
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceSynthesis of mesoporous carbon from banana peels with silica gel 60 as the hard templates†
Dinda P. N. Nahdaa,
Afiten R. Sanjaya a,
Fitria Rahmawatib,
Anne Zulfiac,
Afriyanti Sumbodjad,
Respati K. Pramadewandarue,
Yuni K. Krisnandia,
Zico A. Akbar*a and
Tribidasari A. Ivandini*a
a,
Fitria Rahmawatib,
Anne Zulfiac,
Afriyanti Sumbodjad,
Respati K. Pramadewandarue,
Yuni K. Krisnandia,
Zico A. Akbar*a and
Tribidasari A. Ivandini*a
aDepartment of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Indonesia, Depok 16424, Indonesia. E-mail: zicoalaia@sci.ui.ac.id; ivandini.tri@sci.ui.ac.id
bResearch Group of Solid-State Chemistry & Catalysis, Department of Chemistry, Faculty of Mathematics and Natural Sciences, Universitas Sebelas Maret, Jl. Ir. Sutami 36 A Kentingan, Surakarta 57126, Indonesia
cDepartment of Metallurgical and Materials Engineering, Faculty of Engineering, Universitas Indonesia, Depok 16424, Indonesia
dMaterial Science and Engineering Research Group, Faculty of Mechanical and Aerospace Engineering, Institut Teknologi Bandung, Jl. Ganesha 10, Bandung 40132, Indonesia
eDepartment of Materials and Metallurgical Engineering, INDSYS, Sepuluh Nopember Institute of Technology (ITS), Surabaya 60111, Indonesia
First published on 10th February 2025
Abstract
The synthesis of mesoporous carbon was successfully performed through solid–solid phase reaction employing banana peel powder as the carbon source and silica gel 60 (SG-60) as the hard template. The synthesis was initiated by hydrothermal heating to introduce the banana powder to SG-60 surface, followed by the carbonization process to form the mesoporous carbon. FTIR, Raman, XRF, and XRD characterization confirmed the success of the carbonization step, while N2 physisorption and TEM characterization confirmed the mesoporous structure formation of the synthesized carbon with the template. At an optimum carbon-to-silica precursor ratio of 3![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :1, the synthesized carbon with SG-60 templates proceeds to a specific surface area of 476.97 m2 g−1, which is around 55-fold higher than the one synthesized without any template. Furthermore, evaluation of the capacitance performances was done by creating composite electrodes with nickel foam as the support and polyvinylidene difluoride as the binder. The evaluation was carried out using cyclic voltammetry in 3.0 M KOH, galvanic charge–discharge, and electrochemical impedance spectroscopy confirming a high correlation between the specific surface area and the specific capacitance. The banana peels-derived mesoporous carbon demonstrates a specific capacitance value of 23.1 F g−1, measured using the cyclic voltammetry method. Good stability of the prepared electrode over 2500 voltammetric cycles was also demonstrated, indicating that the use of SG-60 as the hard template is suitable for synthesizing carbon with mesoporous structure from biomass.
:1, the synthesized carbon with SG-60 templates proceeds to a specific surface area of 476.97 m2 g−1, which is around 55-fold higher than the one synthesized without any template. Furthermore, evaluation of the capacitance performances was done by creating composite electrodes with nickel foam as the support and polyvinylidene difluoride as the binder. The evaluation was carried out using cyclic voltammetry in 3.0 M KOH, galvanic charge–discharge, and electrochemical impedance spectroscopy confirming a high correlation between the specific surface area and the specific capacitance. The banana peels-derived mesoporous carbon demonstrates a specific capacitance value of 23.1 F g−1, measured using the cyclic voltammetry method. Good stability of the prepared electrode over 2500 voltammetric cycles was also demonstrated, indicating that the use of SG-60 as the hard template is suitable for synthesizing carbon with mesoporous structure from biomass.
Introduction
Porous carbon materials have been established as a common material for many applications, including pollutant adsorbents,1,2 supporting catalysts,3 and electrode materials4,5 due to their high surface area, good electrical conductivity, high chemical and physical stability as well as their versatility.6 To date, various types of porous carbon materials such as powders, fibers, and tubes have been reported,7 and extensive methods have been developed to produce these types of materials.8 Meantime, mesoporous carbon attracts great attention due to its advantage in high specific surface and porous volume, providing low-resistant pathways for the ions through the porous particles.8 This property is very useful to increase the performance of carbon as adsorbents, catalysts, or electrodes. Moreover, the pore structure of mesoporous carbon is uniform and can be controlled depending on the applications. The optimum pore structures are supposed to possess smaller pores interconnected with larger sets of pores, which could achieve high surface area and efficient ion diffusion pathways simultaneously.9Furthermore, the use of a hard template is one of the promising strategies to produce a mesoporous structure. The high stability and good space-confined effect of the hard template can produce the structure of the final target materials to perfectly replicate the morphology of the template.10 A common example of a hard template for mesoporous carbon synthesis is silica-based materials, such as SBA-15,11 MCM-41,12 and MCM-48.13 These silica templates are popular because of their well-defined and tuneable pore structures, which can be replicated in the carbon structure. Meanwhile, some efforts to use more economical hard templates to synthesize mesoporous carbon have been reported, such as zeolites, clays, and calcium carbonate.14–16 Notably, the use of silica gel 60 (SG-60) as a hard template to synthesize the mesoporous carbon with sucrose as the carbon source can produce a high specific area and pore volume.17 SG-60 refers to a specific grade of silica gel commonly used in thin-layer chromatography and column chromatography. It is a form of amorphous silicon dioxide with a high surface area and uniform particle size. The number 60 refers to the pore size or particle mesh size, typically in angstroms, or in some cases, the mesh size in microns.
On the other hand, biomass materials (i.e. paper, food waste, leaves, wood, and leather products) generally abundant, renewable, inexpensive, and environmentally benign are potentially used as precursors to construct high quality carbon materials. Bananas are the most widely consumed fruits in the world. Based on data from the Food and Agriculture Organization (FAO) in 2021, India, China, and Indonesia are the countries where the highest bananas are produced and consumed. During banana production and consumption, approximately 40% of the total banana fruit, in the form of banana peels, is discarded.18 Banana peels are rich in nutrients, such as carbohydrates, protein, fiber, and fat. Apart from that, banana peels also contain various minerals, such as phosphorus, sodium, calcium, iron, magnesium, and potassium.19 The high carbon content in banana peels has the potential to produce porous carbon materials.
The use of banana peels as the carbon source for producing activated carbon has been reported to yield a specific surface area of 1928 m2 g−1 after activation process using ZnCl2 and FeCl2.4 Meanwhile, the synthesis of porous carbon from banana peels, producing a hierarchical material consists of micropores and mesopores, is reported to yield a specific area of 1362 m2 g−1 after activation process using KOH and ZnCl2.20 However, there are limited reports specifically combining biomass with hard templates for mesoporous carbon synthesis as this synthesis involving solid–solid phase reaction. No record shows the synthesis using hard templates with banana peels as the carbon source, while studies using similar biomass sources, such as coconut shells or rice husks, are available.21,22
Carbon materials have been studied for various applications, including applications in supercapacitors. Numerous publications have reported the development of carbon-based materials for this purpose. The synthesis of porous carbon material is reported to achieve a specific capacitance of 74 F g−1.23 However, the modification of carbon materials, such as mixing with cerium-doped NiO nanocomposite and with MnO2 nanoparticles can provide significant enhancement of the specific capacitance to be comparable with conductive polymer, such as polyaniline (PANI) or metal-based materials, such as NiMoO4 nanorods.24–27 This remains a strong demand for the development of carbon-based supercapacitors, particularly those derived from environmentally friendly precursors, such as biomass waste like banana peels.
In this research, mesoporous carbon was synthesized from banana peel powder using SG-60 as the hard template. The synthesis was performed by hydrothermal reaction of banana peel powder suspension in the presence of SG-60 to introduce the banana peel powder to the SG-60 surface. Afterward, the banana peel powder-coated SG-60 was carbonized to obtain the graphitic phase of carbon. Characterization after removing the hard template carried out using FTIR, Raman, XRD, and XRF confirmed the formation of graphitic carbon, while TEM and N2-physisorption analyses indicated that the mesoporous carbon structure was successfully formed. Without any additional activation, a specific surface area of 476.97 m2 g−1 and a total pore volume of 0.67 cm3 g−1 was achieved, representing an approximately 55-fold increase compared to the synthesized carbon performed without any template. Furthermore, the electrochemical capacitive performance of the synthesized mesoporous was investigated by using cyclic voltammetry (CV), galvanostatic charge–discharge (GCD), and electrochemical impedance spectroscopy (EIS). The result showed that consistent with its higher surface area, the template-used synthesized mesoporous carbon provides a significantly higher specific capacitance than the carbon synthesized without a template. Moreover, good stability of the specific capacitance value was observed. The results suggest that the synthesis of porous carbon materials from biomass with economical hard templates is promising to be developed.
Experimental
Materials
The banana peels (Musa paradisiacal sp.) used in this research were collected from fried banana sellers in Depok, West Java, Indonesia. To synthesize the mesoporous carbon, silica gel 60 (SG-60), sulfuric acid (H2SO4), and ethanol were used, while to perform the electrochemical analysis, polyvinylidene difluoride (PVDF), dimethylacetamide (DMAc), and potassium hydroxide (KOH) were used. Commercial mesoporous carbon was applied to compare the characteristics and performances of synthesized mesoporous carbon as a supercapacitor. All these chemicals were purchased from Merck and used without any purification. Milli-Q water with 18.2 MΩ cm was used to prepare the solution.The synthesized mesoporous carbon was characterized using N2 adsorption and desorption isotherm at 77 K on a Quantachrome Quadrasorb-Evo Surface Area and Pore Size Analyzer. The surface areas were calculated using the Brunauer–Emmett–Teller (BET) method, and the pore size distributions were determined using the Barrett–Joyner–Halenda (BJH) method from the desorption data. X-ray diffraction (XRD) spectra of the samples were obtained using a benchtop X-ray diffractometer (Aeris 600 W with GI-XRD or Heating Stage). Fourier transform infrared (FTIR) spectra were recorded on a Bruker Alpha II FTIR Spectrometer, with samples ground with KBr and pressed into thin wafers. Transmission electron microscopy (TEM) images were acquired on an FEI Tecnai G2 20 S-Twin electron microscope, equipped with a field emission gun at an acceleration voltage of 200 keV using a LaB6 filament and an Eagle™ CCD camera. The Raman spectra were performed using a HORIBA LabRAM HR Evolution Raman Microscope with a 532 nm laser beam and 1800 g mm−1 grating.
Synthesis mesoporous carbon
The banana peels were cleaned and washed with water before dried under the sun. Then, the banana peels were cut into small pieces and pulverized into powder. The powder was filtered using a 300 μm mesh sieve and ready for the synthesis of mesoporous carbon.The synthesis of porous carbon was carried out without a template, whereas the synthesis of mesoporous carbon was conducted using a silica gel template with three different precursor-to-template ratios, including 1:1, 2:1, and 3:1 (w/w) and labelled as MC-S-1, MC-S-2, and MC-S-3, respectively. The synthesis without a template was conducted by carbonizing the banana peel powder in a tubular furnace at 800 °C for 3 hours. Meanwhile, the synthesis using a template involved the addition of 250 mg SG-60 powder into a suspension of carbon precursor in H2SO4 (98 wt%).18,28 This suspension was varied to synthesize MC-S-1, MC-S-2, and MC-S-3 with various amounts of banana peel carbon precursors (250 mg, 500 mg, and 750 mg, respectively) and various amounts of H2SO4 (25 mg, 50 mg, and 75 mg, respectively). The mixture was placed in a 100 mL PTFE autoclave and heated in an oven at 100 °C for 6 hours. The temperature was then increased to 160 °C for an additional 6 hours. Finally, the mixture was carbonized in a tubular furnace at 800 °C for 3 hours in an N2 atmosphere. To remove the templates, the composite was stirred in a reflux system with 4.0 M NH4F for 5 hours at 120 °C, then washed with 80 °C distilled water. The synthesized mesoporous carbon was dried overnight in an oven and characterized using XRD, XRF, Raman, BET, FTIR, and TEM.
Electrochemical study
The electrochemical study was carried out using a three-electrode system. The prepared mesoporous carbon coated on nickel foam (NF) was used as the working electrode, while the Pt spiral and Ag/AgCl system were used as the counter and the reference electrodes, respectively. The working electrodes were prepared by mixing the synthesized mesoporous carbon with polyvinylidene difluoride (PVDF) in a 9:1 ratio and dimethylacetamide (DMAc) as a solvent. The obtained slurry was coated on the NF surface and oven-dried at 70 °C for 12 hours. The electrochemical system used an electrolyte of 3.0 M KOH. This is further supported by the data in Fig. S1,† which compares the use of Na2SO4 and KOH solutions as electrolytes. A significant different in their specific capacitance is observed, with the highest capacitance generated by the use of 3 M KOH as the electrolyte.
The capacitive performance of prepared working electrodes was then studied using cyclic voltammetry (CV), galvanostatic charge–discharge (GCD), and electrochemical impedance spectroscopy (EIS) with Autolab PGSTAT-204 (Metrohm). Additionally, the stability of the performance was measured with CV and GCD over 2500 cycles.
Experimental result
The synthesis and the characterization of porous and mesoporous carbon
Two different types of samples were prepared, including porous carbon without an SG-60 template (PC), and carbon with SG-60 as a template with different carbon to SG-60 ratios (MC-S-1, MC-S-2, MC-S-3 in which the C-to-Si precursor ratio are 1:1, 2:1, 3:1, respectively). FTIR spectroscopy was used to characterize the synthesized mesoporous carbon with and without the SG-60 template before and after carbonization. The FTIR spectrum of mesoporous carbon typically reflects a mix of surface functional groups such as hydroxyls (–OH), aliphatic C–H groups, carbonyls (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O), and graphitic CC bonds.29 Fig. 1 shows that after carbonization, similar IR peak patterns were observed for all carbon types, showing the absence of the absorbance peaks at the wave numbers of 3400–3600 cm−1, 2800–3000 cm−1, and 1600–1000 cm−1, correspond to the OH− stretching vibration of hydroxyl groups, C–H stretching vibrations of aliphatic hydrocarbons, and CO stretching vibration of carbonyl groups, respectively. The disappearance of these functional groups is reasonable as they are commonly available on the surface due to water absorption or residual organic carbon from incomplete carbonization. Meanwhile, the peaks of the C–H bending, C–O stretching, and graphitic carbon remained at around 600–900 cm−1, 100–1300 cm−1, and 1500–1600 cm−1, respectively, confirming the formation of graphitic carbon. In addition, the absorbance peaks at 1100–1150 cm−1, 950–970 cm−1 and 780–820 cm−1 were observed related to the asymmetric stretching vibrations of Si–O–Si, symmetric stretching vibration of Si–O groups on Si–O–H, and symmetric stretching vibration of the group Si–O–Si, respectively.30–32 Although the carbonization step decreased the silica-related peaks, the FTIR indicated that the functional groups containing silica cannot be completely degraded with the carbonization step. Accordingly, the carbon product was then cleaned by using a reflux system in 4.0 M NH4F to dissolve the silica in the template. After removing the template, lower absorbance peaks of the silica groups were detected in the spectrum, especially for MC-S-2 and MC-S-3.
O), and graphitic CC bonds.29 Fig. 1 shows that after carbonization, similar IR peak patterns were observed for all carbon types, showing the absence of the absorbance peaks at the wave numbers of 3400–3600 cm−1, 2800–3000 cm−1, and 1600–1000 cm−1, correspond to the OH− stretching vibration of hydroxyl groups, C–H stretching vibrations of aliphatic hydrocarbons, and CO stretching vibration of carbonyl groups, respectively. The disappearance of these functional groups is reasonable as they are commonly available on the surface due to water absorption or residual organic carbon from incomplete carbonization. Meanwhile, the peaks of the C–H bending, C–O stretching, and graphitic carbon remained at around 600–900 cm−1, 100–1300 cm−1, and 1500–1600 cm−1, respectively, confirming the formation of graphitic carbon. In addition, the absorbance peaks at 1100–1150 cm−1, 950–970 cm−1 and 780–820 cm−1 were observed related to the asymmetric stretching vibrations of Si–O–Si, symmetric stretching vibration of Si–O groups on Si–O–H, and symmetric stretching vibration of the group Si–O–Si, respectively.30–32 Although the carbonization step decreased the silica-related peaks, the FTIR indicated that the functional groups containing silica cannot be completely degraded with the carbonization step. Accordingly, the carbon product was then cleaned by using a reflux system in 4.0 M NH4F to dissolve the silica in the template. After removing the template, lower absorbance peaks of the silica groups were detected in the spectrum, especially for MC-S-2 and MC-S-3.
 | ||
| Fig. 1 FTIR spectra of the synthesized mesoporous carbon with a carbon-to-silica ratio of 1:1, 2:1, and 3:1, before and after carbonization steps as well as after removing the hard template. | ||
To confirm the graphitic levels on the synthesized PC, MC-S-1, MC-S-2, and MC-S-3, Raman characterization was performed after the template removal as displayed in the spectrum in Fig. 2. The spectra show that all the synthesized carbon produces strong Raman peaks at around 1350 cm−1 and 1590 cm−1 corresponding to the D band and G band of the spectrum, respectively. The intensity of the D and G bands in the Raman spectrum was generally used to express the ordered structure and the graphitization levels of the carbon material by analysing the ratio of ID/IG. The lower ID/IG ratio in the spectrum indicates higher graphitization levels and conductivity of the carbon material.33 The intensities of D and G bands of the synthesized PC without template shows the ID/IG ratio of 0.83, while interestingly the ID/IG ratio of the synthesized MC-S-1, MC-S-2, and MC-S-3 were 0.69, 0.70 and 0.73, respectively. This result confirms that the use of SG-60 template not only increases the ordered structure of the mesoporous carbon but also enhances the CC sp2 bond formation leading to higher graphitization levels of the carbon materials.
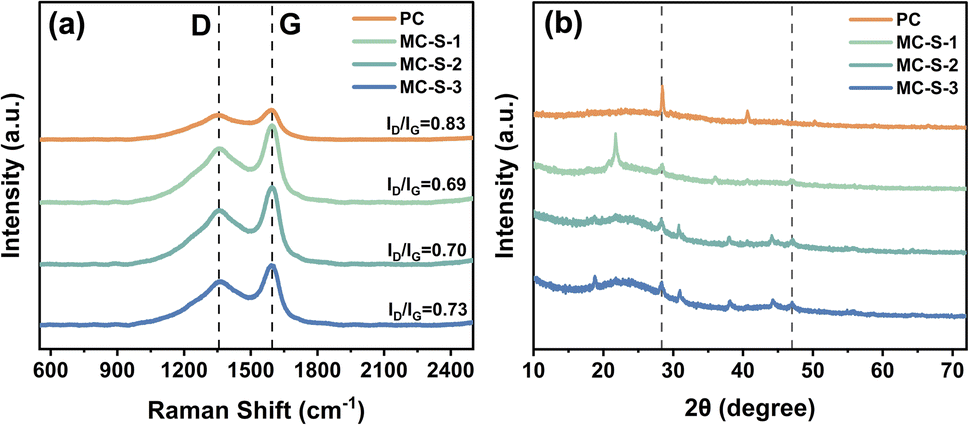 | ||
| Fig. 2 Characterization of the synthesized PC, MC-S-1, MC-S-2, and MC-S-3 is shown in (a) the Raman spectrum and (b) the XRD patterns. | ||
The analysis of the elemental composition in the synthesized carbon after removing the template was performed using XRF as presented in Table 1. The analysis results show that carbon is the main element in the synthesized carbon with 91.52, 96.43, 96.16, and 95.59% content in PC, MC-S-1, MC-S-2, and MC-S-3 samples, respectively. The silica content was found to be exceptionally low (less than 0.2%), indicating that the reflux step was effective. However, all the synthesized carbon materials exhibit lower carbon content compared to the commercial mesoporous carbon (99%). The lower carbon content in the synthesized carbon from banana peels is probably due to the presence of several minerals in the banana peel precursor, such as potassium and manganese, while other minerals including silica, sulfur, and calcium were introduced from the SG-60 template during the carbonization process. It is interesting to note that the potassium content of PC (5.73%) is noticeably higher than the mesoporous samples, which contain only less than 0.5% potassium, while it is completely undetected in the commercial mesoporous carbon. Instead of potassium, sulfur, and calcium content were observed in the MC-S samples. It seems the formation of the mesoporous structure in the synthesized MC facilitates the dissolution process of potassium ions from the carbon matrix, while it causes less effect on sulfur and calcium.
| Materials | Element composition (%wt) | Total (%) | ||||||
|---|---|---|---|---|---|---|---|---|
| Si | S | K | Ca | Mn | O | C | ||
| PC | 0.14 | 0.01 | 5.73 | 0 | 0.07 | 2.54 | 91.52 | 100.00 |
| MC-S-1 | 0.08 | 0.64 | 0.18 | 1.09 | 0.02 | 1.56 | 96.43 | 100.00 |
| MC-S-2 | 0.07 | 1.84 | 0.25 | 1.06 | 0.04 | 1.70 | 96.16 | 100.00 |
| MC-S-3 | 0.19 | 2.16 | 0.45 | 0.93 | 0.04 | 1.99 | 95.59 | 100.00 |
| MC-commercial | 0 | 0 | 0 | 0.03 | 0 | 0.02 | 99.95 | 100.00 |
The XRD patterns of the synthesized PC and MC-S from banana peels are shown in Fig. 2(b). The analysis of the XRD pattern indicated that all the synthesized carbon materials were in an amorphous state, as evidenced by the broad peak features at 2θ angles around 24 and 45°. These peaks are indexed as (002) and (101) planes of the graphitic carbon, confirming the success of the carbonization step.34 However, the XRD patterns of all the synthesized materials also show sharper peaks with relatively high intensity at several 2θ angles, indicating the presence of elements with crystalline structures within the amorphous carbon material. These peaks were observed at 2θ angles of 28.5 and 47.1°, corresponding to JCPDS 00-027-1402 and indicating the presence of cubic silica crystals indexed to the (111) and (220) planes that cannot dissolve with the refluxing process.
The obtained N2-physisorption information was analysed using BET, t-plot, and BJH methods. The isotherm graphs of PC, MC-S-1, MC-S-2, and MC-S-3 are shown in Fig. 3(a). The synthesized PC from the carbonization of banana peel powder without a template shows a Type I isotherm, indicating that PC is dominated by a significant amount of micropores (<1 nm in diameter). On the other hand, all the synthesized mesoporous carbon (MC-S) gives a Type IV isotherm with a hysteresis loop at p/p0 > 0.4, indicating capillary condensation in the mesopores and confirming the formation of mesoporous structure in MC-S-1, MC-S-2, and MC-S-3. Furthermore, the t-plot method is used to evaluate the specific surface area and pore volume of mesopores and micropores as the high specific surface area is advantageous for enhancing the number of active sites during interfacial chemical reactions, such as electrochemical double layer specific capacitance and catalytic activity. The result summarized in Table 2 shows that the synthesis with SG-60 template significantly increases the specific surface areas of the synthesized carbon (SBET and Smeso), especially for the MC-S-3 samples, which shows ∼55-fold increase of SBET (476 m2 g−1) compared to that of PC samples (8 m2 g−1). Considering that the SG-60 template has a surface area of 337.21 m2 g−1, higher surface area of the MC-S-3 with C-to-Si ratio of 3:1 was probably due to the contribution of the microporous structure that has been opened by the formation of the mesoporous structure. Meanwhile, the increasing amount of SG-60 template in the synthesis of MC-S-1 and MC-S-2 (C-to-Si precursor ratios of 1:1 and 2:1) was found to reduce the specific surface area to be around 300 m2 g−1, which is relatively similar to that of the template. This result demonstrates the effectiveness of the hard template method using SG-60 to provide a controlled synthesis of mesoporous carbon from banana peel powder.
 | ||
| Fig. 3 Characterization of the synthesized PC, MC-S-1, MC-S-2, and MC-S-3, shown in (a) the nitrogen adsorption–desorption isotherm graph at 77 K, and (b) its related pore diameter graph calculated based on the BJH method. | ||
| Materials | N2 physisorption | |||||||
|---|---|---|---|---|---|---|---|---|
| SBETa (m2 g−1) | Smicrob (m2 g−1) | Smesob (m2 g−1) | Vtotalb (cm3 g−1) | Vmicrob (cm3 g−1) | Vmesob (cm3 g−1) | SBETa (m2 g−1) | Avg. pore diameterc (nm) | |
| a Analysed using the BET method.b Analysed using the t-plot method.c Analyzed using the BJH method. | ||||||||
| PC | 8.38 | 2.15 | 6.23 | 0.01 | 0.00 | 0.01 | 8.38 | 3.16; 3.87; 4.82; 6.22 |
| MC-S-1 | 320.56 | 33.58 | 286.98 | 0.57 | 0.02 | 0.55 | 320.56 | 3.87; 18.62 |
| MC-S-2 | 300.62 | 64.54 | 236.08 | 0.46 | 0.03 | 0.43 | 300.62 | 3.87; 5.45; 7.24; 18.62 |
| MC-S-3 | 476.97 | 82.04 | 394.93 | 0.67 | 0.04 | 0.63 | 476.97 | 3.87; 8.56; 18.62 |
| Silica gel 60 | 337.21 | 8.47 | 328.74 | 0.71 | 0.00 | 0.71 | 337.21 | 6.24 |
Moreover, the pore diameters of the synthesized carbon were determined based on the BJH method as shown in Fig. 3(b). The SG-60-templated mesoporous carbons (MC-S) show larger average pore diameters (3.87–18.62 nm) than the non-template porous carbon (PC) (3.16–6.22 nm). A wide distribution of pore diameters was exhibited by the MC-S samples, which differs from those of the PC and SG-60 with a pore diameter of around 3–6 nm. It seems the pores in PC were formed during the carbonization process due to the loss of some functional groups in the carbon precursor, while the pores formation in MC-S was also influenced by the structure defects during the carbonization process with the pore formation being driven by the templates.
A comparison of the reported specific surface areas of the porous carbon from banana peels synthesized against different methods is presented in Table 3. Referring to the above statement, all the synthesis methods were commonly done by using soft templates, such as KOH, ZnCl2, and zinc complexes, while the report of the synthesis using hard templates has not been found. In addition, a step for activating the carbon has been commonly added, resulting in a significant specific surface area.8,23,36,38 A remarkable specific surface area was achieved due to its hierarchical structure.40 As the synthesis was carried out using the hard template, a lower specific surface area was expected. However, comparable specific surface area with others in literature could be obtained in this work. The observed slightly lower specific area can be resolved with an additional step to activate the carbon to significantly increase the specific surface area.
| Electrodes | Synthesis methods | SBET (m2 g−1) | Specific capacitance (F g−1) | Ref. |
|---|---|---|---|---|
| Activated carbon | Banana fiber activated in KOH at 110 °C for 5 days and pyrolysis at 800 °C for 1 h under N2 atmosphere | 686 | 74 | 23 |
| Activated carbon in sheet form | Banana peel slurry dried in the mold became paper sheets and carbonized at 500 °C | 581 | 68 | 35 |
| Activated carbon | Banana peels are carbonized at 600 °C and activated in KOH 25% at 700 °C | 540 | 73 | 36 |
| Activated carbon | Banana peels are hydrothermally processed at 200 °C for 24 h and precipitated as porous carbon | 294 | 199 | 37 |
| Hierarchical porous carbon foam | Banana peel marinated in zinc nitrate solution for 7 days at 70 °C, mixed with furfural and 2-aminophenol, then carbonized at 1000 °C for 8 h | 1650 | 206 | 8 |
| Mesoporous carbon | Banana peels are hydrothermally processed at 190 °C for 12 h with colloidal SiO2 and graphene oxide and carbonized at 1000 °C for 3 h | 312 | — | 38 |
| Porous carbon nanospheres | Porous carbon nanospheres prepared using amino acid–urea based on deep eutectic system, PAN, and K2CO3 in solid-phase stirring and carbonization at 700 °C | 1277 | 297 | 39 |
| Mesoporous carbon | Banana peel powder was introduced to silica gel 60 surface and hydrothermally processed at 100 °C and 160 °C for 6 h each, then carbonized at 800 °C under N2 flow | 476.97 | 23.1 | This work |
The various synthesized porous carbons were characterized using TEM. The TEM images with the same magnifications were then analysed to thoroughly investigate the influence of the hard template in the formation of the meso-sized pores in the synthesized carbon. Fig. 4 shows that the TEM images of all the materials exhibit a light–dark pattern, revealing the porous characteristics of the substance.41 The porous carbon synthesized without a silica gel template exhibits an amorphous phase with smaller and irregular pores than other synthesized carbons as shown in Fig. 4(a). This result is in conformance with the N2-physisorption measurements as summarized in Table 2, in which remarkably low surface area and pore volume are shown. The micropores predominantly formed in the absence of a templating agent are responsible for this outcome.42
 | ||
| Fig. 4 TEM images of the synthesized (a) PC, (b) MC-S-1, (c) MC-S-2, and (d) MC-S-3. | ||
Fig. 4(b–d) show TEM images of the synthesized mesoporous carbons using the silica gel 60 template. The use of the template significantly influenced the morphology of the porous structure. All three materials exhibited similar diffraction patterns characterized by well-ordered pore structures and visible parallel stripes.43 The pore size observed in the TEM images also decreased with the addition of silica as the pore-directing agent. This is consistent with the N2 adsorption–desorption results, showing that the specific surface area of the carbon increases. As the amount of silica impregnation in the materials MC-S-1, MC-S-2, and MC-S-3 gradually decreases (50%, 33%, and 25%), the formation of well-ordered porosity becomes more evident. In the inset of Fig. 4(d), carbon material shows packed within the pore space of the silica template, forming nanorod-like 2-D carbon structures with interconnected networks.44
Electrochemical study of the synthesized carbon
To further demonstrate the application of mesoporous carbon derived from banana peels, all the synthesized carbon materials, including the non-template PC and the SG-60 template MC-S, were decorated on the surface of Ni foam by using PVDF as the binder. The prepared electrodes were used as the working electrodes in a three-electrodes electrochemical system and labelled as PC@NF, MC-S-1@NF, MC-S-2@NF, and MC-S-2@NF. Fig. 5(a) shows their related voltammograms in 3.0 M KOH solution applied in the potential range of 0–0.6 V (vs. Ag/AgCl). The voltammograms show a typical oxidation–reduction peak pair at a potential around +0.5 V and +0.2 V (vs. Ag/AgCl), respectively, corresponding to the oxidation reaction of Ni(II) to Ni(III) and the reduction reaction of Ni(III) to Ni(II).40,45,46 This oxidation–reduction peak pair indicates pseudocapacitive properties due to the faradaic reaction of nickel oxidation–reduction, in contrast to the properties of carbon material that exhibits non-faradaic behaviour by forming an electrostatic double layer capacitive (EDLC) rather than undergoing electron transfer reaction. The carbon-modified Ni foam electrodes can be classified as hybrid supercapacitors, which combine the characteristics of EDLC and pseudocapacitors. Although mesoporous carbon does not impart EDLC properties to the electrode, Fig. 5(a) shows that the presence of mesoporous carbon slightly increases the peak currents in the voltammograms. All the synthesized mesoporous carbon shows comparable capacitance. On the contrary, the synthesized carbon without a template (PC) seems to hinder the electron transfer, resulting in EDLC-like voltammograms. It appears that the synthesized mesoporous carbon gives a significant contribution to electron storage sites in supercapacitor systems, resulting in a greater number of electrons participating in oxidation–reduction reactions.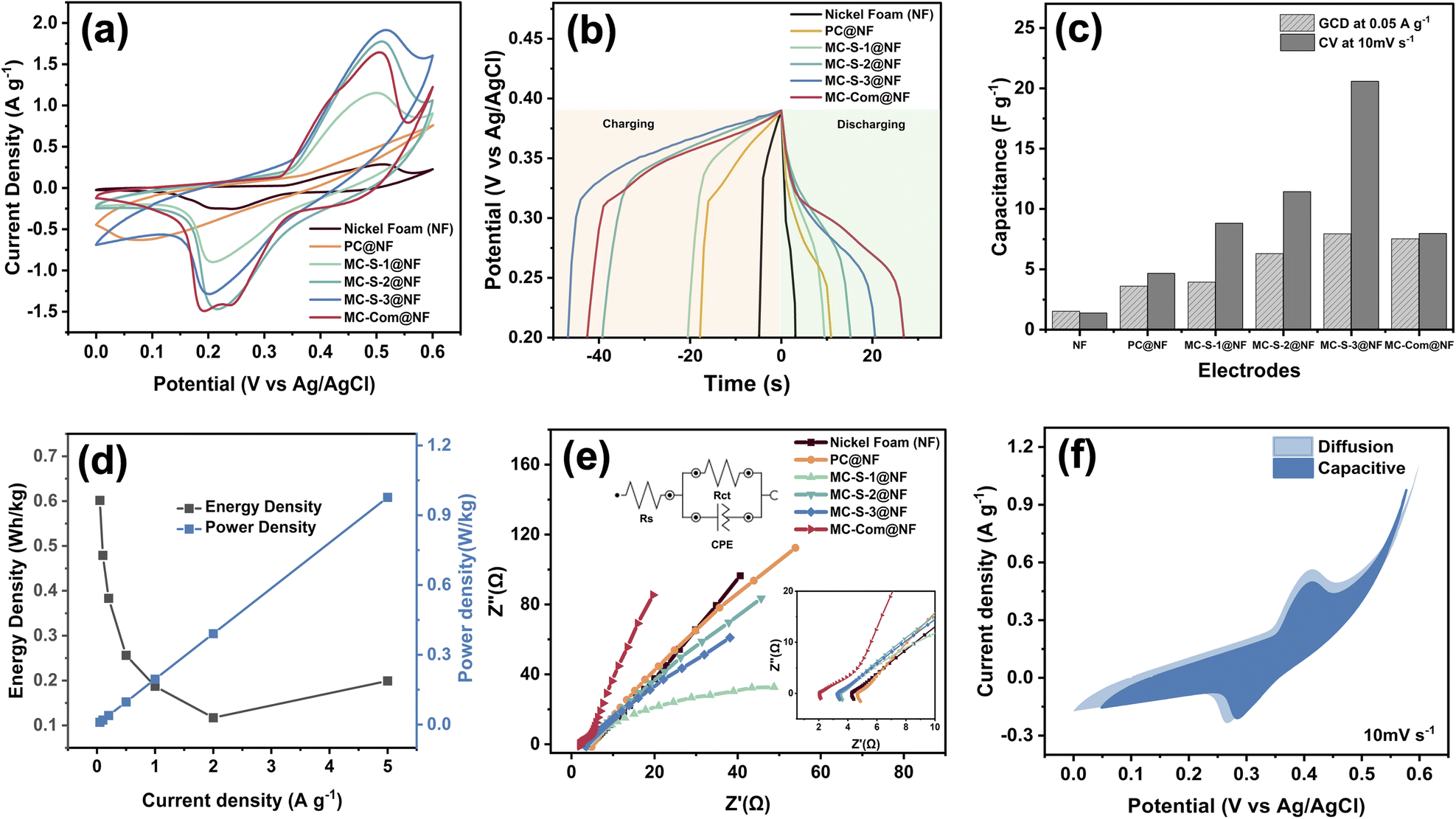 | ||
| Fig. 5 Performance of all the synthesized carbon materials coated on the nickel foam used as the working electrodes in 3.0 M KOH solution in comparison with the unmodified Ni foam and the commercial mesoporous carbon-coated Ni foam shown in (a) voltammograms an applied scan rate of 100 mV s−1, (b) galvanostatic charging–discharging (GCD) curves at current densities of 0.05 A g−1, (c) comparison of the specific capacitance value calculated from voltammetry and GCD data in (a) and (b), (d) energy and power density of MC-S-3 extrapolated from GCD curves at various current applied, (e) Nyquist plots from EIS measurements with the equivalent circuit model of supercapacitors and the magnification of the Nyquist plots at high frequencies shown in the insets, and (f) diffusion and capacitive contributions to the capacitance performance of MC-S-3. | ||
The specific capacitance values of each synthesized carbon were analysed based on CV measurements with the following equation:
 | (1) |
| Electrode | Csp (F g−1) at 100 mV s−1 | Csp (F g−1) at 0.05 A g−1 | Csp (F g−1) at 1 A g−1 | Energy density at 0.05 A g−1 (W h kg−1) | Power density at 0.05 A g−1 (kW kg−1) | Rs (Ω) | Rct (Ω) |
|---|---|---|---|---|---|---|---|
| NF | 6.09 | 1.49 | 0.57 | 0.11 | 0.01 | 4.27 | 9.81 |
| PC@NF | 11.21 | 3.57 | 1.67 | 0.27 | 0.01 | 4.67 | 10.19 |
| MC-S-1@NF | 21.40 | 3.91 | 1.52 | 0.30 | 0.01 | 3.34 | 7.78 |
| MC-S-2@NF | 22.11 | 6.27 | 2.21 | 0.48 | 0.01 | 3.46 | 6.20 |
| MC-S-3@NF | 23.14 | 7.91 | 2.46 | 0.60 | 0.01 | 3.29 | 4.25 |
| MC-Com@NF | 22.01 | 7.50 | 5.78 | 0.57 | 0.01 | 2.08 | 35.80 |
Similar behaviour was also observed when the Ni foam was modified with commercial mesoporous carbon. Furthermore, although the modification with PC increases the capacitance value, the increase is not as high as the modification with MC-S. This behaviour is consistent with the specific surface area data previously presented in Table 2, showing a notably higher specific area of PC than those of MCs. Furthermore, the electrode modified with the synthesized carbon at C-to-Si ratio of 3:1 (MC-S-3@NF) produces the highest specific capacitance at 23.1 F g−1.
The specific capacitance values of the electrodes were also characterized from the GCD curves (Fig. 5(b)), showing the dependence of the currents versus the charging and the discharging times. The specific capacitance values were calculated using the following equation:
 | (2) |
It can be seen from the figure that the modification of Ni foam with carbon significantly increases the discharging time. It is found that the mesoporous carbon electrode synthesized using SG-60 template particularly exhibited relatively longer discharging times than PC. While the discharging time of PC@NF is 28 s at a current density of 0.05 A g−1, MC-S-3@NF can provide discharging time of 62 s.
Theoretically, since the specific capacitance was calculated using eqn (2), a longer discharge time corresponds to a higher capacitance value. Consistent with this statement, the outstandingly higher capacitance value of MC-S-3@NF (7.91 F g−1, at a current density of 0.05 A g−1) than that of PC@NF was observed. This result is also in alignment with the voltammogram results in Fig. 5(a), in which the MC-S-3@NF demonstrated the highest specific capacitance value. A comparison of the resulting specific capacitances from the voltammograms and the GCD curves is shown in Fig. 5(c), displaying a similar tendency of the capacitive performance. This behaviour is greatly correlated to the specific surface area of the synthesized mesoporous carbon, confirming the main contribution of the specific surface area to the value of specific capacitance. It is interesting to note that the banana-derived mesoporous carbons synthesized using silica gel 60 exhibit a relatively comparable surface area and specific capacitance when compared to other banana-derived carbon materials in Table 3. The lower specific capacitance of banana-derived mesoporous carbons synthesized using silica gel 60 is likely due to the low surface area. The banana-derived carbons are synthesized using silica gel 60 produce a mesoporous feature which can be beneficial for reducing ion diffusion resistance of supercapacitor electrolytes.48
EIS is used to analyse the internal impedance of the system at alternating current frequencies.49 Nyquist plot in Fig. 5(e) shows the analysis results for various electrodes modified with the synthesized mesoporous carbon. The measured data on the Nyquist plot, such as series resistance (Rs) and charge transfer resistance (Rct), are simulated using an equivalent circuit as shown in Fig. 5(e), and each value is presented in Table 3. As the Rct values represent the resistance measured during the faradaic reaction, a lower measured Rct indicates a larger contact area at the electrode–electrolyte interface, signifying higher conductivity of the materials. In line with the previous results about the specific capacitance, the data in Table 3 shows the MC-S-3@NF electrode has the lowest Rct value of 6.43 Ω.
The stability of the material was investigated Fig. 6(a) shows the comparison of the voltammograms of MC-S-3@NF in 3.0 M KOH solution at the 1st and the 2500th cycles, while Fig. 6(b) shows the plots of the oxidation peak currents of the voltammograms over 2500 cycles. This figure clearly shows that the peak current gradually increases during the cycles. The probable reason is due to the adsorption of hydroxyl or metal ions in the porous carbon during the cycles causes the increase of the active sites at the porous carbon. It was reported that activation of porous carbon by ion adsorption technique can increase the electrochemical surface area, resulting in an increase of capacitive current.4,20 As shown in the Fig. S2,† the Nyquist plot from electrochemical impedance spectroscopy decreases after 2500 cycles. From the fitting, charge transfer resistance (Rct) in the first cycle is 65.3 Ω and decreases to 32.9 Ω after 2500 cycle of stability test, this is probably due to increase active sites during stability test as previously discussed.50,51 Nevertheless, the results indicate that the MC-S-3@NF electrode can be employed without any fouling in the materials over 2500 cycles.
 | ||
| Fig. 6 The stability of the capacitive performance of the synthesized mesoporous carbon is shown in (a) the voltammograms of 3.0 M KOH at the first and the 2500th cycle, and (b) its related oxidation peak current plots for 2500 cycles. The working electrode was MC-S-3@NF with a scan rate of 100 mV s−1. | ||
Conclusions
Mesoporous carbon has been successfully synthesized from banana peel powder using Silica Gel 60 as a hard template. The formation of the graphitic carbon was confirmed by using FTIR, Raman, XRF, and XRD. Based on N2-physisorption and TEM characterization, the synthesis using the template showed the formation of mesoporous structure, while it was not observed in the product without the template. Additionally, the use of hard template enhanced the specific surface area of the carbon to around 55 times higher than without template. The mesoporous carbon synthesized with a C-to-Si ratio of 3:1 exhibited the highest specific area and consistency to this, the electrode prepared with this mesoporous carbon exhibited the highest specific capacitance of 23.14 F g−1 based on cyclic voltammetry performance in 100 mV s−1 with good stability.
Data availability
All data related to this work is presented in the manuscript and the ESI.†Author contributions
Dinda P. N. Nahda: writing – original draft, visualization, software, investigation, formal analysis, and data curation. Afiten R. Sanjaya: visualization, validation, software, investigation, formal analysis, and data curation. Fitria Rahmawati: validation, investigation, formal analysis, and data curation. Anne Zulfia: writing – review & editing, and supervision. Afriyanti Sumbodja: writing – review & editing, resources, project administration. Respati K. Pramadewandaru: validation, investigation, and formal analysis. Yuni K. Krisnandi: writing – review & editing, supervision, methodology, and conceptualization. Zico A. Akbar: writing – review & editing, visualization, validation, methodology, investigation, data curation, and supervision. Tribidasari A. Ivandini: writing – review & editing, validation, resources, methodology, supervision, conceptualization, and project administration.Conflicts of interest
There are no conflicts of interest to declare.Acknowledgements
This research is funded by the Indonesian Endowment Fund for Education (LPDP) on behalf of the Indonesian Ministry of Education, Culture, Research, and Technology and managed under the INSPIRASI Program (Contract No. 6635/E3/KL.02.02/2023).References
- C. Zhang, L. Yang, L. Zhao, Y. Liu, D. Hong, X. Shi, S. Wang and G. Zhu, Sep. Purif. Technol., 2025, 354, 129236 CrossRef CAS
.
- V. Viena, E. Elvitriana and S. Wardani, IOP Conf. Ser.: Mater. Sci. Eng., 2018, 334, 012037 Search PubMed
- F. K. Tareq, H.-Y. Lee, C.-H. Shin, S. I. Kojo and J.-S. Yu, Electrochim. Acta, 2024, 498, 144627 CrossRef CAS
- A. Tripathy, S. Mohanty, S. K. Nayak and A. Ramadoss, J. Environ. Chem. Eng., 2021, 9, 106398 CrossRef CAS
- L. Wang, W. Li, P. Li, L. Zhang and L. Li, J. Mol. Struct., 2024, 1318, 139380 CrossRef CAS
- Z. Liang, Z. Hong, M. Xie and D. Gu, New Carbon Mater., 2022, 37, 152–179 CrossRef CAS
- Z. Zhai, L. Zhang, T. Du, B. Ren, Y. Xu, S. Wang, J. Miao and Z. Liu, Mater. Des., 2022, 221, 111017 CrossRef CAS
- Y. Lv, L. Gan, M. Liu, W. Xiong, Z. Xu, D. Zhu and D. S. Wright, J. Power Sources, 2012, 209, 152–157 CrossRef CAS
- S. Mehdipour-Ataei and E. Aram, Catalysts, 2022, 13, 2 CrossRef
- M. Inagaki, M. Toyoda, Y. Soneda, S. Tsujimura and T. Morishita, Carbon, 2016, 107, 448–473 CrossRef CAS
- Y. Qu, Z. Zhang, X. Zhang, G. Ren, Y. Lai, Y. Liu and J. Li, Carbon, 2015, 84, 399–408 CrossRef CAS
- C. F. Lin, X. Zhang, H. Lin, N. Wang, J. B. Li and X. Z. Yang, Adv. Mater. Res., 2006, 11–12, 543–546 CAS
- M. Kruk, M. Jaroniec, R. Ryoo and S. H. Joo, J. Phys. Chem. B, 2000, 104, 7960–7968 CrossRef CAS
- T. Kyotani, Z. Ma and A. Tomita, Carbon, 2003, 41, 1451–1459 CrossRef CAS
- C. Santos, M. Andrade, A. L. Vieira, A. Martins, J. Pires, C. Freire and A. P. Carvalho, Carbon, 2010, 48, 4049–4056 CrossRef CAS
- B. Xu, L. Peng, G. Wang, G. Cao and F. Wu, Carbon, 2010, 48, 2377–2380 CrossRef CAS
- J. Galán, A. Rodríguez, J. M. Gómez, S. J. Allen and G. M. Walker, Chem. Eng. J., 2013, 219, 62–68 CrossRef
- Z. Mohd Dom, L. Mujianto, A. Azhar, S. Masaudin and R. Samsudin, Food Res., 2021, 5, 209–215 Search PubMed
- H. F. Hassan, U. F. Hassan, O. A. Usher, A. B. Ibrahim and N. N. Tabe, Int. J. Adv. Res. Chem. Sci., 2018, 5, 10 Search PubMed
- O. Fasakin, J. K. Dangbegnon, D. Y. Momodu, M. J. Madito, K. O. Oyedotun, M. A. Eleruja and N. Manyala, Electrochim. Acta, 2018, 262, 187–196 CrossRef CAS
- M. Panchal, G. Raghavendra, S. Ojha, M. Omprakash and S. K. Acharya, Mater. Res. Express, 2019, 6, 115613 CrossRef
- J. Hou, X. Mao, J. Wang, C. Liang and J. Liang, Chem. Phys., 2021, 551, 111352 CrossRef CAS
- V. Subramanian, C. Luo, A. M. Stephan, K. S. Nahm, S. Thomas and B. Wei, J. Phys. Chem. C, 2007, 111, 7527–7531 CrossRef CAS
- A. M. Al-Enizi, M. Ubaidullah and D. Kumar, Mater. Today Commun., 2021, 27, 102340 CrossRef CAS
- T. Ahamad, M. Naushad, M. Ubaidullah, Y. Alzaharani and S. M. Alshehri, J. Energy Storage, 2020, 32, 101952 CrossRef
- S. F. Shaikh, F. F. M. Shaikh, A. V. Shaikh, M. Ubaidullah, A. M. Al-Enizi and H. M. Pathan, J. Phys. Chem. Solids, 2021, 149, 109774 CrossRef CAS
- J. Ahmed, M. Ubiadullah, M. A. M. Khan, N. Alhokbany and S. M. Alshehri, Mater. Charact., 2021, 171, 110741 CrossRef CAS
- J. M. Gómez, A. Rodríguez, J. García, S. Álvarez and J. Galán, J. Porous Mater., 2015, 22, 1065–1071 CrossRef
- V. Ţucureanu, A. Matei and A. M. Avram, Crit. Rev. Anal. Chem., 2016, 46, 502–520 CrossRef
- G. Das, G. Mariotto and A. Quaranta, J. Electrochem. Soc., 2006, 153, F46 CrossRef CAS
- J. C. Steinbach, M. Schneider, O. Hauler, G. Lorenz, K. Rebner and A. Kandelbauer, Polymers, 2020, 12, 2473 CrossRef CAS PubMed
- W. Yan, D. Liu, D. Tan, P. Yuan and M. Chen, Spectrochim. Acta, Part A, 2012, 97, 1052–1057 CrossRef CAS PubMed
- W. Li and D. Zhao, Chem. Commun., 2013, 49, 943–946 RSC
- Z.-F. Tian, M.-J. Xie, Y. Shen, Y.-Z. Wang and X.-F. Guo, Chin. Chem. Lett., 2017, 28, 863–867 CrossRef CAS
- E. Taer, R. Taslim, Z. Aini, S. D. Hartati and W. S. Mustika, AIP Conf. Proc., 2016, 1801, 040004 CrossRef
- A. Rahmah, A. Zainollah, N. A. Fitriani, D. S. Ramadhan, M. Cahayo and M. Masruroh, Indones. J. Appl. Phys., 2017, 7, 46 CrossRef
- T. N. Nguyen, P. A. Le and V. B. T. Phung, Biomass Convers. Biorefin., 2022, 12, 2407–2416 CrossRef CAS
- F. Li, F. Qin, K. Zhang, J. Fang, Y. Lai and J. Li, J. Power Sources, 2017, 362, 160–167 CrossRef CAS
- J. Wang, Y. Feng, B. Tian, Y. Cheng, E. Ou, H. Li and J. Rong, Mater. Chem. Front., 2024, 8, 3157–3165 RSC
- Y. M. T. A. Putri, J. Gunlazuardi and T. A. Ivandini, Chem. Lett., 2022, 51, 135–138 CrossRef CAS
- O. Teschke, Appl. Phys. Lett., 1996, 68, 2129–2131 CrossRef CAS
- S. R. Hussaini and J. Dvorkin, J. Pet. Sci. Eng., 2021, 206, 108961 CrossRef CAS
- X. Peng, F. Hu, F. L.-Y. Lam, Y. Wang, Z. Liu and H. Dai, J. Colloid Interface Sci., 2015, 460, 349–360 CrossRef CAS PubMed
- M. Kruk, B. Dufour, E. B. Celer, T. Kowalewski, M. Jaroniec and K. Matyjaszewski, J. Phys. Chem. B, 2005, 109, 9216–9225 CrossRef CAS PubMed
- Y. M. T. A. Putri, P. K. Jiwanti, I. Irkham, J. Gunlazuardi, Y. Einaga and T. A. Ivandini, Bull. Chem. Soc. Jpn., 2021, 94, 2922–2928 CrossRef CAS
- D. E. Pissinis, L. E. Sereno and J. M. Marioli, Open J. Phys. Chem., 2012, 02, 23–33 CrossRef CAS
- M. M. Gul, K. S. Ahmad, L. Almanqur, A. G. Thomas, S. A. Alderhami and Y. T. Alharbi, J. Appl. Electrochem., 2024, 78, 519–532 CAS
- C. Ma, Y. Yu, Y. Li, J. Shi, Y. Song and L. Liu, J. Electrochem. Soc., 2014, 161, A1330–A1337 CrossRef CAS
- B. Padha, S. Verma, P. Mahajan and S. Arya, J. Electrochem. Sci. Technol., 2022, 13, 167–176 CrossRef CAS
- S. M. Hanser, Z. Shao, H. Zhao and B. J. Venton, Analyst, 2024, 149, 457–466 RSC
- B. Szubzda, A. Szmaja and A. Halama, Electrochim. Acta, 2012, 86, 255–259 CrossRef CAS
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra08322a |
| This journal is © The Royal Society of Chemistry 2025 |