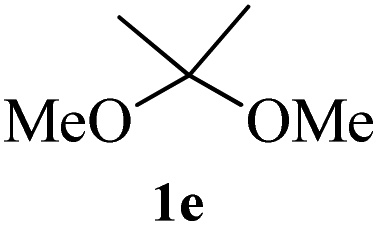DOI:
10.1039/D4RA07809H
(Paper)
RSC Adv., 2025,
15, 5523-5534
FeCl3/SiO2-catalyzed bis-indolylation of acetals and ketals: a highly atom-economical approach to the selective deprotection of protected carbohydrates†
Received
2nd November 2024
, Accepted 3rd February 2025
First published on 18th February 2025
Abstract
A simple and green catalytic system is developed for the synthesis of 3,3′-bisindolyl(methanes) (BIMs) using cyclic/acyclic acetals as the carbon source for the bridging residue between two indole motifs. The reaction occurred under mild and benign conditions using FeCl3/SiO2 as a heterogeneous catalyst without the requirement of any toxic organic solvents. The ready availability and recyclability of the catalytic system allows the reaction to be highly efficient, resulting in very good BIM products. DFT studies were also performed to establish the proposed mechanism and preferential formation of unsymmetrical bisindolylmethanes using equimolar amounts of different indoles. The present protocol is also extended to the bisindolylation-induced selective cleavage of protected carbohydrates to diols in a 100% carbon-preservation and maximized atom-economical manner.
Introduction
Nitrogen heterocycles are one of the most frequent scaffolds in pharmaceuticals. Amongst the various nitrogen heterocycles, the indole moiety is regarded as a privileged structure with potential applications in the field of agro- and medicinal chemistry. When properly functionalized, indoles can exhibit a wide range of pharmacological properties.1 In particular, 3,3′-bis indolylmethanes (BIMs), composed of two indole units, have been isolated from marine and terrestrial natural sources,2 such as plants, sponges, parasitic bacteria, and tunicates that include arundine, vibrindole A, streptindole, arsindoline A and B, turbomycin A and B, barakacin, annonidine B and dalesindole (Fig. 1). Due to the importance of BIM derivatives in the development of novel bioactive molecules, numerous synthetic methods to prepare this class of compounds have been reported.3 The most exploited method for the synthesis of BIMs is the condensation of indoles with various aldehydes and ketones (Ehrlich test of indole), which is largely catalyzed by either protic or Lewis acids and seems to be straightforward and practical.4 Accordingly, various protic or Lewis acid-catalyzed syntheses of bis(indolyl)methanes using indoles (2 equivalents) and carbonyl compounds (1 equivalent) have been reported in the literature.5–25 The use26 of 3-substituted indolyl alcohols and substituted indoles, metal-catalyzed carbonylation and alkylation reactions,27 and metal-free oxidative reactions28 have been adopted for the synthesis of BIMs. In the recent past, several efforts have also been devoted towards the synthesis of BIMs using substituted indoles with aryl amines,29 benzyl amines,30 and benzyl alcohols31 as the second component in the condensation protocols. The synthesis of various symmetrical and unsymmetrical BIMs via the reaction of (3-indolylmethyl)trimethyl-ammonium iodides with a wide range of substituted indole derivatives has been reported.32 The use of C1 and C2 alcohols as the carbon source for the bridging methylene group in BIMs under CuO-peroxymonosulfate (CuO-PMS) catalytic systems33 and N-heterocyclic iodo(azo)lium salt organocatalytic protocols for the promotion of Friedel–Crafts-type reactions between indoles and aldehydes leading to the formation of BIMs has also been reported.34 Despite having some advantages of their own, most of the reported methods indicate that the catalysts commonly used for such transformations are generally associated with one or more disadvantages, such as high toxicity, high cost, difficulty of handling, low thermal stability, and non-recyclability after being used.
 |
| | Fig. 1 Representative examples of biologically active natural/synthetic analogues of BIMs. | |
The protection–deprotection technique is an important and desirable attribute in multi-step or target-oriented organic synthesis to prevent undesired/unwanted reactions. The carbonyls are generally protected as their acetal/ketal formation because of their easy incorporation as well as their survival in a wide range of reaction conditions.35 Conversely, 1,2- and 1,3-diols in carbohydrate chemistry are usually protected by the formation of their isopropylidene or cyclohexylidene derivatives with acetone and cyclohexanone, respectively.35,36 A significant number of strategies for the deprotective cleavage of acetals, ketals, or other 1,3-dioxolanes that work in either acidic or non-acidic conditions are reported in the literature,36 and usually either part of the protecting group goes to waste (Scheme 1, eqn (1) and (2)).
 |
| | Scheme 1 Bis-indolylation-directed cleavage of acetals/ketals using silica-supported ferric chloride (FeCl3/SiO2) as a recyclable catalyst. | |
Iron(III) chloride is extensively used in organic synthesis as an ideal Lewis acid as it is an inexpensive and convenient reagent.37 FeCl3·6H2O in association with ionic liquid or iron with Pd-catalyst one-pot domino reactions for the construction of bis(indolyl)methanes has been reported.38–40 However, this reagent cannot be recycled after its use, which creates pollution issues. In recent years, the use of silica-supported catalysts has received considerable attention in organic syntheses because of enhanced activity, easier handling, recovery of the catalyst, low cost, and simple work-up procedure. Based on these features of supported reagents as heterogeneous catalysts, we23,41 and others42 utilized silica-supported ferric chloride (FeCl3/SiO2) as an activator for functional groups, which was utilized successfully in various organic transformations. It was reported that the acetal group can be activated by ferric chloride43 or other transition metal catalysts44 for the synthesis of heterocycles and other purposes under mild conditions. For several years, we were actively engaged in the development of newer methodologies45a–f for the synthesis of potential bioactive compounds, and in continuation of our efforts towards the development of benign protocols, we hereby report a novel and efficient method for the preparation of bis-indolylmethane derivatives (BIMs) via multi-component assembly of electrophilic substitution reactions of indoles with acetal-protected carbonyls in the presence of silica-supported ferric chloride (FeCl3/SiO2) as a recyclable and eco-friendly catalyst (eqn (3), Scheme 1). We also extended this methodology for the first time towards a selective bis-indolylation-directed deprotection of isopropylidene/cyclohexylidene-protected carbohydrate to the corresponding diol under anhydrous conditions in a highly atom-economical manner (eqn (4), Scheme 1).
Results and discussion
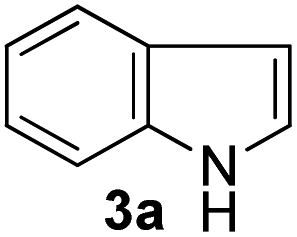
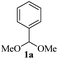
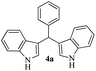
For optimization of reaction conditions and yield of bis-indolylmethanes (4a), we began with benzaldehyde dimethylacetal (1a) and indole (3a) as model substrates using different catalysts that are known to activate acetals under solvent-free conditions. No product was obtained upon grinding of 1a and 3a in a 1![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :2 molar ratio in the absence of catalyst (Table 1, entry 1), but on heating at 80 °C, decomposition of the materials was detected on a TLC plate (Table 1, entry 2). Grinding the mixture of 1a and 3a by keeping their ratios the same with SiO2 (230–400 mesh, 20 mg) for 3 h, 4a was isolated in 40% yield (Table 1, entry 3) along with the recovery of unreacted indole. Considering the Lewis acid character of FeCl3, we employed 5 mol% of FeCl3 while grinding the mixture of 1a and 3a (1:2 molar ratio). Unfortunately, this failed to produce any isolable product from the reaction mixture after 30 min as multiple spots were detected on the TLC plate. Then, based on our previous work,46 we decided to explore the catalytic potentiality of FeCl3/SiO2 (230–400 mesh was used) as an activator of acetal groups in our present work. We were pleased to observe that grinding the mixture with pre-prepared SiO2/FeCl3 (20 mg, containing 2 mol% of FeCl3)23 for 15 min results in a clean transformation of the substrates to the desired BIM 4a in 90% yield (Table 1, entry 5). An increase in the amount of SiO2/FeCl3 (50 mg) does not improve the yield (Table 1, entry 6), but decreasing the amount of catalyst to 10 mg (1 mol% of FeCl3) decreased the yield of 4a to 80% (Table 1, entry 7). To evaluate the catalytic superiority of silica-supported (FeCl3/SiO2), we screened other supported/heterogeneous catalysts such as Nano TS-1, silica-supported perchloric acid, magnetic PANI-FeMnO4, and Amberlite IR 120H+ for the indolylation reaction. However, these catalysts provided inferior results (Table 1, entries 8–11) compared to the SiO2/FeCl3-catalyzed reaction (Table 1, entry 5). In the case of PANI-FeMnO4, a complex mixture was detected using thin-layer chromatographic techniques. We also screened imidazolium-based ionic liquids, such as 1-butyl imidazolium trifluoroacetate and 1-butyl-3-methylimidazolium bromide, under solvent-free homogenous conditions at 80 °C. We have previously reported that the protic ionic liquid 1-butyl imidazolium trifluoroacetate could be an effective medium for the hydrolytic cleavage of acetals/ketals at 70 °C.45a During the investigation, we found that this protic ionic liquid provided a 70% yield of 4a after 2 h. In contrast, the neutral ionic liquid 1-butyl-3-methylimidazolium bromide provided a trace amount of 4a after 3 h.
:2 molar ratio in the absence of catalyst (Table 1, entry 1), but on heating at 80 °C, decomposition of the materials was detected on a TLC plate (Table 1, entry 2). Grinding the mixture of 1a and 3a by keeping their ratios the same with SiO2 (230–400 mesh, 20 mg) for 3 h, 4a was isolated in 40% yield (Table 1, entry 3) along with the recovery of unreacted indole. Considering the Lewis acid character of FeCl3, we employed 5 mol% of FeCl3 while grinding the mixture of 1a and 3a (1:2 molar ratio). Unfortunately, this failed to produce any isolable product from the reaction mixture after 30 min as multiple spots were detected on the TLC plate. Then, based on our previous work,46 we decided to explore the catalytic potentiality of FeCl3/SiO2 (230–400 mesh was used) as an activator of acetal groups in our present work. We were pleased to observe that grinding the mixture with pre-prepared SiO2/FeCl3 (20 mg, containing 2 mol% of FeCl3)23 for 15 min results in a clean transformation of the substrates to the desired BIM 4a in 90% yield (Table 1, entry 5). An increase in the amount of SiO2/FeCl3 (50 mg) does not improve the yield (Table 1, entry 6), but decreasing the amount of catalyst to 10 mg (1 mol% of FeCl3) decreased the yield of 4a to 80% (Table 1, entry 7). To evaluate the catalytic superiority of silica-supported (FeCl3/SiO2), we screened other supported/heterogeneous catalysts such as Nano TS-1, silica-supported perchloric acid, magnetic PANI-FeMnO4, and Amberlite IR 120H+ for the indolylation reaction. However, these catalysts provided inferior results (Table 1, entries 8–11) compared to the SiO2/FeCl3-catalyzed reaction (Table 1, entry 5). In the case of PANI-FeMnO4, a complex mixture was detected using thin-layer chromatographic techniques. We also screened imidazolium-based ionic liquids, such as 1-butyl imidazolium trifluoroacetate and 1-butyl-3-methylimidazolium bromide, under solvent-free homogenous conditions at 80 °C. We have previously reported that the protic ionic liquid 1-butyl imidazolium trifluoroacetate could be an effective medium for the hydrolytic cleavage of acetals/ketals at 70 °C.45a During the investigation, we found that this protic ionic liquid provided a 70% yield of 4a after 2 h. In contrast, the neutral ionic liquid 1-butyl-3-methylimidazolium bromide provided a trace amount of 4a after 3 h.
Table 1 Optimization of BIM (4a) synthesis from benzaldehyde dimethyl acetal (1a) and indole (3a)

|
| Entry |
Catalyst |
Temp. (°C) |
Time (min) |
Yield (%) |
| 1 |
No |
rt |
180 |
No reaction |
| 2 |
No |
80 |
30 |
Decomp |
| 3 |
SiO2 |
rt |
180 |
40 |
| 4 |
FeCl3 |
rt |
30 |
Mixture |
| 5 |
FeCl3–SiO2 (20 mg) |
rt |
15 |
90 |
| 6 |
FeCl3–SiO2 (50 mg) |
rt |
15 |
90 |
| 7 |
FeCl3–SiO2 (10 mg) |
rt |
15 |
80 |
| 8 |
Nano TS 1 (10 mg) |
rt |
15 |
55 |
| 9 |
HClO4–SiO2 (20 mg) |
rt |
15 |
70 |
| 10 |
PANI-FeMnO4 (10 mg) |
rt |
180 |
Mixture |
| 11 |
Amberlite IR120 (20 mg) |
rt |
180 |
83 |
| 12 |
[HBIm]TFA (20 mg) |
70 |
120 |
70 |
| 13 |
[BMIm]Br (20 mg) |
70 |
180 |
Trace |
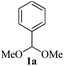
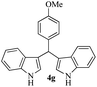
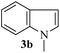
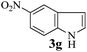
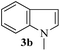
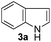
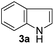
With the optimized conditions in hand (Table 1, entry 5), we decided to explore the substrate scope and efficacy of the present procedure. For this, a wide variety of protected carbonyls (1) were allowed to react with various indoles (3) under standard conditions. Overall, the reaction conditions were found to be general. The reaction of benzaldehyde dimethyl acetal (1a) with N-methyl indole (3b) and 2-methyl indole (3c) in the presence FeCl3/SiO2 under solvent-free conditions at room temperature furnished the desired bisindolylated products 4b–c in high yields (92 and 91%, respectively); however, in the case of an electronically crowded 2-phenyl indole (3d), only 84% yield of the desired product was obtained (Table 2, entries 2–4). The ability to tolerate a halide that is present in the indole ring (3e) also demonstrated the efficacy of the protocol is general, although with a slightly lower yield of the expected product (Table 2, entry 5). The present protocol is compatible with methoxy substituents present in either of the coupling partners. The methoxy group present in the indole ring (3f) provided a better yield with protected benzaldehyde (1a) than the dimethylacetal functionalized anisole (1b) with simple indole (3a) (Table 2, entries 6 and 7). The electron-withdrawing nitro group present in the o- and p-positions of the benzaldehyde diethyl acetal (1c–d) reacted well with a simple indole (3a) or a 1-methyl indole (3b), producing an excellent yield of the expected products 4h–k (Table 2, entries 8–11), but the reaction lasted longer compared to entries 1–7 in Table 2. The reaction works well but slowly when a nitro group is present in the indole moiety (Table 2, entry 12). It was noticed that the acetal generated from ketones such as 2,2-dimethoxy propane (acetone dimethyl acetal; 1e) and cyclohexanone dimethyl acetal (1f) underwent a smooth bisindolylation reaction with the indole/substituted indole, achieving a very good-to-excellent yield of 4m–p (Table 2, entries 13–16). Notably, the treatment of triethyl orthoformate under similar reaction conditions using three equivalents of the indole results in a clean transformation, producing a very high yield of tris-indolyl methane (4q) after 60 minutes (Table 2, entry 17). The present procedure was also shown to proceed well for 2-phenyl 1,3-dioxolane (1h) and 2-(4-chlorophenyl) 1,3-dioxolane (1i) with the indole, producing bis-indolyl derivatives 4a and 4r in good yield (Table 2, entries 18 and 19). In spite of the present methodology for bisindolylation of acetals/ketals having a broad functional group tolerance, the present protocol failed to produce the product when a strong electron-withdrawing group was present in the α-position of the dimethyl acetal-protected aldehyde (Table 2, entries 20 and 21). Based on our recent report46 on the synthesis of multisubstituted imidazole via FeCl3/SiO2-catalyzed activation of acetals, we postulated the mechanism, which is depicted in Fig. 2. We believe that the iron of FeCl3/SiO2 coordinates with both the oxygens of acetal and facilitates the formation of an intermediate oxonium ion (B) after expulsion of the alkoxide (R′O−). Then, the indole acts as a nucleophile to attack the highly reactive oxonium ion B via the C-3 position of the indole ring to produce the intermediate C, which, upon subsequent release of a proton and aromatization, gives another intermediate D. Due to the allylic ether nature of the intermediate D, it is further activated by FeCl3/SiO2 to yield the highly reactive azafulvene-type intermediate E via the indole through an N-triggered elimination of R′O−. This reactive intermediate E invites a second molecule of indole to participate in the Michael addition reaction to afford F. Removal of a proton and release of the catalyst produces bis-indolylmethane 4. Notably, the involvement of the initial formation of the carbonyl compound via hydrolytic cleavage by the catalysis of FeCl3 is excluded, as evident from the NMR experiment. A solution of 1d (10 mg) in DMSO-d6 (0.6 mL) was placed in an NMR tube and then the 1H NMR spectrum was recorded in the presence of FeCl3/SiO2 at ambient temperature after 15 min. No signal of an aldehydic functional group due to hydrolytic cleavage of diethyl acetal group in 1d was detected.
Table 2 Scope and generality of the SiO2/FeCl3-catalyzed synthesis of bisindoles using indoles and protected carbonyl compounds
| Entry |
Protected carbonyl |
Indole |
Product |
Time (min) |
Yield (%) |
| 1 |
 |
 |
 |
15 |
90 |
| 2 |
 |
 |
 |
15 |
92 |
| 3 |
 |
 |
 |
15 |
91 |
| 4 |
 |
 |
 |
30 |
84 |
| 5 |
 |
 |
 |
30 |
85 |
| 6 |
 |
 |
 |
30 |
90 |
| 7 |
 |
 |
 |
30 |
85 |
| 8 |
 |
 |
 |
90 |
93 |
| 9 |
 |
 |
 |
90 |
91 |
| 10 |
 |
 |
 |
90 |
87 |
| 11 |
 |
 |
 |
90 |
82 |
| 12 |
 |
 |
 |
60 |
82 |
| 13 |
 |
 |
 |
30 |
95 |
| 14 |
 |
 |
 |
30 |
86 |
| 15 |
 |
 |
 |
30 |
83 |
| 16 |
 |
 |
 |
30 |
85 |
| 17 |
 |
 |
 |
60 |
91 |
| 18 |
 |
 |
 |
45 |
86 |
| 19 |
 |
 |
 |
45 |
86 |
| 20 |
 |
 |
 |
60 |
NR |
| 21 |
 |
 |
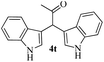 |
60 |
NR |
 |
| | Fig. 2 Proposed reaction mechanism of the FeCl3/SiO2-catalyzed bis-indolylation of an acetal. | |
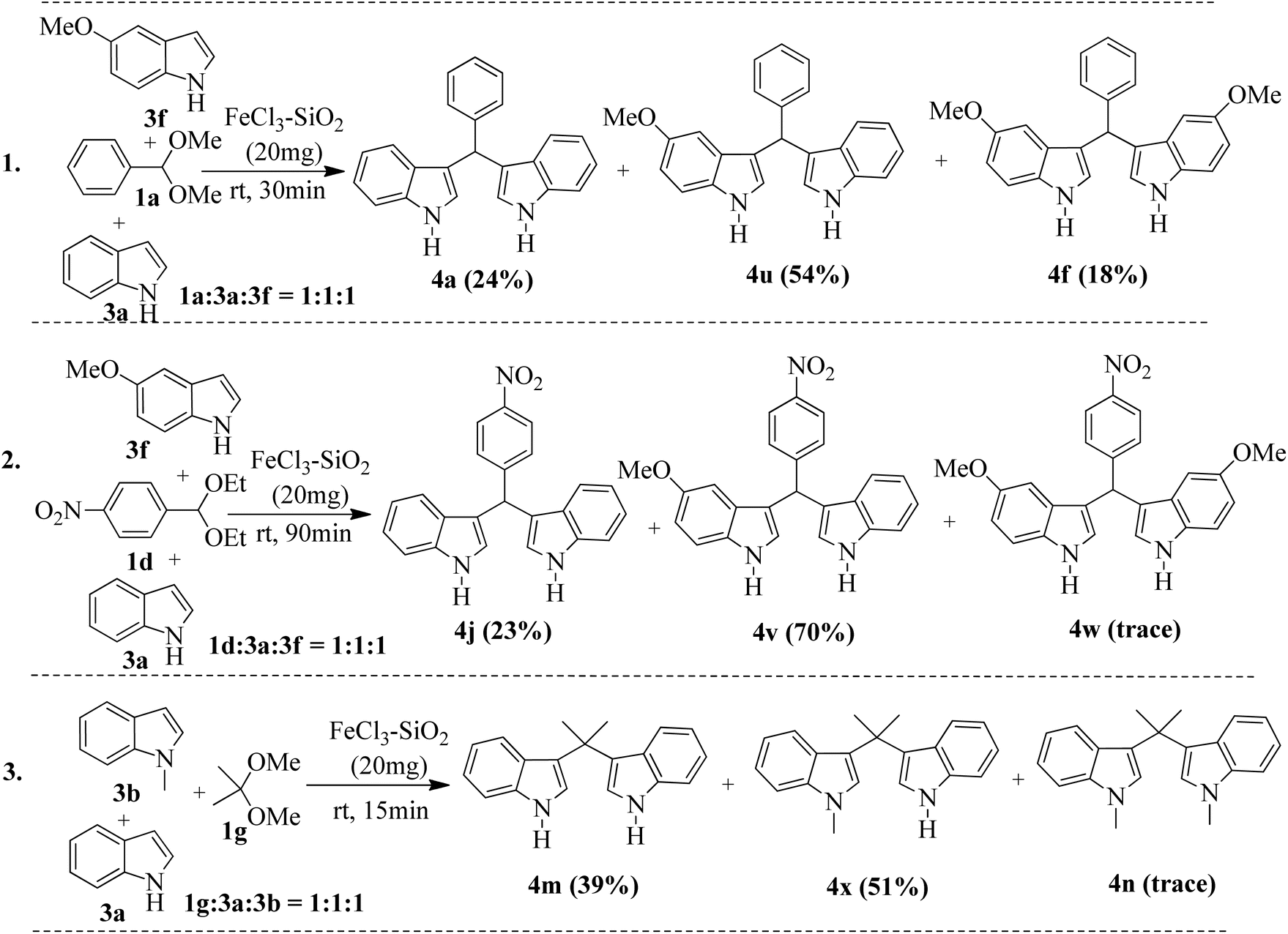
However, the broadening of some signals was detected when such spectra were recorded with a suspension of silica-supported FeCl3. Unsuccessful attempts (Table 2, entries 20 and 21) using benzimidazole 2-carboxaldehyde dimethyl acetal and pyruvaldehyde dimethyl acetal also support our hypothesis, as the corresponding intermediate oxonium (B) is unstable due to the electron-withdrawing ability of the attached residue. It was also reported that pyruvaldehyde itself undergoes a bis-indolylation reaction in the presence of pTSA with indole,11 so if hydrolytic cleavage occurred by FeCl3/SiO2, there would have been the possibility to isolate bisindole 4t from the reaction of pyruvaldehyde dimethyl acetal and indole. The formation and stability of the intermediate E depend on the substituent present in the indole ring. The electron-donating substituents in indole favor the formation of E; in contrast, strong electron-withdrawing groups, such as a nitro group, destabilize it to some extent. By taking advantage of the substituent control formation of E, it is possible to control the preferential synthesis of unsymmetrical BIMs. For example, the reaction of benzaldehyde dimethyl acetal (1a) with indole (3a) and 5-methoxy indole (3f) in a ratio of 1:1:1 under standard conditions produced the unsymmetrical bis-indolyl methane (4u) as the major product (54%), along with symmetrical bisindole 4a (24%) and 4f (18%) (Scheme 2, eqn (1)). Interestingly, the use of 1a:3a:3f in a 1:1:2 ratio produces 4f exclusively, but if we change the ratio to 1:2:1 again, 4u remains the major product (50%). We have also conducted a study using other acetals such as 1d and 1g with a simple indole 3a and reactive indoles 3f or 3b. Using equimolar ratios of 1d/3a/3f provided the cross product 4v (70%) as the major product along with 23% of 4j and a trace amount of 4w. Similarly, unsymmetrical bis-indolyl methane 4x was formed as the major product (51%) when 1g/3a/3b was employed in a 1:1:1 ratio. In all cases, each of the products was purified by column chromatography using ethyl acetate and hexanes (1:9 to 1:1) and then quantified based on the amount of isolated pure products. The preferential formation of unsymmetrical bis-indolylmethanes, as shown in Scheme 2, could be explained by the relative stability of the proposed intermediate E (Fig. 2) generated in situ from indoles and aldehydes. The overall reaction may be considered as a thermodynamically controlled (formation of the intermediate E) first step, followed by a kinetically controlled nucleophilic attack of a second indole molecule, leading to the formation of final products. The electron-donating substituent (–OMe) present in the indole ring of the intermediate E4u stabilizes it over E4a, and thus 4u becomes the major product after the kinetically controlled addition of the second indole molecule to E4u. Symmetrical BIM 4f becomes the minor product due to the low quantity (mostly used in the first step) of 3f [Scheme 2(1)]. Similarly, 4v and 4x are the major products; 4j and 4m are minor, and 4w and 4n are formed in trace amounts, as shown in Scheme 2(2) and 2(3), respectively. Therefore, the results indicated that the stability of E4v > E4j and E4x > E4m. Thus, the outcome of the reaction indicated the highly competitive nature of the condensation process during the course of the reactions. To justify our hypothesis, we performed quantum mechanical calculations to assess the relative stability of all the proposed intermediates for E4a vs. E4u, E4j vs. E4v and E4m vs. E4x (Fig. 3) using Density Functional Theory (DFT) with B3LYP/6311G(d,p) in Gauss 16w software.47 Energy optimization in the DFT study of the proposed intermediates (Fig. 3) predicted that the total energy (TE) of intermediate pairs viz. E4a/E4u, E4j/E4v, and E4m/E4x was −632.4196 and −746.9743, −837.6709 and −952.1623, and −479.9631 and −519.8499 Hartree, respectively, suggesting the thermodynamic stabilities as E4a < E4u, E4j < E4v, and E4m < E4x. Herein, the observed low energy value of the corresponding optimized structures (Fig. 4) of the stable intermediates (E4u and E4v) having an electron-donating group (–OMe) may be attributed to the conjugation of electrons favoring the stabilities. However, in the case of E4m and E4x, hyperconjugative electron delocalization of the –CH3 group attached to the indole nitrogen atom might favor E4x in making it more stable than E4m. In addition, the HOMO–LUMO (HL) gap, as an index of kinetic stability (a low gap indicates greater stability),48,49 supports the stabilities of E4u in E4a/E4u (3.73/3.12 eV) and E4v in E4j/E4v (3.35/3.33 eV) pairs. Moreover, a close HL gap (Fig. 4) of the intermediates in each pair, particularly in E4a/E4u and E4j/E4v, suggests that the second step of the reactions, viz. nucleophilic attack of the second indole, is kinetically controlled and almost equally probable. In the case of E4m and E4x, the comparatively very low HL gap in E4x (0.935 eV) compared to E4m (4.29 eV) indicated that E4x has the highest thermodynamic stability (low TE) as well as kinetic stability compared to E4m. Therefore, this report of bis-indolylation reactions is mainly driven by the thermodynamic stability of the concerned in situ intermediates, which, in turn, is governed by the presence or absence of electron-donating substituents in the intermediates. The optimized structures of intermediates (Fig. 3) and their HOMO–LUMO energy gaps are shown in Fig. 4.
 |
| | Scheme 2 Control experiments for the synthesis of unsymmetrical bis-indolylmethanes. | |
 |
| | Fig. 3 Structure of various proposed intermediates in Scheme 2. | |
 |
| | Fig. 4 Optimized structures of intermediates of Fig. 3 and their HOMO–LUMO energy gaps. | |
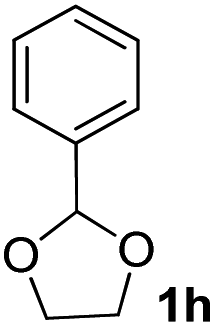
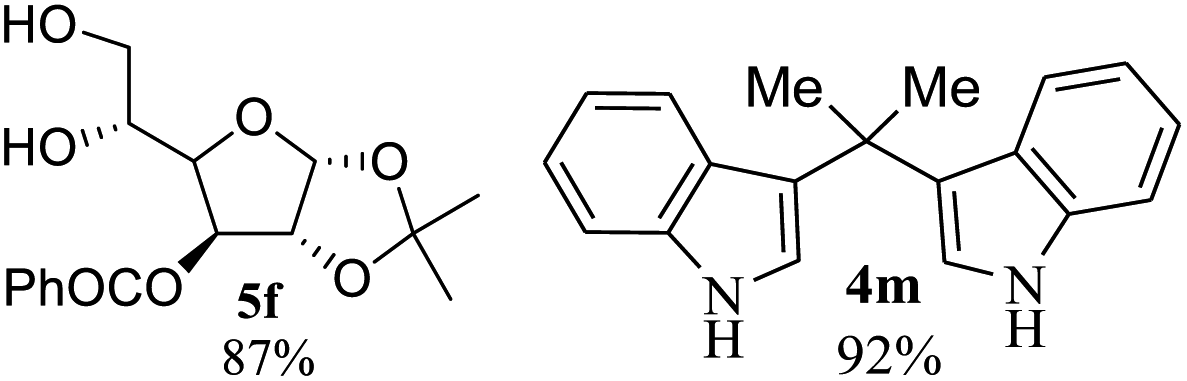
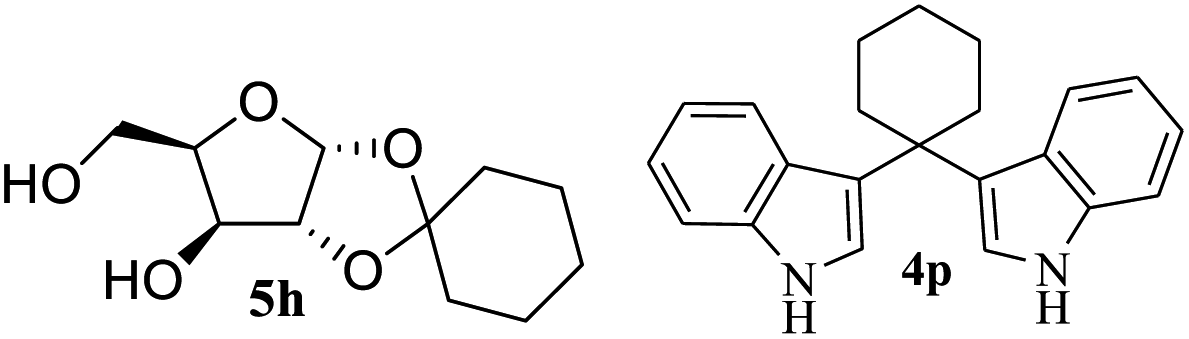
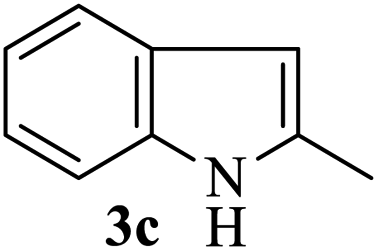
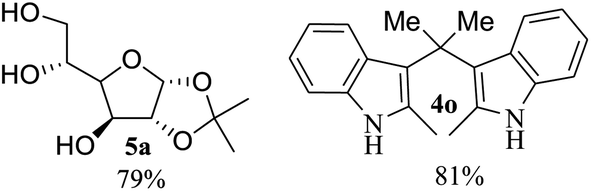
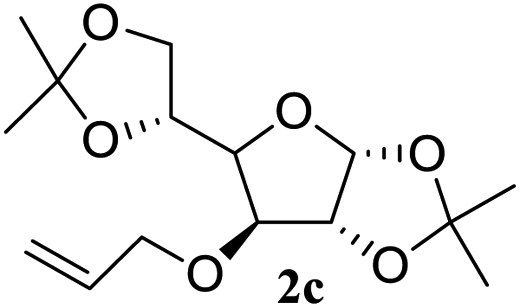
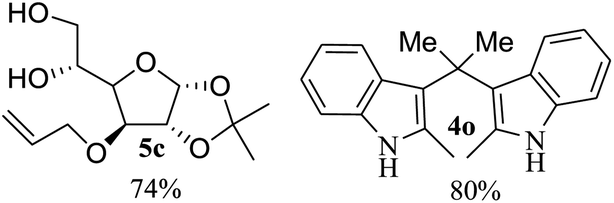
After the successful accomplishment of this new synthetic strategy for the synthesis of bis-indolylmethanes from acetal-/ketal-protected carbonyls with a wide range of indoles using silica-supported ferric chloride, we were curious to observe the reactivity of the same catalyst against the acetal-protected carbohydrates. The hydrolytic deprotection of acetal-protected sugar, generally a ketone (eqn (2) in Scheme 1), goes to waste. Their isolation from the post-reaction mixture is difficult or not valued due to their volatile nature and high solubility in water. The unusual findings assembled in Table 2 indicate that the carbonyl part of acetals/ketals (1) appeared in the bridged carbon of bisindole derivatives (4), while the alcoholic residue (eqn (3), Scheme 1) was eliminated as waste. As the results shown in Table 2 are very interesting, this prompted us to explore the possibility of the selective activation–deprotection of isopropylidene or cyclohexylidene groups present as protecting groups in carbohydrate derivatives, since this could be a viable route for a 100% atom-economical strategy for the deprotection of protected carbohydrates under benign conditions. Thus, we began with 1,2:5,6-di-O-isopropylidene glucofuranose (2a) as a protected substrate and two equivalents of indole (3a) to screen with silica-supported ferric chloride (FeCl3/SiO2) for the cleavage of 1,3-dioxolane in a non-aqueous medium under standard conditions (Table 1, entry 5). Our initial attempt at room temperature failed to produce any cleavage product; however, conducting the reaction at elevated temperatures (95–100 °C) for 1 h in the presence of a small amount of methanol or ethanol (∼0.3 mL) resulted in complete conversion, and bis-indole 4m and the 5,6-deprotected product 5a were produced in 91% and 87% isolated yield, respectively (Table 3, entry 1), after column chromatographic separation. Similarly, the selective deprotective bis-indolylation of 1,2:5,6-di-O-cyclohexylidene glucofuranose (2b) with indole (3a) also afforded the bis indole (4p) and 1,2-O-cyclohexylidene glucofuranose (5b) in 84% and 80% isolated yield, respectively, under similar reaction conditions (Table 3, entry 2). It is worth mentioning here that a large amount of 75% AcOH in water is the reagent of choice for the selective cleavage of 5,6-O-isopropylidene or cyclohexylidene groups in carbohydrate scaffolds,50 which makes the process less economical, laborious, and not eco-friendly, as the use of toluene is essential to removing acetic acid from the product. With this, we realized that the present bis-indolylation of protected carbohydrate could be a general procedure for the selective deprotection of orthogonally protected sugar with 100% atom economy that allows 100% carbon preservation as well. Therefore, to determine the generality of the procedure, each of the protected carbohydrate derivatives 2c–f and different indoles was subjected to treatment with FeCl3/SiO2 (20 mg) in 0.3 mL of ethanol (to facilitate the stirring) at 80 °C (Table 3). Protected carbohydrates 2c–f produced the corresponding 5,6-deprotected products in excellent yield with simple indoles in a regioselective manner. The functional groups present in di-O-isopropylidene or di-O-cyclohexylidene hexose, such as allyl, benzyl, acetyl, and benzoyl groups, survived under the reaction conditions with concomitant formation of bis-indolyl methanes (4m and 4p). The yield of each deprotected sugar and bis-indolyl methanes (4m and 4p) is given in Table 3 (entries 3–6). In all cases, the 1,2-O-isopropylidene or 1,2-O-cyclohexalidene groups remain intact, as revealed by the NMR spectra of the products. However, in the case of 1,2:5,6-di-O-isopropylidene-D-mannitol, complete deprotection was witnessed, and for clean conversion, 4.0 equivalents of indole were needed (Table 3, entry 7). The present protocol was also applicable to 1,2:3,5-di-O-cyclohexylidene xylofuranose. Under the reaction conditions, only the 3,5-O-protecting group was cleaved (Table 3, entry 8). The selective deprotection of protected hexose is general for other indoles as well; for instance, 2-methyl indole can efficiently convert protected sugars 2a and 2c to the corresponding diol 5a and 5c in 79% and 74% yield, respectively. In each entry (Table 3, entries 9 and 10), 3,3′-(propane-2,2-diyl)bis(2-methy-1H-indole) (4o) was formed in approx. 80% yield.
Table 3 Scope and generality of the SiO2/FeCl3-catalyzed bisindolylation-induced selective deprotection of protected sugar at 100 °C
| Entry |
Protected sugar |
Indole |
Time (h) |
Products (yield, %) |
| 1 |
 |
 |
1.0 |
 |
| 2 |
 |
 |
1.0 |
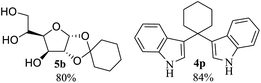 |
| 3 |
 |
 |
1.5 |
 |
| 4 |
 |
 |
1.0 |
 |
| 5 |
 |
 |
1.0 |
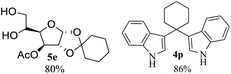 |
| 6 |
 |
 |
1.0 |
 |
| 7 |
 |
 |
3.0 |
 |
| 8 |
 |
 |
1.0 |
 |
| 9 |
 |
 |
1.0 |
 |
| 10 |
 |
 |
1.5 |
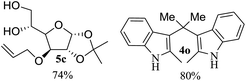 |
Finally, we turned our attention to the recyclability of the catalyst SiO2/FeCl3 used in the bis-indolylation reaction. The recyclability of the catalyst was investigated by recovering the catalyst FeCl3/SiO2 (20 mg) from the reaction of 1-methyl indole (3b, 2 mmol) and benzaldehyde dimethyl acetal (1a, 1.0 mmol) (Table 2, entry 2). After the completion of the reaction (15 min, TLC), the mixture was dissolved in ethyl acetate (5.0 mL) and filtered out through the filter paper. The residue was then washed thoroughly with ethyl acetate (∼5 mL) until no residual product was left. The combined filtrate was concentrated and recrystallized from ethyl acetate–hexane (1:1) to obtain pure product (4b). The residue catalyst was collected from the filter paper, dried under vacuum, weighed, and was used for the next cycle. The weight loss of the catalyst was found to be negligible. The recyclability tests were performed six times in a similar manner, and our results (Fig. 5A) showed that the catalyst retained its activity (92% initially, 92%, 92%, 92%, 91%, 91%, and 89%). We also checked the SEM images and EDAX of the catalyst before and after recycling to see if any morphological changes occurred during its handling in multiple cycles. SEM micrographs of FeCl3/SiO2 (230–400 mesh) show that the particles were random in size and shape and well dispersed. An EDX spectrum of the catalysts confirmed the presence of Si, O, Cl, and Fe elements, suggesting the formation of the FeCl3/SiO2 catalytic system. Similarly, an SEM image of the recycled FeCl3/SiO2 catalyst (after six cycles) was compared with the original images. However, we did not notice any significant differences in morphology and EDAX of the recycled one with the original images (Fig. 5B and C).
 |
| | Fig. 5 (A) Results of the recyclability test of the catalyst FeCl3/SiO2; (B) SEM image and EDAX spectra of the freshly prepared catalyst; (C) SEM image and EDAX spectra of the recycled catalyst (after the 6th cycle). | |
Conclusions
A simple, green, and recyclable catalytic system was developed for the synthesis of 3,3′-bisindolyl(methanes) (BIMs) via the diindolylation of cyclic/acyclic acetals. The reaction occurred under mild and benign conditions using FeCl3/SiO2 as a heterogeneous catalyst without the requirement of any toxic organic solvents. This method relied on a wide range of acetals—aromatic, aliphatic, or carbohydrates—resulting in excellent-to-very-good yields of BIMs. DFT studies were also performed to establish the proposed mechanism and preferential formation of unsymmetrical bisindolylmethanes using equimolar amounts of different indoles. The present protocol was also extended to bisindolylation-induced selective cleavage of protected carbohydrates to diols in a 100% carbon-preservation and maximized atom-economical manner.
Experimental section
General procedure for the indolylation of acetals in the presence of FeCl3/SiO2 (ref. 23 and 46)
To a cone-shaped flask, the acetal (1.0 mmol), indole (2.0 mmol), and FeCl3/SiO2 catalyst (20 mg, 2 mol% of FeCl3) were added (for the carbohydrate substrate, 0.2 to 0.3 mL of alcohol was needed). The reaction mixture was stirred for the stipulated time and temperature mentioned in Tables 2 and 3. After completion of the reaction, the reaction mixture was diluted with EtOAc (5 mL) and filtered. The filtrate was evaporated under vacuum. The desired product was isolated either by crystallization or by column chromatography using ethyl acetate–hexane (1:3 to 3:1).
Physical and spectral data of unknown BIMs
3,3′-((2-Nitrophenyl)methylene) bis(1-methyl-1H-indole) (4i). Yield: 91%, light yellow solid, mp. 160–162 °C; IR (KBr) νmax 3052, 1514, 1467, 1339, 1120, 781 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 7.90 (d, J = 8.0 Hz, 1H), 7.58 (t, J = 8.8 Hz, 1H), 7.48 (t, J = 7.6 Hz, 1H), 7.41 (t, J = 8 Hz, 3H), 7.23 (d, J = 7.6 Hz, 2H), 7.13 (t, J = 7.2 Hz, 2H), 6.94 (t, J = 7.6 Hz, 2H), 6.80 (s, 2H), 6.40 (s, 1H), 3.71 (s, 6H); 13C NMR (100 MHz, DMSO-d6) δ 149.8, 138.1, 137.4, 133.1, 131.0, 128.9, 128.1, 127.0, 124.5, 121.8, 119.2, 119.1, 115.7, 110.3, 34.2, 32.8; HRMS calcd for (C25H21N3O2 + H+) 396.1712, found: 396.1692 (M + H+).
3,3′-(Propane-2,2-diyl)bis(1-methyl-1H-indole) (4n). Yield: 86%, white solid, mp. 130 °C; IR (KBr) νmax 3744, 1463, 1322, 1225, 1049, 734 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.49 (d, J = 8.4 Hz, 1H), 7.30 (d, J = 8.0 Hz, 2H), 7.17 (t, J = 8.4 Hz, 1H), 6.96–6.93 (m, 2H), 3.78 (s, 3H), 1.96 (s, 3H);13C NMR (100 MHz, CDCl3) δ 137.8, 126.7, 125.5, 124.1, 121.5, 120.9, 118.1,109.1, 35.0, 32.7, 31.0, 30.3 HRMS calcd for (C21H22N2 + H+) 303.1861, found: 303.1849 (M + H+).
3,3′-(Propane-2,2-diyl)bis(2-methyl-1H-indole) (4o). Yield: 83%, white solid, mp. 130 °C; IR (KBr) νmax 3378, 2310, 1546, 1453, 1340, 1014, 740 cm−1; 1H NMR (400 MHz, DMSO-d6) δ 10.53 (s, 2H), 7.22 (d, J = 8.0 Hz, 2H), 7.16 (d, J = 8.0 Hz, 2H), 6.84 (t, J = 7.6 Hz, 2H), 6.67 (t, J = 8.0 Hz, 2H), 2.28 (s, 6H), 1.92 (s, 6H); 13C NMR (100 MHz, DMSO-d6) δ 135.4, 130.1, 128.2, 120.2, 119.6, 119.4, 118.0, 110.6, 37.7, 32.2, 14.5; HRMS calcd for C21H22N2 302.1783, found: 302.1747 (M+).
3-((1H-Indol-3-yl)(phenyl)methyl)-5-methoxy-1H-indole (4u). Yield: 61%, off-white solid, mp. 156–158 °C; IR (KBr) νmax 3404, 1484, 1206, 1018 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.93 (s, 1H), 7.83 (s, 1H), 7.42–7.20 (m, 10H), 7.03 (s, 1H), 6.86 (d, J = 8.0 Hz, 2H), 6.67 (d, J = 12.0 Hz, 2H), 5.86 (s, 1H); 13C NMR (100 MHz, CDCl3) δ 153.7, 144.0, 136.8, 131.8, 128.7, 128.2, 127.5, 127.1, 126.2, 124.4, 123.7, 121.9, 120.0, 119.6, 119.4, 119.2, 112.0, 111.7, 111.1, 101.9, 55.9, 40.3; HRMS calcd for (C24H20N2O–H+) 351.1497, found: 351.1516 (M − H+).
3-((1H-Indol-3-yl)(4-nitrophenyl)methyl)-5-methoxy-1H-indole (4v). Yield: 70%, light yellow solid, mp. 180–182 °C; IR (KBr) νmax 3448, 1501, 1341, 1201, 1062, 920, cm−1; 1H NMR (400 MHz, CDCl3) δ 8.17 (d, J = 8.8 Hz, 2H), 8.06 (s, 1H), 7.95 (s, 1H), 7.52 (d, J = 8.8 Hz, 2H), 7.41 (d, J = 8.0 Hz, 1H), 7.42–7.28 (m, 3H), 7.22 (t, J = 8.0 Hz, 1H), 7.06 (d, J = 8.0 Hz, 1H), 6.89 (dd, J = 6.4, 2.4 Hz, 1H), 6.79 (d, J = 2.4 Hz, 1H), 6.71 (s, 1H), 6.67 (s, 1H), 5.96 (s, 1H), 3.73 (s, 3H); 13C NMR (100 MHz, CDCl3) δ 154.0, 151.8, 146.6, 136.8, 131.8, 129.5, 127.1, 126.7, 124.4, 123.7, 123.6, 122.3, 119.6, 118.0, 117.8, 112.3, 112.0, 111.3, 101.6, 76.7, 55.9, 40.2; HRMS calcd for (C24H19N3O3 + H+) 398.1505, found: 398.1513 (M + H+).
3-(2-(1H-Indol-3-yl)propan-2-yl)-1-methyl-1H-indole (4x). Yield: 51%, off-white solid, mp. 102–104 °C; IR (KBr) νmax 3420, 1482, 1326, 1215, 1012 cm−1; 1H NMR (400 MHz, CDCl3) δ 7.90 (s, 1H), 7.47 (q, J = 8.0 Hz, 1H), 7.34 (d, J = 8.0 Hz, 1H), 7.29 (d, J = 8.4 Hz, 1H), 7.18–7.11 (m, 2H), 7.06 (d, J = 2.4 Hz, 1H), 6.96–6.90 (m, 3H), 3.78 (s, 3H), 1.95 (s, 6H); 13C NMR (100 MHz, CDCl3) δ 137.8, 137.1, 128.8, 126.7, 126.4, 125.6, 125.4, 124.0, 121.4, 121.3, 120.9, 120.6, 118.7, 118.1, 111.0, 109.2, 109.1, 100.9, 76.7, 34.9, 32.7, 30.2, 30.0; HRMS calcd for (C20H20N2 + H+) 289.1705, found: 289.1718 (M + H+).
Data availability
The data supporting this article are available as part of the (ESI).†
Author contributions
S. M.—design and conceptualization; B. D.—catalyst preparation, reaction optimization, substrate scope and characterization; K. D.—synthesis of a few starting materials and substrate scope; UCD—theoretical calculations; S. M. and B. D—analysis of the spectral data and writing of the manuscript with input from other authors. All authors reviewed and approved the final version of manuscript.
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
The authors gratefully acknowledge the Central Instrumental Centre (CIC), Tripura University, for the instrumental facility, and the Department of Science and Technology (DST), Government of India, for providing the 400 MHz NMR spectrometer through the DST-FIST programme (No. SR/FST/CSI-263/2015). BD acknowledges Tripura University for the non-NET fellowship.
References
-
(a) T. P. Singh and O. M. Singh, Mini-Rev. Med. Chem., 2018, 18, 9–25 CrossRef CAS PubMed;
(b) Y. Wan, Y. Li, C. Yan, M. Yan and Z. Tang, Eur. J. Med. Chem., 2019, 183, 111691 CrossRef CAS PubMed;
(c) M. Shiri, M. A. Zolfigol, H. G. Kruger and Z. Tanbakouchian, Chem. Rev., 2010, 110, 2250–2293 CrossRef CAS PubMed;
(d) M. Z. Zhang, Q. Chen and G. F. Yang, Eur. J. Med. Chem., 2015, 89, 421–441 CrossRef CAS PubMed.
-
(a) M. Xia, S. Wang and W. B. Yuan, Synth. Commun., 2004, 34, 3175–3182 CrossRef CAS;
(b) M. Damodiran, D. Muralidharan and P. T. Perumal, Bioorg. Med. Chem. Lett., 2009, 19, 3611–3614 CrossRef CAS PubMed;
(c) A. J. K. Karamyan and M. T. Hamann, Chem. Rev., 2010, 110, 4489–4497 CrossRef PubMed;
(d) M. Mari, A. Tassoni, S. Lucarini, M. Fanelli, G. Piersanti and G. Spadoni, Eur. J. Org Chem., 2014, 2014, 3822–3830 CrossRef CAS.
-
(a) K. P. Pandey, M. T. Rahman and J. M. Cook, Molecules, 2021, 26, 3459 CrossRef CAS PubMed;
(b) L. Khanna, Mansi, S. Yadav, N. Misra and P. Khanna, Synth. Commun., 2021, 51, 2892–2923 CrossRef CAS.
-
(a) K. Rathi, O. S. Tiwari, V. Rawat, J. L. Jat, D. K. Yadav and V. P. Verma, Org. Biomol. Chem., 2024, 22, 3287–3298 RSC;
(b) A. Nasreen, R. Varala and K. S. Rao, Org. Commun., 2017, 10, 104–113 CrossRef CAS;
(c) H. Veisi, R. Gholbedaghi, J. Malakootikhah, A. Sedrpoushan, B. Maleki and D. Kordestani, J. Heterocycl. Chem., 2010, 47, 1398–1405 CrossRef CAS;
(d) Z. Zheng, D. Zha, P. Cui, H. Zhang, C. Li, J. Shi and B. Han, Results Chem., 2021, 3, 100247 CrossRef CAS.
- M. Chakrabarty, N. Ghosh, R. Basak and Y. Harigaya, Tetrahedron Lett., 2002, 43, 4075–4078 CrossRef CAS.
- M. M. Heravi, K. Bakhtiari, A. Fatehi and F. F. Bamoharram, Catal. Commun., 2008, 9, 289–292 CrossRef CAS.
- R. S. Balaskar, B. B. Shingate, M. S. Shingare and D. V. Mane, Arab. J. Chem., 2016, 9, S120–S123 CrossRef CAS.
- P. J. Das and J. Das, Tetrahedron Lett., 2012, 53, 4718–4720 CrossRef CAS.
- B. L. Tornquist, D. P. G. Bueno, J. C. M. Willig, I. M. D. Oliveira, A. S. Helio, J. Rafique, S. Saba, B. A. Iglesias, G. V. Botteselle and F. Manarin, ChemistrySelect, 2018, 3, 6358–6363 CrossRef CAS.
- S.-J. Ji, M.-F. Zhou, D.-G. Gu, S.-Y. Wang and T.-P. Loh, Synlett, 2003, 2077–2079 CrossRef CAS.
- A. Suárez, F. Martíne, S. Suárez-Pantiga and R. Sanz, ChemistrySelect, 2017, 1, 1–5 Search PubMed.
- J. R. Satam, K. D. Parghi and R. V. Jayaram, Catal. Commun., 2008, 9, 1071–1078 CrossRef CAS.
- A. Z. Halimehjani and V. Barati, ChemistrySelect, 2018, 3, 3024–3028 CrossRef CAS.
- R. M. N. Kalla, S. C. Hong and I. Kim, ACS Omega, 2018, 3, 2242–2253 CrossRef CAS PubMed.
- P. Thirupathi and S. S. Kim, J. Org. Chem., 2010, 75, 5240–5249 CrossRef CAS PubMed.
- N. C. Ganguly, P. Mondal and S. K. Barik, Green Chem. Lett. Rev., 2012, 5, 73–81 CrossRef CAS.
- H. Lin, Y. Zang, X. Sun and G. Lin, Chin. J. Chem., 2012, 30, 2309–2314 CrossRef CAS.
- K. A. Chavan, M. Shukla, A. N. Singh Chauhan, S. Maji, G. Mali, S. Bhattacharyya and R. D. Erande, ACS Omega, 2022, 7, 10438–10446 CrossRef CAS PubMed.
- S. R. Mendes, S. Thurow, F. Penteado, M. S. da Silva, R. A. Gariani, G. Perin and E. J. Lenardão, Green Chem., 2015, 17, 4334–4339 RSC.
- M. Esmaielpour, B. Akhlaghinia and R. Jahanshahi, J. Chem. Sci., 2017, 129, 313–328 CrossRef CAS.
- N. A. Khalafi, M. Nourisefat and F. Panahi, RSC Adv., 2014, 4, 22497–22500 RSC.
- C. Ramesh, J. Banerjee, R. Pal and B. Das, Adv. Synth. Catal., 2003, 345, 557–559 CrossRef CAS.
- B. Deb, S. Debnath, A. Chakraborty and S. Majumdar, RSC Adv., 2021, 11, 30827–30839 RSC.
- Y.-S. Zhao, H.-L. Ruan, X.-Y. Wang, C. Chen, P.-F. Song, C.-W. Lü and L.-W. Zou, RSC Adv., 2019, 9, 40168–40175 RSC.
- Z. B. Xie, D. Z. Sun, G. F. Jiang and Z. G. Le, Molecules, 2014, 19, 19665–19677 CrossRef PubMed.
- J. Xiao, H. Wen, L. Wang, L. Xu, Z. Hao, C.-L. Shao and C.-Y. Wang, Green Chem., 2016, 18, 1032–1037 RSC.
-
(a) J.-B. Peng, X. Qi and X.-F. Wu, Synlett, 2016, 28, 175–194 CrossRef;
(b) J.-B. Peng, X. Qi and X.-F. Wu, ChemSusChem, 2016, 9, 2279–2283 CrossRef CAS PubMed;
(c) Y. Bai, D. C. Davis and M. Dai, J. Org. Chem., 2017, 82, 2319–2328 CrossRef CAS PubMed.
-
(a) J. Jin, Y. Li, S. Xiang, W. Fan, S. Guo and D. Huang, Org. Biomol. Chem., 2021, 19, 4076–4081 RSC;
(b) V. D. Kadu, S. N. Chandrudu, M. G. Hublikar, D. G. Raut and R. B. Bhosale, RSC Adv., 2020, 10, 23254–23262 RSC.
- F. Stanek, R. Pawlowski, P. Morawska, R. Bujok and M. Stodulski, Org. Biomol. Chem., 2020, 18, 2103–2112 RSC.
- P. Saini, P. Kumari, S. Hazra and A. J. Elias, Chem.–Asian J., 2019, 14, 4154–4159 CrossRef CAS PubMed.
- N.-K. Nguyen, M.-T. Ha, H. Y. Bui, Q. T. Trinh, B. N. Tran, V. T. Nguyen, T. Q. Hung, T. T. Dang and X. H. Vu, Catal. Commun., 2021, 149, 106240 CrossRef CAS.
- T. Pillaiyar, E. Gorska, G. Schnakenburg and C. E. Müller, J. Org. Chem., 2018, 83, 9902–9913 CrossRef CAS PubMed.
- A. Devi, M. M. Bharali, S. Biswas, T. J. Bora, J. K. Nath, S. Lee, Y.-B. Park, L. Saikia, M. J. Baruah and K. K. Bania, Green Chem., 2023, 25, 3443–3448 RSC.
- E. M. Galathri, T. J. Kuczmera, B. J. Nachtsheim and C. G. Kokotos, Green Chem., 2024, 26, 825–831 RSC.
- P. G. M. Wuts, Greene's Protective Groups in Organic Synthesis, John Wiley & Sons, Inc., Hoboken, New Jersey, 5th edn, 2014 Search PubMed.
- A. G. Volbeda, G. A. van der Marel and J. D. C. Codée, Protecting Group Strategies in Carbohydrate Chemistry, in Protecting Groups – Strategies and Applications in Carbohydrate Chemistry, ed. S. Vidal, Wiley-VCH, Weinheim, 2019, pp. 1–28 Search PubMed.
-
(a) D. D. Diaz, P. O. Miranda, J. I. Padron and V. S. Martin, Curr. Org. Chem., 2006, 10, 457–476 CrossRef;
(b) M. Mishra, S. Mohapatra, N. P. Mishra, B. K. Jena, P. Panda and S. Nayak, Tetrahedron Lett., 2019, 60, 150925 CrossRef CAS;
(c) I. Bauer and H.-J. Knolker, Chem. Rev., 2015, 115, 3170–3387 CrossRef CAS PubMed;
(d) A. A. O. Sarhan and C. Bolm, Chem. Soc. Rev., 2009, 38, 2730–2744 RSC;
(e) S. Ruengsangtongkul, P. Taprasert, U. Sirion and J. Jaratjaroonphong, Org. Biomol. Chem., 2016, 14, 8493–8502 RSC;
(f) C. Chantana and J. Jaratjaroonphong, J. Org. Chem., 2021, 86, 2312–2327 CrossRef CAS PubMed.
- S.-J. Ji, M.-F. Zhou, D.-G. Gu, Z.-Q. Jiang and T.-P. Loh, Euro. J. Org. Chem., 2004, 1584–1587 CrossRef CAS.
- V. Terrasson, J. Michaux, A. Gaucher, J. Wehbe, S. Marque, D. Prim and J.-M. Campagne, Euro. J. Org. Chem., 2007, 5332–5335 CrossRef.
- C. Fan, R. Li, J. Duan, K. Xu, Y. Liu, D. Wang and X. He, Synth. Commun., 2022, 52, 1155–1164 CrossRef CAS.
- S. Majumdar, A. Chakraborty, S. Bhattacharjee, S. Debnath and D. K. Maiti, Tetrahedron Lett., 2016, 57, 4595–4598 CrossRef CAS.
-
(a) D. Habibi and M. Nasrollahzadeh, Synth. Commun., 2010, 40, 3159–3167 CrossRef CAS;
(b) M. A. Taher, C. Karami, M. S. Arabi, H. Ahmadian and Y. Karami, Int. Nano Lett., 2016, 6, 85–90 CrossRef CAS;
(c) E. Keinan and Y. Mazur, J. Org. Chem., 1978, 43, 1020–1022 CrossRef CAS.
-
(a) S. E. Sen, S. L. Roach, J. K. Boggs, G. J. Ewing and J. Magrath, J. Org. Chem., 1997, 62(19), 6684–6686 CrossRef CAS;
(b) T. Xu, Q. Yang, D. Li, J. Dong, Z. Yu and Y. Li, Chem.–Eur. J., 2010, 16, 9264–9272 CrossRef CAS PubMed.
-
(a) S. Ghosh, S. Khamarui, K. S. Gayen and D. K. Maiti, Sci. Rep., 2013, 3, 2987 CrossRef PubMed;
(b) S. Ghosh, S. Debnath, U. K. Das and D. K. Maiti, Ind. Eng. Chem. Res., 2017, 56(42), 12056–12069 CrossRef CAS.
-
(a) S. Majumdar, M. Chakraborty, D. K. Maiti, S. Chowdhury and J. Hossain, RSC Adv., 2014, 4, 16497–16502 RSC;
(b) S. Majumdar, M. Chakraborty, N. Pramanik and D. K. Maiti, RSC Adv., 2015, 5, 51012–51018 RSC;
(c) K. Das and S. Majumdar, RSC Adv., 2022, 12, 21493–21502 RSC;
(d) A. Rudra Paul, S. Debnath and S. Majumdar, ChemistrySelect, 2023, 8, e202300007 CrossRef CAS;
(e) A. Rudra Paul, S. Sarkar, J. Hossain, S. A. Hussain and S. Majumdar, Res. Chem. Intermed., 2022, 48, 4963–4985 CrossRef CAS;
(f) K. Das, B. Das, B. Paul, R. Natarajan and S. Majumdar, Silicon, 2024, 16, 967–977 CrossRef CAS.
- B. Das, A. Bhattacharyya, B. Paul, R. Natarajan and S. Majumdar, RSC Adv., 2024, 14, 33512–33523 RSC.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, et al, Gaussian 16, Revision C.01, Gaussian, Inc., Wallingford CT, 2019 Search PubMed.
- J.-I. Aihara, J. Phys. Chem. A, 1999, 103, 7487–7495 CrossRef CAS.
- N. Blankevoort, P. Bastante, R. J. Davidson, R. J. Salthouse, A. H. S. Daaoub, P. Cea, S. M. Solans, A. S. Batsanov, S. Sangtarash, M. R. Bryce, N. Agrait and H. Sadeghi, ACS Omega, 2024, 9, 8471–8477 CAS.
-
(a) S. Majumdar, A. Bhattacharjya and A. Patra, Tetrahedron, 1999, 55, 12157–12174 CrossRef CAS;
(b) G. V. M. Sharma, I. S. Reddy, V. G. Reddy and A. V. Rama Rao, Tetrahedron: Asymmetry, 1999, 10, 229–235 CrossRef CAS.
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ,
Kamal Das
,
Kamal Das