FMOs), which are the Highest Occupied Molecular Orbital (HOMO) and Lowest Unoccupied Molecular Orbital (LUMO), along with their energy gap, are highly helpful characteristics. One way to interpret HOMO is as the outermost orbital with electrons, signifying the ability to give an electron, and LUMO as the innermost orbital with free spaces, signifying the ability to accept an electron. Both intramolecular charge transfer (ICT) within the molecule and reduced chemical reactivity, which suggests stronger kinetic stability, are indicated by a lower HOMO–LUMO energy gap value.73
Acarbose has an energy gap of 0.23029 eV since its HOMO energy is −0.21573 eV and its LUMO energy is −0.01456 eV. Its strong stability and mild reactivity are reflected in this comparatively large energy gap, which is in line with its biological function as an enzyme inhibitor. However, there are significant differences in the FMO energies of the compounds.
The HOMO values of molecules compounds 1, 2, 8, 9, and 10 ranged from −0.23222 eV (compound 1) to −0.19685 eV (compound 8), suggesting that their electron-donating potential differed little from that of acarbose. All of the derivatives had negative LUMO values, but compound 10 had the lowest −0.07028 eV, indicating a higher capacity to receive electrons than acarbose. In comparison to acarbose, compound 1 has the smallest energy gap (0.31547 eV), suggesting more chemical reactivity and decreased stability.
As compounds 8 and 10 were the lead compounds in in vitro and in vivo studies so they both have better electron-donating and accepting capacities than acarbose, as seen by their lower HOMO values (−0.22072 eV and −0.19694 eV, respectively) and negative LUMO energies (−0.07861 eV and −0.07028 eV, respectively) as shown in Fig. 5.
 | ||
| Fig. 5 Optimized structure and HOMO–LUMO diagram of acarbose in comparison with 8 and 10. | ||
With compound 8 at 0.29933 eV and compound 10 at 0.26722 eV, the derivatives' energy gaps are also noticeably smaller, suggesting more chemical reactivity and less stability. Because of their decreased stability, these compounds might be more suited for particular uses that call for higher reactivity, including enzyme inhibition or redox reactions. The variations in HOMO–LUMO energies and energy gaps between acarbose and its derivatives demonstrate how structural changes affect their electrical characteristics and point to possible improvements in their functional performance as a result of increased reactivity.74
3.6.7.3. Optimized structures. The optimized structures of compounds 1, 2, 8, 9, 10, and acarbose were determined using DFT analysis at the basic set of 6-311G/B3LYP (Fig. 5). The distribution of electrons and arrangement of atoms in space are revealed by these optimized geometries, and this information directly affects the compound's chemical and biological behavior.75 The reference, acarbose, has a complicated, highly branching structure with several functional groups that contain hydroxyl and oxygen. These characteristics help it interact with biological targets like proteins in the body to treat diabetes because of its high polarity and notable dipole moment of 9.201717 debye. Compounds 8 and 10 have altered structures with substitutions that change their electrical characteristics and geometries. For example, the addition of heteroatoms, like nitrogen in compound 10, alters the distribution of electron densities and dipole moments. In comparison to acarbose, these compounds are more reactive due to their enhanced electronic delocalization caused by the higher conjugation.
3.6.7.4. Electron affinity (A) and ionization potential (I). The electron affinity (A) shows the energy shift that occurs when an electron is introduced, whereas the ionization potential (I) shows the energy needed to remove an electron. Acarbose has an electron affinity of 0.01456 eV and an ionization potential of 0.21573 eV.
Compound 1 has the highest ionization potential (0.23222 eV) of all the derivatives, indicating that it is more resilient to electron removal than acarbose. The strongest electron affinities are shown by compound 1 (0.08325 eV) and compound 9 (0.08672 eV), suggesting that they may be electron acceptors.
3.6.7.5. Global reactivity descriptors. The chemical reactivity and stability of the molecules are shown by the global reactivity descriptors, which include chemical potential (μ), electronegativity (χ), chemical hardness (η), and softness (S):
Chemical potential (μ): the chemical potential (μ) of compound 1 is the greatest at 0.157735 eV, suggesting that it is more stable than acarbose (0.115145 eV).
Electronegativity (χ): in comparison to compound 1 (−0.157735 eV) and the other derivatives, acarbose has a lower electronegativity (−0.115145 eV), indicating a diminished propensity to attract electrons.
Chemical hardness (η): acarbose is comparatively softer (0.100585 eV), suggesting lesser stability and more reactivity, while compound 1 is the hardest chemical (0.074485 eV), followed by compound 10 (0.063405 eV).
Chemical softness (S): in comparison to the derivatives, where compound 2 (16.8535 eV) and compound 10 (15.7903 eV) are less reactive, acarbose has the highest softness value (9.941840 eV), indicating that it is more reactive.
3.6.7.6. Electrophilicity and nucleophilicity indices. Acarbose (0.263625 eV) has a lower electrophilicity index (ω) than its derivatives, compound 1 (0.66806 eV) and compound 9 (0.7091 eV), which indicate a stronger propensity to take electrons. Acarbose (7.165634) has a higher nucleophilicity index (N) than its derivatives, suggesting a stronger capacity for electron donation.
3.6.7.7. MEP scale and structure. A visual tool for comprehending positive, negative, and neutral electrostatic potential regions in relation to color grading and a molecule's relative polarity is the molecular electrostatic potential (MEP). These surfaces also show the charge density, molecular size, molecular structure, and chemically reactive sites of molecules. The various hues on the molecule's MESP surface correspond to different electrostatic potential regions, such as light blue, slightly electron deficient, yellow, slightly electron-rich, green neutral, and red, electron-rich, partially negative charge. The electron density distribution and reactivity patterns for acarbose (reference), compounds 8, and 10 are depicted in the molecular electrostatic potential (MEP) maps.76
The MEP map for acarbose shows areas of electrophilic activity (blue) and very electron-rich areas (red) near oxygen-containing functional groups, which may indicate potential sites for nucleophilic interactions.77 Compound 8 shows an even distribution with slightly fewer electron density peaks due to modifications in the molecular structure which restrict reactivity. However, compound 10 shows localized electron-rich regions near oxygen atoms and electron-deficient regions surrounding specific structural features, suggesting the possibility of selective interaction. The MEP scale illustrates the gradual transition from electron-rich (red) to electron-deficient (blue), which aids in understanding molecular polarity and chemical reactivity.
Acarbose has the maximum electron density around hydroxyl groups, compound 8 has a more balanced electrical profile, and compound 10 has a more polarized distribution as shown in Fig. 6. Overall, these compounds' MEP structures show their reactive regions. These differences are essential for comprehending their biological activity, ability to inhibit enzymes, and general molecular behavior in certain applications.78
 | ||
| Fig. 6 MEP structure and scale of acarbose, 8 and 10 and also the MEP scale illustration. | ||
In contrast to acarbose, the derivatives exhibit distinct electrical and molecular properties that influence their stability and reactivity. Compound 10 has a moderate chemical potential and a strong dipole moment, making it most similar to acarbose in terms of polarity. While compound 1, which has the smallest energy gap, has the most chemical reactivity, compound 2, with its high hardness and ionization potential, has the best stability. The compounds may be used in specific applications, according to our conclusions about their stability and reactivity profiles.
| Compound no. | Mol. formula | Mol. wt (g mol−1) | No. HBAa | No. HBDb | M log Pc | Mol. log Sd (mg L−1) | Mol. vol A3 | Drug-likeness model score | Violation | Lipinski's RO5 |
|---|---|---|---|---|---|---|---|---|---|---|
| a The table above depicts all Lipinski's RO5 components, i.e. anumber of hydrogen-bond acceptors. bNumber of hydrogen-bond donors. cOctanol–water partition coefficient. dMeasured solubility. | ||||||||||
| 1 | C22H16O3 | 328.11 | 3 | 0 | 3.08 | 1.68 | 333.12 | 0.00 | 0 | Yes |
| 2 | C34H25NO3 | 495.18 | 3 | 0 | 5.00 | 0.33 | 497.20 | −0.07 | 1 | Yes |
| 3 | C23H18O3 | 342.13 | 3 | 0 | 3.29 | 1.01 | 354.06 | 0.22 | 0 | Yes |
| 4 | C20H14O3S | 334.07 | 4 | 0 | 2.67 | 2.35 | 326.88 | −0.05 | 0 | Yes |
| 5 | C22H15ClO3 | 362.07 | 3 | 0 | 3.56 | 0.37 | 350.32 | 0.58 | 0 | Yes |
| 6 | C22H15NO5 | 373.36 | 5 | 0 | 2.07 | 1.87 | 358.19 | −0.35 | 0 | Yes |
| 7 | C22H15NO5 | 373.36 | 5 | 0 | 2.07 | 2.08 | 358.11 | −0.23 | 0 | Yes |
| 8 | C23H18O4 | 358.12 | 4 | 0 | 2.71 | 2.25 | 364.97 | 0.44 | 0 | Yes |
| 9 | C20H14O4 | 318.32 | 4 | 0 | 1.82 | 8.46 | 318.56 | −0.11 | 0 | Yes |
| 10 | C24H21NO3 | 371.43 | 3 | 0 | 2.92 | 2.70 | 382.68 | 0.13 | 0 | Yes |
4. Conclusions
This study successfully identified 3-benzyloxyflavone derivatives (1–10) as potent β-glucosidase inhibitors through a multidisciplinary approach encompassing experimental and computational evaluations. The in vitro and in vivo assessments demonstrated that all synthesized compounds (1–10) exhibited remarkable inhibitory activity against β-glucosidase, with IC50 values ranging from 0.17 μM to 1.02 μM. This performance significantly surpassed the reference standard, acarbose (IC50 = 34.09 μM). Among the tested derivatives, compounds 3 (IC50 = 0.28 μM), 8 (IC50 = 0.17 μM), and 10 (IC50 = 0.25 μM) exhibited the highest potency. Acute toxicity evaluations confirmed that the compounds were well-tolerated at doses up to 100 mg kg−1. Subsequent in vivo investigations focused on the antihyperglycemic potential of six selected compounds (3, 5, 6, 7, 8, and 10), chosen based on their superior β-glucosidase inhibitory activity. The antidiabetic efficacy of these compounds 3, 5, 6, 7, 8, and 10 was further validated using a PGLT-induced diabetic rat model, where they demonstrated a spectrum of good to excellent antihyperglycemic effects. Kinetic analysis indicated that compounds 8 and 10 acted as competitive inhibitors of β-glucosidase. The SAR study revealed that the nature and position of electron-donating substituents were critical for enhancing inhibitory potency. Molecular docking studies of compounds 8 and 10, complemented by MD simulations for compound 8, demonstrated robust and stable interactions with the enzyme's active site, corroborating their molecular binding capabilities. DFT calculations for key compounds (1, 2, 8, 9, and 10) provided deeper insights into their electronic properties and reactivity, further validating their potential as inhibitors. Drug-likeness assessments indicated favorable pharmacokinetic profiles for all synthesized derivatives (1–10), supporting their viability as drug candidates. This comprehensive investigation provides a strong foundation for the design and optimization of β-glucosidase inhibitors, offering valuable insights for the development of novel antidiabetic agents. Future studies will focus on advancing the most promising compounds toward preclinical and clinical evaluations.Ethical statement
The treatment of the animals adhered to the guidelines outlined in the “Animal Bylaws 2008 of the University of Malakand (Scientific Procedures Issue I).” Ethical approval for the study was obtained from the Ethical Committee of the Department of Pharmacy, in accordance with the Animal Bylaws 2008 of the University of Malakand, as per notification no. Pharm/EC-Thzp/41-08/21.Data availability
All the data is provided in the manuscript and ESI files.†Author contributions
Nafeesa Naeem: experimental work performance, data analysis, and collection, performed enzyme inhibition activity, software, first-draft preparation; Ehsan Ullah Mughal: supervision, main idea, methodology, resources, reviewing and editing, final writing the manuscript.Conflicts of interest
Authors have no conflict of interest to declare.Acknowledgements
The authors acknowledge the Department of Chemistry, University of Gujrat, for providing the necessary laboratory facilities for the successful completion of this research.References
- E. Karakılıç, S. Durmuş, S. Sevmezler, O. Şahin and A. Baran, Bioorg. Med. Chem., 2018, 26, 4276–4287 CrossRef PubMed
.
- Y. Bhatia, S. Mishra and V. Bisaria, Crit. Rev. Biotechnol., 2002, 22, 375–407 CrossRef CAS PubMed
- J. R. K. Cairns and A. Esen, Cell. Mol. Life Sci., 2010, 67, 3389 CrossRef PubMed
- E. Beutler, Science, 1992, 256, 794–799 CrossRef CAS
- Y. Gao, J. Duan, X. Dang, Y. Yuan, Y. Wang, X. He, R. Bai, X.-Y. Ye and T. Xie, J. Enzyme Inhib. Med. Chem., 2023, 38, 2195991 CrossRef
- R. R. Singhania, A. K. Patel, R. K. Sukumaran, C. Larroche and A. Pandey, Bioresour. Technol., 2013, 127, 500–507 CrossRef CAS PubMed
- A. V. Morant, K. Jørgensen, C. Jørgensen, S. M. Paquette, R. Sánchez-Pérez, B. L. Møller and S. Bak, Phytochemistry, 2008, 69, 1795–1813 CrossRef CAS PubMed
- M. Bhat, Biotechnol. Adv., 2000, 18, 355–383 CrossRef CAS PubMed
- Y. Chen, Y. Deng, Y. Li, Y. Qin, Z. Zhou, H. Yang and Y. Sun, ACS Appl. Mater. Interfaces, 2024, 16, 21546–21556 CrossRef CAS PubMed
- R. Li, P. Luo, Y. Guo, Y. He and C. Wang, Expert Opin. Drug Saf., 2024, 1–5 Search PubMed
- J. Zhu, X. Jiang, X. Luo, R. Zhao, J. Li, H. Cai, X. Y. Ye, R. Bai and T. Xie, Drug Dev. Res., 2023, 84, 718–735 CAS
- R. Kumar, S. Singh and O. V. Singh, J. Ind. Microbiol. Biotechnol., 2008, 35, 377–391 CAS
- A. Esen, Glucosidases: Overview, in ACS Symposium Series, American Chemical Society, 1993, vol. 533, p. 1 Search PubMed
- Q. Q. Lu, Y. M. Chen, H. R. Liu, J. Y. Yan, P. W. Cui, Q. F. Zhang, X. H. Gao, X. Feng and Y. Z. Liu, Drug Dev. Res., 2020, 81, 1037–1047 CrossRef CAS PubMed
- E. U. Mughal, J. Ashraf, E. M. Hussein, Y. Nazir, A. S. Alwuthaynani, N. Naeem, A. Sadiq, R. I. Alsantali and S. A. Ahmed, ACS Omega, 2022, 7, 17444–17461 CrossRef CAS PubMed
- A. T. Rufino, V. M. Costa, F. Carvalho and E. Fernandes, Med. Res. Rev., 2021, 41, 556–585 CAS
- R. J. Obaid, E. U. Mughal, N. Naeem, A. Sadiq, R. I. Alsantali, R. S. Jassas, Z. Moussa and S. A. Ahmed, RSC Adv., 2021, 11, 22159–22198 CAS
- K. Wen, X. Fang, J. Yang, Y. Yao, K. S. Nandakumar, M. L. Salem and K. Cheng, Curr. Med. Chem., 2021, 28, 1042–1066 CAS
- A. Ullah, S. Munir, S. L. Badshah, N. Khan, L. Ghani, B. G. Poulson, A.-H. Emwas and M. Jaremko, Molecules, 2020, 25, 5243 CrossRef CAS PubMed
- S. J. Maleki, J. F. Crespo and B. Cabanillas, Food Chem., 2019, 299, 125124 CrossRef CAS PubMed
- I. Górniak, R. Bartoszewski and J. Króliczewski, Phytochem. Rev., 2019, 18, 241–272 CrossRef
- L. Ciumărnean, M. V. Milaciu, O. Runcan, Ş. C. Vesa, A. L. Răchişan, V. Negrean, M.-G. Perné, V. I. Donca, T.-G. Alexescu and I. Para, Molecules, 2020, 25, 4320 CrossRef PubMed
- E. U. Mughal, A. Sadiq, M. Ayub, N. Naeem, A. Javid, S. H. Sumrra, M. N. Zafar, B. A. Khan, F. P. Malik and I. Ahmed, J. Biomol. Struct. Dyn., 2021, 39, 6154–6167 CrossRef CAS PubMed
- J. Ashraf, E. U. Mughal, A. Sadiq, M. Bibi, N. Naeem, A. Ali, A. Massadaq, N. Fatima, A. Javid and M. N. Zafar, J. Biomol. Struct. Dyn., 2021, 39, 7107–7122 CAS
- L. Chen, H. Teng, Z. Xie, H. Cao, W. S. Cheang, K. Skalicka-Woniak, M. I. Georgiev and J. Xiao, Crit. Rev. Food Sci. Nutr., 2018, 58, 513–527 CAS
- P. Kachlicki, A. Piasecka, M. Stobiecki and Ł. Marczak, Molecules, 2016, 21, 1494 Search PubMed
- L. Chen, Z. Jiang, L. Yang, Y. Fang, S. Lu, O. U. Akakuru, S. Huang, J. Li, S. Ma and A. Wu, Chin. J. Chem., 2023, 41, 199–206 CAS
- L. Chepeleva, D. Tarasenko, A. Chumak, O. Demidov, A. Snizhko, O. Kolomoitsev, V. Kotliar, E. Gladkov, A. Tatarets and A. Kyrychenko, Funct. Mater., 2023, 30, 495 Search PubMed
- V. Gopal, J. Sudhakaran, N. Ramachandran, T. K. Mana, A. R. Kana, A. O. Nair, P. Mohan, T. Madhusudhan, S. Shanmugaraju and P. Nanjan, Sens. Diagn., 2024, 3, 1263–1271 RSC
- S. Tang, B. Wang, X. Liu, W. Xi, Y. Yue, X. Tan, J. Bai and L. Huang, Food Front., 2025, 218–247 CrossRef CAS
- L. Kang, X.-H. Gao, H.-R. Liu, X. Men, H.-N. Wu, P.-W. Cui, E. Oldfield and J.-Y. Yan, Mol. Divers., 2018, 22, 893–906 CrossRef CAS PubMed
- R. Nath, S. Manna, S. Panda, A. Maity, K. Bandyopadhyay, A. Das, S. A. A. Khan, B. Debnath and M. J. Akhtar, Chem. Biodiversity, 2024, e202401899 Search PubMed
- G. Zeng, Z. Wu, W. Cao, Y. Wang, X. Deng and Y. Zhou, Nat. Prod. Res., 2020, 34, 1041–1045 CrossRef CAS PubMed
- M. E. Erkanli, K. El-Halabi and J. R. Kim, Enzyme Microb. Technol., 2023, 110363 Search PubMed
- Y. Cheng, L. Wang, S. Zhang, W. Jian, B. Zeng, L. Liang and Z. Deng, Curr. Med. Chem., 2024 DOI:10.2174/0109298673309489240816063313
- A. M. Dirir, M. Daou, A. F. Yousef and L. F. Yousef, Phytochem. Rev., 2022, 21, 1049–1079 CrossRef CAS PubMed
- N. Agrawal, M. Sharma, S. Singh and A. Goyal, Curr. Top. Med. Chem., 2022, 22, 2069–2086 CrossRef CAS PubMed
- E. B. de Melo, A. da Silveira Gomes and I. Carvalho, Tetrahedron, 2006, 62, 10277–10302 CrossRef
- H. Saleem, A. Yaqub, R. Rafique, T. Ali Chohan, D.-e.-S. Malik, M. I. Tousif, U. Khurshid, N. Ahemad, R. Ramasubburayan and K. R. Rengasamy, Crit. Rev. Food Sci. Nutr., 2024, 64, 9805–9828 CrossRef CAS PubMed
- J. Zhu, C. Chen, B. Zhang and Q. Huang, Crit. Rev. Food Sci. Nutr., 2020, 60, 695–708 CrossRef CAS PubMed
- Y. Cahyana and T. Adiyanti, Indones. J. Chem., 2021, 21, 512–526 CrossRef CAS
- V. Jaitak, Mini-Rev. Med. Chem., 2019, 19, 762–786 CrossRef PubMed
- Y. Ji, B. Li, M. Qiao, J. Li, H. Xu, L. Zhang and X. Zhang, Appl. Microbiol. Biotechnol., 2020, 104, 6587–6600 CrossRef CAS PubMed
- X. H. Gao, J. J. Tang, H. R. Liu, L. B. Liu and Y. Z. Liu, Drug Dev. Res., 2019, 80, 438–445 CrossRef CAS PubMed
- K. Kytidou, M. Artola, H. S. Overkleeft and J. M. Aerts, Front. Plant Sci., 2020, 11, 357 CrossRef PubMed
- F. Sok Yen, C. Shu Qin, S. Tan Shi Xuan, P. Jia Ying, H. Yi Le, T. Darmarajan, B. Gunasekaran and S. Salvamani, Evidence-Based Complementary Altern. Med., 2021, 2021, 2057333 Search PubMed
- J. Ashraf, E. U. Mughal, A. Sadiq, N. Naeem, S. A. Muhammad, T. Qousain, M. N. Zafar, B. A. Khan and M. Anees, J. Mol. Struct., 2020, 1218, 128458 CrossRef CAS
- R. Mehmood, E. U. Mughal, E. B. Elkaeed, R. J. Obaid, Y. Nazir, H. A. Al-Ghulikah, N. Naeem, M. M. Al-Rooqi, S. A. Ahmed and S. W. A. Shah, ACS Omega, 2022, 7, 30215–30232 CrossRef CAS PubMed
- H. A. Al-ghulikah, E. U. Mughal, E. B. Elkaeed, N. Naeem, Y. Nazir, A. Y. A. Alzahrani, A. Sadiq and S. W. A. Shah, J. Mol. Struct., 2023, 1275, 134658 CrossRef CAS
- M. Alamzeb, S. W. A. Shah, H. Hussain, M. Zahoor, S. Ahmad, E. U. Mughal, S. Ahmad, I. Ullah, S. Khan and A. Ullah, ACS Omega, 2024, 9, 9813–9822 CrossRef CAS PubMed
- R. Mehmood, A. Sadiq, R. I. Alsantali, E. U. Mughal, M. A. Alsharif, N. Naeem, A. Javid, M. M. Al-Rooqi, G.-e.-S. Chaudhry and S. A. Ahmed, ACS Omega, 2022, 7, 3775–3795 CrossRef CAS PubMed
- V. R. Shah, J. D. Bhaliya and G. M. Patel, J. Basic Clin. Physiol. Pharmacol., 2021, 32, 197–214 CrossRef CAS PubMed
- S. Bouchagra, F. Benamia and Z. Djeghaba, Res. J. Pharm., Biol. Chem. Sci., 2016, 7, 2493–2505 CAS
- C. E. Cassidy and W. N. Setzer, J. Mol. Model., 2010, 16, 311–326 CrossRef CAS PubMed
- W. Jorgensen and J. Chandrasekhar, J. Chem. Phys., 1983, 79, 926 CrossRef CAS
- K. A. Qureshi, I. Al Nasr, W. S. Koko, T. A. Khan, M. Q. Fatmi, M. Imtiaz, R. A. Khan, H. A. Mohammed, M. Jaremko and A.-H. Emwas, Antibiotics, 2021, 10, 887 CrossRef CAS PubMed
- B. Hess, H. Bekker, H. J. Berendsen and J. G. Fraaije, J. Comput. Chem., 1997, 18, 1463–1472 CrossRef CAS
- H. Grubmüller, H. Heller, A. Windemuth and K. Schulten, Mol. Simul., 1991, 6, 121–142 CrossRef
- U. Essmann, L. Perera, M. L. Berkowitz, T. Darden, H. Lee and L. G. Pedersen, J. Chem. Phys., 1995, 103, 8577–8593 CrossRef CAS
- B. J. Grant, L. Skjærven and X. Q. Yao, Protein Sci., 2021, 30, 20–30 CrossRef CAS PubMed
- J. Huang and A. D. MacKerell Jr, J. Comput. Chem., 2013, 34, 2135–2145 CrossRef CAS PubMed
- R. Srivastava, F. A. Al-Omary, A. A. El-Emam, S. K. Pathak, M. Karabacak, V. Narayan, S. Chand, O. Prasad and L. Sinha, J. Mol. Struct., 2017, 1137, 725–741 CrossRef CAS
- W. Shoukat, M. Hussain, A. Ali, N. Shafiq, A. H. Chughtai, B. Shakoor, A. Moveed, M. N. Shoukat, M. Milošević and M. Mohany, J. Mol. Struct., 2025, 1320, 139614 CrossRef CAS
- K. Yamamoto, H. Miyake, M. Kusunoki and S. Osaki, FEBS J., 2010, 277, 4205–4214 CrossRef CAS PubMed
- Y. Xu, Y. Wang, K. Liu, Y. Peng, D. Yao, H. Tao, H. Liu and G. Song, IUCrJ, 2019, 6, 996–1006 CrossRef CAS PubMed
- K. Lisina and S. Piramanayagam, World. J. Pharm. Sci., 2014, 283–293 CAS
- M. E. Gondokesumo and I. M. Kurniawan, J. Basic Clin. Physiol. Pharmacol., 2019, 30, 20190282 CrossRef CAS PubMed
- N. Shafiq, B. Shakoor, N. Yaqoob, S. Parveen, S. Brogi, A. Mohammad Salamatullah, M. Rashid and M. Bourhia, J. Biomol. Struct. Dyn., 2024, 1–22 CrossRef PubMed
- F. Sezer Senol, M. Tareq Hassan Khan, G. Orhan, E. Gurkas, I. Erdogan Orhan, N. Subutay Oztekin and F. Ak, Curr. Top. Med. Chem., 2014, 14, 1469–1472 CrossRef PubMed
- W. J. Hehre, R. Ditchfield and J. A. Pople, J. Chem. Phys., 1972, 56, 2257–2261 CrossRef CAS
- C. Lee, W. Yang and R. G. Parr, Phys. Rev. B:Condens. Matter Mater. Phys., 1988, 37, 785 CrossRef CAS PubMed
- S. Böhm and O. Exner, Phys. Chem. Chem. Phys., 2004, 6, 510–514 RSC
- V. Choudhary, A. Bhatt, D. Dash and N. Sharma, J. Comput. Chem., 2019, 40, 2354–2363 CrossRef CAS PubMed
- J. Singh, M. S. Khan and S. Uddin, Polym. Bull., 2023, 80, 3055–3083 CrossRef CAS PubMed
- L. Fan and T. Ziegler, J. Chem. Phys., 1991, 95, 7401–7408 CrossRef CAS
- P. Govindasamy and S. Gunasekaran, Spectrochim. Acta, Part A, 2015, 136, 1543–1556 CrossRef CAS PubMed
- Ö. Mıhçıokur and T. Özpozan, J. Mol. Struct., 2017, 1149, 27–41 CrossRef
- E. S. Marinho and M. M. Marinho, Int. J. Sci. Eng. Res., 2016, 7, 1264 Search PubMed
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra08558b |
| This journal is © The Royal Society of Chemistry 2025 |