
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceNew 8-hydroxyquinoline-based azo dyes as highly sensitive colorimetric chemosensors for nickel cation†
Sefiu Olalekan Olaleye ab,
Sunday Olakunle Idowu*a,
Shakil Ahmedc and
Maria Aqeel Khan*b
ab,
Sunday Olakunle Idowu*a,
Shakil Ahmedc and
Maria Aqeel Khan*b
aDepartment of Pharmaceutical Chemistry, Faculty of Pharmacy, University of Ibadan, Orita UI, Ibadan, 200284, Nigeria. E-mail: olakunleid@yahoo.com
bThird World Center for Science and Technology, H. E. J. Research Institute of Chemistry, International Center for Chemical and Biological Sciences, University of Karachi, Karachi, 75270, Pakistan. E-mail: drmaria.aqeel@iccs.edu
cIndustrial Analytical Center, International Center for Chemical and Biological Sciences, University of Karachi, Karachi, 75270, Pakistan
First published on 4th February 2025
Abstract
A new series of sensitive 8-hydroxyquinoline azo-compounds (S1–S8) as sensors was synthesized by diazotization and diazo-coupling, and these compounds were characterized through UV, FTIR, 1H-NMR, 13C-NMR and ESI-MS investigations. The UV-visible spectral data revealed that Ni2+ was sensed by compounds S2, S3, S4, and S6, producing colour changes ranging from violet to orange, and exhibited bathochromism of +8, +67, +63, and +85 nm, respectively. Optimized conditions included a mixture of ethanol/water (4![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1) solvent system; pH 4 for compounds S3, S4, and S6, and pH 8 for compound S2; reaction time of 2 min; and a stoichiometric ratio of 3:1. The limits of detection for the Ni2+ cation demonstrated by the dyes were in the range of 0.012–0.038 μM using UV-visible spectroscopy, which is lower than the permissible value of Ni2+ (1.2 μM) in drinking water specified by the United States Environmental Protection Agency. Intra-day and inter-day accuracies of the Ni2+ cation spiked in tap and underground water (in terms of relative errors) were less than 5%, while precisions were less than 4.0% (RSD). The accuracy and precision of chemosensor-based analyses were comparable to those of the inductively coupled plasma mass spectrometry (ICP-MS) method. Colour changes of paper strips with sensors S2, S3, S4, and S6 ranged from orange to violet in the presence of nickel. Infrared analysis confirmed that sensors interacted with Ni2+ through nitrogen and hydroxyl moieties of quinoline. Thus, an easy-to-use, highly sensitive, and low-cost method for analyzing nickel in water samples was established.
:1) solvent system; pH 4 for compounds S3, S4, and S6, and pH 8 for compound S2; reaction time of 2 min; and a stoichiometric ratio of 3:1. The limits of detection for the Ni2+ cation demonstrated by the dyes were in the range of 0.012–0.038 μM using UV-visible spectroscopy, which is lower than the permissible value of Ni2+ (1.2 μM) in drinking water specified by the United States Environmental Protection Agency. Intra-day and inter-day accuracies of the Ni2+ cation spiked in tap and underground water (in terms of relative errors) were less than 5%, while precisions were less than 4.0% (RSD). The accuracy and precision of chemosensor-based analyses were comparable to those of the inductively coupled plasma mass spectrometry (ICP-MS) method. Colour changes of paper strips with sensors S2, S3, S4, and S6 ranged from orange to violet in the presence of nickel. Infrared analysis confirmed that sensors interacted with Ni2+ through nitrogen and hydroxyl moieties of quinoline. Thus, an easy-to-use, highly sensitive, and low-cost method for analyzing nickel in water samples was established.
1. Introduction
Nickel is an essential element with daily requirements within the range of 25 to 35 μg,1 and living cells are harmed upon its homeostatic imbalance. It is required in the active sites of many crucial enzymes, including catalytic processes, carbon monoxide dehydrogenases, hydrogenases, acetyl-coenzymes, superoxide dismutases, and acireductone dioxygenases.2,3Nickel enters into water bodies through manufacturing and metallurgical activities, mining, incineration and combustion of fossil fuels; catalyst and chemical production; and effluents from industry (EFSA, 2015). Excessive exposure to Ni can raise the plasma level above the normal range of 12–85 μg L−1,4 which can lead to acute pneumonitis, dermatitis, asthma, cancer of the lung and sinus, adverse effects on blood and kidneys, and other disorders of the respiratory and central nervous systems.5 Hence, there is a need to rapidly monitor the nickel levels in environmental samples.
Instrumental methods, such as inductively coupled plasma-mass spectrometry (ICP-MS), inductively coupled plasma-optical emission spectrometry (ICP-OES), inductively coupled plasma-atomic emission spectrometry (ICP-AES), atomic absorption spectrometry (AAS), atomic absorption spectrometry (AES), flame atomic absorption (FAA), flame atomic emission (FAE), and graphite furnace atomic absorption, that are used to determine nickel ion concentration suffer from several drawbacks. Although the ICP methods offer the advantages of multiple element analysis, a large analytical range, and low detection limits, they are not suitable for low-resource economies because of their high equipment cost, high operating cost (argon), complex operation, and high level of staff expertise.6 On the other hand, AAS, AES, FAE, and FAA are relatively cost-effective, but they are single-element procedures that are poorly selective and sensitive.6 Therefore, there is a need for the development of chemosensors that offer simple instantaneous detection, low cost, high sensitivity, and selectivity in the analysis of Ni.7
Azo compounds have found applications in different endeavours, especially in chemosensor designs. Sancenon and co-workers (2003)8 reported that the introduction of an azo bridge in chemosensors enhances their chromogenic characteristics, which enhances their suitability as sensors. Azo derivatives of 8-hydroxyquinoline have been reported as chemosensors for Pb2+,9 Cd2+,10 Hg2+,10 CN−,11 and F− ions.12 Prior evidences revealed that there are very few studies in the literature on sensors for nickel ion detection, as depicted in Fig. 1,13–15 and the use of azo-based derivatives of 8-hydroxyquinoline has not been widely explored for sensing nickel ions. In addition, some of the reported prior chemosensor-based methods utilized milieus, such as dimethyl sulfoxide (DMSO),16,17 N,N-dimethylformamide (DMF),18 acetonitrile (ACN),19,20 and tetrahydrofuran (THF).21 These solvents are not only eco-unfriendly, but are also relatively expensive and not readily available for chemosensing experiments. Hence, this study reports a new class of 8-hydroxyquinoline-based azo dyes as highly sensitive colorimetric chemosensors for the simple, inexpensive, and rapid analysis of nickel in ethanol/water mixtures.
 | ||
| Fig. 1 Chemical structures of some prior chemosensors for nickel. | ||
2. Experimental
2.1 General information, material and methods
Sulfanilamide, 2-methoxy-4-nitroaniline, 4-methoxy-2-nitroaniline, aniline, 2-amino-4-nitrophenol, 2,3-dimethylaniline, 8-amino-2-naphthol, sodium nitrite, 8-hydroxyquinoline, hexane, ethyl acetate, dichloromethane, acetone, acetonitrile, ethanol, methanol, DMF, DMSO, glacial acetic acid, concentrated hydrochloric acid, concentrated trioxonitrate(V) acid, concentrated sulphuric acid, potassium hydroxide, potassium acetate, copper sulphate, lead nitrate, zinc sulphate, calcium sulphate, mercuric acetate, lithium chloride, magnesium sulphate, sodium sulphate, potassium sulphate, silver nitrate, cobalt(II) chloride, ferric chloride, ferrous sulphate, aluminium chloride, nickel(II) acetate, and pH buffers were used in this study. All the reagents were obtained from BDH, Poole, England. Thin-layer chromatography (TLC) was developed on pre-coated silica gel aluminum plates (Kieselgel 60F254, E. Merck, Germany). A hotplate magnetic stirrer purchased from Thermo Fisher Scientific, US, was used. 1H-NMR and 13C-NMR spectra were recorded on Bruker Avance spectrometers (UK) at 500, 600, and 800 MHz. UV-Vis spectral data were obtained on a Nicolet Evolution 300 UV-visible spectrophotometer (Thermo Electron Corporation, US). FTIR spectral data were obtained on a Nicolet iS20 FTIR spectrometer (Thermo Fisher Scientific, US). ESI-MS measurements were recorded on an AmaZon speed ESI-ion trap mass spectrometer (Bruker, UK). The ICP-MS 7700 Series (Agilent Technologies, US) was used. The pH of the solution was obtained on a pH meter (Hanna, US). The melting point was obtained on an M-560 (BUCHI, Switzerland) instrument.2.2 Synthesis
:concentrated hydrochloric acid (10:1). The reaction was allowed to stay for 30 min in an ice bath. Afterwards, 1.05 mmol of nitroxoline dissolved in 20 mL of glacial acetic acid was introduced into the diazonium solutions, while maintaining the reaction at a temperature of less than 5 °C for 3 h. The reaction progress was monitored with TLC, and the reaction pH was adjusted to 6–7 with 6 M potassium acetate solution after a reaction time of 2 h. The ensuing colored solid was filtered, washed with water, and dried at room temperature. Pure compound S1 was obtained by dissolution of the crude product in ethyl acetate and recrystallization by adding n-hexane until turbidity was observed. Compound S2 was purified by column chromatography through gradient elution with hexane and EtOAc.4′-[2-(8-Hydroxy-5-nitroquinolin-7-yl)diazen-1-yl]benzene-1-sulfonamide (S1).
Orange solid; yield: 74%; FTIR (ATR, ν, cm−1): 3400, 3300, 3200, 2981, 1609 (C

![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N), 1574, 1506 (NN), 1396 (C–O); 1H-NMR (500 MHz, DMSO-d6, δ, ppm): 9.37 (d, J2,3 = 8.7 Hz, 1H, H-2), 9.04 (d, J4,3 = 4.4 Hz, 1H, H-4), 8.89 (s, 1H, H-6), 8.12 (d, J2′,3′/6′,5′ = 8.2 Hz, 2H, H-2′/H-6′), 8.06–8.02 (m, 3H, overlapped, H-3, H-3′/H-5′), 7.54 (s, 2H, sulfonamide NH2, exchanges with D2O); 13C-NMR (600 MHz, DMSO-d6, δ, ppm): 162.5 (C), 153.3 (C), 147.1 (CH), 145.5 (C), 138.9 (C), 135.9 (C), 134.4 (CH), 132.1 (CH), 127.1 (CH), 126.5 (C), 124.8 (C), 123.0 (CH), 117.8 (CH); MS (ESI, m/z): 374 [M++H].
N), 1574, 1506 (NN), 1396 (C–O); 1H-NMR (500 MHz, DMSO-d6, δ, ppm): 9.37 (d, J2,3 = 8.7 Hz, 1H, H-2), 9.04 (d, J4,3 = 4.4 Hz, 1H, H-4), 8.89 (s, 1H, H-6), 8.12 (d, J2′,3′/6′,5′ = 8.2 Hz, 2H, H-2′/H-6′), 8.06–8.02 (m, 3H, overlapped, H-3, H-3′/H-5′), 7.54 (s, 2H, sulfonamide NH2, exchanges with D2O); 13C-NMR (600 MHz, DMSO-d6, δ, ppm): 162.5 (C), 153.3 (C), 147.1 (CH), 145.5 (C), 138.9 (C), 135.9 (C), 134.4 (CH), 132.1 (CH), 127.1 (CH), 126.5 (C), 124.8 (C), 123.0 (CH), 117.8 (CH); MS (ESI, m/z): 374 [M++H].
7-[(2-(2′-Methoxy-4′-nitrophenyl)diazen-1-yl]-5-nitroquinolin-8-ol) (S2).
Red solid; yield: 71%; FTIR (ATR, ν, cm−1): 3300, 3081, 2850, 1610 (C
 N), 1569, 1506 (NN), 1384 (C–O); 1H NMR (500 MHz, DMSO-d6, δ, ppm): 9.23 (d, J2,3 = 8.5 Hz, 1H, H-2), 8.87 (s, 1H, H-6), 8.68 (dd, J4,3 = 4.8 Hz, J4,2 = 1.3 Hz, 1H, H-4), 7.94 (d, J6′,5′ = 2.4 Hz, 1H, H-6′), 7.91 (dd, J5′,6′ = 8.7 Hz, J5′,3′ = 2.4 Hz, 1H, H-5′), 7.72–7.58 (m, 2H, overlapped, H-3, H-6′), 4.07 (s, 3H, OCH3-7′); 13C-NMR (800 MHz, DMSO-d6, δ, ppm): 174.6 (C), 155.3 (C), 147.4 (CH), 146.9 (C), 146.7 (C), 138.1 (C), 132.7 (CH), 126.5 (CH), 125.1 (CH), 125.0 (C), 119.2 (CH), 117.2 (CH), 116.4 (CH), 107.9 (C), 55.9 (CH3); MS (ESI, m/z): 370 [M++H].
:water (1:1), was introduced dropwise into the diazonium solutions. The reaction was allowed to stay for 3 h at the temperature. The progress of the reaction was continuously monitored with TLC. The pH was adjusted to 6–7 with 1 M KOH solution. The colored solid formed was filtered, washed with water, and dried at room temperature. Dyes S3–S5, S7 and S8 were purified in an acetone/water mixture, while pure compound S6 was obtained from an ethyl acetate/n-hexane system.
N), 1569, 1506 (NN), 1384 (C–O); 1H NMR (500 MHz, DMSO-d6, δ, ppm): 9.23 (d, J2,3 = 8.5 Hz, 1H, H-2), 8.87 (s, 1H, H-6), 8.68 (dd, J4,3 = 4.8 Hz, J4,2 = 1.3 Hz, 1H, H-4), 7.94 (d, J6′,5′ = 2.4 Hz, 1H, H-6′), 7.91 (dd, J5′,6′ = 8.7 Hz, J5′,3′ = 2.4 Hz, 1H, H-5′), 7.72–7.58 (m, 2H, overlapped, H-3, H-6′), 4.07 (s, 3H, OCH3-7′); 13C-NMR (800 MHz, DMSO-d6, δ, ppm): 174.6 (C), 155.3 (C), 147.4 (CH), 146.9 (C), 146.7 (C), 138.1 (C), 132.7 (CH), 126.5 (CH), 125.1 (CH), 125.0 (C), 119.2 (CH), 117.2 (CH), 116.4 (CH), 107.9 (C), 55.9 (CH3); MS (ESI, m/z): 370 [M++H].
:water (1:1), was introduced dropwise into the diazonium solutions. The reaction was allowed to stay for 3 h at the temperature. The progress of the reaction was continuously monitored with TLC. The pH was adjusted to 6–7 with 1 M KOH solution. The colored solid formed was filtered, washed with water, and dried at room temperature. Dyes S3–S5, S7 and S8 were purified in an acetone/water mixture, while pure compound S6 was obtained from an ethyl acetate/n-hexane system.5-[2-(2′-Methoxy-4′-nitrophenyl)diazen-1-yl]quinolin-8-ol (S3).
Red solid; yield: 80%; FTIR (ATR, ν, cm−1): 3300, 3112, 2820, 1614 (C
 N), 1506 (NN), 1360 (C–O); 1H-NMR (500 MHz, DMSO-d6, δ, ppm): 9.33 (d, J2,3 = 8.4 Hz, 1H, H-2), 9.01 (d, J5′,6′ = 4.1 Hz, 1H, H-5′), 8.03 (s, 1H, H-3′), 7.99 (d, J6,7 = 2.8 Hz, 1H, H-6), 7.94 (d, J4,3 = 2.0 Hz, 1H, H-4), 7.89 (d, J6′,5′ = 8.8 Hz, 1H, H-6′), 7.80 (dd, J3,2 = 8.5 Hz, J3,4 = 4.1 Hz, 1H, H-3), 7.28 (d, J7,6 = 10.2 Hz, 1H, H-7), 4.10 (s, 3H, OCH3-7′); 13C-NMR (800 MHz, DMSO-d6, δ, ppm): 158.7 (C), 156.1 (C), 149.1 (C), 149.0 (CH), 145.8 (C), 145.1 (C), 139.4 (CH), 137.5 (C), 136.4 (CH), 136.1 (C), 107.5 (CH), 56.7 (CH3); MS (ESI, m/z): 325 [M++H].
N), 1506 (NN), 1360 (C–O); 1H-NMR (500 MHz, DMSO-d6, δ, ppm): 9.33 (d, J2,3 = 8.4 Hz, 1H, H-2), 9.01 (d, J5′,6′ = 4.1 Hz, 1H, H-5′), 8.03 (s, 1H, H-3′), 7.99 (d, J6,7 = 2.8 Hz, 1H, H-6), 7.94 (d, J4,3 = 2.0 Hz, 1H, H-4), 7.89 (d, J6′,5′ = 8.8 Hz, 1H, H-6′), 7.80 (dd, J3,2 = 8.5 Hz, J3,4 = 4.1 Hz, 1H, H-3), 7.28 (d, J7,6 = 10.2 Hz, 1H, H-7), 4.10 (s, 3H, OCH3-7′); 13C-NMR (800 MHz, DMSO-d6, δ, ppm): 158.7 (C), 156.1 (C), 149.1 (C), 149.0 (CH), 145.8 (C), 145.1 (C), 139.4 (CH), 137.5 (C), 136.4 (CH), 136.1 (C), 107.5 (CH), 56.7 (CH3); MS (ESI, m/z): 325 [M++H].
5-[2-(4′-Methoxy-2′-nitrophenyl)diazen-1-yl]quinolin-8-ol (S4).
Red solid; yield: 75%; FTIR (ATR, ν, cm−1): 3244, 2821, 1611 (C
 N), 1507 (NN), 1361 (C–O); 1H-NMR (500 MHz, DMSO-d6, δ, ppm): 9.20 (d, J2,3 = 8.5 Hz, 1H, H-2), 8.99 (d, J4,3 = 4.0 Hz, 1H, H-4), 8.05 (d, J6′,5′ = 9.0 Hz, 1H, H-6′), 7.86 (d, J6,7 = 8.5 Hz, 1H, H-6), 7.77 (dd, J3,2 = 8.6, J3,4 = 4.1 Hz, 1H, H-3), 7.66 (d, J3′,5′ = 2.7 Hz, 1H, H-3′), 7.37 (dd, J5′,6′ = 9.0 Hz, J5′,3′ = 2.8 Hz, 1H, H-5′), 7.24 (d, J7,6 = 8.6 Hz, 1H, H-7), 3.93 (s, 3H, OCH3-7′); 13C-NMR (600 MHz, DMSO-d6, δ, ppm): 161.0 (C), 158.4 (C), 149.2 (CH), 148.8 (C), 138.7 (C), 137.9 (C), 137.5 (C), 131.7 (CH), 127.5 (CH), 123.5 (C), 120.6 (CH), 118.9 (CH), 115.8 (CH), 112.0 (CH), 108.3 (CH), 56.6 (CH3); MS (ESI, m/z): 325 [M++H].
N), 1507 (NN), 1361 (C–O); 1H-NMR (500 MHz, DMSO-d6, δ, ppm): 9.20 (d, J2,3 = 8.5 Hz, 1H, H-2), 8.99 (d, J4,3 = 4.0 Hz, 1H, H-4), 8.05 (d, J6′,5′ = 9.0 Hz, 1H, H-6′), 7.86 (d, J6,7 = 8.5 Hz, 1H, H-6), 7.77 (dd, J3,2 = 8.6, J3,4 = 4.1 Hz, 1H, H-3), 7.66 (d, J3′,5′ = 2.7 Hz, 1H, H-3′), 7.37 (dd, J5′,6′ = 9.0 Hz, J5′,3′ = 2.8 Hz, 1H, H-5′), 7.24 (d, J7,6 = 8.6 Hz, 1H, H-7), 3.93 (s, 3H, OCH3-7′); 13C-NMR (600 MHz, DMSO-d6, δ, ppm): 161.0 (C), 158.4 (C), 149.2 (CH), 148.8 (C), 138.7 (C), 137.9 (C), 137.5 (C), 131.7 (CH), 127.5 (CH), 123.5 (C), 120.6 (CH), 118.9 (CH), 115.8 (CH), 112.0 (CH), 108.3 (CH), 56.6 (CH3); MS (ESI, m/z): 325 [M++H].
5-[2-(7′-Hydroxynaphthalen-1-yl)diazen-1-yl]quinolin-8-ol (S5).
Brown solid; yield: 78%; FTIR (ATR, ν, cm−1): 3400, 2980, 1623 (C
 N), 1504 (NN), 1380 (C–O); 1H-NMR (600 MHz, C3D6O, δ, ppm): 9.47 (dd, J2,3 = 8.5 Hz, J2,4 = 1.6 Hz, 1H, H-2), 8.98 (dd, J4,3 = 4.1 Hz, J4,2 = 1.6 Hz, 1H, H-4), 8.36 (d, J8′,6′ = 2.4 Hz, 1H, H-8′), 8.20 (d, J6,7 = 8.4 Hz, 1H, H-6), 7.99 (d, J4′,3′ = 4.2 Hz, 1H, H-4′), 7.98 (d, J2′,3′ = 3.8 Hz, 1H, H-2′), 7.93 (d, J5′,6′ = 8.9 Hz, 1H, H-5′), 7.79 (dd, J3,2 = 8.5 Hz, J3,4 = 4.1 Hz, 1H, H-3), 7.44 (t, J3′,(2′,4′) = 7.8 Hz, 1H, H-3′), 7.34 (d, J7,6 = 8.4 Hz, 1H, H-7), 7.27 (dd, J6′,5′ = 8.8, J6′,8′ = 2.5 Hz, 1H, H-6′); 13C NMR (600 MHz, C3D6O, δ, ppm): 157.3 (C), 157.2 (C), 149.9 (CH), 147.7 (C), 141.2 (C), 138.8 (C), 134.3 (CH), 133.3 (CH), 131.9 (CH), 130.8 (C), 130.6 (C), 128.3 (C), 124.1 (CH), 123.4 (CH), 119.9 (CH), 116.0 (CH), 113.2 (CH), 111.3 (CH), 105.3 (CH); MS (ESI, m/z): 316 [M++H].
N), 1504 (NN), 1380 (C–O); 1H-NMR (600 MHz, C3D6O, δ, ppm): 9.47 (dd, J2,3 = 8.5 Hz, J2,4 = 1.6 Hz, 1H, H-2), 8.98 (dd, J4,3 = 4.1 Hz, J4,2 = 1.6 Hz, 1H, H-4), 8.36 (d, J8′,6′ = 2.4 Hz, 1H, H-8′), 8.20 (d, J6,7 = 8.4 Hz, 1H, H-6), 7.99 (d, J4′,3′ = 4.2 Hz, 1H, H-4′), 7.98 (d, J2′,3′ = 3.8 Hz, 1H, H-2′), 7.93 (d, J5′,6′ = 8.9 Hz, 1H, H-5′), 7.79 (dd, J3,2 = 8.5 Hz, J3,4 = 4.1 Hz, 1H, H-3), 7.44 (t, J3′,(2′,4′) = 7.8 Hz, 1H, H-3′), 7.34 (d, J7,6 = 8.4 Hz, 1H, H-7), 7.27 (dd, J6′,5′ = 8.8, J6′,8′ = 2.5 Hz, 1H, H-6′); 13C NMR (600 MHz, C3D6O, δ, ppm): 157.3 (C), 157.2 (C), 149.9 (CH), 147.7 (C), 141.2 (C), 138.8 (C), 134.3 (CH), 133.3 (CH), 131.9 (CH), 130.8 (C), 130.6 (C), 128.3 (C), 124.1 (CH), 123.4 (CH), 119.9 (CH), 116.0 (CH), 113.2 (CH), 111.3 (CH), 105.3 (CH); MS (ESI, m/z): 316 [M++H].
5-[2-(2′-Hydroxy-5′-nitrophenyl)diazen-1-yl]quinolin-8-ol (S6).
Reddish-yellow solid; yield: 83%; FTIR (ATR, ν, cm−1): 3300, 2971, 1622 (C
 N), 1502 (NN), 1348 (C–O); 1H-NMR (500 MHz, C3D6O, δ, ppm): 9.24 (d, J2,3 = 7.9 Hz, 1H, H-2), 9.01 (s, 1H, H-4, partly split), 8.78 (d, J6′,4′ = 2.6 Hz, 1H, H-6′), 8.35 (d, J6,7 = 7.9 Hz, 1H, H-6), 8.32 (d, J4′,3′ = 6.9 Hz, 1H, H-4′), 7.88–7.83 (m, 1H, H-3), 7.34 (d, J7,6 = 7.9 Hz, 1H, H-7), 7.31 (d, J3′,4′ = 8.7 Hz, 1H, H-3′); 13C-NMR (800 MHz, C3D6O, δ, ppm): 156.7 (C), 150.2 (CH), 150.1 (C), 138.7 (C), 138.4 (C), 132.8 (C–H), 128.1 (C), 124.9 (CH), 119.5 (CH), 118.9 (CH), 111.6 (CH); MS (ESI, m/z): 311 [M++H].
N), 1502 (NN), 1348 (C–O); 1H-NMR (500 MHz, C3D6O, δ, ppm): 9.24 (d, J2,3 = 7.9 Hz, 1H, H-2), 9.01 (s, 1H, H-4, partly split), 8.78 (d, J6′,4′ = 2.6 Hz, 1H, H-6′), 8.35 (d, J6,7 = 7.9 Hz, 1H, H-6), 8.32 (d, J4′,3′ = 6.9 Hz, 1H, H-4′), 7.88–7.83 (m, 1H, H-3), 7.34 (d, J7,6 = 7.9 Hz, 1H, H-7), 7.31 (d, J3′,4′ = 8.7 Hz, 1H, H-3′); 13C-NMR (800 MHz, C3D6O, δ, ppm): 156.7 (C), 150.2 (CH), 150.1 (C), 138.7 (C), 138.4 (C), 132.8 (C–H), 128.1 (C), 124.9 (CH), 119.5 (CH), 118.9 (CH), 111.6 (CH); MS (ESI, m/z): 311 [M++H].
5-[2-(2′,3′-Dimethylphenyl)diazen-1-yl]quinolin-8-ol (S7).
Red solid; yield: 73%; FTIR (ATR, ν, cm−1): 3600, 2950, 1577 (C
 N), 1510 (NN), 1379 (C–O); 1H-NMR (500 MHz, DMSO-d6, δ, ppm): 9.26 (dd, J2,3 = 8.6 Hz, J2,4 = 1.7 Hz, 1H, H-2), 8.93 (dd, J4,3 = 4.2 Hz, J4,2 = 1.7 Hz, 1H, H-4), 7.95 (d, J6,7 = 8.5 Hz, 1H, H-6), 7.71 (dd, J3,2 = 8.6 Hz, J3,4 = 4.1 Hz, 1H, H-3), 7.59 (d, J6′,5′ = 8.0 Hz, 1H, H-6′), 7.29 (d, J4′,5′ = 7.3 Hz, 1H, H-4′), 7.22 (t, J5′,(4′,6′) = 7.8 Hz, 1H, H-5′), 7.21 (d, J7,6 = 7.8 Hz, 1H, H-7), 2.61 (s, 3H, CH3-7′), 2.33 (s, 3H, CH3-8′); 13C-NMR (600 MHz, DMSO-d6, δ, ppm): 158.1 (C), 150.7 (C), 148.8 (CH), 138.8 (C), 138.2 (C), 138.2 (C), 136.0 (C), 131.8 (CH), 131.6 (CH), 127.6 (C), 125.9 (CH), 123.1 (CH), 115.1 (CH), 113.1 (CH), 111.9 (CH), 19.6 (CH3), 13.0 (CH3); MS (ESI, m/z): 278 [M++H].
N), 1510 (NN), 1379 (C–O); 1H-NMR (500 MHz, DMSO-d6, δ, ppm): 9.26 (dd, J2,3 = 8.6 Hz, J2,4 = 1.7 Hz, 1H, H-2), 8.93 (dd, J4,3 = 4.2 Hz, J4,2 = 1.7 Hz, 1H, H-4), 7.95 (d, J6,7 = 8.5 Hz, 1H, H-6), 7.71 (dd, J3,2 = 8.6 Hz, J3,4 = 4.1 Hz, 1H, H-3), 7.59 (d, J6′,5′ = 8.0 Hz, 1H, H-6′), 7.29 (d, J4′,5′ = 7.3 Hz, 1H, H-4′), 7.22 (t, J5′,(4′,6′) = 7.8 Hz, 1H, H-5′), 7.21 (d, J7,6 = 7.8 Hz, 1H, H-7), 2.61 (s, 3H, CH3-7′), 2.33 (s, 3H, CH3-8′); 13C-NMR (600 MHz, DMSO-d6, δ, ppm): 158.1 (C), 150.7 (C), 148.8 (CH), 138.8 (C), 138.2 (C), 138.2 (C), 136.0 (C), 131.8 (CH), 131.6 (CH), 127.6 (C), 125.9 (CH), 123.1 (CH), 115.1 (CH), 113.1 (CH), 111.9 (CH), 19.6 (CH3), 13.0 (CH3); MS (ESI, m/z): 278 [M++H].
5-(Phenyldiazenyl)quinolin-8-ol (S8).
Yellow solid; yield: 76%; FTIR (ATR, ν, cm−1): 3433, 2930, 1621 (C
 N), 1496 (NN), 1404 (C–O); 1H NMR (500 MHz, DMSO-d6): δ 9.51 (d, J2,3 = 8.5 Hz, 1H, H-2), 9.06 (dd, J4,3 = 4.4 Hz, J4,2 = 1.6 Hz, 1H, H-4), 8.06 (d, J6,7 = 8.5 Hz, 1H, H-6), 8.01 (d, J2′,3′/6′,5′ = 7.0 Hz, 2H, H-2′/H-6′), 7.90 (dd, J3,2 = 8.6 Hz, J3,4 = 4.4 Hz, 1H, H-3), 7.61 (t, J3′,2′/5′,6′ = 8.2 Hz, J3′,4′/5′,4′ = 6.7 Hz, 2H, H-3′/H-5′), 7.55 (t, J4′,(3′,5′) = 7.5 Hz, 1H, H-4′), 7.40 (d, J7,6 = 8.6 Hz, 1H, H-7); MS (ESI, m/z): 250 [M++H].
N), 1496 (NN), 1404 (C–O); 1H NMR (500 MHz, DMSO-d6): δ 9.51 (d, J2,3 = 8.5 Hz, 1H, H-2), 9.06 (dd, J4,3 = 4.4 Hz, J4,2 = 1.6 Hz, 1H, H-4), 8.06 (d, J6,7 = 8.5 Hz, 1H, H-6), 8.01 (d, J2′,3′/6′,5′ = 7.0 Hz, 2H, H-2′/H-6′), 7.90 (dd, J3,2 = 8.6 Hz, J3,4 = 4.4 Hz, 1H, H-3), 7.61 (t, J3′,2′/5′,6′ = 8.2 Hz, J3′,4′/5′,4′ = 6.7 Hz, 2H, H-3′/H-5′), 7.55 (t, J4′,(3′,5′) = 7.5 Hz, 1H, H-4′), 7.40 (d, J7,6 = 8.6 Hz, 1H, H-7); MS (ESI, m/z): 250 [M++H].
2.3 Stock solution preparations for solvatochromic assessment and chemosensing studies
2.4 Solvatochromic behaviours of compounds S1–S8
An aliquot of 100 μL of each dye stock solution was pipetted into eight different vials. The volume was made up to 5 mL with dichloromethane, methanol, ethanol, ethyl acetate, dichloromethane, acetonitrile, dimethylformamide, and dimethyl sulfoxide. The resultant concentrations of solutions of compounds S1, S2, S3, S4, S5, S6, S7, and S8 were 26.8, 27.2, 30.8, 30.8, 31.8, 32.2, 36.2, and 40 μM, respectively. Baseline corrections of the UV-visible spectrometer were carried out with the aforementioned solvents (i.e., methanol, ethanol, ethyl acetate, dichloromethane, acetonitrile, DMF, and DMSO). Then, the UV-visible scans of the solutions were recorded at 200 to 700 nm on the Thermo Electron Corporation Nicolet Evolution 300 UV-visible spectrophotometer with Vision Pro software.2.5 Chemosensing screening
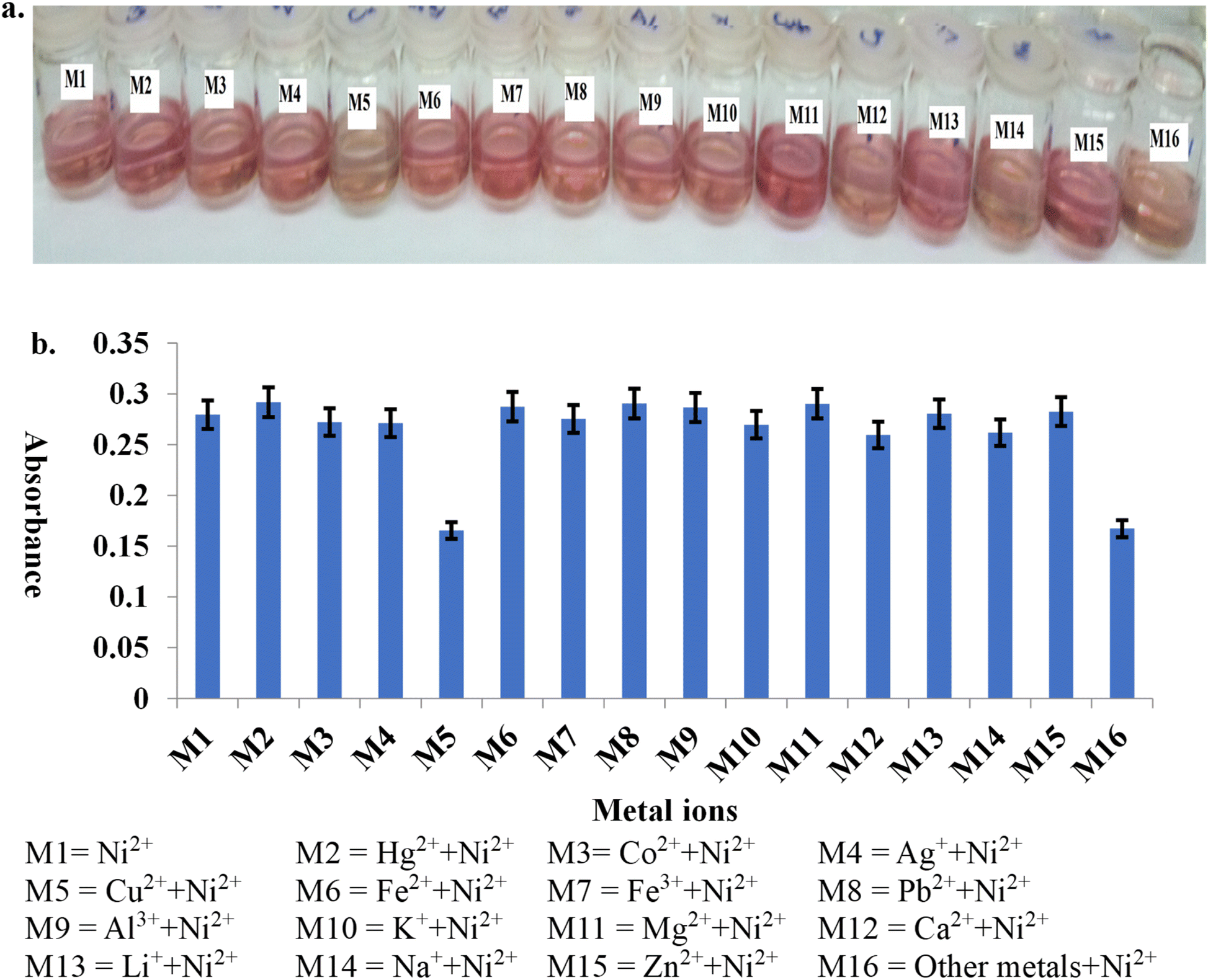
A volume of 500 μL of each metal ion solution (Cu2+, Pb2+, Zn2+, Ca2+, Hg2+, Li+, Mg2+, Na+, K+, Ag+, Co2+, Fe3+, Fe2+, Al3+, Ni2+) (concentration of all salts is 200 ppm) was added into fifteen vials, followed by 100 μL of phosphate buffer and 100 μL of each dye. The mixtures were swirled gently for 10 seconds, and the vials were allowed to stay for 2 minutes before they were made up to 5 mL with ethanol and water (80:20, v/v). Baseline corrections of the UV-visible spectrometer were done with a mixture of ethanol, water, and phosphate buffer. The absorption spectra of the mixtures were measured from 200 to 700 nm, and overlaid for each dye. An ideal chemosensor was selected, along with metal ions that gave different colouration from others. The working analytical wavelength was determined by inspection of the overlaid spectra for wavelength, where there was a maximum difference in absorptivity in terms of hyperchromism and bathochromism.
Diluting the solvent for the sensor–nickel(II) reaction. An aliquot of 500 μL Ni2+, 100 μL buffers (pH 8 for compound S2 and pH 4 for compounds S3, S4, and S6), and 100 μL dye solution were added to vials and swirled for 10 seconds. This was then diluted to 5 mL with ethanol. The UV spectrum of the resulting solution between 200 and 700 nm was acquired. The procedure was repeated with methanol, acetone, DMSO, and deionized water as dilution solvents.
A mixture of 500 μL Ni2+, 100 μL buffer, and 100 μL dye was made up to 5 mL with ethanol and water in the ratio of 20 to 80. The ethanol:water proportion was replaced with 40%, 50%, 60%, and 80% ethanol. The absorption scan was read at 200–700 nm after the baseline correction of the spectrophotometer.
Chelating time of the sensor–nickel(II) reaction. A volume of 500 μL of Ni2+ solution was added to an empty vial. This was followed by introduction of 100 μL of buffer, and then 100 μL of the dye stock solution. After gently stirring the mixture, the effect of different times allowed for chelation to occur between nickel and dyes at 30 °C, was tested at 0, 2, 5, 10, 15, 20, 25, and 30 minutes. Then, ethanol and water (80
:20) were added to the mixture to make a final volume of 5 mL. The absorbance was read at wavelengths of maximum absorption (λmax); 448, 513, 470, and 497 nm for compounds S2, S3, S4, and S6, respectively. This was repeated with replicate samples.
:water (80:20). The absorbance was read at λmax: 448, 513, 470, and 497 nm for compounds S2, S3, S4, and S6, respectively. A duplicate determination was made in each case.:20). The absorbance was read at a maximum wavelength of 448 nm. The determination was done in replicates. The entire procedure was repeated with working concentrations of compounds S6, S3, and S4, and their absorbance measurements were taken at analytical wavelengths of 497, 513, and 470 nm, respectively.:water (80:20). Determination was done in replicates, and the absorbance was read at 448 nm. Blank determinations with Ni2+ without other metal ions were carried out. The entire procedure was repeated with compounds S6, S3, and S4, and their absorbance measurements were taken at analytical wavelengths of 497, 513, and 470 nm, respectively.Linearity of the response and construction of the calibration curve. Into vials, each containing 100 μL of phosphate buffer at pH 8, volumes of 0, 10, 20, 25, 30, 35, 40, and 50 μL from the Ni2+ stock solution were added, respectively. Then, 150 μL of compound S2 solution was added to each vial. The mixtures were swirled for 10 seconds and allowed to stay for 2 minutes, then the ethanol
:water (80:20, v/v) solvent system was added to make a 5 mL mixture. Absorbance readings were recorded at 448 nm, and three replicate determinations were done. A calibration line equation was generated from the average absorbance values, and a regression coefficient was obtained from the curve. The procedure was repeated for compounds S3 (513 nm), S4 (470 nm), and S6 (497 nm).
Limit of detection (LOD), limit of quantitation (LOQ) and binding constant. A 150 μL volume of each dye solution (S2, S3, S4, and S6) was measured into vials containing 100 μL of phosphate buffer. The mixture was made up to 5 mL with ethanol
:water (80:20, v/v). The procedure was done six times for each dye, and absorbance readings were recorded at 448, 513, 470, and 497 nm for compounds S2, S3, S4, and S6, respectively. The LOD was determined as  and LOQ as
and LOQ as 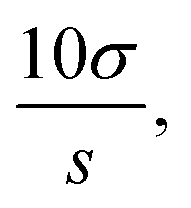 where σ was the standard deviation from six blank determinations and s was the slope.
where σ was the standard deviation from six blank determinations and s was the slope.The binding constant was estimated from Benesi–Hildebrand's plot.  was plotted against
was plotted against  A was the absorbance reading at different concentrations of Ni2+ solution, and Ao was the absorbance of the azo dye alone.
A was the absorbance reading at different concentrations of Ni2+ solution, and Ao was the absorbance of the azo dye alone.

Preparation of water sample for analysis. Water samples were collected from two sources: tap water from the Third World Center for Science and Technology, the International Center for Chemical and Biological Sciences (ICCBS), University of Karachi, Pakistan; and underground water from Ayub Goth (Suparco Road), Gulshan-e-Hijri, Karachi East, Pakistan. Samples were transferred into thoroughly cleaned bottles and filtered twice to remove particulate matter.
Recovery studies. Accuracy and precision were the analytical parameters determined through recovery studies. Into quadruplicate samples of tap and underground water at two concentration levels of 1.607 and 3.215 μM of spiked Ni2+ solution, 100 μL buffer pH 4 and 150 μL of compound S3 solution were added. The reaction mixture was swirled and set aside for 2 minutes. Thereafter, it was made up to 5 mL with an ethanol
:water solution (80:20, v/v). This was repeated four times for each volume and water sample on three different days. The absorbance readings were recorded at 513 nm with a mixture of phosphate buffer, ethanol, and water as blank solvent. A three day recovery study was carried out, and the accuracy and precision were determined from the regression equation. Recovery studies using compounds S2 and S4 were carried out by spiking water samples with 3.215 and 5.626 μM of Ni2+ solution.
ICP-MS analysis. The level of Ni2+ present in the blank tap and underground water was assessed. A calibration curve was constructed using a standard nickel solution. Nickel ion concentrations of 1.607, 3.2147, and 5.626 μM were added into empty vials. The vials were made up to 5 mL of tap and underground water. The concentration of Ni2+ was determined by extrapolating on the calibration line of the standard Ni2+ solution prepared. This was performed in duplicate.
Test of equivalence of chemosensor probes and ICP-MS. The accuracy and precision of the two methods were compared with the Student's t-test and F-test, respectively, at p < 0.05, using the same water samples from tap and underground sources.
:1 ratio.3. Results and discussion
3.1 Synthesis of compounds S1–S8
From the illustration in Scheme 1, it can be observed that two different sets of chemosensors are prepared: set-I comprises compounds S1 and S2, while set-II consists of compounds S3–S8. Products are obtained by utilizing a typical diazotization reaction, where corresponding diazonium salts are initially prepared in situ via treating substituted anilines or aminonaphthol (I) with sodium nitrite in acidic solution. Upon the addition of the coupling components (nitroxoline and 8-hydroquinoline), this preparation gave the corresponding diazo product. The formation of a colored product was indicated by colour changes of the diazonium solution from yellow upon adding the solution of coupling components. For set-II, the electrophilic substitution of diazonium ions (derived from substituted anilines) on the quinoline moiety occurred para to the hydroxyl group in compounds S3–S8. However, in compounds S1 and S2 (set-I), substitution took place at the carbon ortho to the phenolic group, with the para position being occupied by the nitro substituent (of nitroxoline). Nitroxoline (II) in turn was obtained by nitration reaction (electrophilic aromatic substitution) on 8-hydroquinoline (III). | ||
| Scheme 1 Synthetic routes of compounds S1–S8. | ||
3.2 Solvatochromism
N–).
| Compound | λmax (nm), εmax × 105 M−1 cm−1 (log of εmax) | ||||||
|---|---|---|---|---|---|---|---|
| MeOH | EtOAc | EtOH | DMSO | DMF | CH2Cl2 | CH3CN | |
| s – shoulder. | |||||||
| S1 | 420, 0.11(4.05) | 420, 0.10(3.99) | 426, 0.14(4.13) | 435, 0.12(4.07) | 437, 0.13(4.11) | 420, 0.08(3.92) | 431, 0.17(4.24) |
| 517, 0.09(3.97)s | 525, 0.09(3.94)s | ||||||
| S2 | 430, 0.09(3.97) | 438, 0.13(4.11) | 426, 0.13(4.13) | 445, 0.12(4.07) | 442, 0.14(4.16) | 437, 0.15(4.17) | 436, 0.17(4.22) |
| 502, 0.09(3.93) | 495, 0.08(3.92) | 555, 0.12(4.09)s | 554, 0.13(4.11)s | ||||
| S3 | 430, 0.09(3.95) | 419, 0.16(4.20) | 439, 0.10(4.01) | 654, 0.29(4.46) | 651, 0.34(4.54) | 421, 0.15(4.18) | 628, 0.29(4.46) |
| 576, 0.07(3.82)s | |||||||
| S4 | 400, 0.14(4.15) | 403, 0.23(4.37) | 409, 0.18(4.25) | 419, 0.20(4.30) | 413, 0.16(4.22) | 400, 0.19(4.27) | 400, 0.16(4.19) |
| 517, 0.13(4.10)s | 530, 0.19(4.27)s | 514, 0.08(3.90)s | |||||
| S5 | 420, 0.07(3.84) | 419, 0.13(4.10) | 424, 0.10(3.99) | 448, 0.13(4.12) | 430, 0.12(4.07) | 421, 0.13(4.10) | 416, 0.11(4.04) |
| S6 | 442, 0.10(3.97) | 413, 0.08(3.91) | 413, 0.10(4.01) | 413, 0.11(4.05) | 425, 0.09(3.93) | 413, 0.10(3.99) | 409, 0.12(4.09) |
| 525, 0.16(4.19)s | 512, 0.08(3.88)s | ||||||
| S7 | 380, 0.14(4.15) | 380, 0.17(4.23) | 383, 0.13(4.12) | 394, 0.14(4.14) | 390, 0.11(4.05) | 379, 0.16(4.19) | 376, 0.12(4.09) |
| 479, 0.08(3.89)s | |||||||
| S8 | 382, 0.07(3.86) | 380, 0.07(3.85) | 385, 0.07(3.84) | 394, 0.08(3.91) | 393, 0.08(3.89) | 380, 0.08(3.90) | 376, 0.08(3.88) |
| 497, 0.04(3.56)s | |||||||
 | ||
| Scheme 2 Tautomeric transformation of azo to hydrazone form. | ||
Further indication of tautomerism was evident from the 13C-NMR data of compounds S2 and S5, recorded in deuterated dimethyl sulfoxide (Fig. S6 and S15, given in ESI†). Compound S2 showed a peak at δ 174.6, while compound S5 exhibited a peak at δ 174.0 for C-8 in both compounds. However, all the other azo compounds produced peaks in the range of δ 157.2 to 162.0 when characterized in the same deuterated solvent. Thomas and Adegoke (2022)24 demonstrated that the carbon atom holding the hydroxyl group has a chemical shift of around δ 160.0 in azo form, while the shift occurs at δ 170.0 for ketonic carbon in the hydrazone form. The ketonic carbon peak was completely absent when compound S5 was analyzed in deuterated acetone (Fig. S14, ESI†). This indicates that the compound exists mainly in the azo form in acetone.
Typically, the hydrazone tautomer is associated with the low-field signal in the 1H-NMR spectrum.25 The peak at δ 17.3, with an integral of 0.95 in the 1H-NMR spectrum of compound S5 in deuterated DMSO (Fig. S15†), was attributed to the β-hydrogen of N–H of the hydrazone tautomer of compound S5. This lends credence to the fact that compound S5 exists predominantly as a hydrazone tautomer. On the other hand, the downfield peak was conspicuously absent in the 1H-NMR spectrum of compound S2; this might be due to the rapid equilibrium shift to the azo form.
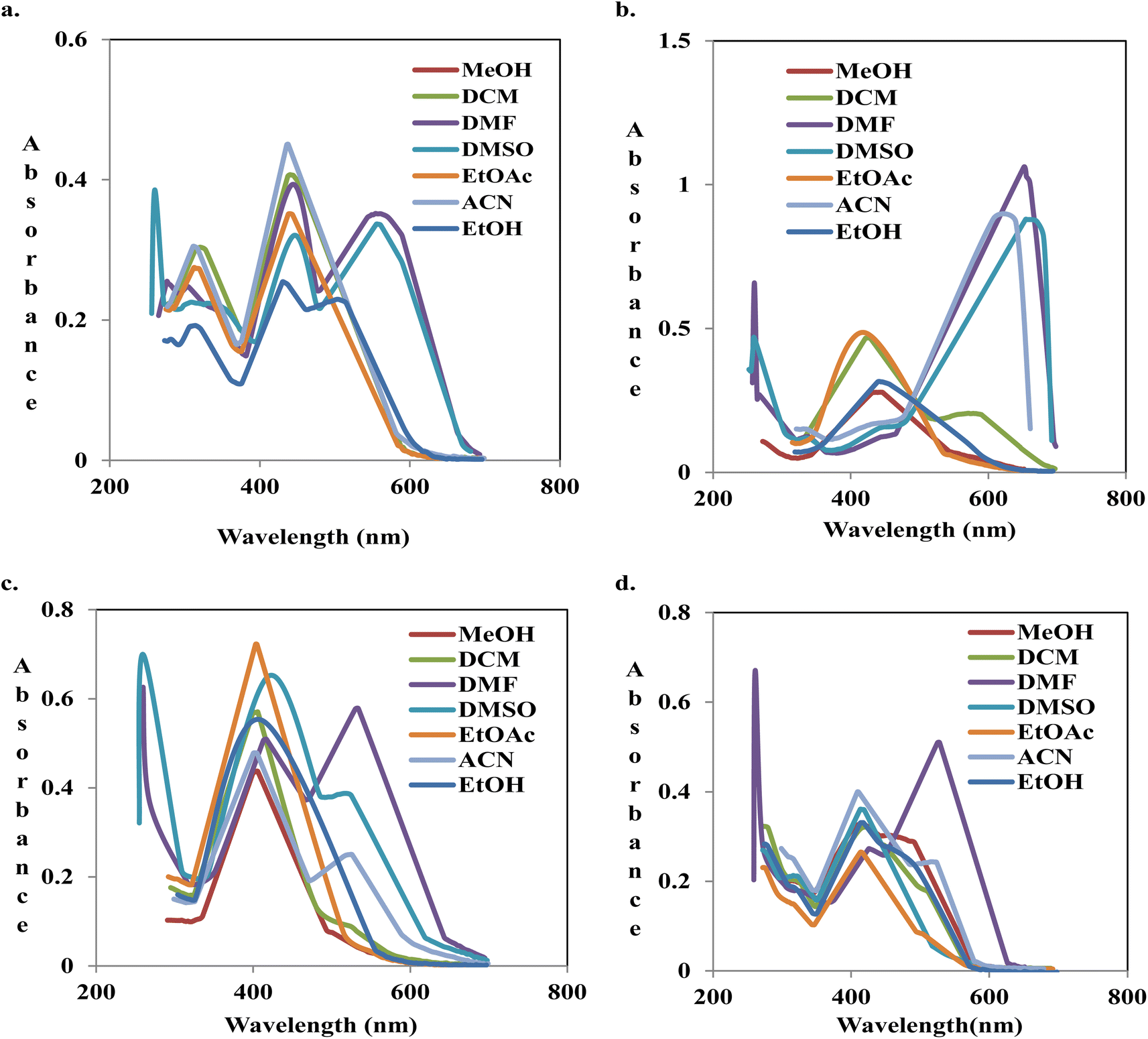 | ||
| Fig. 2 Overlaid spectra of compounds S2 (a), S3 (b), S4 (c) and S6 (d) in different solvents. | ||
| Compound | Colours in solvents | ||||||
|---|---|---|---|---|---|---|---|
| EtOAc | MeOH | EtOH | CH2Cl2 | DMSO | ACN | DMF | |
| S1 | Yellow | Yellow | Yellow | Yellow | Orange | Yellow | Orange |
| S2 | Orange | Pink | Orange | Orange | Purple | Yellow | Purple |
| S3 | Yellow | Yellow | Orange | Green | Blue | Blue | Blue |
| S4 | Yellow | Yellow | Yellow | Yellow | Orange | Pink | Deep pink |
| S5 | Yellow | Yellow | Yellow | Yellow | Orange | Pink | Pink |
| S6 | Yellow | Orange | Yellow | Yellow | Yellow | Orange | Pink |
| S7 | Yellow | Yellow | Yellow | Orange | Orange | Pink | Pink |
| S8 | Yellow | Yellow | Yellow | Yellow | Yellow | Yellow | Yellow |
Absorption of compound S8 was negligible in all the solvents in the visible region due to the absence of a substituent. However, there was a shoulder at 484 nm in DMF. Moving from ethyl acetate to hydrogen bond acceptor (HBA) solvents such as DMSO and DMF, there was a positive solvatochromism of 13 nm, but a blue shift (−4 nm) was observed in acetonitrile. This might be a result of weak interaction with the acetonitrile. The addition of weak activators, methyl groups, at position-2 and -3 in compound S7 did not reveal a noticeable difference in the spectral pattern when compared to compound S8. Meanwhile, the shoulder band in the visible region of DMF was more pronounced. The observed shoulder in the spectra of compounds S7 and S8 might be a result of enol formation.
The dielectric constant of solvents is in the following order: EtOH > CH2Cl2 > EtOAc. There was no difference in the absorption maxima of compound S6, but the absorptivity decreased steadily. Moving to a more polar solvent, MeOH, there was bathochromism (Δλ = +29 nm) and hyperchromicity. A non-hydrogen bond donor solvent, e.g., DMF, enhances the hydrazone formation, although compound S6 exists primarily in the azo form in the solvent. The absorption spectrum indicated Δλ = +83 nm, relative to what was acquirable in EtOH. The ortho-hydroxyl group of the benzenoid could be involved in intramolecular interaction with the α-nitrogen of the azo-linkage due to its proximity to the group (Fig. 3A).
 | ||
| Fig. 3 Intramolecular hydrogen bond formation in hydrazones of compounds (A) S6, (B) S4, and (C) S2. | ||
Hence, it is not likely to be free for hydrogen bond formation with non-hydroxylic solvents. The long-wavelength absorption observed in DMF was probably due to interaction via the β-proton in the hydrazone (Fig. 4A).
 | ||
| Fig. 4 Intermolecular hydrogen bond formation (in red line) between DMF (in green color) and the hydrazone moiety of compounds (A) S6, (B) S3, and (C) S2. | ||
In moving from methanol to DMSO and DMF, compound S5 indicated a red shift of +28 and +10 nm, respectively. DMSO, a solvent with a greater dielectric constant than DMF, interacted strongly via inter-charge transfer (ICT) in terms of hydrogen bonding with β-hydroxyl in the naphthalene moiety.
Compounds S3 and S4 are structural isomers; the two azo dyes differ in the position of –NO2 and –OCH3 group on the benzenoid nucleus. In EtOAc, compound S3 revealed an absorption band at 419 nm. Moving to CH2Cl2, a slight positive solvatochromism of +2 nm, with a shoulder peak at 576 nm, was observed. There was bathochromism of Δλ = +9 nm in methanol, a more polar solvent. A noticeable red shift of +209, +232, and +235 nm with marked hyperchromicity was indicated in ACN, DMF, and DMSO, respectively. With the spectral pattern, equilibrium was shifted towards hydrazone formation in these solvents. The hydrogen bonding with DMF and other non-hydroxylic solvents through hydrazone β-proton likely accounted for the observation (Fig. 4B). The pull effect of para-nitro to azo and the push phenomenon of the hydroxyl group of the quinoline might also be a contributing factor to the chromophoric elongation. There was only a slight difference in the absorption maxima on increasing polarity from EtOAc to MeOH.
On the contrary, in hydrogen bond acceptor (HBA) solvents, compound S4 indicated an absorption maximum at 530 nm in DMF and shoulder peaks at 514 and 507 nm in DMSO and ACN, respectively. Compound S4 has a NO2 group ortho to the NN linkage and OCH3 at the para-position. The proximity of NO2 and azo-linkage results in intramolecular hydrogen bonding to give a six-membered ring, thus stabilizing the hydrazone form, as shown in Fig. 3B. A similar trend was observed by Rashidnejad and co-workers when they compared the solvatochromism of arylazo-5HQ with 8HQ derivatives.26 Interaction with hydrogen bond donor solvents (MeOH, EtOH, and CH2Cl2) might result in dipole–dipole interaction, unlike in compound S3.
There was a slight difference in the spectral behaviours of compound S1 in CH2Cl2, EtOAc, MeOH, and EtOH. There was a bathochromic shift moving to DMF (+15 nm), ACN (+11 nm), and DMSO (+17 nm). As the polarity increased in the order of EtOAc > CH2Cl2 > MeOH > DMF > DMSO, there was a proportional shift of maximum absorption of compound S2 to longer wavelengths. Phenolic hydroxyl, in compound S2, was not available for bonding with HBA due to intramolecular hydrogen bonding (Fig. 3C). In each of the solvents, the wavelength of the maximum absorption in compound S2 was more than that in compound S1. This might be connected to the pull–pull effect of the two NO2 moieties on the structure of compound S2.
It is noteworthy that the presence of a strong electron-withdrawing substituent like NO2 stabilizes the hydrazone tautomer by reducing the electron density of the aromatic ring structure. Compounds S2, S3, S4, and S6 indicated prominent tautomerism in DMSO and DMF (Fig. 2). All the 8-hydroxyquinoline azo dyes except compounds S7 and S8 revealed a significant decline in the absorption intensity of almost nine-fold in CH2Cl2 in the visible region. This could have been due to the strong solvent–solute interaction of dyes (S7 and S8) with CH2Cl2 via hydrogen bonding formation with –NN– (Fig. 5).
 | ||
| Fig. 5 Hydrogen bond formation (in red line) between CH2Cl2 (in green color) and compounds (A) S7 and (B) S8. | ||
3.3 Chemosensor screening
The addition of nickel to the pink solution of compound S2 and the yellow solutions of compounds S3, S4, and S6 gave colour changes to yellowish orange, deep violet, and orange, respectively. The overlaid absorption spectra of the resulting nickel–dye solutions of compounds S3 (Fig. 6), S2, S4, and S6 (Fig. S28, S29 and S31 ESI†) gave red shifts of +8, +67, +63 and +85 nm, respectively, relative to other cations: Cu2+, Pb2+, Zn2+, Ca2+, Hg2+, Li+, Mg2+, Na+, K+, Ag+, Co2+, Fe3+, Fe2+, and Al3+. In the presence of compounds S1, S5 and S7, none of the metal ions, including nickel ions, showed a peak where the difference in absorptivity was maximal (Fig. S27, S30 and S32 ESI†). For further analysis of nickel using compounds S2, S3, S4, and S6, analytical wavelengths of 448, 513, 470, and 497 nm were selected, respectively, while further work could not be done with the other azo dyes. | ||
| Fig. 6 Colour changes (a) and overlaid absorption spectra of nickel and other cations (b) in the presence of compound S3 in ethanol:water (80:20, v/v). | ||
From the chemosensor screening using S1–S7, compounds S1, S5, and S7 were not suitable as chemosensors for cation detection and quantitation. On the other hand, compounds S2, S3, S4, and S6 could serve as chemosensors for nickel ions. Complex solutions of the dyes and nickel gave different colours and distinct spectral patterns compared with other interfering metal ions. Enhanced conjugation due to Ni2+ coordination to 8-hydroxyquinoline-based azo dyes might be responsible for the observed colour change and the emergence of an absorption peak at a long wavelength. In previous research, the Goswami group (2014)20 reported that the sensor–nickel complex showed π-electron delocalization, thus reducing the energy gap between LUMO and HOMO. It is noteworthy that the presence of a nitro group on the benzenoid residue of the diazonium component, which was absent from compounds S1, S5, and S7, was a common characteristic of the chemical structures of compounds S2, S3, S4, and S6. The nitro group could have promoted conjugation in the chemical structures; therefore, this structural feature should be noted when designing sensors for colorimetric measurement of nickel.
Diluting the solvent for the sensor–nickel(II) reaction. Absorption spectra of the nickel complex of compounds S2–S4, and S6 in acetone, DMSO, ethanol, methanol, and water are shown in Fig. S33–S37 (ESI).† High absorption intensity was shown by all chemosensors in acetone and low absorptivity in water. This might be due to the stronger interaction of the complex with acetone than in water. Although acetone gave the highest absorptivity, ethanol was used for the chemosensing experiment in this study. Ethanol is cost-effective, readily available, and environmentally friendly.28–32
Sharma and co-workers (2016)27 reported limited ionization of metallic salts in organic solvents. This might be the reason for the use of organic solvent–water binary mixtures as sensing milieus by the majority of methods.16,17,33–36 Similarly, a binary chemosensing medium of organic solvent and water, comprising ethanol and water, was utilized in this work to solvate the cations. Ethanol/water (20%, 40%, 50%, 60%, and 80%) was added to replace the solvents, and UV-visible scans were recorded to select the best ethanol/water mixture.
As shown in Fig. 7, the absorption decreased as the water content rose. The increased polarity limited the solubility of the dye–nickel complex; and consequently, weak interaction with water. It was observed that complexes aggregated and precipitated on standing. Therefore, for chemosensing measurement, the proportion of 80% ethanol and 20% water that gave the highest absorbance intensity was selected.
 | ||
| Fig. 7 Absorption spectra of compounds (a) S2, (b) S3 in the presence of nickel in different proportions of ethanol:water (80:20, v/v). | ||
Chelating time. The difference in the reaction time intervals on chelation between nickel and azo dyes was carried out at an incubation room temperature of 30 °C. Generally, as shown in Fig. 8, it was observed that the absorbance reached the maximum value at 2 minutes and declined steadily afterwards. This indicated that the chelating reaction was fast and completed after swirling the sensor/Ni2+ mixture for 10 seconds. Two minutes was used as the optimal chelating time.
 | ||
| Fig. 8 Absorbance of compounds (a) S6 (0.0161 mM) and (b) S2 (0.0136 mM) in the presence of nickel ion at different time intervals in ethanol:water (80:20, v/v). | ||
 | ||
| Fig. 9 Absorbance of compounds (a) S3 (0.0154 mM) and (b) S4 (0.0154 mM) in the presence of nickel ion at different pH values in ethanol:water (80:20, v/v). | ||
:1 ratio of the sensor to Ni2+.
 | ||
| Fig. 10 Plot of the absorbance readings against the mole ratio of (a) S4 and (b) S6 in ethanol:water (80:20, v/v). | ||
With 3 mol of the ligands, oxygen and nitrogen in each mol of the sensors, electron pairs could be donated into the empty orbitals of nickel ions. Nickel has six coordination numbers in this case, and it is expected to be octahedral in shape. Fig. 11 shows the proposed product of sensor S4 with nickel.
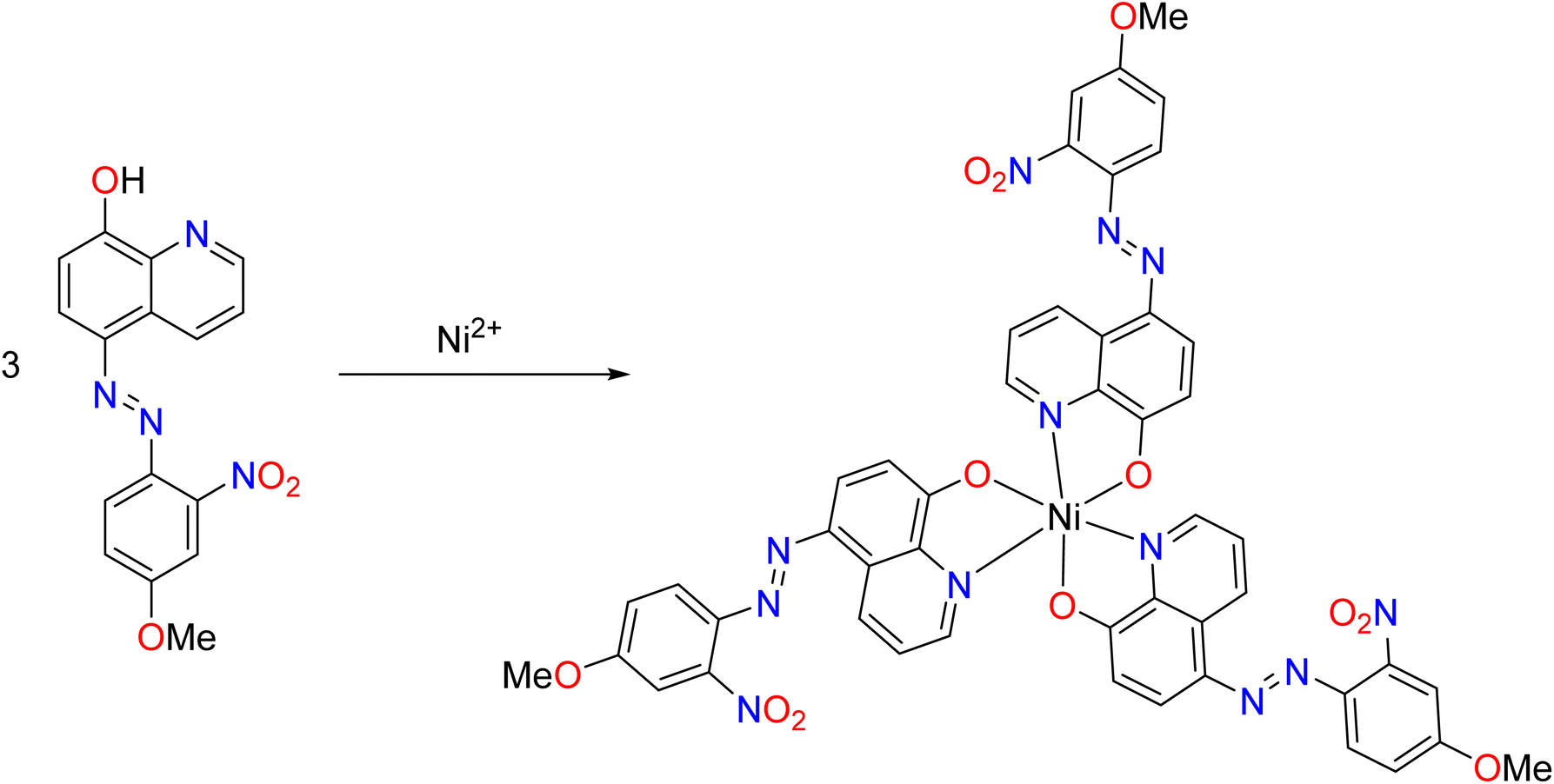 | ||
| Fig. 11 Proposed reaction of the sensor with nickel ions. | ||
| Parameter | Sensors | |||
|---|---|---|---|---|
| S2 | S3 | S4 | S6 | |
| a y = mx + c, where y is the absorbance for concentration × μM.b Three replicate determinations. | ||||
| Analytical wavelength (λmax, nm) | 448 | 513 | 470 | 497 |
| Range (μM) | 1.35–5.40 | 1.54–4.63 | 1.54–6.17 | 1.61–6.45 |
| Limit of detection (μM) | 0.014 | 0.0376 | 0.0117 | 0.0116 |
| Limit of quantitation (μM) | 0.0459 | 0.1252 | 0.0389 | 0.0388 |
| Sandell's sensitivity (nM per 0.001 absorbance unit) | 45.30 | 6.18 | 12.41 | 12.26 |
|
||||
| Regression equationa | ||||
| Slopeb, m | 0.0089 | 0.0652 | 0.0325 | 0.0329 |
| 95% confidence interval of slope | 0.0012 | 0.0068 | 0.0080 | 0.0070 |
| Interceptb, c | 0.3484 | 0.2332 | 0.1326 | 0.1011 |
| 95% confidence interval of slope | 0.0206 | 0.0087 | 0.0100 | 0.0012 |
| Correlation coefficient | 0.9904 | 0.9959 | 0.9971 | 0.9953 |
Furthermore, the LODs of the new sensors were far less than the limit of 1.2 μM set for nickel ion detection by the United States Environmental Protection Agency.40 This showed the high sensitivity of the sensor method.
 against
against  gave 4.181 × 104, 2.816 × 103, 1.151 × 104 and 1.09 × 103 M−1, respectively. The k values follow the trend S6 < S3 < S4 < S2. The highest value of K for compound S2 is connected to the electron delocalization by the two nitro groups on the sensor. The binding constants were in the order of 103 to 104 M−1. These values are in agreement with previously reported methods.
gave 4.181 × 104, 2.816 × 103, 1.151 × 104 and 1.09 × 103 M−1, respectively. The k values follow the trend S6 < S3 < S4 < S2. The highest value of K for compound S2 is connected to the electron delocalization by the two nitro groups on the sensor. The binding constants were in the order of 103 to 104 M−1. These values are in agreement with previously reported methods.The closeness of the absorbance values using compound S2 revealed that nickel could be determined in the presence of other cations. The visual inspection of colour changes alluding to this property is shown in Fig. S43 (ESI).† On the contrary, Cu2+ ions interfered significantly with the determination of Ni2+ using sensors S6 and S3, while a slight interference occurred for compound S4. Also, the colour changes affirmed this finding, as illustrated in Fig. 12, S44 and S45.† A similar interference in nickel determination in the presence of copper has been reported previously in the literature.15,33,41,42 A rhodamine-based chemosensor developed by Abebe and co-workers (2011)43 could only determine nickel in the absence of cobalt.
 | ||
| Fig. 12 Colours of sensor S3 in the presence of Ni2+ and other metal ions (a), and the absorbance reading of mixtures at 513 nm (b). | ||
Table 7 shows the t-test and F-test values for sensors (S2, S3, S4, and S6), respectively. The t-test for the tap water sample ranged from 0.093 to 0.277, while the underground sample ranged from 0.051 to 0.088. The results of the t-test from both samples showed that there was no significant difference in accuracy, with P greater than 0.05. Therefore, the sensor and ICP-MS methods were equivalent in terms of the mean.
| Water source | T-test | F-test |
|---|---|---|
| S2 method | ||
| Tap | 0.277 | 0.657 |
| Underground | 0.071 | 0.543 |
|
||
| S3 method | ||
| Tap | 0.246 | 0.139 |
| Underground | 0.088 | 0.941 |
|
||
| S4 method | ||
| Tap | 0.116 | 0.178 |
| Underground | 0.051 | 0.158 |
|
||
| S6 method | ||
| Tap | 0.093 | 0.568 |
| Underground | 0.060 | 0.855 |
The F-test values for tap and underground water samples were 0.139–0.657 and 0.158–0.941 respectively. In both samples, these values were greater than 0.05. This indicated that there was no significant difference in the precision of the methods. Hence, they were equivalent in terms of variance.
N peak in S2 + Ni2+ appeared at 1594 cm−1, while in S2, the peak was at 1610 cm−1. The C–O vibrational frequencies at 1384 cm−1 and 1402 cm−1 appeared for S2 and complex, respectively (Fig. 13). Other sensors interacted with nickel ions in a similar version. It could be deduced that the sensors interacted with Ni2+ through metal-to-ligand charge transfer via the hydroxyl and nitrogen groups of the hydroxyquinoline.
 | ||
| Fig. 13 Superimposed FTIR spectra of (A) sensor S2 on (B) sensor S2 + Ni2+. | ||
 | ||
| Fig. 14 Colours of the 1 cm × 5 cm filter paper (a) immersed in S3 solution, (b) immersed in Ni2+ solution, and (c) air-dried paper coated with S3 in Ni2+ solution in (b). | ||
(2) The Li group (2012)46 and Wang and co-workers (2012)47 utilized coumarin as a structural motif to sense nickel, while the Ghosh group (2006)48 and Maisonneuve and co-workers (2008)49 reported the use of dipyrrin and benzothiazole, respectively. These structural motifs were synthesized through multiple steps and lengthy procedures. However, the azo dyes employed in this research were synthesized by two-step reactions: diazotization and diazocoupling.
(3) The 8-hydroxyquinoline-based azo dyes are simple and rapid sensors, with 0–2 min for the determination of nickel ions. Analytical time was gained compared to the methods of Lv and Luo (2012)39 and Shrivas group (2017),4 who reported incubation times of 30 and 5 min for the chelation of sensors and nickel ions, respectively.
(4) The use of a binary mixture of water and organic solvent as a chemosensing medium was common to the 8-hydroxyquinoline-based azo dyes and all the other chemosensor techniques. However, the Annaraj group (2016)50 and Li and co-workers (2009)42 reported the use of water as a medium for the chemosensing determination of nickel. Li et al.'s method used a glutathione-stabilized silver nanoparticle, and Annaraj et al.'s sensor has carboxyl as a water-solubilizing group. In some other methods, monosolvents were used; Chowdhury and co-workers (2018),51 Xiang et al. (2014),21 and the Gupta group (2016)37 utilized acetonitrile, tetrahydrofuran, and dimethylsulfoxide, respectively. These solvents are not environmentally friendly, and they are more expensive relative to the ethanol used in this study.
4. Conclusion
Eight 8-hydroxyquinoline azo sensors were synthesized and characterized, of which compounds S2, S3, S4, and S6 were suitable chemosensors for nickel. The optimized conditions were ethanol/water (80:20, v/v); pH 4 for compounds S3, S4, and S6, and pH 8 for compound S2; a reaction time of 2 minutes; and a stoichiometric ratio of 3:1. Infrared spectral analysis confirmed that sensors interacted with Ni2+ through quinoline nitrogen and hydroxyl moieties. Copper(II) ion interfered with Ni2+ determination using the sensors, except with compound S2. The sensor-based methods were of equivalent accuracy and precision to the ICP-MS method. The presence of a nitro group on the benzenoid residue of the diazonium component conferred selectivity on the sensors for nickel ion detection. The crystalline structure of the nickel complex of the azo dyes could not be obtained for X-ray crystallography. Furthermore, the detection and determination of nickel using the sensors could be limited by the high water content, as the complexes formed tend to aggregate and separate out on standing. Despite this, a simple, highly sensitive, and low-cost method of nickel analysis in water samples was established. The use of azo-based 8-hydroxyquinoline herein is the first described method for nickel ion detection and quantitation.
Data availability
The data used to support the findings of the study are available in the article.Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors are grateful to the Third World Academy of Sciences (TWAS) for the “Award of ICCBS-TWAS Sandwich Postgraduate Fellowship” (FR number: 3240316612) to S. O. O., and the Third World Center for Science and Technology (TWC), H. E. J. Research Institute of Chemistry, International Center for Chemical and Biological Sciences (ICCBS), Karachi, Pakistan.References
- B. Kaur, N. Kaur and S. Kumar, Coord. Chem. Rev., 2018, 358, 13–69 CrossRef CAS.
- B. Zambelli, F. Musiani, S. Benini and S. Ciurli, Acc. Chem. Res., 2011, 44, 520–553 CrossRef CAS PubMed.
- R. J. Maier, Biochem. Soc. Trans., 2005, 33, 83–85 CrossRef CAS PubMed.
- K. Shrivas, P. Maji and K. Dewangan, Spectrochim. Acta, Part A, 2017, 173, 630–636 CrossRef CAS PubMed.
- S. C. Dodani, Q. He and C. J. Chang, J. Am. Chem. Soc., 2009, 131(50), 18020–18021 CrossRef CAS PubMed.
- S. C. Wilschefski and M. R. Baxter, Clin. Biochem. Rev., 2019, 40(3), 115–133 Search PubMed.
- D. Maity and T. Govindaraju, Inorg. Chem., 2011, 50, 11282–11284 CrossRef CAS PubMed.
- F. Sancenon, R. Martinez-Manez, M. A. Miranda, M. J. Segui and J. Soto, Angew. Chem., Int. Ed., 2003, 42(6), 647–650 CrossRef CAS PubMed.
- L. Zhao, D. Suib and Y. Wang, RSC Adv., 2015, 5(21), 16611–16617, 10.1039/C5RA00696A.
- Y. Cheng, M. Zhang, H. Yang, F. Li, T. Yi and C. Huang, Dyes Pigm., 2008, 76(3), 775–783 CrossRef CAS.
- O. Arslan, B. Aydıner, E. Yalçın, B. Babür, N. Seferoğlu and Z. Seferoğlu, J. Mol. Struct., 2017, 1149, 499–509 CrossRef CAS.
- Y.-F. Cheng, Z.-Q. Liu, M. Shi, Q. Zhao, F.-Y. Li, T. Yi and C.-H. Huang, Chin. J. Chem., 2007, 25(5), 616–622 CrossRef CAS.
- M. T. Shah, A. Balouch and E. Alveroglu, J. Mater. Chem., 2018, 6, 1105–1115 CAS.
- L. Lin, S. Hu, Y. Yan, D. Wang, L. Fan, Y. Hu and G. Yin, Res. Chem. Intermed., 2017, 43, 283–295 CrossRef CAS.
- I. Zhang, Y. Wang, C. Wan, Z. Xing, W. W. Li, M. Li and S. Zhang, RSC Adv., 2015, 5, 66416–66419 RSC.
- A. Ashraf, M. Islam, M. Khalid, A. P. Davis, M. T. Ahsan, M. Yaqub, A. Syed, M. Abdallah, A. M. Elgorban, A. H. Bahkali and Z. Shafiq, Sci. Rep., 2021, 11(1), 1–13 CrossRef PubMed.
- X. Liu, Q. Lin, T. Wei and Y. Zhang, New J. Chem., 2014, 38(4), 1418–1423 RSC.
- G. Yang, X. Meng, S. Fang, L. Wang, Z. Wang, F. Wang, H. Duan and A. Hao, New J. Chem., 2018, 42, 14630–14641 RSC.
- G. V. Kumar, M. P. Kesavan, M. Sankarganesh, K. Sakthipandi, J. Rajesh and G. Sivaraman, New J. Chem., 2018, 42, 2865–2873 RSC.
- S. Goswami, S. Chakraborty, A. K. Das, A. Manna, A. Bhattacharyya, C. K. Quah and H. K. Fun, RSC Adv., 2014, 4, 20922–20926 RSC.
- G. Xiang, L. Wang, W. Cui, X. An, L. Zhou, L. Li and D. Cao, Sens. Actuators, 2014, 196, 495–503 CrossRef CAS.
- V. G. Voronin, I. D. Petrova, A. N. Leksin and B. V. Shemeryankin, Pharm. Chem. J., 1976, 10(9), 1215–1217 CrossRef.
- J. S. Renny, L. L. Tomasevich, E. H. Tallmadge and D. B. Collum, Angew. Chem., Int. Ed., 2013, 52(46), 11998–12013 CrossRef CAS PubMed.
- O. E. Thomas and O. A. Adegoke, J. Taibah Univ. Sci., 2022, 16(1), 451–462 CrossRef.
- J. Cai, Z. Li, Y. Qiu, Z. OuYang, W. Lin, L. Yang, W. Feng, X. Yu and W. Dong, New J. Chem., 2016, 40(11), 9370–9379 RSC.
- H. Rashidnejad, M. Ramezanitaghartape, N. Pesyan, P. J. Mahon, M. M. Raposo, P. J. Coelho, A. Lup and A. Soltani, J. Mol. Struct., 2021, 1223, 129323 CrossRef CAS.
- H. Sharma, N. Kaur, A. Singh, A. Kuwar and N. Singh, J. Mater. Chem. C, 2016, 4, 5154–5194 RSC.
- D. R. Joshi and N. Adhikari, J. Pharm. Res. Int., 2019, 28(3), 1–18 Search PubMed.
- S. Kilo, T. Goen and H. Drexier, Int. Arch. Occup. Environ. Health, 2016, 89(8), 1309–2320 CrossRef CAS PubMed.
- J. V. Ashurst and T. M. Nappe, Methanol Toxicity, Treasure Island (FL): StatPearls, 2023 Search PubMed.
- G. A. Burdock and I. G. Carabin, Toxicol. Lett., 2004, 150(1), 3–18 CrossRef CAS PubMed.
- Food and Drug Administration, 2018 Code of Federal Regulation, Title 21–Food and Drugs, retrieved May 31, 2023, from, https://www.fda.gov Search PubMed.
- A. K. Manna, J. Mondal, K. Rout and G. K. Patra, Sens. Actuators, 2018, 275, 350–358 CrossRef CAS.
- G. Dhaka, N. Kaur and J. Singh, Supramol. Chem., 2015, 27(10), 654–660 CrossRef CAS.
- M. R. Ganjali, M. Hosseini, M. Motalebi, M. Sedaghat, F. Mizani, F. Faridbod and P. Norouzi, Spectrochim. Acta, Part A, 2015, 140, 283–287 CrossRef CAS PubMed.
- M. Sahu, A. K. Manna, S. Chowdhury and G. K. Patra, RSC Adv., 2020, 10, 44860–44875 RSC.
- V. K. Gupta, A. K. Singha, L. K. Kumawata and N. Mergu, Sens. Actuators, 2016, 222, 468–482 CrossRef CAS.
- Y. Lei, H. Li, W. Gao, M. Liu, J. Chen, J. Ding, X. Huang and H. Wu, J. Mater. Chem., 2014, 2, 7402–7410 RSC.
- X.-L. Lv and S.-Z. Luo, Anal. Bioanal. Chem., 2012, 402, 2999–3002 CrossRef CAS PubMed.
- M. Dhanushkodi, G. G. Kumar, B. K. Balachandar, S. Sarveswari, S. Gandhi and J. Rajesh, Dyes Pigm., 2020, 173, 1–29 CrossRef.
- R. Joseph, B. Ramanujam, H. Pal and C. P. Rao, Tetrahedron Lett., 2008, 49, 6257–6261 CrossRef CAS.
- H. Y. Li, S. Gao and Z. Xi, Inorg. Chem. Commun., 2009, 12(4), 300–303 CrossRef CAS.
- F. Abebe, C. Eribal, G. Ramakrishna and E. Sinn, Tetrahedron Lett., 2011, 52, 5554–5558 CrossRef CAS.
- J. H. Kang, Y. S. Lee, H. M. Ahn and C. Kim, Sens. Actuators, 2017, 242, 25–34 CrossRef CAS.
- S. Babaee, S. G. Pakdehi and A. S. Nabavi, J. New Dev. Chem., 2016, 1, 58–69 Search PubMed.
- H. Li, L. Cai and Z. Chen, Adv. Chem. Sens., 2012, 1, 121–150 Search PubMed.
- L. Wang, D. Ye and D. Cao, Spectrochim. Acta, Part A, 2012, 90, 40–44 CrossRef PubMed.
- S. K. Ghosh and T. Pal, Chem. Rev., 2007, 107(11), 4797–4862 CrossRef CAS PubMed.
- S. Maisonneuve, Q. Fang and J. Xie, Tetrahedron, 2008, 64(37), 8716–8720 CrossRef CAS.
- B. Annaraj, L. Mitu and M. A. Neelakantan, J. Mol. Struct., 2016, 1104, 1–6 CrossRef CAS.
- B. Chowdhury, M. Karar, S. Paul, M. Joshi, A. Choudhury and B. Biswas, Sens. Actuators, 2018, 276, 560–566 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra08510h |
| This journal is © The Royal Society of Chemistry 2025 |