
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynergistic effect of scattered rare metals on Pt/CeO2 for propane oxidative dehydrogenation with CO2†
Jiulong Wanga,
Fang Wanga,
Longyang Lia,
Weihao Zhaoa,
Shiyuan Wanga,
Zelin Maa,
Yan Kongb,
Yazhou Shuanga,
Weiwei Xiaa,
Jie Jiana,
Pengfei Guoa and
Hongqiang Wang *a
*a
aState Key Laboratory of Solidification Processing, Center for Nano Energy Materials, School of Materials Science and Engineering, Northwestern Polytechnical University (NPU), Shaanxi Joint Laboratory of Graphene, Xi'an, 710072, China. E-mail: hongqiang.wang@nwpu.edu.cn
bMinistry of Education Engineering Research Center of Water Resource Comprehensive Uti-lization in Cold and Arid Regions, School of Environmental and Municipal Engineering, Lanzhou Jiaotong University, Lanzhou, 730070, China
First published on 23rd January 2025
Abstract
The oxidative dehydrogenation of propane with CO2 (CO2-ODP) is a green industrial process for producing propene. Cerium oxide-supported platinum-based (Pt/CeO2) catalysts exhibit remarkable reactivity toward propane and CO2 due to the unique delicate balance of C–H and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bond activation. However, the simultaneous activation and cleavage of C–H, C–C, and C–O bonds on Pt/CeO2-based catalysts may substantially impede the selective activation of C–H bonds during the CO2-ODP process. Here, we report that the scattered rare metal oxide (SROx, SR = Ga, In) overlayer on Pt/CeO2 exhibits extraordinary activity and selectivity for the CO2-ODP reaction. With the assistance of Pt, the SROx-Pt/CeO2 could achieve a propane conversion of 38.13% and a CO2 conversion of 67.72%. More importantly, the selectivity of the product propene has increased from 33.28% to 88.24%, a level that is even comparable to the outstanding performance of currently reported PtSn/CeO2 catalysts. A mechanistic study reveals that the strong affinity of the overlayer SROx to the propane reduces the barrier of C–H bond activation and balances the C–H cleavage rates and the C–O bond groups, accounting for the excellent selective CO2-ODP performance of SROx-Pt/CeO2 catalysts. The SROx-modified Pt/CeO2 strategy offers a novel approach to modulating CO2-ODP, thereby facilitating the highly selective preparation of propene.
O bond activation. However, the simultaneous activation and cleavage of C–H, C–C, and C–O bonds on Pt/CeO2-based catalysts may substantially impede the selective activation of C–H bonds during the CO2-ODP process. Here, we report that the scattered rare metal oxide (SROx, SR = Ga, In) overlayer on Pt/CeO2 exhibits extraordinary activity and selectivity for the CO2-ODP reaction. With the assistance of Pt, the SROx-Pt/CeO2 could achieve a propane conversion of 38.13% and a CO2 conversion of 67.72%. More importantly, the selectivity of the product propene has increased from 33.28% to 88.24%, a level that is even comparable to the outstanding performance of currently reported PtSn/CeO2 catalysts. A mechanistic study reveals that the strong affinity of the overlayer SROx to the propane reduces the barrier of C–H bond activation and balances the C–H cleavage rates and the C–O bond groups, accounting for the excellent selective CO2-ODP performance of SROx-Pt/CeO2 catalysts. The SROx-modified Pt/CeO2 strategy offers a novel approach to modulating CO2-ODP, thereby facilitating the highly selective preparation of propene.
Introduction
Propene, a vital building block in the chemical industry, is widely used to produce numerous chemicals. Conventional methods for propene production, such as steam cracking and fluid catalytic cracking, are facing challenges in meeting the escalating demand and improving yields.1,2 As a result, alternative technologies for propene production are being actively explored. The oxidative dehydrogenation of propane with CO2 (CO2-ODP) has emerged as an attractive approach. This process offers a route to produce propene and enables the reduction of CO2 to CO, presenting an environmentally friendly solution.3–5 The presence of CO2 provides an O atom that reacts with H atoms from propane to produce H2O, enhancing the equilibrium conversion of propane.6 Additionally, CO2 acts as a mild oxidant, effectively suppressing the undesired deep oxidation of propane, resulting in a high propene yield. Although the CO2-ODP process can suppress the undesired deep oxidation of propane, the pronounced competitive adsorption between propane and CO2 constrains the high selectivity for the desired product, propene. Consequently, the design of highly selective catalysts for the efficient modulation of reactivity represents a paramount scientific challenge that warrants urgent attention.To date, the investigated catalysts for CO2-ODP are based on metal oxides and zeolites have been explored as catalysts.7–16 Among these, metal oxides have garnered considerable attention due to their capacity to function as active sites while simultaneously mitigating carbon deposition, thereby enhancing the stability and longevity of catalysts.8,17–19 For instance, CeO2, Al2O3, TiO2, and ZrO2, are extensively utilized in propane oxidative dehydrogenation on account of their outstanding thermal stability, specific redox properties, and well-defined acidity/alkalinity.8,18–20 In particular, CeO2 is employed as a support owing to its exceptional capabilities in oxygen storage, CO2 capture, and coke combustion.21 The presence of abundant oxygen vacancies facilitates robust anchoring for the loading of active metals, thereby synergistically enhancing both catalytic activity and selectivity.22 Nonetheless, these materials continue to encounter difficulties in achieving efficient synergistic activation of propane and CO2. Pt metal is widely acknowledged as the primary active site in the propane dehydrogenation reaction, capable of activating propane and CO2 molecules. Of these, the Pt/CeO2 system has demonstrated particular promise in terms of its ability to cleave C–C bonds in propane and to reduce CO2 emissions.19,23 To design an effective CO2-ODP catalyst, a delicate balance in activating the CO bonds in CO2 and the C–H bonds in propane is required. Bifunctional catalysts that can achieve this dual activation have been investigated, with the combination of Pt-scattered rare metals emerging as a promising strategy, even comparable to Pt–Sn bimetals.17,18,24–26 This is attributed to the oxides of scattered rare metals (Ga, In) exhibiting a greater reactivity toward the cleavage of C–H bonds compared to C–C bonds in propane. Although some insight has been gained into the role of Pt-scattered rare metal in enhancing Pt dispersion and electron transfer, the understanding of synergistic mechanism between the carrier and multiple active sites in the CO2-ODP process remain limited.
Here, we report that the scattered rare metal oxide (SROx) and SnOx overlayer formed over Pt/CeO2 exhibits extraordinary activity and selectivity for the CO2-ODP reaction. Specifically, the traditional wetness impregnation and calcination method are adopted, CeO2 is selected as the carrier, Pt and scattered rare metal (Ga, In) and Sn species are prone to form an oxide overlayer, while the oxygen vacancies formed by the conversion of Ce4+ to Ce3+ provide abundant anchoring sites for the Pt and scattered rare metal species. As a result, the scattered rare metal, Sn and Pt species can be firmly anchored on CeO2 carriers. The incorporation of SROx and SnOx facilitates pronounced electronic interactions with the Pt species, leading to a synergistic effect that restrains the activation and cleavage of C–C bonds in propane by the highly active Pt sites. In turn, this restraint promotes the cleavage of C–H bonds and enhances the dehydrogenation process. Moreover, the presence of SROx and SnOx serves to optimize the surface properties of the catalysts, thereby improving CO2 activation and the selectivity for propene products. Additionally, systematic investigations into the CO2-ODP reveal that the introduction of optimal quantities of scattered rare metals establishes a delicate equilibrium among the activation and cleavage of C–H, CO, and C–C bonds during the CO2-ODP process. This balance enhances the activation of the CO bond and facilitates the cleavage and dehydrogenation of C–H bonds, while concurrently suppressing the activation of C–C bonds. Consequently, this approach enables efficient conversion of CO2 while achieving a high selectivity for propene production. Therefore, by modulating the electronic structure and surface properties of the catalytic system, the scattered rare metal facilitates a nuanced balance between the selective activation and cleavage of C–H, C–C, and CO bonds. This study offers a novel perspective for the advancement of high-performance Pt-based catalysts for CO2-ODP by exploiting the modulation of primary active site strategies via the incorporation of scattered rare metal species.
Results and discussion
The structure and characterization of the catalysts
The hybrid materials of scattered rare metals and Sn species on Pt/CeO2 were prepared via successive synthesis procedures illustrated in Fig. 1a. CeO2 is obtained by the urea-assisted hydrothermal method and high-temperature calcination. The synthesized CeO2 as support has a cubic fluorite structure, which can provide abundant oxygen vacancies, promote the adsorption and activation of oxygen and thereby participate in the oxidative dehydrogenation reaction of propane.27 The supported PtGa/CeO2, PtIn/CeO2, and PtSn/CeO2 were prepared using the incipient wetness impregnation method. The obtained PtGa/CeO2, PtIn/CeO2, PtSn/CeO2, and Pt/CeO2 powder catalysts appear in dark brown, khaki, gray-black, and light brown colors, respectively, forming a sharp contrast with the light yellow CeO2 (Fig. S1†). ICP-OES was employed to quantify the concentrations of Pt, Ga, Sn, and In in the synthesized catalyst, yielding values of 0.94–1.28 wt%, 0.31–5.2 wt%, 3.10 wt%, and 2.97 wt%, respectively (Table S1†). The obtained loads exhibited a general consistency with the theoretically calculated values. Further investigation of the morphology and chemistry of the catalyst via scanning electron microscopy (SEM) and energy-dispersive X-ray spectroscopy (EDX) mapping. As shown in Fig. 1b–e, the deposited metallic species do not change the irregular microstructure of the CeO2. In addition, SEM and SEM-EDX with different magnifications show that PtGa, PtIn and PtSn are uniformly loaded on the CeO2 support (Fig. S2–S7†). Transmission electron microscopy (TEM) images show that PtGa/CeO2 (Fig. 1f and g), PtIn/CeO2 (Fig. 1l–m), and PtSn/CeO2 (Fig. 1r) exhibit similar unique nanostructures. High-resolution transmission electron microscopy (HR-TEM) images reveal that Pt, Ga, In, and Sn are mainly uniformly dispersed on the CeO2 support in their oxidized states (Fig. 1h–k, n–p and s–v). Specifically, the distances between the diffraction fringes of different components were found to be 0.31, 0.30, 0.26, 0.49, and 0.47 nm, respectively, identical to the CeO2 (111), PtO (100), Ga2O3 (111), In2O3 (200), and SnO2 (001) crystal planes (Fig. 1i, j, p, u and v).TEM-EDX elemental mapping further confirmed that Pt, Ce, Ga, In, and Sn elements are uniformly distributed throughout the CeO2 support in the catalyst (Fig. 1k, q and w). These results preliminarily demonstrate that the rare metals and Pt are mainly loaded on the CeO2 support in an oxidized state, forming a unique metal oxide coverage layer. This unique microstructure will provide abundant active sites and good electron transfer channels for the propane oxidative dehydrogenation reaction catalyst. | ||
| Fig. 1 (a) A schematic illustration of the synthesis procedure of M/CeO2 composites. (b–e) SEM images of CeO2, PtGa/CeO2, PtIn/CeO2, PtSn/CeO2. TEM, HR-TEM, and corresponding mapping of PtGa/CeO2 (f–k), PtIn/CeO2 (l–q), and PtSn/CeO2 (r–w). | ||
Textural and crystal properties
The Raman spectra of synthesized CeO2, Pt/CeO2, PtGa/CeO2, PtIn/CeO2 and PtSn/CeO2 are presented in Fig. (2a and b). These figures demonstrate that the Raman signal at ∼461 cm−1 correlates with the symmetric stretching Ce–O F2g mode in CeO2.23 After loading with metal, the intensity of the Ce–O peak is weakened, indirectly proving that the metal has been successfully loaded onto the CeO2 support, weakening the symmetric stretching peak of Ce–O. The F2g vibrational mode of CeO2 is highly sensitive to lattice distortions induced by the incorporation of metal ions. As shown in Fig. 2a, the Raman peaks corresponding to the F2g vibrational mode of CeO2 weaken upon metal loading, with those for Pt/CeO2, PtGa/CeO2, and PtIn/CeO2 shifting to higher wave numbers. This shift is attributed to the interaction between the metal species and oxygen, which leads to a reduction in the concentration of oxygen vacancies. In contrast, PtSn/CeO2 causes minimal lattice distortion, suggesting that the introduction of Sn prevents Pt from depleting the oxygen vacancies in CeO2.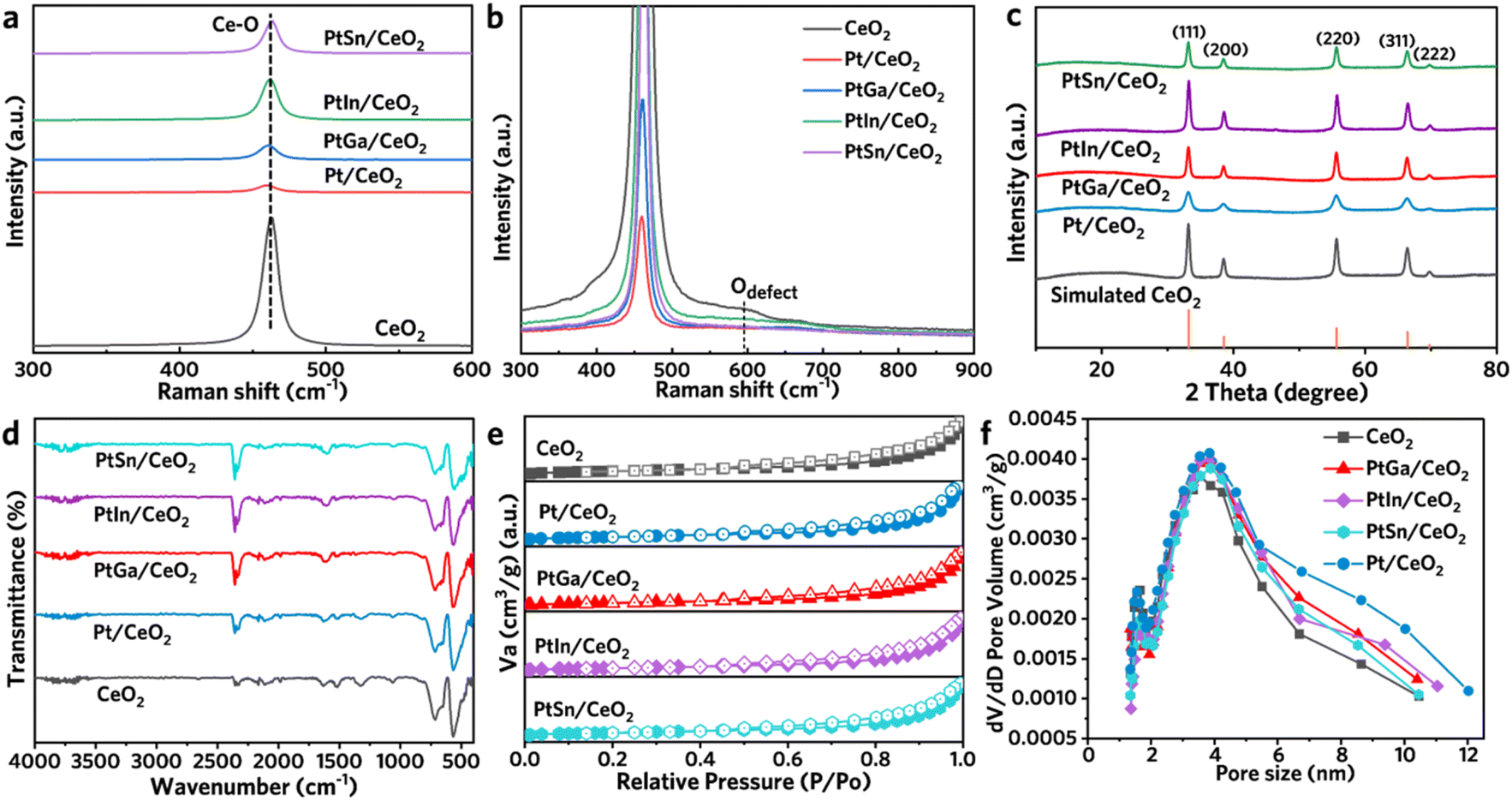 | ||
| Fig. 2 (a and b) Raman spectra at different regions of resh CeO2, Pt/CeO2, PtGa/CeO2, PtIn/CeO2 and PtSn/CeO2. (c) XRD patterns, (d) FTIR spectra, (e) N2 adsorption/desorption curves and (f) pore size distribution plots ofof fresh CeO2, Pt/CeO2, PtGa/CeO2, PtIn/CeO2 and PtSn/CeO2. | ||
As shown in Fig. 2b, the Raman band at 595 cm−1 is assigned to oxygen defects of catalysts. It reveals that abundant oxygen vacancies can promote the adsorption and activation of oxygen molecules, thereby participating in the oxidative dehydrogenation reaction of propane. The powder X-ray diffraction (XRD) patterns of CeO2, Pt/CeO2, PtGa/CeO2, PtIn/CeO2 and PtSn/CeO2 both showed the diffraction of the typical cubic structured CeO2 phase (Fig. S8 and 2c). As shown in Fig. 2c, the characteristic reflections at 2θ of 33.3°, 38.6°, 55.7°, 66.5° and 69.9 corresponding to (111), (200), (220), (311), and (222) of cubic CeO2 phase (PDF # 34-0394). No crystalline metallic Pt0, PtOx, GaOx, InOx, or SnOx were detected, suggesting that these species are highly dispersed on the surface of CeO2. Fourier transform infrared (FTIR) spectroscopy further confirmed that the composite of loaded Pt and scattered rare metals and Sn still exhibits the same surface functionalized structure as CeO2 (Fig. 2d).
As revealed from N2 adsorption–desorption isotherms, the CeO2, Pt/CeO2, PtGa/CeO2, PtIn/CeO2 and PtSn/CeO2 samples showed a type-IV isotherm (Fig. 2e). The specific surface area of CeO2 is 14.9 m2 g−1, which slightly decreases after loading with metals. The specific surface areas of Pt/CeO2, PtGa/CeO2, PtIn/CeO2, and PtSn/CeO2 are 13.8, 13.1, 12.3, and 12.5 m2 g−1, respectively (Table S2†). The average pore diameter ranges from 3.7 to 4.4 nm (Fig. 2f). These results indicate that the loaded metal species hardly occupy the pores of the CeO2 support, but instead dispersed uniformly on the surface of the support.
Surface chemistry
X-ray photoelectron spectroscopy (XPS) experiments were conducted to determine the surface compositions and oxidation state of metals on the catalyst, aiming to understand their roles in the catalytic process and to identify the active sites. As illustrated in Fig. 3a, the Ce 3d XPS profiles of Pt/CeO2, PtGa/CeO2, PtIn/CeO2, and PtSn/CeO2 exhibit distinct signals designated as v, v2, and v3, and u, u2,and u3 corresponding to the Ce4+ 3d5/2 and Ce4+ 3d3/2 states, respectively.22,28 In contrast, the signals labeled v1 and u1 are attributed to the Ce3+ 3d5/2 and Ce3+ 3d3/2 states, respectively. This result aligns with the Ce 3d spectra of pure CeO2 (Fig. S8†), suggesting that the superior redox properties of CeO2 are preserved following the incorporation of metal species. It is well established that the redox characteristics of CeO2 promote the formation of a significant concentration of oxygen vacancies, which serve as highly favorable sites for metal bonding.29 The O 1 s XPS spectra of the catalysts illustrated in Fig. S9† provide definitive evidence for the presence of lattice oxygen (OL), oxygen vacancies (Ov), and various oxygen species that are chemically adsorbed on the surface (Oa, such as O22−, O−, OH−), respectively. Furthermore, our findings indicate that the concentrations of oxygen vacancies in the PtGa/CeO2 (3.46%), PtIn/CeO2 (1.93%), and PtSn/CeO2 (3.76%) catalysts, following the incorporation of scattered rare metals and Sn, exhibited a notable decrease compared to those observed in the Pt/CeO2 (6.09%) catalysts (Fig. S9†). This trend suggests a significant interaction between the scattered rare metals and the oxygen vacancies. Similarly, the concentrations of oxygen vacancies in the Ga/CeO2, In/CeO2, and Sn/CeO2 catalysts exhibited a decreasing trend relative to those in the CeO2 catalysts (Fig. S10†). This observation further implies that the interaction between the scattered rare metals and oxygen vacancies may significantly influence the electronic structure and the performance of CO2-ODP. Concurrently, the analogous electronic structural characteristics of scattered rare metals and Sn suggest their significant potential to substitute Sn in modulating the catalytic activity of Pt/CeO2. | ||
| Fig. 3 (a–e) Ce 3d, Pt 4f, Ga 3d, In 3d and Sn 3d XPS of catalysts. | ||
The Pt 4f5/2 and Pt 4f7/2 spectra of Pt/CeO2 and PtSn/CeO2 have been deconstructed into two distinct peaks, whereas the Pt 4f5/2 and Pt 4f7/2 spectra of PtGa/CeO2 and PtIn/CeO2 remain unresolved into additional peaks (Fig. 3b). In comparison to Pt/CeO2 (72.82 eV), the binding energies of Pt2+ exhibit a negative shift in the cases of PtGa/CeO2 (72.74 eV), PtIn/CeO2 (72.78 eV), and PtSn/CeO2 (72.73 eV). This observation suggests a charge transfer interaction between the scattered rare metal species (Ga, In, and Sn) and Pt. This phenomenon is further elucidated by the 3d XPS spectra of Ga, In, and Sn metals, as depicted inFig. (3c and d). The Ga 3d spectra of the Ga/CeO2 and PtSGa/CeO2 catalysts exhibit four distinct peaks corresponding to Ga0, Ga1+, Ga3+ and O 2 s (Fig. 3c).30 Furthermore, the Ga2+ 3d spectra of PtGa/CeO2 (20.33 eV) tend to move in the direction of higher binding energy in comparison to Ga/CeO2 (20.2 eV). Similarly, the binding energy peaks of In 3d and Sn 3d in PtIn/CeO2 and PtSn/CeO2 exhibit a notable shift towards higher binding energies when compared to those observed in In/CeO2 and Sn/CeO2, respectively (Fig. 3d and e). Furthermore, In and Sn exhibit comparatively simpler electronic structures than Ga, which predominantly exists in an oxidized state within the catalyst. The deconvolution analysis of these peaks, conducted using the Gaussian curve fitting method, presents challenges in accurately identifying the In/Sn species in the zerovalent state. This difficulty suggests that the In/Sn species predominantly exists in an oxidized state within the catalyst. Specifically, the In 3d spectra for PtIn/CeO2 and In/CeO2 are presented in Fig. 3d. Notably, the In 3d5/2 peak appears at 444.78–444.83 eV, indicative of the oxidation state of In within the catalyst.31 Similarly, the Sn 3d spectra of PtSn/CeO2 and Sn/CeO2 indicated that the Sn species predominantly exist in the Sn2+/Sn4+ oxidation states within the catalyst (Fig. 3e).32 The aforementioned analyses furnish robust evidence for the predominant presence of the scattered rare metal in a specific oxidation state on the catalysts. Meanwhile, the potent electronic interactions between the Pt2+ active sites and the scattered rare metal species are likely to contribute to the augmented selectivity of propene in the CO2-ODP process. Consequently, this modification in electronic structure was associated with a significant enhancement in catalytic performance, suggesting a potential correlation between the chemical states of the scattered rare metals (Ga, In) and their respective catalytic activities.
Catalytic performance of CO2-ODP
To understand the synergistic regulation of C–H bond cleavage and CO bond activation behavior of the scattered rare metal and Pt sites on the CeO2 support, detailed propane dehydrogenation experiments were conducted. The CeO2 catalyst exhibits only modest average conversion for propane and CO2 were approximately 5.16% and 6.4%, respectively, with a propylene selectivity of about 34.31% (Fig. S11†). This implies that CeO2 activates CO2 slightly more effectively than propane, with the low selectivity of propene due to the presence of by-products. However, upon the addition of scattered rare metals (Ga, In), the selectivity of propene markedly increased to 88.24%, which is almost comparable to that of PtSn/CeO2 (93.91%) (Fig. 4). This indicates that the introduction of these scattered rare metals facilitates the activation and cleavage of C–H bonds. Additionally, the conversion for CO2 has also increased by 10–14% relative to CeO2, suggesting that the catalyst also facilitates enhanced CO2 conversion. The detailed experimental results showed that Pt1/CeO2 exhibited high activity for the conversion of propane and CO2, which were 38.45% and 75.42%, respectively (Fig. 4a and c). The poor ability of its C–H cleavage resulted in a C3H6 selectivity of only 33.04% (Fig. 4b). However, the introduction of the scattered rare metal Ga and In into Pt1/CeO2 results in a notable increase in the selectivity of the corresponding C3H6, reaching 83.89 and 79.53% respectively. This phenomenon can be attributed to the capacity of the scattered rare metals' oxide coverings to modulate the catalysts' surface properties, thereby enhancing their selectivity for C3H6. Furthermore, we have also found that Ga and In species interact with CO2 during the prolonged CO2-ODP reaction, thereby strengthening the oxidative dehydrogenation ability of the catalyst (Fig. 4d–f). This is achieved by modulating the surface properties of the catalyst and inhibiting the occurrence of side reactions, which ultimately leads to an improvement in the selectivity of C3H6 and its relatively stable retention. Besides, the Pt/CeO2 exhibited remarkably high conversion of propane and CO2, but the propylene selectivity was lower than that of the SROx-Pt/CeO2 catalysts (Fig. 4e). This may be predominantly attributed to the facile accumulation of carbon on the surface and the over-oxidation of propylene caused by the high activity in the CO2-ODP reaction.33
 | ||
| Fig. 4 (a–c) The initial conversion and selectivity of C3H8, CO2 and C3H6, (d–f) the conversion and selectivity of C3H8, CO2 and C3H6 with time varies over catalysts. (g–i) The conversion of C3H8 and yields of C3H6 and CO2 with time varies over Pt1Gax/CeO2. The initial performance was calculated from data collected by gas chromatography after the reaction gas was passed into the system and held for 20 minutes before the reaction. | ||
To enhance the understanding of the modulation behavior of the CO2-ODP reaction through the synergistic interaction of scattered rare metals and Pt/CeO2. Here, Ga is taken as an example, and its influence behavior on the catalytic activity and target product yield is investigated by precisely modulating the loading of Ga with fixed Pt content. As shown in Fig. 4g, the conversion of C3H8 is close to that of Pt/CeO2 when the loading of Ga is in the range of 0.3–0.7 wt%, and the conversion of C3H8 decreases from 38.13% to 28.38% with a slight increase in Ga content. It is evident that the yield of C3H6 from Pt1Ga0.5/CeO2 significantly surpasses that of Pt1Ga0.3/CeO2 and Pt1Ga0.7/CeO2, achieving approximately 20% (Fig. 4h and S12a†). Surprisingly, Pt1Ga0.5/CeO2 has a high CO2 yield comparable to that of Pt1Ga0.3/CeO2 and Pt1Ga0.7/CeO2 (Fig. 4i and S12b†). This indicates that the interaction of moderate amounts of Ga with Pt/CeO2 can effectively modulate catalytic activity and selectivity when the Ga content remains lower than that of Pt, in accordance with the Sabatier principle. To further investigate the effects of high levels of the scattered rare metal Ga on the reactivity of propane dehydrogenation over Pt/CeO2, we conducted experiments using Pt1Ga3–5/CeO2. As shown in Fig. 4g, the conversion of C3H8 decreases with increasing Ga content. Notably, when the Ga loading exceeds 3 wt%, a further decline in C3H8 conversion is observed, reaching a minimum of 3.16% for the Pt1Ga5/CeO2 catalyst (Fig. 4g). Moreover, the high Ga loading results in the occupation of an excess of active sites, thereby leading to a significant reduction in the cleavage of C–H bonds and the activation of CO bonds (Fig. 4h and i). Notably, Pt1Ga3/CeO2 exhibited the highest C3H6 selectivity of 83.89% within the system (Fig. S12a†). This was a consequence of the intricate equilibrium between C–H bond cleavage and CO bond activation in systems. This is corroborated by the H2-TPR curves, which indicate that both groups of samples undergo two primary stages of reduction reactions during the programmed heating process. Both Pt1/CeO2 and Pt1Ga3/CeO2 exhibit weak hydrogen reduction peaks around 370 °C, attributed to the reduction of adsorbed oxygen on the surface of CeO2 by hydrogen (Fig. S13†). Notably, the intensity of the hydrogen reduction peak for Pt/CeO2 is greater than that for Pt1Ga3/CeO2, owing to its relatively loose surface structure and higher porosity, which facilitate the adsorption of more oxygen molecules, consistent with the BET test results (Table S2†). The second peak corresponds to the reduction of bulk lattice oxygen in CeO2 by hydrogen. Upon the addition of the second metal, Ga, to Pt1/CeO2, the reduction temperature shifts from 787 °C to 797 °C, accompanied by a slight decrease in peak intensity.34 This change is attributed to the interaction between Ga species and CeO2, which reduces the mobility of bulk lattice oxygen and the availability of flowable lattice oxygen.
To gain a deeper understanding of the factors influencing the CO2-ODP performance of Pt1Gax/CeO2, we characterized the series of catalysts using Raman spectroscopy, TEM, and XRD spectra. The Raman spectroscopy analysis results show that with the increase of Ga content, the Raman vibration peak intensity of Ce–O bond gradually decreases, indicating that the more Ga species content, the stronger the interaction with the CeO2 carrier (Fig. S14†). TEM images show that the level of Ga content in Pt1Gax/CeO2 does not affect the cubic fluorite structure of CeO2 (Fig. S15†). Fig. S16† presents the XRD patterns, which indicate that an increase in Ga content correlates with a decrease in the intensity of the characteristic XRD peaks of CeO2. This suggests that higher Ga loading results in reduced crystallinity of the carrier. Overall, these findings further substantiate that incorporating Ga into Pt/CeO2 enhances the interaction with CeO2, diminishing the activation ability of CO bonds while providing additional active sites for C–H bond cleavage, thereby significantly improving the selectivity for propylene.
To gain insights into coke formation, Raman spectroscopy and TG-DSC experiments were conducted on spent catalysts following a time on stream (TOS) of 80 minutes. As illustrated in Fig. S17a,† the Raman spectra of the spent catalyst after the propane dehydrogenation reaction exhibit distinct carbon D and G peaks at 1380 and 1600 cm−1, respectively, indicating the presence of coke on the catalyst surface. Notably, the extent of carbon deposition increases with rising Ga content, a trend that correlates well with the observed changes in reactivity during the CO2-ODP process. Moreover, the TG analysis of the catalysts after the reaction also showed that the weight loss in the range of 200–450 °C was more pronounced for Pt1Ga3/CeO2 and Pt1Ga5/CeO2 compared to that of Pt1Ga0.5/CeO2 (Fig. S17b†), which further confirmed that the increase in the Ga content would lead to a gradual decrease in the catalytic performance due to the large amount of carbon deposited on the catalyst surface. Additionally, we compared the morphology of Pt1Ga0.5/CeO2 catalysts before and after the propane dehydrogenation reaction. As illustrated in Fig. S18,† the catalysts exhibited no significant changes in their microscopic morphology before and after the reaction, with Pt and Ga elements uniformly dispersed across the CeO2 matrix. This observation suggests that the catalyst structure remained largely intact following the propane dehydrogenation reaction, indicating that the observed decline in catalytic performance is likely attributable to the accumulation of carbon on the catalyst surface. Furthermore, the XPS analysis indicates that an increased Ga content in the spent catalyst enhances the transfer of electrons from Pt to the Ga species.35 This transfer results in a reduced number of active electrons at the Pt sites, ultimately leading to diminished CO2-ODP performance (Fig. S19†). Therefore, the decrease in the catalytic performance of Pt1Gax/CeO2 over TOS is due to the poisoning of the Pt active sites by the coke formed in the dehydrogenation reaction.
Importantly, from an industrial perspective, Pt1Ga0.5/CeO2 displays more significant potential for converting C3H8 and the high-value utilization of CO2. This is because the yield of C3H6 is still maintained at approximately 20% while maintaining relatively high reactivity for C3H8 and CO2. It is important to note that the performance of propane dehydrogenation gradually declines with increasing Ga content, with a particularly pronounced decrease observed for the Pt1Ga5/CeO2 catalyst. This is attributed to the fact that when the Ga metal content significantly exceeds that of Pt, it can alter the microenvironment of the catalytic system. These changes intensify side reactions, such as deep dehydrogenation and carbon–carbon bond cleavage, leading to carbon build-up.36 This carbon accumulation negatively impacts the optimization of active sites, ultimately reducing the catalytic performance. The phenomena above serve to confirm the primary role of the appropriately covered dilute metal oxides in the CO2-ODP reaction, namely to modulate the surface properties, act in synergy with Pt/CeO2 to promote CO2 reduction, lower the bond energy of the CO bond, and accelerate the C–H bond breakage.
Reaction mechanism
Based on the characterization and catalytic activity results, a possible reaction mechanism was proposed (Fig. 5). The addition of oxides of scattered rare metals (Ga and In) to Pt/CeO2 might change the electronic structure of the catalyst, which in turn affects the adsorption and activation of propane and CO2 molecules. The modified electronic structure could enhance the interaction between the catalyst and the reactants, leading to a more efficient CO2-ODP process. Specifically, the CeO2 carrier in Pt/CeO2 could facilitate the capture and activation of CO2 due to its exceptional oxygen release capability. The Pt sites act as active centers, adsorbing and activating propane molecules and promoting the cleavage of C–H bonds. The scattered rare metal oxides regulate the surface properties of the catalysts, allowing CO2 to undergo chemical adsorption on the catalyst surface, forming adsorbed CO2 molecules. Subsequently, electron transfer enables carbon atoms in CO2 molecules to gain electrons, thereby increasing their electron cloud density and reducing the bond energy of CO bonds. Ultimately, under the influence of the catalyst, the CO bond is broken, resulting in the formation of an active intermediate that participates in the oxidative dehydrogenation reaction of propane.
 | ||
| Fig. 5 A possible CO2-ODP mechanism based on SROx-Pt/CeO2. | ||
In the end, this process occurs efficiently as a result of the synergistic cooperation of the various components of the catalyst, which effectively reduces the activation energy barrier. Pt sites primarily facilitate the activation and cleavage of CO bonds, while scattered rare metal oxides promote the activation and acceleration of C–H bond cleavage, thereby synergistically reducing the energy barrier for propane oxidative dehydrogenation within the catalytic system. Moreover, the presence of CO2 prompts the reaction to proceed via an oxidizing mechanism, whereby hydrogen is oxidized to water. This facilitates the overall dehydrogenation process. Additionally, CO2 interacts with the catalyst, modulating the surface properties and enhancing the selectivity towards C3H6.
Conclusions
In this work, we investigated the effect of modulation of scattered rare metals on Pt/CeO2 catalyst for CO2-ODP. The results showed that adding scattered rare metal oxides significantly influenced the reaction activity and selectivity. The catalytic activity and selectivity of a catalyst are influenced by scattered rare metals, which regulate the charge density and valence state of Pt sites. The cooperative interaction of the constituent elements of the SROx-Pt/CeO2 catalysts markedly enhanced the cleavage of C–H bonds and activation of CO bonds, while simultaneously inhibiting C–C bond rupture. This resulted in a notable acceleration of the propane oxidative dehydrogenation reaction, facilitating the production of propene. This work offers novel insights into the design of highly efficient catalysts for CO2-assisted propane oxidative dehydrogenation, potentially contributing to the development of more sustainable and efficient processes for propene production and CO2 utilization.
Experimental
Catalyst preparation
Characterization
Scanning Electron Microscopy (SEM)-Mapping images were obtained on a ZEISS Sigma 300. XRD (Bruker D8 Discover A25, Germany) analysis was conducted using a PANalytical X'pert PPR diffractometer equipped with CuKα radiation source (λ = 1.5418 Å) and operated at 40 mA and 40 kV. Transmission Electron Microscopy (TEM) images were obtained from an FEI Talos F200X equipped with a field emission gun (FEG) operating at 200 kV. STEM and HR-TEM were carried out on an FEI Talos F200X instrument at 400 kV. X-ray Photoelectron Spectroscopy (XPS) spectra were recorded using a PHI 5000 Versa Probe II spectrometer with monochromatic Al-Kα excitation. Fourier Transform Infrared Spectroscopy (FT-IR) analyses were performed with a Nicolet 560 IR spectrometer (USA), covering a spectral range from 400 to 4000 cm−1. Hydrogen Temperature-Programmed Reduction (H2-TPR) experiments were conducted using a chemisorption analyzer (Micromeritics, Model AutoChem II 2920). Before analysis, 50 mg of the sample was pretreated under an Argon (Ar) flow at 500 °C for 1 hour to eliminate any adsorbed impurities. Subsequently, the treated samples were cooled to 50 °C, and a reducing gas (10 vol% H2 in Ar) was introduced into the reactor at a flow rate of 50 mL min−1. The temperature of the reaction system was then ramped from 50 to 1000 °C at a heating rate of 10 °C min−1. Nitrogen adsorption–desorption isotherms of the catalysts were obtained at −77 K using a NOVA4000 instrument (Quantachrome, USA). The average pore size and total pore volume were analyzed using the Barrett–Joyner–Halenda (BJH) method at the maximum relative pressure. The metal content was determined via Inductively Coupled Plasma Atomic Emission Spectrometry (ICP-OES) using an Agilent 5110 spectrometer. Thermogravimetric and differential scanning calorimetry analyses (TG-DSC) of spent catalysts were conducted using a Thermo-analyzer System (USA TA TGA 550).Catalytic activity tests
The catalytic activity of the catalysts for CO2-assisted propane oxidative dehydrogenation was evaluated in a fixed-bed reactor. The reactor was equipped with appropriate temperature control and gas inlet-outlet systems. A mixture of propane, CO2, and an inert gas (such as Ar) was introduced into the reactor with a controlled flow rate. The reaction temperature was varied from a specific range to study its effect on the reaction. Specifically, a total of 0.3 g of catalyst was loaded into a tubular quartz fixed-bed microreactor with an inner diameter of 8 mm, equipped with three gas mass flow meters and a heating unit. The reactor was first heated to 600 °C under an Ar flow before introducing the reaction feed gas consisting of C3H8 (8 mL min−1), CO2 (8 mL min−1), and Ar (24 mL min−1), resulting in a weight hourly space velocity (WHSV) of 3.1 h−1 for C3H8. The reaction products were analyzed using an online gas chromatograph (Fuli GC 9790 II), with sampling taken at 20 minutes intervals. Hydrocarbon products were separated and detected using an RB-Al2O3 packed column (30 m × 0.53 mm × 15 μm) coupled with a flame ionization detector (FID), while other products were separated and detected using a PorapakN capillary column (2 m × 4 mm, 80–100 mesh), a TDX-01 packed column (2 m × 4 mm, 60–80 mesh), and a thermal conductivity detector (TCD). Catalytic tests were also performed at different WHSVs for C3H8: WHSV = 1.6 h−1 (0.6 g catalyst, C3H8/CO2/Ar = 8/8/24 mL min−1), WHSV = 5.9 h−1 (0.3 g catalyst, C3H8/CO2/Ar = 15/15/45 mL min−1), and WHSV = 11.8 h−1 (0.2 g catalyst, C3H8/CO2/Ar = 20/20/60 mL min−1). The regeneration performance was evaluated by calcining the spent catalyst (CO2-ODP for 200 minutes: C3H8/CO2/Ar = 1/1/2, WHSV(C3H8) = 3.1 h−1) in air at 550 °C for 3 hours to remove the coke, and this process was repeated three times.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Jiulong Wang: investigation, methodology, validation, writing –original draft. Fang Wang: supervision and methodology. Longyang Li: methodology. Weihao Zhao: methodology. Shiyuan Wang: data curation. Zelin Ma: data curation. Yankong: investigation, formal analysis. Yazhou Shuang: data curation. Weiwei Xia: data curation. Jie Jian: supervision. Pengfei Guo: supervision. Hongqiang Wang: conceptualization, funding acquisition, formal analysis, writing – review & editing, resources.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by Key Projects in Fields of Intergovernmental International Cooperation of the National Key Research and Development Program of China (No. 2021YFE0115100), Shaanxi Province Key Research and Development Program (No. 2022KWZ-04), the Shaanxi Science and Technology Innovation Team (No. 2023-CX-TD-44). The authors would like to thank the Analytical & Testing Center of Northwestern Polytechnical University and the Shaanxi Materials Analysis and Research Center for XPS, XRD, SEM and TEM characterizations. The authors would also like to thank Dr Dan Feng and Dr Xiubo Yang from the Analytical & Testing Center of Northwestern Polytechnical University for the TEM and SEM measurement. In addition, the authors would like to thank Shiyanjia Lab (http://www. http://shiyanjia.com) for the H2-TPR measurement.Notes and references
- T. Otroshchenko, G. Jiang, V. A. Kondratenko, U. Rodemerck and E. V Kondratenko, Chem. Soc. Rev., 2021, 50, 473–527 RSC.
- M. Monai, M. Gambino, S. Wannakao and B. M. Weckhuysen, Chem. Soc. Rev., 2021, 50, 11503–11529 RSC.
- M. A. Atanga, F. Rezaei, A. Jawad, M. Fitch and A. A. Rownaghi, Appl. Catal., B, 2018, 220, 429–445 CrossRef CAS.
- E. Gomez, S. Kattel, B. Yan, S. Yao, P. Liu and J. G. Chen, Nat. Commun., 2018, 9, 1398 CrossRef.
- E. Gomez, B. Yan, S. Kattel and J. G. Chen, Nat. Rev. Chem, 2019, 3, 638–649 CrossRef CAS.
- G. Li, C. Liu, X. Cui, Y. Yang and F. Shi, Green Chem., 2021, 23, 689–707 RSC.
- N. Mimura, I. Takahara, M. Inaba, M. Okamoto and K. Murata, Catal. Commun., 2002, 3, 257–262 CrossRef CAS.
- R. Wu, P. Xie, Y. Cheng, Y. Yue, S. Gu, W. Yang, C. Miao, W. Hua and Z. Gao, Catal. Commun., 2013, 39, 20–23 CrossRef CAS.
- P. Michorczyk and J. Ogonowski, Appl. Catal., A, 2003, 251, 425–433 CrossRef CAS.
- K. Nakagawa, C. Kajita, K. Okumura, N. Ikenaga, M. Nishitani-Gamo, T. Ando, T. Kobayashi and T. Suzuki, J. Catal., 2001, 203, 87–93 CrossRef CAS.
- O. Ovsitser, M. Cherian, A. Brückner and E. V Kondratenko, J. Catal., 2009, 265, 8–18 CrossRef CAS.
- S. Vajda, M. J. Pellin, J. P. Greeley, C. L. Marshall, L. A. Curtiss, G. A. Ballentine, J. W. Elam, S. Catillon-Mucherie, P. C. Redfern, F. Mehmood and P. Zapol, Nat. Mater., 2009, 8, 213–216 CrossRef CAS PubMed.
- C. Yu, Q. Ge, H. Xu and W. Li, Catal. Lett., 2006, 112, 197–201 CrossRef CAS.
- M. A. Botavina, Y. A. Agafonov, N. A. Gaidai, E. Groppo, V. Cortés Corberán, A. L. Lapidus and G. Martra, Catal. Sci. Technol., 2016, 6, 840–850 RSC.
- P. Michorczyk, P. Pietrzyk and J. Ogonowski, Microporous Mesoporous Mater., 2012, 161, 56–66 CrossRef CAS.
- E. V Kondratenko, M. Cherian, M. Baerns, D. Su, R. Schlögl, X. Wang and I. E. Wachs, J. Catal., 2005, 234, 131–142 CrossRef.
- L. Cao, P. Yan, S. Wen, W. Bao, Y. Jiang, Q. Zhang, N. Yu, Y. Zhang, K. Cao, P. Dai and J. Xie, J. Am. Chem. Soc., 2023, 145, 6184–6193 CrossRef CAS PubMed.
- H. Yan, K. He, I. A. Samek, D. Jing, M. G. Nanda, P. C. Stair and J. M. Notestein, Science, 2021, 371, 1257–1260 CrossRef CAS.
- M. A. Salaev, A. A. Salaeva, T. S. Kharlamova and G. V Mamontov, Appl. Catal., B, 2021, 295, 120286 CrossRef CAS.
- Z. Lu, G. Sun, S. Chen, D. Fu, C. Pei and J. Gong, Chem. Eng. Sci., 2024, 297, 120304 CrossRef CAS.
- F. Xing, Y. Nakaya, S. Yasumura, K. Shimizu and S. Furukawa, Nat. Catal., 2022, 5, 55–65 CrossRef CAS.
- L. Wang, G.-Q. Yang, X. Ren and Z.-W. Liu, Nanomaterials, 2022, 12, 417 CrossRef CAS.
- H. Wang, G.-Q. Yang, Y.-H. Song, Z.-T. Liu and Z.-W. Liu, Catal. Today, 2019, 324, 39–48 CrossRef CAS.
- Y.-L. Shan, H.-L. Sun, S.-L. Zhao, K.-X. Li, K.-H. Xia, J.-W. Ding and W.-L. Yu, Chem. Eng. J., 2022, 450, 137969 CrossRef CAS.
- Q. Liu, M. Luo, Z. Zhao and Q. Zhao, Chem. Eng. J., 2020, 380, 122423 CrossRef CAS.
- Q.-Y. Chang, K.-Q. Wang, Z.-J. Sui, X.-G. Zhou, D. Chen, W.-K. Yuan and Y.-A. Zhu, ACS Catal., 2021, 11, 5135–5147 CrossRef CAS.
- X. Huang, K. Zhang, B. Peng, G. Wang, M. Muhler and F. Wang, ACS Catal., 2021, 11, 9618–9678 CrossRef CAS.
- H. Wang, F.-X. Cao, Y.-H. Song, G.-Q. Yang, H.-Q. Ge, Z.-T. Liu, Y.-Q. Qu and Z.-W. Liu, J. CO2 Util., 2019, 34, 99–107 CrossRef CAS.
- J. Wang, S. Liu, M. Tang, W. Fu, Y. Wang, K. Yin and Y. Dai, Small, 2023, 19, 2300547 CrossRef CAS PubMed.
- S. Moon, D. Lee, J. Park and J. Kim, ACS Appl. Mater. Interfaces, 2023, 15, 37687–37695 CrossRef CAS PubMed.
- L.-L. Shen, K. Xia, W.-Z. Lang, L.-F. Chu, X. Yan and Y.-J. Guo, Chem. Eng. J., 2017, 324, 336–346 CrossRef CAS.
- B. Wu, Y. Shi, J. Zhang, Z. Wang, S. Sun, L. Wang, H. Huang, L. Zhang and Y. Wu, Chem. Eng. J., 2024, 498, 155205 CrossRef CAS.
- X. Li, S. Li, Y. Cheng, L. Zheng, L. Song, X. Zi and H. Dai, Catalysts, 2024, 14, 778 CrossRef CAS.
- Q. Yang, L. Du, X. Wang, C. Jia and R. Si, Chin. J. Catal., 2016, 37, 1331–1339 CrossRef CAS.
- G.-Q. Yang, X. Ren, V. A. Kondratenko, H.-B. Zhang, E. V Kondratenko and Z.-W. Liu, Nano Res., 2023, 16, 6237–6250 CrossRef CAS.
- X. Chang, Z. Lu, R. Luo, X. Wang, G. Sun, D. Fu, Z.-J. Zhao and J. Gong, Chem, 2024, 11, 102294 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra08481k |
| This journal is © The Royal Society of Chemistry 2025 |