
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceHeterobifunctional cross-linker with dinitroimidazole and azide modules for protein and oligonucleotide functionalization†
Qunfeng Luo *,
Shuli Liu,
Yaoguang Hua,
Chunqiu Long,
Sijia lv,
Juncheng Li and
Yuzhi Zhang
*,
Shuli Liu,
Yaoguang Hua,
Chunqiu Long,
Sijia lv,
Juncheng Li and
Yuzhi Zhang
School of Basic Medical Sciences, Jiangxi Medical College, Nanchang University, Nanchang, Jiangxi 330006, People's Republic of China. E-mail: luoqunfeng@ncu.edu.cn
First published on 10th February 2025
Abstract
Dinitroimidazole (DNIm) was recently identified as a powerful bioconjugation agent that could selectively modify thiol over amine on biomolecules at an ultrahigh speed in an aqueous buffer. However, its derivative containing a DNIm module and a terminal alkyne module failed to construct functional agents bearing a DNIm warhead via the CuAAC reaction. To solve this problem, a heterobifunctional cross-linker was designed and synthesized by linking a DNIm module with an azide module via an oxoaliphatic amido bond spacer arm. Its two modules, DNIm and azide, reacted with a thiol and cyclooctyne, respectively, in an orthogonal way. The cross-linker facilitated the preparation of various functional agents bearing a DNIm warhead via SPAAC reaction and was further applied to protein functionalization (including biotinylation and fluorescence labeling) and oligonucleotide functionalization (including PEGylation, oligonucleotide–peptide and oligonucleotide–protein conjugate). Thus, the cross-linker not only provided convenient access to those functional agents bearing a DNIm warhead but also combined DNIm chemistry with click chemistry of SPAAC to enlarge their respective application range in the bioconjugation field.
Introduction
Heterobifunctional cross-linkers comprise two different reactive modules connected by a linker chain. There are many such reactive modules, such as N-hydroxysuccinimide (NHS)1,2 (1c; Fig. 1A), maleimide (1a),3 the α-haloacetamide group4 (1b), alkynyl groups (1a, 1b, 1e),4 azide group (1c)4 and recently developed dinitroimidazole (1e).5 Cross-linkers are the centerpiece of bioconjugation chemistry for development of peptide- or antibody–drug conjugates6,7 and therapeutic oligonucleotide drugs.8 | ||
| Fig. 1 (A) Chemical structures of heterobifunctional cross-linkers from previous work and this work. (B) Chemical synthesis of DNIm-N3. (C) SPAAC reaction between the cross-linker 1 and cyclooctynes generated functional agents bearing a DNIm warhead, but CuAAC reaction between 1e and azides failed to. | ||
The strain-promoted alkyne azide cycloaddition (SPAAC) is an outstanding reaction in the bioorthogonal conjugation field owing to its splendid chemical stability, non-toxicity, mild reaction conditions, and high efficiency.9 SPAAC reaction utilizes two reaction modules, namely, azide and cyclooctyne groups. Many research groups have focused on expanding the pool of cyclooctynes and enhancing their reactivity toward azides. So far, the most commonly used cyclooctynes are dibenzocyclooctyne (DBCO)10 and bicyclononyne (BCN)11 owing to their relatively simple synthesis with sufficient yield and great coupling efficiency.
Recently, it was identified that dinitroimidazole (DNIm) could selectively modify a thiol over an amine on biomolecules at an ultrahigh speed in an aqueous buffer.5 DNIm and its thiol adduct generated via bioconjugation were extremely stable under tested conditions. DNIm revealed a few superiorities in stability and water solubility aspects compared to those of the maleimide and perfluoroaryl reagents. Besides, the adduct core fragment thiol-(4-nitroimidazole) was harmless to 293T cells under 10 μM, which endowed it with great potential to be utilized in live cells or pharmaceutical development. Considering these advantages, DNIm is promising to be a valuable addition to the current bioconjugation toolbox.
We envisioned combining DNIm chemistry with click chemistry of SPAAC to enlarge their respective application range in the bioconjugation field. With this in mind, we tried to incorporate two components (DNIm and azide group) into one to generate heterobifunctional cross-linker 1 (DNIm-N3) via an oxoaliphatic amido bond spacer arm (Fig. 1). Although a similar cross-linker containing DNIm and terminal alkyne modules has been reported5 (1e; Fig. 1A), it has failed to construct functional agents bearing a DNIm warhead via the CuAAC reaction (Fig. 1C and ESI Fig. 7†) resulting in the destruction of DNIm. We speculate that side reactions might happen owing to the additives in the click reaction mixture, such as copper, sodium ascorbate and THPTA. If the side reaction results from copper, it is rational and necessary to design and synthesize cross-linker 1, which employs copper-free SPAAC chemistry, which could overcome the obstacle existing in 1e. Furthermore, the azide group in 1 endows it with more power in bioconjugation owing to the reaction versatility of azides.12 Notably, we only focused on the SPAAC reaction in this work.
Results and discussion
CuAAC reaction between cross-linker 1e and d2
A model reaction of CuAAC was conducted by incubating compounds 1e and d2 (an azide, Fig. 1B) under routine conditions, CuSO4/VcNa/THPTA in this work (ESI Fig. 7†). Compound 1e (1 mM) and azide d2 (1 mM) were dissolved in neutral HEPES buffer (50 mM). Then, CuSO4 (1 mM), THPTA (1 mM) and VcNa (2 mM) were added successively. The cycloaddition product ed2 was formed in high yield as showed by HPLC analysis, but lost one –NO2 group at the N1 position of the imidazole ring, which was confirmed by HRMS and NMR data (ESI Fig. 8†). The detailed mechanism for the formation of the side product ed2 is still under exploration. In other words, the CuAAC reaction between 1e and azide failed to generate functional agents bearing a DNIm warhead, which prompted us to probe other alternative approaches to achieve this goal.Design and chemical synthesis of cross-linker DNIm-N3
As is known, organic azides are potentially explosive substances that decompose with the release of nitrogen through the slightest input of external energy, for example, pressure, impact, or heat. Those manipulable or nonexplosive organic azides conform to the rule that the number of nitrogen atoms must not exceed that of carbon and that (NC + NO)/NN ≥ 3 (N = number of atoms).13 On the other hand, considering the reported synthetic route to various DNIm derivatives and the commercial availability of azide materials, we decided to synthesize cross-linker 1.The key precursor m1 was synthesized first referring to the reported method.5 Next, compound m1 was condensed with NHS in the presence of DCC in dry DCM to afford compound m2 confirmed by HRMS and NMR in 74% yield. Compound m2 has two reaction modules, the DNIm module for sulfhydryl, and the NHS ester module for amine. Compound d3 containing an amine group and an azide group was designed to react with the NHS ester module of compound m2. To obtain compound d3, compound d1 was mixed with DPPA and DBU in dry THF under an N2 atmosphere and 0 °C to give compound d2 in which the hydroxyl group in d1 was replaced with an azide group. Then, compound d2 was treated with HCl in 1,4-dioxane solution to release the amine group to afford compound d3. Lastly, compound m2 was mixed with compound d3 in slightly basal HEPES buffer, which afforded the desired compound 1 as confirmed by HRMS and NMR in 89% yield (Fig. 1B and ESI Fig. 1–6†).
Orthogonal reaction of DNIm and azide modules in DNIm-N3 with Cys and (DBCO or BCN) derivatives
With the cross-linker 1 in hand, we then tested its reaction characteristics with Cys and (BCN or DBCO) derivatives. We started with the reaction between cross-linker 1 and L-cysteine c1. In neutral HEPES buffer, cross-linker 1 reacted with c1 to give the desired product mc1 identified by HPLC and HRMS (Fig. 2A and ESI Fig. 9†). Byproduct 2 was also observed with a 19% percentage in which the –NO2 group at the N1 position was lost compared to that in 1. c2 is a pentapeptide with amino acid sequence CWHKM containing one free cysteine residue and was chosen to react with cross-linker 1 (Fig. 2A). In the same condition as that between 1 and c1, only DNIm module in 1 reacted with the sulfhydryl group in peptide c2 to give compound mc2 in high yield as confirmed by HPLC and HRMS (Fig. 2A and ESI Fig. 9†).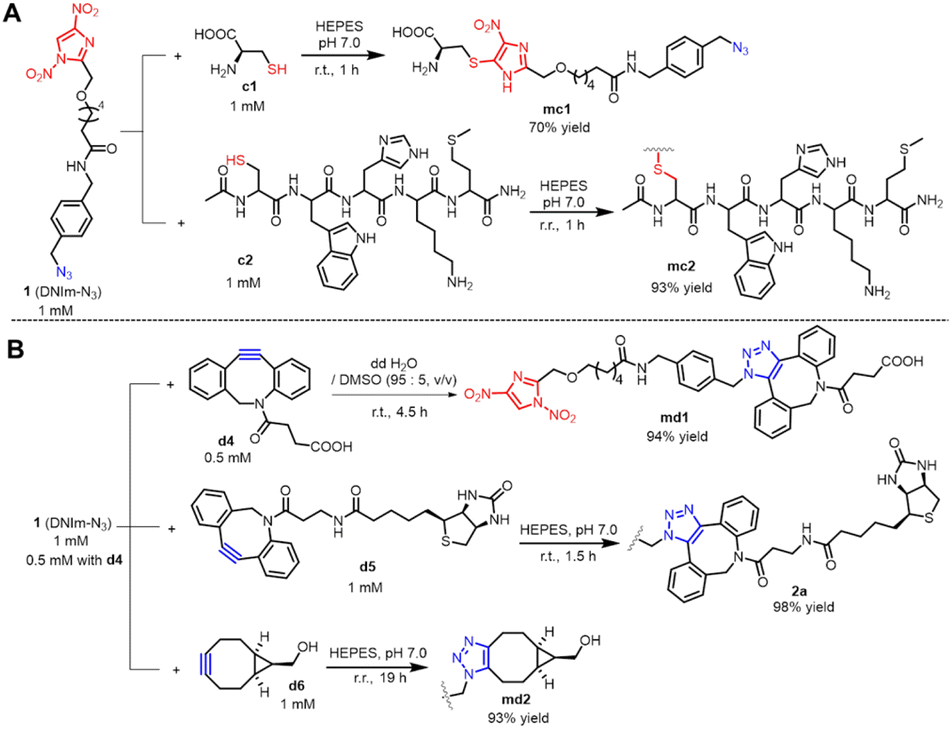 | ||
| Fig. 2 Orthogonal reaction of DNIm and azide modules in DNIm-N3 with Cys and (DBCO or BCN) derivatives: (A) reaction of DNIm module with Cys; (B) reaction of the azide module with DBCO or BCN derivatives. | ||
Then, we explored the reaction of cross-linker 1 with DBCO or BCN derivatives. In an aqueous solution with 5% DMSO; incubation of the cross-linker 1 with d4 for 4.5 h at r. t. produced the desired product md1 as determined by HPLC and HRMS. However, the HPLC spectrum presented two adjacent peaks with nearly the same integral area, which may be attributed to two regioisomeric triazoles formed in approximately equal amounts as reported by Agard et al.14 (Fig. 2B and ESI Fig. 10†). Another DBCO derivative bearing a biotin group, d5 was adopted to react with cross-linker 1. In neutral HEPES buffer, after 1.5 h of stirring, the desired product 2a formed nearly quantitatively as shown by HPLC and HRMS (Fig. 2B, ESI Fig. 11A and C†). To further explore the orthogonality between the azide module in DNIm-N3 and the DBCO group, incubation of compound DNIm with d4 in neutral HEPES buffer was performed. The reaction proceeded for 52 h at r. t. and no product was formed as demonstrated by HPLC analysis, which convinced us that DNIm did not interfere with the reaction between the azide module and the DBCO group (ESI Fig. 11B and D†). Likewise, BCN derivative d6 was stirred with cross-linker 1 in neutral HEPES buffer to give the title compound md2 in high yield (Fig. 2B and ESI Fig. 12†). Briefly, DNIm and azide modules in cross-linker 1 demonstrated good orthogonality toward Cys and cyclooctyne derivatives (DBCO and BCN), respectively, which makes it a good heterobifunctional cross-linker.
Preparation of functional agents bearing a DNIm warhead with DNIm-N3 via SPAAC reaction
The orthogonal reaction characteristics of the two modules in cross-linker 1 prompted us to utilize it to modify proteins. Attaching various functionalities, such as fluorophore and biotin, to proteins is valuable in the fields of biomedical research.15 We initiated this application by preparing several functional agents (3a, 4a, and 5a; Fig. 3) bearing a DNIm warhead for Cys modification. To obtain compound 3a, the intermediate BCN-biotin was prepared first through acylation of biotin-NH2 by compound BCN-NHS in a weakly basal methanol solution. Then, the incubation of compound BCN-biotin with cross-linker 1 in neutral HEPES buffer produced compound 3a via the SPAAC reaction in high yields as determined by HPLC analysis and HRMS (Fig. 3 and ESI Fig. 13†). Likewise, to obtain compound 4a, the intermediate BCN-dansyl was synthesized first through acylation of dansyl-NH2 by compound BCN-NHS (Fig. 3 and ESI Fig. 14†). Then, the incubation of compound BCN-dansyl with cross-linker 1 in MeOH produced compound 4a via SPAAC reaction. Before being applied to protein bioconjugation, the solvent MeOH was removed in a vacuum. Nearly in the same synthetic route as in 4a; compound furazan–NH2 was condensed with BCN-NHS to form the intermediate BCN-furazan, which further proceeded through the reaction of SPAAC with DNIm-N3 to give compound 5a (Fig. 3 and ESI Fig. 15†). All the functional agents 3a, 4a and 5a were applied directly for biomolecule modification without further purification.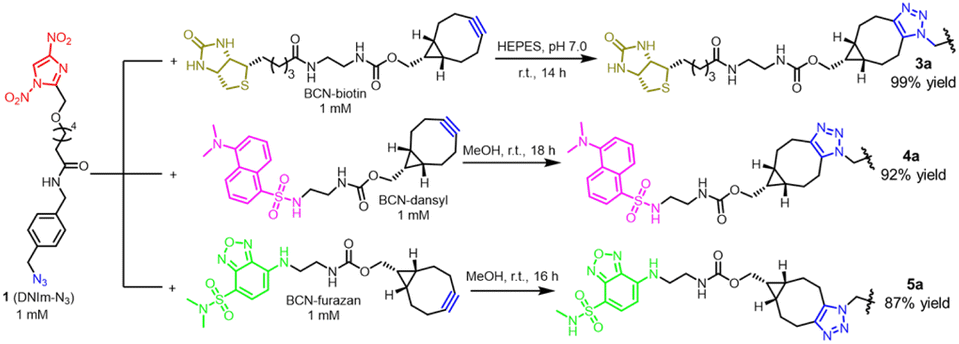 | ||
| Fig. 3 Preparation of functional agents bearing a dinitroimidazole warhead with cross-linker 1 via the SPAAC reaction. | ||
Modification of protein by functional dinitroimidazole agents bearing biotin or fluorescence group
As is known, the binding affinity of biotin to streptavidin is one of the highest reported for non-covalent interactions to date, with KD ∼ 0.01 pM. Thus, biotinylation is widely used in biotechnological fields, such as protein purification16 and drug target fishing.17 In this work, the biotin group was incorporated into the cross-linker 1 to generate functional molecules 2a and 3a near quantitatively (Fig. 2B, 3, ESI Fig. 11 and 13†). Bovine serum albumin (BSA) was chosen as a model protein. It contains one free cysteine (Cys34) and 59 lysines. Treatment of BSA with compound 2a or 3a for 1–2 h in a neutral buffer successfully attached the biotin group to BSA as determined by western blot analysis (Fig. 4A, C, E, and 6A) and LC-MS (Fig. 5E and F). Notably, in Fig. 5F, a minor product of BSA doubly modified by 2a was detected (BSA-2a*), one at –SH, another at –NH2. It might result from the organic solvent DMSO added in the solution (Fig. 4E), since we have reported that in a neutral aqueous buffer, DNIm attacks –SH rather than –NH2, however, in organic solution, DNIm attacks –NH2 at a high rate.5 Also, the remaining high concentration of the labelling reagent (2a) might bring about the unexpected modification at –NH2. In comparison with the experiment in lane 2 of Fig. 4C, BSA was treated with cross-linker 1 and compound BCN-biotin, successively (Fig. 4C lane 1, Fig. 4D lane 4 and lane 5, Fig. 4B). The desired protein bands were also observed by western blot analysis and the molecular weight of the two bioconjugates (BSA-N3, BSA-3a) were confirmed by LC-MS (Fig. 5B and C), although the minor amount of BSA remained. All the biotinylation on BSA showed a high conversion rate as determined by the software Thermo Scientific BioPharmaFinder™ 5.3 (Fig. 5; F3. Data analysis, 2. Experimental procedures in the ESI file†). Similarly, 3CL was also selected as a model protein for biotinylation. 3CL used in this work is a variant of the main protease (Mpro) of SARS-COV-2 with Ser46 changed to Phe (S46F), and a purification tag with 33 amino acids was added at its N terminal. It is composed of 339 amino acids and contains twelve free cysteines and eleven lysines, while only three cysteines are exposed to the solvent.18 Incubation of 3CL with compound 2a or 3a for 1 h also generated the biotinylated products as demonstrated by western blot data (Fig. 4A, F, and 6A). Unexpectedly, the two bioconjugates soon precipitated after desalting using the PD MiniTrap™ G-25 column, and therefore no signal of mass spectroscopy was detected in the supernatant. | ||
| Fig. 4 Modification of protein by functional dinitroimidazole agents bearing the biotin group. (A) Reaction scheme for BSA and 3CL modified by compound 2a; (B) reaction scheme for BSA modified by DNIm-N3 and BCN-biotin successively. BSA was modified by DNIm-N3 first to attach an azide group to BSA, followed by linking to the BCN-biotin via SPAAC reaction; (C) western blot analysis of biotinylated BSA. Lane 1: BSA + 1, then BCN-biotin. Conditions (refer to (B)): incubation of 30 μM of BSA with 60 μM of DNIm-N3 in a neutral Na2HPO4 buffer for 0.5 h. Then, the excess DNIm-N3 was removed through PD MiniTrap™ G-25 columns (Cytiva). The intermediate (15 μM) was then mixed with BCN-biotin (30 μM) in dd H2O for 26 h to produce the biotinylated BSA bioconjugate. Lane 2: BSA + compound 3a. Conditions: incubation of 35 μM of BSA with 285 μM of compound 3a in phosphate buffer at pH 7.0. (D) Western blot analysis of biotinylated BSA. Lane 4: BSA + 1, then BCN-biotin. Conditions: refer to lane 1 in (C). Lane 5: BSA + 1. Conditions: refer to the first step of lane 4. (E) Western blot analysis of biotinylated BSA by compound 2a. Lane 7: BSA + compound 2a. Conditions: BSA (30 μM), compound 2a (120 μM), 3% DMSO. (F) Western blot analysis of biotinylated 3CL by compound 2a or 3a. Lane 9: 3CL + compound 3a. Conditions: 3CL (30 μM), compound 3a (400 μM). Lane 10: 3CL + compound 2a. Conditions: 3CL (30 μM), compound 2a (240 μM), 3% DMSO. The uncropped versions for (C–F) are available in ESI Fig. 21.† | ||
 | ||
Fig. 5 Deconvoluted mass spectrum of BSA and its bioconjugates. (A) Deconvoluted mass spectrum of BSA. Mass for BSA: Calc. 66![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 429.98 Da, Obs. 66434.55 Da. (B) Deconvoluted mass spectrum of BSA-N3 that formed by the modification of BSA by DNIm-N3 with a conversion rate of 89.2%. Mass for conjugate BSA-N3, Calc. 66829.15 Da, Obs. 66830.27 Da. The hydrate of BSA was also detected as shown in the mass spectrum: BSA + H2O, Calc. 66447.99 Da, Obs. 66451.80 Da. (C) Deconvoluted mass spectrum of BSA-3a formed by the modification of BSA by 1 and BCN-biotin, successively. Mass for BSA-3a: Calc. 67291.38 Da, Obs. 67294.02 Da. The conversion rate for the step (from BSA-N3 to BSA-3a) is 100%. Mass for the hydrate of BSA (BSA + H2O): Calc. 66447.99 Da, Obs. 66452.44 Da. (D) Deconvoluted mass spectrum of BSA in another test. Mass for BSA: Calc. 66429.98 Da, Obs. 66427.23 Da. BSA′ (66597.27) is a gradient with low abundance in the sample. (E) Deconvoluted mass spectrum of BSA-3a* formed by the modification of BSA by 3a with a conversion rate of 96.6%. Mass for BSA-3a* Calc. 67291.38 Da, Obs. 67290.37 Da. Theoretically, BSA-3a* and BSA-3a (C) are the same ones with identical molecular weights. The mass signal 67459.24 (BSA′-3a*) is also a reasonable adduct formed by the modification of BSA′ (66597.27, (D)) by 3a. Mass for BSA′-3a*: Calc. 67458.67 Da, Obs. 67459.24 Da. When calculating the conversion rate, it was also included. (F) Deconvoluted mass spectrum of BSA-2a formed by the modification of BSA by 2a with a conversion rate of 83.7%. Mass for BSA-2a: Calc. 67331.35 Da, Obs. 67331.70 Da. The mass signal 68219.70 (BSA-2a*) is the product of BSA doubly modified by 2a, one at –SH, another at –NH2. When calculating the conversion rate, the single-modified and dual-modified products were considered as a whole. Mass for BSA-2a*: Calc. 68217.71 Da, Obs. 68219.70 Da. Mass for methanol adduct of BSA (BSA + MeOH): Calc. 66459.26 Da, Obs. 66460.48 Da. 429.98 Da, Obs. 66434.55 Da. (B) Deconvoluted mass spectrum of BSA-N3 that formed by the modification of BSA by DNIm-N3 with a conversion rate of 89.2%. Mass for conjugate BSA-N3, Calc. 66829.15 Da, Obs. 66830.27 Da. The hydrate of BSA was also detected as shown in the mass spectrum: BSA + H2O, Calc. 66447.99 Da, Obs. 66451.80 Da. (C) Deconvoluted mass spectrum of BSA-3a formed by the modification of BSA by 1 and BCN-biotin, successively. Mass for BSA-3a: Calc. 67291.38 Da, Obs. 67294.02 Da. The conversion rate for the step (from BSA-N3 to BSA-3a) is 100%. Mass for the hydrate of BSA (BSA + H2O): Calc. 66447.99 Da, Obs. 66452.44 Da. (D) Deconvoluted mass spectrum of BSA in another test. Mass for BSA: Calc. 66429.98 Da, Obs. 66427.23 Da. BSA′ (66597.27) is a gradient with low abundance in the sample. (E) Deconvoluted mass spectrum of BSA-3a* formed by the modification of BSA by 3a with a conversion rate of 96.6%. Mass for BSA-3a* Calc. 67291.38 Da, Obs. 67290.37 Da. Theoretically, BSA-3a* and BSA-3a (C) are the same ones with identical molecular weights. The mass signal 67459.24 (BSA′-3a*) is also a reasonable adduct formed by the modification of BSA′ (66597.27, (D)) by 3a. Mass for BSA′-3a*: Calc. 67458.67 Da, Obs. 67459.24 Da. When calculating the conversion rate, it was also included. (F) Deconvoluted mass spectrum of BSA-2a formed by the modification of BSA by 2a with a conversion rate of 83.7%. Mass for BSA-2a: Calc. 67331.35 Da, Obs. 67331.70 Da. The mass signal 68219.70 (BSA-2a*) is the product of BSA doubly modified by 2a, one at –SH, another at –NH2. When calculating the conversion rate, the single-modified and dual-modified products were considered as a whole. Mass for BSA-2a*: Calc. 68217.71 Da, Obs. 68219.70 Da. Mass for methanol adduct of BSA (BSA + MeOH): Calc. 66459.26 Da, Obs. 66460.48 Da. | ||
 | ||
| Fig. 6 Modification of protein by functional dinitroimidazole molecules bearing biotin or a fluorescent group. All the fluorescence images were converted to black and white. (A) Bioconjugation of protein by functional dinitroimidazole derivatives. (B) SDS-PAGE analysis of the fluorescent labeling of RhoA by compound 4a. Conditions: RhoA (18 μM), compound 4a (100 μM), 5% DMSO. The fluorescence was irradiated at 302 nm. (C) SDS-PAGE analysis of the fluorescent labeling of 3CL by compound 4a. Condition: 3CL (25 μM), compound 4a (100 μM), 5% DMSO. (D) SDS-PAGE analysis of the fluorescent labeling of RhoA by compound 5a. Condition: RhoA (16 μM), compound 5a (50 μM), 5% DMSO. The fluorescence was irradiated with blue light (440–485 nm). (E) SDS-PAGE analysis of the fluorescent labeling of 3CL by compound 5a. Conditions: 3CL (30 μM), compound 5a (100 μM), and 3% DMSO. | ||
Then, we tested the activity of functional DNIm agents bearing the fluorescence group. Compound 4a contains a dansyl fluorophore that can be efficiently excited by ultraviolet (UV) light,19 while 302 nm light was used in this work. Incubation of 3CL with compound 4a for 1 h in neutral phosphate buffer resulted in strong fluorescent labelling compared to that in the control in which 4a was not added (Fig. 6C and ESI Fig. 18C†). RhoA is a fusion protein that is composed of 422 amino acids and contains 10 cysteine residues and 38 lysine residues20 and was also selected as a model protein. 18 μM RhoA was mixed with 100 μM compound 4a for 1 h in neutral buffer. Sodium dodecyl sulfate-polyacrylamide gel electrophoresis (SDS-PAGE) analysis and irradiation with 302 nm light demonstrated evident fluorescence (Fig. 6B and ESI Fig. 18A†). Inspired by the labelling effect of compound 4a, we then designed compound 5a (Fig. 3) bearing a benzofurazan skeleton that is suitable to be excited with visible light at 450 nm.21 Incubation of RhoA with compound 5a in a neutral buffer formed the desired fluorescent band demonstrated by SDS-PAGE analysis and irradiation with blue light (440–485 nm) (Fig. 6D and ESI Fig. 18B†). 3CL was also applied to explore the modification power of compound 5a. 3CL was treated with compound 5a in a neutral buffer for 1 h affording a bright band excited by blue light (Fig. 6E and ESI Fig. 18D†). Unfortunately, we did not obtain the mass spectra of the four samples due to precipitation after desalting.
When carrying out protein expression and purification, precipitation is a common issue. In bioconjugation chemistry, new tags are attached to the protein, which may change the active spatial structure and induce precipitation. Though precipitation prevented us from obtaining mass spectra of partial bioconjugation products, SDS-PAGE and western blot analysis manifested the desired products. Overall, cross-linker 1 enabled us to perform facile functionalization of protein using dinitroimidazole and azide chemistry.
Modification of oligonucleotide by DNIm-N3
Natural oligonucleotides is polyanionic macromolecules with poor drug-like properties. However, the conjugation of specific molecules to oligonucleotides is a promising approach to developing nucleic acid-based drugs. Over the past two decades, medicinal chemists have identified many conjugation strategies, e.g., conjugation with polyethylene glycol, which can improve the nuclease stability, with antibody, which can improve biodistribution to a specific region or cell type, and/or with cholesterol, which increase lipophilicity.22 The significance of bioconjugation of oligonucleotides inspired us to probe the applicability of the cross-linker 1 in oligonucleotide modification. An oligonucleotide (o1) with 22 bases and one click group DBCO at a 5′ terminal was designed and ordered. Next, o1 was mixed with cross-linker 1 in neutral HEPES buffer to give the intermediate DNA-D-DNIm via SPAAC reaction, followed by the addition of cysteine-containing chemicals, e.g., SH-PEG-OH (2000 Da), peptide c2 (AcNH-CWHKM-CONH2) and BSA (Fig. 7A). The resulting oligonucleotide-PEG and oligonucleotide–peptide conjugate were analyzed by Urea-PAGE (Fig. 7B, lane 3 and 4) and the data showed that o1 was successfully conjugated by PEG and peptide in 44% and 47% yield in two reaction steps, respectively, processed using ImageJ. In contrast to the reaction order “o1 + DNIm-N3 + BSA” shown in Fig. 7C lane 7, BSA was first conjugated with DNIm-N3, and subsequently linked to o1 via the SPAAC reaction (Fig. 7C lane 8). Again, we performed a one-pot reaction in which o1 and BSA were mixed immediately followed by the addition of DNIm-N3. All these three samples (lanes 7–9 of Fig. 7C) were tested by SDS-PAGE and demonstrated 36%, 47% and 33% yield in two steps, respectively, as analysed using ImageJ. We found that the labelling efficiency on oligonucleotide seemed evidently lower than that on the protein (Fig. 5). However, it is reasonable considering that: first, an equal amount of labelling reagent was used in the reaction with oligonucleotide (Experimental procedures 2C in the ESI file†), while several equivalents of reagents were used for protein; secondly, the yield for oligonucleotide covered two steps, especially one step involving a reaction between two biomacromolecules, while that for protein covered only one. | ||
| Fig. 7 Modification of the oligonucleotide by cross-linker DNIm-N3. (A) Reaction scheme for preparation of oligonucleotide conjugates. Oligonucleotide o1 was first incubated with DNIm-N3 to attach a DNIm group onto it, then conjugated with cysteine-containing PEG or peptide (protein) via DNIm chemistry. Please see Experimental procedures 2C in the ESI file† for detailed profiles of the reaction; (B) Urea-PAGE analysis of oligonucleotide-PEG and oligonucleotide–peptide conjugates; (C) SDS-PAGE analysis of oligonucleotide–BSA conjugate. These five samples (lanes 3, 4, 7, 8 and 9) were quantitatively analyzed by ImageJ (ImageJ 1.52n, Wayne Rasband, National Institutes of Health, USA). Calculation equation: the intensity of the product band/the intensity of the control. The uncropped versions for (B) and (C) are available in ESI Fig. 22.† | ||
The conjugate oligonucleotide–peptide in lane 4 of Fig. 7B was confirmed by MALDI-TOF mass analysis (ESI Fig. 19†). Likewise, the conjugate oligonucleotide–BSA in lane 9 of Fig. 7C was confirmed by MALDI-TOF mass analysis. The three samples in lanes 7–9 of Fig. 7C showed the same peak around 74150, while only one was presented here (ESI Fig. 20†).
Conclusions
We developed a practical heterobifunctional cross-linker bearing two orthogonal reaction modules, namely, DNIm and the azide group. Various functional agents bearing a DNIm warhead were successfully prepared with a cross-linker 1 via the SPAAC reaction, which provided us a solution to the side reaction that appeared in the CuAAC reaction of cross-linker 1e. Therefore, our previous speculation seems reasonable that the presence of copper might be one of the factors that leads to the side reaction. These functional agents were utilized to modify protein, including biotinylation and fluorescence labeling. We also extended the application range to oligonucleotide functionalization, including PEGylation, oligonucleotide–peptide and oligonucleotide–protein conjugate. Above all, cross-linker 1 was compatible with protein as well as oligonucleotide biomolecules, which makes it a versatile tool for bioconjugation.Data availability
The data supporting this article have been included as part of the ESI.†Author contributions
Qunfeng Luo carried out all the chemical synthesis work and prepared the manuscript. Shuli Liu carried out most biochemical experiments. Yaoguang Hua conducted partial biochemical experiments. Chunqiu Long assisted Shuli Liu in completing the biochemical experiments. Juncheng Li and Sijia Lv and Yuzhi Zhang took charge of the picture collection and process.Conflicts of interest
There are no conflicts to declare.Acknowledgements
This work was financially supported by the National Natural Science Foundation of China (22007077). The authors gratefully acknowledge the Institute of Biomedical Sciences, Nanchang University, for providing good experimental instruments and equipment. The authors are grateful to the Biomedical Testing Center of Nanchang University and Dr Wuxin You and Prof. Ansgar Poetsch for technical support in corroboration of protein adducts with mass spectrometry. The authors also thank Prof. Jin Zhang (Nanchang University) for supporting the biochemical experiments and Experimentalists Juan Liang and Ye He (Nanchang University) for their help with ultraviolet transmission reflectometry. We also thank Prof. Sai-Sai Xie (Jiangxi University of Traditional Chinese Medicine) for supporting the chemical synthesis experiments and Dr Qi Chen (Nanchang University) for his instructive advice about the usage of horseradish peroxidase-labelled streptavidin.Notes and references
- J. Konč, L. Brown, D. R. Whiten, Y. Zuo, P. Ravn, D. Klenerman and G. J. L. Bernardes, Angew Chem. Int. Ed. Engl., 2021, 60, 25905–25913 CrossRef PubMed.
- G. T. Hermanson, in Bioconjugate Techniques, ed. G. T. Hermanson, Academic Press, Boston, 3rd edn, 2013, pp. 299–339, DOI:10.1016/B978-0-12-382239-0.00006-6.
- N. L. Kjærsgaard, R. A. Hansen and K. V. Gothelf, Bioconjugate Chem., 2022, 33, 1254–1260 CrossRef PubMed.
- A. M. ElSohly, J. I. MacDonald, N. B. Hentzen, I. L. Aanei, K. M. El Muslemany and M. B. Francis, J. Am. Chem. Soc., 2017, 139, 3767–3773 CrossRef CAS PubMed.
- Q. Luo, Y. Tao, W. Sheng, J. Lu and H. Wang, Nat. Commun., 2019, 10, 142 CrossRef PubMed.
- K. Tsuchikama and Z. An, Protein Cell, 2016, 9, 33–46 CrossRef PubMed.
- M. Alas, A. Saghaeidehkordi and K. Kaur, J. Med. Chem., 2021, 64, 216–232 CrossRef CAS PubMed.
- T. C. Roberts, R. Langer and M. J. A. Wood, Nat. Rev. Drug Discovery, 2020, 19, 673–694 CrossRef CAS PubMed.
- R. Dudchak, M. Podolak, S. Holota, O. Szewczyk-Roszczenko, P. Roszczenko, A. Bielawska, R. Lesyk and K. Bielawski, Bioorg. Chem., 2023, 143, 106982 CrossRef PubMed.
- M. F. Debets, S. S. van Berkel, S. Schoffelen, F. P. J. T. Rutjes, J. C. M. van Hest and F. L. van Delft, Chem. Commun., 2010, 46, 97–99 RSC.
- J. Dommerholt, S. Schmidt, R. Temming, L. J. A. Hendriks, F. P. J. T. Rutjes, J. C. M. van Hest, D. J. Lefeber, P. Friedl and F. L. van Delft, Angew Chem. Int. Ed. Engl., 2010, 49, 9422–9425 CrossRef CAS PubMed.
- C. I. Schilling, N. Jung, M. Biskup, U. Schepers and S. Bräse, Chem. Soc. Rev., 2011, 40, 4840–4871 RSC.
- S. Bräse, C. Gil, K. Knepper and V. Zimmermann, Angew. Chem., Int. Ed., 2005, 44, 5188–5240 CrossRef PubMed.
- N. J. Agard, J. A. Prescher and C. R. Bertozzi, J. Am. Chem. Soc., 2004, 126, 15046–15047 CrossRef CAS PubMed.
- E. Holz, M. Darwish, D. B. Tesar and W. Shatz-Binder, Pharmaceutics, 2023, 15, 600 CrossRef CAS PubMed.
- J.-N. Rybak, S. B. Scheurer, D. Neri and G. Elia, Proteomics, 2004, 4, 2296–2299 CrossRef CAS PubMed.
- X. Chen, Y. Wang, N. Ma, J. Tian, Y. Shao, B. Zhu, Y. K. Wong, Z. Liang, C. Zou and J. Wang, Signal Transduction Targeted Ther., 2020, 5, 72 CrossRef PubMed.
- Q. Hu, Y. Xiong, G. H. Zhu, Y. N. Zhang, Y. W. Zhang, P. Huang and G. B. Ge, MedComm, 2020, 3(3), e151 CrossRef PubMed.
- B. Patrascu, S. Mocanu, A. Coman, A. M. Madalan, C. Popescu, A. Paun, M. Matache and P. Ionita, Int. J. Mol. Sci., 2020, 21, 3559 CrossRef CAS PubMed.
- H. Zhou, X. Yue, Z. Wang, S. Li, J. Zhu, Y. Yang and M. Liu, Protein Expr. Purif., 2021, 180, 6 CrossRef PubMed.
- T. Santa, Biomed. Chromatogr., 2014, 28, 760–766 CrossRef CAS PubMed.
- S. Benizri, A. Gissot, A. Martin, B. Vialet, M. W. Grinstaff and P. Barthélémy, Bioconjug. Chem., 2019, 30, 366–383 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4ra07987f |
| This journal is © The Royal Society of Chemistry 2025 |