
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSymmetrical di-substituted phenylamino-s-triazine derivatives as anticancer agents: in vitro and in silico approach†
Em Canh Pham ab,
Bich-Ngoc Thi Lea,
Anh Minh Ngoa,
Long Binh Vongc and
Tuyen Ngoc Truong*a
ab,
Bich-Ngoc Thi Lea,
Anh Minh Ngoa,
Long Binh Vongc and
Tuyen Ngoc Truong*a
aFaculty of Pharmacy, University of Medicine and Pharmacy at Ho Chi Minh City, 700000 Ho Chi Minh City, Vietnam. E-mail: truongtuyen@ump.edu.vn
bFaculty of Pharmacy, Hong Bang International University, 700000 Ho Chi Minh City, Vietnam
cSchool of Biomedical Engineering, International University, Vietnam National University Ho Chi Minh City, Ho Chi Minh City 700000, Vietnam
First published on 1st April 2025
Abstract
A series of symmetrical tri-substituted s-triazine derivatives were designed and synthesized by two different methods (reflux and microwave-assisted methods). The structures of compounds were determined by infrared (IR), nuclear magnetic resonance (1H NMR and 13C NMR), and mass spectrometry. The yield of the microwave-assisted method (91–98%) was significantly higher (about 10%) than that of the reflux method (80–88%) meanwhile the reaction time was significantly shorter (15–30 min). Compound 3b showed good cytotoxic activity against the MCF7 (human breast cancer) cell line with an IC50 value of 6.19 μM. Compounds 3a and 2e showed strong cytotoxic activity against the C26 (colon carcinoma) cell line with IC50 values of 1.21 and 8.28 μM, respectively. Compound 3e showed good cytotoxic activity against both MCF7 and C26 cell lines with IC50 values of 13.74, and 14.66 μM respectively. In particular, compound 2d exhibited the best potent cytotoxic activity among the synthesized compounds against both MCF7 and C26 cell lines with IC50 values of 6.54 and 0.38 μM, respectively. Moreover, compounds 2e, 3a, and 3e showed higher selectivity on cancer cell lines and lower toxicity on BAEC (bovine aorta endothelial) normal cells compared to compounds 2d, 3a, paclitaxel, and doxorubicin. In silico studies revealed five potent compounds with good physicochemical and ADMET profiles and potent interactions with key anticancer targets (EGFR, DHFR, VEGFR2, CDK2, mTOR, and PI3K) compared to reference drugs. This work paved the way for the synthesis of more potent compounds based on the phenylamino-s-triazine scaffold and the exploration of their diverse and potential biological activities as well as their mechanisms of action.
1. Introduction
Cancer is a dangerous disease that greatly affects human health because it is the second leading cause of death in the world.1 According to Globocan 2022 statistics, there are about 9.7 million deaths from cancer worldwide annually, second only to deaths from cardiovascular disease, and about 20 million new cases of cancer.2 Breast cancer and colorectal cancer are two common and aggressive cancers. Breast cancer is the most common malignant cancer in women, while colorectal cancer is the third most common cancer worldwide, accounting for about 10% of all cancer cases.3,4 Furthermore, the incidence and mortality from breast and colon cancers are projected to increase rapidly at a rate of millions of cases per year in the future.3,4 This is a huge socio-economic burden for the world, especially low- and middle-income countries. Therefore, new cancer drug development for breast and colorectal cancer treatment needs more attention with small molecule orientation to reduce costs and increase treatment efficacy.Five-membered and six-membered heterocyclic nuclei exhibit a wide range of potential pharmacological activities.5–9 The s-triazine (1,3,5-triazine) nucleus is an important pharmacophore in the development of new drugs with diverse biological activities such as antiviral,10 antibacterial,11,12 antifungal,11,12 anti-inflammatory,13,14 antimalarial,15,16 and anticancer.17–24 However, the anticancer activity of s-triazine derivatives has received the most research attention because these derivatives have shown great potential in the treatment of many different types of cancer including leukemia, breast cancer, colon cancer, cervical cancer, etc.25–27 In particular, many cancer drugs containing s-triazine nucleus have been developed and have made great contributions to cancer treatment worldwide such as Altretamine (anti-ovarian cancer),28 Tretamine (antineoplastic),29 Enasidenib (antileukemia),30 Gedatolisib (treatment of neoplasm, ovary cancer, breast cancer, advanced cancer, and endometrial cancer),31 Decitabine and Azacitidine (treat myelodysplastic syndrome and acute myeloid leukemia),32,33 and Bimiralisib (anti-breast cancer)26 (Fig. 1).
 | ||
| Fig. 1 Some marketed anticancer drugs contain s-triazine. | ||
Potential s-triazine derivatives have demonstrated mechanisms of action for anticancer activity by inhibiting various enzymes, such as dihydrofolate reductase (DHFR),34,35 vascular endothelial growth factor receptor 2 (VEGFR2),36 phosphoinositide 3-kinase (PI3K),37 cyclin-dependent kinase (CDK),38 epidermal growth factor receptor (EGFR),39,40 mammalian target of rapamycin (mTOR),41 DNA topoisomerase,42 and carbonic anhydrase.43 Many drug discovery studies have also screened through these targets to find s-triazine derivatives with potent anticancer activity.25–27
There are various methods for the synthesis of s-triazine derivatives, but the direct synthesis from simple starting materials such as cyanuric chloride (2,4,6-trichlorotriazine) is a popular and simple approach with cost-effective, and easily applicable at an industrial scale.44 The three chlorine substituents of cyanuric chloride can be sequentially substituted at different temperatures by different N-nucleophiles (saturated and aromatic amines) to construct new drug candidates with optimum physicochemical properties and potential pharmacological activities.45
1.1 Rational drug design
Tretamine, Altretamine, Dioxadet, Bimiralisib, and Gedatolisib drugs are symmetrical s-triazine derivatives with potent antitumor activity. The designed s-triazine derivatives showed common characteristics with these drugs by containing an s-triazine nucleus with two saturated cyclic amino groups (Fig. 2). The present study converted the amino group (cyclic–NH–) of Dioxadet to the aromatic amino groups (4-substituted aniline). Furthermore, the remaining two positions of the s-triazine nucleus were supplemented with 6-membered cyclic aliphatic amino groups (morpholine and piperidine) similar to Bimiralisib and Gedatolisib (dual inhibition of PI3K/mTOR).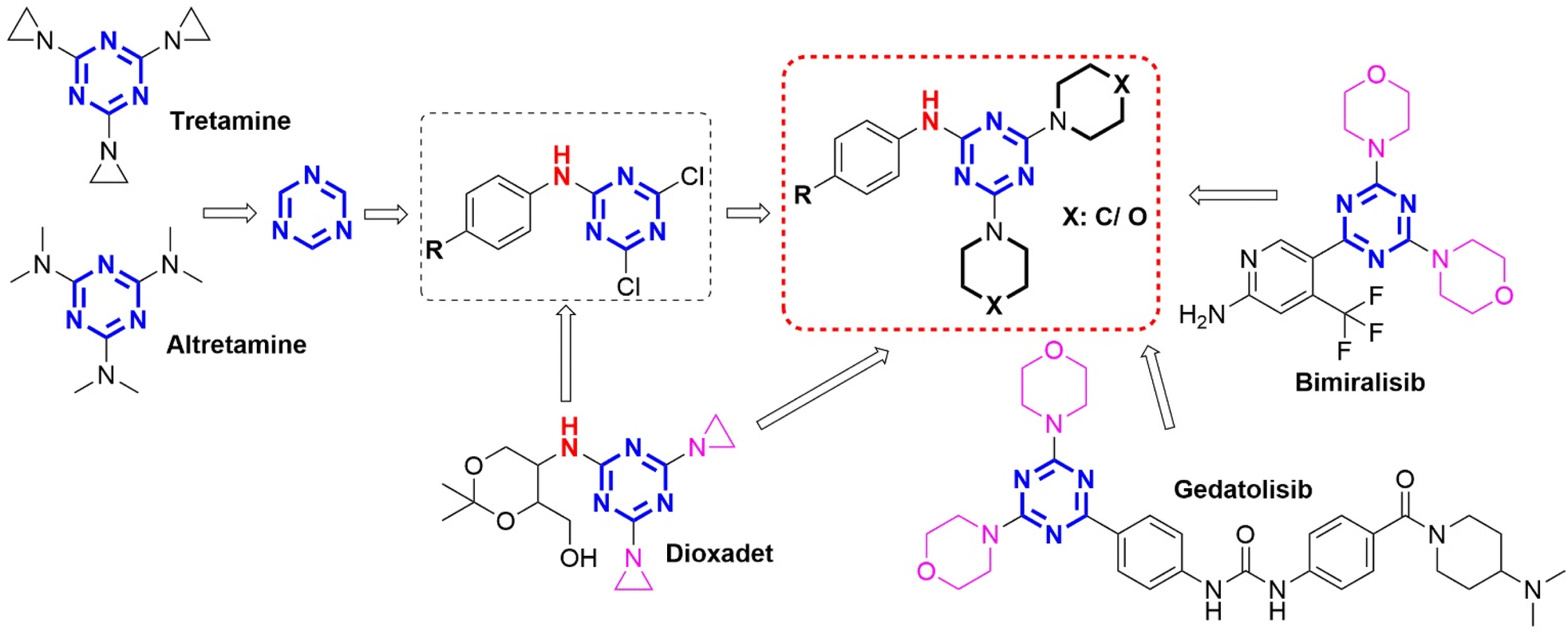 | ||
| Fig. 2 Rational study design of phenylamino-s-triazine derivatives from anticancer drugs. | ||
The aim of this study was to synthesize different symmetrical phenylamino-s-triazine derivatives containing one aromatic amine group and two hexagonal saturated cyclic amine groups and evaluate their anticancer activity against two cancer cell lines (MCF7 – human breast cancer cell line and C26 – colon carcinoma cell line) and one normal cell line (BAEC – bovine aorta endothelial cell line). The potential compounds will be further in silico molecular docking and ADMET (absorption, distribution, metabolism, excretion, and toxicity) studies to understand their potential drug–receptor interactions and pharmacokinetic parameters.
2. Results and discussion
2.1. Chemistry
Mono-substituted s-triazine derivatives were synthesized by a nucleophilic substitution reaction between 4-substituted aniline derivatives and cyanuric chloride in THF solvent with the presence of solid K2CO3 at low temperature of 0–5 °C to ensure the substitution of only the first chlorine atom and control the reaction selectivity (Scheme 1). The reaction occurred rapidly within 30–40 min and the reaction yield was high (91–96%). In addition, the reaction yield depended on the basicity of the aniline derivatives. More basic aniline derivatives with electron-donating substituents (4-OCH3 and 4-CH3, 95–96%) showed higher yields than less basic aniline derivatives with electron-withdrawing substituents (4-F and 4-NO2, 91–92%). The yields of the mono-substituted s-triazine derivatives in this study showed similarities with some published studies (4-COOH – 95.4% (CH2Cl2/solid Na2CO3);46 4-Br, 4-Cl, 4-OCH3 – 92–95% (acetone/aqueous solution of NaHCO3)47) despite different reaction solvents and alkalines. However, the reaction time was recorded to be shorter than other studies (1–3 h).46,47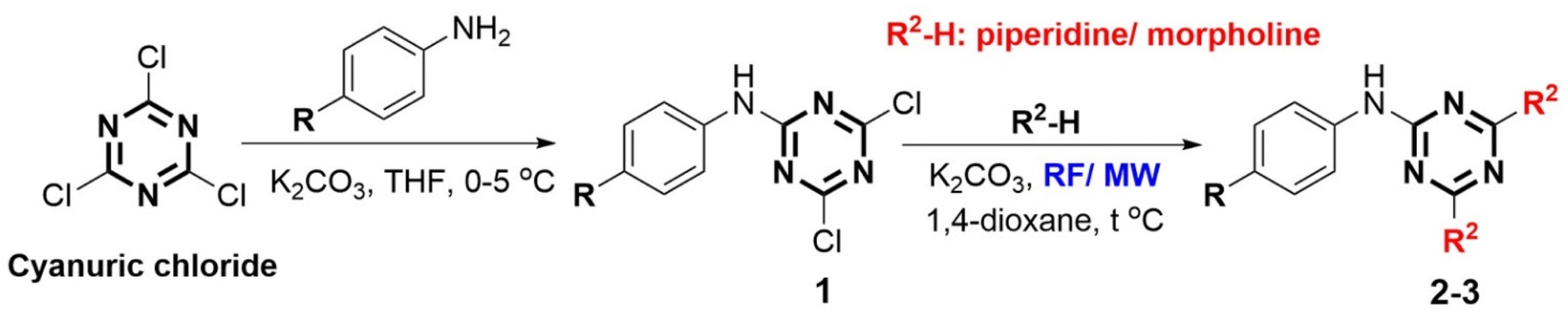 | ||
| Scheme 1 Construction of phenylamino-s-triazine derivatives (MW: microwave irradiation, RF: reflux, THF: tetrahydrofuran). | ||
The tri-substituted s-triazine derivatives were prepared by nucleophilic reaction of the remaining two chlorine groups of mono-substituted s-triazine derivatives with saturated cyclic amines (piperidine and morpholine) in the presence of 1,4-dioxane/solid K2CO3 by two different methods including reflux (RF) and microwave-assisted (MW) methods. The reaction yields, presented in Table 1, showed a difference between the two methods RF and MW. In general, the synthesis reaction of tri-substituted s-triazine derivatives exhibited high yields (>80%). The MW method (300 W, 15–30 min, 91–98%) exhibited shorter reaction times and about 10% higher yields than the RF method (12–24 h, 80–88%). Furthermore, the MW method used less 1,4-dioxane solvent than the RF method. These results are similar to those in the study of Al-Zaydi et al., 2017 with the starting material of 4-COOH aniline for a tri-substituted reaction with saturated cyclic amines (piperidine and morpholine) in 1,4-dioxane/water (1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1)/Na2CO3 (RF – 74.5–85.8%, 8–10 h; MW (400 W) – 88–93.1%, 10 min).46 This demonstrated that MW as the “green” method has great advantages in terms of time and yield in the synthesis of tri-substituted s-triazine derivatives, especially in limiting the use of solvents and protecting the environment.
:1)/Na2CO3 (RF – 74.5–85.8%, 8–10 h; MW (400 W) – 88–93.1%, 10 min).46 This demonstrated that MW as the “green” method has great advantages in terms of time and yield in the synthesis of tri-substituted s-triazine derivatives, especially in limiting the use of solvents and protecting the environment.
|
|||||||
|---|---|---|---|---|---|---|---|
| Entry | R Group | Code | Physicochemical parametersa | Yield | |||
| R | R2 | RF | MW | ||||
| a calculated using SwissADME, Piper – piperidinyl, Mor – morpholino, RF – reflux method (/conventional heating), MW – microwave-assisted methods, MWt – molecular weight, nHA – number of hydrogen bond acceptor, nHD – number of hydrogen bond donor, nRot – number rotatable bond, TPSA – polar surface area (Angstroms squared), MR – Molar Refractivity, logP − logPo/w (XLOGP3), logS − logS (ESOL). |
|||||||
| 1 | –Cl | Piper | 2a | MWt: 372.90 nHA: 3 | MR: 113.28 | 83 | 97 |
| nHD: 1 | LogP: 5.08 TPSA: 57.18 |
||||||
| nRot: 4 | LogS: −5.43 |
||||||
| 2 | –F | Piper | 2b | MWt: 356.44 nHA: 4 | MR: 108.23 | 85 | 92 |
| nHD: 1 | LogP: 4.55 |
||||||
| nRot: 4 | TPSA: 57.18 | ||||||
| LogS: −4.99 |
|||||||
| 3 | –OCH3 | Piper | 2c | MWt: 368.48 nHA: 4 | MR: 114.76 | 81 | 98 |
| nHD: 1 | LogP: 4.42 |
||||||
| nRot: 5 | TPSA: 66.41 | ||||||
| LogS: −4.91 |
|||||||
| 4 | –CH3 | Piper | 2d | MWt: 352.48 nHA: 3 | MR: 113.23 | 82 | 97 |
| nHD: 1 | LogP: 4.82 |
||||||
| nRot: 4 | TPSA: 57.18 | ||||||
| LogS: −5.14 |
|||||||
| 5 | –NO2 | Piper | 2e | MWt: 383.45 nHA: 5 | MR: 117.09 | 88 | 96 |
| nHD: 1 | LogP: 4.28 |
||||||
| nRot: 5 | TPSA: 100.32 | ||||||
| LogS: −4.90 |
|||||||
| 6 | –Cl | Mor | 3a | MWt: 376.84 nHA: 5 | MR: 105.83 | 81 | 98 |
| nHD: 1 | LogP: 2.64 |
||||||
| nRot: 4 | TPSA: 75.64 | ||||||
| LogS: −3.92 |
|||||||
| 7 | –F | Mor | 3b | MWt: 360.39 nHA: 6 | MR: 100.78 | 84 | 91 |
| nHD: 1 | LogP: 2.11 |
||||||
| nRot: 4 | TPSA: 75.64 | ||||||
| LogS: −3.48 |
|||||||
| 8 | –OCH3 | Mor | 3c | MWt: 372.42 nHA: 6 | MR: 107.32 | 80 | 98 |
| nHD: 1 | LogP: 1.99 |
||||||
| nRot: 5 | TPSA: 84.87 | ||||||
| LogS: −3.40 |
|||||||
| 9 | –CH3 | Mor | 3d | MWt: 356.42 nHA: 5 | MR: 105.79 | 81 | 97 |
| nHD: 1 | LogP: 2.38 |
||||||
| nRot: 4 | TPSA: 75.64 | ||||||
| LogS: −3.63 |
|||||||
| 10 | –NO2 | Mor | 3e | MWt: 387.39 nHA: 7 | MR: 109.65 | 86 | 93 |
| nHD: 1 | LogP: 1.84 |
||||||
| nRot: 5 | TPSA: 118.78 | ||||||
| LogS: −3.39 |
|||||||

The chemical structures of all the s-triazine derivatives were suitably elucidated by IR, 1H NMR, 13C NMR, and MS spectroscopy. The IR spectra of all compounds showed a medium absorbance band in the ν 3381–2900 cm−1 region characteristic of the –NH– group and a strong absorbance band in the ν 1539–1389 cm−1 region characteristic of the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) N groups of the s-triazine ring. In addition, the 1H NMR spectra of the tri-substituted s-triazine derivatives showed a characteristic –NH– proton of Ar–NH-s-triazine as a singlet in the δ 9.85–8.48 ppm region and four aromatic proton signals with two types of doublet proton (ortho coupling) in the δ 8.16–6.83 ppm region. Besides, the proton signals of the piperidine and morpholine-saturated rings showed good agreement including Hpiperidine δ 3.73–1.48 ppm (8H δ 3.73–3.68 ppm, 4H δ 1.65–1.59 ppm, and 8H δ 1.56–1.48 ppm) and Hmorpholine δ 3.71–3.61 ppm (8H δ 3.71–3.68 ppm and 8H δ 3.64–3.61 ppm). Furthermore, CAr (δ 164.7–113.5 ppm), Cpiperidine δ 43.6–23.8 ppm (1C δ 43.6–43.3 ppm, 1C δ 25.4–24.9 ppm and 1C δ 24.4–23.8 ppm) and Cmorpholine δ 66.0–43.2 ppm (1C δ 66.0–65.6 ppm and 1C δ 43.4–43.2 ppm) were identified in the 13C NMR spectrum. The molecular ion peak M (m/z) of compounds 2–3 was observed in the mass spectrum, confirming the hypothesized structure. In particular, all tri-substituted s-triazine derivatives showed physicochemical properties of fragments (MWt (molecular weight) < 500) that follow Lipinski's rules which could lead to potent compounds for further development (Table 1).
N groups of the s-triazine ring. In addition, the 1H NMR spectra of the tri-substituted s-triazine derivatives showed a characteristic –NH– proton of Ar–NH-s-triazine as a singlet in the δ 9.85–8.48 ppm region and four aromatic proton signals with two types of doublet proton (ortho coupling) in the δ 8.16–6.83 ppm region. Besides, the proton signals of the piperidine and morpholine-saturated rings showed good agreement including Hpiperidine δ 3.73–1.48 ppm (8H δ 3.73–3.68 ppm, 4H δ 1.65–1.59 ppm, and 8H δ 1.56–1.48 ppm) and Hmorpholine δ 3.71–3.61 ppm (8H δ 3.71–3.68 ppm and 8H δ 3.64–3.61 ppm). Furthermore, CAr (δ 164.7–113.5 ppm), Cpiperidine δ 43.6–23.8 ppm (1C δ 43.6–43.3 ppm, 1C δ 25.4–24.9 ppm and 1C δ 24.4–23.8 ppm) and Cmorpholine δ 66.0–43.2 ppm (1C δ 66.0–65.6 ppm and 1C δ 43.4–43.2 ppm) were identified in the 13C NMR spectrum. The molecular ion peak M (m/z) of compounds 2–3 was observed in the mass spectrum, confirming the hypothesized structure. In particular, all tri-substituted s-triazine derivatives showed physicochemical properties of fragments (MWt (molecular weight) < 500) that follow Lipinski's rules which could lead to potent compounds for further development (Table 1).
2.2. In vitro anticancer activity
The IC50 results (μM) on two cancer cell lines (MCF7 and C26) and one normal cell line (BAEC) are presented in Table 2. The comparison of IC50 values of the s-triazine derivatives with potential anticancer activity on the cancer cell lines MCF7 and C26 compared to the reference drugs Paclitaxel (PTX) and Doxorubicin (DOX) is shown in Fig. 3. The mono-substituted s-triazine derivatives (1a–1c) showed strong anticancer activity on two cell lines MCF7 and C26 with IC50 values in the range of 1.77–13.46 μM compared to the reference drugs PTX (IC50 = 2.35–4.32 μM) and DOX (IC50 = 8.06–10.52 μM). Compound 1e exhibited strong anticancer activity on the MCF7 cell line (IC50 = 10.28 μM) but weak anticancer activity on the C26 cell line (IC50 = 48.34 μM). In particular, compound 1d showed the strongest anticancer activity among the synthesized mono-substituted s-triazine derivatives with IC50 = 0.2 μM on the MCF7 cell line and is stronger than the reference drugs PTX and DOX. However, the strong toxicity on cancer cell lines of mono-substituted s-triazine derivatives may be due to the chemical reaction of the Cl group on the s-triazine nucleus. The di-substituted s-triazine derivatives can be formed at room temperature and even at low temperatures in the presence of base groups (-NH2) in the cell.48 Therefore, mono-substituted s-triazine derivatives are likely to have non-selective anticancer activity and high toxicity to normal cells and the human body.| Entry | Code | Cancer cell line | Normal cell line | |
|---|---|---|---|---|
| MCF7 | C26 | BAEC | ||
| a DOX – Doxorubicin, PTX – paclitaxel, MCF7 – human breast cancer cell line, C26 – colon carcinoma cell line, BAEC – bovine aorta endothelial cell line, IC50 ± SEM (μM, SEM – standard error of the mean). The values in bold highlight the best compounds with the best IC50 values compared to positive controls, * – statistically significant (p < 0.05) compared to reference drug PTX. | ||||
| 1 | 1a | 5.35 ± 1.05 | 13.46 ± 0.24 | — |
| 2 | 1b | 7.89 ± 1.80 | 9.10 ± 0.61 | — |
| 3 | 1c | 1.77 ± 0.05 | 9.89 ± 0.17 | — |
| 4 | 1d | 0.20 ± 0.11 | 9.99 ± 0.59 | — |
| 5 | 1e | 10.28 ± 1.51 | 48.34 ± 1.94 | — |
| 6 | 2a | 23.60 ± 2.56 | 47.52 ± 2.86 | 48.99 ± 3.16 |
| 7 | 2b | 32.93 ± 4.23 | 33.63 ± 1.86 | 21.24 ± 2.40 |
| 8 | 2c | 50.93 ± 8.54 | 99.40 ± 14.07 | 15.09 ± 1.98 |
| 9 | 2d | 6.54± 1.16* | 0.38± 0.23* | 1.84± 0.64* |
| 10 | 2e | 26.11 ± 1.86 | 8.28± 1.90* | 315.1 ± 9.53 |
| 11 | 3a | 42.40 ± 4.48 | 1.21± 0.47* | 41.23 ± 2.55 |
| 12 | 3b | 6.19± 1.20* | 24.53 ± 0.86 | 7.98± 0.97* |
| 13 | 3c | 43.52 ± 4.23 | 76.82 ± 8.45 | 2.01 ± 0.59 |
| 14 | 3d | 22.32 ± 1.49 | 16.34 ± 2.22 | 0.57 ± 0.24 |
| 15 | 3e | 13.74± 1.96* | 14.66± 1.70 | 86.34 ± 6.79 |
| 16 | DOX | 10.52 ± 0.86 | 8.06 ± 1.02 | 4.79 ± 0.53 |
| 17 | PTX | 2.35 ± 0.14 | 4.32 ± 0.52 | 2.67 ± 0.41 |
 | ||
| Fig. 3 The IC50 values (μM) of potential s-triazine derivatives compared to Paclitaxel (PTX) and Doxorubicin (DOX) (MCF7 – human breast cancer cell line, C26 – colon carcinoma cell line, BAEC – bovine aorta endothelial cell line). | ||
The tri-substituted s-triazine derivatives (2a, 3a, 2b, 2c, and 3c) exhibited weak to moderate anticancer activity against the MCF7 cell line with IC50 ranging from 23.60 to 50.93 μM. Compound 3b (4-F-Ar, dimorpholino) exhibited strong anticancer activity with IC50 = 6.19 μM against the MCF7 cell line compared to the reference drugs PTX (IC50 MCF7 = 2.35 μM) and DOX (IC50 MCF7 = 10.52 μM). Meanwhile, compounds 2a, 2b, 3b, 2c, and 3c exhibited weak to moderate anticancer activity against the C26 cell line with IC50 ranging from 24.53 to 99.40 μM compared to the reference drugs PTX (IC50 C26 = 4.32 μM) and DOX (IC50 C26 = 8.06 μM). Compounds 2e (4-NO2–Ar, dipiperidinyl) and 3a (4-Cl–Ar, dimorpholino) showed potent anticancer activity on the C26 cell line with IC50 values of 8.28 and 1.21 μM, respectively. Notably, compounds 2d (4-CH3–Ar, dipiperidinyl) and 3e (4-NO2–Ar, dimorpholino) showed strong anticancer activity on both MCF7 and C26 cell lines compared to the reference drugs PTX (IC50 = 2.35–4.32 μM) and DOX (IC50 = 8.06–10.52 μM). Compound 2d had IC50 values on the MCF7 and C26 cell lines of 6.54 and 0.38 μM, respectively, and exhibited stronger anticancer activity on the C26 cell line than PTX and DOX. In contrast, compound 3e had IC50 values on MCF7 and C26 cell lines of 13.74 and 14.66 μM, respectively, and exhibited weaker anticancer activity on the C26 cell line than PTX and DOX (Fig. 3A). Therefore, the study results showed that tri-substituted s-triazines 2d and 3e have the most potential among the synthesized s-triazine derivatives due to their strong anticancer activity against both MCF7 and C26 cell lines.
In addition, all s-triazine derivatives were also tested for cytotoxicity against the BAEC normal cell line to evaluate their selectivity towards cancer cell lines and systemic toxicity. The potentially active compounds 2d (IC50 = 1.84 μM) and 3b (IC50 = 7.98 μM) exhibited high cytotoxicity against BAEC normal cells similar to the reference drugs PTX (IC50 = 2.67 μM) and DOX (IC50 = 4.79 μM). Meanwhile, the potentially active s-triazines 2e (IC50 = 315.1 μM), 3a (IC50 = 41.23 μM), and 3e (IC50 = 86.24 μM) exhibited low cytotoxicity against BAEC normal cells compared to PTX and DOX. These results demonstrated that compounds 2e, 3a, and 3e have higher selectivity on cancer cell lines and lower toxicity to the human body compared to compounds 2d and 3b (Fig. 3B). However, the development of targeted drug delivery systems (liposome, transferosome, niosomes, gold nanoparticles, nanotube, etc) has significantly reduced the side effects and toxicity of anticancer drugs.49 Therefore, good anticancer s-triazine derivatives are potential candidates for the development of new anticancer drugs.
2.3. Structure–activity relationships (SAR)
The s-triazine derivatives with low molecular weight (MWt < 500) according to Lipinski's rule are receiving research interest worldwide. 4-Bromophenylamino-s-triazine derivatives combined with mono-pyrazolyl and morpholino groups on the s-triazine ring showed good anticancer activity against MCF7 (IC50 = 4.53 μM), colon carcinoma (HCT-116, IC50 = 0.50 μM), and liver carcinoma (HepG2, IC50 = 3.01 μM).47 Similarly, 4-bromo/4-chlorophenylamino-s-triazine derivatives combined with indol-3-ylpyrazolyl and morpholino groups exhibited strong anticancer activity against MCF7, A549 (non-small cell lung cancer) and HDFs (non-cancerous human dermal fibroblasts) with IC50 values in the range of 2–4 μM. The IC50 values of these compounds were significantly higher than doxorubicin (IC50 = 0.42 μM). This result indicates that the toxicity of these derivatives is significantly lower than that of doxorubicin towards healthy cells and may exhibit a better therapeutic index.20 Meanwhile, the sulfaguanidine-triazine derivatives with 4-sulfamoylphenylamino group showed IC50 values ranging from 14.8 to 33.2 μM against MCF7 and A549 cancer cell lines, especially the s-triazine derivative containing 4-sulfamoylphenylamino, diethylamine and morpholine could be a promising lead compound for developing new target-selected anticancer compounds (% inhibition against PI3Kα enzyme = 68%).23 Besides, the 4,6-dimorpholino-s-triazine derivatives with a 4-acylphenylamino group (bearing a pyridyl group) exhibited potent cytotoxic activity against SW620 (human colon cancer cells), A549, HeLa (human cervical cancer cells), and MCF7 with IC50 values of 8.71, 9.55, 15.67, and 21.77 μM, respectively.22 Similar to these studies, most of the potential active s-triazines contained electron-withdrawing groups such as 4-halogeno (4-Cl and 4-F) and 4-nitro (4-NO2) on the benzene ring of the phenylamino-s-triazine scaffold. In addition, the morpholino group was also a common substituent present in the structure of lead compounds for strong anticancer activity. Furthermore, substituting dipiperidinyl and dimorpholino groups on the 4-substituted phenylamino-s-triazine scaffold in the present study may enhance the anticancer activity, increase selectivity, and limit toxicity against normal cells. The symmetrical s-triazine compounds 2e, 3a, and 3e were shown to be approximately 15–118 times less toxic than PTX and indol-3-yl-pyrazolyl-s-triazine,20 and approximately 8.6–65.8 times less toxic than DOX (Fig. 3B). These compounds have great potential in the development of anticancer drugs with low toxicity, high selectivity, and better therapeutic index.2.4. In silico ADMET profile
The ADMET profile of the potent compounds and reference drug Gedatolisib (Ged) is shown in Table 3.| Parameter | 2d | 2e | 3a | 3b | 3e | Ged | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| a DOX – doxorubicin (MWt: 543.17, nHA: 12, nHD: 7, nRot: 5, Flex: 0.167, logP: 1.208, TPSA: 206.07, logS: −3.326), PTX – paclitaxel (MWt: 853.33, nHA: 15, nHD: 4, nRot: 15, Flex: 0.341, logP: 2.85, TPSA: 221.29, logS: −5.133), Ged – Gedatolisib (MW: 615.33, nHA: 13, nHD: 2, nRot: 10, Flex: 0.263, logP: 2.305, TPSA: 128.29, logS: −2.824), Caco-2 permeability (optimal: higher than −5.15 log unit), MDCK permeability (low permeability: < 2 × 10−6 cm s−1, medium permeability: 2–20 × 10−6 cm s−1, high passive permeability: > 20 × 10−6 cm s−1), PAMPA – the experimental data for Peff was logarithmically transformed (logPeff < 2: low-permeability, logPeff > 2.5: high-permeability), Pgp – P-glycoprotein, HIA – Human Intestinal Absorption (–: ≥ 30%, +: < 30%), F: Bioavailability (+: < percent value, –: ≥ percent value), PPB: Plasma Protein Binding (optimal: < 90%), VD: Volume Distribution (optimal: 0.04–20 L kg−1), BBB: Blood–Brain Barrier Penetration, Fu: The fraction unbound in plasms (low: < 5%, middle: 5–20%, high: > 20%), CL: Clearance (low: < 5 mL min−1 kg−1, moderate: 5–15 mL min−1 kg−1, high: > 15 mL min−1 kg−1), T1/2 (ultra-short half-life drugs: 0.5 – < 1 h; short half-life drugs: 1–4 h; intermediate short half-life drugs: 4–8 h; long half-life drugs: > 8 h), hERG blockers (IC50 ≤ 10 μM or ≥ 50% inhibition at 10 μM were classified as hERG +, IC50 > 10 μM or < 50% inhibition at 10 μM were classified as hERG –), DILI: Drug-Induced Liver Injury, Rat Oral Acute Toxicity (0: low-toxicity > 500 mg kg−1, 1: high-toxicity < 500 mg kg−1), FDAMDD – Maximum Recommended Daily Dose, BCF – Bioconcentration Factors, IGC50 – Tetrahymena pyriformis 50 percent growth inhibition Concentration, LC50FM – 96 h fathead minnow 50 percent lethal concentration, LC50DM – 48 h daphnia magna 50 percent lethal concentration. The output value is the probability of being inhibitor/substrate/active/positive/high-toxicity/sensitizer/carcinogens/corrosives/irritants (category 1) or non-inhibitor/non-substrate/inactive/negative/low-toxicity/non-sensitizer/non-carcinogens/noncorrosives/nonirritants (category 0). For the classification endpoints, the prediction probability values are transformed into six symbols: 0–0.1(−−−), 0.1–0.3(−−), 0.3–0.5(−), 0.5–0.7(+), 0.7–0.9(++), and 0.9–1.0 (+++). Additionally, the corresponding relationships of the three labels are as follows: E – excellent, M – medium, P – poor. |
||||||||||||
| Absorption | ||||||||||||
| Caco-2 permeability | −4.745 | E | −4.713 | E | −4.769 | E | −4.756 | E | −4.801 | E | −5.322 | P |
| MDCK permeability | 0.0 | E | 0.0 | E | 0.0 | E | 0.0 | E | 0.0 | E | 0.0 | E |
| PAMPA | −−− | E | −−− | E | −−− | E | −−− | E | −−− | E | −−− | E |
| Pgp-inhibitor | +++ | M | +++ | P | +++ | P | +++ | P | ++ | P | +++ | P |
| Pgp-substrate | −−− | E | −−− | E | −−− | E | −−− | E | −−− | E | −−− | E |
| HIA | −−− | E | −−− | E | −−− | E | −−− | E | −−− | E | −−− | E |
| F20% | −−− | E | −−− | E | −−− | E | −−− | E | −−− | E | −−− | E |
| F30% | −−− | E | −−− | E | −−− | E | −−− | E | −−− | E | −−− | E |
| F50% | −−− | P | −−− | E | ++ | P | + | M | ++ | P | +++ | P |
|
||||||||||||
| Distribution | ||||||||||||
| PPB (%) | 99.2 | P | 99.2 | P | 97.1 | P | 94.6 | P | 94.7 | P | 76.5 | E |
| VDss (L kg−1) | 4.428 | E | 3.151 | E | 1.342 | E | 2.032 | E | 1.636 | E | 2.66 | E |
| BBB penetration | −−− | E | −−− | E | −−− | E | −−− | E | −−− | E | −−− | E |
| Fu (%) | 0.4 | P | 0.4 | P | 3.0 | P | 6.2 | E | 6.4 | E | 18.0 | E |
| OATP1B1 inhibitor | +++ | P | +++ | P | +++ | P | +++ | P | +++ | P | +++ | P |
| OATP1B3 inhibitor | +++ | P | +++ | P | +++ | P | +++ | P | +++ | P | +++ | P |
| BCRP inhibitor | −−− | P | −−− | P | −−− | P | −−− | P | −−− | P | −−− | P |
| MRP1 inhibitor | +++ | P | +++ | P | + | M | + | M | +++ | P | −−− | M |
| BSEP inhibitor | +++ | P | +++ | P | +++ | P | +++ | P | +++ | P | +++ | P |
|
||||||||||||
| Metabolism | ||||||||||||
| CYP1A2 inhibitor | +++ | +++ | +++ | +++ | +++ | +++ | ||||||
| CYP1A2 substrate | +++ | +++ | +++ | ++ | +++ | −−− | ||||||
| CYP2C19 inhibitor | +++ | +++ | ++ | −−− | + | −−− | ||||||
| CYP2C19 substrate | +++ | +++ | −−− | −−− | −−− | +++ | ||||||
| CYP2C9 inhibitor | ++ | −−− | ++ | ++ | + | −−− | ||||||
| CYP2C9 substrate | −−− | −−− | −−− | −−− | −−− | −−− | ||||||
| CYP2D6 inhibitor | −−− | +++ | −−− | −−− | −−− | −−− | ||||||
| CYP2D6 substrate | ++ | + | −−− | −−− | + | −−− | ||||||
| CYP3A4 inhibitor | −−− | −−− | ++ | + | ++ | −−− | ||||||
| CYP3A4 substrate | +++ | +++ | −−− | −−− | −−− | ++ | ||||||
| CYP2B6 inhibitor | +++ | +++ | −−− | −−− | −−− | +++ | ||||||
| CYP2B6 substrate | −−− | −−− | −−− | −−− | −−− | −−− | ||||||
| CYP2C8 inhibitor | +++ | + | +++ | +++ | ++ | −−− | ||||||
| HLM stability | +++ | E | +++ | E | −−− | P | −−− | P | −−− | P | −−− | P |
|
||||||||||||
| Excretion | ||||||||||||
| CLplasma (mL min−1 kg−1) | 4.995 | E | 4.479 | E | 5.307 | M | 5.254 | M | 4.902 | E | 5.329 | M |
| T1/2 | 0.226 | 0.464 | 0.229 | 0.254 | 0.54 | 0.297 | ||||||
|
||||||||||||
| Toxicity | ||||||||||||
| hERG blockers | 0.555 | M | 0.569 | M | 0.564 | M | 0.411 | M | 0.43 | M | 0.949 | P |
| hERG blockers (10 μm) | 0.801 | P | 0.795 | P | 0.717 | P | 0.576 | M | 0.599 | M | 0.337 | M |
| DILI | 0.647 | M | 0.974 | P | 0.991 | P | 0.958 | P | 0.999 | P | 0.998 | P |
| AMES toxicity | 0.19 | E | 0.697 | M | 0.3 | M | 0.517 | M | 0.883 | P | 0.545 | M |
| Rat oral acute toxicity | 0.119 | E | 0.242 | E | 0.117 | E | 0.189 | E | 0.179 | E | 0.289 | E |
| FDAMDD | 0.525 | M | 0.624 | M | 0.067 | E | 0.128 | E | 0.09 | E | 0.457 | M |
| Skin sensitization | 0.359 | M | 0.72 | P | 0.775 | P | 0.467 | M | 0.926 | P | 0.635 | M |
| Carcinogenicity | 0.658 | M | 0.71 | P | 0.966 | P | 0.978 | P | 0.98 | P | 0.947 | P |
| Eye corrosion | 0.001 | E | 0.001 | E | 0.0 | E | 0.002 | E | 0.001 | E | 0.0 | E |
| Eye irritation | 0.771 | P | 0.873 | P | 0.625 | M | 0.763 | P | 0.915 | P | 0.0 | E |
| Respiratory toxicity | 0.538 | M | 0.748 | P | 0.287 | E | 0.358 | M | 0.594 | M | 0.791 | P |
| Human hepatotoxicity | 0.773 | P | 0.829 | P | 0.906 | P | 0.916 | P | 0.936 | P | 0.986 | P |
| Drug-induced nephrotoxicity | 0.615 | M | 0.5 | M | 0.977 | P | 0.988 | P | 0.917 | P | 0.997 | P |
| Drug-induced neurotoxicity | 0.781 | P | 0.178 | E | 0.937 | P | 0.968 | P | 0.439 | M | 0.991 | P |
| Ototoxicity | 0.354 | M | 0.314 | M | 0.524 | M | 0.492 | M | 0.431 | M | 0.864 | P |
| Hematotoxicity | 0.227 | E | 0.364 | M | 0.169 | E | 0.183 | E | 0.267 | E | 0.475 | M |
| Genotoxicity | 0.672 | M | 1.0 | P | 0.955 | P | 0.958 | P | 1.0 | P | 1.0 | P |
| RPMI-8226 immunotoxicity | 0.072 | E | 0.078 | E | 0.138 | E | 0.159 | E | 0.162 | E | 0.624 | M |
| A549 cytotoxicity | 0.451 | M | 0.57 | M | 0.162 | E | 0.076 | E | 0.103 | E | 0.051 | E |
| Hek293 cytotoxicity | 0.75 | P | 0.822 | P | 0.756 | P | 0.509 | M | 0.601 | M | 0.749 | P |
| BCF | 1.679 | 1.168 | 1.387 | 0.532 | 0.482 | 0.755 | ||||||
| IGC50 | 3.759 | 3.772 | 3.765 | 3.06 | 3.274 | 3.379 | ||||||
| LC50DM | 4.558 | 4.802 | 5.118 | 4.709 | 4.307 | 5.31 | ||||||
| LC50FM | 4.432 | 4.479 | 4.65 | 3.774 | 3.943 | 4.206 | ||||||
logP ≤ 4.15, nHA ≤ 10, and nHD ≤ 5),6 Ghose (160 ≤ MWt ≤ 480, −0.4 ≤ WlogP ≤ 5.6, 40 ≤ MR ≤ 130, and 20 ≤ atoms ≤ 70), Veber (nRot ≤ 10 and TPSA ≤ 140), Egan (WlogP ≤ 5.88 and TPSA ≤ 131.6), and Muegge (200 ≤ MWt ≤ 600, −2 ≤ XlogP ≤ 5, TPSA ≤ 150, num. rings ≤ 7, num. carbon > 4, num. heteroatoms > 1, nRot ≤ 15, nHA ≤ 10, and nHD ≤ 5) (Table 1). Moreover, the absorption of these compounds is predicted to be good to excellent through parameters such as high MDCK permeability (>20 × 10−6 cm s−1), PAMPA (the experimental data for Peff was logarithmically transformed), Pgp-substrate, HIA (human intestinal absorption), F20%, and F30% compared to the reference drug Ged. In particular, all compounds showed Caco-2 permeability higher than the reference drug Ged and higher than the −5.15 log unit. All compounds are most likely not Pgp inhibitors similar to Ged. The study results predicted that these compounds exhibited high gastrointestinal absorption and have great potential for new drug development.2.5. In silico molecular docking study
Molecular docking is a powerful computational tool for predicting protein-ligand interaction energy scores. However, the reliability of predictions regarding non-covalent interactions varies depending on several factors such as scoring function,50,51 protein and ligand flexibility,52,53 water molecules and solvent effects,54,55 and ligand binding diversity.52,56 The Autodock Vina tool can address these aspects to significantly improve the accuracy of molecular docking predictions. Specifically, molecular docking using AutoDock Vina offers several key benefits that make it a widely used tool in computational drug discovery including high speed, accuracy, automatic calculation of the grid maps, flexibility, support for multiple ligands and receptors, compatibility, and support for different force fields.57The binding affinity and hydrogen bond formation of the s-triazine derivatives with 6 targets including dihydrofolate reductase (DHFR), vascular endothelial growth factor receptor 2 (VEGFR2), phosphoinositide 3-kinase (PI3K), cyclin-dependent kinase 2 (CDK2), epidermal growth factor receptor (EGFR), mammalian target of rapamycin (mTOR) are shown in Table 4.25–27 The interactions of the symmetrical tri-substituted s-triazine with amino acid residues at the active site of anticancer targets are shown in Fig. 4–6.
| Entry | Compound | EGFR | DHFR | CDK2 | VEGFR2 | mTOR | PI3K | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| BA | Type | BA | Type | BA | Type | BA | Type | BA | Type | BA | Type | ||
| a BA – binding affinity (kcal mol−1), HB–hydrogen bond (conventional/strong hydrogen bond), CHB – carbon–hydrogen bond, DHB – π–donor hydrogen bond, EGFR – epidermal growth factor receptor, DHFR – dihydrofolate reductase, CDK2 – cyclin-dependent kinase 2, VEGFR2 – vascular endothelial growth factor receptor 2, mTOR – mammalian target of rapamycin, PI3K – phosphoinositide 3-kinase. | |||||||||||||
| 1 | 2d | −9.8 | 1 CHB | −9.1 | 1 CHB | −9.1 | 1 CHB | −9.5 | 1 HB, 1 CHB | −9.4 | 1 HB, 1 CHB | −10.2 | 1 HB |
| 2e | −9.7 | 1 HB | −9.6 | 2 HB, 1 CHB | −9.3 | 1 HB, 1 DHB | −9.5 | 1 HB, 1 CHB | −9.1 | 1 HB | −9.8 | 3 HB | |
| 2 | 3a | −9.2 | 2 HB, 1 CHB | −8.5 | 2 CHB | −8.8 | — | −9 | 1 HB, 2 CHB | −8.9 | 1 HB, 2 CHB | −9.4 | 1 CHB |
| 3 | 3b | −9.3 | 1 HB, 1 CHB | −8.5 | 1 HB, 1 CHB | −8.9 | 1 HB, 2 CHB | −8.8 | 1 HB, 4 CHB | −8.8 | 2 HB, 4 CHB | −9.6 | 2 HB, 1 CHB |
| 3e | −9.1 | 1 HB, 1 CHB | −9.3 | 3 HB, 3 CHB | −8.7 | 1 HB | −8.6 | 1 HB, 3 CHB | −8.7 | 1 HB, 2 CHB | −9.0 | 2 HB, 1 CHB | |
| 4 | Co-crystallized ligand | −7.4 | — | −8.7 | — | −8 | — | −9.4 | — | −7.3 | — | −8.7 | — |
| 5 | Erlotinib | −7.7 | 4 HB, 2 CHB | — | — | — | — | — | — | — | — | — | — |
| 6 | Methotrexate | — | — | −8.3 | 6 HB, 2 CHB | — | — | — | — | — | — | — | — |
| 7 | Seliciclib | — | — | — | — | −7.8 | 1 HB, 1 CHB | — | — | — | — | — | — |
| 8 | Axitinib | — | — | — | — | — | — | −9.7 | 1 HB, 1 CHB | — | — | — | — |
| 9 | Gedatolisib | — | — | — | — | — | — | — | — | −9.4 | 3 HB, 3 CHB | −10.6 | 4 HB, 5 CHB |
 | ||
| Fig. 4 2D and 3D representation of the interaction of s-triazine derivatives and reference drugs with epidermal growth factor receptor (EGFR) and dihydrofolate reductase (DHFR) targets. | ||
 | ||
| Fig. 5 2D and 3D representation of the interaction of s-triazine derivatives and reference drugs with cyclin-dependent kinase 2 (CDK2) and vascular endothelial growth factor receptor 2 (VEGFR2) targets. | ||
 | ||
| Fig. 6 2D and 3D representation of the interaction of s-triazine derivatives and reference drugs with mammalian target of rapamycin (mTOR) and phosphoinositide 3-kinase (PI3K) targets. | ||
Compound 2d (4-CH3-phenylamino and piperidinyl) exhibited binding affinities ranging from −9.1 to −10.2 kcal mol−1 and formed strong hydrogen, or carbon-hydrogen bonds, with all six tested anticancer targets. In addition, this compound showed greater binding affinities than the reference drugs Erlotinib, Methotrexate, and Seliciclib on three targets, namely EGFR, DHFR, and CDK2, and exhibited a binding affinity close to that of the reference drugs Axitinib and Gedatolisib on three targets, namely VEGFR2, mTOR, and PI3K. Meanwhile, compound 2e (4-NO2-phenylamino and piperidinyl) exhibited binding affinities ranging from −9.1 to −9.8 kcal mol−1 and formed strong hydrogen, or π–donor hydrogen bonds, or carbon-hydrogen bonds with all six tested anticancer targets. Similar to compound 2d, this compound exhibited greater binding affinity than reference drugs Erlotinib, Methotrexate, and Seliciclib on 3 targets such as EGFR, DHFR, and CDK2, and nearly equal to the binding affinity of reference drug Axitinib on VEGFR2 target. However, compound 2e exhibited lower binding affinity than Gedatolisib and 2d on two targets mTOR and PI3K. Compounds 3a (4-Cl-phenylamino and morpholino), 3b (4-F-phenylamino and morpholino) and 3e (4-NO2-phenylamino and morpholino) exhibited binding affinities ranging from −8.5 to −9.3 kcal mol−1 and were greater than the reference drugs Erlotinib, Methotrexate and Seliciclib on the three targets EGFR, DHFR, and CDK2. In contrast, these compounds exhibited lower binding affinities than the reference drugs Axitinib and Gedatolisib on three targets VEGFR2, mTOR, and PI3K. Therefore, the study results suggested that all potent compounds have a multitarget anticancer mechanism of action and have a great advantage in limiting resistance to current cancer drugs.
At the EGFR active site, reference drug Erlotinib formed four strong hydrogen bonds (HBs) with amino acid LYS721 (2.05–2.92 Å), two carbon-hydrogen bonds (CHB) with amino acids THR766 (3.75 Å) and ASP831 (3.48 Å), one electrostatic (π–anion) interaction with ASP831 (3.85 Å), and hydrophobic interactions (π–σ, π–π stacked, alkyl, and π–alkyl) with amino acid residues LEU694, PHE699, LEU694, VAL702, ALA719, and LEU820 (3.47–5.56 Å). Like Erlotinib, compounds 3a and 3e formed HBs with amino acid LYS721 (2.42–2.53 Å). Meanwhile, compounds 3b and 2e formed electrostatic (π–cation) interactions with bond lengths ranging from 4.14 to 4.37 Å, and 2d exhibited hydrophobic (alkyl) interaction with amino acid LYS721 with a bond length of 5.10 Å. In addition, compounds 2d, 3a and 3e exhibited electrostatic (π–anion) interactions with ASP831 (3.37–4.24 Å) similar to Erlotinib. Furthermore, compounds 2d, 3a and 3e exhibited 6–7 hydrophobic interactions with amino acids similar to Erlotinib such as PHE699, LEU694, LYS721, LEU820, PHE699, VAL702, and LEU820 (3.67–5.28 Å). Docking analysis also revealed that LYS721 and ASP831 located in the EGFR binding pocket play a key role in the binding mode of the ligand to the receptor. Therefore, compounds 3a and 3e showed the most similar interactions compared to the reference drug Erlotinib at the EGFR active site.
At the DHFR active site, the reference drug Methotrexate formed many interactions including 6 HBs with amino acid residues ALA7 (2.80 Å), GLU17 (2.18 Å), ASN18 (2.08 Å), ALA19 (2.41 Å), ILE94 (2.79 Å), and LEU28 (3.05 Å); 2 CHBs with amino acids ASN23 (2.89 Å) and ILE94 (3.43 Å) and hydrophobic interactions (π–π T-shaped and π–alkyl) with amino acids PHE31 (5.54 Å), ILE50 (4.39 Å) and LEU28 (5.49 Å). Similar to Methotrexate, all five compounds formed CHBs with LEU28 or/and ASN23 (3.16–3.52 Å) and exhibited hydrophobic interactions with amino acids PHE31, ILE50, and LEU28 (3.80–5.48 Å). Additionally, compounds 2e and 3b formed one HB with ALA7 (2.61–2.81 Å), and 3e formed two HBs with ALA7 (2.71 and 2.84 Å). However, all five compounds only showed hydrophobic interactions (alkyl and π–alkyl) with ALA19 (3.83–5.04 Å) and these interactions were weaker than the HB of Methotrexate at ALA19.
At the CDK2 active site, the reference drug Seliciclib exhibited some interactions such as one HB (LEU83, 1.93 Å), one CHB (ASP145, 3.38 Å), and hydrophobic interactions (π–σ, alkyl, and π–alkyl) with amino acids PHE80, ILE10, PHE82, VAL18, LEU134, VAL18, and LYS33 (3.70–5.09 Å). Compounds 2d, 2e, 3d, and 3e exhibited hydrophobic interactions (π–σ or π–alkyl) with amino acids VAL18, ILE10, and LEU134 (3.76–5.36 Å), whereas compound 3b exhibited hydrophobic interactions (alkyl and π–alkyl) with amino acids LEU134 and PHE80 (4.62–5.40 Å) similar to Seliciclib. Additionally, compound 2d exhibited the highest number of hydrophobic interactions similar to Seliciclib at amino acids such as PHE80, ILE10, VAL18, LEU134, VAL18, and LYS33 (4.24–5.46 Å). However, this compound did not form HB with LEU83 compared to 3b, 3e, and Seliciclib.
At the VEGFR2 active site, the reference drug Axitinib showed some interactions such as one HB (CYS919, 2.22 Å), one CHB (LYS920, 3.59 Å), and hydrophobic interactions (π–σ, π–π T-shaped, and π–alkyl) with the amino acids LEU840, VAL848, PHE1047, LEU1035, ALA866, CYS1045, and LYS838 (3.58–5.42 Å). Compounds 2d, 2e, 3a, and 3b showed π–sulfur hydrophobic interactions with CYS1045 (5.51–5.79 Å) similar to the reference drug Axitinib. Moreover, compound 3e exhibited the most similar interactions with amino acid residues against VEGFR2 compared to Axitinib including one HB (CYS919, 2.41 Å), 1 CHB (LEU840, 3.76 Å), and hydrophobic interactions (π–σ and π–alkyl) with LEU840, ALA866, CYS919, and LEU1035 (3.81–5.38 Å).
At the PI3K active site, the reference drug Gedatolisib formed many interactions including four HBs with amino acids ARG277 (2.56 and 2.41 Å), HIS658 (2.48 Å), and GLU880 (2.28 Å); five CHBs with GLN846 (3.50 Å), CYS869 (3.56 Å), TYR787 (3.35 Å), GLU301 (3.71 Å), and CYS275 (3.56 Å); five electrostatic (π–cation and π–anion) interactions with amino acids ARG277 (4.64 Å), ARG690 (4.21 Å), ARG849 (3.85 Å), GLU826 (4.82 Å), and GLU880 (4.15 Å) and hydrophobic interactions (alkyl and π–alkyl) with amino acids LEU823, TRP201, HIS658 and PHE694 (4.50–5.31 Å). Compounds 2d, 3b, and 3e exhibited the highest number of interactions (6–8 interactions) with amino acids similar to Gedatolisib such as PHE694, TYR787, CYS869, ARG690, TRP201, GLU880, ARG849, and HIS658 (2.07–5.45 Å). In addition, compound 2e formed two HBs with ARG277 with bond lengths ranging from 2.67 to 2.94 Å similar to Gedatolisib, however, this compound only formed hydrophobic interactions (alkyl and π–alkyl) with amino acids ARG849 and HIS658 (4.43–4.80 Å) compared to Gedatolisib. Meanwhile, all tested compounds did not show interactions with amino acids similar to Gedatolisib at the mTOR active site.
In summary, in terms of hydrogen bond formation, compound 3b formed the most hydrogen bonds with 5–6 hydrogen bonds (1–2 HBs and 4 CHBs) with bond lengths ranging from 2.17 to 3.76 Å on the VEGFR2 and mTOR targets compared to other compounds and reference drugs. Compound 2e formed three HBs (2.52–2.94 Å) at the PI3K active site. Notably, compound 3e formed three HBs (2.39–2.84 Å) and 3 CHBs (3.17–3.57 Å) on the DHFR target compared to other compounds (1–3 HBs, 2.35–5.48 Å) and reference drug Methotrexate (6 HBs and 1 CHB, 2.08–3.43 Å). Furthermore, all the potential derivatives exhibited interactions (hydrogen bond, electrostatic (π–ion), hydrophobic interactions (π–σ, π–π T-shaped, alkyl, π–alkyl)) with some crucial amino acid residues similar to the reference drugs at the action site of the five targets, except for mTOR. In addition, the 4-phenylamino group of the s-triazine nucleus showed many strong interactions with the 6 tested anticancer targets. Therefore, this group is an important structural part that enhances the anticancer activity of s-triazine derivatives and exhibits multitarget interaction properties. In conclusion, five compounds 2d, 2e, 3a, 3b, and 3e exhibited potential anticancer activity as demonstrated through in vitro and in silico studies.
3. Conclusion
In summary, ten tri-substituted s-triazine derivatives have been designed, synthesized, and evaluated for in vitro and in silico anticancer activities. The microwave-assisted method showed excellent yields (>90%) and significantly reduced reaction time compared to the reflux method. In addition, five compounds showed potent anticancer activity with IC50 < 15 μM against two tested cell lines (MCF7 and C26) compared to reference drugs PTX and DOX. From the structure–activity relationship, the most potent s-triazine derivatives contained electron-withdrawing groups such as 4-halogeno and 4-nitro on the benzene ring and morpholino group of the phenylamino-s-triazine scaffold. Three symmetrical s-triazines 2e, 3a, and 3e showed low toxicity on BAEC normal cell lines compared to PTX and DOX. Molecular docking demonstrated that five potent compounds showed good hydrogen and hydrophobic interactions against multi-targets such as DHFR, VEGFR2, PI3K, CDK2, EGFR, and mTOR that resemble the co-crystallization ligand, and reference drugs. Furthermore, the obtained in silico results predicted that these compounds may show a good ADMET profile. Therefore, these s-triazines are potential candidates for further research in the development of new anticancer drugs.4. Experimental section
4.1. Materials
All chemicals and solvents were purchased from commercial suppliers Merck and Acros. All the reactions were carried out under an inert atmosphere of nitrogen. TLC was performed on pre-coated aluminum sheets of silica (60 F254 nm) and visualized by shortwave UV light at λ 254. Column chromatography uses 0.040–0.063 mm granular silica gel (Merck).The microwave reactor used was the Microwave synthesizer – CEM Discover, USA, fitted with a magnetic stirrer for continuous stirring and an infrared temperature sensor, which enabled and controlled the temperature. Melting points (mp) were determined using a Sanyo-Gallenkamp melting point apparatus. IR spectra were recorded on an IRAffinity-1S. NMR spectra were recorded on a Bruker Avance 500 NMR Spectrometer. Chemical shifts were measured in δ (ppm). Mass spectrometry was measured on 1100 series LC-MS Trap Agilent.
4.2. Experimental procedures
:water (1:1, v/v) solvent. Reaction yields range from 91 to 96%.N), 2978 (ArCH), 806 (C–N, s-triazine), 791 (Ar–Cl). 1H NMR (500 MHz, DMSO-d6, δ ppm): 10.91 (1H, s, NH), 7.60 (2H, d, J = 9 Hz, HAr), 7.42 (2H, d, J = 8.5 Hz, HAr). 13C NMR (125 MHz, DMSO-d6, δ ppm): 163.8, 154.1, 136.0, 135.9, 128.9, 128.8, 128.4, 123.1, 122.9. LC-MS (m/z) [M + H]+ calcd for C9H6Cl3N4 274.9653, found 274.9405.N), 1207 (Ar–F), 1011 (C–N, s-triazine), 791 (Ar–Cl). 1H NMR (500 MHz, DMSO-d6, δ ppm): 11.13 (1H, s, NH), 7.58 (2H, d, J = 9.0 Hz, HAr), 7.20 (2H, d, J = 9.0 Hz, HAr). 13C NMR (125 MHz, DMSO-d6, δ ppm): 164.0 (>C); 161.19 và 159.26 (>C, JC–F = 241.25 Hz); 154.0 (>C); 133.40 và 133.38 (>C, JC–F = 2.5 Hz); 125.53 và 125.47 (>C, JC–F = 7.5 Hz); 116.33 và 116.15 (>C, JC–F = 22.5 Hz). LC-MS (m/z) [M − H]− calcd for C9H4Cl2FN4 256.9803, found 257.0161.N), 2985 (ArCH), 802 (C–N, s-triazine), 790 (Ar–Cl). 1H NMR (500 MHz, DMSO-d6, δ ppm): 10.97 (1H, s, NH), 7.42 (2H, d, J = 9.0 Hz, HAr), 6.95 (2H, d, J = 9.0 Hz, HAr), 3.10 (3H, s, –OCH3). 13C NMR (125 MHz, DMSO-d6, δ ppm): 157.8, 153.7, 127.8, 125.3, 123.6, 114.6, 114.2, 55.5. LC-MS (m/z) [M − H]− calcd for C10H7Cl2N4O 269.0002, found 268.9938.N), 1013 (C–N, s-triazine), 791 (Ar–Cl). 1H NMR (500 MHz, DMSO-d6, δ ppm): 11.03 (1H, s, NH), 7.46 (2H, d, J = 8.5 Hz, HAr), 7.17 (2H, d, J = 8.5 Hz, HAr), 2.27 (3H, s, –CH3). 13C NMR (125 MHz, DMSO-d6, δ ppm): 163.8, 153.8, 134.4, 130.4, 129.7, 121.8, 20.6. LC-MS (m/z) [M − H]− calcd for C10H7Cl2N4 253.0053, found 253.0039.N), 1315 (NO), 1113 (C–N, s-triazine), 800 (Ar–Cl). 1H NMR (500 MHz, DMSO-d6, δ ppm): 10.89 (1H, s, NH), 8.25 (2H, d, J = 9.0 Hz, HAr), 7.91 (2H, d, J = 9.0 Hz, HAr). 13C NMR (125 MHz, DMSO-d6, δ ppm): 160.7, 154.2, 144.2, 142.5, 125.0, 119.8. LC-MS (m/z) [M − H]− calcd for C9H4Cl2N5O2 283.9748, found 283.9722.4.2.7.1 The reflux method. A mixture of mono-substituted s-triazine derivatives 1a–e (5 mmol), saturated cyclic amine (15 mmol), and potassium carbonate (10 mmol) in 1,4-dioxane (30 mL) was refluxed until the reaction was complete (10–12 h). Then, 1,4-dioxane was removed by a Heidolph rotary evaporator. The crude solid was purified by recrystallization with ethanol
:water (2:8, v/v) solvent or column chromatography on silica gel using hexane/ethyl acetate as eluent. Reaction yields range from 80 to 88%.
4.2.7.2 The microwave-assisted method. A mixture of mono-substituted s-triazine derivatives 1a–e (5 mmol), saturated cyclic amine (15 mmol), and potassium carbonate (10 mmol) in 1,4-dioxane (10 mL) was placed in a microwave synthesizer and irradiated at a fixed power of 300 W at 107 °C until the reaction was complete (15–30 min). Then, 1,4-dioxane was removed by a Heidolph rotary evaporator. The crude solid was purified by recrystallization with ethanol
:water (2:8, v/v) solvent or column chromatography on silica gel using hexane/ethyl acetate as eluent. Reaction yields range from 91 to 98%.
4.2.7.3 Solubility. The synthesized compounds are well soluble in DMSO, 1,4-dioxane, and methanol; moderately soluble in ethanol; poorly soluble in non-polar solvents (hexane and ethyl acetate) and water.
4.2.7.4 Stability. The tri-substituted s-triazine derivatives are stable at room temperature and can be stored long-term at low temperatures of 4–8 °C.
N), 1240 (C–N, s-triazine), 804 (Ar–Cl). 1H NMR (500 MHz, DMSO-d6, δ ppm): 8.96 (1H, s, NH), 7.69 (2H, d, J = 8.5 Hz, HAr), 7.07 (2H, d, J = 8.5 Hz, HAr), 3.69 (8H, t, J = 4.5 Hz, –CH2–), 1.59 (4H, t, J = 4.5 Hz, –CH2–), 1.48 (8H, s, –CH2–). 13C NMR (125 MHz, DMSO-d6, δ ppm): 164.39, 164.35, 164.01, 163.99, 157.8, 155.9, 139.6, 136.96, 136.94, 128.0, 124.6, 120.8, 120.7, 120.6, 114.7, 114.5, 43.5, 25.3, 24.3. LC-MS (m/z) [M + H]+ calcd for C19H26ClN6 373.1902, found 373.1898.N), 1109 (C–N, s-triazine), 802 (Ar–Cl). 1H NMR (500 MHz, DMSO-d6, δ ppm): 8.82 (1H, s, NH), 7.65 (2H, d, J = 9.0 Hz, HAr), 7.01 (2H, d, J = 9.0 Hz, HAr), 3.70 (8H, t, J = 5.5 Hz, –CH2–), 3.63 (8H, t, J = 5.0 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6, δ ppm): 164.7, 163.8, 158.0, 156.1, 136.3, 121.2, 121.1, 114.4, 114.2, 65.6, 43.2. LC-MS (m/z) [M + H]+ calcd for C17H22ClN6O2 377.1487, found 377.1477.N), 1207 (Ar–F), 1107 (C–N, s-triazine). 1H NMR (500 MHz, DMSO-d6, δ ppm): 8.66 (1H, s, –NH–), 7.68 (2H, d, J = 9.0 Hz, HAr), 7.04 (2H, d, J = 9.0 Hz, HAr), 3.70 (8H, t, J = 5.5 Hz, –CH2–), 1.62 (4H, t, J = 5.0 Hz, –CH2–), 1.52 (8H, t, J = 4.0 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6, δ ppm): 164.4, 164.0, 157.86 and 155.98 (JC–F = 235 Hz), 136.99 and 136.98 (JC–F = 1.25 Hz), 120.78 and 120.72 (JC–F = 7.5 Hz), 114.78 and 114.61 (JC–F = 21.25 Hz), 43.6, 25.3, 24.3. LC-MS (m/z) [M + H]+ calcd for C19H26FN6 357.2197, found 357.2124.N), 1253 (Ar–F), 1109 (C–N, s-triazine). 1H NMR (500 MHz, DMSO-d6, δ ppm): 9.11 (1H, s, –NH–), 7.66 (2H, d, J = 9.0 Hz, HAr), 7.08 (2H, d, J = 9.0 Hz, HAr), 3.68 (8H, t, J = 5.0 Hz, –CH2–), 3.61 (8H, t, J = 5.0 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6, δ ppm): 164.7, 163.9, 158.09 and 156.20 (JC–F = 236.25 Hz), 136.6, 121.19 and 121.13 (JC–F = 7.5 Hz), 114.90 and 114.73 (JC–F = 21.25 Hz), 66.0, 43.3. LC-MS (m/z) [M + H]+ calcd for C17H22FN6O2 361.1783, found 361.1778.N), 1033 (C–N, s-triazine). 1H NMR (500 MHz, DMSO-d6, δ ppm): 8.72 (1H, s, NH), 7.57 (2H, d, J = 9.0 Hz, HAr), 6.83 (2H, d, J = 9.0 Hz, HAr), 3.70 (3H, s, –OCH3), 3.68 (8H, t, J = 5.0 Hz, –CH2-), 1.59 (4H, t, J = 4.5 Hz, –CH2–), 1.48 (8H, d, J = 3.5 Hz, –CH2). 13C NMR (125 MHz, DMSO-d6, δ ppm): 164.5, 164.0, 154.0, 133.8, 120.8, 113.5, 55.1, 43.5, 25.4, 24.4. LC-MS (m/z) [M + H]+ calcd for C20H29N6O 369.2397, found 369.2299.N), 1109 (C–N, s-triazine), 800 (Ar–Cl). 1H NMR (500 MHz, DMSO-d6, δ ppm): 8.90 (1H, s, NH), 7.55 (2H, d, J = 9.0 Hz, HAr), 6.84 (2H, d, J = 9.0 Hz, HAr), 3.70 (3H, s, –OCH3), 3.68 (8H, t, J = 5.0 Hz, –CH2–), 3.61 (8H, t, J = 4.5 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6, δ ppm): 164.7, 163.9, 154.2, 133.3, 121.1, 113.6, 66.3, 66.0, 55.1, 43.3. LC-MS (m/z) [M + H]+ calcd for C18H25N6O3 373.1983, found 373.1981.N), 1101 (C–N, s-triazine). 1H NMR (500 MHz, DMSO-d6, δ ppm): 8.48 (1H, s, NH), 7.55 (2H, d, J = 8.5 Hz, HAr), 7.04 (2H, d, J = 8.0 Hz, HAr), 3.70 (8H, t, J = 5.5 Hz, –CH2–), 2.24 (3H, s, –CH3), 1.63 (4H, t, J = 5.5 Hz, –CH2–), 1.52 (8H, t, J = 5.5 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6, δ ppm): 164.4, 163.9, 137.7, 129.6, 128.2, 119.3, 43.3, 24.9, 24.0, 19.8. LC-MS (m/z) [M + H]+ calcd for C20H29N6 353.2448, found 353.2369.N), 1109 (C–N, s-triazine). 1H NMR (500 MHz, DMSO-d6, δ ppm): 8.95 (1H, s, NH), 7.53 (2H, d, J = 8.5 Hz, HAr), 7.05 (2H, d, J = 8.0 Hz, HAr), 3.69 (8H, t, J = 5.0 Hz, –CH2–), 3.61 (8H, t, J = 4.5 Hz, –CH2–), 2.23 (3H, s, –CH3). 13C NMR (125 MHz, DMSO-d6, δ ppm): 164.7, 163.9, 137.6, 130.2, 128.7, 119.7, 66.0, 43.3, 20.3. LC-MS (m/z) [M + H]+ calcd for C18H25N6O2 357.2304, found 357.2304.N), 1109 (C–N, s-triazine). 1H NMR (500 MHz, DMSO-d6, δ ppm): 9.46 (1H, s, NH), 8.13 (2H, d, J = 9.5 Hz, HAr), 7.95 (2H, d, J = 9.5 Hz, HAr), 3.73 (8H, t, J = 5.0 Hz, –CH2–), 1.65 (4H, t, J = 5.0 Hz, –CH2-), 1.56–1.51 (8H, m, –CH2–). 13C NMR (125 MHz, DMSO-d6, δ ppm): 164.2, 163.7, 147.0, 140.2, 124.1, 118.0, 43.5, 24.9, 23.8. LC-MS (m/z) [M + H]+ calcd for C19H26N7O2 384.2142, found 384.2064.N), 1109 (C–N, s-triazine). 1H NMR (500 MHz, DMSO-d6, δ ppm): 9.85 (1H, s, NH), 8.16 (2H, d, J = 9.5 Hz, HAr), 7.93 (2H, d, J = 9.5 Hz, HAr), 3.71 (8H, t, J = 4.5 Hz, –CH2–), 3.64 (8H, t, J = 4.5 Hz, –CH2–). 13C NMR (125 MHz, DMSO-d6, δ ppm): 164.6, 163.9, 147.1, 140.4, 124.8, 118.5, 65.9, 43.4. LC-MS (m/z) [M + H]+ calcd for C17H20N7O4 386.1582, found 386.1509.4.3. In vitro anticancer activity
The cytotoxic activity of the s-triazine derivatives was evaluated against the two cancer cell lines (MCF7 – ATCC HTB-22 and C26 – ATCC CRL-2638) and one normal cell line (BAEC – CSC 2B2, Cell Systems) using the methyl thiazolyl tetrazolium (MTT) method conducted according to the MTT assay protocol. PTX and DOX were used as the positive control. The assay detects the reduction of yellow tetrazolium by metabolically active cells to purple formazan, which is measured using spectrophotometry. The cell lines were seeded into 96-well plates at a density of 5000 cells per well and incubated at 37 °C in 5% CO2 for 24 h with growth media consisting of 2 mM L-glutamine, 100 IU mL−1 penicillin, 100 μg mL−1 streptomycin, Eagle's minimum essential medium, and 10% fetal calf serum. After that, a series of concentrations of the tested compounds and the reference drugs (PTX and DOX) in DMSO was added to each well of the plate and incubated for 24 h. The 10 μL fresh solution of MTT reagent was added to each well, and the plate was incubated in a CO2 incubator at 37 °C for 4 h until a purple precipitate appeared. Finally, the cells were solubilized in ethanol and their optical density was recorded at 570 nm.5,7 The percent of proliferation inhibition was calculated using the following formula:At = absorption of test compound, Ab = absorption of blank, Ac = absorption of control.

4.4. ADME-Tox predictions
The physicochemical and in silico ADMET properties were performed using ADMETlab 3.0 descriptors algorithm protocol.584.5. In silico molecular docking studies
The structure of ligands and reference drugs were drawn in ChemBioDraw Ultra 19.0 and the energy of each ligand was minimized using ChemBio3D Ultra 19.0.5–9 The ligands with minimized energy were then used as input for AutoDock Vina, in order to carry out the docking simulation. Anticancer targets of DHFR, VEGFR2, PI3K, CDK2, EGFR, and mTOR were retrieved from the protein data bank (Table 5). The targets were removed all the water molecules and added only polar hydrogen and Kollman charges. The AutoDock Tools was used to set the grid box for docking simulations. In addition, the docking protocol was validated by extracting and re-docking the co-crystallized ligand into the active site. Then, synthesized compounds and reference drugs were docked with the target to determine the docking parameters with the help of grid-based ligand docking (Table 6). Finally, the interaction information (bond type, length, amino acid residue, etc.) and the pictorial representation of the interaction between the ligands and the target were processed using Discovery Studio 2021 software.| Entry | Target | Symbol | PDB ID | Organism | Expression system | Ref drug |
|---|---|---|---|---|---|---|
| 1 | Dihydrofolate reductase | DHFR | 1RG7 | Escherichia coli | Escherichia coli | Methotrexate |
| 2 | Vascular endothelial growth factor receptor 2 | VEGFR2 | 4AG8 | Homo sapiens | Spodoptera frugiperda | Axitinib |
| 3 | Phosphoinositide 3-kinase | PI3K | 5JHB | Homo sapiens | Spodoptera frugiperda | Gedatolisib |
| 4 | Cyclin-dependent kinase 2 | CDK2 | 2 A4L | Homo sapiens | Spodoptera frugiperda | Seliciclib |
| 5 | Epidermal growth factor receptor | EGFR | 1M17 | Homo sapiens | Spodoptera frugiperda | Erlotinib |
| 6 | Mammalian target of rapamycin | mTOR | 4JSV | Homo sapiens | Homo sapiens | Gedatolisib |
| Target | Size | Center | ||||
|---|---|---|---|---|---|---|
| x | y | z | x | y | z | |
| DHFR | 40 | 40 | 40 | −1.65679 | 22.0285 | 23.0763 |
| VEGFR2 | 40 | 40 | 40 | 20.8237 | 25.5351 | 39.4596 |
| PI3K | 40 | 40 | 40 | 21.3492 | −4.07429 | 20.8696 |
| CDK2 | 40 | 40 | 40 | 100.865 | 101.747 | 79.8926 |
| EGFR | 40 | 40 | 40 | 22.0137 | 0.252828 | 52.794 |
| mTOR | 40 | 40 | 40 | 50.1244 | −1.7953 | −45.9034 |
4.6. Statistical analysis
All values are expressed in Mean ± SEM (Standard Error of Mean). The difference in IC50 value between tested compounds and reference drug was analyzed by one-way analysis of variance (ANOVA) with Tukey's Honestly Significant Difference (Tukey HSD) post hoc test using SPSS 26 software. The results were considered statistically significant if p < 0.05. The chart is drawn using Microsoft Excel software.Data availability statements
The data supporting this article have been included as part of the ESI.†Author contributions
Em Canh Pham: conceptualization, methodology, investigation, data curation, supervision, writing-original draft preparation, writing – review & editing. Bich-Ngoc Thi Le: investigation. Anh Minh Ngo: investigation. Long Binh Vong: investigation, supervision. Tuyen Ngoc Truong: data curation, supervision, writing-original draft preparation, writing – review & editing.Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.Acknowledgements
[Em Canh Pham] was funded by the PhD Scholarship Programme of Vingroup Innovation Foundation (VINIF), code [VINIF.2023.TS.025].References
- F. Bray, J. Ferlay, I. Soerjomataram, R. L. Siegel, L. A. Torre and A. Jemal, Ca-Cancer J. Clin., 2018, 68, 394–424 CrossRef PubMed.
- F. Bray, M. Laversanne, H. Sung, J. Ferlay, R. L. Siegel, I. Soerjomataram and A. Jemal, Ca-Cancer J. Clin., 2024, 74, 229–263 CrossRef PubMed.
- WHO, Breast cancer [13 March 2024]. Available online: https://www.who.int/news-room/fact-sheets/detail/breast-cancer.
- WHO, Colorectal cancer [11 July 2023]. Available online: https://www.who.int/news-room/fact-sheets/detail/colorectal-cancer.
- P. C. Em, L. T. Tuong Vi, L. H. Huong Ha, V. T. Bich Ngoc, B. V. Long, V. T. Thao, D. V. Duy, T. N. Ngoc Vi, N. L. Bao Khanh and N. T. Tuyen, RSC Adv., 2023, 13, 399–420 RSC.
- P. C. Em and N. T. Tuyen, ACS Omega, 2022, 7, 33614–33628 Search PubMed.
- P. C. Em, L. T. Tuong Vi and N. T. Tuyen, RSC Adv., 2022, 12, 21621–21646 Search PubMed.
- P. C. Em, L. T. Tuong Vi, T. P. Long, T. N. Huong-Giang, N. L. Bao Khanh and N. T. Tuyen, Arabian J. Chem., 2022, 15, 103682 CrossRef.
- P. C. Em, N. T. Tuyen, D. Hanh Nguyen, D. V. Duy and D. T. Hong Tuoi, Med. Chem., 2022, 18, 558–573 CrossRef PubMed.
- B. Viira, A. Selyutina, A. T. García-Sosa, M. Karonen, J. Sinkkonen, A. Merits and U. Maran, Bioorg. Med. Chem., 2016, 24, 2519–2529 Search PubMed.
- R. Kumar, A. D. Singh, J. Singh, H. Singh, R. K. Roy and A. Chaudhary, Mini-Rev. Med. Chem., 2014, 14, 72–83 CrossRef PubMed.
- H. R. Bhat, A. Masih, A. Shakya and S. Kumar, J. Heterocycl. Chem., 2020, 57, 390–399 Search PubMed.
- P. Asadi, M. Alvani, V. Hajhashemi, M. Rostami and G. Khodarahmi, J. Mol. Struct., 2021, 1243, 130760 Search PubMed.
- L. Marín-Ocampo, L. A. Veloza, R. Abonia and J. C. Sepúlveda-Arias, Eur. J. Med. Chem., 2019, 162, 435–447 Search PubMed.
- A. Shakya and J. Heterocyclic, Chem, 2020, 57, 2389–2399 Search PubMed.
- N. Adhikari, A. Kashyap, A. Shakya, S. K. Ghosh, D. R. Bhattacharyya, H. R. Bhat and U. P. Singh, J. Heterocycl. Chem., 2020, 57, 2389–2399 CrossRef.
- J. Hu, Y. Zhang, N. Tang, Y. Lu, P. Guo and Z. Huang, Bioorg. Med. Chem., 2021, 32, 115997 CrossRef PubMed.
- Y. Mehmood, F. Anwar, U. Saleem, S. Hira, B. Ahmad, M. Bashir, M. T. Imtiaz, S. Najm and T. Ismail, Life Sci., 2021, 285, 119994 CrossRef PubMed.
- M. Tayyab Imtiaz, F. Anwar, U. Saleem, B. Ahmad, S. Hira, Y. Mehmood, M. Bashir, S. Najam and T. Ismail, Front. Pharmacol, 2021, 12, 686614 CrossRef PubMed.
- I. Shawish, M. S. Nafie, A. Barakat, A. Aldalbahi, H. H. Al-Rasheed, M. Ali, W. Alshaer, M. Al Zoubi, S. Al Ayoubi, B. G. De la Torre, F. Albericio and A. El-Faham, Front. Chem., 2022, 10, 1078163 Search PubMed.
- S. Oggu, P. Akshinthala, N. K. Katari, L. K. Nagarapu, S. Malempati, R. Gundla and S. B. Jonnalagadda, Heliyon, 2023, 9, e15935 Search PubMed.
- J. Li, L. Li, Y. Liu, J. Zhang, C. Shi, S. Zhou and H. Qiu, Heterocycl. Commun., 2023, 29, 20220152 Search PubMed.
- M. A. Alelaimat, M. A. Al-Sha'er and H. A. Basheer, ACS Omega, 2023, 8, 14247–14263 Search PubMed.
- R. Dona, R. A. Rizki, J. Jasril, N. Frimayanti and R. Hendra, Pharm. Edu., 2024, 24, 172–178 Search PubMed.
- D. Maliszewski and D. Drozdowska, Pharmaceuticals, 2022, 15, 221 Search PubMed.
- Q. Dai, Q. Sun, X. Ouyang, J. Liu, L. Jin, A. Liu, B. He, T. Fan and Y. Jiang, Molecules, 2023, 28, 4278 Search PubMed.
- H. Y. Lim and A. V. Dolzhenko, Eur. J. Med. Chem., 2024, 276, 116680 Search PubMed.
- N. Keldsen, H. Havsteen, I. Vergote, K. Bertelsen and A. Jakobsen, Gynecol. Oncol., 2003, 88, 118–122 Search PubMed.
- B. A. Chabner, P. C. Amrein, B. Druker, M. D. Michaelson, C. S. Mitsiades, P. E. Goss, D. P. Ryan, S. Ramachandra, P. G. Richardson and J. G. Supko, Antineoplastic Agents, Mcgraw-Hill (Tx), New York, 9th ed, 2006 Search PubMed.
- M. I. Del Principe, G. Paterno, R. Palmieri, L. Maurillo, F. Buccisano and A. Venditti, Expert Opin. Pharmacother., 2019, 20, 1935–1942 Search PubMed.
- R. E. Shor, J. Dai, S.-Y. Lee, L. Pisarsky, I. Matei, S. Lucotti, D. Lyden, M. J. Bissell and C. M. Ghajar, Mol. Oncol., 2022, 16, 130–147 Search PubMed.
- H. Guan, B. Mi, Y. Li, W. Wu, P. Tan, Z. Fang, J. Li, Y. Zhang and F. Li, Cell. Signalling, 2015, 27, 969–977 Search PubMed.
- O. Salim, T. Toptas, E. Avsar, O. K. Yucel, E. Ozturk, B. Ferhanoglu, A. Geduk, O. Mehtap, A. Tombak, E. N. Tiftik, B. Deveci, E. Kurtoglu, O. Kara, I. K. Atagunduz, T. F. Tuglular and L. Undar, Leuk. Res., 2016, 45, 82–89 Search PubMed.
- H.-L. Ng, X. Ma, E.-H. Chew and W.-K. Chui, J. Med. Chem., 2017, 60, 1734–1745 Search PubMed.
- X. Zhou, K. Lin, X. Ma, W.-K. Chui and W. Zhou, Eur. J. Med. Chem., 2017, 125, 1279–1288 Search PubMed.
- P. Pathak, P. K. Shukla, V. Kumar, A. Kumar and A. Verma, Inflammopharmacology, 2018, 26, 1441–1453 Search PubMed.
- M. A. Alelaimat, M. A. Al-Sha’er and H. A. Basheer, ACS Omega, 2023, 8, 14247–14263 CrossRef PubMed.
- G.-H. Kuo, A. DeAngelis, S. Emanuel, A. Wang, Y. Zhang, P. J. Connolly, X. Chen, R. H. Gruninger, C. Rugg, A. Fuentes-Pesquera, S. A. Middleton, L. Jolliffe and W. V. Murray, J. Med. Chem., 2005, 48, 4535–4546 CrossRef PubMed.
- M. S. Raghu, C. B. P. Kumar, M. K. Prashanth, K. Y. Kumar, B. S. Prathibha, G. Kanthimathi, S. A. Alissa, H. A. Alghulikah and S. M. Osman, New J. Chem., 2021, 45, 13909–13924 RSC.
- W. Yan, Y. Zhao and J. He, Mol. Med. Rep., 2018, 18, 4175–4185 Search PubMed.
- B. Zhang, Q. Zhang, Z. Xiao, X. Sun, Z. Yang, Q. Gu, Z. Liu, T. Xie, Q. Jin, P. Zheng, S. Xu and W. Zhu, Bioorg. Chem., 2020, 95, 103525 CrossRef PubMed.
- K. Bergant, M. Janežič, K. Valjavec, I. Sosič, S. Pajk, M. Štampar, B. Žegura, S. Gobec, M. Filipič and A. Perdih, Eur. J. Med. Chem., 2019, 175, 330–348 CrossRef PubMed.
- A. I. Zain-Alabdeen, T. F. El-Moselhy, N. Sharafeldin, A. Angeli, C. T. Supuran and M. H. El-Hamamsy, Sci. Rep., 2022, 12, 16756 CrossRef PubMed.
- P. Singla, V. Luxami and K. Paul, Eur. J. Med. Chem., 2015, 102, 39–57 Search PubMed.
- M. I. Ali and M. M. Naseer, RSC Adv., 2023, 13, 30462–30490 RSC.
- K. M. Al-Zaydi, H. H. Khalil, A. El-Faham and S. N. Khattab, Chem. Cent. J., 2017, 11, 39 CrossRef.
- I. Shawish, A. Barakat, A. Aldalbahi, A. M. Malebari, M. S. Nafie, A. A. Bekhit, A. Albohy, A. Khan, Z. Ul-Haq, M. Haukka, B. G. de la Torre, F. Albericio and A. El-Faham, ACS Omega, 2022, 7, 24858–24870 CrossRef PubMed.
- P. Singla, V. Luxami and K. Paul, Bioorg. Med. Chem., 2015, 23, 1691–1700 CrossRef PubMed.
- V. V. Veselov, A. E. Nosyrev, L. Jicsinszky, R. N. Alyautdin and G. Cravotto, Cancers, 2022, 14, 622 Search PubMed.
- J. Li, A. Fu and L. Zhang, Interdiscip. Sci.:Comput. Life Sci., 2019, 11, 320–328 Search PubMed.
- D. Barradas-Bautista, I. H. Moal and J. Fernández-Recio, Proteins, 2017, 85, 1287–1297 Search PubMed.
- J. A. Erickson, M. Jalaie, D. H. Robertson, R. A. Lewis and M. Vieth, J. Med. Chem., 2004, 47, 45–55 Search PubMed.
- S. Y. Huang and X. Zou, Int. J. Mol. Sci., 2010, 11, 3016–3034 Search PubMed.
- S. E. Wong and F. C. Lightstone, Expert Opin. Drug Discovery, 2011, 6, 65–74 Search PubMed.
- N. Huang and B. K. Shoichet, J. Med. Chem., 2008, 51, 4862–4865 Search PubMed.
- M. Totrov and R. Abagyan, Curr. Opin. Struct. Biol., 2008, 18, 178–184 Search PubMed.
- O. Trott and A. J. Olson, J. Comput. Chem., 2010, 31, 455–461 Search PubMed.
- L. Fu, S. Shi, J. Yi, N. Wang, Y. He, Z. Wu, J. Peng, Y. Deng, W. Wang, C. Wu, A. Lyu, X. Zeng, W. Zhao, T. Hou and D. Cao, Nucleic Acids Res., 2024, 52, W422–W431 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: ADMET and docking information, and NMR spectra. See DOI: https://doi.org/10.1039/d4ra08508f |
| This journal is © The Royal Society of Chemistry 2025 |