DOI:
10.1039/D4RA08300H
(Paper)
RSC Adv., 2025,
15, 5737-5749
Mechanism study of a novel modified mesoporous silicon-based NiT50/MCM-41 adsorbent for the selective adsorption of methylene blue from water†
Received
22nd November 2024
, Accepted 30th January 2025
First published on 20th February 2025
Abstract
In this study, a new silica-based adsorbent strategy was developed to achieve a high adsorption capacity and high MB adsorption selectivity by impregnating a cationic [Ni(tepa)]2+ complex on a negatively charged silicon carrier, MCM-41. When the [Ni(tepa)]2+ loading was 50%, the MB adsorption capacity of the silicon-based adsorbent (NiT50/MCM-41) could reach 130 mg g−1, which was much higher than that of the original silicon-based MCM-41 adsorbent (23 mg g−1). Interestingly, NiT50/MCM-41 exhibited excellent MB adsorption selectivity in binary mixed dye solutions (MB/R6G, MB/RhB, and MB/MO). The adsorption kinetics and adsorption isotherm followed a second-order kinetic model and the Langmuir adsorption model, respectively. Experimental results showed that the aqueous solution of [Ni(tepa)]2+ can provide an alkaline environment to promote the deprotonation of Si–OH on the surface of silicon dioxide to form SiO−, which makes the surface negatively charged, and [Ni(tepa)]2+ effectively occupies these negatively charged sites through Coulomb interactions to better accommodate the cationic complex [Ni(tepa)]2+. Structural characterization and analysis confirmed that the NiOx and NiCx components were successfully introduced in the mesoporous channels of the silicon-based adsorbent through heat treatment, enabling the cationic dye to be immobilized on the silicon-based adsorbent through Lewis acid–base interaction, which effectively improved the adsorption capacity and adsorption selectivity towards MB.
1 Introduction
With the rapid industrialization, urbanization and population growth, water pollution has become increasingly severe worldwide.1 Dyes, produced in large quantities and widely applied in textiles, paper, printing, food, cosmetics, hair dyes, wood staining, pharmaceuticals, and leather manufacturing as well as in agricultural, biological, and chemical research,2,3 are a significant contributor to water contamination. Dyes are carcinogenic, teratogenic, and mutagenic. Thus, their presence can not only cause severe damage to the natural environment, disrupting ecosystems and contaminating resources, but also have grave impacts on industrial activities and the daily life of people, posing significant risks to human health and overall well-being.4–7 Methylene blue (MB) is considered one of the most common aromatic cationic dyes, and related studies have proven that exposure to MB can cause skin damage and eye burns, while its direct ingestion can cause an increased heart rate, causing indigestion and methemoglobinemia.8 Therefore, preventing the destructive effects of harmful dye wastewater on water, soil, animals, plants, and humans through the effective treatment of harmful dye wastewater is highly important.
To eliminate the contamination of dyes, various technologies have been developed and applied, such as physical,9 chemical,10–12 biological treatment,13 and photodegradation14–16 methods. Among the widely adopted solutions, adsorption stands out for its simple operation, low energy consumption, and ease of large-scale application, showing great potential for wastewater treatment. The efficiency of the adsorption process primarily relies on the selection of appropriate adsorbents.17 Numerous adsorbents have been studied for dye removal from wastewater. Recently, the development of selective adsorbents for the adsorption of specific dyes has attracted considerable interest. These adsorbents enable the selective separation of dye mixtures in industrial wastewater treatment. Numerous adsorbents with selective adsorption capabilities have been reported to date, such as metal–organic frameworks, nitrogen-doped nanoporous carbon, and LnCu–polyoxometalates.18–20 However, given the complexity of real industrial wastewater treatment environments, creating novel sorbent materials with high capacity, high selectivity, and high energy efficiency for the adsorption of dyes from water remains a significant challenge.
Compared with traditional solid adsorbents, mesoporous molecular sieve MCM-41 is increasingly used as an adsorbent in environmental remediation due to its highly ordered pore structure, large pore volume and adjustable size, and high specific surface area.21 However, pure MCM-41 lacks active adsorption sites.22,23 The surface of the pores in MCM-41 has easy-to-modify silica hydroxyl groups, and thus functionalized groups can be firmly bonded to its pore surface by a silylation reaction. Therefore, it is essential to develop functionalized silicon-based materials through surface modification with silica to serve as adsorbents for the selective adsorption of dyes in wastewater.
Given the complexity of the actual industrial wastewater environment, this study aimed to develop a novel and highly efficient adsorbent for the selective removal of methylene blue (MB) from various ionic wastewater. By exploring a unique “[Ni(tepa)]2+ modification-thermal treatment” strategy, the limitations of traditional adsorbents and pure MCM-41 could be overcome. Tetraethylenepentamine (tepa) is used to form a coordination cation [Ni(tepa)]2+ with nickel. This complex not only provided an alkaline environment to promote the interaction with the silica surface but also occupied the negative charge sites through Coulomb interactions. Additionally, through thermal treatment, NiOx and NiCx components were successfully introduced onto the surface of the silicon-based adsorbent, enhancing its selective adsorption capacity for MB. Thus, this study provides a new perspective and method for the development of high-performance adsorbents in the field of wastewater treatment, addressing the urgent need for effective solutions to deal with dye-contaminated wastewater.
2 Materials and methods
2.1 Material synthesis
2.1.1 Preparation of silicon support MCM-41. The silicon support MCM-41 was prepared using hexadecyl trimethyl ammonium bromide (CTAB, AR) and tetraethyl orthosilicate (TEOS, AR) under alkaline conditions following the Stöber method reported previously.24 Briefly, CTAB (0.73 g), ethanol (114.8 g) and H2O (238.8 g) were mixed at 35 °C to obtain a clear solution. Then, NH3 (4.35 g) was added, and the mixture was stirred for 30 min. Subsequently, TEOS (4.5 g) was rapidly added to the solution, and the mixture was stirred for 24 h. The obtained suspension was centrifuged to obtain a white solid, which was subsequently washed with ethanol three times and washed with deionized water once. Finally, the obtained white solid was dried overnight and transferred to a muffle furnace, and calcination removed the templating agent (the roasting conditions were heating at a rate of 2 °C min−1 to 550 °C for 6 h to remove the templating agent).
2.1.2 Preparation of silicon-based adsorbents. The nickel precursor solution was created by dissolving 5 mmol of Ni(NO3)2·6H2O (Sinopharm, AR) in 12 mL of distilled water. Then, 10 mmol of TEPA (Sinopharm, CP) was added dropwise to the nickel solution, and the mixture was stirred slowly at 30 °C for 1 h to form an [Ni(tepa)]2+ complex solution. Then, 0.1 g of silica support was added to the prepared [Ni(tepa)]2+ solution and mixed using ultrasonication for 1 h. Subsequently, this mixture was dried at 80 °C for 12 h, and then ground into a fine powder. Powders with different [Ni(tepa)]2+ loadings (10–70 wt%) were produced. Following our previous work,25 the powder was placed in a 50 mL covered crucible and heated in static air to 400 °C. The temperature was increased at a rate of 4 °C min−1 and maintained for 2 h. The final adsorbents were labeled NiTX/MCM-41, where X represents the loading amount of [Ni(tepa)]2+ in the silica support.
2.2 Details of the experiment
2.2.1 Dye adsorption experiment. Firstly, 12 mg of NiTX/MCM-41 was mixed with 30 mL of an aqueous solution of organic dye (MB, R6G, RhB, or MO) at a certain concentration. The beaker was shaken at 30 °C at a constant shaking speed of 300 rpm for a specified duration. After the adsorption process, the supernatant was collected from the stock solution using a syringe filter (hydrophobic, 0.22 μm). The residual dye concentration was measured using UV-vis spectroscopy after dilution (if necessary) at the maximum wavelengths. The above-mentioned steps were repeated multiple times.The adsorption capacities Qt and Qe were calculated using eqn (1) and (2), as follows:
where
C0,
Ct, and
Ce represent the initial concentration, the concentration at a given time (
t), and the equilibrium concentration of the dye (mg L
−1), respectively;
m is the mass of NiT
X/MCM-41 (g); and
V is the volume of the solution (L).
2.2.2 Effect of pH on dye adsorption. The pH of the MB solution was adjusted to 2–10 with 0.1 M NaOH or HCl aqueous solution. Then, 12 mg absorbent was mixed with 25 mL of a 25 mg per L MB solution with a pH in the range of 2–10. Next, 5 mL of phosphate-buffered saline (PBS) was added to ensure that there was no significant change in the pH value. As before, the beakers were shaken (300 rpm) for 30 min at 30 °C. The above-mentioned steps were repeated multiple times.
2.2.3 Effect of the contact time and initial concentration. The adsorption experiments were performed by mixing 12 mg of adsorbent with 25 mL of MB solutions of different concentrations (30, 40, 50, or 100 mg L−1), and then 5 mL of phosphate-buffered saline (PBS) added to ensure that the differences in adsorption were caused by the concentration. Shaking was performed for different contact times (2, 5, 10, 15, 20, 30, 45, or 60 min). The above-mentioned steps were repeated multiple times.
2.2.4 Kinetic studies and adsorption isotherm. Two kinetic models (pseudo-first-order26 and pseudo-second-order27) and two isotherm models (Langmuir28 and Freundlich29) were investigated.The two kinetic models can be described in linear form as follows:
| | |
ln(Qe − Qt) = ln(Qe) − k1t
| (3) |
| | |
t/Qt = 1/k2Qe2 + t/Qe
| (4) |
where
Qe and
Qt (mg g
−1) represent the amount of
MB adsorbed at equilibrium and a specific time (min), respectively.
k1 (min
−1) is the rate constant of the pseudo-first-order model and
k2 (g mg
−1 min
−1) is the pseudo-second-order rate constant.
Also, the Langmuir and Freundlich model can be described in linear form as follows:
| | |
Ce/Qe = 1/Q0KL + 1/Q0Ce
| (5) |
| |
ln![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) Qe = lnKF + 1/nlnCe Qe = lnKF + 1/nlnCe
| (6) |
where
KL (L mg
−1) and
Q0 (mg g
−1) are the Langmuir constants involved in adsorption rate and adsorption capacity.
KF and
n are Freundlich constants.
2.2.5 Selective separation of mixed solutions of MB/R6G, MB/RhB, and MB/MO. A solution of MB was combined (1:1, v/v) with a solution of identical concentration (25 mg L−1) of R6G, RhB, or MO to produce mixtures of MB/R6G, MB/RhB, and MB/MO. In these experiments, 12 mg of absorbent was added to 30 mL of the mixed MB/R6G, MB/RhB, or MB/MO solution. The adsorption process was observed using UV-vis spectroscopy.
2.3 Characterization
UV-vis spectroscopic studies were performed on a UV1800 spectrophotometer. The crystal phase of the catalyst powder was characterized using a Rigaku Smart Lab (Japan) X-ray diffractometer (XRD) with Cu Kα (40 kV, 100 A) radiation at a test wavelength of 0.1542 nm, scanning rate of 10° min−1, and scanning 2θ range of 5–80°. The surface morphologies and sizes of the samples were examined by scanning electron microscopy (SEM) on a HITACHI SU8010 instrument with an accelerating voltage of 5.0 kV, and all the samples were sputtered with a thin gold film. X-ray photoelectron spectroscopy (XPS) was tested using a Thermo Scientific K-Alpha instrument in the United States to determine the surface composition of the catalyst and the valence state of the elements. The results were corrected for the charge effect based on the position of the C (1s) reference peak at 284.8 eV. N2 isotherms were measured using a Quantachrome autosorb-iQ2 gas adsorption analyzer at −196 °C. The zeta potential values of RSA and the composites were measured using a Zetasizer Nano S90. Fourier transform infrared (FTIR) spectroscopy (Nicolet Nexus 410) was used to record the infrared spectra (IR) in the range of 4000 to 400 cm−1.
3 Results and discussion
3.1 Characterization
3.1.1 SEM images analysis. The morphological characteristics of MCM-41 and NiT50/MCM-41 were observed by scanning electron microscopy (SEM) and transmission electron microscopy (TEM), as shown in Fig. 1a–c. The SEM and TEM images both show the morphological structures as spherical particles with a uniform size (close to 500 nm), featuring a certain degree of stacking and aggregation. There was no significant change in the morphology of the sample before and after modification. This indicates that the morphology of the mesoporous silicon support was preserved after loading [Ni(tepa)]2+ and undergoing appropriate thermal treatment modification.
 |
| | Fig. 1 SEM images of (a) MCM-41 and (b) NiT50/MCM-41; (c) HRTEM images of NiT50/MCM-41; (d) XRD results for MCM-41 and NiT50/MCM-41; (e) N2 adsorption isotherms of MCM-41 and NiT50/MCM-41 and (f) pore size distribution analysis for MCM-41 and NiT50/MCM-41. | |
3.1.2 XRD spectral analysis. Small-angle X-ray diffraction (SAXRD) can be used to characterize the structure of ordered porous samples. The SAXRD patterns of the mesoporous silicon support MCM-41 and as-prepared NiT50/MCM-41 are shown in Fig. 1d. In this study, the mesoporous silica MCM-41 prepared exhibited weak peaks at the 2θ values of 2.52° and 3.5–5°, corresponding to the (100) and (110), and (200) crystal planes of the hexagonal structure, respectively (no. 01-073-0626).30,31 The diffraction peak for the prepared NiT50/MCM-41 remained unchanged, signifying that the ordered hexagonal mesoporous pore structure was maintained after loading [Ni(tepa)]2+ on the mesoporous silicon support MCM-41 and subsequent heat treatment.32 Moreover, a decrease in peak intensity was observed, indicating the partial loss of the periodicity of the MCM-41 pore structure, which is likely due to the [Ni(tepa)]2+ loading within the pores of the mesoporous silica MCM-41. The inset figure in Fig. 1d shows the wide-angle XRD (WAXRD) patterns of NiT50/MCM-41, which feature a broad diffusion peak at 20–30°, suggesting that the primary component of NiT50/MCM-41 is the amorphous silica phase.33 The absence of characteristic peaks for nickel oxide or other Ni species in NiT50/MCM-41 implies that the Ni species may exist as finely dispersed nanoparticles and/or as amorphous particles within the framework of NiT50/MCM-41.34
3.1.3 N2 physical adsorption analysis. The textural features (surface area, pore volume, and pore size) were characterized via physical nitrogen adsorption. Fig. 1e and f show the nitrogen adsorption isotherms and pore size distribution curves for the MCM-41 and NiT50/MCM-41 samples, respectively. Fig. 1e reveals a type IV isotherm with a capillary condensation step, which is the typical pattern for mesoporous materials according to the IUPAC classification, indicating the presence of mesoporous structures in both MCM-41 and NiT50/MCM-41.31 These results indicate that the silicon-based adsorbent prepared by the “[Ni(tepa)]2+ modification-thermal treatment” method retained the ordered hexagonal mesoporous structure of the mesoporous silica support (MCM-41), which is consistent with the XRD results. Fig. 1f indicates that both MCM-41 and NiT50/MCM-41 have narrow mesopores with a size in the range of 2–3 nm. However, the pore size distribution of NiT50/MCM-41 shifted slightly towards a smaller size. The surface area, pore volume, and average pore size of MCM-41 are 1558 m2 g−1, 0.79 cm3 g−1, and 2.37 nm, respectively. After modification, these values for NiT50/MCM-41 decreased to 958 m2 g−1, 0.52 cm3 g−1, and 2.15 nm, respectively, due to the incorporation of Ni species within the channels of the MCM-41 support. These results indicate that Ni species were successfully introduced into MCM-41.
3.2 Unary adsorption performance for MB
The adsorption properties of the as-prepared silicon-based materials with different loading masses of [Ni(tepa)]2+ (10–70 wt%) for MB, followed by thermal treatment at a temperature of 400 °C were investigated (Fig. 2a). The adsorption of MB by NiTX/MCM-41 was, in all cases, much better than the adsorption by the MCM-41 support (0% in Fig. 2a). As the [Ni(tepa)]2+ loading mass gradually increased, the MB adsorption capacity of NiTX/MCM-41 first increased rapidly, but then decreased gradually. In the loading range of 0–20%, the adsorption capacity of the as-prepared adsorbents increased rapidly with an increase in loading mass. The MB adsorption capacity of NiT20/MCM-41 was 102 mg g−1, which was much greater than that of the MCM-41 support (23 mg g−1). When the [Ni(tepa)]2+ loading was 50%, the adsorption of MB by NiT50/MCM-41 was 130 mg g−1, which is the maximum adsorption performance. Also, it exhibited a better adsorption performance compared to other silica-based adsorbents (Table 1). With a further increase in the loading amount of [Ni(tepa)]2+, an excessive amount of [Ni(tepa)]2+ will aggregate within the pore channels, resulting in an uneven distribution of active sites, increase in steric hindrance, and reduction in the effective contact of adsorbate molecules, thus decreasing the adsorption capacity. The NiT50/MCM-41 catalyst was tested for multiple cycles under the same conditions, and the results are shown in Fig. S1,† which shows that its adsorption capacity did not significantly decrease.
 |
| | Fig. 2 (a) Adsorption of MB by NiTX/MCM-41 and (b) adsorption of MB by MCM-41 and NiT50/MCM-41 at different pH values under the following conditions: 50 mL of 30 mg per L MB; adsorbent mass (10 mg); temperature (30 °C); and contact time (30 min). (c) Adsorption of MB by NiT50/MCM-41 with different contact times under different conditions: 50 mL of 30 mg per L MB; adsorbent mass (10 mg); and temperature (30 °C). (d) Different initial concentrations; conditions: 50 mL of MB; adsorbent mass (10 mg); temperature (30 °C); contact time (30 min). Error bars represent the degree of data dispersion. | |
Table 1 Comparison of the dye adsorption capacities of different adsorbents
| Absorbent |
C0 (mg L−1) |
Absorbent mass (mg) |
pH |
Qmax (mg g−1) |
Ref. |
| MCM-41 |
100 |
5 g L−1 |
— |
16 |
31 |
| Co–Fe@Al2O3–MCM-41 |
100 |
20 |
10 |
100 |
35 |
| MCM-41 |
50 |
20 |
6.32 |
5.28 |
36 |
| Mn/MCM-41 |
45.38 |
| MCM-41 |
100 |
50 |
9 |
149.5 |
34 |
| Fe–MCM-41 |
194 |
| MCM-41 |
100 |
1.6 g L−1 |
7 |
19 |
37 |
| TiO2@mSiO2 |
37 |
| TiON@mSiO2 |
60 |
| MCM-41 |
36 |
20 |
7 |
65.7 |
38 |
| CuO/MCM-41 |
87.8 |
| MCM-41 |
25 |
50 |
— |
10.95 |
32 |
| NiO/MCM-41 |
23.26 |
| MCM-41 |
40 |
20 |
6 |
41.6 |
30 |
| GDMA/MCM-41 |
77.6 |
| MCM-41 |
30 |
10 |
7 |
23 |
This work |
| NiT50-400/MCM-41 |
130 |
Fig. 2b illustrates the influence of the pH of the MB aqueous solution on the adsorption of MB by MCM-41 and NiT50/MCM-41, with pH values in the range of 2 to 10. The MB adsorption capacities of both MCM-41 and NiT50/MCM-41 increased with an increase in pH. However, notable differences between MCM-41 and NiT50/MCM-41 were also evident. When the pH value was between 7 and 10, the adsorption capacity of MCM-41 rapidly increased to 100 mg g−1 from 20 mg g−1. This may be because with an increase in the concentration of OH− ions, the negative charge on the surface of MCM-41 increased, which is more favorable for the adsorption of MB via electrostatic interactions. However, in the case of NiT50/MCM-41, the adsorption capacity for MB increased significantly between pH 3 and 7 but changed minimally between pH 7 and 10. These findings indicate that the synthesized silicon-based adsorbent, NiT50/MCM-41, possesses strong adsorption capacity under both acidic and basic conditions. Additionally, the considerable differences between MCM-41 and NiT50/MCM-41 suggest that electrostatic interactions alone may not account for the adsorption behavior of NiT50/MCM-41 toward MB.
The effects of varying contact times and initial concentrations of MB aqueous solution on the adsorption of MB by NiT50/MCM-41 were examined, and the results are presented in Fig. 2c and d, respectively. According to Fig. 2c, the adsorption capacity was 50 mg g−1 within the first 30 s, and after 20 min, it began to stabilize at about 130 mg g−1, showing only slight changes with further contact time. Fig. 2d demonstrates that the adsorption capacity increased with higher initial MB concentrations. When the initial concentration of the MB aqueous solution reached 80 mg L−1, the adsorption capacity of NiT50/MCM-41 was 200 mg g−1. The minimum adsorption capacity is also an important index for evaluating the adsorption performance of an adsorbent. The MB dye has a characteristic absorption peak in the UV-vis spectrum, which can reflect the concentration of dye in aqueous solutions. Fig. S6† displays the UV-vis spectra of MB aqueous solutions with varying initial concentrations (15 mg L−1, 10 mg L−1, and 5 mg L−1) before and after adsorption by NiT50/MCM-41. For all three tested concentrations, the characteristic peaks of MB in the samples nearly vanished post-adsorption. The inset photograph demonstrates that the solution turned colorless after the adsorption process.
3.3 Kinetic studies and adsorption isotherm
The adsorption kinetic data were fitted using pseudo-first-order and pseudo-second-order kinetic models. The fitting results are shown in Fig. 3a and b, respectively, and the fitting data are given in Table 2. Compared to the Qe (exp.) of 130 mg g−1, it can be seen that the Qe (cal.) obtained using the second-order kinetic fit is closer to the experimental value, and the R2 value is also better for the second-order kinetic fit. These results show that the adsorption kinetics of the adsorption process follow a second-order kinetic model. The above-mentioned results indicate that the chemical adsorption between the adsorbent and the dye dominates the adsorption of the dye on NiT50/MCM-41, and multiple active sites of NiOx and NiCx jointly participate in the dye-adsorption process.
 |
| | Fig. 3 (a) Pseudo-first-order model, (b) pseudo-second-order model, (c) Langmuir isotherm, and (d) Freundlich isotherm fitting of MB adsorption by NiT50/MCM-41. | |
Table 2 Pseudo-first order and pseudo-second order kinetic parameters for MB adsorption by NiT50/MCM-41
| Model |
Qe (cal.) |
k |
R2 |
| Pseudo-first-order |
12.02 |
0.13 |
0.945 |
| Pseudo-second-order |
138.80 |
0.0072 |
0.993 |
The Langmuir and Freundlich equations were used to fit the adsorption isotherms to further investigate the adsorption mechanism. The fitting results are shown in Fig. 3c and d, respectively, and the fitting data are presented in Table 3. The adsorption of the isotherm models followed the order of Langmuir > Freundlich. Thus, the results indicate that the adsorption isotherm is more consistent with the Langmuir adsorption model (the linear-fitting correlation coefficient R2 is close to 1). The Langmuir adsorption isotherm shows that the active sites on the surface of the NiT50/MCM-41 adsorbent are occupied in the form of single-molecular-layer adsorption, and no multi-layer stacking adsorption behavior occurs. Moreover, in the NiT50/MCM-41 system, the active sites formed after the loading of [Ni(tepa)]2+ exhibit similar adsorption energies during the adsorption process.39 In addition, the Freundlich isotherm indicates that multi-layer adsorption may also occur on NiT50/MCM-41, with an R2 of 0.878.40
Table 3 Langmuir and Freundlich isotherm parameters for NiT50/MCM-41
| Model |
k |
R2 |
| Langmuir |
0.00485 |
0.981 |
| Freundlich |
0.423 |
0.878 |
3.4 Selective adsorption of mixed solutions of MB/R6G, MB/RhB, and MB/MO
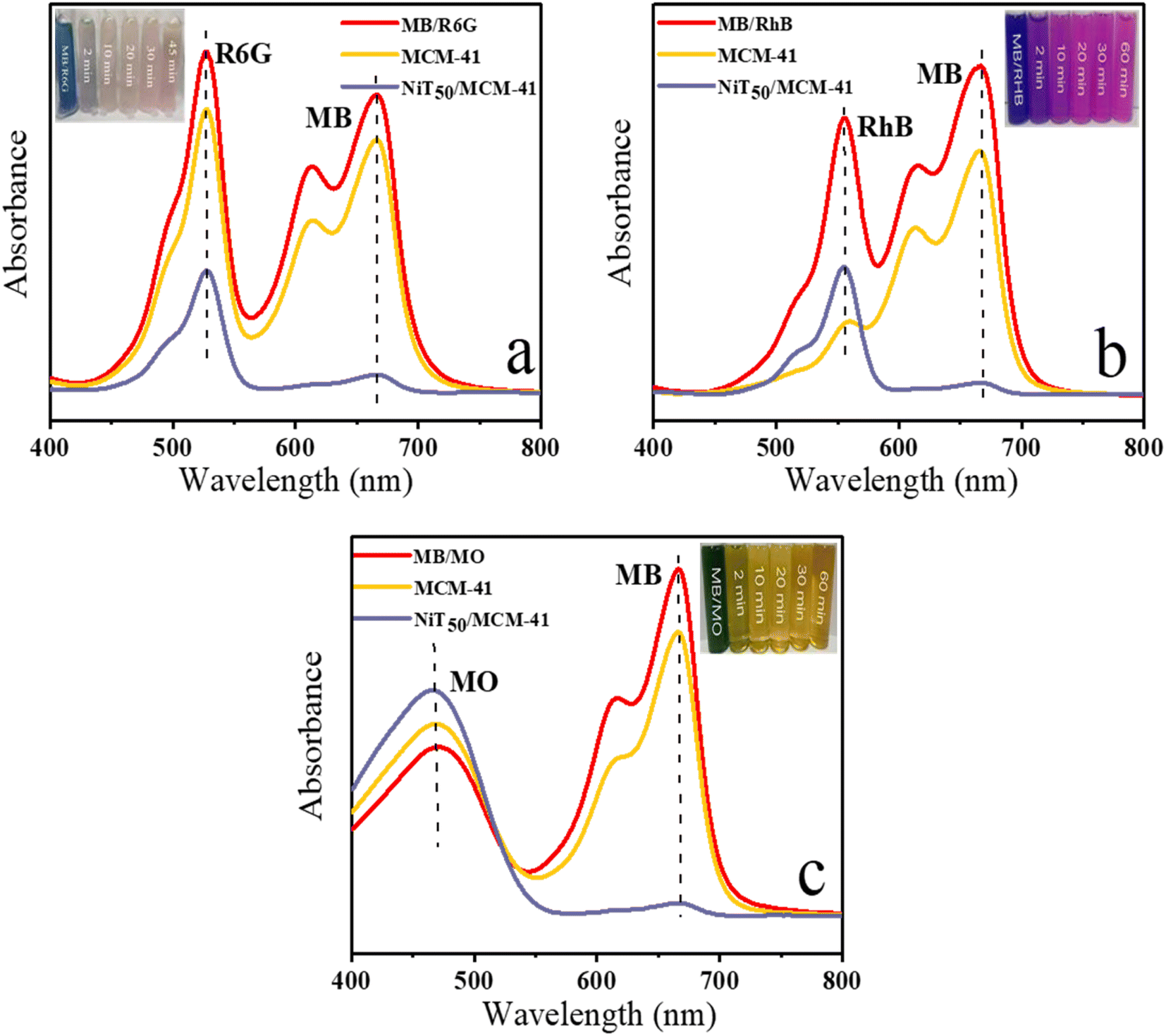
Organic dyes (MB, R6G, RhB, and MO) with various charges and sizes were chosen to evaluate the adsorption and separation properties of MCM-41 and NiT50/MCM-41. This study utilized three mixed dye solutions of MB/R6G, MB/RhB, and MB/MO. Fig. 4 illustrates the UV-vis spectra of these binary mixture dye solutions before and after adsorption by MCM-41 and NiT50/MCM-41. As depicted in Fig. 4a, after adsorption by MCM-41, the peaks for MB and R6G showed minimal reduction, maintaining about 80% of their initial values, indicating the weak adsorption capacity of MCM-41 for MB/R6G. In contrast, after adsorption by NiT50/MCM-41, the peaks for both MB and R6G significantly decreased. The corresponding peak of MB almost completely disappeared, and the corresponding peak of R6G was only about 40% of that before adsorption. The results showed that the MB and R6G adsorption ability of the as-prepared NiT50/MCM-41 was obviously greater than that of the original MCM-41 support. The selective adsorption ability for MB in a binary mixture (MB/R6G) was shown based on the difference in the ability of NiT50/MCM-41 to adsorb MB and R6G. The inset clearly shows that after adsorption by NiT50/MCM-41, the color of the corresponding adsorbed solution changed from that of the original mixture to the color of R6G. However, the solution adsorbed by MCM-41 has no such change for MB/RhB (in Fig. 4b), and the characteristic peak corresponding to RhB was obviously reduced after adsorption by MCM-41, maintaining only about 25% of that before adsorption, whereas the adsorption of MB was much weaker, still maintaining about 75%. These results indicate that MCM-41 prefers the adsorption of RhB from the mixture. Nevertheless, after adsorption by NiT50/MCM-41, the corresponding peak of MB almost completely disappeared, and the corresponding peak of RhB was still about 40% of that before adsorption. Compared with that of the original MCM-41 support, the adsorption ability of the as-synthesized silicon-based adsorbent NiT50/MCM-41 is obviously greater, and the adsorption of RhB is also suppressed, which shows the selective adsorption ability of NiT50/MCM-41 for MB in binary mixtures. On the one hand, the adsorption capacity for MB is significantly enhanced; on the other hand, the adsorption of RhB is inhibited, leading to the selective adsorption of MB in the MB/RhB binary mixture by NiT50/MCM-41. The inset image shows that after adsorption by NiT50/MCM-41, the adsorbed solution turned red, indicating the presence of RhB. Similarly, for the MB/MO mixture, the MB peak remained around 80% after adsorption by MCM-41, whereas it nearly vanished after adsorption by NiT50/MCM-41 (Fig. 4c). The inset image clearly demonstrates that following adsorption by NiT50/MCM-41, the solution changed from green to orange, highlighting the superior selective adsorption performance for MB in the MB/MO binary mixture by NiT50/MCM-41.
 |
| | Fig. 4 Selective adsorption capability of MCM-41 and NiT50/MCM-41 toward mixed dyes: (a) MB and R6G; (b) MB and RhB; (c) MB and MO. The inset shows the color changes over time of three mixed dye solutions adsorbed using NiT50/MCM-41. Conditions: 50 mL of mixed dye mixture; adsorbent mass (10 mg); temperature (30 °C). | |
These results suggest that based on the “[Ni(tepa)]2+ modification-thermal treatment” strategy, the cationic [Ni(tepa)]2+ complex was impregnated onto the negatively charged silica support, followed by suitable thermal treatment, and the silicon-based adsorbent was successfully prepared and showed excellent adsorption capacity and selective adsorption performance for MB.
3.5 The mechanism of the new strategy
The mechanism for preparing the silicon-based adsorbent using the “[Ni(tepa)]2+ modification-thermal treatment” strategy was further examined using different samples related to MCM-41. MCM-41 was used as the silicon support. Ni-tepa/MCM-41 is a sample with a 50% [Ni(tepa)]2+ loading mass but without any thermal treatment. NiT50/MCM-41, the silicon-based adsorbent developed in this study, has a 50% [Ni(tepa)]2+ loading mass, followed by thermal treatment at 400 °C. Ni/MCM-41 was prepared using nickel nitrate and MCM-41, ensuring that both the nickel loading and the thermal treatment temperature matched that used for NiT50/MCM-41.
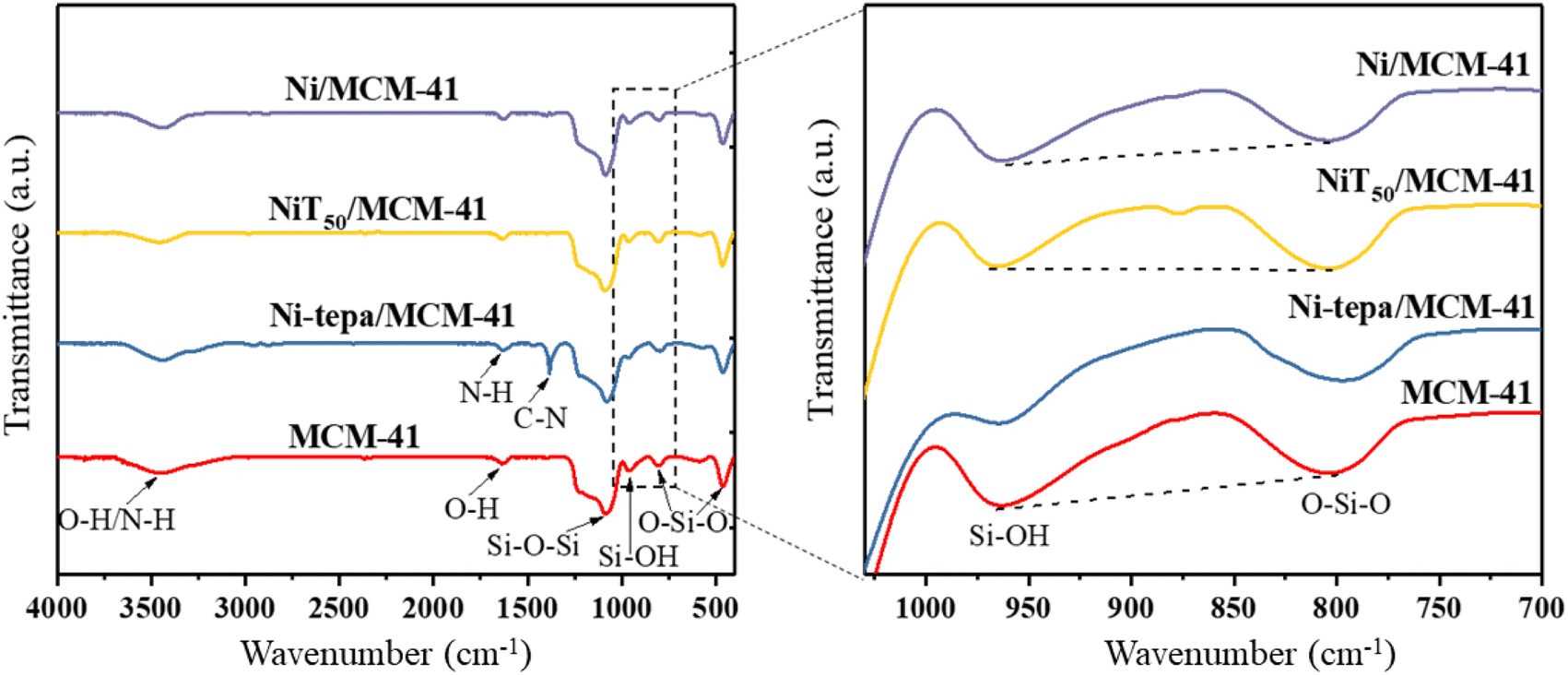
The FTIR characterization of the MCM-41, Ni-tepa/MCM-41, NiT50/MCM-41, and Ni/MCM-41 samples is presented in Fig. 5. In the spectrum of MCM-41, the peaks at 1100, 813, and 472 cm−1 are attributed to the stretching and deformation vibrations of Si–O–Si.33,41 The peak at 965 cm−1 is associated with the asymmetric vibration of Si–OH, while the broad absorption peak near 3440 cm−1 is due to the O–H stretching vibration. In Ni-tepa/MCM-41, the peaks at 1602 and 1510 cm−1 correspond to the N–H bending vibration and C–N resonance vibration from tepa, indicating the effective introduction of the cationic complex [Ni(tepa)]2+ on the silicon support surface.42 The significant decrease in the intensity of the peak 965 cm−1 suggests an interaction between the silicon hydroxyl groups and [Ni(tepa)]2+ during the modification process. The peak at 965 cm−1 was reduced in all three samples, viz. Ni-tepa/MCM-41, NiT50/MCM-41, and Ni/MCM-41, compared to the original MCM-41 support. It is noteworthy that the Si–OH peaks in Ni-tepa/MCM-41 and NiT50/MCM-41, prepared using [Ni(tepa)]2+, were considerably weaker than that in Ni/MCM-41, which was prepared from nickel nitrate.
 |
| | Fig. 5 FTIR spectra of different samples: MCM-41, Ni-tepa/MCM-41, NiT50/MCM-41, and Ni/MCM-41. | |
The XPS spectra were further analyzed to investigate the species present in the samples. Fig. 6 displays the O 1s XPS spectra of MCM-41, Ni-tepa/MCM-41, NiT50/MCM-41, and Ni/MCM-41. After curve fitting, two oxide components (Oα and Oβ) were identified in the spectra of MCM-41 and Ni-tepa/MCM-41. In the case of NiT50/MCM-41 and Ni/MCM-41, three oxide components (Oα, Oβ, and Oγ) were distinguished. The Oα, Oβ, and Oγ components correspond to Si–O–Si, Si–OH, and NiOx,43,44 respectively. The low ratios of the C–O and C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O bonds led to their exclusion from this study.43 The contents of different states of O atoms based on the peak ratio analysis in the O 1s spectrum are shown in Table 4. The proportion of Si–OH in Ni/MCM-41 was 16.1%, showing little change from the raw silicon support MCM-41 (17.7%). However, the Si–OH proportions in Ni-tepa/MCM-41 and NiT50/MCM-41 were substantially lower, at 7.5% and 4.7%, respectively, which is consistent with the FTIR findings.
O bonds led to their exclusion from this study.43 The contents of different states of O atoms based on the peak ratio analysis in the O 1s spectrum are shown in Table 4. The proportion of Si–OH in Ni/MCM-41 was 16.1%, showing little change from the raw silicon support MCM-41 (17.7%). However, the Si–OH proportions in Ni-tepa/MCM-41 and NiT50/MCM-41 were substantially lower, at 7.5% and 4.7%, respectively, which is consistent with the FTIR findings.
 |
| | Fig. 6 O 1s spectra of different samples: (a) MCM-41, (b) Ni-tepa/MCM-41, (c) NiT50/MCM-41, and (d) Ni/MCM-41. | |
Table 4 Contents of different O states in MCM-41, Ni-tepa/MCM-41, NiT50/MCM-41 and Ni/MCM-41
| Sample |
Oα |
Oβ |
Oγ |
| MCM-41 |
82.3 |
17.7 |
— |
| Ni-tepa/MCM-41 |
92.5 |
7.5 |
— |
| NiT50/MCM-41 |
81.1 |
4.7 |
14.2 |
| Ni/MCM-41 |
66.0 |
16.1 |
17.9 |
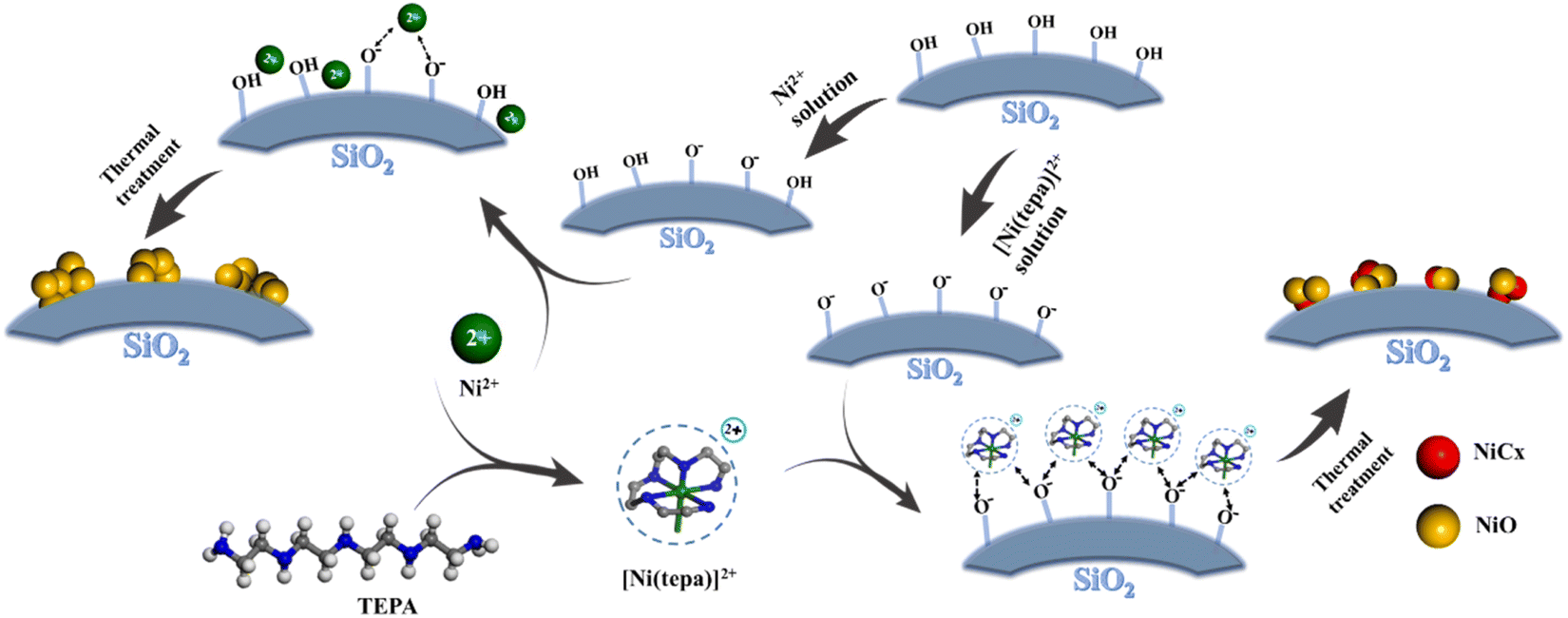
Based on the above-mentioned results, the process for the preparation of silicon-based adsorbents using the “[Ni(tepa)]2+ modification-thermal treatment” strategy can be explained as follows: the surface of silica contains many Si–OH groups, which can lose protons to form SiO−, resulting in negatively charged surfaces. When a cation is introduced, it attaches to the negatively charged SiO− on the silica surface via electrostatic interactions. However, only a certain number of Si–OH groups can deprotonate to form SiO− under specific conditions. The pH of the [Ni(tepa)]2+ aqueous solution used was close to 10, providing an alkaline environment, which promotes the deprotonation of Si–OH on the silica surface, increasing the formation of SiO−. This environment facilitates the accommodation of the cationic complex [Ni(tepa)]2+. This explains why the proportion of Si–OH decreased from 17.7% in the MCM-41 support to 7.5% in Ni-tepa/MCM-41 after loading [Ni(tepa)]2+. Comparing NiT50/MCM-41 and Ni/MCM-41, which were prepared by impregnating the cations ([Ni(tepa)]2+ and Ni2+) followed by thermal treatment, it was observed that the proportion of Si–OH in Ni/MCM-41 changed slightly to 16.1% from the raw silicon support (17.7%), which is more than three times that of NiT50/MCM-41. This is because the nickel nitrate solution is acidic (pH 4), while the [Ni(tepa)]2+ solution is alkaline (pH 10), making the alkaline environment more conducive to Si–OH deprotonation. In Ni/MCM-41, the many black spots (Fig. S7†) correspond to the NiO particles loaded on the silicon support by nickel nitrate, consistent with the wide-angle XRD results for Ni/MCM-41 (Fig. S8†).45 However, in the TEM image of NiT50/MCM-41, no distinct black spots were observed, indicating highly dispersed nickel species introduced through the “[Ni(tepa)]2+ modification-thermal treatment” strategy, which is consistent with the XRD results. The C 1s spectra (Fig. S9†) and HRTEM image of NiT50/MCM-41 (Fig. 1c) suggest that the highly dispersed nickel species in NiT50/MCM-41 are NiO and NiCx.46–48 The pore size distribution of Ni/MCM-41 was 2.30 nm, which is only 0.7 nm less than the raw MCM-41 support, while that for NiT50/MCM-41 shifted from 2.37 to 2.15 nm. These results indicate that the “[Ni(tepa)]2+ modification-thermal treatment” strategy allows more nickel species to enter the mesoporous channels.
Therefore, in the “[Ni(tepa)]2+ modification-thermal treatment” process, on the one hand, the [Ni(tepa)]2+ aqueous solution can provide an alkaline environment, which can promote the deprotonation of Si–OH on the silica surface and lead to the formation of SiO−. On the other hand, the introduced cationic complex [Ni(tepa)]2+ effectively occupies these formed negatively charged sites via Coulomb interactions. Then, with suitable thermal treatment, the NiO and NiCx components are successfully introduced into the mesoporous channels of the as-prepared silicon-based adsorbent. Fig. 7 is a simple preparation schematic diagram.
 |
| | Fig. 7 Schematic of the new strategy. | |
3.6 Adsorption mechanism
XPS analysis was conducted to further elucidate the adsorption mechanism. X-ray photoelectron spectroscopy (XPS) was performed using a Thermo Scientific K-Alpha instrument in the United States to determine the surface composition of the adsorbent as well as the valence state of the elements. The test results were corrected for the charge effect based on the position of the C (1s) reference peak at 284.8 eV. Also, a peak-fitting analysis was performed on the N 1s spectrum. Fig. 8 displays the N 1s and Ni 2p XPS spectra of NiT50/MCM-41, NiT50/MCM-41 with adsorbed MB (denoted as NiT50/MCM-41–MB), and pure MB. The Ni 2p spectrum of NiT50/MCM-41 (Fig. 8a) could be deconvoluted into three distinct peaks, indicating three different states of Ni species in the silicon-based adsorbent prepared using the “[Ni(tepa)]2+ modification-thermal treatment” process. The XPS signals at 856.6 eV and 853.3 eV correspond to NiO and NiCx, respectively.46,49 This observation is consistent with the C 1s spectra (Fig. S9†) and HRTEM image of NiT50/MCM-41 (Fig. 1c). Following the adsorption process (NiT50/MCM-41–MB), the Ni species showed a new peak at a binding energy of 854.5 eV. The plausible mechanism was proposed, where cationic dyes such as MB typically contain organic ammonium cations, which can accept electrons and function as Lewis acids. The NiO and NiCx components were distributed in the mesopores of MCM-41, resulting in differences in the surface charge density of the nickel species within different states. The electron-withdrawing ability of the O atom provides the electron-attracting ability of the Ni atom in NiO. Therefore, it is not conducive to the adsorption of MB, which tends to accept electrons. However, in the case of NiCx, its low binding energy suggests that it can easily provide electrons as a Lewis base. Consequently, based on the Lewis acid–base interaction between MB and NiT50/MCM-41, there was a significant change in its Ni 2p spectrum before and after adsorption. This unique subject–object interaction explains the low MB adsorption capacity of Ni/MCM-41, which contains only NiO. The high-resolution N 1s spectra of NiT50/MCM-41–MB and pure MB are shown in Fig. 8. The peak at 399.9 eV (Fig. 8c) is assigned to the N atoms of adsorbed MB,50 which is slightly greater than the binding energy of pure MB (399.6 eV) (Fig. 8d), demonstrating the strong interaction of MB with NiT50/MCM-41 through N atoms, which is attributed to Lewis acid–base interactions.
 |
| | Fig. 8 XPS spectra of the samples. (a) Ni 2p spectrum of NiT50/MCM-41, (b) Ni 2p spectrum of NiT50/MCM-41 with adsorbed MB (denoted as NiT50/MCM-41–MB), (c) N 1s spectrum of NiT50/MCM-41–MB, and (d) N 1s spectrum of MB. | |
The reasons for the excellent selective adsorption of MB by the silicon-based adsorbent prepared via the “[Ni(tepa)]2+ modification-thermal treatment” strategy were further investigated. Fig. 9 illustrates the adsorption capacities of individual dyes MB, R6G, RhB, and MO by the different samples including MCM-41, Ni-tepa/MCM-41, NiT50/MCM-41, and Ni/MCM-41. In the case of the cationic dyes MB and R6G, NiT50/MCM-41 exhibited significantly higher adsorption capacities (130 and 78 mg g−1) compared to MCM-41 (22 and 23 mg g−1), respectively. This improvement is attributed to the successful introduction of NiO and NiCx components after the [Ni(tepa)]2+ modification-thermal process, which facilitated the fixation of cationic dyes on the silicon-based adsorbent via Lewis acid–base interactions, resulting in efficient adsorption (reaction eqn (7)). In contrast, Ni-tepa/MCM-41 and Ni/MCM-41 lacked these adsorption sites, leading to much lower adsorption capacities for MB and R6G. Moreover, because the R6G space volume is larger than that of MB and the branched-chain structure of R6G is longer, the diffusion and transfer restrictions of R6G are greater, which results in the adsorption of R6G being lower than that of MB. Although RhB is also a cationic dye, it contains –COOH groups that are easily deprotonated, making RhB electrically neutral under the experimental conditions (pH = 7).51 The adsorption capacity of MCM-41 for RhB reached 96 mg g−1. This high capacity is due to the large specific surface area and pore volume of the raw silicon support, which provides ample adsorption space. Additionally, the COO- groups formed by the deprotonation of –COOH in RhB can easily form hydrogen bonds with Si–OH, fixing RhB to MCM-41. In contrast, the adsorption capacities of Ni-tepa/MCM-41 and NiT50/MCM-41 for RhB were much lower, at only 60 and 50 mg g−1, respectively. This reduction is because the [Ni(tepa)]2+ loading modification process promotes the deprotonation of Si–OH (reaction eqn (8)), hindering the adsorption of RhB. [Ni(tepa)]2+ effectively occupies these negatively charged sites through Coulomb interactions, resulting in its good accommodation (reaction eqn (9)). The zeta potentials of MCM-41, NiT50/MCM-41, and Ni/MCM-41 are −25, −18, and −23 mV, respectively, limiting the adsorption of anionic MO dye due to charge repulsion. However, the zeta potential of Ni-tepa/MCM-41 is close to 0 mV, resulting in the much higher adsorption of MO of about 25 mg g−1. In summary, the above-mentioned reasons explain the significantly increased adsorption capacity and excellent selective adsorption of MB by the silicon-based adsorbent prepared using the “[Ni(tepa)]2+ modification-thermal treatment” strategy.
| | |
NiO + MB → NiO–MB+, NiCx + MB → NiCx–MB+
| (7) |
| | |
[Ni(tepa)]2+ + H2O → [Ni(tepa–H)]+ + OH−, Si–OH + OH− → SiO− + H2O
| (8) |
| | |
SiO− + [Ni(tepa)]2+ → SiO–Ni(tepa)+
| (9) |
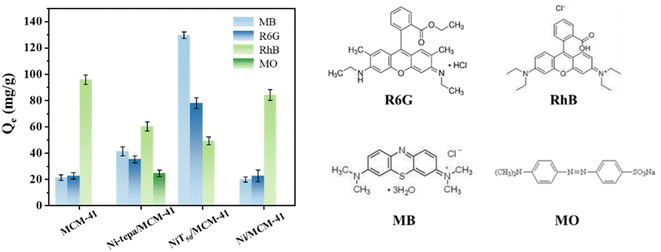 |
| | Fig. 9 Adsorption of different dyes (MB, RhB, R6G, and MO) by different MCM-41, Ni-tepa/MCM-41, NiT50/MCM-41, and Ni/MCM-41 samples. Error bars represent the degree of data dispersion. | |
4 Conclusions
In summary, a new silicon-based material was successfully created using the “[Ni(tepa)]2+ modification-thermal treatment” approach and was applied for the selective adsorption of MB from aqueous solutions. Using MCM-41 as the silicon support, when the [Ni(tepa)]2+ loading was 50%, followed by thermal treatment at 400 °C, the adsorption capacity of MB by the silicon-based adsorbent (NiT50/MCM-41) reached 130 mg g−1. This value is significantly higher compared to the raw silicon support MCM-41 (23 mg g−1). Additionally, NiT50/MCM-41 exhibited excellent selective adsorption properties for MB from binary mixed dye solutions (MB/R6G, MB/RhB, and MB/MO). Its adsorption kinetics and adsorption isotherm follow a second-order kinetic model and the Langmuir adsorption model, respectively. During the “[Ni(tepa)]2+ modification-thermal treatment”, the [Ni(tepa)]2+ aqueous solution provided an alkaline environment, which promoted the deprotonation of Si–OH on the silica surface, increasing the formation of SiO−. In addition, the introduced cationic complex [Ni(tepa)]2+ effectively occupied these negatively charged sites through Coulomb interactions, resulting in its good accommodation. The successful introduction of NiO and NiCx components on the surface of the prepared silica-based adsorbent through appropriate heat treatment led to the immobilization of the cationic dye on the silica-based adsorbent through Lewis acid–base interaction, resulting in the excellent selective adsorption of MB from mixed solutions (MB/R6G, MB/RhB, and MB/MO).
Data availability
The authors declare that they did not use databases or AI for the creation of the article.
Author contributions
Baojuan Luo: conceptualization, formal analysis, investigation, validation, writing – original draft. Menghan Zhang: resources, data curation. Nana Zhou: conceptualization, methodology, investigation. Yongxin Fu: resources, project administration. Xiaoxun Ma: supervision, project administration. Chengyi Dai: supervision, project administration, funding acquisition.
Conflicts of interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Acknowledgements
This work was partly supported by financial support from the National Key Research and Development Program of China (2022YFB4101800), the National Natural Science Foundation of China (22278337), and the Youth Innovation Team Construction Scientific Research Program of Education Department of Shaanxi Provincial Government, China (23JP171).
References
- J. McLaren and I. D. Williams, Sci. Total Environ., 2015, 538, 478–491 CrossRef CAS PubMed.
- M. A. Ackacha, K. Połeć-Pawlak and M. Jarosz, J. Sep. Sci., 2003, 26, 1028–1034 CrossRef CAS.
- D. Chen, S. Chen, Y. Jiang, S. Xie, H. Quan, L. Hua, X. Luo and L. Guo, RSC Adv., 2017, 7, 49024–49030 RSC.
- G. Mahmoud, S. Abdel-Aal, N. El-Kelesh and E. Alshafei, Environ. Ecol. Res., 2017, 5, 480–488 Search PubMed.
- M. A. M. Salleh, D. K. Mahmoud, W. A. W. A. Karim and A. Idris, Desalination, 2011, 280, 1–13 CrossRef CAS.
- C. Xie, Mater. Today Commun., 2024, 41, 110843–110853 CrossRef CAS.
- P. V. Nidheesh, RSC Adv., 2015, 5, 40552–40577 RSC.
- M. Li, Y. Li, X. Zhang, H. Zheng, A. Zhang, T. Chen, W. Liu, Y. Yu, J. Liu, Q. Du, D. Wang and Y. Xia, RSC Adv., 2020, 10, 37757–37765 RSC.
- N. Al-Bastaki, Chem. Eng. Process., 2004, 43, 1561–1567 CrossRef CAS.
- E. R. Bandala, M. A. Peláez, A. J. García-López, M. D. J. Salgado and G. Moeller, Chem. Eng. Process., 2008, 47, 169–176 CrossRef CAS.
- S. Wang, Dyes Pigm., 2008, 76, 714–720 CrossRef CAS.
- Z. Qiusheng, L. Xiaoyan, Q. Jin, W. Jing and L. Xuegang, RSC Adv., 2014, 5, 2100–2112 RSC.
- Y. Pan, Y. Wang, A. Zhou, A. Wang, Z. Wu, L. Lv, X. Li, K. Zhang and T. Zhu, Chem. Eng. J., 2017, 326, 454–461 CrossRef CAS.
- M. Samir, H. H. Hassan, H. H. El-Maghrabi and A. A. Nada, Desalin. Water Treat., 2022, 255, 110–119 CrossRef CAS.
- E. A. Nada, H. H. El-Maghrabi, H. R. Ali, S. A. El-Wahab, D. Y. Sabry, Y. M. Moustafa and A. A. Nada, Desalin. Water Treat., 2022, 255, 236–241 CrossRef CAS.
- A. A. Nada, B. O. Orimolade, H. H. El-Maghrabi, B. A. Koiki, M. Rivallin, M. F. Bekheet, R. Viter, D. Damberga, G. Lesage, I. Iatsunskyi, E. Coy, M. Cretin, O. A. Arotiba and M. Bechelany, Appl. Mater. Today, 2021, 24, 101129 CrossRef.
- S. De Gisi, G. Lofrano, M. Grassi and M. Notarnicola, Sustainable Mater. Technol., 2016, 9, 10–40 CrossRef CAS.
- Y.-F. Hui, C.-L. Kang, T. Tian, S. Dang, J. Ai, C. Liu, H.-R. Tian, Z.-M. Sun and C.-Y. Gao, CrystEngComm, 2017, 19, 1564–1570 RSC.
- S. Kundu, I. H. Chowdhury and M. K. Naskar, ACS Omega, 2018, 3, 9888–9898 CrossRef CAS PubMed.
- F.-Y. Yi, W. Zhu, S. Dang, J.-P. Li, D. Wu, Y. Li and Z.-M. Sun, Chem. Commun., 2015, 51, 3336–3339 RSC.
- S. Tian, W. Liu, B. Liu, H. Li, G. Deng and X. Zhang, Mater. Today Commun., 2023, 35, 105740 CrossRef CAS.
- M. Algarra, M. V. Jiménez, E. Rodríguez-Castellón, A. Jiménez-López and J. Jiménez-Jiménez, Chemosphere, 2005, 59, 779–786 CrossRef CAS PubMed.
- Z. A. ALOthman, Materials, 2012, 5, 2874–2902 CrossRef CAS.
- Z. Teng, X. Su, Y. Zheng, J. Sun, G. Chen, C. Tian, J. Wang, H. Li, Y. Zhao and G. Lu, Chem. Mater., 2013, 25, 98–105 CrossRef CAS.
- C. Dai, M. Zhang, X. Guo and X. Ma, Chem. Eng. J., 2021, 407, 127181 CrossRef CAS.
- Y. S. Ho, G. McKay, D. A. J. Wase and C. F. Forster, Adsorpt. Sci. Technol., 2000, 18, 639–650 CrossRef CAS.
- Y. S. Ho and G. McKay, Chem. Eng. J., 1998, 70, 115–124 CrossRef CAS.
- I. Langmuir, J. Am. Chem. Soc., 1918, 40, 1361–1403 CrossRef CAS.
- K. Y. Foo and B. H. Hameed, Chem. Eng. J., 2010, 156, 2–10 CrossRef CAS.
- Z. Cherifi, B. Boukoussa, A. Mokhtar, M. Hachemaoui, F. Z. Zeggai, A. Zaoui, K. Bachari and R. Meghabar, React. Funct. Polym., 2020, 153, 104611 CrossRef CAS.
- R. E. Morsi and R. S. Mohamed, R. Soc. Open Sci., 2018, 5, 172021 CrossRef PubMed.
- X. Xiao, F. Zhang, Z. Feng, S. Deng and Y. Wang, Phys. E, 2015, 65, 4–12 CrossRef CAS.
- H. Han, W. Wei, Z. Jiang, J. Lu, J. Zhu and J. Xie, Colloids Surf., A, 2016, 509, 539–549 CrossRef CAS.
- J. P. Dhal, T. Dash and G. Hota, J. Porous Mater., 2020, 27, 205–216 CrossRef CAS.
- A. C. Pradhan, A. Paul and G. R. Rao, J. Chem. Sci., 2017, 129, 381–395 CrossRef CAS.
- Y. Shao, X. Wang, Y. Kang, Y. Shu, Q. Sun and L. Li, J. Colloid Interface Sci., 2014, 429, 25–33 CrossRef CAS PubMed.
- A. A. Nada, M. F. Bekheet, S. Roualdes, A. Gurlo and A. Ayral, J. Mol. Liq., 2019, 274, 505–515 CrossRef CAS.
- Z. Liang, Z. Zhao, T. Sun, W. Shi and F. Cui, J. Colloid Interface Sci., 2017, 485, 192–200 CrossRef CAS PubMed.
- S. Chander, S. Yadav, H. R. Sharma and A. Gupta, J. Alloys Compd., 2024, 986, 174088 CrossRef CAS.
- S. Chander, S. Yadav, R. Singh, H. R. Sharma and A. Gupta, J. Ind. Eng. Chem., 2024 DOI:10.1016/j.jiec.2024.10.027.
- H. Du, X. Ma, P. Yan, M. Jiang, Z. Zhao and Z. C. Zhang, Fuel Process. Technol., 2019, 193, 221–231 CrossRef CAS.
- Z. Wu, C. Zhang, L. Peng, X. Wang, Q. Kong and X. Gu, ACS Appl. Mater. Interfaces, 2018, 10, 3175–3180 CrossRef CAS PubMed.
- W. Bao, X. Liang, Y. Liu, Y. Gao and J. Wang, IEEE Trans. Dielectr. Electr. Insul., 2017, 24, 2911–2919 CAS.
- M.-G. Jeong, E. J. Park, B. Jeong, D. H. Kim and Y. D. Kim, Chem. Eng. J., 2014, 237, 62–69 CrossRef CAS.
- P. M. Carraro, A. A. G. Blanco, C. Chanquía, K. Sapag, M. I. Oliva and G. A. Eimer, Microporous Mesoporous Mater., 2017, 248, 62–71 CrossRef CAS.
- W. Zhang, W. Li, Y. Li, S. Peng and Z. Xu, Catal. Today, 2019, 335, 326–332 CrossRef CAS.
- W. Xiong, Q. Guo, Z. Guo, H. Li, R. Zhao, Q. Chen, Z. Liu and X. Wang, J. Mater. Chem. A, 2018, 6, 4297–4304 RSC.
- C. Zhang, H. Yue, Z. Huang, S. Li, G. Wu, X. Ma and J. Gong, ACS Sustain. Chem. Eng., 2013, 1, 161–173 CrossRef CAS.
- C. Yang, L. Fu, R. Zhu and Z. Liu, Phys. Chem. Chem. Phys., 2016, 18, 4635–4642 RSC.
- P. Chen, Z. Cao, X. Wen, J. Wang, F. Yang, P. Qiu, Y. Yue, G. Liu, S. Wang and H. Zhong, J. Taiwan Inst. Chem. Eng., 2017, 80, 128–136 CrossRef CAS.
- R. W. Ramette and E. B. Sandell, J. Am. Chem. Soc., 1956, 78, 4872–4878 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence *
*