
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceBioactive polyketides from cultures of the entomopathogenic fungus Hypocrella luteola TBRC-BCC 76666†
Jittra Kornsakulkarn,
Patchanee Auncharoen,
Wasana Noisripoom,
Suchada Mongkolsamrit and
Chawanee Thongpanchang *
*
National Center for Genetic Engineering and Biotechnology (BIOTEC), National Science and Technology Development Agency (NSTDA), 111 Thailand Science Park, Phahonyothin Road, Khlong Nueng, Khlong Luang, Pathum Thani 12120, Thailand. E-mail: chawanee@biotec.or.th
First published on 17th February 2025
Abstract
Entomopathogenic fungi of the genus Hypocrella are known to produce bioactive compounds, in particular polyketides. Here, a chemical investigation of the entomopathogenic fungus Hypocrella luteola strain TBRC-BCC 76666 was performed to identify possible novel compounds indicated from HPLC and 1H NMR spectra profiles of culture extracts. Eight novel compounds were isolated, including six new polyketides (1–6) and two other naturally occurring compounds (7–8), along with seven known compounds. The compound structures were established by spectroscopic analysis, the application of modified Mosher's method, and electronic circular dichroism. Compounds 1 and 5 were cytotoxic against both NCI-H187 and Vero cells (IC50 = 9.9 and 58.9 μM, respectively), whereas compound 4 was active only against MCF-7 cells (IC50 = 42.5 μM). Ascherxanthone B (9) exhibited antiproliferative activity against all cell lines tested (Plasmodium falciparum, mammalian, and fungal), with IC50 and MIC values in the range 0.9–30.5 μM.
Introduction
Entomopathogenic fungi are considered as a potential source of structurally diverse and biologically active compounds that may be useful as starting points in drug discovery. Hypocrella (teleomorphs) and Aschersonia (anamorphs) are genera of entomopathogenic fungi in the order Hypocreales, family Clavicipitaceae. They specifically infect scale insects and whiteflies and have been reported to produce a wide variety of bioactive secondary metabolites including polyketides.1,2 Polyketides are a group of compounds with diverse structures and biological activities.3,4 Many of these compounds have been widely used in clinical therapies, such as the antibiotics erythromycin, tylosin, rifamycin, tiacumicin B, and tetracyclines, the antifungal agents amphotericin B and nystatin, the antiparasitic agent avermectin, the immunosuppressants FK506 and rapamycin, and the antitumor agents doxorubicin and mithramycin. Several bioactive polyketides from isolates of Hypocrella and Aschersonia have been reported from the Thailand Bioresource Research Center-BIOTEC Culture Collection (TBRC-BCC), including (+)-rugulosin,5 ascherxanthones A–G,6–8 confluxanthones A–G,8 ascherlactones A and B,9 ascherchromanone A,9 and xanthone and anthraquinone-type mycotoxins.10,11 As part of our ongoing research on the discovery of bioactive compounds from entomopathogenic fungi, we selected the fungus Hypocrella luteola TBRC-BCC 76666 for chemical investigation, because the HPLC and 1H NMR spectra of the crude extracts showed chemical profiles suggestive of novel compounds. Details of the isolation, structure elucidation and biological activities of the chemical constituents are reported herein.Results and discussion
The fungus Hypocrella luteola TBRC-BCC 76666 was isolated from scale insects (Hemiptera) found on the underside of leaves of dicotyledons. The fungus was cultivated in potato dextrose agar (PDA) and then inoculated into potato dextrose broth (PDB). The fermentation was performed under static conditions before extraction. Chemical composition separation of the fungal crude extract led to the isolation of eight new naturally occurring compounds (1–8; Fig. 1), together with seven known compounds.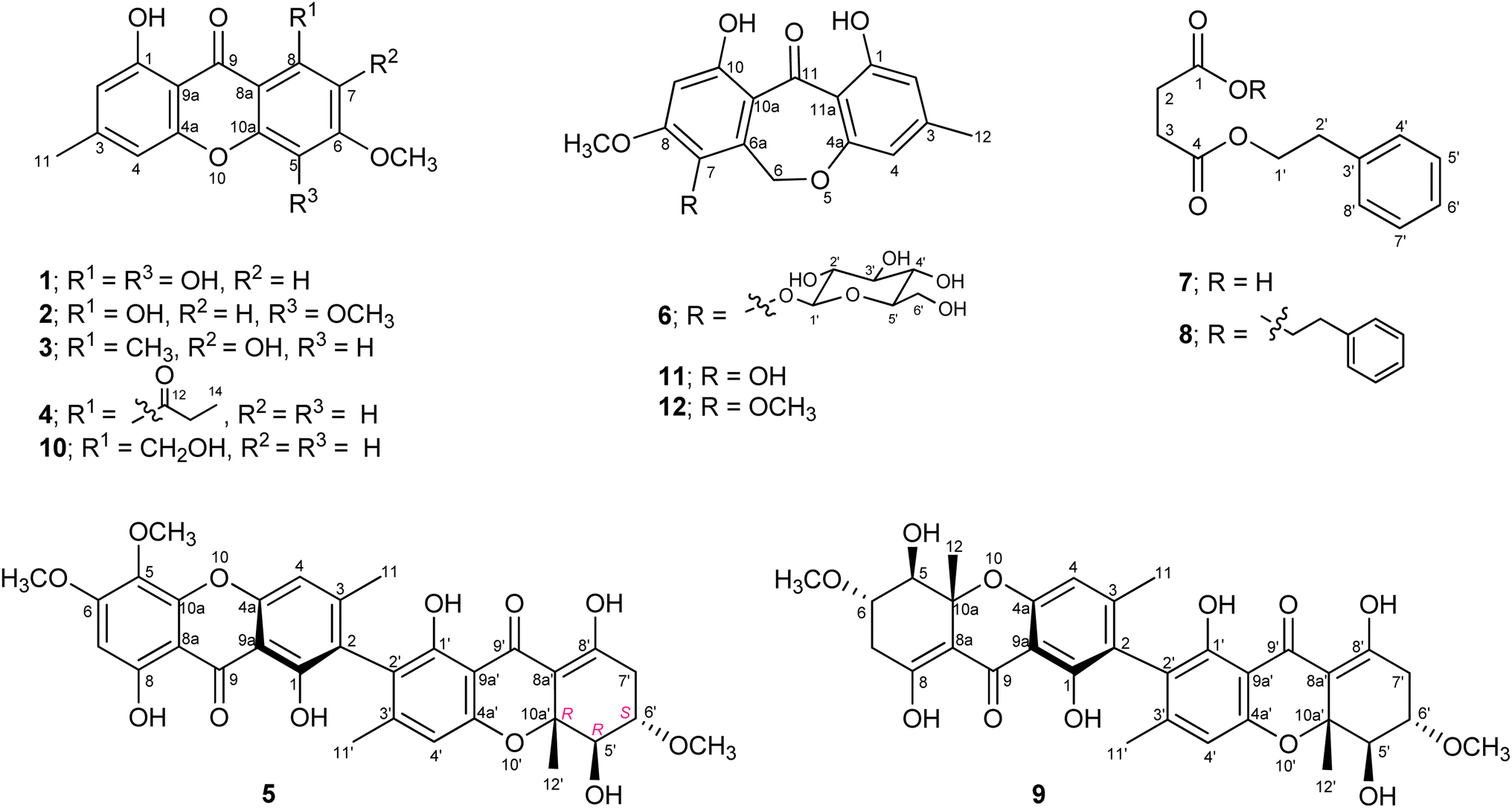 | ||
| Fig. 1 Structure of compounds 1–12. | ||
Compound 1 was obtained as a yellow solid with the molecular formula C15H12O6, as deduced from HRESIMS. The IR spectrum showed the absorption bands at 3397 and 1637 cm−1 for a hydroxyl group and a γ-pyrone carbonyl group, respectively. Analysis of the NMR spectroscopic data (Table 1) revealed the presence of two chelated phenolic protons (δH 11.84, 11.70), one methyl group, one methoxy group, three aromatic protons, twelve aromatic carbons with six oxygenated, and one carbonyl carbon. These data accounted for all the NMR resonances. These data in combination with 10 degrees of unsaturation required from its molecular formula, suggested a tricyclic compound structure for 1. The HMBC correlations from H-2 to C-1/C-4, C-9a, H-4 to C-2/C-4a/C-9a/C-11, H-7 to C-5/C-8/C-8a, H3-11 to C-2/C-3/C-4, OCH3-12 to C-6, 1-OH to C-1/C-2/C-9a and 8-OH to C-7/C-8/C-8a (Fig. 2) and the cross peak between OCH3 and H-7 in the NOESY spectrum established the structural feature of 1 as shown in Fig. 1. Therefore, compound 1 was identified as 1,5,8-trihydroxy-6-methoxy-3-methylxanthone.
| Position | 1a | 2a | 3a | 4b | ||||
|---|---|---|---|---|---|---|---|---|
| δC | δH mult. (J in Hz) | δC | δH mult. (J in Hz) | δC | δH mult. (J in Hz) | δC | δH mult. (J in Hz) | |
| a 500 MHz for 1H and 125 MHz for 13C.b 400 MHz for 1H and 100 MHz for 13C. | ||||||||
| 1 | 161.1 | — | 161.1 | — | 161.7 | — | 161.4 | — |
| 2 | 111.8 | 6.61 s | 111.8 | 6.61 d (0.5) | 110.9 | 6.56 d (1.9) | 111.7 | 6.62 s |
| 3 | 149.3 | — | 149.3 | — | 147.3 | — | 148.7 | — |
| 4 | 107.8 | 6.81 s | 107.9 | 6.82 d (0.5) | 106.4 | 6.63 d (1.9) | 107.2 | 6.72 s |
| 4a | 156.0 | — | 156.1 | — | 155.6 | — | 155.9 | — |
| 5 | 125.6 | — | 128.9 | — | 96.7 | 6.76 s | 100.7 | 6.87 d (2.4) |
| 6 | 153.9 | — | 160.4 | — | 151.8 | — | 165.0 | — |
| 7 | 94.2 | 6.41 s | 94.9 | 6.39 s | 140.5 | — | 110.4 | 6.65 d (2.4) |
| 8 | 155.6 | — | 158.6 | — | 124.2 | — | 144.8 | — |
| 8a | 102.2 | — | 102.1 | — | 113.3 | — | 111.4 | — |
| 9 | 184.7 | — | 184.5 | — | 183.6 | — | 180.1 | — |
| 9a | 105.2 | — | 105.3 | — | 106.4 | — | 106.6 | — |
| 10a | 152.8 | — | 149.3 | — | 152.9 | — | 158.2 | — |
| 11 | 22.6 | 2.42 s | 22.6 | 2.42 s | 22.4 | 2.40 s | 22.6 | 2.42 s |
| 12 | — | — | — | — | 13.4 | 2.82 s | 207.1 | — |
| 13 | — | — | — | — | — | — | 37.2 | 2.80 q (7.2) |
| 14 | — | — | — | — | — | — | 8.3 | 1.29 t (7.2) |
| 1-OH | — | 11.84 s | — | 11.79 s | — | 13.14 s | — | 12.22 s |
| 5-OH/OCH3 | — | — | 56.4 | 3.98 s | — | — | — | — |
| 6- OCH3 | 56.6 | 4.00 s | 61.7 | 3.91 s | 56.4 | 4.03 s | 56.1 | 3.94 s |
| 7-OH | — | — | — | — | — | 5.68 s | — | — |
| 8-OH | — | 11.70 s | — | 11.96 s | — | — | — | — |
 | ||
| Fig. 2 Key HMBC correlations of compound 1. | ||
The 1H and 13C NMR spectra of compound 2 (Table 1) matched those of compound 1 except for the presence of one additional methoxy signal. The MS data gave a molecular weight 14 Da higher than that of 1 and the molecular formula was determined to be C16H14O6 on the basis of HRESIMS. The correlations from an additional methoxy proton to C-5 in the HMBC spectrum indicated the placement of the methoxy group at C-5 in 2 instead of a hydroxyl group in 1. The NOESY correlations between 5-OCH3 and 6-OCH3 and between 6-OCH3 and H-7 also supported this assignment. Compound 2 was thus identified as 1,8-dihydroxy-5,6-dimethoxy-3-methylxanthone.
Compound 3 with the molecular formula C16H14O5 from HRESIMS was obtained as a yellow solid. Comparison of the 1H and 13C NMR spectra of 3 (Table 1) with those of 1 suggested that both compounds were closely related. Detailed analysis of 2D NMR spectroscopic data revealed the same xanthone skeleton with a difference in the substituents on one aromatic ring. The presence of the methoxy, hydroxy, and methyl groups at C-6, C-7, and C-8, respectively, indicated by the correlations from OCH3 to C-6, H-5 to C-7/C-8a/C-10a, and H3-12 to C-7/C-8/C-8a in the HMBC spectrum and the basis of their chemical shifts. Compound 3 was, therefore, identified as 1,7-dihydroxy-6-methoxy-3,8-dimethylxanthone.
The molecular formula of compound 4 was deduced as C18H16O5 from HRESIMS. The 1H and 13C NMR spectra of 4 (Table 1) showed the signal for one chelated phenolic proton, four aromatic protons, one methoxy, one methylene, two methyl, and two carbonyl groups. The HMBC correlations from H-2 to C-1/C-4/C-9a, H-4 to C-2/C-4a/C-9a/C-11, H-5 to C-6/C-7/C-8a/C-10a, H-7 to C-5/C-6/C-8a/C-12, H3-11 to C-2/C-3/C-4, H2-13 to C-12/C-14, H3-14 to C-12/C-13, OCH3-12 to C-6 established the structure of 4 as shown in Fig. 1. Compound 4 was thus identified as 1-hydroxy-6-methoxy-3-methyl-8-propionylxanthone.
Compound 5 was obtained as a yellow solid. The similar UV absorption (λmax 233, 265, 342 nm) to that of 2 indicated a xanthone-type compound. The molecular formula C32H30O12, as deduced from HRESIMS, and the 1H and 13C NMR spectra (Table 2) suggested that 5 was an unsymmetrical dimeric xanthone. Analysis of 2D NMR spectroscopic data revealed that one subunit had the same structure as 2 except that the C-2 protonated aromatic carbon in 2 was replaced by a non-protonated one in 5. Another subunit of 5 was tetrahydroxanthone, which resembled that found in the known co-metabolite ascherxanthone B (9).6 The establishment of the tetrahydroxanthone unit was deduced based on the 1H–1H COSY and the HMBC correlations as shown in Fig. 3. The remaining non-protonated aromatic carbons of both subunits suggested the connection of two subunits via C-2 and C-2′. The relative configuration of 5 was assigned by analysis of the 1H–1H coupling constants and the NOESY correlations. The large coupling constants observed between H-5′ and H-6′ (9.9 Hz) indicated the trans-diaxial orientation of these two protons. The cross peak between H-6′ and H3-12′ in the NOESY spectrum led to the conclusion that H3-12′ and H-6′ are on the same face of the ring system. Application of modified Mosher's method12 with 5 established the (5′R) configuration (Fig. 4); therefore, the absolute configurations at C-6′ and C-10a′ were assigned as S and R, respectively. The ECD spectrum of 5 (Fig. 5) showed positive exciton coupling at 224 nm and negative exciton coupling at 257 nm, indicating an R configuration of the chiral axis. Compound 5 was named ascherxanthone H.
| Position | 5 | |
|---|---|---|
| δC, type | δH mult. (J in Hz) | |
| 1 | 158.4, C | — |
| 2 | 117.7, C | — |
| 3 | 150.0, C | — |
| 4 | 108.5, CH | 7.01 d (0.4) |
| 4a | 155.4, C | — |
| 5 | 128.8, C | — |
| 6 | 160.3, C | — |
| 7 | 94.9, CH | 6.40 s |
| 8 | 158.6, C | — |
| 8a | 102.1, C | — |
| 9 | 184.5, C | — |
| 9a | 105.4, C | — |
| 10a | 149.2, C | — |
| 11 | 20.9, CH3 | 2.15 s |
| 1-OH | — | 12.13 s |
| 5-OCH3 | 61.7, CH3 | 3.91 s |
| 6-OCH3 | 56.4, CH3 | 3.97 s |
| 8-OH | — | 11.94 s |
| 1′ | 159.4, C | — |
| 2′ | 116.0, C | — |
| 3′ | 149.3, C | — |
| 4′ | 110.0, CH | 6.53 d (0.2) |
| 4a′ | 157.5, C | — |
| 5′ | 75.9, CH | 4.12 d (9.9) |
| 6′ | 75.5, CH | 3.49 ddd (9.9, 9.5, 6.9) |
| 7′ | 34.2, CH2 | (a) 3.0 dd (18.6, 6.9) |
| (b) 2.51 dd (18.6, 9.5) | ||
| 8′ | 170.4, C | — |
| 8a′ | 106.4, C | — |
| 9′ | 187.7, C | — |
| 9a′ | 104.8, C | — |
| 10a′ | 80.5, C | — |
| 11′ | 20.8, CH3 | 2.04 s |
| 12′ | 21.5, CH3 | 1.56 s |
| 1′-OH | — | 11.64 s |
| 5′-OH | — | 2.94 br s |
| 6′-OCH3 | 57.5, CH3 | 3.52 s |
| 8′-OH | — | 13.67 s |
 | ||
| Fig. 3 Key COSY, HMBC and NOESY correlations of compound 5. | ||
 | ||
| Fig. 4 Δδ-Values (δS − δR) of (S)- and (R)-MTPA esters 5a and 5b. | ||
 | ||
| Fig. 5 ECD spectrum of compound 5. | ||
The molecular formula of compound 6 was determined to be C22H24O11 on the basis of HRESIMS. The typical absorptions for hydroxyl (3404 cm−1) and conjugated carbonyl (1620 cm−1) were observed in the IR spectrum. The 1H NMR spectrum showed six hydroxyl protons, three aromatic protons, two methylenes, five methines, one methoxy, and one methyl group (Table 3). Analysis of the 2D NMR spectroscopic data (Fig. 6) revealed a dibenzo[b,e]oxepinone skeleton similar to that of the known co-metabolite chaetone G (11)13 except for the presence of a sugar unit in 6. The cross peaks from the anomeric proton H-1′ to H-2′, H-2′ to H-3′, H-3′ to H-4′, H-4′ to H-5′, and H-5′ to H-6′ in the COSY spectrum established the sugar moiety. The large vicinal coupling constants (J1′,2′ = 7.8 Hz, J2′,3′ = 8.5 Hz, J3′,4′ = 8.8 Hz, J4′,5′ = 9.1 Hz) proved the axial orientation of protons 1′–5′ of the sugar unit. The sugar unit was then assigned as β-glucopyranose. The attachment of the sugar unit at C-7 was indicated by the HMBC correlation from the anomeric proton H-1′ to C-7. Comparison of the specific rotations of the aqueous layer of its hydrolysate ([α]26D +38.3) with that of β-D-glucopyranose ([α]20D +18.7) established the D configuration of β-glucopyranose. The 1H NMR spectroscopic data of the aglycone unit, obtained by acid hydrolysis of 6, were identical to those of chaetone G.13 Compound 6 was therefore identified as chaetone G-7-β-D-glucopyranose.
| Position | 6 (in DMSO-d6) | |
|---|---|---|
| δC, type | δH mult. (J in Hz) | |
| 1 | 164.3, C | — |
| 2 | 112.0, CH | 6.50 d (0.5) |
| 3 | 148.9, C | — |
| 4 | 110.2, CH | 6.37 d (0.5) |
| 4a | 161.4, C | — |
| 6 | 67.4, CH2 | (a) 5.39 d (13.4) |
| (b) 5.13 d (13.4) | ||
| 6a | 132.8, C | — |
| 7 | 133.8, C | — |
| 8 | 158.1, C | — |
| 9 | 101.4, CH | 6.66 s |
| 10 | 159.7, C | — |
| 10a | 114.9, C | — |
| 11 | 194.6, C | — |
| 11a | 110.7, C | — |
| 12 | 21.3, CH3 | 2.25 s |
| 1-OH | — | 13.18 br s |
| 8-OMe | 56.3, CH3 | 3.87 s |
| 10-OH | — | 11.22 br s |
| 1′ | 103.6, CH | 4.50 d (7.8) |
| 2′ | 73.9, CH | 3.25 ddd (8.5, 7.8, 4.5) |
| 3′ | 76.2, CH | 3.20 ddd (8.8, 8.5, 4.3) |
| 4′ | 69.8, CH | 3.12 ddd (9.1, 8.8, 5.0) |
| 5′ | 76.8, CH | 3.00 ddd (9.1, 5.8, 2.1) |
| 6′ | 60.9, CH2 | (a) 3.57 ddd (11.5, 5.9, 2.1) |
| (b) 3.40 ddd (11.5, 5.9, 5.8) | ||
| 2′-OH | — | 5.20 d (4.5) |
| 3′-OH | — | 5.04 d (4.3) |
| 4′-OH | — | 4.96 d (5.0) |
| 6′-OH | — | 4.15 t (5.9) |
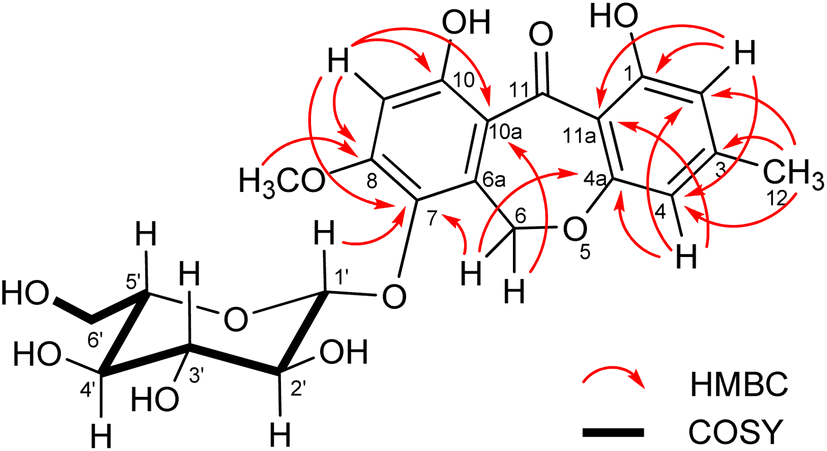 | ||
| Fig. 6 Key COSY and HMBC correlations of compound 6. | ||
Compound 7 was obtained as a brown solid. The 1H and 13C NMR spectra together with the HSQC data revealed the presence of five aromatic protons, four methylene, and two carbonyl groups. The spin systems of C-2-C-3, C-1′-C-2′, and C-4′-C-8′ were determined from the 1H–1H COSY spectroscopic data. In the HMBC spectrum, correlations from H-2 and H-3 to C-1/C-4, H-1′ to C-4/C-3′, H-2′ to C-3′/C-4′, and H-4′ to C-2′ were observed. Considering the chemical shift of C-1 (δC 176.9) and the molecular formula C12H14O4, as deduced by HRESIMS, carboxylic acid was assigned for C-1. The NMR spectroscopic data of compound 7 were in good agreement with the previously reported data for synthetic 4-oxo-4-phenethoxybutanoic acid.14 To our knowledge, this is the first known natural product isolation of this compound.
The presence of eight carbons in the 13C NMR spectrum and the molecular formula C20H22O4, as deduced by HRESIMS, led to the suggestion that compound 8 was a symmetrical dimeric compound. The NMR spectroscopic data showed good correlation to those of the synthetic diphenethyl succinate.15 The HMBC correlations from H-2 to C-1, H-1′ to C-4/C-2′/C-3′, H-2′ to C-1′/C-3′/C-4′, H-4′ to C-2′/C-8′, H-5′ to C-3′/C-7′, and H-6′ to C-4′ supported this assignment. To our knowledge, this is the first report for the isolation of 8 from a natural source.
The structures of the known compounds were dereplicated from HRESIMS and NMR (1H and 13C) spectroscopic data. The known compounds were identified as ascherxanthone B (9),6 1-hydroxy-8-(hydroxymethyl)-6-methoxy-3-methylxanthone (10),16 chaetone G (11),13 1,10-dihydroxy-7,8-dimethoxy-3-methyldibenzo[b,e]oxepin-11(6H)-one (12),13 pestalafuranones B17 and F,18 and zeorin.19
Compounds 1–7 and ascherxanthone B (9) were evaluated for bioactivity against Plasmodium falciparum (K1), phytopathogenic fungi (Alternaria brassicicola and Colletotrichum acutatum), cancerous cells, MCF-7 (human breast cancer) and NCI-H187 (human small-cell lung cancer), and non-cancerous Vero cells (African green monkey kidney) (Table 4). Compounds 1–7 showed no activity as anti-plasmodial and anti-fungal agents. Compounds 1 and 5 were active against NCI-H187 with IC50 values of 19.0 and 9.9 μM, respectively, whereas compound 4 was active against MCF-7 cells with an IC50 value of 42.5 μM. Ascherxanthone B (9) has been reported to possess antifungal activity against rice blast pathogen Magnaporthe grisea.6 In this study, it was shown to be active against all tested cell lines with IC50 and MIC values in the range 0.9–30.5 μM. However, all these active compounds also showed cytotoxicity against Vero cells with quite a low selectivity index (SI < 10).
| Compounds | Antimalarial P. falciparum, K1 (IC50, μM) | Cytotoxicity (IC50, μM) | Anti-phytopathogenic fungal (MIC, μM) | |||
|---|---|---|---|---|---|---|
| NCI-H187 | MCF-7 | Vero | C. acutatum | A. brassicicola | ||
| a Positive control for antimalarial assay.b Positive control for cytotoxicity assay.c Positive control for antifungal assay. | ||||||
| 1 | >34.7 | 19.0 | >173.5 | 58.91 | >173.5 | >173.5 |
| 2 | >33.1 | >165.4 | >165.4 | >165.4 | >165.4 | >165.4 |
| 3 | >34.9 | >174.6 | >174.6 | >174.6 | >174.6 | >174.6 |
| 4 | >32.0 | >160.1 | 42.5 | >160.1 | >160.1 | >160.1 |
| 5 | >16.5 | 9.9 | >82.4 | 21.6 | >82.4 | >82.4 |
| 6 | >21.5 | >107.7 | >107.7 | >107.7 | >107.7 | >107.7 |
| 7 | >45.0 | >225.1 | >225.1 | >225.1 | >225.1 | >225.1 |
| Ascherxanthone B (9) | 13.5 | 0.9 | 30.5 | 4.7 | 5.1 | 5.1 |
| Dihydroartemisinina | 0.002 | — | — | — | — | — |
| Chloroquine diphosphatea | 0.4 | — | — | — | — | — |
| Doxorubicinb | 0.2 | 11.9 | — | — | — | |
| Ellipticineb | 10.7 | — | 3.9 | — | — | |
| Tamoxifenb | — | 18.2 | — | — | — | |
| Amphotericin Bc | — | — | — | 3.4 | 1.7 | |
Conclusions
This study demonstrated that Hypocrella luteola TBRC-BCC 76666 is a rich source of bioactive polyketides, especially for the xanthone-type compounds and their dimeric forms. Five xanthones (1–4 and 10) and two xanthone dimers (5 and 9) were isolated. Xanthones 1 and 4 and ascherxanthone H (5) were active only against mammalian cell lines, whereas ascherxanthone B (9) exhibited antiproliferative activity against all cell lines tested (P. falciparum, mammalian, and fungal). Although the data set of these isolated compounds was too small to demonstrate a structure–activity relationship (SAR), the compound structures and biological activities can be combined with data of related compounds from other studies to guide the design and synthesis of derivatives with improved biological activity.Experimental
General experimental procedures
Melting points were measured using a Metler MP90 melting point apparatus and reported as uncorrected. Optical rotation measurements were conducted by using a JASCO P-2000 digital polarimeter. UV and FT-IR spectra were recorded on an JASCO V-730 spectrophotometer and a Bruker Alpha spectrometer, respectively. Circular dichroism (CD) spectra were recorded on a JASCO J-180 spectropolarimeter. NMR spectra were recorded on a Bruker AV500D spectrometer. ESITOF MS data were obtained using a Bruker micrOTOF mass spectrometer. Column chromatography was performed using silica gel 60 (70-230 Mesh ASTM, Merck). HPLC experiments were performed using a Dionex-Ultimate 3000 series instrument equipped with a binary pump, an autosampler, and a diode array detector.Fungal material
Hypocrella sp. was found attached to a scale insect (Hemiptera) host on the underside of leaves of dicotyledonous plants. The specimen was collected from Chanthaburi Agricultural Research and Development Center in Chanthaburi Province, by Artit Khonsanit et al., on 19 December 2014. The axenic fungal culture was deposited in the Thailand Bioresource Research Center under the accession number TBRC-BCC 76666. The specimen was deposited at the BIOTEC Bangkok Herbarium (BBH) under the accession number BBH 39507. The DNA sequences of the large subunit ribosomal DNA (LSU) and the translation elongation factor-1α gene (TEF1), genes of this isolate were obtained using standard methods. The sequences are available from GenBank with LSU, TEF1 sequence accession numbers PQ538629 and PQ585791, respectively. The sequences of this strain were generated and incorporated into a phylogenetic analysis using a combined dataset of 31 taxa with multi-locus sequences. The resulting phylogenetic tree strongly supported the fungal strain TBRC-BCC 76666 as part of the ‘Hypocrella luteola’ clade (Fig. S75†), leading to its identification as H. luteola (Clavicipitaceae, Hypocreales, Hypocreomycetidae, Sordariomycetes, Pezizomycotina, Ascomycota, Fungi).Fermentation and isolation
The fungus, TBRC-BCC 76666, was maintained on potato dextrose agar at 25 °C. The agar was cut into pieces (1 × 1 cm) and inoculated into 8 × 250 mL Erlenmeyer flasks containing 25 mL of potato dextrose broth (PDB, potato starch 4.0 g L−1, dextrose 20.0 g L−1). After incubation at 25 °C for 7 days on a rotary shaker (200 rpm), each primary culture was transferred into 1 L Erlenmeyer flask containing 250 mL of the same liquid medium (PDB) and incubated under the same conditions for 7 days. Each 25 mL portion of the secondary culture was transferred into 80 × 1 L Erlenmeyer flasks containing 250 mL of the same liquid medium (PDB) and the fermentation was carried out under static conditions at 25 °C for 50 days.After filtration of the culture, the filtrate of the cultures (broth) was extracted with EtOAc (3 × 20 L) and evaporated to dryness, leaving a dark brown gum (extract A, 2.06 g). The mycelia were macerated in MeOH (2 L) for three days, and then in CH2Cl2 (2 L) for three days. MeOH and CH2Cl2 extracts were combined and evaporated under reduced pressure. The residue was diluted with H2O (800 mL) and the mixture was repeatedly extracted with hexane (3 × 800 mL), followed by EtOAc (3 × 800 mL). The combined EtOAc extract was concentrated under reduced pressure to obtain a brown solid (extract B, 1.28 g).
Extract A (broth extract) was fractionated using Sephadex LH-20 and eluted with MeOH to give ten fractions (A1–A10) and compound 11 (20.5 mg) was obtained from fraction A10. Fraction A3 was subjected to preparative HPLC using a reverse-phase column (gradient elution with MeCN–H2O, 25–85%) to yield compounds pestalafuranone B (3.4 mg), 7 (6.9 mg), pestalafuranone F (4.6 mg), and 8 (1.1 mg). Compounds 4 (1.8 mg) and 6 (13.3 mg) were obtained from fractions A5 and A6, respectively, after purification by preparative HPLC (gradient elution with MeCN–H2O, 30–100%). Further purification of fraction A7 by preparative HPLC (gradient elution with MeCN–H2O, 33–100%) afforded compounds 12 (12.1 mg) and 2 (1.6 mg). Compounds 1 (2.0 mg), 10 (0.9 mg), 3 (1.5 mg), and 12 (2.4 mg) were obtained from fraction A8 after consecutive purification by preparative HPLC. Purification of fractions A9 using preparative HPLC (gradient elution with MeCN–H2O, 20–100%) furnished compound 1 (1.9 mg), 11 (7.9 mg) and 3 (2.9 mg).
Extract B (mycelial extract) was triturated with MeOH and filtered to obtain zeorin (120 mg). The filtrate (0.84 g) was fractionated using a Sephadex LH-20 column and eluted with MeOH to obtain six fractions (B1–B6). Fraction B1 was purified by preparative HPLC (gradient elution with MeCN–H2O, 40–100%), which yielded compound 9 (4.4 mg). Consecutive purification of fraction B3 using preparative HPLC gave compounds 1 (5.4 mg), 12 (9.4 mg), 2 (4.5 mg), and 5 (6.5 mg). Compounds 1 (2.3 mg) and 11 (3.3 mg) were obtained from fraction B4 after purification by preparative HPLC (gradient elution with MeCN–H2O, 30–100%).
![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) ε) 234 (4.35), 266 (4.45), 340 (4.18), 397 (3.63) nm; IR (ATR) νmax 3397, 2924, 2854, 1637, 1606, 1585, 1519, 1491, 1451, 1322, 1283, 1207, 1158, 1109, 1069 cm−1; 1H and 13C NMR data, see Table 1; HRMS (ESITOF) m/z 287.0565 [M − H]+ (calcd for: C15H12O6 − H, 287.0561).ε) 233 (4.12), 257 (4.29), 337 (4.04), 379 (3.50) nm; IR (ATR) νmax 3398, 2925, 2854, 1633, 1607, 1518, 1494, 1374, 1331, 1270, 1210, 1157, 1117, 1072 cm−1; 1H and 13C NMR data, see Table 1; HRMS (ESITOF) m/z 325.0688 [M + Na]+ (calcd for: C16H14O6 + Na, 325.0683).ε) 238 (4.40), 254 (4.60), 270 (4.38), 298 (4.31), 369 (4.05) nm; IR (ATR) νmax 3369, 2924, 2855, 1654, 1605, 1485, 1272, 1215, 1154, 1124, 1046 cm−1; 1H and 13C NMR data, see Table 1; HRMS (ESITOF) m/z 309.0742 [M + Na]+ (calcd for: C16H14O5 + Na, 309.0733).ε) 235 (4.24), 253 (4.14), 268 (3.95), 305 (3.99), 352 (3.64) nm; IR (ATR) νmax 3351, 2924, 2853, 1694, 1655, 1604, 1492, 1459, 1420, 1380, 1288, 1214, 1166, 1094, 1022 cm−1; 1H and 13C NMR data, see Table 1; HRMS (ESITOF) m/z 335.0885 [M + Na]+ (calcd for: C18H16O5 + Na, 335.0890).ε) 233 (4.36), 265 (4.51), 342 (4.50) nm; CD (CH3CN) Δε (nm) +37.08 (224), −13.65 (257), −0.31 (295), −1.19 (307), +1.69 (325), −6.19 (350); IR (ATR) νmax 3432, 2924, 2853, 1615, 1584, 1512, 1458, 1370, 1278, 1259, 1170, 1108, 1038 cm−1; 1H and 13C NMR data, see Table 2; HRMS (ESITOF) m/z 629.1626 [M + Na]+ (calcd for: C32H30O12 + Na, 629.1629).ε) 199 (4.66), 223 (4.29), 312 (3.96), 373 (4.17) nm; IR (ATR) νmax 3404, 2923, 1620, 1581, 1561, 1491, 1460, 1387, 1284, 1244, 1209 cm−1; 1H and 13C NMR data, see Table 3; HRMS (ESITOF) m/z 487.1215 [M + Na]+ (calcd for: C22H24O11 + Na, 487.1211).ε) 208 (3.93), 247 (3.37) nm; IR (ATR) νmax 3500, 3029, 2962, 1730, 1716, 1454, 1393, 1357, 1209, 1166 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.66 (1H, br s, 1-OH), 7.30 (2H, t, J = 7.2 Hz, H-5′, H-7′), 7.20–7.25 (3H, m, H-4′, H-6′, H-8′), 4.32 (2H, t, J = 7.0 Hz, H-1′), 2.94 (2H, t, J = 7.0 Hz, H-2′), 2.66 (2H, m, H-3), 2.61 (2H, m, H-2); 13C-NMR (100 MHz, CDCl3) δ 176.9 (C-1), 172.1 (C-4), 137.6 (C-3′), 128.9 (C-4′, C-8′), 128.5 (C-5′, C-7′), 126.6 (C-6′), 65.3 (C-1′), 35.0 (C-2′), 28.9 (C-2), 28.7 (C-3); HRMS (ESITOF) m/z 245.0782 [M + Na]+ (calcd for: C12H14O4 + Na, 245.0784).ε) 211 (5.14), 230 (4.84) nm; IR (ATR) νmax 2959, 2928, 1735, 1618, 1454, 1411, 1357, 1262, 1214, 1161 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.20–7.32 (10H, m, Ar-H), 4.29 (4H, t, J = 7.1 Hz, H-1′, H-1′′), 2.93 (4H, t, J = 7.1 Hz, H-2′, H-2′′), 2.59 (4H, s, H-2, H-3); 13C-NMR (125 MHz, CDCl3) δ 172.2 (C-1, C-4), 137.7 (C-3′, C-3′′), 128.9 (C-4′, C-8′, C-4′′, C-8′′), 128.5 (C-5′, C-7′, C-5′′, C-7′′), 126.6 (C-6′, C-6′′), 65.2 (C-1′, C-1′′), 35.1 (C-2′, C-2′′), 29.1 (C-2, C-3); HRMS (ESITOF) m/z 349.1417 [M + Na]+ (calcd for: C20H22O4 + Na, 349.1410).
ε) 234 (4.35), 266 (4.45), 340 (4.18), 397 (3.63) nm; IR (ATR) νmax 3397, 2924, 2854, 1637, 1606, 1585, 1519, 1491, 1451, 1322, 1283, 1207, 1158, 1109, 1069 cm−1; 1H and 13C NMR data, see Table 1; HRMS (ESITOF) m/z 287.0565 [M − H]+ (calcd for: C15H12O6 − H, 287.0561).ε) 233 (4.12), 257 (4.29), 337 (4.04), 379 (3.50) nm; IR (ATR) νmax 3398, 2925, 2854, 1633, 1607, 1518, 1494, 1374, 1331, 1270, 1210, 1157, 1117, 1072 cm−1; 1H and 13C NMR data, see Table 1; HRMS (ESITOF) m/z 325.0688 [M + Na]+ (calcd for: C16H14O6 + Na, 325.0683).ε) 238 (4.40), 254 (4.60), 270 (4.38), 298 (4.31), 369 (4.05) nm; IR (ATR) νmax 3369, 2924, 2855, 1654, 1605, 1485, 1272, 1215, 1154, 1124, 1046 cm−1; 1H and 13C NMR data, see Table 1; HRMS (ESITOF) m/z 309.0742 [M + Na]+ (calcd for: C16H14O5 + Na, 309.0733).ε) 235 (4.24), 253 (4.14), 268 (3.95), 305 (3.99), 352 (3.64) nm; IR (ATR) νmax 3351, 2924, 2853, 1694, 1655, 1604, 1492, 1459, 1420, 1380, 1288, 1214, 1166, 1094, 1022 cm−1; 1H and 13C NMR data, see Table 1; HRMS (ESITOF) m/z 335.0885 [M + Na]+ (calcd for: C18H16O5 + Na, 335.0890).ε) 233 (4.36), 265 (4.51), 342 (4.50) nm; CD (CH3CN) Δε (nm) +37.08 (224), −13.65 (257), −0.31 (295), −1.19 (307), +1.69 (325), −6.19 (350); IR (ATR) νmax 3432, 2924, 2853, 1615, 1584, 1512, 1458, 1370, 1278, 1259, 1170, 1108, 1038 cm−1; 1H and 13C NMR data, see Table 2; HRMS (ESITOF) m/z 629.1626 [M + Na]+ (calcd for: C32H30O12 + Na, 629.1629).ε) 199 (4.66), 223 (4.29), 312 (3.96), 373 (4.17) nm; IR (ATR) νmax 3404, 2923, 1620, 1581, 1561, 1491, 1460, 1387, 1284, 1244, 1209 cm−1; 1H and 13C NMR data, see Table 3; HRMS (ESITOF) m/z 487.1215 [M + Na]+ (calcd for: C22H24O11 + Na, 487.1211).ε) 208 (3.93), 247 (3.37) nm; IR (ATR) νmax 3500, 3029, 2962, 1730, 1716, 1454, 1393, 1357, 1209, 1166 cm−1; 1H NMR (400 MHz, CDCl3) δ 9.66 (1H, br s, 1-OH), 7.30 (2H, t, J = 7.2 Hz, H-5′, H-7′), 7.20–7.25 (3H, m, H-4′, H-6′, H-8′), 4.32 (2H, t, J = 7.0 Hz, H-1′), 2.94 (2H, t, J = 7.0 Hz, H-2′), 2.66 (2H, m, H-3), 2.61 (2H, m, H-2); 13C-NMR (100 MHz, CDCl3) δ 176.9 (C-1), 172.1 (C-4), 137.6 (C-3′), 128.9 (C-4′, C-8′), 128.5 (C-5′, C-7′), 126.6 (C-6′), 65.3 (C-1′), 35.0 (C-2′), 28.9 (C-2), 28.7 (C-3); HRMS (ESITOF) m/z 245.0782 [M + Na]+ (calcd for: C12H14O4 + Na, 245.0784).ε) 211 (5.14), 230 (4.84) nm; IR (ATR) νmax 2959, 2928, 1735, 1618, 1454, 1411, 1357, 1262, 1214, 1161 cm−1; 1H NMR (500 MHz, CDCl3) δ 7.20–7.32 (10H, m, Ar-H), 4.29 (4H, t, J = 7.1 Hz, H-1′, H-1′′), 2.93 (4H, t, J = 7.1 Hz, H-2′, H-2′′), 2.59 (4H, s, H-2, H-3); 13C-NMR (125 MHz, CDCl3) δ 172.2 (C-1, C-4), 137.7 (C-3′, C-3′′), 128.9 (C-4′, C-8′, C-4′′, C-8′′), 128.5 (C-5′, C-7′, C-5′′, C-7′′), 126.6 (C-6′, C-6′′), 65.2 (C-1′, C-1′′), 35.1 (C-2′, C-2′′), 29.1 (C-2, C-3); HRMS (ESITOF) m/z 349.1417 [M + Na]+ (calcd for: C20H22O4 + Na, 349.1410).(S)-MTPA ester 5a. White solid; 1H NMR (500 MHz, acetone-d6) δ 7.91 (2H, m, ArH of MTPA), 7.78 (2H, m, ArH of MTPA), 7.71 (1H, m, ArH of MTPA), 7.50–7.60 (10H, m, ArH of MTPA), 7.28 (2H, m, ArH of MTPA), 7.06 (1H, d, J = 0.7 Hz, H-4), 7.04 (2H, m, ArH of MTPA), 6.95 (1H, s, H-7), 6.77 (1H, d, J = 0.5 Hz, H-4′), 6.70 (1H, m, ArH of MTPA), 5.90 (1H, d, J = 11.0 Hz, H-5′), 4.12 (3H, s, 6-OCH3), 4.04 (3H, s, 5-OCH3), 4.01 (1H, m, H-6′), 3.88 (3H, s, OCH3 of MTPA), 3.85 (3H, s, OCH3 of MTPA), 3.77 (3H, s, OCH3 of MTPA), 3.76 (3H, s, OCH3 of MTPA), 3.44 (3H, s, 6′-OCH3), 3.03 (1H, dd, J = 18.4, 6.5 Hz, Ha-7′), 2.69 (1H, dd, J = 18.4, 8.9 Hz, Hb-7′), 2.12 (3H, s, H-11′), 2.11 (3H, s, H-11), 1.48 (3H, s, H-12); HRMS (ESITOF) m/z 1493.3259 [M + Na]+ (calcd for: C72H58O20F12 + Na, 1493.3222).
(R)-MTPA ester 5b. White solid; 1H NMR (500 MHz, acetone-d6) δ 7.84 (2H, m, ArH of MTPA), 7.72 (2H, m, ArH of MTPA), 7.68 (2H, m, ArH of MTPA), 7.45–7.58 (10H, m, ArH of MTPA), 7.18–7.22 (4H, m, ArH of MTPA), 7.08 (1H, d, J = 0.7 Hz, H-4), 7.06 (1H, d, J = 0.5 Hz, H-4′), 6.87 (1H, s, H-7), 5.89 (1H, d, J = 10.9 Hz, H-5′), 4.08 (3H, s, 6-OCH3), 3.99 (3H, s, 5-OCH3), 3.89 (1H, m, H-6′), 3.87 (3H, s, OCH3 of MTPA), 3.78 (3H, s, OCH3 of MTPA), 3.66 (3H, s, OCH3 of MTPA), 3.39 (3H, s, OCH3 of MTPA), 3.31 (3H, s, 6′-OCH3), 3.14 (1H, dd, J = 18.6, 6.7 Hz, Ha-7′), 2.53 (1H, dd, J = 18.6, 8.9 Hz, Hb-7′), 2.17 (3H, s, H-11′), 2.15 (3H, s, H-11), 1.78 (3H, s, H-12); HRMS (ESITOF) m/z 1493.3259 [M + Na]+ (calcd for: C72H58O20F12 + Na, 1493.3222).
Biological assays
Antimalarial activity against P. falciparum K1 was evaluated by using the microculture radioisotope technique.20 The assays was performed in 96-well plates in duplicate. Test compounds were two-fold diluted to obtain at least six different concentrations, and 25 μL of each compound dilution was then added to wells containing 200 μL of parasite suspension. After incubation for 24 h, 25 μL of a medium containing 0.5 μCi [3H] hypoxanthine was added and further incubated for 18–20 h. Levels of incorporated radioactive labeled hypoxanthine, which served as an indicator of parasite growth, were determined by using a TopCount microplate scintillation counter.The resazurin microplate assay (REMA)21 was used to evaluate cytotoxicity against cancerous cells, including MCF-7 (human breast cancer, ATCC HTC-22) and NCI-H187 (human small-cell lung cancer, ATCC CRL-5804). The assay was performed in 384-well plate in triplicate. Each well was added with 5 μL of test compound, followed by the addition of 45 μL of cell suspension. The plate was incubated at 37 °C in a humidified atmosphere of 5% CO2 for 3 d (MCF-7) or for 5 d (NCI-H187). Then, 12.5 μL of 62.6 μg mL−1 resazurin solution was applied to each well, and the plate was further incubated at 37 °C for 4 h. The fluorescent signal was measured at an emission wavelength of 590 nm and excitation wavelength of 530 nm, using the bottom-reading mode of fluorometer. The blank was subtracted from the signal to obtain background-corrected values.
Non-cancerous Vero cells (African green monkey kidney fibroblast, ATCC CCL-81) was evaluated using the green fluorescent protein microplate assay (GFPMA).22,23 The assay was done in 384-well plates in triplicate. To each well, 5 μL of compound dilution and 45 μL of the cell suspension were added, and the plate was incubated at 37 °C in a humidified incubator in an atmosphere with 5% CO2 for 4 d. The fluorescent signal was recorded in the bottom reading mode of the fluorometer with excitation and emission wavelengths of 485 and 535 nm, respectively. The day-0 signal was subtracted from the day-4 signal to obtain the proliferation signal.
The 5(6)-carboxyfluorescein diacetate (CFDA)24–26 fluorometric assay was used to evaluate anti-phytopathogenic fungal activity against C. acutatum (BCC 58146) and A. brassicicola (BCC 42724). The assay was done in 384-well plates in triplicate. To each well, 25 μL of spore-suspension was added and the plate was incubated at room temperature for 2 h (C. acutatum) or 3 h (A. brassicicola) to allow spore adhesion and germination. Then, 25 μL of test compound dilution was added to each well and the plate was incubated at 25 °C for 16–18 h. For the detection of fluorescent signal, 25 μL of a mixture containing 2 μL of 0.9 μg mL−1 CFDA in 70% DMSO and 25 μL of 40% v/v glycerol was added to each well and kept in the dark for 5–10 min. After that, the plate was washed with tap water, blotted dry on paper towels, and 25 μL of distilled water added to each well. Fluorescence measurement was recorded at excitation and emission wavelengths of 485 and 535 nm, respectively, by using the bottom-reading mode of fluorometer. The signal of blank wells was subtracted from test wells to obtain the proliferation signal.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
All authors declare that they have no conflicts of interest.Acknowledgements
This research was supported by the National Science, Research and Innovation Fund, Thailand Science Research and Innovation (TSRI) (Grant No. FFB680075/0337) through the National Science and Technology Development Agency (P 2451270). We thank Dr Philip J. Shaw for manuscript editing.References
- I. Molnár, D. M. Gibson and S. B. Krasnoff, Nat. Prod. Rep., 2010, 27, 1241–1275 RSC.
- L. Zhang, O. E. Fasoyin, I. Molnár and Y. Xu, Nat. Prod. Rep., 2020, 37, 1181–1206 RSC.
- A. A. Malico, L. Nichols and G. J. Williams, Curr. Opin. Chem. Biol., 2020, 58, 45–53 CrossRef CAS PubMed.
- H. L. Robertsen and E. M. Musiol-Kroll, Antibiotics, 2019, 8, 157 CrossRef CAS PubMed.
- M. Isaka, R. Haritakun, S. Mongkolsamrit, S. Supothina, T. Feng and J.-K. Liu, Tetrahedron Lett., 2018, 59, 620–623 CrossRef CAS.
- C. Chutrakul, T. Boonruangprapa, R. Suvannakad, M. Isaka, P. Sirithunya, T. Toojinda and K. Kirtikara, J. Appl. Microbiol., 2009, 107, 1624–1631 CrossRef CAS PubMed.
- M. Isaka, S. Palasarn, K. Kocharin and J. Saenboonrueng, J. Nat. Prod., 2005, 68, 945–946 CrossRef CAS PubMed.
- K. Sadorn, S. Saepua, N. Boonyuen, W. Choowong, P. Rachtawee and P. Pittayakhajonwut, J. Nat. Prod., 2021, 84, 1149–1162 CrossRef CAS PubMed.
- K. Sadorn, S. Saepua, W. Punyain, W. Saortep, W. Choowong, P. Rachtawee and P. Pittayakhajonwut, Fitoterapia, 2020, 144, 104606 CrossRef CAS PubMed.
- J. Kornsakulkarn, S. Saepua, P. Laksanacharoen, P. Rachtawee and C. Thongpanchang, Tetrahedron Lett., 2013, 54, 3813–3815 CrossRef CAS.
- J. Kornsakulkarn, S. Saepua, K. Srichomthong, S. Supothina and C. Thongpanchang, Tetrahedron, 2012, 68, 8480–8486 CrossRef CAS.
- J. A. Dale and H. S. Mosher, J. Am. Chem. Soc., 1973, 95, 512–519 CrossRef CAS.
- J. Kornsakulkarn, S. Saepua, P. Laksanacharoen, P. Rachtawee and C. Thongpanchang, Tetrahedron Lett., 2016, 57, 305–307 CrossRef CAS.
- S.-S. Weng, C.-S. Ke, F.-K. Chen, Y.-F. Lyu and G.-Y. Lin, Tetrahedron, 2011, 67, 1640–1648 CrossRef CAS.
- Y.-P. Lam, X. Wang, F. Tan, W.-H. Ng, Y.-L. S. Tse and Y.-Y. Yeung, ACS Catal., 2019, 9, 8083–8092 CrossRef CAS.
- J.-H. Pan, J.-J. Deng, Y.-G. Chen, J.-P. Gao, Y.-C. Lin, Z.-G. She and Y.-C. Gu, Helv. Chim. Acta, 2010, 93, 1369–1374 CrossRef CAS.
- H. Liu, S. Liu, L. Guo, Y. Zhang, L. Cui and G. Ding, Molecules, 2012, 17, 14015–14021 CrossRef CAS PubMed.
- H. Zhang, Z. Deng, Z. Guo, X. Tu, J. Wang and K. Zou, Molecules, 2014, 19, 819–825 CrossRef PubMed.
- Y. Tsuda, K. Isobe, S. Fakushima, H. Ageta and K. Iwata, Tetrahedron Lett., 1967, 8, 23–28 CrossRef PubMed.
- R. E. Desjardins, C. J. Canfield, J. D. Haynes and J. D. Chulay, Antimicrob. Agents Chemother., 1979, 16, 710–718 CrossRef CAS PubMed.
- J. O'Brien, I. Wilson, T. Orton and F. Pongnan, Eur. J. Biochem., 2000, 267, 5421–5426 CrossRef PubMed.
- C. Changsen, S. G. Franzblau and P. Palittapongarnpim, Antimicrob. Agents Chemother., 2003, 47, 3682–3687 CrossRef CAS PubMed.
- L. Hunt, M. Jordan, M. De Jesus and F. M. Wurm, Biotechnol. Bioeng., 1999, 65, 201–205 CrossRef CAS PubMed.
- E. A. Aremu, T. Furumai, Y. Igarashi, Y. Sato, H. Akamatsu, M. Kodama and H. Otani, J. Gen. Plant Pathol., 2003, 69, 211–217 CrossRef CAS.
- J. Guarro, I. Pujol, C. Aguilar, C. Llop and J. Fernández-Ballart, J. Antimicrob. Chemother., 1998, 42, 385–387 CrossRef CAS PubMed.
- R. P. Haugland, in Handbook of fluorescent probes and research products, ed. J. Gregory, Molecular Probes, Inc., Oregon, USA, 2022, p. 966 Search PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra08431d |
| This journal is © The Royal Society of Chemistry 2025 |