
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceRecent advances on anticancer activity of benzodiazine heterocycles through kinase inhibition
Mohamed S. Nafie
 *ab,
Sherif Ashraf Fahmy
c,
Shaima H. Kahwashb,
Mohamed K. Diabd,
Kamal M. Dawood
e and
Ashraf A. Abbas
e
*ab,
Sherif Ashraf Fahmy
c,
Shaima H. Kahwashb,
Mohamed K. Diabd,
Kamal M. Dawood
e and
Ashraf A. Abbas
e
aDepartment of Chemistry, College of Sciences, University of Sharjah, Sharjah, 27272, United Arab Emirates. E-mail: mohamed.elsayed@sharjah.ac.ae
bDepartment of Chemistry, Faculty of Science, Suez Canal University, Ismailia, 41522, Egypt. E-mail: mohamed_nafie@science.suez.edu.eg
cDepartment of Pharmaceutics and Biopharmaceutics, University of Marburg, Robert-Koch-Str. 4, 35037, Marburg, Germany
dPest Physiology Department, Plant Protection Research Institute, Agricultural Research Center, Giza, 12311, Egypt
eChemistry Department, Faculty of Science, Cairo University, Giza, 12613, Egypt
First published on 19th February 2025
Abstract
The benzodiazines (phthalazine, quinazoline, quinoxaline, and cinnoline) have emerged as attractive scaffolds for creating novel anticancer drugs. These nitrogen-containing heterocycles are intriguing because they have a variety of configurations and can change chemically, allowing us to tailor their pharmacokinetic and pharmacodynamic features. Numerous studies have found that derivatives of these compounds have potent anticancer properties via inhibiting topoisomerases, protein kinases, and receptor tyrosine kinases. These compounds impair critical processes that control cancer proliferation and survival. Most benzodiazine derivatives have achieved clinical success, demonstrating the heterocycles' therapeutic potential. The use of phthalazine, cinnoline, and quinazoline derivatives should open new avenues in developing better and more targeted cancer treatments. In this overview, we summarize recent advances in synthesizing these compounds and illustrate how they serve as promising chemotherapeutic agents. Therefore, current research organizes the latest information to provide a clearer picture of design strategies that boost efficacy and selectivity, allowing the identification of potential anticancer drug candidates down the line. This research study also highlights the need to establish heterocyclic derivatives as a promising source of new molecules for cancer treatment with improved efficacy and decreased effects.
 Mohamed S. Nafie | Mohamed S. Nafie (Ph.D., MRSC). Assistant Prof. of Bioorganic Chemistry, College of Sciences, University of Sharjah, United Arab Emirates (UAE). In 2024, he is recognized as one of the top 2% World Scientists' List by Stanford University Published 16 September 2024, Version 7, DOI: https://doi.org/10.17632/btchxktzyw.7. In 2022, he was awarded with the best Scholar in Suez Canal University, Egypt. He is a member of Member of the Royal Society of Chemistry (MRSC, since 2022), American Chemical Society member (ACS, since 2021) and the European Society for Medical Oncology (ESMO, since 2024). He got his Ph.D. in Bioorganic Chemistry (2018), M.Sc. in Biochemistry 2015, and B.Sc. in Chemistry from Suez Canal University (Egypt). In 2017 he was awarded the Erasmus+KA107 scholarship at the Faculty of Pharmacy, University of Pisa, Italy. In 2020, he was awarded the Daniel Turnberg Travel Fellowship at the Institute of Medical Sciences, University of Aberdeen, UK. His research interest is to design and synthesize novel target-oriented chemotherapeutic anti-cancer agents using the Computer-Aided Drug Design (CADD), medicinal chemistry, biochemical, and molecular biology assays. He established and led several research groups and international collaborations funded by prestigious grants and institutions. His research work was awarded as Top cited articles in WILEY for 2021–2022 and 2022–2023. He published more than 138 peer-reviewed articles in international journals with Scopus H-index 23. Additionally, he served as scientific editor in Frontiers in Chemistry, Metabolites (MDPI) and as reviewers in more than 225 papers in distinguished international journals (Clarivate analytics). |
 Sherif Ashraf Fahmy | Dr. Sherif Ashraf Fahmy is Visiting Assistant Professor at the Department of Pharmaceutics and Biopharmaceutics, Faculty of Pharmacy, Philipps-Universität Marburg, Germany. Furthermore, he is the Head of the Herbal Medicine and Nanotherapeutics Research Group. Dr Sherif Ashraf Fahmy obtained his B.Pharm. with Honors from the Faculty of Pharmacy, Cairo University, and his M.Sc. and Ph.D. from the American University in Cairo, in collaboration with the Department of Pharmaceutics and Biopharmaceutics, Philipps-Universität Marburg, Germany. Dr Fahmy was selected as one of the first recipients of the prestigious fellowship offered by the Alfi Foundation for Ph.D. students. He has received several other awards and recognitions, most notably the Alexander von Humboldt Georg Forster Research Fellowship, the Royal Society of Chemistry Research Fund, and the Fulbright Scholarship at Ohio University. Also, he has been listed in the top 2% World Scientists' List by Stanford University published 16 September 2024, Version 7, DOI: https://doi.org/10.17632/btchxktzyw.7. His research interests revolve around natural products, inorganic complexes, nanomedicine, and drug delivery. Dr Fahmy has published several peer-reviewed articles in international journals and many abstracts at local and international conferences. Furthermore, he serves as a reviewer and editor for several international journals and serves as a member of the topical advisory panel for the Pharmaceutics journal, member of the Royal Society of Chemistry (MRSC), American Chemical Society member and the European Society for Medical Oncology (ESMO). |
 Kamal M. Dawood | Kamal M. Dawood graduated from Cairo University, Egypt in 1987, and received his Ph.D. in 1995 from Cairo University. In 1997 he was awarded the UNESCO Fellowship at TIT for one year and in 1999 he was awarded the JSPS Fellowship for two years and in both fellowships he worked with Professor T. Fuchigami at Tokyo Institute of Technology (TIT) in the field of “Anodic Selective Fluorination of Heterocycles”. Further, he was awarded the Alexander von Humboldt (AvH) Fellowship at Hanover University in 2004–2005 with Prof. A. Kirschning (in the area of polymer supported palladium catalyzed cross coupling reactions) and as AvH three short visits in 2007, 2008 and 2012 with Prof. P. Metz at TU-Dresden (in the field of Metathesis Reactions in Domino Processes). Since May 2007 – to date, he has been appointed as full Professor of Organic Chemistry, Faculty of Science, Cairo University. He worked as Professor of Organic Chemistry at Chemistry Department, Kuwait University from September 2013 till August 2017. He received a number of National Awards: Cairo University Award in Chemistry (2002), the State-Award in Chemistry (2007), Cairo University Award for Academic Excellence (2012) and Cairo University Merit Award (2017). He published about 160 scientific papers, reviews and book chapters in distinguished international journals. There are about 4000 citations of his work (Scopus h-index 33). |
 Ashraf A. Abbas | Ashraf A. Abbas was born in Egypt in September 1968 and he is presently Professor of Organic Chemistry, Department of Chemistry, Faculty of Science, Cairo University, Giza, Egypt since 2009. He graduated from Cairo University in 1990. He received his M.Sc. and Ph.D. degrees in 1994 and 1997, respectively, from Cairo University. He spent six months in the Fakultat fur Chemie, Universitat of Konstanz (Germany) on a DAAD fellowship (1996) to finish his Ph.D. thesis and one year in Tokyo Institute of Technology (TIT, Japan) as UNESCO fellow (postdoctoral fellowship) from October 2001 to September 2002. He received several research prizes in chemistry; (1) he was awarded the prize of Prof. Dr Mohamed Abdel Salam in chemistry (2001) for young scientists provided by Academy of Scientific Research and Technology (Egypt). (2) He was awarded the Cairo University encouragement prize in chemistry in 2004. (3) He was awarded the Third Word Academy of Science (TWAS) prize in chemistry for young scientists in 2004 provided by ICTP-Strada, Triesta-Italy, (4) he was awarded the State Award in Chemistry, Egypt 2005. He published many papers in the field of synthesis and applications of macrocycles and bis-heterocycles chemistry. |
1. Introduction
Cancer is among the leading causes of morbidity and mortality globally and imposes major healthcare and economic burdens.1 The high incidence of acquired resistance, in addition to serious side effects associated with contemporary therapeutic agents, sustains the search for new anticancer agents despite the fact that many chemotherapeutic agents were introduced and progress with targeted therapy was made.2 Heterocyclic compounds have attracted considerable interest in this context owing to a structural diversity that can interact with diverse targets of biological significance.3 Different heterocyclic systems have been studied in this respect, and phthalazine, cinnoline, and quinazoline derivatives appear to be good candidates for building blocks for potent anticancer drugs.4Organic compounds with a ring structure of at least two distinct elements are known as heterocyclic compounds. Nitrogen, oxygen, and sulfur are the most common heteroatoms.5 These are used for many medicines, and their importance in medicinal chemistry is well established. These heteroatoms in the structures provide other interactions with proteins and nucleic acids (NAs), which make them candidates for drug design.6 In addition, the potential for chemical manipulation of these scaffolds facilitates the balancing of pharmacokinetic parameters, such as solubility, bioavailability, and metabolic stability, which are key determinants for therapeutic success.7 Nitrogen-containing heterocycles, such as phthalazine, cinnoline, quinazoline, and quinoxaline, all are isomeric forms of benzodiazine with the molecular formula C8H6N2 (Fig. 1), each of them has unique structural features and can interact with cancer cells in distinct ways.4 Because these heterocyclic frameworks are adaptable, much research has been conducted on how to create them and how they work in living organisms.3 This has resulted in the discovery of several derivatives with potent anticancer activity. Recent studies have proven their ability to disrupt various oncogenic pathways, making them potential templates for future drug development efforts.8
 | ||
| Fig. 1 Isomeric forms of benzodiazines with the molecular formula C8H6N2. | ||
Phthalazine (2,3-benzodiazine) has a bicyclic structure featuring a benzene ring fused to a pyridazine ring.9 This rigid scaffold can hold multiple substituents, allowing for diverse biological functions.10 The derivatives of phthalazines have been extensively screened for their antibacterial, anticonvulsant, anti-inflammatory, and anticancer actions. Among them, some of the FDA-approved phthalazine anticancer drugs are shown in Fig. 2, known for their capacity to alter critical biological targets implicated in cancer growth.11 Phthalazine derivatives with anticancer properties frequently work through their interactions with enzymes, including topoisomerases and kinases. These were associated with DNA replication, transcription , and cell cycle regulation.12 Topoisomerase II activity is inhibited by phthalazine-based inhibitors, for example. Normal cells need this enzyme to copy their DNA when they divide rapidly. Topoisomerase II inhibitors promote DNA damage and apoptosis, effectively treating various cancer types.13 Several phthalazine derivatives are also known to function as potent tyrosine kinase inhibitors, key regulators of cell growth, and survival-supporting signal transduction pathways.13 Such characteristics highlighted the possibility of phthalazine derivatives being chemotherapeutics.14
 | ||
| Fig. 2 Some FDA-approved phthalazine-based anticancer drugs.15 Molecular targets are; phosphodiesterase 5 (PDE5) inhibitor, tyrosine kinase inhibitor, vascular endothelial growth factor receptor 2, Hedgehog (Hh) signaling pathway, DNA methyltransferase (DNMT), cAMP-dependent protein kinase (PKA), and phosphodiesterase (PDE) inhibitor. | ||
Quinazoline (1,3-benzodiazine) has been the subject of extensive research for therapeutic purposes.16 The adaptability of the quinazoline scaffold allowed for a wide range of functional alterations that increase activity and selectivity.17 Quinazoline derivatives showed many biological activities such as antibacterial, anti-inflammatory, and anticancer.18 Some FDA-approved quinazoline anticancer drugs are depicted in Fig. 3. Quinazoline compounds target essential cellular pathways cancer cells rely on for growth and survival.19 Gefitinib, a selective epidermal growth factor receptor (EGFR) inhibitor, established one of the most convincing examples of quinazoline derivatives for non-small cell lung cancer therapy.20 It was, therefore, possible to identify small molecules that inhibit distinct receptor tyrosine kinases (RTKs) and pathways that drive tumorigenesis. Biomedicinally, it is found that the quinazoline derivatives inhibited the activity of EGFR, VEGFR, and other kinases, in turn inhibiting critical processes such as cell division, migration, and angiogenesis.21 This inhibited critical processes such as cell division, migration, and angiogenesis.22 These results demonstrate the possibilities of quinazoline derivatives as a therapeutic target for generating new anticancer approaches.
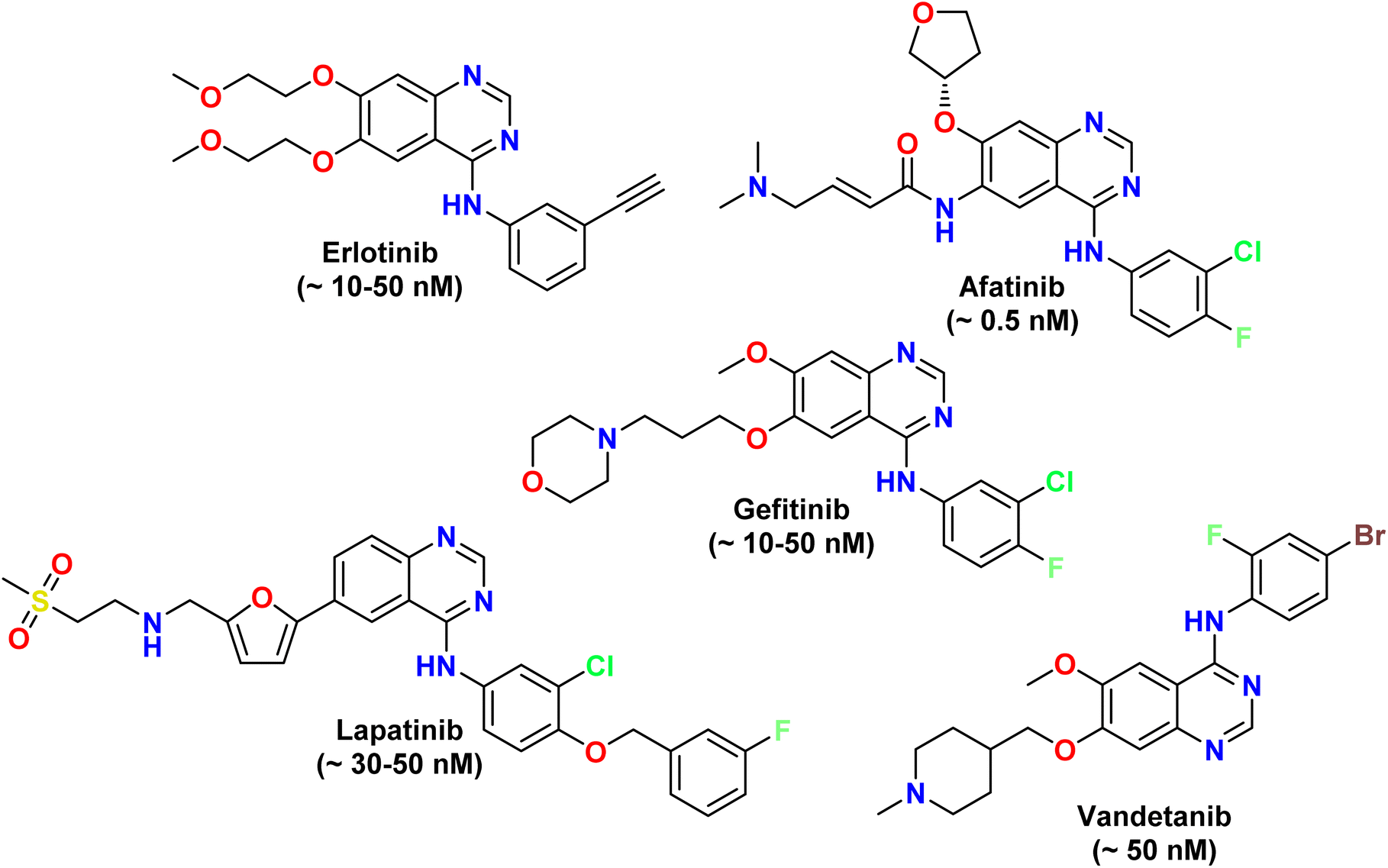 | ||
| Fig. 3 Some FDA-approved quinazoline-based anticancer drugs.23 Most of these drugs as mainly EGFR inhibitors, while Vandetanib is a multi-targeted tyrosine kinase inhibitor (TKI). | ||
Quinoxaline (1,4-benzodiazine) derivatives have received great attention as anticancer agents because of their unique structural properties and activity.24 Their core structure surfaces a benzene ring, which forms the solid base for numerous modifications of pyrazine ring that withstands many chemical reactions.25 As shown in Fig. 4, with some quinoxaline anticancer drugs. Various quinoxaline derivatives can interact with different molecular targets in cancer cells. These include kinases, topoisomerases, and other proteins that help control the cell cycle and kill cells.26 According to studies, these compounds effectively block tyrosine kinases, interfere with DNA synthesis, and cause oxidative stress in tumor cells, ultimately leading to cell death.27 They also control many signaling pathways, such as the PI3K/AKT/mTOR and MAPK pathways, which makes them more likely to work as multi-targeted cancer treatments.28 Recent studies have highlighted the importance of quinoxaline derivatives in inhibiting cell proliferation, angiogenesis, and metastasis, making them promising for further development as chemotherapeutic medicines.29 The ability to synthesize these molecules using a variety of catalytic and microwave-assisted procedures increased their appeal, allowing the development of derivatives with greater efficacy and lower toxicity.30 As a result, quinoxaline derivatives provided an essential framework for creating new anticancer drugs with broad therapeutic applications.
 | ||
| Fig. 4 Some quinoxaline-based anticancer drugs.31 | ||
Cinnoline (1,2-benzodiazine) is a structural analog of phthalazine in which the nitrogen atoms are adjacent in the diazine ring.32 Such a relatively minor structural modification can lead to dramatic variances in biological activities that enable selective inhibition of distinct cancer pathways. Derivatives of 3H-cinnoline derivatives have gained immense interest in medicinal chemistry due to their diverse pharmacological applications, principally as anticancer agents, showcasing lucrative physicochemical properties and potential to be modified via various synthetic strategies.33 The synthesis of cinnoline derivatives was carried out through catalytic methods.34 Such synthetic improvements have been beneficial in producing drugs based on the structure of cinnoline with improved potency against cancer cells.35 Cinnoline derivatives have been shown to induce apoptosis, reduce cell proliferation , and inhibit angiogenesis in vitro and in vivo.36 They typically do this by inhibiting the actions of enzymes like cyclin-dependent kinases (CDKs) and protein kinases.37 These enzymes are necessary for cancer cells to cycle through the cell cycle and to transmit signals.38 These characteristics suggested cinnoline derivatives as promising candidates for further exploration in oncology.39–41
Advances in molecular biology and computational chemistry have helped us better grasp how these heterocyclic scaffolds interact with their biological targets.42 Researchers have used high-throughput screening, molecular docking studies, and structure–activity relationship analysis to find important structural features needed to fight cancer. This knowledge has been helpful in guiding the design and synthesis of novel derivatives with increased effectiveness.43 Additionally, recent research demonstrates that phthalazine, cinnoline, and quinazoline derivatives can simultaneously alter multiple signaling pathways, enabling their multifaceted application in cancer treatment.8 This multi-targeted activity is beneficial in the case of heterogenous cancers, where a single-target medication may fail to deliver long-term therapeutic effects.44 By targeting several elements of cancer cell survival, proliferation, and metastasis, these compounds have the potential to produce more effective and long-lasting responses in cancer patients.6
Recently, our research target has been to build up a wide array of simple- and bis-organic molecules incorporating various heterocyclic nuclei and different functionalities with promising anticancer inhibitory activities against many human cell lines.34,45–49 Encouraged by the facts and in continuation of our longstanding interest in the synthesis of biologically active heterocycles, we are engaged in a new, interesting protocol. The purpose of this work is to provide a comprehensive update on the synthetic pathways of fused 6-membered N-heterocyclic scaffolds with two N-atoms, particularly phthalazines, quinoxalines, cinnolines, and quinazolines, that have been documented in the most recent literature between 2020 and 2024 and emphasizing their potentials as promising anticancer agents.
2. Phthalazine derivatives
The reaction of 1-chloro-4-phenylphthalazine 1 with the methyl 4-aminobenzoate under reflux resulted in the production of methyl 4-[(4-phenylphthalazin-1-yl)amino]benzoate (2) in 81% yield. Heating of the ester 2 with hydrazine hydrate produced the hydrazide 3, which upon heating with the appropriate isocyanate and/or isothiocyanate produced the corresponding semicarbazide 4a, b and/or thiosemicarbazide 4c–g, derivatives respectively in 81–87% yields (Scheme 1).50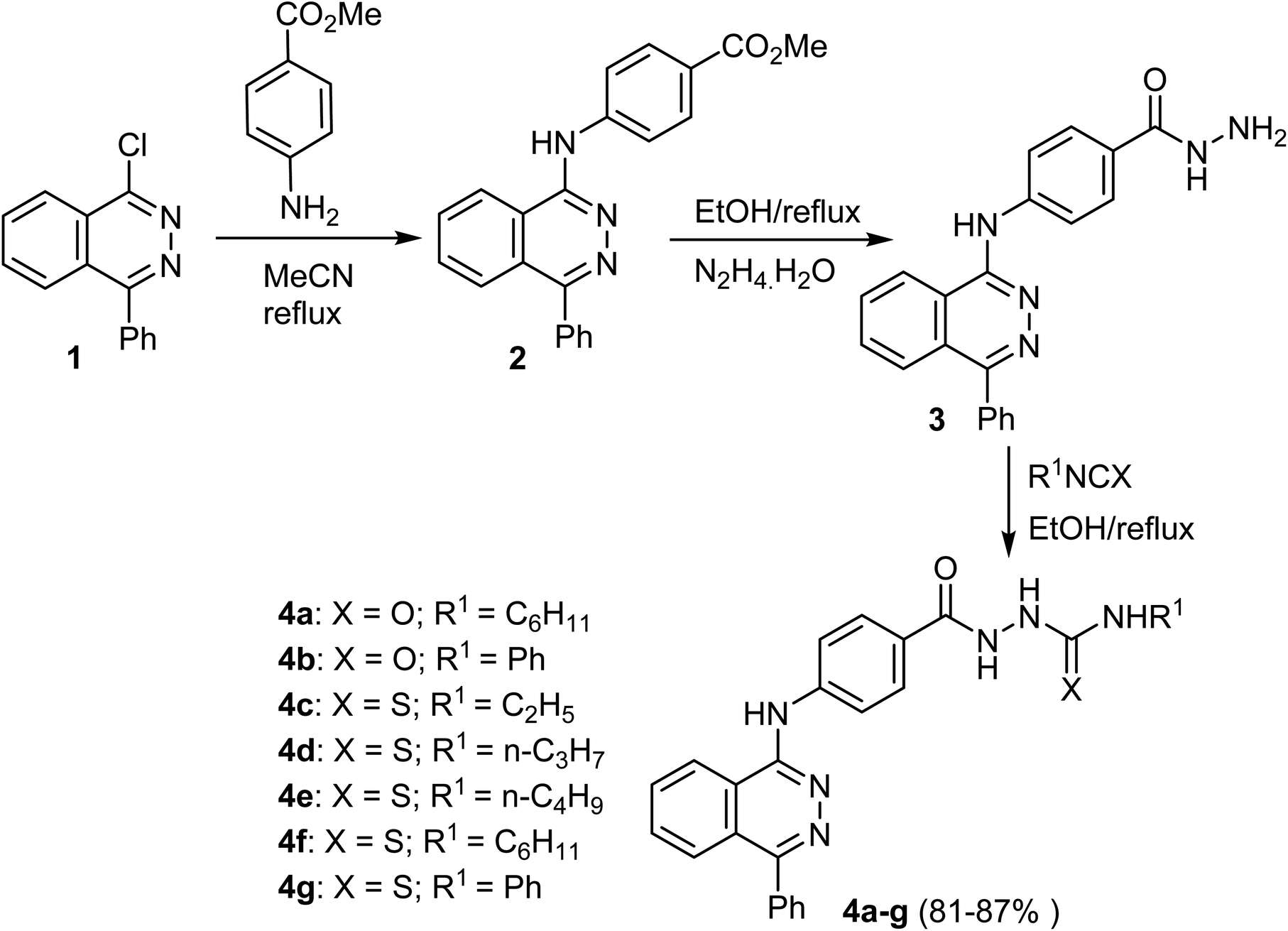 | ||
| Scheme 1 Synthetic route for the preparation of phthalazine derivatives 4a–g. | ||
Khedr et al. reported the synthesis of novel phthalazine scaffolds 7a–g as depicted in Scheme 2. Thus, heating of 1-chloro-4-phenylphthalazine 1 with the 4-aminoacetophenone gave the acetyl derivative 5, which, upon condensation with the suitable aromatic benzaldehyde produced the chalcone derivatives 6a–g in 81–88% yields. Cyclocondensation of 6a–g with hydrazine hydrate yielded corresponding pyrazoline derivatives 7a–g in 70–78% yields (Scheme 2).51
 | ||
| Scheme 2 Synthetic route for the preparation of target compounds 5–7. | ||
The acetyl derivative 9 was obtained by refluxing the chloro derivative 8 with 4-aminoacetophenone. The matching hydrazine derivatives 10a–g were produced in 65–79% yields by condensation of 8 with the suitable acid hydrazides. Additionally, reaction of urea or thiourea derivatives with 8 yielded the corresponding phthalazine derivatives 11a, b in 72–75% yields (Scheme 3).52
 | ||
| Scheme 3 Synthetic route for the preparation of phthalazine derivatives 10 and 11. | ||
Hydrazinolysis of the methyl(2-(4-benzyl-1-oxophthalazin-2(1H)-yl)acetyl)glycinate ester (12) produced the corresponding hydrazide derivative 13. Condensation of the hydrazide 13 with some active methylene compounds furnished the corresponding hydrazones 14a–c in 84–91% yields, as shown in Scheme 4.53
 | ||
| Scheme 4 Preparation of phthalazine derivatives 14a–c. | ||
Scheme 5 describes the synthetic pathway to 2-phenyl-2,3-dihydrophthalazine-1,4-dione-1,2,3-triazole hybrids 20a–l. Firstly, the reaction of phthalic anhydride (15) with phenylhydrazine (16) in acetic acid containing sodium acetate produced 2-phenyl-2,3-dihydrophthalazine-1,4-dione (17). Then, using potassium tert-butoxide as a base, compound 17 was alkylated with propargyl bromide in DMF to yield the 2,3-dihydrophthalazine derivative 18. Next, the 1,3-dipolar cycloaddition reaction between aryl azides 19a–l and terminal alkyne 18 in the presence of copper(I) iodide in THF produced the 2-phenyl-2,3-dihydrophthalazine-1,4-dione-1,2,3-triazole hybrids 20a–l in moderate to good yields, 76–88%. It's crucial to note that aryl azides 19 with electron-withdrawing groups produced higher yields than those with electron-donating groups.54
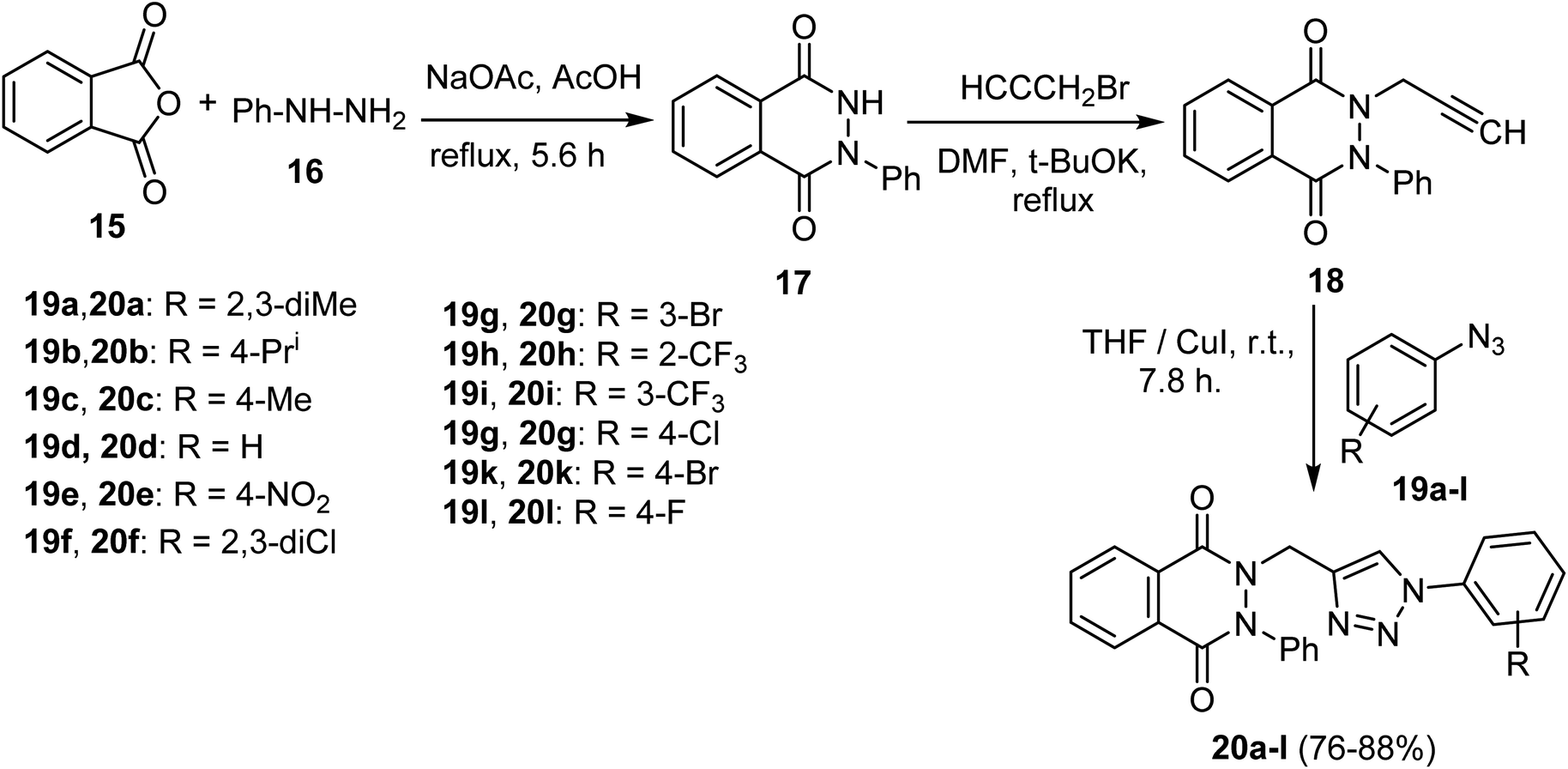 | ||
| Scheme 5 Preparation of phthalazine derivatives 20a–l. | ||
Most compounds 20a–l exhibited promising physiochemical properties through ADME pharmacokinetics regarding obeying Lipinski's rule of drug-likeness.55 Using Molsoft web-based software, compounds exhibited a good scale of obeyable parameters, particularly number of H-bond acceptors (HBA = 4 lower than 10), H-bond doner (HBD = 0, lower than 5), MW ≤ 500, log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) p ≤ 4.25, and in a summary a drug-likeness score is 0.40 (positive value as a drug-like). The incorporation of hydrogen bonding is critically important in the binding of drugs to their targets due to its ability to provide specificity, stability, and strength to the drug–target complex.56
p ≤ 4.25, and in a summary a drug-likeness score is 0.40 (positive value as a drug-like). The incorporation of hydrogen bonding is critically important in the binding of drugs to their targets due to its ability to provide specificity, stability, and strength to the drug–target complex.56
2.1 Anticancer activity of phthalazine-based derivatives
Synthesis of 4f and evaluation of its cytotoxic activity against three human cancer cell lines (HepG2, HCT-116, and MCF-7) was reported. Compound 4f exhibited notable cytotoxicity against tested cell lines “HepG2, HCT-116, and MCF-7” with IC50 values of 3.97 μM, 4.83 μM, and 4.58 μM, respectively. The inhibitory effects of 4f against VEGFR-2 were also significant with IC50 0.08 μM, compared to the reference drug Sorafenib (IC50 = 0.10 μM). The ability of 4f to interact with VEGFR-2 was also confirmed by docking studies.50The antiproliferative activity of compound 6e was evaluated in vitro against three human cancer cell lines: HepG2, HCT-116, and MCF-7. Compound 6e showed significant anticancer activity against all the tested cell lines HepG2, HCT-116, and MCF-7with IC50 values of 11.23 μM, 10.12 μM, and 13.92 μM, respectively, compared with Sorafenib (IC50 = 9.18 μM, 5.47 μM, and 7.26 μM, respectively) and Doxorubicin (IC50 = 7.94 μM, 8.07 μM, and 6.75 μM, respectively). The inhibitory effects of 6e against VEGFR-2 were also significant with IC50 0.11 μM, compared to the reference drug Sorafenib (IC50 = 0.10 μM). The ability of 6e to interact with VEGFR-2 was also confirmed by docking studies.51
The antiproliferative activity of another series of phthalazine derivatives 10g and 11a was evaluated against two human cancer cell lines, MCF-7 (breast cancer) and HepG-2 (hepatocellular carcinoma), compared with Sorafenib as a reference drug. The two derivatives, 10g and 11a demonstrated, exhibited notable activity against MCF-7 and HepG-2, with IC50 values (0.15 μM and 0.12 μM for 10g) and IC50 values (0.18 μM and 0.09 μM for 11a), respectively. They showed significant anticancer activities against the tested cancer cell lines compared to the reference drug. The ability of 10g and 11a to inhibit VEGFR-2 was evaluated in vitro, and these two compounds showed significant activity with IC50 values 0.148 μM and 0.196 μM, respectively, compared to Sorafenib (IC50 = 0.059 μM). Docking studies supported the obtained results and demonstrated the ability of these derivatives to interact with VEGFR-2 active sites.52
Compound 14b was screened for anticancer activity in vitro against two human cancer cell lines, MCF-7 and MDA-MB-231, using Erlotinib as a reference drug and tested against a normal cell line (MCF-10A) to determine their specificity. Compound 14b showed significant cytotoxic activity against the cell lines MCF-7 and MDA-MB-231with IC50 values of 1.9 μM and 0.57 μM, respectively, compared to Erlotinib with IC50 1.32 μM and 1.02 μM, respectively. In addition, the IC50 value of 14b against MCF-10A was 41.6 μM. The inhibition activity of this 14b against EGFR was evaluated; the inhibitory effects of 14b against EGFR were also significant with IC50 21.4 nM, compared to reference drug Erlotinib (IC50 = 80.1 nM) and the percentage of inhibition of EGFR with 10 μM of 14b was 97.6%, compared to the Erlotinib 93.9%. The ability of 14b to interact with EGFR was also confirmed by docking studies.53
The antiproliferative activity of compound 20e was evaluated in vitro against three human cancer cell lines, A-375, A-549, and MCF-7, and Doxorubicin was used as a reference drug. 20e displayed significant cytotoxicity against the three cell lines, A-375, A-549, and MCF-7, with IC50 values of 2.33 μM, 7.21 μM, and 3.96 μM, respectively. The ability of these novel derivatives to interact with EGFR active sites and to inhibit its activity was also confirmed by docking studies (Table 1).54
| Entry | Structure | Kinase inhibition activity | Anticancer activity | Ref. | |||||
|---|---|---|---|---|---|---|---|---|---|
| 1 | 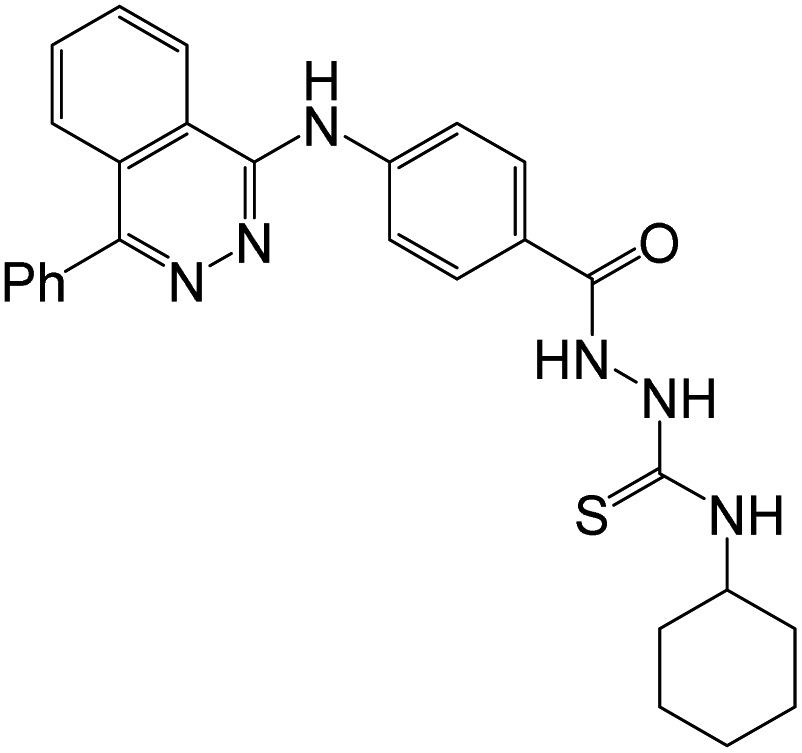 |
Enzymes | IC50 [μM] | Cell lines | IC50 [μM] | 50 | |||
| (4f) | Sorafenib | (4f) | Sorafenib | Doxorubicin | |||||
| VEGFR-2 | 0.08 ± 0.01 | 0.10 ± 0.02 | HepG2 | 3.97 ± 0.2 | 9.18 ± 0.6 | 7.94 ± 0.6 | |||
| HCT116 | 4.83 ± 0.2 | 5.47 ± 0.3 | 8.07 ± 0.8 | ||||||
| MCF-7 | 4.58 ± 0.3 | 7.26 ± 0.3 | 6.75 ± 0.4 | ||||||
| 2 |  |
Enzymes | IC50 [μM] | Cell lines | IC50 [μM] | 51 | |||
| (6e) | Sorafenib | (6e) | Sorafenib | Doxorubicin | |||||
| VEGFR-2 | 0.11 ± 0.02 | 0.10 ± 0.02 | HepG2 | 11.23 ± 1.1 | 9.18 ± 0.6 | 7.94 ± 0.6 | |||
| HCT116 | 10.12 ± 1.0 | 5.47 ± 0.3 | 8.07 ± 0.8 | ||||||
| MCF-7 | 13.92 ± 1.2 | 7.26 ± 0.3 | 6.75 ± 0.4 | ||||||
| 3 |  |
Enzymes | IC50 [μM] | Cell lines | IC50 [μM] | 52 | |||
| (10g) | Sorafenib | (10g) | Sorafenib | ||||||
| VEGFR-2 | 0.148 ± 0.01 | 0.059 ± 0.01 | MCF-7 Hep G2 | 0.15 ± 01 | 0.05 ± 0.01 | ||||
| 0.12 ± 0.01 | 0.03 ± 0.01 | ||||||||
| 4 |  |
Enzymes | IC50 [μM] | Cell lines | IC50 [μM] | 52 | |||
| (11a) | Sorafenib | (11a) | Sorafenib | ||||||
| VEGFR-2 | 0.196 ± 0.01 | 0.059 ± 0.01 | MCF-7 Hep G2 | 0.18 ± 01 | 0.05 ± 0.01 | ||||
| 0.09 ± 0.01 | 0.03 ± 0.01 | ||||||||
| 5 | 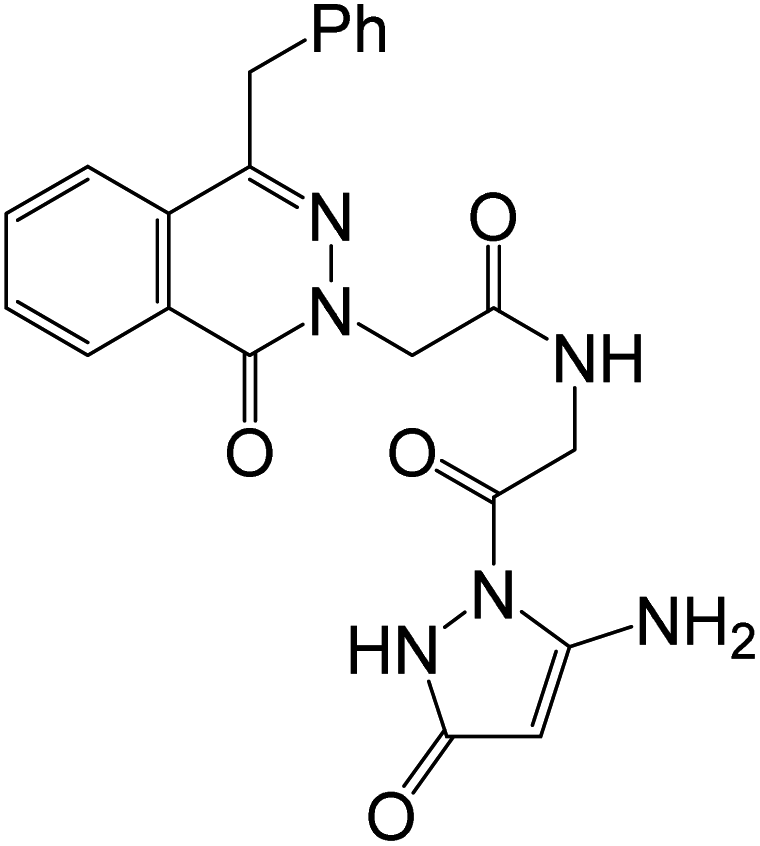 |
Enzymes | IC50 [μM] | Cell lines | IC50 [μM] | 53 | |||
| (14b) | Erlotinib | (14b) | Erlotinib | ||||||
| EGFR | 21.4 ± 0.67 | 80.1 ± 1.21 | MCF-7 | 1.9 ± 0.01 | 1.32 ± 0.04 | ||||
| Kinase inhibition (%) at 10 μM | MDA-MB-231 | 0.57 ± 0.09 | 1.02 ± 0.1 | ||||||
| (21) | Erlotinib | ||||||||
| 97.6 ± 2.49 | 93.9 ± 2.68 | MCF-10A | 41.6 ± 1.8 | 30.9 ± 1.8 | |||||
| 6 |  |
Enzymes | Docking study | Cell lines | IC50 [μM] | 54 | |||
| (20e) | (20e) | Doxorubicin | |||||||
| EGFR | Compound 20e displayed the best docking score of −11.16 | A-375 | 2.33 ± 0.43 | 2.18 ± 0.18 | |||||
| A-549 | 7.21 ± 0.61 | 5.51 ± 0.039 | |||||||
| MCF-7 | 3.96 ± 0.41 | 2.02 ± 0.17 | |||||||
3. Quinazoline derivatives
Synthesis of the quinazoline-based molecular hybrids 24a–g was reported by Xu et al. by applying the Buchwald–Hartwig coupling of 22 with aminoindazole derivative 23 to afford the targeted N-(1H-indazol-3-yl)quinazolin-4-amine products 24a–g in 65–96% yields (Scheme 6).57 | ||
| Scheme 6 Synthetic routes of quinazolin-4-amine derivatives 24a–g. | ||
Liu et al. described a synthetic route for the quinazoline compounds 31a–g starting with 7-methoxyquinazolin-4(3H)-one (25). Thus, chlorination of 25 with thionyl chloride followed by subsequent nucleophilic displacement with aniline derivatives 27 gave products 28a–e. Reduction of 28a–e with stannous chloride dihydrate afforded 29a–e, which upon treatment with the isocyanates 30a–c yielded the target compounds 31a–g in 70–89% yields (Scheme 7).58
 | ||
| Scheme 7 Synthetic routes of quinazoline derivatives 31a–g. | ||
The synthetic route to the 3-phenylquinazolin-2,4(1H,3H)-dione derivatives 33a–j was described by Hassan et al. These compounds were synthesized, in 65–82% yields, via a single pot three-component reaction of 4-(2,4-dioxo-1,4-dihydro-2H-quinazolin-3-yl)-benzoyl chloride 32 with ammonium thiocyanate followed by aryl(hetaryl)amines at reflux condition (Scheme 8).59
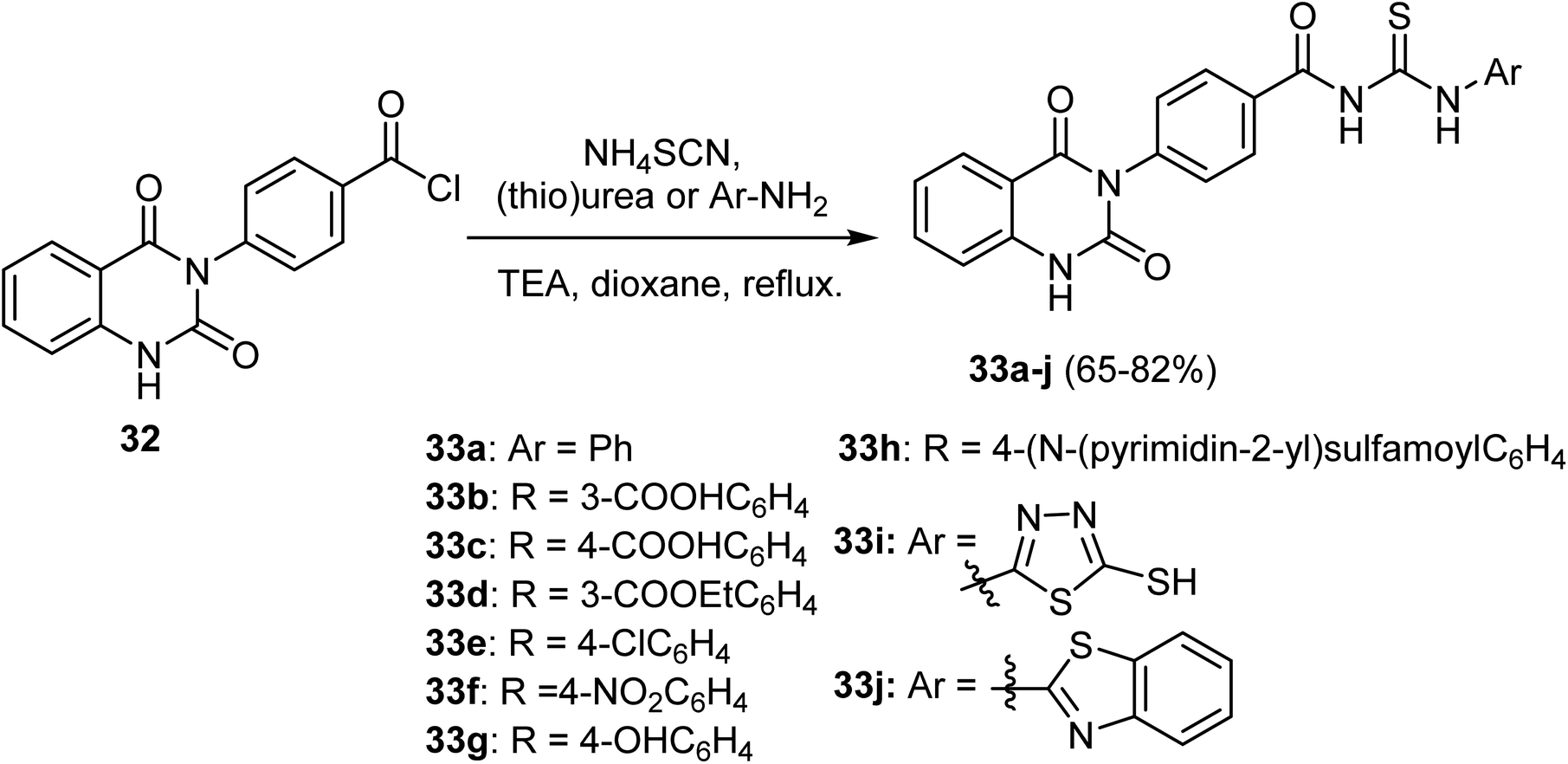 | ||
| Scheme 8 Synthetic pathway of 3-phenylquinazolin-2,4-dione derivatives 33a–j. | ||
Mortazavi et al. designed and synthesized a series of quinazoline-triazole molecular hybrids 38a–i as targeted anticancer agents. The reaction of 4-(prop-2-ynyloxy)benzaldehyde (35) with quinazolin-4-yl-hydrazine (34) in ethanol afforded 4-(2-(4-(prop-2-ynyloxy)benzylidene)-hydrazinyl)quinazoline 36. The reaction of 36 with the intermediate benzyl azide 37a–i at room temperature afforded the targeted compounds 38a–i (Scheme 9).60
 | ||
| Scheme 9 Synthesis of quinazoline triazole derivatives 38a–i. | ||
El-Hamaky et al. designed and synthesized a series of quinazoline-1,2,3-triazole hybrids 45a–r (in 74–92% yields) via a step-wise protocol starting with anthranilic acid (39) according to the described procedure in Scheme 10.61
 | ||
| Scheme 10 Quinazoline-1,2,3-triazole hybrids 45a–r. | ||
A series of 4-(3-1H-indazolyl)aminoquinazoline derivatives were described by Han et al. as shown in Scheme 11. Thus, protecting the NH group of indazole ring in 4-((1H-indazol-3-yl)amino)-quinazoline (46) with Boc moiety followed by hydrolysis of the ester function produced the quinazoline-2-carboxylic acid derivative 48. Acylchlorination of compound 48 with oxalyl chloride, then amidation with various amines, resulted in the formation of compounds 49a–51j in 27–43% yields.62
 | ||
| Scheme 11 Preparation of 4-(3-1H-indazolyl)amino quinazoline derivatives 49a–51j. | ||
The reaction of 4-(6-iodo-2-mercapto-4-oxoquinazolin-3(4H)-yl)benzenesulfonamide 52 with 2-chloro-N-substituted acetamides 53a–o in the presence of K2CO3 at room temperature, afforded a series of alkyl quinazolyl sulfides hybrids 54a–o in 60–91% yield (Scheme 12).20
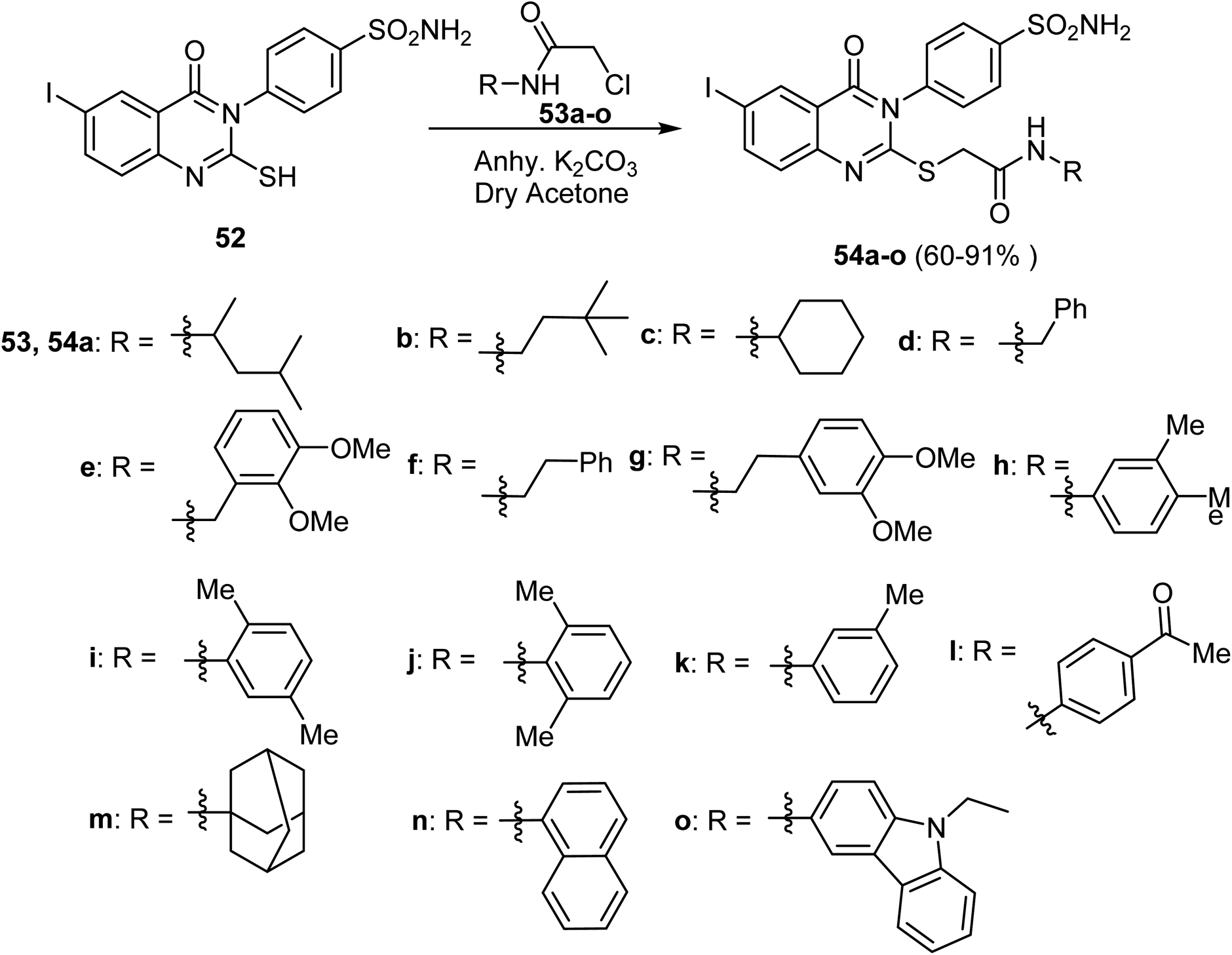 | ||
| Scheme 12 Synthesis of the quinazolinone derivatives 54a–o. | ||
Suzuki cross-coupling reaction of the boronic acid pinacol ester 55 with the bromoquinazoline derivative 56 afforded the corresponding cross-coupled product 57. The latter compound 57 underwent Buchwald coupling reaction with aryl(heteroaryl) halides to afford compounds 58a–k. Deprotections of the protecting groups of 58 resulted in the formation of the pyrido[2,3-b][1,4]oxazin-7-yl)quinazoline derivatives 59 and 60 in 42–93% yields (Scheme 13).63
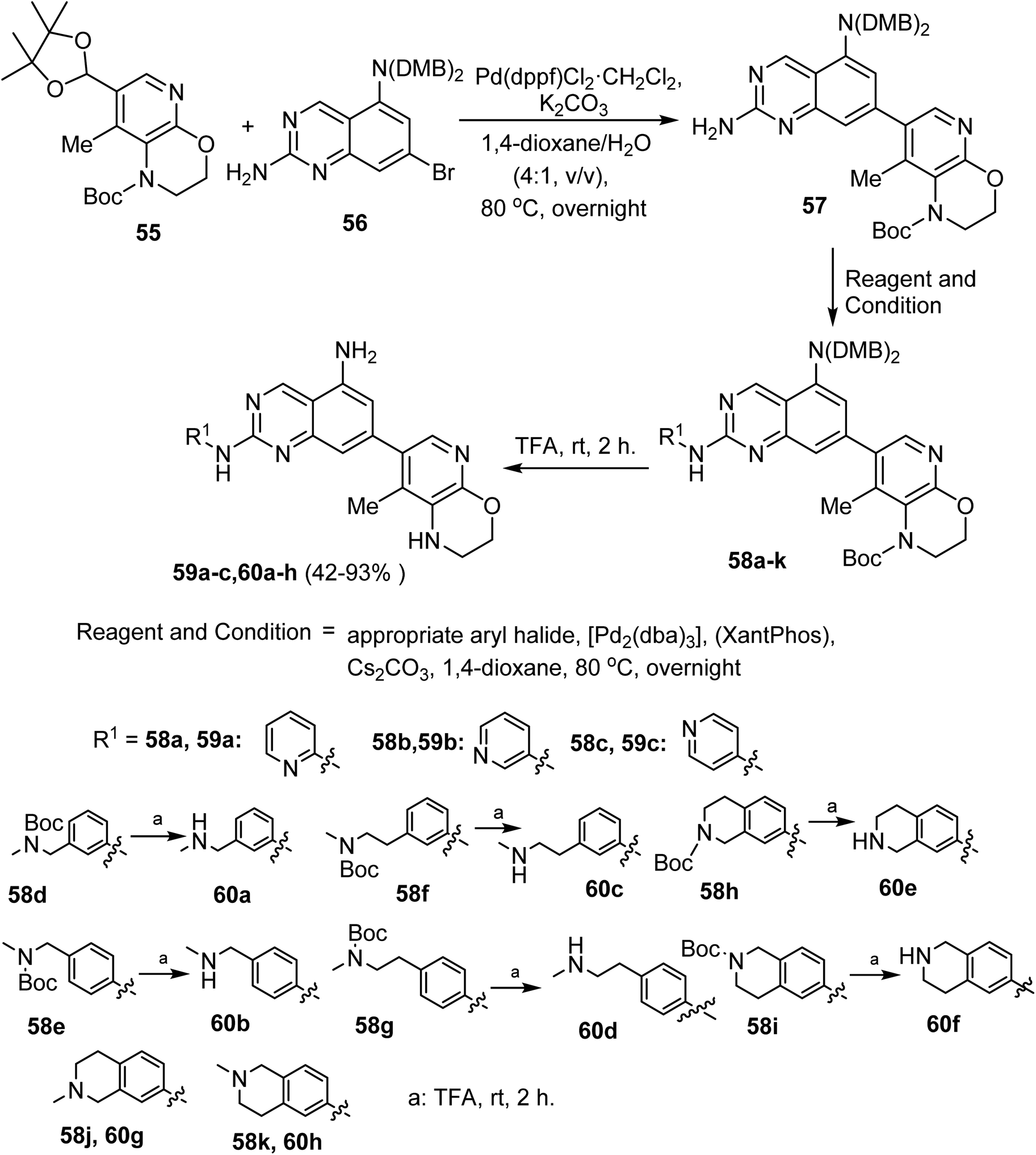 | ||
| Scheme 13 Synthesis of compounds 59a–c and 60a–h. | ||
Ahmed et al. designed and synthesized some quinazoline compounds aiming to discover new anticancer agents. Thus, the reaction of 6-bromo-2-mercapto-3-phenylquinazolin-4(3H)-one (61a) with ethyl 2-chlorooacetate in DMF afforded 62a, which was then heated with hydrazine hydrate in ethanol to afford the hydrazide derivative 63a. The condensation reaction of 63a and appropriate aromatic aldehydes gave the corresponding hydrazones 64a–d in 66–80% yields (Scheme 14).64
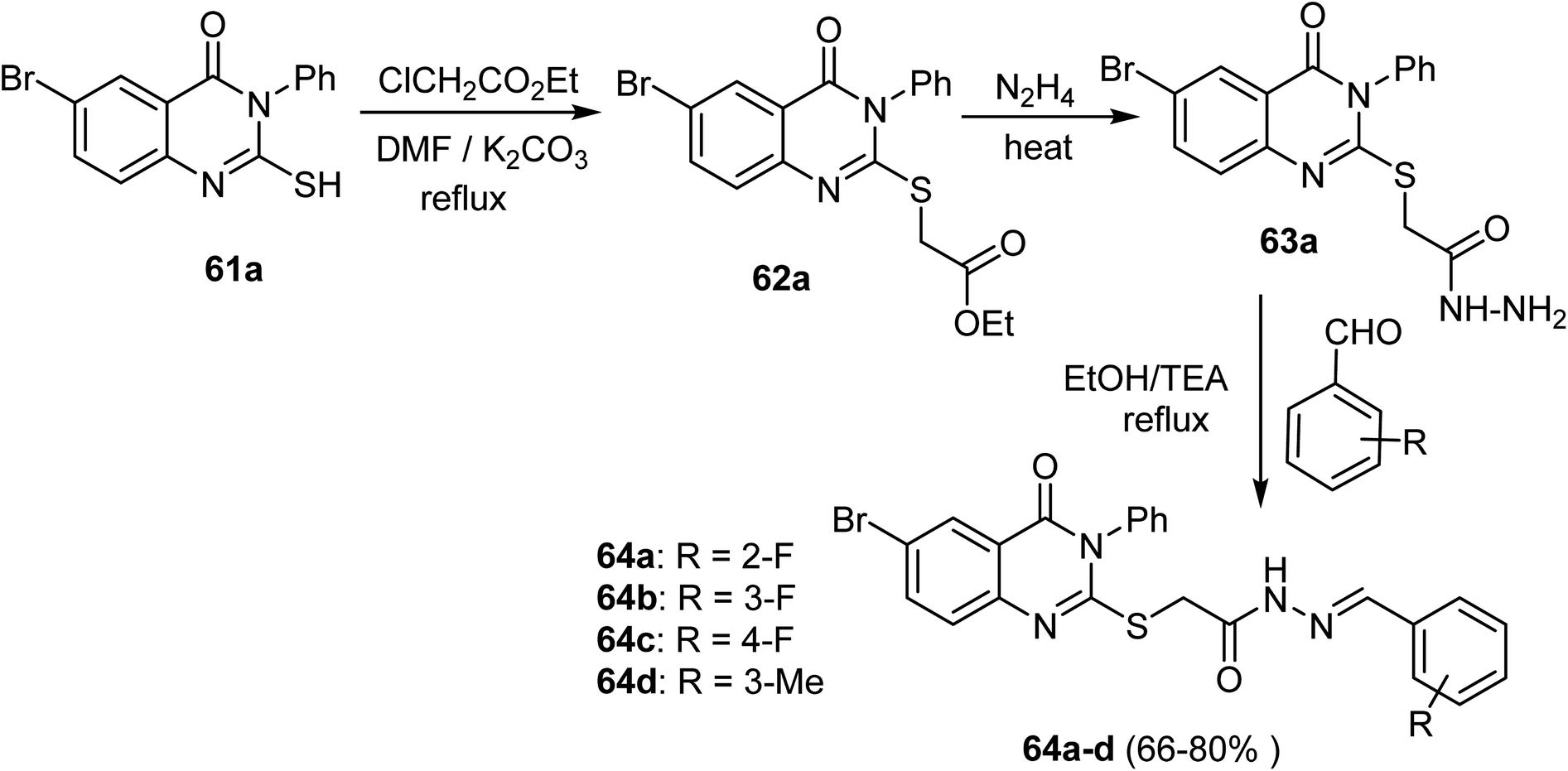 | ||
| Scheme 14 Synthetic route of compounds 64a–d. | ||
Treatment of 6-chloro-2-mercapto-3-phenylquinazolin-4(3H)-one (61b) with ethyl 2-chlorooacetate in DMF afforded compound 62b, which was then refluxed with hydrazine hydrate in ethanol to give hydrazide derivative 63b. Condensation of 63b with the appropriate aromatic aldehyde afforded the corresponding target hydrazones 64e–h in 74–82% yields (Scheme 15).65
 | ||
| Scheme 15 Synthesis of the target quinazolines 64e–h. | ||
Wang et al. described a synthetic route for the 4-amino-quinazoline derivatives 68a–o via Buchwald coupling of aryl amines with 4-chloro-6-iodoquinazoline (65) to give the intermediates 66a–o. Suzuki–Miyaura cross-coupling reaction with 2-aminopyridine-5-boronic pinacol acid ester 67 with 67 gave the corresponding products 68a–o as depicted in Scheme 16.66
 | ||
| Scheme 16 Preparation of 4-amino-quinazoline derivatives 68a–o. | ||
Heating the quinazoline derivatives 68a–o with the bromopyruvate esters 69 gave the target products 70a–s in 37–63% yields, as shown in (Scheme 17). Treatment of the intermediate 67 with alpha-bromoketones 71 afforded compounds 72, which upon coupling with intermediates 66 yielded the target products 6-(2-phenylimidazo[1,2-a]pyridin-6-yl)quinazolin-4-amine derivatives 73a–i in 55–73% yields.66
 | ||
| Scheme 17 Preparation of quinazoline derivatives 70a–s and 73a–i. | ||
Allam et al. described the synthesis of 6-bromo-2-(pyridin-3-yl)-4-substituted quinazolines 76a–c (in 55–63% yields) from the reaction of 4-chloro derivative 74 with 2-aminobenzothiazoles 75 in refluxing DMF as depicted in Scheme 18.67
 | ||
| Scheme 18 Synthesis of quinazoline derivatives 76a–c. | ||
A series of quinazoline-thioacetamide derivatives 79a–j and quinazoline-thioacetamides constituting sulfonamide tail 79k–q, were designed and synthesized by Ghorab et al. according to Scheme 19. Thus, 2-mercapto-3-phenylquinazolinone 77 reacted with 2-chloro-N-substituted acetamide 78 in dry acetone and K2CO3, afforded the 2-((4-oxo-3-phenyl-3,4-dihydro-quinazolin-2-yl)thio)acetamide derivatives 79a–q in 61–89% yields.19
 | ||
| Scheme 19 Synthesis of quinazoline-thioacetamide derivatives 79a–q. | ||
A series of 8-methoxy-2-trimethoxyphenyl-3-substituted quinazoline-4(3)-one derivative 81–84 were reported by Altamimi et al. and screened for antitumor activity. Thus, synthesis of the target compounds started with hydrazinolysis of 8-methoxy-2-trimethoxyphenyl-4H-benzo[d][1,3]oxazin-4-one (80) with hydrazine to afford 3-amino-quanazoline-4(3H)-one derivative (81) in 81% yield. Treatment of compound 81 with benzene sulphonyl chloride yielded the benzenesulfonylaminoquinazoline derivative 82 in 80% yield. Treatment of compound 80 with ammonia produced the benzamide derivative 83 in 92% yield. When compound 80 was refluxed with 3,4,5-trimethoxy aniline, it afforded the quinazoline-4(3H)-one 84 derivatives in 83% yields (Scheme 20).68
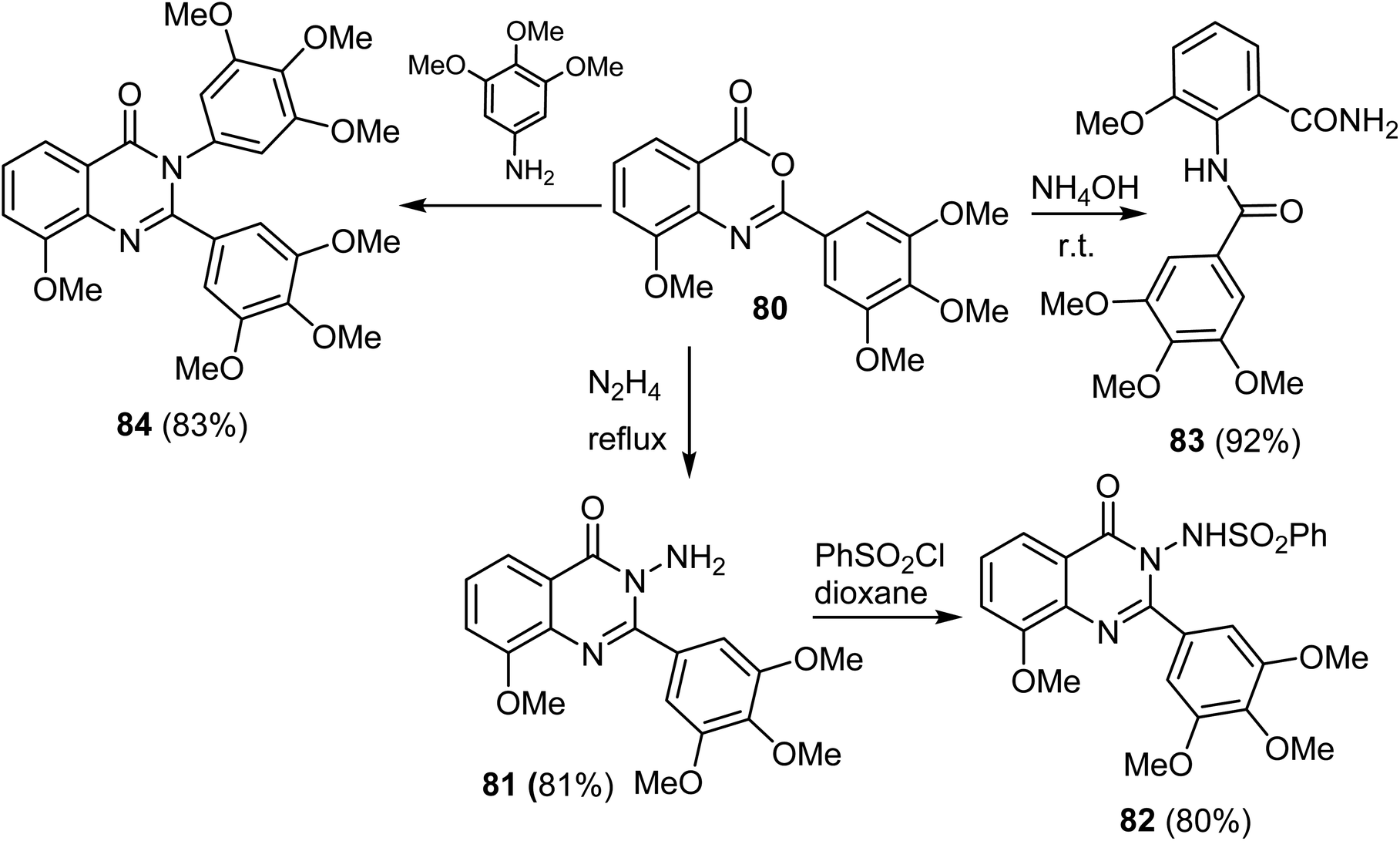 | ||
| Scheme 20 Synthesis of 8-methoxy-2-trimethoxyphenyl-3-substituted quinazoline-4(3)-one 81–84. | ||
The synthesis of the target quinazoline heterocycles 88 and 89 was achieved by the route depicted in Scheme 21. Thus, the reaction of 3-(4-hydroxyphenyl)propanoic acid (85) and ethyl 2-amino-5-chlorobenzoate (86) in xylene, in the presence of phosphorus trichloride under reflux condition furnished the corresponding ester 87 in 73% yield. Cyclocondensation of the benzoate ester 87 with hydrazine hydrate in n-butanol afforded the quinazolin-4-one scaffold 88 in 65% yield. Finally, condensation of N-amino derivative 88 with 4-hydroxy-3-methoxybenzaldehyde in refluxing n-butanol afforded the corresponding Schiff base 89 in 84% yield.69
 | ||
| Scheme 21 Synthesis of quinazolin-4-one scaffold 89. | ||
3.1 Anticancer activity of quinazoline-based derivatives
Compound 24f was designed, synthesized, and tested against tropomyosin receptor kinases (TRKs); the compound showed good inhibitory activity against TRKWT (IC50 = 0.55 nM), TRKG595R (IC50 = 25.1 nM) and TRKG667C (IC50 = 5.1 nM), compared to Larotrectinib with IC50 values 1.1 nM, 81.7 nM, and 51.1 nM respectively. The compound also demonstrated potent superior to Larotrectinib antiproliferative activity against a panel of Ba/F3 cell lines transformed with NTRK wild type and mutant fusions (IC50 = 10–200 nM).57The antiproliferative activity of another series of quinazoline derivatives was evaluated against ten human cancer cell lines A375, HeLa, Eca-109, H1975, MDA-MB-453, SW1353, Sgc7901, A549, HT-29, and MCF-7 compared with Gefitinib as reference drugs. Compound 31c demonstrated exhibited notable activity against A375, HeLa, Eca-109, H1975, MDA-MB-453, SW1353, Sgc7901, A549, HT-29, and MCF-7, with IC50 values (3.36, 2.48, 6.27, 7.66, 2.77, 2.01, 3.78, 1.04, 2.57 and 2.26 μM) respectively. It showed significant anticancer activities against the tested cancer cell lines compared to the reference drug Gefitinib. The ability of 31c to inhibit EGFR was evaluated in vitro, and this compound showed significant activity with IC50 values of 10.66 nM, compared to Gefitinib (IC50 = 25.42 nM). Docking studies supported the obtained results and demonstrated the ability of these derivatives to interact with EGFR active sites.58
The antiproliferative activity of a novel series of 3-phenylquinazolin-2,4(1H,3H)-diones was evaluated against HCT-116 (colorectal carcinoma) compared with Cabozantinib as a reference drug. Compound 33e demonstrated exhibited notable activity against HCT-116, with IC50 value 3.403 μM. It showed significant anticancer activities against the tested cancer cell line compared to the reference drug. The ability of 33e to inhibit both VEGFR-2/c-MetTKs was evaluated in vitro, and the compound showed significant activity with IC50 values 83 nM and 48 nM, respectively, compared to Cabozantinib with IC50 values 59 nM, 30 nM, respectively. Docking studies supported the obtained results and demonstrated the ability of this compound to interact with VEGFR-2/c-MetTKs active sites.59
A novel series of quinazoline derivatives with 1,2,3-triazole moiety was synthesized and then evaluated against the following cancer cell lines: AsPC-1 and Mia-Paca-2 (pancreatic cancer), HT-29 (colorectal cancer cells), MKN-45 (gastric cancer), EBC-1 (lung cancer) and K562 (leukemia). Compound 38c demonstrated exhibited notable activity against AsPc-1, EBC-1, MKN-45, Mia-Paca-2, HT-29, and K562, with IC50 values (15.3, 19.0, 22.0, 25.6, 21.0 and 31.5 μM) respectively. Compound 38c showed the highest inhibition activity against MET among the tested derivatives, which was also confirmed by the results of the western blot test. Compound 38c also inhibited PDGFRA activity by 58% at 10 μM concentration.60
The antiproliferative activity of another series of 4-(3-1H-indazolyl)amino quinazoline derivatives were evaluated against a human cancer cell line A549 compared with PF-3758309 as a reference drug. The structural–activity relationship (SAR) studies showed that a hydrophobic group at the 1′ or 2′-position of the ethylenediamine side chain of 51a–j (Scheme 11) enhanced their binding affinity. PAK4 inhibition test demonstrated that both 51c and 51d with 2′-methyl group displayed higher activity than that of 51a–b with 1′-methyl group. Larger hydrophobic substituents; i-propyl group at 2′-position of the ethylenediamine side chain of 51e–f resulted in a significant improvement of enzymatic activities. Compounds with 2′-large substituents e.g. phenyl (51g–h) or benzyl (51i–j) showed similar inhibitory potency to 51e–f. Thus, the IC50 values of 51e–j were determined against PAK4. The derivative 51e demonstrated notable activity against A549, with IC50 value (0.61 μM). It showed significant anticancer activities against the tested cancer cell line compared to reference drugs. The ability of 51e to inhibit PAK4 was evaluated in vitro, and this compound showed significant activity with an IC50 value 10 nM, compared to PF-3758309 (IC50 = 9 nM). Docking studies supported the results obtained and demonstrated the ability of these derivatives to interact with PAK4 active sites.70
The antiproliferative activity of compound 54n was evaluated in vitro against four human cancer cell lines HepG2, MCF-7, HCT-116, and A549, and Sorafenib and Erlotinib were used as reference drugs. Compound 54n displayed significant cytotoxicity against the four cell lines, HepG2, MCF-7, HCT-116, and A549, with IC50 values of 0.3425 μM, 0.0977 μM, 0.2000 μM, and 0.5134 μM, respectively. Compound 54n also showed good in vitro inhibition potency of EGFR and VEGFR-2 with IC50 values 0.0728 μM and 0.0523 μM, respectively. With almost similar potency as the reference drugs Sorafenib (IC50 = 0.1400 μM against VEGFR) and Erlotinib (IC50 = 0.2420 μM against EGFR). The ability of these novel derivatives to interact with EGFR and VEGFR-2 active sites and to inhibit their activities was also confirmed by docking studies.20 The SAR study showed that the impact of substituting the pyridine and two methoxy moieties, the urea, and amino linkers of 54 derivatives (Scheme 12) played a significant influence in the anticancer action. The impact of substituents of varied electronic and lipophilic natures on anticancer activity was also examined. It was found that compounds 54g, n (having neohexyl and naphthyl groups) showed the most significant anticancer activity compared to the other derivatives against MCF-7, HepG2, A549, and HCT116 cell lines. Both derivatives 54g, n, also displayed the highest inhibition of EGFRT790M and VEGFR-2 enzymes. Compound 8 has the hydrophobic 3,4-dimethoxyphenethyl group, showed more significant activity against the MCF-7 and HCT-116 cell lines than compound 54d (having benzyl group), 54e (having 2,3-dimethoxybenzyl moiety), and 54f (with phenethyl moiety). Furthermore, the naphthalene-containing compound 54n was more active against MCF-7, HCT116, and A549 cell lines than those having a carbazole or an adamantyl group.
Compound 45d was screened for anticancer activity in vitro against four human cancer cell lines, HeLa, HePG2, MCF7, and HCT116, using Doxorubicin as a reference drug, and also tested against a normal Caucasian fibroblast-like fetal lung cell line (WI-38) to determine their specificity. Compound 45d showed significant cytotoxic activity against the cell lines HeLa, HePG2, MCF7, and HCT116 with IC50 values of 2.57 μM, 5.96 μM, 6.41 μM, and 10.63 μM, respectively, compared to Doxorubicin with IC50 5.57 μM, 4.50 μM, 4.17 μM, and 5.23 μM, respectively. In addition, the IC50 value of 45d against WI-38 was 40.53 μM. The inhibition activity of this 45d against EGFR and VEGFR-2 was evaluated, with IC50 values 0.103 μM and 0.069 μM, respectively, with almost similar potency as the reference drugs Sorafenib (IC50 = 0.031 μM against VEGFR) and Erlotinib (IC50 = 0.049 μM against EGFR). The ability of these novel derivatives to interact with EGFR and VEGFR-2 active sites and to inhibit their activities was also confirmed by docking studies.61 The structure–activity correlation of the synthesized derivatives 45a–r (Scheme 10) against HeLa, HePG-2, MCF-7, and HCT-116 cell lines showed that quinazolinone derivative 45d having 4-methylphenyl moiety as an electron-donating group demonstrated the best inhibitory activity against the four cell lines. As shown in Scheme 10, for compounds 45 (R![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) H), replacing XMe with bulky groups (X = MeO, CF3, Br, or Cl) resulted in a decrease in the inhibitory activity against all cell lines. However, for compounds 45 (RCl), the data exhibited that the best activity was obtained for XBr against all cell lines; however, replacing XBr with (X = MeO, CF3, NO2, or Cl) resulted in moderate to weak activity against all cell lines.
H), replacing XMe with bulky groups (X = MeO, CF3, Br, or Cl) resulted in a decrease in the inhibitory activity against all cell lines. However, for compounds 45 (RCl), the data exhibited that the best activity was obtained for XBr against all cell lines; however, replacing XBr with (X = MeO, CF3, NO2, or Cl) resulted in moderate to weak activity against all cell lines.
A novel series of quinazoline derivatives with 2,5-diamine moiety was synthesized. The inhibition activity of these compounds against HPK1 was tested. Compound 60h showed good inhibition potency of HPK1 with IC50 value 2.7 nM.63
The antiproliferative activity of 64b was evaluated in vitro against two human cancer cell lines MCF-7 and HCT116, and Doxorubicin was used as a reference drug. Compound 64b displayed significant cytotoxicity against the two cell lines, MCF-7 and HCT116, with IC50 values of 4.43 μM and 8.23 μM, respectively. Compound 64b also showed good in vitro inhibition potency of VEGFR-2, and the percentage of inhibition of VEGFR-2 was 71.28%; Sorafenib was utilized as a positive control.64
The antiproliferative activity of 64f was evaluated in vitro against two human cancer cell lines MCF-7 and HCT116, and Doxorubicin was used as a reference drug. Compound 64f displayed significant cytotoxicity against the two cell lines, MCF-7 and HCT116, with IC50 values of 9.63 μM and 1.58 μM, respectively, compared to Doxorubicin with IC50 8.09 μM and 11.26 μM, respectively. Compound 64f also showed good in vitro inhibition potency of VEGFR-2 with IC50 value 3.19 μM, with almost similar potency as the reference drug Sorafenib (IC50 = 3.24 μM).65
Compound 73i was screened for anticancer activity in vitro against five human cancer cell lines HCC827, A549, SH-SY5Y, HEL, and MCF-7, using HS-173 as a reference drug, and also tested against normal cell line (MRC-5) to determine their specificity. Compound 73i showed significant cytotoxic activity against the cell lines HCC827, A549, SH-SY5Y, HEL and MCF-7with IC50 values of 0.09 μM, 0.18 μM, 0.37 μM, 0.19 μM, and 0.43 μM, respectively, compared to HS-173 with IC50 3.90 μM, 5.91 μM, 10.71 μM, 5.24 μM, and 4.36 μM, respectively. In addition, the IC50 value of 73i against MRC-5 was 1.98 μM. The inhibition activity of this derivative 73i against PI3Kα was evaluated, with IC50 value 1.94 nM with higher potency than the reference drug HS-173 (IC50 = 3.72 nM). The ability of 73i to interact with PI3Kα active sites and to inhibit their activity was also confirmed by docking studies.66
Compound 76a was screened for its antiproliferative activity in vitro against four human cancer cell lines A549, MCF-7, PC9, and HCC827, using Gefitinib as a reference drug, and also tested against normal cell line (WI38) to determine their specificity. The bioassay results disclosed that 76a had a promising cytotoxic activity with IC50 values of 178.34 μM, 2.49 μM, 1.05 μM, and 3.43 μM against A549, MCF-7, PC9 and HCC827, respectively, compared to Gefitinib with IC50 values of 34.389 μM, 4.972 μM, 1.36 μM, and 3.99 μM, respectively. In addition, the IC50 value of compound 76a against WI38 was 82.8 μM. The ability of 76a to inhibit EGFR was evaluated in vitro and showed a significant IC50 value of 0.096 μM, compared to Gefitinib (IC50 = 0.166 μM). Docking studies supported the obtained results and demonstrated the ability to interact with EGFR active sites.67
Synthesis of compound 79g and evaluation of its cytotoxic activity against MCF-7 was reported, using Doxorubicin as a reference drug. 79g exhibited notable cytotoxicity against tested cell line “MCF-7” with IC50 value of 38.42 μM, compared to Doxorubicin with IC50 value of 32.02 μM. The inhibitory effects of 79g against VEGFR were also significant with IC50 0.176 μM, compared to reference drug Sorafenib (IC50 0.042 μM). The ability of 79g to interact with VEGFR was also confirmed by docking studies.19
A series of trimethoxy quinazolines derivatives were designed, synthesized, and evaluated for antiproliferative activity using MTT assay against three cancer cell lines: HeLa, A549 and MDA, and Docetaxel was employed as a reference drug. Particularly, compound 83 exhibited potent cytotoxicity with IC50 values of 2.8 μM, 81 μM, and 0.79 μM against the tested cell lines HeLa, A549, and MDA, respectively, compared to the reference drugs Docetaxel with IC50 values of 9.65 μM, 10.8 μM, and 3.98 μM, respectively. The inhibitory activity of 83 against VEGFR and EGFR was evaluated, and Docetaxel was used as a reference drug. Compound 83 showed the most potent VEGFR and EGFR inhibitory activity, with IC50 values 98.1 nM and 106 nM, respectively, compared to the reference drugs Docetaxel (IC50 = 89.3 nM and 56.1 nM against VEGFR and EGFR, respectively). The ability of these novel derivatives to interact with EGFR and VEGFR-2 active sites and to inhibit their activities was also confirmed by docking studies (Table 2).68
| Entry | Structure | Kinase inhibition activity | Anticancer activity | Ref. | ||||||
|---|---|---|---|---|---|---|---|---|---|---|
| 7 | 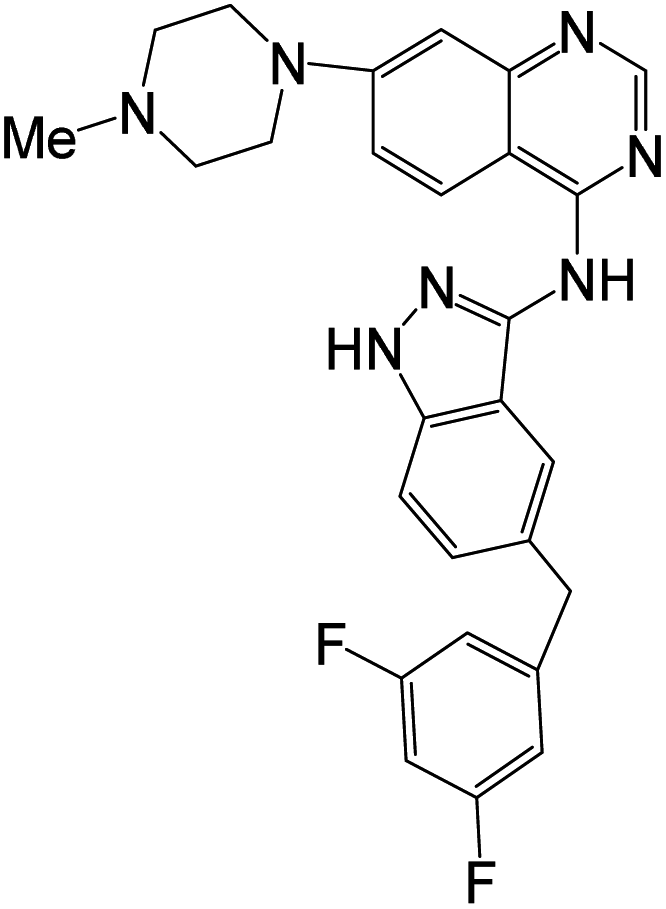 |
Enzymes | IC50[nM] | Cell lines | IC50 [nM] | 57 | ||||
| (24f) | Larotrectinib | (24f) | Larotrectinib | |||||||
| TRKAWT | 0.55 | 1.1 | Ba/F3-ETV6-TRKAWT | 14.6 | 9.5 | |||||
| TRKG595R | 25.1 | 81.7 | Ba/F3-ETV6-TRKBWT | 4.9 | 3.7 | |||||
| TRKG667C | 5.4 | 51.1 | Ba/F3-LMNA-TRKAG595R | 2886.4 | 205.0 | |||||
| Ba/F3-LMNA-TRKAG667C | 755.0 | 48.3 | ||||||||
| 8 |  |
Enzymes | IC50 [nM] | Cell lines | IC50 [μM] | 58 | ||||
| (31c) | Gefitinib | (31c) | Gefitinib | |||||||
| EGFR | 10.66 | 25.42 | A375 | 3.36 ± 0.24 | 8.46 ± 0.65 | |||||
| Hela | 2.48 ± 0.22 | 20.82 ± 1.43 | ||||||||
| Eca-109 | 6.27 ± 0.81 | 43.82 ± 5.66 | ||||||||
| H1975 | 7.66 ± 0.87 | 27.80 ± 2.09 | ||||||||
| MDA-MB-453 | 2.77 ± 0.16 | 21.55 ± 2.36 | ||||||||
| SW1353 | 2.01 ± 0.34 | 37.02 ± 4.21 | ||||||||
| Sgc7901 | 3.78 ± 0.92 | 22.30 ± 5.55 | ||||||||
| L02 | 21.03 ± 1.16 | 25.42 ± 1.65 | ||||||||
| A549 | 1.04 ± 0.32 | 12.08 ± 0.88 | ||||||||
| HT-29 | 2.57 ± 0.18 | 8.06 ± 0.53 | ||||||||
| MCF-7 | 2.26 ± 0.46 | 13.87 ± 0.68 | ||||||||
| 9 |  |
Enzymes | IC50 [nM] | Cell lines | IC50 [μM] | 59 | ||||
| (33e) | Cabozantinib | (33e) | Cabozantinib | |||||||
| VEGFR-2 | 83 ± 5 | 59 ± 3 | HCT116 | 3.403 ± 0.18 | 16.350 ± 0.86 | |||||
| c-Met | 48 ± 3 | 30 ± 2 | WI38 | 11.61 ± 0.69 | 44.71 ± 2.65 | |||||
| 10 |  |
Enzyme | Kinase inhib. (%) at 10 μM | Cell lines | IC50 [μM] | 60 | ||||
| (38c) | (38c) | Crizotinib | Cabozantinib | |||||||
| PDGFRA | 58 | AsPc-1 | 15.3 ± 2.3 | 2.45 ± 1.3 | 1.4 ± 0.1 | |||||
| EBC-1 | 19.0 ± 0.7 | 0.006 ± 0.001 | 0.059 ± 0.014 | |||||||
| MKN-45 | 22.0 ± 4.8 | 0.05 ± 0.003 | 1.04 ± 0.06 | |||||||
| Mia-Paca-2 | 25.6 ± 2.4 | 2.36 ± 1.2 | 3.8 ± 0.6 | |||||||
| HT-29 | 21.0 ± 4.7 | ND | 3.7 ± 0.67 | |||||||
| K562 | 31.5 ± 6.4 | ND | 4.0 ± 1.0 | |||||||
| 11 |  |
Enzymes | IC50 [nM] | Cell lines | IC50 [μM] | 70 | ||||
| (51e) | PF-3758309 | (51e) | PF-3758309 | |||||||
| PAK4 | 10 | 9 | A549 | 0.61 ± 0.03 | 0.67 ± 0.13 | |||||
| T293 | >10 | >10 | ||||||||
| Enzymes | Kinase inhibition (%) at 100 nM | |||||||||
| (51e) | PF-3758309 | |||||||||
| PAK4 | 73 | 81 | ||||||||
| 12 | 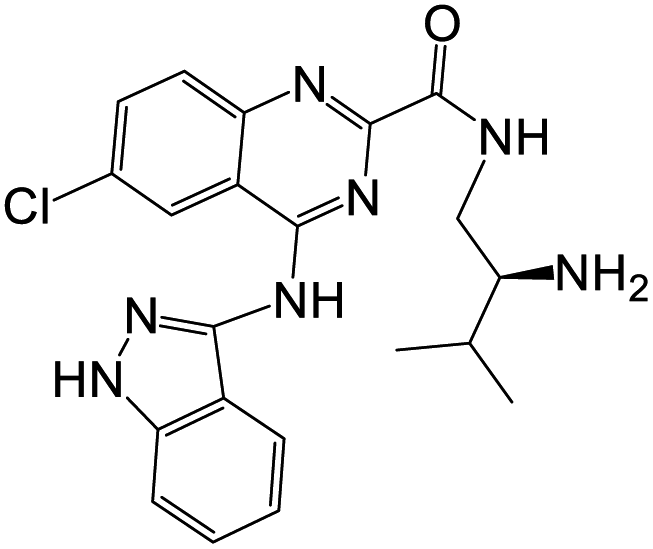 |
Enzymes | IC50 [μM] | Cell lines | IC50 [μM] | 20 | ||||
| (54n) | Sorafenib | Erlotinib | (54n) | Sorafenib | Erlotinib | |||||
| EGFR | 0.0728 ± 0.01 | — | 0.2420 ± 0.02 | HepG2 | 0.3425 ± 0.02 | 0.400 ± 0.03 | 0.773 ± 0.07 | |||
| MCF-7 | 0.0977 ± 0.01 | 0.404 ± 0.03 | 0.549 ± 0.05 | |||||||
| VEGFR | 0.0523 ± 0.01 | 0.1400 ± 0.01 | — | |||||||
| HCT-116 | 0.2000 ± 0.02 | 0.558 ± 0.05 | 0.820 ± 0.06 | |||||||
| A549 | 0.5134 ± 0.05 | 0.505 ± 0.05 | 0.1391 ± 0.01 | |||||||
| 13 |  |
Enzymes | IC50 [ μM] | Cell lines | IC50 [μM] | 61 | ||||
| (45d) | Erlotinib | Sorafenib | (45d) | DOX | ||||||
| EGFR | 0.103 ± 0.005 | 0.049 ± 0.002 | — | Hela | 2.57 ± 0.1 | 5.57 ± 0.4 | ||||
| HePG2 | 5.96 ± 0.3 | 4.50 ± 0.2 | ||||||||
| VEGFR | 0.069 ± 0.003 | — | 0.031 ± 0.001 | |||||||
| MCF7 | 6.41 ± 0.4 | 4.17 ± 0.2 | ||||||||
| HCT116 | 10.63 ± 0.8 | 5.23 ± 0.3 | ||||||||
| WI38 | 40.53 ± 2.2 | 6.72 ± 0.65 | ||||||||
| 14 |  |
Enzymes | IC50 [ nM] | Cell lines | IC50 [μM] | 63 | ||||
| (60h) | — | — | ||||||||
| HPK1 | 2.7 ± 2.2 | — | — | — | ||||||
| 15 |  |
Enzymes | Kinase inhibition (%) | Cell lines | IC50 [μM] | 64 | ||||
| (64b) | (64b) | DOX | ||||||||
| VEGFER | 71.28 | MCF-7 | 4.43 ± 0.006 | 8.09 ± 0.006 | ||||||
| HCT116 | 8.23 ± 0.009 | 11.26 ± 0.007 | ||||||||
| 16 |  |
Enzymes | IC50 [ μM] | Cell lines | IC50 [μM] | 65 | ||||
| (64f) | Sorafenib | (64f) | DOX | |||||||
| VEGFR | 3.19 | 3.24 | MCF-7 | 9.63 ± 0.016 | 8.09 ± 0.007 | |||||
| HCT116 | 1.58 ± 0.008 | 11.26 ± 0.009 | ||||||||
| 17 |  |
Enzymes | IC50 [ nM] | Cell lines | IC50 [μM] | 66 | ||||
| (73i) | HS-173 | (73i) | HS-173 | |||||||
| PI3Kα | 1.94 ± 0.66 | 3.72 ± 0.93 | HCC827 | 0.09 ± 0.01 | 3.90 ± 0.34 | |||||
| A549 | 0.18 ± 0.01 | 5.91 ± 0.19 | ||||||||
| SH-SY5Y | 0.37 ± 0.08 | 10.71 ± 1.92 | ||||||||
| HEL | 0.19 ± 0.01 | 5.24 ± 1.28 | ||||||||
| MCF-7 | 0.43 ± 0.04 | 4.36 ± 0.90 | ||||||||
| MRC-5 | 1.98 ± 0.89 | — | ||||||||
| 18 |  |
Enzymes | IC50 [ μM] | Cell lines | IC50 [μM] | 67 | ||||
| (76a) | Gefitinib | (76a) | Gefitinib | |||||||
| EGFR | 0.09 ± 0.00278 | 0.166 ± 0.00638 | A549 | 178.34 ± 8.9 | 4.389 ± 0.21 | |||||
| MCF-7 | 2.49 ± 0.12 | 4.972 ± 0.24 | ||||||||
| WI38 | 82.8 ± 4.14 | 34.95 ± 1.72 | ||||||||
| PC9 | 1.05 ± 0.02 | 1.36 ± 0.02 | ||||||||
| HCC827 | 3.43 ± 0.066 | 3.99 ± 0.07 | ||||||||
| 19 |  |
Enzymes | IC50 [ μM] | Cell lines | IC50 [μM] | 19 | ||||
| (79g) | Sorafenib | (79g) | Doxorubicin | |||||||
| VEGFR | 0.176 ± 0.007 | 0.042 ± 0.002 | MCF-7 | 38.42 ± 1.45 | 32.02 ± 0.06 | |||||
| 20 | 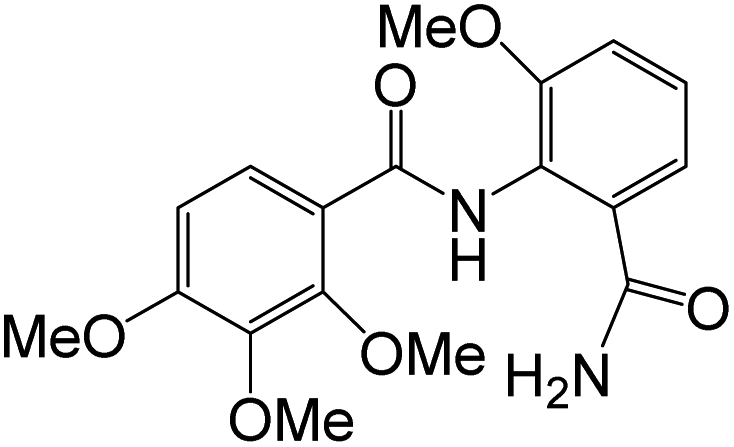 |
Enzymes | IC50 [ nM] | Cell lines | IC50 [μM] | 68 | ||||
| (83) | Docetaxel | (83) | Docetaxel | |||||||
| VEGFR2 | 98.1 ± 2.93 | 89.3 ± 2.67 | Hela | 2.8 ± 0.07 | 9.65 ± 0.2 | |||||
| EGFR | 106 ± 2.22 | 56.1 ± 1.17 | A549 | 81 ± 2.1 | 10.8 ± 0.23 | |||||
| MDA | 0.79 ± 0.04 | 3.98 ± 0.08 | ||||||||
4. Quinoxaline derivatives
A series of [1,2,4]triazolo[4,3-a]quinoxaline derivatives 94a–j were designed, synthesized, and biologically tested for their antiproliferative activities against two selected tumor cell lines, MCF-7 and HepG2. Thus, acetylation of p-aminobenzoic acid 90 using chloroacetyl chloride in DMF followed by thionyl chloride provided 4-(2-chloroacetamido)benzoyl chloride 91. Stirring of 91 with different amines in acetonitrile in the presence of TEA afforded the target intermediate 92a–j. Finally, reaction of potassium salts 93 with the intermediates 92a–j in dry DMF afforded the anticipated triazolo-quinoxalines 94a–j in 60–83% yields (Scheme 22).70 | ||
| Scheme 22 Preparation of triazolo quinoxalines 94a–j. | ||
Similarly, Alsaif et al. reported the synthesis of a new series of triazolo-quinoxaline hybrids derivatives, 94k–p, as VEGFR-2 inhibitors, as depicted in Scheme 23. Thus, heating the potassium salt 93 with the intermediates 92k–p in dry DMF in the presence of KI as a catalyst afforded the triazolo-quinoxaline derivatives 94k–p in 60–70% (Scheme 23).71
 | ||
| Scheme 23 Synthesis of triazoloquinoxaline-based derivatives, 94k–p. | ||
A series of quinoxaline-2(1H)-one derivatives were synthesized from the reaction of the potassium salt 96 with the appropriate 4-(2-chloroacetamido)-N-(substituted)benzamides 97 and 99a–c. The reaction occurred in dry DMF at reflux, in the presence of a catalytic amount of KI to yield the target quinoxaline derivatives 98 in 70% yield and 100a, b in 73–93% yield, respectively (Scheme 24).72
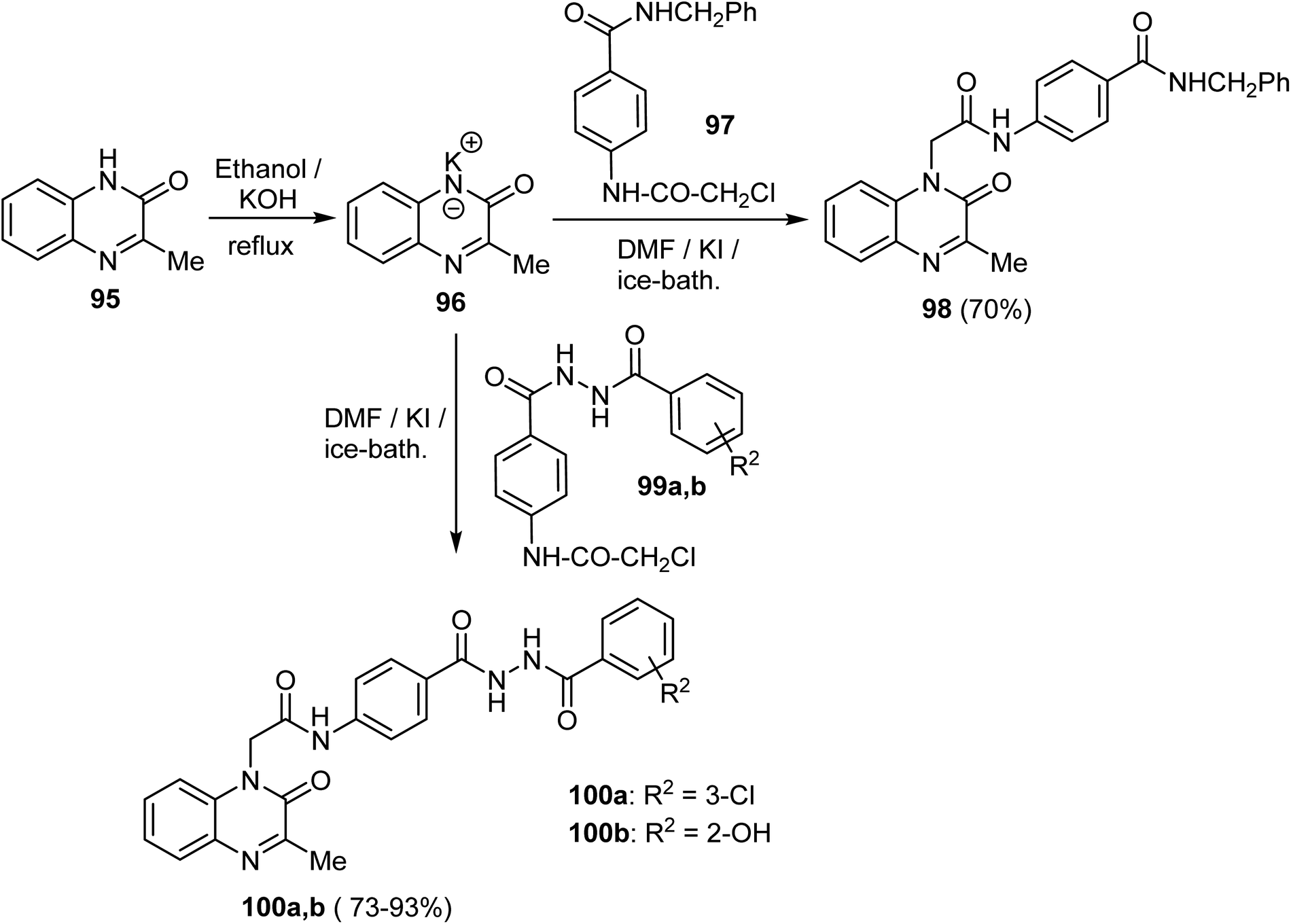 | ||
| Scheme 24 Synthesis of quinoxaline-2(1H)-one derivatives, 98, 100. | ||
Synthesis of a series of condensed bis([1,2,4]triazolo)[4,3-a:3′,4′-c]quinoxaline derivatives 102a–n were reported and investigated for their cytotoxic activities against HepG2 and MCF-7 and examined in vitro for their VEGFR-2 inhibitory activity. Thus, the synthetic pathway of the target structures 102a–n started with heating the potassium salt 101 with the intermediates 92. The was carried out in dry DMF using KI as a catalyst to furnish the triazolo-quinoxaline scaffolds 102a–n in 63–80% yields (Scheme 25).73
 | ||
| Scheme 25 Synthesis of quinoxaline scaffolds 102a–n. | ||
Alanazi et al. described a synthetic route to the bis([1,2,4]triazolo)[4,3-a:3′,4′-c]quinoxaline derivatives and examined them as VEGFR-2 inhibitors. Therefore, the bis([1,2,4]triazolo)[4,3-a:3′,4′-c]quinoxalin-3-ylthio)acetamido)-N-phenylbenzamide derivatives 102o–aa were obtained in 68–80% yield by refluxing the potassium salt 101 with the intermediates 92 in dry DMF in the presence of KI catalyst (Scheme 26).74
 | ||
| Scheme 26 Synthesis of bis([1,2,4]triazolo)[4,3-a:3′,4′-c]quinoxaline derivatives 102o–aa. | ||
Synthesis of a series of [1,2,4]triazolo[4,3-a]quinoxaline derivatives 104a–i was achieved by treatment of the potassium salt 103 with the intermediates 92 under typical reaction conditions as above to produce the derivatives 104a–i in 75–85% yields (Scheme 27).75
 | ||
| Scheme 27 Synthesis of [1,2,4]triazolo[4,3-a]quinoxalin-4-ylthio)acetamides 104a–i. | ||
The [1,2,4]triazolo[4,3-a]quinoxaline molecular hybrids 94q–v and 104k–p were reported as anticancer agents with potential effects against VEGFR-2, the synthesis of compounds 94q–v took place smoothly upon treatment of the potassium salt 93 with the chloroacetyl derivatives 92 under similar conditions as above to produce the products 94q–v in 55–70% yields. Analogously, the target triazolo[4,3-a]quinoxalin compounds 104k–p were produced in 55–70% yields by heating the potassium salt 103 with 92 in DMF in the presence of KI in a water-bath for 10 hours (Scheme 28).76
 | ||
| Scheme 28 Synthesis of triazolo[4,3-a]quinoxaline 94q–v and 104k–p. | ||
As illustrated in Scheme 29 the quinoxaline products 106a–d and 107a–c were obtained in satisfactory yields 50–60% by reacting the starting compound quinoxalin-2(1H)-one (105) with 4,4-(2-chloroacetamido)-N-substituted arylamide derivatives 92 and 99 in the presence of K2CO3 and catalytic quantity of KI (Scheme 29).77
 | ||
| Scheme 29 Synthesis of quinoxaline derivatives 106–107. | ||
In a similar protocol, a new series of 3-methylquinoxalin-2(1H)-one derivative 108a–j was constructed employing 3-methylquinoxalin-2(1H)-one 95. Thus, heating 95 with alcoholic KOH gave the corresponding potassium salt 96, which, upon treatment with the key intermediates 92 resulted in the production of the 2-oxoquinoxaline 108a–j in 70–76% (Scheme 30).78
 | ||
| Scheme 30 Synthesis of the 3-methylquinoxaline derivatives 108a–j. | ||
Under eco-friendly reaction conditions, synthesis of the benzoxazolyl quinoxalines 111a, b was accomplished as shown in Scheme 31. Thus, a mixture of substituted 2-aminophenol 109a, b and quinoxaline-2-carbaldehyde (110) was added to silica chloride (1 eq.), then the mixture was heated on a sand bath at 120 °C for 4 h to furnish the target (1,3-benzoxazol-2-yl)quinoxaline scaffolds 111a, b in 74–80% yields (Scheme 31).79
 | ||
| Scheme 31 Synthesis of (1,3-benzoxazol-2-yl)quinoxalines scaffolds 111a, b. | ||
Kumar et al. reported the synthesis of a library of the imidazo[1,2-a]quinoxaline derivatives as inhibitors of epidermal growth factor receptor (EGFR).80 As per Scheme 32, panel of imidazo[1,2-a]quinoxalines 113–115 were prepared via microwave-assisted ring-cyclization of 5-amino-1-(2-amino-phenyl)-1H-imidazole-4-carbonitrile (112) with variety of aromatic aldehydes in methanolic solution containing p-TSA (p-toluene sulphonic acid). The reaction resulted in the formation of the imidazo[1,2-a]quinoxaline-2-carbonitriles 113a–d in 70–93% yield and 4,5-dihydroimidazo[1,2-a]quinoxaline-2-carbonitriles 114a–k in 70–92%, respectively. Analogously, the reaction of compound 112 with aryl ketones furnished the dihydroimidazo[1,2-a]quinoxaline scaffolds 115a, c in 88–89% yields.80
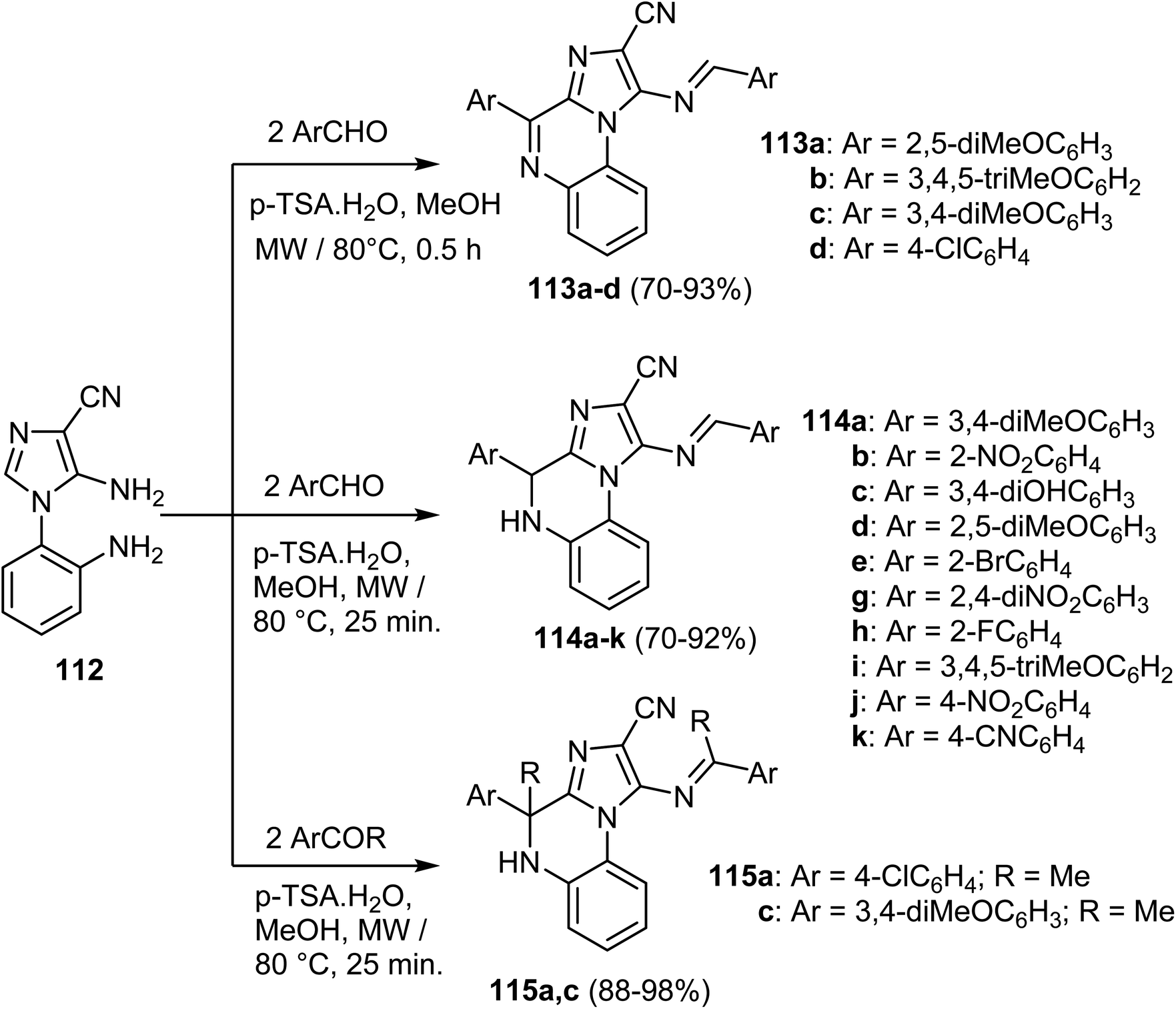 | ||
| Scheme 32 Synthesis of the imidazo[1,2-a]quinoxaline scaffolds 113–115. | ||
Alswah et al. reported the synthesis of a series of triazolo-quinoxaline-chalcone hybrids 119a–k as described in Scheme 33. Therefore, the fused chlorotriazoloquinoxaline 117 was produced in good yield by cyclizing hydrazinylquinoxaline 116 with excess triethylorthopropionate under heating. Next, the preparation of compound 118 involved dissolving 117 in acetonitrile, followed by heating with 4-aminoacetophenone under reflux using a catalytic quantity of TEA to produce the triazoloquinoxaline 118. Finally, condensation of compound 118 with aromatic aldehydes led to the construction of the substituted triazoloquinoxaline chalcone derivatives 119a–k in 11–86% yields.81
 | ||
| Scheme 33 Synthesis of the triazoloquinoxaline-chalcone derivatives 119a–k. | ||
Conjugated polymer nanoparticles (CPNs) consisting of fluorinated thiophene-quinoxaline type conjugated polymers were reported by Koralli et al. The conjugated polymers were used to create nano-precipitated and encapsulated aqueous CPNs. As shown in Scheme 34, the thiophene-quinoxaline based polymers with fluorine atoms were constructed by reaction of one equiv. of 3,4-difluorothiophene-2,5-diyl)bis(trimethylstannane) (124) with one equiv. of either 5,8-dibromo-6-fluoro-2,3-bis(3-(octyloxy)phenyl)quinoxaline (125a) or 5,8-dibromo-6,7-difluoro-2,3-bis(3-(octyloxy)phenyl)quinoxaline (125b) in dry toluene in the presence of tris(dibenzylideneacetone)dipalladium(0) [Pd2(dba3)] (0.02 equiv.) and tri(o-tolyl)phosphine [P(o-tol)3] to produce the polymeric material 126a, b (T2fQf and T2fQ2f).82
 | ||
| Scheme 34 Synthesis of thiophene-quinoxaline type conjugated polymers T2fQf and T2fQ2f. | ||
4.1 Anticancer activity of quinoxaline-based derivatives
The synthesized series of [1,2,4]triazolo[4,3-a]quinoxalin-4(5H)-one and [1,2,4]triazolo-[4,3-a]quinoxaline derivatives were also examined for their anticancer activity against two cancer cell lines MCF-7 and HepG-2 using Sorafenib and MTT bioassay method was employed. Compound 94d showed the most potent activity against tested cancer cell lines MCF-7 and HepG2 with IC50 values of 7.2 and 4.1 μM, respectively. The ability of this derivative 94d to inhibit VEGFR-2 was evaluated in vitro and showed a significant IC50 value of 3.4 nM when compared to Sorafenib, with an IC50 of 3.2 nM. Molecular docking studies showed that compound (24) had binding modes and interactions against VEGFR-2 like Sorafenib.70The antiproliferative activity of another series of quinoxaline-2(1H)-one derivatives 98 and 100b was evaluated against three human cancer cell lines HepG-2 (hepatocellular carcinoma), MCF-7 (breast cancer) and HCT-116 (colorectal carcinoma) compared with Doxorubicin and Sorafenib as reference drugs. The two derivatives, 98 and 100b demonstrated, exhibited notable activity against HepG-2, HCT-116, and MCF-7, with IC50 values (5.30 μM, 2.20 μM, and 5.50 μM for 98) and IC50 values (6.60 μM, 4.70 μM, and 6.90 μM for 100b) respectively. It showed significant anticancer activities against the tested cancer cell lines compared to reference drugs. The ability of 98 and 100b to inhibit VEGFR-2 was evaluated in vitro, and these two compounds showed significant activity with IC50 values 1.09 and 1.19 μM, respectively, compared to Sorafenib (IC50 = 1.27 μM). Docking studies supported the obtained results and demonstrated the ability of these derivatives to interact with VEGFR-2 active sites.72
Next, a series of bis([1,2,4]triazolo[4,3-a:3′,4′-c]quinoxaline) derivatives were evaluated for their cytotoxicity against MCF-7 and HepG2 cancer cell lines using MTT assay and Sorafenib as reference drug. Compound 102j showed an interesting anticancer activity against the tested cancer cell lines MCF-7 and HepG2 with IC50 values of 10.3 μM and 6.4 μM, respectively. The ability of this derivative 102j to inhibit VEGFR-2 was evaluated (in vitro) and showed a significant IC50 value of 3.7 nM, compared to Sorafenib (IC50 = 3.12 nM). The docking studies supported these results and demonstrated the ability of the 102j to interact with VEGFR-2 active sites.73
The triazoloquinoxaline-based molecular hybrid 94k was screened for its antiproliferative activity (in vitro) against two human cancer cell lines MCF-7 and HepG-2, using Sorafenib as a reference drug. The bioassay results disclosed that compound 94k had a promising cytotoxic activity with IC50 values of 6.2 μM and 4.9 μM against MCF-7 and HepG2, respectively, compared to Sorafenib with IC50 values of 3.53 and 2.18 μM, respectively. The ability of 94k to inhibit VEGFR-2 was evaluated (in vitro) and showed a significant IC50 value of 3.9 nM, compared to Sorafenib (IC50 = 3.13 nM). Docking studies supported the obtained results and demonstrated the ability to interact with VEGFR-2 active sites.71
The antiproliferative activity of 102h was evaluated (in vitro) against two human cancer cell lines HepG-2 and MCF-7, and Sorafenib was used as a reference drug. Compound 102h displayed significant cytotoxicity against the two cell lines, HepG-2 and MCF-7, with IC50 values of 3.3 μM and 4.4 μM, respectively. Compound 102h also showed good in vitro inhibition potency of VEGFR-2 with IC50 value 3.2 μM, with almost similar potency as the reference drug Sorafenib (IC50 = 3.1 μM). The ability of these novel derivatives to interact with VEGFR-2 active sites and to inhibit its activity was also confirmed by docking studies.74
Compound 104a showed an interesting anticancer activity with IC50 values 8.2 and 5.4 μM, respectively, against MCF-7 and HepG2. The ability of these derivatives to inhibit VEGFR-2 was evaluated (in vitro), and 104a showed a significant IC50 value of 3.4 nM compared to Sorafenib (IC50 = 3.12 nM). Molecular docking studies showed the binding mode and interaction of compound 30 against VEGFR-2.75
Compound 94q was synthesized and tested in vitro for its anticancer activity against two human cancer cell lines (MCF-7 and HepG2), and the results were compared to Sorafenib. 94q showed significant cytotoxic activity against both MCF-7 and HepG2 with IC50 values 5.8 μM and 4.3 μM, respectively, that were very close to Sorafenib drug (IC50 = 3.51 and 2.17 μM, respectively). The in vitro inhibitory activity of 94q against VEGFR-2 was evaluated and showed significant IC50 = 3.2 nM, compared to the reference Sorafenib (IC50 = 3.12 nM). Molecular docking studies also showed binding mode and interaction against VEGFR-2.76
Compound 107b was designed, synthesized, and screened in vitro for its anticancer activity against three cancer cell lines “HepG2, HCT-116, and MCF-7”. Compound 107b exhibited good cytotoxicity with (IC50 7.45, 3.04, and 7.85 μM) against HepG2, HCT-116, and MCF-7, respectively. The inhibitory effect of compound 32 against VEGFR-2 (in vitro) showed significant inhibitory activity against VEGFR-2 with an IC50 value of 0.493 μM, compared to Sorafenib (IC50 = 0.589 μM).77
Synthesis of 108e was achieved, and its antiproliferative activity was evaluated by MTT assay against two cancer cell lines, MCF-7 and HepG2, using Sorafenib as a reference drug. The bioassay results showed that 108e exhibited a high potent cytotoxicity with IC50 values of 2.7 and 2.1 μM against MCF-7 and HepG2, respectively. The in vitro inhibition activity of compound 108e against VEGFR-2 showed significant activities with an IC50 value of 2.6 nM, compared with Sorafenib (IC50 = 3.1 nM).78
Some benzoxazole-based quinoxaline heterocyclic hybrids were synthesized, and their cytotoxicity was evaluated using MTT bioassay against five cancer cell lines (MDA-MB-231, MCF-7, A549, KB, and HEK293). Compound 111b was the most potent cytotoxic compound against all the tested cancer cell lines MDA-MB-231, MCF-7, A549, KB, and HEK293 with IC50 values of 0.53, 0.50, 0.82, 0.90 and 14.39 μM, respectively. In addition, the inhibitory effect of 111b against tyrosine kinase exhibited potent activity with IC50 = 0.10 μΜ.79
Compound 113b was tested for anticancer activity in vitro against A549 “non-small cell lung cancer”, HCT-116 “colorectal carcinoma”, MDA-MB-231 “breast cancer” and H1975 “Gefitinib-resistant non-small cell lung cancer”. Compound 113b showed significant anticancer activity against the tested cancer cell lines A549, HCT-116, MDA-MB-231, and H1975 with IC50 values of 2.7, 5.1, 4.1 and 3.65 μM, respectively. The inhibitory effects of 113b against EGFR were determined and exhibited promising inhibitory activity. Docking studies also supported the results and demonstrated the ability to interact with EGFR active sites.80
Some triazoloquinoxaline-chalcone molecular hybrids were synthesized and screened in vitro for their anticancer activity against three human cancer cell lines: MCF-7, HCT-116, and HEPG-2, and compared with Doxorubicin. As an example, 119g showed the most potent cytotoxic activity with IC50 values of 1.65, 3.61, 8.58 μM, against MCF-7, HCT-116, and HEPG-2, respectively. The inhibitory effects of compound 119g against EGFR TK were significant, with an IC50 value of 0.039 μM, compared to the reference drug Staurosporine (IC50 = 0.054 μM). The molecular docking studies showed binding modes with the EGFR TK.81
The study of Koralli et al. assessed the biological activity of thiophene-quinoxaline-based conjugated polymer nanoparticles (CPNs) in breast cancer cells (T-47D, MDA-MB-231) and healthy epithelial cells (MCF10A). Nanoprecipitated T2fqf CPNs labeled 80% of MDA-MB-231 cells and 30% of T-47D cells but showed low uptake in MCF10A (<0.15%). Nanoparticles of T2fqf (1.59 mg mL−1) significantly showed the growth of MDA-MB-231 cells and increased late apoptosis tenfold after four days. The encapsulated T2fqf and T2fqf CPNs, on the other hand, didn't kill cancer cells very much. However, at doses as high as 4 mg mL−1, the encapsulated T2fqf caused MCF10A cells to die. Healthy cells tolerated nanoprecipitated T2fqf well at low dosages, while it preferentially induced apoptosis in cancer cells. Nanoparticle of T2fqf may be able to work as a selective theragnostic agent, especially in the treatment of triple-negative breast cancer (Table 3).82
| Entry | Structure | Kinase inhibition activity | Anticancer activity | Ref. | ||||
|---|---|---|---|---|---|---|---|---|
| 21 |  |
Enzymes | IC50 [nM] | Cell lines | IC50 [μM] | 70 | ||
| (94d) | Sorafenib | (94d) | Sorafenib | |||||
| VEGFR-2 | 3.4 ± 0.3 | 3.2 ± 0.1 | MCF-7 | 7.2 ± 0.6 | 3.51 ± 0.21 | |||
| HepG-2 | 4.1 ± 0.4 | 2.17 ± 0.13 | ||||||
| 22 | 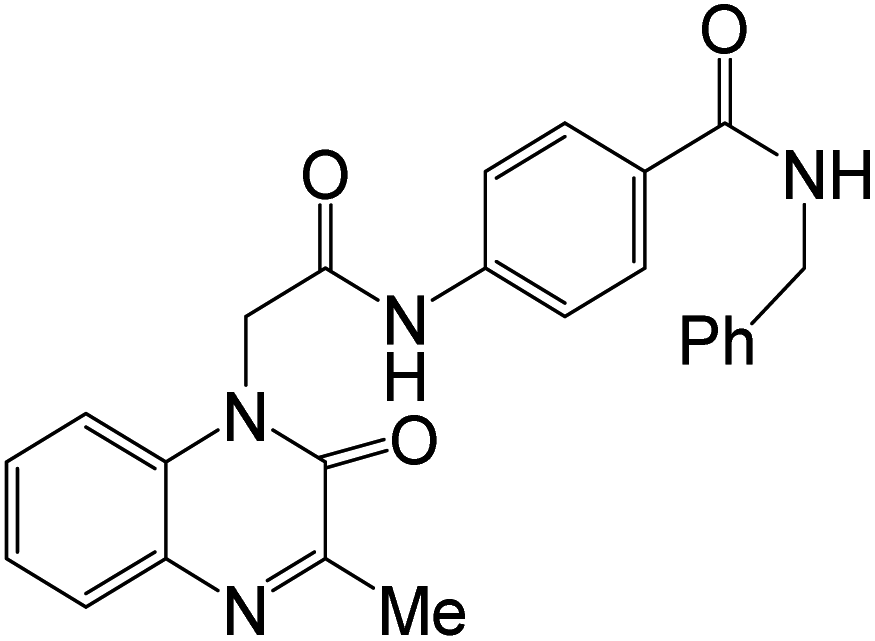 |
Enzymes | IC50 [μM] | Cell lines | IC50 [μM] | 72 | ||
| (98) | Sorafenib | (98) | Doxorubicin | |||||
| VEGFR-2 | 1.09 | 1.27 | HepG-2 | 5.30 ± 0.21 | 8.28 ± 0.33 | |||
| HCT-116 | 2.20 ± 0.08 | 9.63 ± 0.39 | ||||||
| MCF-7 | 5.50 ± 0.22 | 7.67 ± 0.31 | ||||||
| VERO | 11.82 ± 0.14 | — | ||||||
| 23 |  |
Enzymes | IC50 [μM] | Cell lines | IC50 [μM] | 72 | ||
| (100b) | Sorafenib | (100b) | Doxorubicin | |||||
| VEGFR-2 | 1.19 | 1.27 | HepG-2 | 6.60 ± 0.26 | 8.28 ± 0.33 | |||
| HCT-116 | 4.70 ± 1.88 | 9.63 ± 0.39 | ||||||
| MCF-7 | 6.90 ± 0.28 | 7.67 ± 0. 31 | ||||||
| VERO | 22.12 ± 0.24 | — | ||||||
| 24 | 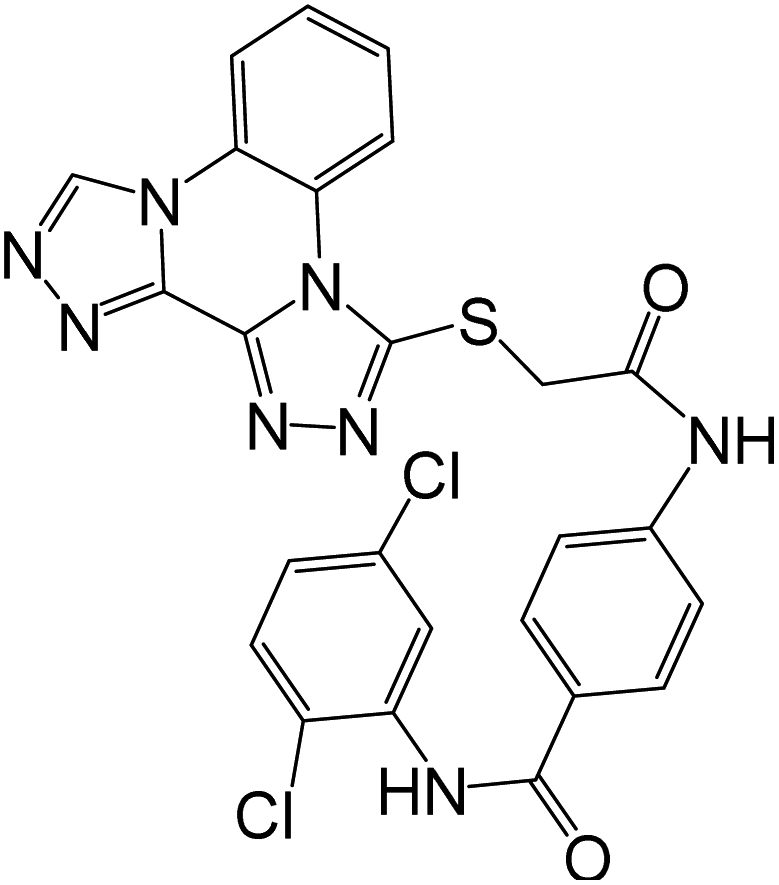 |
Enzymes | IC50 [μM] | Cell lines | IC50 [μM] | 73 | ||
| (102j) | Sorafenib | (102j) | Sorafenib | |||||
| VEGFR-2 | 3.7 | 3.12 | MCF-7 | 10.3 ± 0.8 | 3.51 ± 0.22 | |||
| HepG-2 | 6.4 ± 0.5 | 2.17 ± 0.14 | ||||||
| 25 |  |
Enzymes | IC50 [nM] | Cell lines | IC50 [μM] | 71 | ||
| (94k) | Sorafenib | (94k) | Sorafenib | |||||
| VEGFR-2 | 3.9 | 3.13 | MCF-7 | 6.2 | 3.53 | |||
| HepG-2 | 4.9 | 2.18 | ||||||
| 26 |  |
Enzymes | IC50 [μM] | Cell lines | IC50 [μM] | 74 | ||
| (102v) | Sorafenib | (102h) | Sorafenib | |||||
| VEGFR-2 | 3.2 | 3.1 | HepG-2 | 3.3 | 2.17 | |||
| MCF-7 | 4.4 | 3.43 | ||||||
| 27 |  |
Enzymes | IC50 [nM] | Cell lines | IC50 [μM] | 75 | ||
| (104a) | Sorafenib | (104a) | Sorafenib | |||||
| VEGFR-2 | 3.4 | 3.12 | MCF-7 | 8.2 | 3.51 | |||
| HepG-2 | 5.4 | 2.17 | ||||||
| Normal hepatocytes | 31.34 | 16.55 | ||||||
| 28 |  |
Enzymes | IC50 [nM] | Cell lines | IC50 [μM] | 76 | ||
| (94q) | Sorafenib | (94q) | Sorafenib | |||||
| VEGFR-2 | 3.2 ± 0.4 | 3.12 ± 0.3 | MCF-7 | 5.8 ± 0.7 | 3.51 ± 0.2 | |||
| HepG-2 | 4.3 ± 0.5 | 2.17 ± 0.7 | ||||||
| 29 |  |
Enzymes | IC50 [μM] | Cell lines | IC50 [μM] | 77 | ||
| (107b) | Sorafenib | (107b) | Sorafenib | |||||
| VEGFR2 | 0.493 | 0.589 | HepG-2 | 7.45 ± 0.30 | 7.31 ± 0.29 | |||
| HCT-116 | 3.04 ± 0.12 | 9.40 ± 0.38 | ||||||
| MCF-7 | 7.85 ± 0.31 | 7.21 ± 0.29 | ||||||
| 30 | 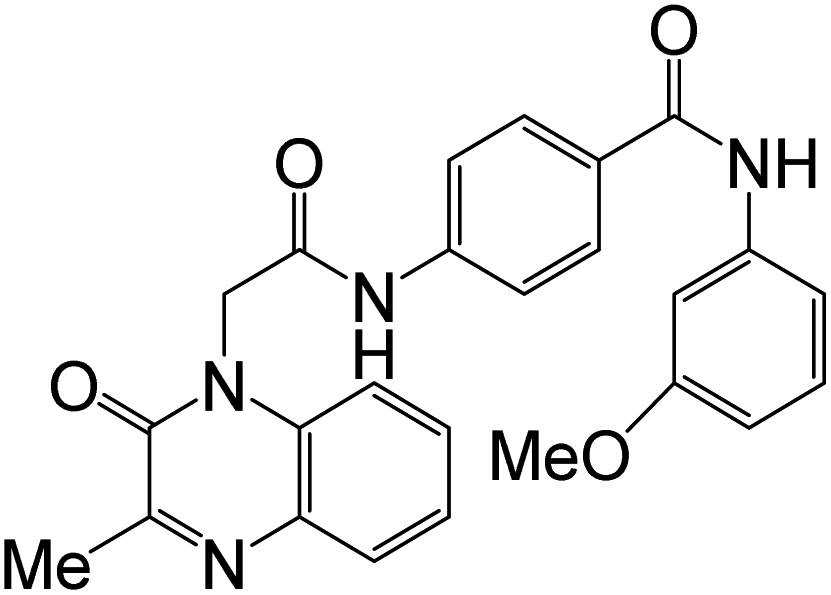 |
Enzymes | IC50 [nM] | Cell line | IC50 [μM] | 78 | ||
| (108e) | Sorafenib | (108e) | Sorafenib | |||||
| VEGFR-2 | 2.6 | 3.1 | MCF-7 | 2.7 | 3.4 | |||
| HepG-2 | 2.1 | 2.2 | ||||||
| 31 |  |
Enzymes | IC50 [μM] | Cell lines | IC50 [μM] | 79 | ||
| (111b) | Sorafenib | (111b) | Paclitaxel | |||||
| Tyrosine kinase | 0.10 ± 0.16 is it correct | MDA-MB-231 | 0.53 ± 0.02 | 0.3 ± 0.02 | ||||
| MCF-7 | 0.50 ± 0.08 | ___ | ||||||
| A549 | 0.82 ± 0.20 | ___ | ||||||
| KB | 0.90 ± 0.05 | ___ | ||||||
| HEK293 | 14.39 ± 0.28 | ___ | ||||||
| 32 |  |
Enzymes | IC50 [nM] | Cell lines | IC50 [μM] | 80 | ||
| (113b) | Erlotinib | (113b) | Erlotinib | |||||
| EGFR | 211.22 ± 0.027 is it correct | A549 | 2.7 ± 0.032 | 4.56 ± 0.019 | ||||
| HCT-116 | 5.1 ± 0.029 | 2.98 ± 0.023 | ||||||
| MDA-MB-231 | 4.1 ± 0.031 | 3.33 ± 0.018 | ||||||
| H1975 | 3.65 | ___ | ||||||
| 33 |  |
Enzymes | IC50 [μM] | Cell lines | IC50 [μM] | 81 | ||
| (119g) | Staurosporine | (119g) | Doxorubicin | |||||
| EGFR | 0.039 ± 0.16 | 0.054 ± 0.10 | HCT-116 | 3.61 ± 0.18 | 1.55 ± 0.03 | |||
| MCF-7 | 1.65 ± 0.13 | 0.27 ± 0.08 | ||||||
| HepG-2 | 8.58 ± 0.06 | 0.22 ± 0.01 | ||||||
| 34 |  |
— | Nanoparticles of T2fqf (1.59 mg mL−1) significantly showed the growth of MDA-MB-231 cells and increased late apoptosis tenfold after four days | 82 | ||||
5. Cinnoline derivatives
As illustrated in Scheme 35, Nazmy et al. reported an effective one-pot multicomponent reaction for synthesizing the functionalized cinnolines. The anticipated tetrahydrocinnoline-4-carbonitrile derivatives 130 were thus produced in 88–94% yields by reacting cyano-pyridazinone-carboxylates 127 with aromatic aldehydes 128 and nitromethane 129 in dioxane/piperidine under controlled microwave heating at 100 °C for 7–20 min.36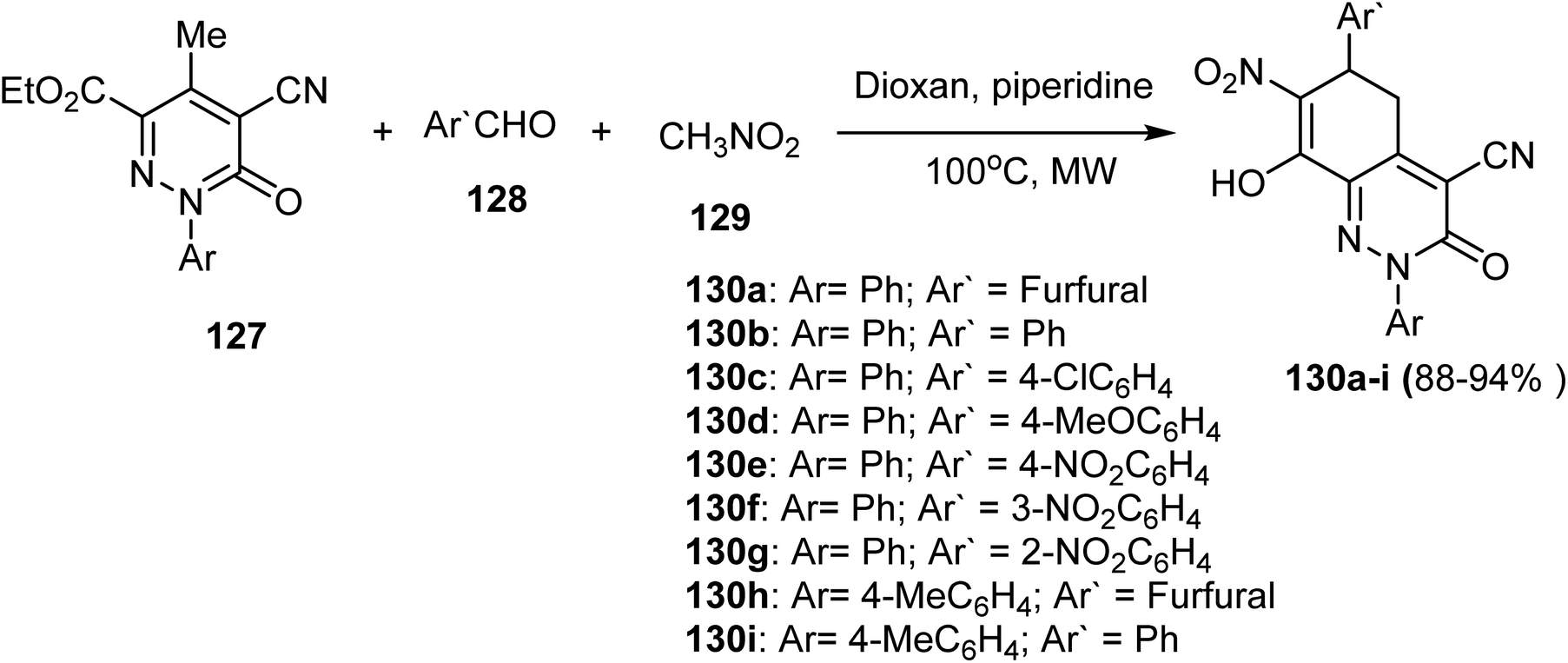 | ||
| Scheme 35 Synthesis of densely functionalized cinnoline 130a–i derivatives. | ||
Tian et al. have screened a series of cinnoline derivatives for anticancer activity, targeting PI3K, a key pathway that dominates cancer cell proliferation and survival. Several of these cinnoline-derived compounds were strongly inhibitory against PI3K enzymes, several in the nanomolar range of inhibition. Among the synthesized compounds, 134q exhibited the most potent antiproliferative activity with an IC50 of 0.264, 2.04, and 1.14 μM against U87MG, HeLa, and HL60 cells, respectively. This compound exhibited a potent inhibitory activity of PI3K/Akt pathway based on decreased phosphorylation levels and could represent a lead compound for further development as PI3K inhibitor for cancer therapy (Scheme 36).83
 | ||
| Scheme 36 Synthesis of the cinnoline derivatives 134a–r. | ||
El-Dhaibi et al. described access to products of the cinnoline-4-carbonitrile derivative 138–139, employing very simple substrates. Thus, 2-(2-nitrophenyl)-2-(3-oxoisoindolin-1-yl)acetonitrile 137 was first synthesized from the reaction between 2-cyanobenzaldehyde 135 and 2-(2-nitrophenyl)acetonitrile 136, followed by cyclization and subsequent rearrangement through a one-pot process mechanism in methanolic solution containing TEA as outlined in Scheme 37. Then, the isoindolin-1-one derivative 137 was treated with a solution of 5% KOH in MeOH under reflux for 30 minutes to furnish the anticipated products methyl 2-(4-cyanocinnolin-3-yl)benzoate (138) along with its corresponding acid 139 in 60% and 34% yields, respectively.84
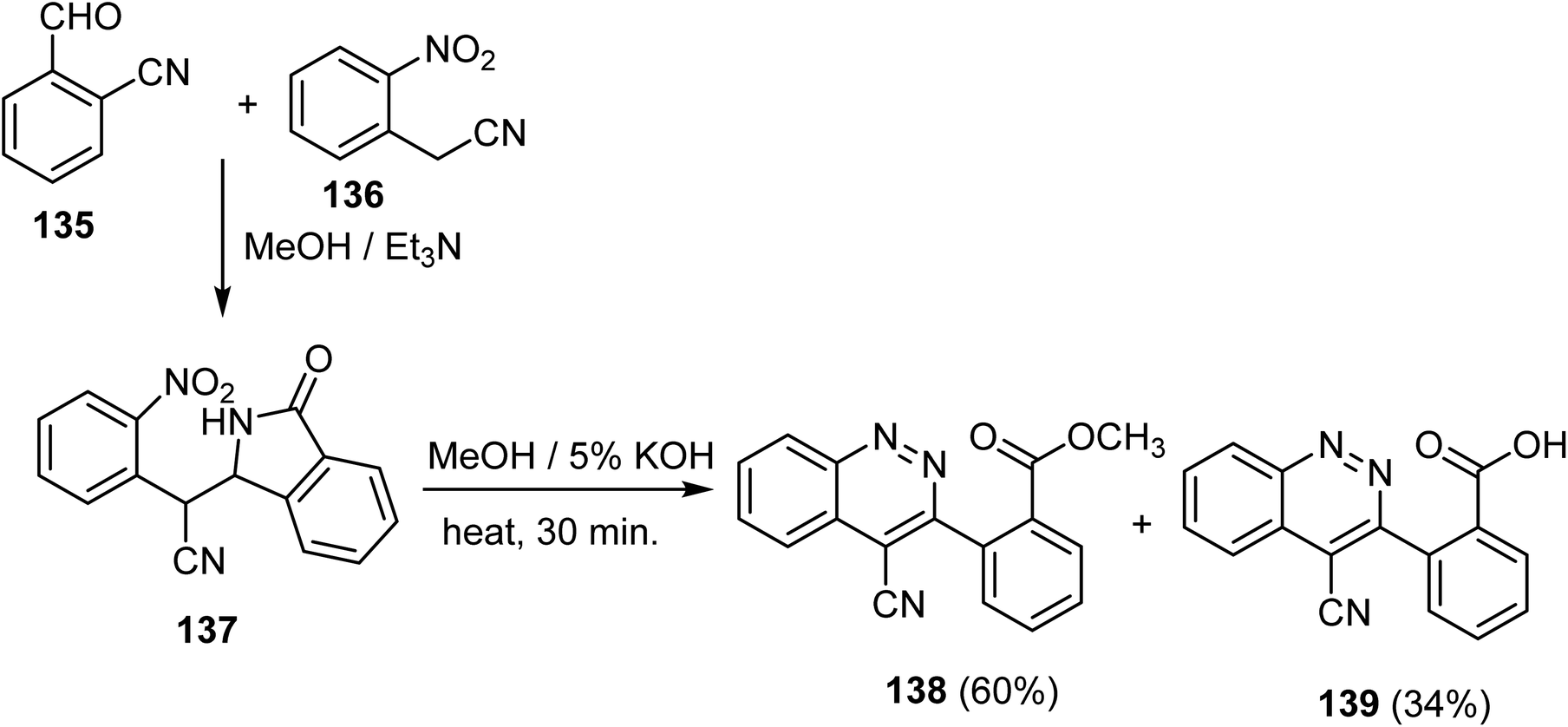 | ||
| Scheme 37 Synthesis of the cinnoline derivatives 14 and 15. | ||
An efficient approach towards synthesizing 6-aryl-4-azidocinnolines 142 was developed by Danilkina et al., starting from 3-phenyltriaz-1-ene derivatives 140. Thus, Richter cyclization of the triazenes 140a–g using HBr (20 equiv.) in acetone gave the corresponding 4-bromo-3-pentyl-6-arylcinnoline 141a–g in a good yield. Subsequent nucleophilic substitution of bromine atom by an azido group using sodium azide for the 4-bromo-cinnoline derivatives 141a–g proceeded smoothly, to provide the 6-aryl(heteroaryl)-4-azidocinnoline derivatives 142a–g in high yields 69–91% (Scheme 38). Finally, using copper(II) sulfate/sodium ascorbate catalytic system, the reaction of azidocinnolines 142a–g both with terminal aromatic alkynes bearing EWG, EDG, and aliphatic alkynes 143a–g in the mixture of THF/H2O resulted in the corresponding 4-triazolylcinnolines 144a–g mostly in good outcome (28–86%).85
 | ||
| Scheme 38 Synthesis of the 6-aryl-4-triazolylcinnolines 144a–g. | ||
5.1 Anticancer activity of cinnoline-based derivatives
Evaluation of the cytotoxic activity of compound 130b against two human cancer cell lines (RPMI-8226 and LOX IM VI) and WI-38 (normal cell line) was reported. Compound 130b exhibited notable cytotoxicity against tested cell line “RPMI-8226 and LOX IM VI” with IC50 values of 17.12 μg mL−1 and 12.32 μg mL−1, respectively, and the IC50 value for the WI-38 cell line was 23.62 μg mL−1. The values of IC50 for 130b against the normal and malignant cell lines confirmed its specificity and selectivity.36Tian et al., in their study, explored cinnoline derivatives as potential anticancer agents by targeting the PI3K pathway, which is essential in cancer cell proliferation and survival. Various cinnoline-based compounds exhibited potent inhibitory effects on PI3K enzymes, with many achieving nanomolar potencies. Compound 134q showed the highest antiproliferative activity across multiple cancer cell lines, with notable efficacy against U87MG, HeLa, and HL60 cells (IC50 values of 0.264 μM, 2.04 μM, and 1.14 μM, respectively). This compound effectively inhibited the PI3K/Akt pathway by reducing phosphorylation levels, thus demonstrating potential as a leading structure for further development as a PI3K inhibitor in cancer therapy (Table 4).83
| Entry | Structure | Kinase inhibition activity | Anticancer activity | Ref. | ||
|---|---|---|---|---|---|---|
| 36 |  |
— | Cell lines | IC50 [μg mL−1] | 36 | |
| 130b | Staurosporine | |||||
| RPMI-8226 | 17.12 ± 1.31 | 24.97 ± 1.47 | ||||
| LOX IM VI | 12.32 ± 0.75 | 8.45 ± 0.42 | ||||
| WI38 | 23.62 ± 8.57 | 14.21 ± 0.34 | ||||
| 37 |  |
Enzymes | IC50 [nM] | Cell lines | IC50 [μM] | 83 |
| 134q | ||||||
| PI3K | 0.65 nM | U87MG | 0.264 | |||
| HeLa | 2.04 | |||||
| HepG2 | 16.6 | |||||
| MCF-7 | 31.1 | |||||
| HL60 | 1.14 | |||||
6. Conclusion
In conclusion, phthalazine, quinazoline, quinoxaline, and cinnoline derivatives have been shown to have considerable anticancer activity. Their unique structural properties, together with their ability to interact with a variety of oncogenic targets, propel these compounds to the forefront of new chemotherapeutic development. These compounds not only inhibit important proteins kinases, but they also have multi-targeted effects, which may lead to more effective and long-lasting cancer treatments. Regarding the physio-chemical properties, compounds were investigated for their ADME pharmacokinetics and drug-likeness properties. As illustrated in the citing references whenever it applied, most active compounds exhibited obeyable values following Lipinsiki's rule of five of “molecular weight, number of rotatable bonds, H-bond donor, and acceptors along with a number of violations”. The knowledge gained from biological findings and synthetic development provides intriguing avenues for future research. The increasing need for specific and effective anticancer therapeutics ensures a wide field of study for these heterocycles with promising potential to address the limitations of presently existing cancer therapies. This will be important to guide the development of next-generation anticancer drugs. Increasing understanding of their molecular mechanisms will be critical for the development of next-generation anticancer drugs.Data availability
All data associated with this manuscript will be available upon reasonable request from the corresponding authors.Conflicts of interest
The authors declare that they have no financial or personal interests.Acknowledgements
Dr Mohamed S. Nafie appreciates to the Seed Research Project No. (24021440154) funded by the Research and Graduate Studies at the University of Sharjah, United Arab Emirates. Also, he acknowledges the electronic library sources at the University of Sharjah, providing full access to published papers and searching on Chemistry databases. Dr Sherif Ashraf Fahmy acknowledges the financial support and sponsorship from the Alexander von Humboldt Foundation, Germany.References
- R. J. Klement, Global Transitions, 2024, 6, 45–65 CrossRef.
- D. C. Joshi, A. Sharma, S. Prasad, K. Singh, M. Kumar, K. Sherawat, H. S. Tuli and M. Gupta, Discover Oncol., 2024, 15, 342 Search PubMed.
- K. Taruneshwar Jha, A. Shome, Chahat and P. A. Chawla, Bioorg. Chem., 2023, 138, 106680 CrossRef CAS PubMed.
- J. Bariwal, V. Kumar, Y. Dong and R. I. Mahato, Med. Res. Rev., 2019, 39, 1137–1204 CrossRef CAS PubMed.
- A. M. Fahim, E. H. I. Ismael and H. E. M. Tolan, Polycyclic Aromat. Compd., 2023, 1–42 Search PubMed.
- R. Pal, G. S. P. Matada, G. Teli, M. Saha and R. Patel, Bioorg. Chem., 2024, 152, 107696 CrossRef CAS.
- R. J. Obaid, N. Naeem, E. U. Mughal, M. M. Al-Rooqi, A. Sadiq, R. S. Jassas, Z. Moussa and S. A. Ahmed, RSC Adv., 2022, 12, 19764–19855 RSC.
- A. Mushtaq, P. Wu and M. M. Naseer, Pharmacol. Ther., 2024, 254, 108579 CrossRef CAS PubMed.
- P. Taslimi, F. Türkan, K. Turhan, H. S. Karaman, Z. Turgut and İ. Gulcin, J. Heterocycl. Chem., 2020, 57, 3116–3125 CrossRef CAS.
- O. Mammoliti, K. Jansen, S. El Bkassiny, A. Palisse, N. Triballeau, D. Bucher, B. Allart, A. Jaunet, G. Tricarico, M. De Wachter, C. Menet, J. Blanc, V. Letfus, R. Rupčić, M. Šmehil, T. Poljak, B. Coornaert, K. Sonck, I. Duys, L. Waeckel, L. Lecru, F. Marsais, C. Jagerschmidt, M. Auberval, P. Pujuguet, L. Oste, M. Borgonovi, E. Wakselman, T. Christophe, N. Houvenaghel, M. Jans, B. Heckmann, L. Sanière and R. Brys, J. Med. Chem., 2021, 64, 14557–14586 Search PubMed.
- R. Samala, S. K. Nukala, N. S. Thirukovela, G. Dasari and S. Bandari, Polycyclic Aromat. Compd., 2023, 43, 9175–9192 Search PubMed.
- J. Sangshetti, S. K. Pathan, R. Patil, S. Akber Ansari, S. Chhajed, R. Arote and D. B. Shinde, Bioorg. Med. Chem., 2019, 27, 3979–3997 CrossRef CAS.
- M. M. Khalifa, A. A. Al-Karmalawy, E. B. Elkaeed, M. S. Nafie, M. A. Tantawy, I. H. Eissa and H. A. Mahdy, J. Enzyme Inhib. Med. Chem., 2022, 37, 299–314 CrossRef CAS.
- K. Budipramana and F. Sangande, Chem. Biol. Drug Des., 2024, 103, e14534 CrossRef CAS PubMed.
- S. Zaib and I. Khan, Bioorg. Chem., 2020, 105, 104425 CrossRef CAS PubMed.
- S. Zare, L. Emami, Z. Faghih, F. Zargari, Z. Faghih and S. Khabnadideh, Sci. Rep., 2023, 13, 14461 CrossRef CAS PubMed.
- S. N. Murthy Boddapati, H. Babu Bollikolla, K. Geetha Bhavani, H. Singh Saini, N. Ramesh and S. Babu Jonnalagadda, Arabian J. Chem., 2023, 16, 105190 CrossRef CAS.
- I. A. Bala, A. M. Asiri and R. M. El-Shishtawy, Med. Chem. Res., 2024, 33(12), 2372–2419 CrossRef CAS.
- W. M. Ghorab and M. M. Ghorab, J. Mol. Struct., 2024, 1317, 139060 Search PubMed.
- M. M. Ghorab, A. M. Soliman, K. El-Adl and N. S. Hanafy, Bioorg. Chem., 2023, 140, 106791 CrossRef CAS PubMed.
- S. Hu, C. Jiang, M. Gao, D. Zhang, N. Yao, J. Zhang and Q. Jin, J. Mol. Struct., 2024, 1299, 137224 CrossRef CAS.
- F. H. Al-Ostoot, S. Salah and S. A. Khanum, Cancer Invest., 2024, 42, 559–604 CrossRef CAS PubMed.
- J. Dhuguru and O. A. Ghoneim, Molecules, 2022, 27, 2294 CrossRef CAS.
- S. K. Suthar, N. S. Chundawat, G. P. Singh, J. M. Padrón and Y. K. Jhala, Eur. J. Med. Chem. Rep., 2022, 5, 100040 Search PubMed.
- G. Yashwantrao and S. Saha, Org. Chem. Front., 2021, 8, 2820–2862 Search PubMed.
- M. G. Salem, S. A. Abu El-ata, E. H. Elsayed, S. N. Mali, H. A. Alshwyeh, G. Almaimani, R. A. Almaimani, H. A. Almasmoum, N. Altwaijry, E. Al-Olayan, E. M. Saied and M. F. Youssef, RSC Adv., 2023, 13, 33080–33095 Search PubMed.
- V. Montero, M. Montana, M. Carré and P. Vanelle, Eur. J. Med. Chem., 2024, 271, 116360 CrossRef CAS.
- N. Bhusare and M. Kumar, Oncol. Res., 2024, 32, 849–875 Search PubMed.
- M. A. Ismail, M. S. Abusaif, M. S. A. El-Gaby, Y. A. Ammar and A. Ragab, RSC Adv., 2023, 13, 12589–12608 Search PubMed.
- D. Zeleke and T. Damena, Results Chem., 2024, 7, 101283 CrossRef CAS.
- S. K. Suthar, N. S. Chundawat, G. P. Singh, J. M. Padrón and Y. K. Jhala, Eur. J. Med. Chem. Rep., 2022, 5, 100040 CAS.
- P. Saini, K. Kumar, S. Meena, D. K. Mahawar, A. Dandia, K. L. Ameta and V. Parewa, in N-Heterocycles, ed. K. L. Ameta, R. Kant, A. Penoni, A. Maspero and L. Scapinello, Springer Nature Singapore, Singapore, 2022, pp. 331–354 Search PubMed.
- J. R. Yerrabelly, M. B. Bommagani, H. Yerrabelly, S. C. Mullaguri and R. K. Kancha, J. Heterocycl. Chem., 2024, 61, 958–970 CrossRef CAS.
- H. M. Al-Matar, K. M. Dawood and W. M. Tohamy, RSC Adv., 2018, 8, 34459–34467 RSC.
- B. Kumaraswamy, K. Hemalatha, R. Pal, G. S. P. Matada, K. R. Hosamani, I. Aayishamma and N. V. S. S. Aishwarya, Eur. J. Med. Chem., 2024, 275, 116561 CrossRef CAS PubMed.
- M. H. Nazmy, R. A. Mekheimer, M. E. Shoman, M. Abo-Elsebaa, M. Abd-Elmonem and K. U. Sadek, Bioorg. Chem., 2020, 101, 103932 CrossRef CAS PubMed.
- L. Zhang, C. Cheng, J. Li, L. Wang, A. A. Chumanevich, D. C. Porter, A. Mindich, S. Gorbunova, I. B. Roninson, M. Chen and C. McInnes, J. Med. Chem., 2022, 65, 3420–3433 CrossRef CAS PubMed.
- M. Zabihi, R. Lotfi, A.-M. Yousefi and D. Bashash, J. Cancer Res. Clin. Oncol., 2023, 149, 1585–1606 CrossRef CAS PubMed.
- M. Szumilak and A. Stanczak, Molecules, 2019, 24, 2271 CrossRef CAS PubMed.
- R. K. Tonk, S. Bawa and D. Kumar, Mini-Rev. Org. Chem., 2025, 22(2), 162–176 CrossRef.
- M. M. Kandeel, A. M. Kamal, B. H. Naguib and M. S. A. Hassan, Anticancer Agents Med. Chem., 2018, 18(8), 1208–1217 CrossRef CAS PubMed.
- R. Pal, G. Teli, G. S. P. Matada and P. S. Dhiwar, J. Mol. Struct., 2023, 1291, 136021 CrossRef CAS.
- F. Sangande, E. Julianti and D. H. Tjahjono, Int. J. Mol. Sci., 2020, 21, 7779 CrossRef CAS PubMed.
- A. Doostmohammadi, H. Jooya, K. Ghorbanian, S. Gohari and M. Dadashpour, Cell Commun. Signaling, 2024, 22, 228 CrossRef PubMed.
- A. A. Abbas, T. A. Farghaly and K. M. Dawood, RSC Adv., 2024, 14, 19752–19779 RSC.
- K. M. Dawood, M. A. Raslan, A. A. Abbas, B. E. Mohamed and M. S. Nafie, Anticancer Agents Med. Chem., 2023, 23(3), 328–345 CrossRef CAS PubMed.
- D. M. Mohamed, N. A. Kheder, M. Sharaky, M. S. Nafie, K. M. Dawood and A. A. Abbas, RSC Adv., 2024, 14, 24992–25006 RSC.
- M. E. Salem, E. M. Mahrous, E. A. Ragab, M. S. Nafie and K. M. Dawood, ACS Omega, 2023, 8, 35359–35369 CrossRef CAS PubMed.
- F. M. Thabet, K. M. Dawood, E. A. Ragab, M. S. Nafie and A. A. Abbas, RSC Adv., 2022, 12, 23644–23660 RSC.
- K. El-Adl, M. K. Ibrahim, F. Khedr, H. S. Abulkhair and I. H. Eissa, Arch. Pharm., 2022, 355, 2100278 CrossRef CAS PubMed.
- F. Khedr, M. Ibrahim, I. H. Eissa, H. S. Abulkhair and K. El-Adl, Arch. Pharm., 2021, 354, 2100201 CrossRef CAS PubMed.
- H. H. Bayoumi, M. K. Ibrahim, M. A. Dahab, F. Khedr and K. El-Adl, RSC Adv., 2024, 14, 21668–21681 RSC.
- S. M. Emam, S. M. E. Rayes, I. A. I. Ali, H. A. Soliman and M. S. Nafie, BMC Chem., 2023, 17, 90 CrossRef CAS PubMed.
- U. Boda, V. Guguloth, S. Mood and H. Guguloth, Russ. J. Org. Chem., 2023, 59, 1064–1070 CrossRef CAS.
- C. A. Lipinski, Drug Discovery Today: Technol., 2004, 1, 337–341 CrossRef CAS PubMed.
- R. Patil, S. Das, A. Stanley, L. Yadav, A. Sudhakar and A. K. Varma, PLoS One, 2010, 5, e12029 CrossRef PubMed.
- Y. Xu, W. Zhao, X. Zhang, X. Yu, Y. Chen, Z. Wang, Y. Chu, X. Zhu and P. Zhang, Bioorg. Med. Chem., 2024, 99, 117608 CrossRef CAS PubMed.
- Y. Liu, Q. Ma, X. Kong, X. Huo, Z. Dong, Y. Ma, K. Yang, W. Niu and K. Zhang, Bioorg. Chem., 2024, 143, 107087 Search PubMed.
- A. Hassan, A. M. Mosallam, A. O. A. Ibrahim, M. Badr and A. H. Abdelmonsef, Sci. Rep., 2023, 13, 18567 Search PubMed.
- M. Mortazavi, M. Eskandari, F. Moosavi, T. Damghani, M. Khoshneviszadeh, S. Pirhadi, L. Saso, N. Edraki and O. Firuzi, Sci. Rep., 2023, 13, 14685 CrossRef CAS PubMed.
- N. F. M. El Hamaky, A. Hamdi, W. A. Bayoumi, A. A. Elgazar and M. N. A. Nasr, Bioorg. Chem., 2024, 148, 107437 CrossRef CAS PubMed.
- W. Han, Y. Yang, F. Yu, Q. Li, A. Liu, W. Xu, J. Li and X. Xue, Bioorg. Med. Chem., 2023, 95, 117501 CrossRef CAS PubMed.
- H. Shi, H. Tang, Y. Li, D. Chen, T. Liu, Y. Chen, X. Wang, L. Chen, Y. Wang, H. Xie and B. Xiong, Eur. J. Med. Chem., 2023, 248, 115064 CrossRef CAS PubMed.
- M. F. Ahmed, A. S. Khalifa and E. M. Eed, Russ. J. Bioorg. Chem., 2022, 48, 739–748 CrossRef CAS.
- M. F. Ahmed, E. Y. Santali and R. I. Alsantali, Russ. J. Gen. Chem., 2022, 92, 2047–2057 CrossRef CAS.
- M. Li, D. Wang, Q. Li, F. Luo, T. Zhong, H. Wu, L. Xiong, M. Yuan, M. Su and Y. Fan, Int. J. Mol. Sci., 2023, 24, 6851 CrossRef CAS.
- H. A. Allam, E. E. Aly, A. K. B. A. W. Farouk, A. M. El Kerdawy, E. Rashwan and S. E. S. Abbass, Bioorg. Chem., 2020, 98, 103726 CrossRef CAS PubMed.
- A. S. Altamimi, A. S. El-Azab, S. G. Abdelhamid, M. A. Alamri, A. H. Bayoumi, S. M. Alqahtani, A. B. Alabbas, A. I. Altharawi, M. A. Alossaimi and M. A. Mohamed, Molecules, 2021, 26, 2992 Search PubMed.
- R. M. Borik and M. A. Hussein, Curr. Pharm. Biotechnol., 2022, 23, 1179–1203 Search PubMed.
- N. A. Alsaif, M. A. Dahab, M. M. Alanazi, A. J. Obaidullah, A. A. Al-Mehizia, M. M. Alanazi, S. Aldawas, H. A. Mahdy and H. Elkady, Bioorg. Chem., 2021, 110, 104807 Search PubMed.
- N. A. Alsaif, A. Elwan, M. M. Alanazi, A. J. Obaidullah, W. A. Alanazi, A. F. Alasmari, H. Albassam, H. A. Mahdy and M. S. Taghour, Mol. Diversity, 2022, 26, 1915–1932 Search PubMed.
- K. El-Adl, H. M. Sakr, R. G. Yousef, A. B. M. Mehany, A. M. Metwaly, M. A. Elhendawy, M. M. Radwan, M. A. ElSohly, H. S. Abulkhair and I. H. Eissa, Bioorg. Chem., 2021, 114, 105105 CrossRef CAS PubMed.
- N. A. Alsaif, M. S. Taghour, M. M. Alanazi, A. J. Obaidullah, A. A. Al-Mehizia, M. M. Alanazi, S. Aldawas, A. Elwan and H. Elkady, J. Enzyme Inhib. Med. Chem., 2021, 36, 1093–1114 CrossRef CAS PubMed.
- M. M. Alanazi, H. A. Mahdy, N. A. Alsaif, A. J. Obaidullah, H. M. Alkahtani, A. A. Al-Mehizia, S. M. Alsubaie, M. A. Dahab and I. H. Eissa, Bioorg. Chem., 2021, 112, 104949 Search PubMed.
- N. A. Alsaif, M. S. Taghour, M. M. Alanazi, A. J. Obaidullah, W. A. Alanazi, A. Alasmari, H. Albassam, M. A. Dahab and H. A. Mahdy, Bioorg. Med. Chem., 2021, 46, 116384 Search PubMed.
- N. A. Alsaif, H. A. Mahdy, M. M. Alanazi, A. J. Obaidullah, H. M. Alkahtani, A. M. Al-Hossaini, A. A. Al-Mehizi, A. Elwan and M. S. Taghour, Arch. Pharm., 2022, 355, 2100359 Search PubMed.
- K. El-Adl, H. M. Sakr, R. G. Yousef, A. B. M. Mehany, H. S. Abulkhair and I. H. Eissa, Arch. Pharm., 2022, 355, 2200048 Search PubMed.
- M. M. Alanazi, A. Elwan, N. A. Alsaif, A. J. Obaidullah, H. M. Alkahtani, A. A. Al-Mehizia, S. M. Alsubaie, M. S. Taghour and I. H. Eissa, J. Enzyme Inhib. Med. Chem., 2021, 36, 1732–1750 CrossRef CAS PubMed.
- S. Desai, V. Desai and S. Shingade, Bioorg. Chem., 2020, 94, 103382 CrossRef CAS PubMed.
- M. Kumar, G. Joshi, S. Arora, T. Singh, S. Biswas, N. Sharma, Z. R. Bhat, K. Tikoo, S. Singh and R. Kumar, Molecules, 2021, 26, 1490 CrossRef CAS PubMed.
- M. Alswah, A. Bayoumi, K. Elgamal, A. Elmorsy, S. Ihmaid and H. Ahmed, Molecules, 2017, 23, 48 CrossRef PubMed.
- P. Koralli, S. Tsikalakis, M. Goulielmaki, S. Arelaki, J. Müller, A. D. Nega, F. Herbst, C. R. Ball, V. G. Gregoriou, A. Dimitrakopoulou-Strauss, S. Wiemann and C. L. Chochos, Mater. Chem. Front., 2021, 5, 4950–4962 RSC.
- C. Tian, C. Yang, T. Wu, M. Lu, Y. Chen, Y. Yang, X. Liu, Y. Ling, M. Deng, Y. Jia and Y. Zhou, Bioorg. Med. Chem. Lett., 2021, 48, 128271 CrossRef CAS PubMed.
- F. B. El Dhaibi, A. Youssef, J. C. Fettinger, M. J. Kurth and M. J. Haddadin, ACS Omega, 2022, 7, 26871–26880 Search PubMed.
- N. A. Danilkina, N. S. Bukhtiiarova, A. I. Govdi, A. A. Vasileva, A. M. Rumyantsev, A. A. Volkov, N. I. Sharaev, A. V. Povolotskiy, I. A. Boyarskaya, I. V. Kornyakov, P. V. Tokareva and I. A. Balova, Molecules, 2019, 24, 2386 CrossRef PubMed.
| This journal is © The Royal Society of Chemistry 2025 |