
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceMetal nanoparticles in neuroinflammation: impact on microglial dynamics and CNS function
Masood Alaei†
ab,
Khadijeh Koushki†
c,
Kimia Taebi†
ab,
Mahdieh Yousefi Taba
d,
Samaneh Keshavarz Hedayati
e and
Sanaz Keshavarz Shahbaz‡
 *fb
*fb
aStudent Research Committee, Qazvin University of Medical Sciences, Qazvin, Iran
bUSERN Office, Qazvin University of Medical Science, Qazvin, Iran. E-mail: Sanaz.ks_023@yahoo.com; sanaz.keshavarzshahbaz@jax.org
cDepartment of Neurosurgery, University of Texas Houston Health Science Center (UTHealth), Houston, TX, USA
dDepartment of Anatomy and Cell Biology, Faculty of Medicine, Mashhad University of Medical Sciences, Mashhad, Iran
eDepartment of Pharmacology, Faculty of Medicine, Qazvin University of Medical Science, Qazvin, Iran
fCellular and Molecular Research Center, Research Institute for Prevention of Non-Communicable Disease, Qazvin University of Medical Sciences, Qazvin, 34197-59811, Iran
First published on 18th February 2025
Abstract
Microglia, the primary immune cells of the central nervous system (CNS), are crucial in maintaining brain homeostasis and responding to pathological changes. While they play protective roles, their activation can lead to neuroinflammation and the progression of neurodegenerative diseases. Metal nanoparticles (NPs), due to their unique ability to cross the blood–brain barrier (BBB), have emerged as promising agents for drug delivery to the CNS. In this way, we aim to review the dual role of metal-containing NPs, gold (AuNPs), silver (AgNPs), iron oxide (IONPs), zinc oxide (ZnONPs), cobalt (CoNPs), titanium dioxide (TiO2NPs), and silica (SiO2NPs) in modulating microglial activity. Some NPs promote anti-inflammatory effects, while others exacerbate neuroinflammation. We examine how these NPs influence microglial activation, focusing on their potential therapeutic benefits and risks. A deeper understanding of NP-microglia interactions is crucial for developing safe and efficient treatments for neuroinflammatory and neurodegenerative disorders.
 Masood Alaei | Masood Alaei holds a Bachelor's degree in Medical Laboratory Sciences from Qazvin University of Medical Sciences. As a member of the Universal Scientific Education and Research Network (USERN), he has been actively involved in research, authoring review articles on nanotechnology and conducting original laboratory-based research projects. |
 Khadijeh Koushki | Khadijeh koushki is a researcher in the field of immunology, who works as a PostDoc researcher at The University of Texas Health Science Center at Houston (UTHealth). She has published several articles on topics such as asthma and allergy immunotherapy, drug delivery systems and autoimmune disorders. She has also received a PhD degree in immunology from Mashhad University of Medical Science. |
 Kimia Taebi | Kimia Taebi earned a Bachelor's degree in Medical Laboratory Sciences from Qazvin University of Medical Sciences. She is a member of the Universal Scientific Education and Research Network (USERN) and has contributed to various scientific publications. Her work includes review articles in the field of nanotechnology as well as original research articles in laboratory sciences. |
 Mahdieh Yousefi Taba | Mahdieh Yoosefi Taba earned a Master's degree in Anatomical Sciences and Cell Biology from Mashhad University of Medical Sciences. Her research focuses on aging models, with a particular emphasis on investigating the protective effects of therapeutic agents against oxidative stress in infertility and hippocampal dark neurons. |
 Samaneh Keshavarz Hedayati | Samaneh Keshavarz Hedayati is a researcher in the field of pharmacology and microbiology, who works as a research demonstrator in Qazvin University of Medical Science in Iran. She has also received a MSc. degree in microbiology. |
 Sanaz Keshavarz Shahbaz | Sanaz Keshavarz Shahbaz is a researcher in the field of immunology, who works as a research assistant professor at Qazvin University of Medical Science in Iran and PostDoc research associates at The Jackson laboratory for genomic medicine, Farmington, CT, USA. She has published several articles on topics such as asthma and allergy immunotherapy, transplantation immunology, cancer, drug delivery systems and autoimmune disorders, neuroimmunology. She has also received a PhD degree in immunology from Mashhad University of Medical Science. |
1. Introduction
Neuroinflammation is increasingly recognized as a pivotal factor in the pathogenesis of various central nervous system (CNS) disorders, including neurodegenerative diseases such as Alzheimer's and Parkinson's. This inflammatory response, while essential for maintaining homeostasis and responding to injury, can become detrimental when dysregulated, leading to neuronal damage and exacerbation of disease progression. Microglia, the resident immune cells of the CNS, play a dual role in neuroinflammation; they are crucial for both protective responses and the development of inflammatory processes.1Recent advancements in nanomedicine have highlighted the potential of polymer and metal nanoparticles (NPs) as innovative therapeutic agents capable of modulating microglial activity and influencing neuroinflammatory responses. Recently, we reviewed polymeric NP carriers applied to deliver microglial inhibition in neurological disorders with remarkable results, showcasing the promising role of these NPs in microglial modulation during drug delivery.2
In addition, due to the unique physicochemical properties—such as high surface area, tunable size, and functional versatility—Metal NPs can effectively cross the blood–brain barrier (BBB),3 presenting new opportunities for targeted drug delivery in treating CNS disorders. Various metal-containing nanoparticles, including gold (AuNPs),4 silver (AgNPs), iron oxide (IONPs), zinc oxide (ZnONPs), cobalt (CoNPs), titanium dioxide (TiO2NPs), and silica (SiO2NPs), have been shown to interact with microglia, either promoting anti-inflammatory effects or exacerbating neuroinflammation.5
In this way, this review aims to provide a comprehensive analysis of metal nanoparticles' impact on microglial dynamics and their implications for CNS function. By examining the complex interplay between these nanomaterials and microglial cells, we seek to elucidate their potential therapeutic benefits and risks in addressing neuroinflammatory conditions. Understanding these interactions is crucial for developing safe and effective treatments for neurodegenerative diseases and advancing the field of nanomedicine.
2. Microglia and their neuroinflammatory roles
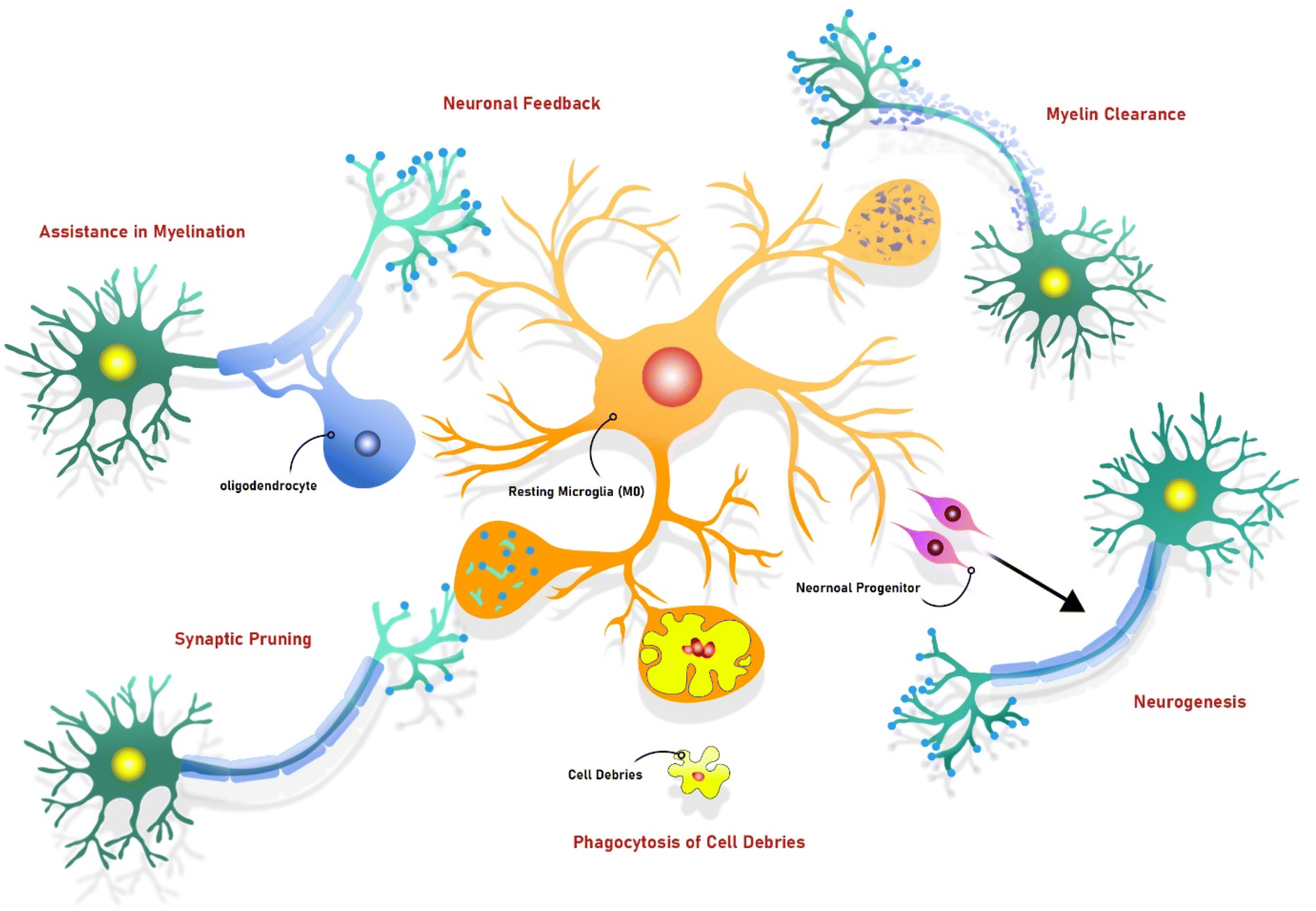
Microglia are macrophage-like innate immune cells in the central nervous system (CNS), serving as the brain's primary effector cells that monitor the CNS for infections and injuries.6–8 As the first responders to foreign pathogens and harmful particles in the brain, microglia act as key indicators of brain damage.9–11 Constituting 20% of glial cells located in the brain, microglia originate from hematopoietic stem cells.12 During brain development, these cells enter the brain via circulation and can exhibit neurotoxic or neuroprotective responses in their microenvironment.13Neuroprotective functions of microglia include maintaining homeostasis by regulating the brain's internal environment, metabolic regulation, and facilitating immune responses.14,15 In their resting state, known as the M0 phenotype, microglia clear apoptotic debris, remove dysfunctional synapses, and support pruning in developing brains. This process involves synaptic remodeling, phagocytosis of cells with intracellular inclusions, neuronal feedback,16 facilitating myelination,17 neurogenesis, and trophic maintenance of neurons.18,19 These functions are crucial for preserving a healthy brain environment.
Microglia also have two activated states: the “M2” anti-inflammatory phenotype and the “M1” proinflammatory phenotype. M2 microglia are responsible for healing-related actions, such as maintaining homeostasis and promoting anti-inflammatory processes. They contribute to the generation of anti-inflammatory cytokines and neurotrophic agents.10,20,21 In contrast, M1 microglia are the first line of defense, responsible for homeostasis and pro-killing functions. They can produce proinflammatory cytokines interleukin-1b (IL-1b), IL-17, IL-12, IL-6, IL-18, IFN-c, IL-23, inducible nitric oxide synthase (iNOS), tumour necrosis factor-alpha (TNF-α), cyclooxygenase-2 (COX-2), reactive oxygen species (ROS), prostaglandin E2 (PGE2), and (MHC-II).22,23
Although the pro-inflammatory functions of the M1 phenotype are protective in certain situations, excessive release of cytotoxic substances has been attributed to the development of neuroinflammatory disorders.24 Many studies have shown that microglial activation plays a pivotal role in the pathogenesis of neurodegenerative disorders, including Parkinson's disease, Alzheimer's disease (AD), psychiatric disease, ischemic disease, traumatic brain injury, and stroke.10,25–27 Microglia become activated in response to pathogen-associated molecular patterns (PAMPs), danger-associated molecular patterns (DAMPs), and certain nanostructures, triggering a pro-inflammatory response.
To perform their surveillance functions, microglia are equipped with different signaling immunoreceptors, such as Toll-like receptors (TLR4 and TLR2), complement phagocytic receptors (CR4 and CR3), and scavenger receptors (Cluster of Differentiation-36 (CD36) and CD204) to interact with extracellular species.25,28,29 When neurons are exposed to harmful stimuli, they begin to generate “help me” signals, including fractalkine, interleukin-34 (IL-34), and CX3C chemokine.30,31 In response, microglia become activated and release proinflammatory cytokines, including TNF-α and IL-1β, along with neurotoxic molecules, such as ROS.32 Moreover, activated microglia can release glutamate, which causes an increase in the neuronal and neurite number, thereby contributing to neurodegenerative disorder exacerbation.25,33
Upon stimulation of microglial surface receptors, several signaling pathways become activated. They induce the production of inflammatory cytokines, the NLRP inflammasome activation, and beta-secretase enzyme (BACE) expression,29 ultimately driving neuroinflammation and neuronal death. Microglia can also accidently damage neurons while attempting to limit infections by producing cathepsin B, superoxide, nitric oxide, and derivative oxidants.34 Furthermore, microglia-mediated reduction in insulin-like growth factor 1 (IGF-1) and nutritional brain-derived neurotropic factor (BDNF) contributes to neuronal death35,36 (Fig. 1). These mechanisms highlight the potential of microglia M1 in the beginning and progression of neurodegenerative diseases.
 | ||
| Fig. 1 Normal functions of microglia in the CNS. | ||
Given the crucial role of microglia, particularly M1 microglia, in neuroinflammatory processes, potent inhibitors that target microglial immune activity may offer promising strategies for addressing these issues.
Resting microglia, known as M0 microglia, perform multiple health-promoting functions. They have the potential to support neuronal progenitors in the neurogenesis process. Additionally, they can appropriately eliminate cell debris and dead cell bodies through phagocytosis. Moreover, M0 microglia can phagocytose unnecessary synaptic connections (called synaptic pruning) to leave more space to form new connections between neurons. An oligodendrocyte is a cell that is responsible for forming myelin around axons. Interestingly, M0 microglia participate in this process by assisting oligodendrocytes. Microglia M0 also contribute to the neuronal feedback mechanism. When neurons are activated, microglia use negative neuronal feedback to prevent neuronal overactivation, thus leading to a balanced neuronal environment. Additionally, resting microglia can play a role in myelin clearance by attempting to phagocytose myelin debris to prevent impaired neurogenesis.
3. Inhibitors of microglial immune activity
According to microglia's vital role in neuroinflammation and the progression of neurodegenerative diseases, inhibiting their activity is a prominent therapeutic solution. Several compounds, including resveratrol, curcumin, cannabidiol,37 ginsenosides, flavonoids, sulforaphane,38 candesartan cilexetil,39 propentofylline, luteolin,40 quercetin,41 and minocycline,42 are available as conventional and potent inhibitors of microglial activity. Bellow, some of these compounds and their inhibitory effects on microglia are briefly discussed.Resveratrol, a polyphenol abundant in peanuts, raisins, red grapes, and berries, possesses anti-inflammatory, antioxidant, and antiapoptotic properties.43,44 Studies have illustrated that resveratrol can limit microglial activity and mitigate rotenone-induced neurotoxicity, CD11 (a microglial activity marker), TNF- α, and IL-1β.45 Curcumin, another well-known inhibitor, is a potent anti-inflammatory and antioxidant agent found in turmeric. Interestingly, curcumin plays a suppressive role in microglial activity and decreases microglia-induced inflammatory cytokines.46 It can also block the MAPK signaling pathway, which further inhibits the activation of NF-κβ.47 Curcumin suppresses the production of COX-2 and other inflammatory cytokines by modulating TLR4 signaling.48 Quercetin, a natural flavonoid present in vegetables and fruits, namely green tea, onions, apples, red grapes, and berries, is also a potent inhibitor of microglial activity due to its antioxidant and anti-inflammatory properties.41 The treatment with quercetin reduces TLR-4 expression in the hippocampus and cortex, a key receptor involved in microglial activation.49 In addition, it can reduce the expression of ionized calcium-binding adapter molecule 1 (Iba-1), IL-1β, TNFα, and COX2.50 Minocycline, a well-known antibiotic, has also been extensively studied for its ability to inhibit microglial function, reducing the number of microglial inflammatory mediators.51,52 Minocycline can also reduce MHC-II expression in microglia.42
While these drugs offer significant potential in mitigating microglial immune responses, drug delivery to the CNS is still a considerable challenge because of the restrictive nature of the blood–brain barrier (BBB). Its specific structure, surrounding cells, and molecular transport mechanisms limit the efficient delivery of many therapeutic agents. Therefore, the following section further discusses these challenges and potential solutions.
4. CNS drug delivery through the BBB and its challenges
BBB, along with extracellular matrix (ECM) and nonfenestrated monolayer of cells, significantly controls the microenvironment of CNS neurons. These structures protect the CNS from circulating toxins, infectious agents, and harmful substances, such as foreign microorganisms.53–56 The BBB is formed by endothelial cells, whose proliferation is stimulated by neighboring cells such as astrocytes (which surround brain vessels) and pericytes (which help maintain the integrity of the BBB)57Various transport systems control the traffic of substances across the BBB, including fenestra, transendothelial channels, pinocytotic vesicles, active efflux transport proteins, and breast cancer resistance proteins.58 These systems are crucial in controlling the flow of specific drugs and essential nutrients into the CNS. Passive distribution via a paracellular or transcellular pathway for low molecular weight or lipophilic substances (the majority of CNS-targeting drugs); vesicular trafficking, such as adsorptive-mediated transcytosis (for positively-charged substances); receptor-mediated transcytosis (an energy-dependent pathway for proteins hormones and proteins); and carrier-mediated transport (for amino acids and glucose) are some examples.59–61 The BBB's relative impermeability is primarily due to tight junctions between endothelial cells.62,63 The mentioned mechanisms and complexes are potential obstacles for most drugs to enter the CNS, making drug delivery challenging with high failure rates and increased costs.64 Even if drugs manage to cross the BBB, achieving therapeutic concentrations within the CNS can be difficult. Ensuring that drugs are effective without causing adverse side effects and neurotoxicity remains a critical challenge.65 Also, CNS disorders often require targeted treatments that are tailored to individual patients. Achieving this specificity in drug delivery systems is complex and ongoing.66 Moreover, many conventional therapeutic agents suffer from low bioavailability due to rapid metabolism or elimination, leading to degradation. Therefore, encapsulating drugs can protect them from degradation and improve their pharmacokinetic profiles.67 The ability to control drug release is also vital for maintaining therapeutic concentrations over time and avoiding excessive drug dosages, which many conventional drugs lack this ability.68 These problems can lead to ineffective treatment for CNS disorders such as cerebral malignancies due to low brain penetration. Over the last decade, researchers have developed various technologies to overcome these challenges. One promising approach is using targeted vectors (peptides, proteins, antibodies, or specific formulations) to aid the transport of drugs across the BBB.69 Additionally, nano-delivery systems have gained attention as a novel strategy. These systems offer the possibility to improve therapeutic precision and maintain medicine efficacy while minimizing toxicity.23,70
5. Nanoparticles for drug delivery to the brain
Advances in biotechnology have shifted therapeutic approaches from conventional medicine to nanotherapeutic agents.71 Due to the challenges linked to macromolecular drugs crossing the BBB and their inability to target specific sites and ineffective dosages, nanotherapeutics have gained significant attention for drug delivery to the CNS and targeting microglia-related neuroinflammation.72 Nanomaterials include nonpolymeric, lipid-based, polymer-based, and metal-based nanoparticles. These NPs range in diameter from 1 to 100 nanometers (nm). These ultrafine particles exhibit unique chemical and physical features that differ significantly from larger particles of the same substance because of their high surface area-to-volume ratio.73,74 Some NPs exist in the environment, while others are engineered in industrial settings.75Some NPs can traverse vessel walls and enter brain tissue. This ability is mainly attributed to their physiochemical characteristics, including size, shape, surface chemistry, surface charge, and surface traits.76 Therefore, engineering the NPs to modify their morphological features enhances their ability to cross the BBB. One strategy involves designing the NPs in combination with elements specific to pathological sites and BBB-penetrating molecules, such as the spontaneous exploitation of NPs with trans-Golgi network (TGN) peptides and the cancer cell-specific aptamer AS1411. Such modification can remarkably improve BBB penetration and target delivery.77 The shape and surface characteristics of NPs also influence how microglia internalize them;78 for instance, spiky “urchin-shaped” gold NPs(AuNps) show higher levels of microglial uptake in contrast to rod or spherical-shaped AuNPs.72,79 Hence, the way microglia take NPs up is highly dependent on the design of the NP surface and properties.80 Microglia internalize NPs primarily through active processes like invagination and endocytosis.69 However, certain NPs may also passively diffuse across the cell membrane. Furthermore, the uptake of NPs varies between microglial phenotypes. Compared with resting microglia, lipopolysaccharide (LPS)-activated microglia display greater dendrimer uptake, which is associated with increased endocytosis.81
Once NPs reach the brain tissue, they can release their therapeutic cargo at the target location in a time-dependent way and navigate the drugs (genes, small molecular agents, and biomolecules) to the target organelle without being trapped in endo/lysosomes. Various strategies have been developed to facilitate lysosomal escape. One common approach is the proton sponge effect, where pH-sensitive nanoparticles swell and disrupt the lysosomal membrane upon exposure to the acidic environment, leading to the release of their contents into the cytosol. Other methods include osmotic lysis, which results from the disassembly of nanoparticles in response to low pH, and mechanical disruption techniques that utilize nanomechanical actions or photochemical processes to destabilize lysosomal membranes.82 This controlled release mechanism decreases the required drug dosage and side effects of using nanomaterials.56,83,84 Engineering NPs is also critical for drug delivery. One example involves using stimulus-sensitive bonds to ensure the accurate release of cargo in the expected areas in response to spatial variations in redox capacity.85 Additionally, NPs designed with epidermal growth factor (EGF) and two types of bioresponsive bonds that enhance vascular permeability86 can deliver drugs directly to subcellular organelles, improving drug efficacy in the brain.87,88 Such designs have been employed to deliver DNA-binding agents and ROS-generating drugs into mitochondria,76,89 facilitating the treatment of stroke, glioma, epilepsy, and AD. Besides, the unique electrical and optical traits of some nanoparticles enable them to treat CNS disorders.71
Put simply, NPs favor the treatment of neurodegenerative disorders by delivering essential drugs to the brain, which is partially impossible for macromolecular drugs to reach alone. Nonetheless, metal-containing NPs hold promise for enhancing drug delivery performance compared to NPs alone. However, they also present side effects such as toxicity in the brain by switching M0 microglia to the M1 phenotype. Alternatively, they may alleviate the neuroinflammation by promoting the M2 phenotype. Therefore, the following section further discusses how metal-containing NPs, as multifaceted substances, influence brain health and neuroinflammatory conditions in detail.
6. Metal-containing NPs: a double-edged sword
Inorganic NPs, including metals (such as iron, silver, and gold) and metal oxides (including zinc oxide, iron oxide, titanium dioxide, cerium oxide, etc.), have gained considerable interest in recent years because of their diverse usages across medical, sunscreen, cosmetic, and industrial fields.90 These metal-containing NPs possess unique physicochemical features, making them helpful in diagnosing and treating CNS disorders.91 Their ability to translocate into the CNS through the BBB, eye-to-brain, nerve signaling pathways, and cell uptake opens new possibilities for therapeutic interventions.75Once inside the brain, metal-containing NPs are immediately internalized by microglia and astrocyte-like (ALT) cells. M1 microglia produce CCL2 and proinflammatory cytokines, for instance, NO, IL-12, IL-1β, and IL-6, which result in acute neuroinflammatory reactions. On the contrary, the M2 phenotype releases anti-inflammatory cytokines such as TGF-β, IL-10, and IL-4, aiding the resolution of neuroinflammation and repairing the damaged brain.92 Metal-containing NPs can influence this polarization, with some promoting M1 activation and exacerbating inflammation, while others encourage the M2 phenotype, facilitating neuroprotection and recovery.93
Metal NP-induced M1 phenotype can exert neurotoxic effects through various mechanisms, including the generation of ROS, which leads to oxidative stress, inducing neuronal apoptosis and necrosis and contributing to neurodegenerative diseases.94 Furthermore, metal NPs can trigger the activation of microglia, leading to the release of pro-inflammatory cytokines, exacerbating neuroinflammation, and compromising neuronal health.95 The neurotoxic potential of metal NPs is significantly influenced by their physicochemical properties. Smaller NPs generally exhibit higher reactivity and greater cellular uptake, which can enhance their therapeutic efficacy but also increase the risk of toxicity. Surface modifications, such as PEGylation, can alter NP-cell interactions, improve circulation time, and reduce immunogenicity, potentially mitigating some toxic effects.96 The neurotoxicity of metal NPs is often dose-dependent, with low concentrations potentially eliciting beneficial effects by modulating microglial activity and promoting anti-inflammatory responses. Higher concentrations can overwhelm cellular defense mechanisms, leading to toxicity. This highlights the crucial need to optimize dosages in therapeutic applications to maximize efficacy while minimizing adverse effects.97 Chronic exposure to metal NPs may lead to cumulative neurotoxic effects that are not immediately apparent. NP prolonged exposure potentially results in significant alterations in microglial function and neuronal integrity, leading to long-term cognitive deficits or the exacerbation of existing neurological conditions.98
For instance, silver nanoparticles (AgNPs) are able to promote M1 polarization and induce neurotoxicity. Duffy et al. reported that AgNPs triggered the production of proinflammatory cytokines like TNF-α in BV2 microglial cells, leading to neuroinflammatory responses.99 Additionally, AgNPs have been shown to enhance the level of the proinflammatory chemokine CXCL13 in microglia, astrocytes, and Neuro2a (N2a) cells, further elevating the levels of IL-1β.100 Besides silver, other metal-based NPs, like titanium dioxide nanoparticles (TiO2NPs), have been linked to neuroinflammatory responses. TiO2NPs can activate inflammasomes and nuclear factor-κB (NF-κB), activating microglia and subsequent inflammation.75
Despite these proinflammatory effects, some metal-containing NPs also exhibit anti-inflammatory properties. AuNPs, for instance, have been evidenced to prevent the proinflammatory reactions in microglia by promoting M2 phenotype, thereby contributing to CNS repair.101 Similarly, exposure to AgNPs has been found in another article to be associated with a reduction in inflammation. This could be possibly due to the detoxification of silver ions through sulfidation and the activation of hydrogen sulfide-synthesizing enzymes.102 Moreover, iron oxide nanoparticles (IONPs) have been demonstrated to inhibit the production of IL-1β in LPS-stimulated microglia, further underscoring the potential neuroprotective role of metal-based NPs.103 (Fig. 2).
 | ||
| Fig. 2 The possible functions of metal-containing nanoparticles in microglial activation. | ||
Although remarkable endeavors have been made to understand the neurotoxic and neuroprotective effects of metal-containing NPs, much remains to be learned about their role in neuroinflammation. Additionally, since studies have shown that microglia activation can serve as an early warning and defensive response against exogenous NPs that invade and accumulate in the CNS, it is crucial to investigate the interaction between various metal-containing NPs conjugated with specific drugs and microglia. Hence, additional exploration is needed to fully exploit the ability of nanotechnology to treat CNS disorders.
In the following section, we aim to comprehensively discuss some well-known metal-containing NPs and their conjugation with certain drugs, highlighting how NP uptake by microglia influences treatment outcomes (Table 1).
| NP type | The surface coating | NP properties | Cell type/animal models | Mechanisms & outcomes | Ref |
|---|---|---|---|---|---|
| a Abbreviations: Au, gold; Ag, silver; IO, iron oxide; Co, cobalt; ZnO, zinc oxide; TiO2, titanium dioxide; SiO2, silica; Si, silicon. | |||||
| Au | Gold-quercetin NPs | 27 nm, 100, 200, 400 μg mL−1 | LPS-stimulated microglia | Significant decrease in both the transcriptional and translational levels of inducible NO synthase, cyclooxygenase-2; COX-2 and iNOS | 110 |
| Inhibiting the release of NO and proinflammatory PGE2 from LPS-stimulated microglia without causing any cytotoxic effect | |||||
| Au | AuNP-FIB-BS hard corona | 50–3 nm, dose: 26 μg mL−1 | Murine BV2 microglia | Significant decrease in oxidative stress and ROS generation | 111 |
| Significant increase in cellular uptake | |||||
| Au | Ephedra sinica Stapf extract extract-capped AuNPs | 57.6 ± 3.07 nm | Microglia | Decrease in the production of the proinflammatory cytokines, including IL-1β, IL-6, and TNF-α in LPS-stimulated microglia through the downregulation of JAK/STAT, NF-κB, IKK-α/β, JNK, and p38 MAPK signaling pathways | 112 |
| Upregulation of NQO1 and HO-1 | |||||
| Activation of AMPK and Nrf2 | |||||
| Au | Polyethyleneglycol-coupled GNPs | 8.09 ± 3.60 nm (85 × 106 nL−1) | Microglia | Self-limited, transient, and predominantly localized cellular response of microglia at the injection site within 3 to 90 days following intracerebral injection | 113 |
| No chronic microglial response by NPs | |||||
| Au | Paeonia moutan-functionalized GNPs | 100 nm, 5, 10 and 20 μg mL−1 | BV2 microglia and C57BL/6 mice/mouse model of parkinsonian | Inhibiting the inflammatory cytokines (IL-1β, IL-6, and TNFα) and NO synthesis | 115 |
| Trapping the reactive oxygen in LPS-stimulated BV2 murine microglia suffering from PD | |||||
| Significant reduction in the expression of inflammatory COX2 and iNOS | |||||
| Increase in the expression of tyrosine hydroxylase | |||||
| Improving motor coordination and alleviating the neuroinflammation in the Parkinson model | |||||
| Au | Paeonia moutan-functionalized GNPs | 190–450 nm | BV2 microglia | Decrease in formation of intracellular α-syn oligomers, pro-inflammatory cytokines (IL-6 and TNF-α) secretion, and α-syn internalization, in vitro | 116 |
| Lowered α-syn-induced production of ROS and NO in microglia | |||||
| Nuclear translocation of NF-κB | |||||
| Suppression the expression of Iba-1 by α-syn-challenged microglia | |||||
| Au | Anthocyanin-loaded poly (ethylene glycol)-AuNPs | 135 ± 5 nm | Aβ1-42-induced mouse model and BV2 microglia | Reduction in Aβ1-42-induced neuroapoptotic markers and neuroinflammation by restricting the p-GSK3β/NF-κB/p-JNK pathways in both in vivo and in vitro AD models | 118 |
| Significant mitigation in Aβ-induced apoptosis in both BV2 microglia and the mouse hippocampus by reducing Cyt. c release, Bax protein expression, and increasing Bcl2 protein levels | |||||
| Reduction in the production of Iba-1 and GFAP in microglia | |||||
| Mitigating the expression of iNOS and p-NF-κB proteins | |||||
| Ag | AgNPs | 20 nm, 50 μg mL−1 | Mouse BV-2 microglia | Decrease in microglial growth by AgNPs and CdTe-QDs by stopping the cells in the G1 phase (CdTe-QDs) or S phase (AgNPs and CeO2NPs) of the cell cycle | 129 |
| Ce | Cerium oxide NPs | 25 nm, 100 μg mL−1 | Significant reduction in Aβ uptake by BV-2 microglia with AgNPs and CeO2NPs, but not CdTe-QDs | ||
| Cadmium telluride quantum dots | 3.8 nm, 3 or 10 μg mL−1 | No impacts on the secretion of IL-6, IL-1b, and IFNg by Aβ, nor NPs or their combinations | |||
| Significant increase in TNFa secretion by CeO2NPs | |||||
| Ag | — | 20 nm, dose: 50 μg mL−1 | Mouse BV-2 microglia | Efficiently blocking the Aβ uptake by microglia | 130 |
| Ag | — | 20 nm | Mouse BV-2 microglia | Impairing Aβ clearance by BV-2 microglia by competing with Aβ for scavenger receptors | 131 |
| Ag | — | 10 nm, 6, 3, and 1 μg mL−1 | BV2 microglia | Releasing soluble factors like NO and H2O2 from glial cells | 121 |
| Significantly inhibiting the induced ROS and cytokines (TNF-α, MCP-1, and IL-6) from LPS-activated BV-2 | |||||
| Decreasing cell viability of BV-2 by releasing H2O2 from ALT cells through indirect AgNP exposure | |||||
| Ag | — | 23 nm diameter | Glial cells | Destruction of the cerebellum granular layer, causing cerebellar ataxia-like symptoms in rats | 124 |
| Ag | — | 23.44 ± 4.92 nm, 5 μg mL−1 | BV2 microglia cell lines of mouse | AgNPs-induced M1 polarization of microglia in a time- and dose-dependent way by inhibiting the fusion of autophagosomes with lysosomes | 126 |
| Increasing the expression of pro-inflammatory genes such as IL-1β, TNF-α, Iba-1, NF-κB, and MCP-1 in BV2 cells | |||||
| Reducing the mRNA expression of anti-inflammatory cytokines | |||||
| Ag | — | 3–5 nm, dose: 5–12.5 μg mL−1 | Murine BV-2 microglia | Inducing pro-inflammatory cytokine secretion such as IL-1β secretion and gene expression of CXCL13, GSS, and macrophage MARCO | 100 |
| Altering protein and gene expressions of Aβ deposition by inducing the expression of amyloid precursor protein (APP) gene | |||||
| Ag | — | 49.7 ± 10.5 nm | Microglia | Reducing LPS-stimulated NO, TNF-α, and ROS production | 133 |
| Significant anti-inflammatory effects | |||||
| Reducing microglial toxicity to dopaminergic neurons | |||||
| Ag | — | Human microglia cells (HMC3) | M1 to M2 phenotype switch | 134 | |
| Enhancing the expressions of anti-inflammatory markers including transforming TGF-β and IL-10 | |||||
| A significant reduction in mRNA expressions of TNF-α and IL-6 | |||||
| Biogenic AgNPs were protected against oxidative stress and neuroinflammation by targeting Nrf2/HO-1 and TLR4/MyD88 signaling pathways | |||||
| Ag | — | 18 ± 1.8 nm | Microglia | Neurobehavioral alterations in offspring | 172 |
| Body fat increase | |||||
| Long-term gut dysbiosis | |||||
| Reducing the microglial counts | |||||
| IO | — | ∼65 nm | Cultured rat microglia | Time- and concentration-dependent uptake | 136 |
| Longer incubation periods of exposure or higher concentrations or severely attenuated cell viability | |||||
| IO | — | 58.7 nm, 1–510 μ Fe/mL | Primary murine microglia | Attenuation of the IL-1β production, but not TNF-α, mediated by their accumulation in lysosomes and affecting the secretory lysosomal pathway of cytokine recessing | 137 |
| Suppression of IL-1β converting enzyme in IONP-treated murine microglia by decreasing the activity of cathepsin B | |||||
| Magnetic iron oxide (γ-Fe2O3) | — | 11 ± 3.5 nm | rTg4510 tau-mutant mice | A significant decrease in the number of activated microglia in comparison with the same concentration of the free peptides by stabilizing the peptide to the γ-Fe2O3 NPs | 138 |
| Fe2O3 | — | γ -Fe2O3: (31 ± 17) nm | BV2 microglia | Proliferation of microglia | 139 |
| α-Fe2O3NP: (22 ± 5) nm | Increased phagocytosis | ||||
| Dose: 0.02, 0.2, 2 mol Fe/L of Fe2O3-NP suspensions or FeCl3 solution | Higher release of ROS and NO by microglia | ||||
| Co | — | 50 nm | C57BL/6J mice brain | Toxic effects and inflammatory responses in BV2 microglia and mice by activating the NOX2 (NADPH oxidase 2) | 142 |
| Dose: 1.25, 2.5 and 5 μg mL−1 | BV2 microglia | Catalyzing ROS production, IL-1β, NLRP3, accompanied by tau phosphorylation | |||
| Co | — | 96 and 123 nm | Microglia | Microglial activation | 143 |
| ZnO | — | 38.52 ± 2.82 nm, dose: 6.6 μg mL−1 | Mouse microglia N9 cell line | Disrupting the MMP activity and subsequently inducing the apoptotic pathway in the microglia by NADPH oxidase-independent ROS and ATP depletion | 146 |
| Disrupting mitochondrial membrane potential | |||||
| Microglia apoptosis, involving altered intracellular calcium (Ca2+) level, mitochondrial ROS production, caspase-9 and -3 activation, ERK and p38 phosphorylation, and cytochrome-c release | |||||
| ZnO | — | 50 nm, dose: 10 μg mL−1 | Murine BV-2 microglia | Increase in the ROS levels and oxidative stress in BV-2 cells in a time-dependent manner through autophagy and PINK1/parkin-mediated mitophagy | 150 |
| Increasing count of swollen mitochondria and autophagosomes | |||||
| ZnO | — | 20 nm (5, 10, 20, 40, and 80 μg mL−1) | Murine BV-2 microglia | Influencing the lysosomal destabilization | 151 |
| Inducing extensive cellular and organelle (mitochondria, lysosome), ROS accumulation, and consequently nonapoptotic cell death, leading to the release of lysosomal enzymes | |||||
| Promoting inflammation by cell debris and accumulating ROS at the CNS level | |||||
| ZnO | — | 42.31 ± 17.94 nm, dose: 30 μg mL−1 | BV2 microglia cell line | Driving microglia and inflammatory responses in the CNS by activating the Ca2+-dependent ERK, p38, NF-κB pathways | 152 |
| ZnO | — | 26.4 ± 2.3 nm, 5 μg mL−1 | BV2 microglia | Microglial activation and proliferation by ERK and Akt signaling pathways | 153 |
| ZnO | — | 50 nm | Microglia | Induction of tau protein expression, microglia activation, and oxidative stress in the brain, resulting in neurotoxicity | 154 |
| ZnO | Luteolin/ZnO NPs | 17 nm | Microglia | Regulating microglia polarization by targeting C/EBPA and alleviating inflammatory injury by modulating the redox-sensitive signal transduction pathways | 155 |
| TiO2 | TiO2 NPs (Degussa P25) | 330 nm, dose: 2.5–120 ppm | BV2 microglia | Upregulation of NF-κB and ERK/MAPK | 148 |
| Stimulating BV2 microglia to have an prolonged and immediate release of ROS | |||||
| Damaging neurons at low concentrations in cultures of the brain striatum, probably by microglial-generated ROS | |||||
| Influencing genomic pathways linked to cell cycling, upregulation of apoptotic pathways, inflammation, mitochondrial bioenergetics, and downregulation of energy metabolism | |||||
| TiO2 | — | 20–30 nm, 0.1 to 200 μg mL−1 | BV2 microglia | TiO2NP accumulation in BV-2 cells | 157 |
| Induction of oxidative stress and mitochondrial dysfunctions | |||||
| Damaging the permeability of cell membranes, inhibiting cell adhesion with a loss of mitochondrial transmembrane potential. Induction of ROS overproduction and reducing cell viability | |||||
| TiO2 | — | 1–100 nm | Microglia | Producing excessive ROS via the oxidative burst | 158 |
| Interfering with mitochondrial energy production in vitro | |||||
| Damaging membrane integrity | |||||
| TiO2 | TiO2 NPs (Degussa P25) | 30 nm | BV2 microglia | Formation of free radicals in cellular and morphological expressions | 159 |
| TiO2 | — | 21 nm, 25–200 μg mL−1 | Male C57BL/6 mice and murine BV2 microglia cell line | Stimulating inflammatory mediators in the brain and neurons in vitro | 160 |
| Significantly elevating pro-inflammatory cytokine (TNF-α and IL-1β) mRNAs and IL-1β protein levels in the brains of LPS-exposed mice | |||||
| Enhancing TNF-α production and NF-κB binding activity by LPS-stimulated BV2 microglia | |||||
| Causing neuroinflammatory responses by enhancing microglial activation in the preinflamed brain and leading microglia N9 to apoptosis | |||||
| TiO2 | TiO2NPs | 20–60 nm/0.25 mg mL−1 and 0.5 mg mL−1 | Primary microglia | Inducing a significant expression of iNOS and subsequent NO secretion | 162 |
| HAP-NPs | Upregulating the expression levels of MIP-1 and MCP-1 from NP-stimulated microglia by inducing NF-κB activation | ||||
| Increasing the production of TNF-α, IL-6, and IL-1β by TiO2-NPs and HAP-NPs | |||||
| TiO2 | — | 35 nm, dose: 4–125 μg mL−1 | Microglia N9 | Inducing TiO2-induced apoptosis | 173 |
| TiO2 | — | 6 nm, dose: 100–5 μg mL−1 | BV-2 microglia | Inducing IL-1β production and ROS production | 174 |
| Clathrin-dependent endocytosis, phagocytosis, and a slow translocation to the lysosome in BV2 cells | |||||
| More TiO2NP uptake in LPS-activated BV-2 than normal BV-2, resulting in more released ROS, IL-6, IL-1β, and MCP-1 levels | |||||
| SiO2 | Fluorescein isothiocyanate -tagged SiO2 NPs | 115 nm | Male C57BL/6N mice & microglia | Increasing Iba-1 antibody in the hippocampus | 166 |
| SiO2 | Silica-coated magnetic NPs containing rhodamine B isothiocyanate dye | 50 nm | Murine BV2 microglia | Increasing the expression of Iba1 | 169 |
| Increasing the serine protein, especially excitotoxic D-serine secretion in the growth medium of activated microglia from primary rat microglia | |||||
| Activation of primary microglia | |||||
| Accumulation of ubiquitinated proteins and increasing the inclusion bodies in primary cortical and dopaminergic neurons, cocultured with activated primary microglia | |||||
| Reduction of intracellular ATP levels and proteasome activity in cocultured neuronal cells, especially in primary cortical neurons, by D-serine secretion | |||||
| Si | — | 48.53 ± 3.12 nm | Murine BV2 microglia | Significantly increasing caspase-1, ASC, and NLRP3 after stimulation by LPS and SiNPs | 170 |
| Raising the production of inflammatory factors, including IL-6, IL-1β, and TNF-α | |||||
| Decreasing the cell viability by increasing the concentration of NP | |||||
| Changing the ultrastructure | |||||
| Invading the cytoplasm | |||||
| Activating the NLRP3 inflammasome | |||||
| Releasing a large number of inflammatory factors | |||||
| Disrupting cellular antioxidant function | |||||
| Inducing ferroptosis | |||||
| Increasing intracellular ferrous ion levels | |||||
| SiO2 | — | — | Microglia | Elevating oxidative stress levels | |
| Activation of microglial functions | |||||
| Si | — | 150–200 nm | Primary rat microglia | Significantly increasing intracellular RNS and ROS productions | 149 |
| Decreasing TNF-α gene expression | |||||
| Increasing the expression of COX-2 gene | |||||
| Inducing a small but detectable IL-1β release | |||||
Metal-containing nanoparticles have serious impacts on microglia in the CNS. They can directly influence resting microglia (M0) to switch them to the M1 or M2 phenotype. They can also induce this effect on the M1 and M2 phenotypes, resulting in a phenotypic switch between M2 and M1. In case of a switch to the M1 phenotype, proinflammatory and inflammatory cytokines are likely to be released, resulting in an inflammatory state in the CNS and, consequently, neuronal death. On the other hand, if a phenotype switch occurs to the M2 phenotype, it is probable that anti-inflammatory cytokines will be released, leading to a protective state in the CNS and healthy neurons.
6.1. Gold nanoparticles (AuNPs)
AuNPs possess distinctive physicochemical characteristics, including surface plasmon resonance and high biocompatibility, making them notably attractive for several biomedical uses, such as photothermal and photodynamic therapies. These properties enable targeted heating of tissues and enhanced imaging capabilities, making them versatile tools for both treatment and diagnosis of CNS disorders.104,105 Gold nanoparticles possess inherent antibacterial properties that make them effective against a range of pathogens, including antibiotic-resistant strains.106 Their ability to amplify signals also makes them valuable tools in clinical settings as potent biosensors.107,108 They have been explored in fields like pharmacology, drug delivery, cancer therapy, biosensing, and bio-imaging because of their suitable shape and size.109 This section summarizes the results of some studies on the effects of these NPs on microglia-mediated neuroinflammation during drug delivery to CNS.Several researchers have confirmed the anti-inflammatory function of AuNPs in the CNS by influencing microglia. For example, Ozdal et al. revealed that gold-quercetin NPs exhibited superior anti-inflammatory and therapeutic effects compared to free quercetin. These NPs notably reduced the translational and transcriptional levels of inflammation-associated enzymes, PGE2, and nitric oxide (NO) in LPS-stimulated microglia without causing cytotoxic effects. This highlights the potential of AuNPs as carriers to address solubility issues of therapeutic compounds like quercetin.110 Another example involves dihydrolipoic acid (DHLA)-functionalized AuNPs, which act as neuroprotective antioxidants. These GNPs polarize microglia to M2-like phenotype, effectively reducing oxidative stress and NF-κB signaling. Additionally, they support microglial survival by preventing apoptosis.101 Moreover, Kuschnerus and colleagues reported that AuNPs coated with a hard corona composed of fibrinogen (FIB) and bovine serum (BS), in vitro, significantly enhanced cellular uptake and lowered oxidative stress and ROS production in microglia more effectively than AuNPs-FIB, protein corona (PC), and BS-T120W3-AuNPs each alone. This selective formation of AuNP-corona complexes may offer a promising strategy for controlling oxidative stress and improving cellular uptake.111 Additionally, in an in vitro study, E. sinica Stapf extract (ES)-functionalized AuNPs lowered proinflammatory cytokine levels by downregulating the NF-κB and Janus kinase/signal transducers and activators of transcription (JAK/STAT) in LPS-stimulated microglia, thereby suggesting that ES-functionalized AuNPs may mitigate neuroinflammation and neurodegenerative disorders.112 Also, Diaz and colleagues113 evaluated microglial responses to intracerebrally injected PEGylated AuNPs (polyethylene glycol-coupled AuNPs). The results indicated a transient and predominantly localized cellular response of microglia and astrocytes at the injection site with minimal harmful effects on the brain for 3 to 90 days. It was suggested that neural tissues could tolerate PEGylated GNPs well. Interestingly, Hutter et al. discovered that AuNP exposure caused a limited and transient upregulation of TLR-2 and inflammatory markers like IL-1α, NO, and GM-CSF in microglia. Notably, microglial activation occurred in a limited number at a slow pace, highlighting their ability for long-term drug delivery to the CNS.114
AuNPs have also shown privileges in treating neurodegenerative diseases like Parkinson's disease (PD). In this regard, a study utilizing Paeonia moutan-functionalized AuNPs (PM-AuNPs) in a PD mouse model showed that these NPs significantly inhibited the production of NO and inflammatory cytokines and scavenged ROS in LPS-stimulated BV2 microglia all without causing cytotoxic effects. PM-AuNPs reduced levels of COX2 and iNOS, key markers of inflammation, while improving motor coordination in the PD model.115 Consistently, Zhao et al. observed that PM-AuNPs reduced α-synuclein internalization and oligomer formation, as well as decreasing TNF-α and IL-6 levels in vitro. This supports PM-AuNPs’ role in managing PD-related neuroinflammation.116 Additionally, AuNPs have been used to treat Alzheimer's disease (AD). As a study has shown, anthocyanins-loaded GNPs successfully crossed the BBB. They inhibited a key inflammatory pathway in microglia-induced neuroinflammation, p-GSK3β, without harming neurons. GSK-3β mainly regulates the balance between proinflammatory and anti-inflammatory agents in microglia. Intriguingly, it activates the JNK and NF-κB pathways, resulting in enhanced chemokine and cytokine production117 by microglia in AD models. Therefore, anthocyanins-loaded GNPs could also indirectly inhibit NF-κB and JNK pathways. Anthocyanins conjugated with PEG-GNPs (AnPEG-GNPs) could also reduce the expression of Aβ1–42-escalated neuroapoptotic markers in BV2 microglia in mouse AD models, making it a promising therapeutic approach.118
Overall, AuNPs offer considerable advantages, including their anti-inflammatory properties and ability to cross the BBB, making them promising carriers for drug delivery in neurological disorders like AD and PD, with minimal microglia-mediated side effects for neurons and the brain. Nonetheless, more investigation is necessary to address the potential for even mild and transient neuroinflammatory responses associated with AuNPs, ensuring their safety and efficacy in CNS treatments.
6.2. Silver nanoparticles (AgNPs)
Silver nanoparticles (Ag nanoparticles) have been studied for their capability of crossing the BBB and are valuable for addressing challenges linked to the delivery of therapeutic agents to the CNS.119 Microglia mainly take up these NPs,120,121 suggesting that AgNPs can polarize these cells toward either M1 or M2 phenotypes.122 This section discusses various studies exploring the effects of AgNPs on microglia during drug delivery to the CNS.Several studies have shown that AgNPs can induce neurotoxic effects through microglial activation. For instance, one study evidenced that prenatal AgNP exposure led to cognitive dysfunctions and abnormal behaviors in adults, which were linked to microglial activation.123 Another survey by Hsiao et al. found the toxic effects of AgNPs on neurons were indirectly mediated by the release of NO and H2O2 from glial cells. Although, cytokines, namely IL-6 and TNF-α, were not involved in this process.121 Additionally, in an animal study, intranasal administration of 23 nm AgNPs resulted in microglial activation, destructing the cerebellum granular layer. This process caused cerebellar ataxia-like symptoms in rats, as well as motor dysfunction and impaired locomotor activity.124 Autophagy significantly affects microglial inflammation and phenotype transformation.125 Shang et al.126 explained that AgNPs promoted M1 polarization and inflammation in microglia in a time- and dose-dependent manner by avoiding the fusion of autophagosomes with lysosomes, thereby altering the lysosomal function and impairing autophagy. This finding provides insights into the molecular mechanisms behind AgNP-induced neurotoxicity.127 Moreover, Huang et al. reported that AgNPs promoted neuroinflammation, oxidative stress, and Aβ deposition in microglia, which was mediated by the secretion of IL-1β, the production of CXCL13 (C-X-C motif chemokine 13), macrophage receptor with collagenous structure (MARCO), and glutathione synthetase (GSS).100 One of the key concerns about AgNPs is their potential to exacerbate neurodegenerative diseases like AD. Aβ deposits cause toxicity to neurons as they cause proinflammatory responses and oxidative stress in the CNS.128 Since AgNPs and Aβ both are taken up by microglia via the scavenger receptor 1 (Scara1), AgNPs may compete with Aβ for uptake, potentially impairing Aβ clearance and worsening AD pathology.129–131 Sikorska et al. also observed that the AgNPs accompanied by cerium oxide nanoparticles (CeO2NPs) reduced microglial phagocytic activity and amyloid-β (Aβ) uptake by BV-2 microglia, which may assist in the AD pathogenesis. AgNPs also attenuated the microglial viability once combined with cadmium telluride quantum dots (CdTe-QDs), favoring the pathogenesis of AD.129
On the contrary, some studies suggest that AgNPs can exhibit anti-inflammatory and neuroprotective properties. For example, AgNPs, in combination with CdTe-QDs or CeO2NPs, even at relatively nontoxic concentrations, could decrease microglial growth by arresting the cell cycle at the G1 phase or S phase, respectively. This suggests a new approach to alleviate neuroinflammation and further disorders.129 Likewise, Lyu et al. confirmed that using AgNPs reduced the number of microglia, supporting their anti-inflammatory role.132 Moreover, citrate-capped AgNPs have demonstrated both anti-inflammatory and antioxidant effects in microglia. These AgNPs were specifically absorbed by microglia and further reduced LPS-stimulated NO, ROS, and TNFα production, leading to less neurotoxicity of microglia for dopaminergic neurons. Also, LDH release, following AgNP treatment, showed a significant reduction, underscoring the role of AgNPs in heightening neuronal cell viability (Fig. 3).133 Furthermore, the inhibitory role of biogenic AgNPs in LPS-induced neuro-inflammation by HMC3 microglial cells was studied. Cotreatment with AgNPs significantly decreased the production of inflammatory markers while increasing anti-inflammatory markers, facilitating a shift from M1 to M2 phenotype in microglia. Therefore, biogenic AgNPs are able to defend CNS against oxidative stress and neuroinflammation.134
 | ||
| Fig. 3 Anti-inflammatory effects of AgNPs on microglial cells (a and b) N9 microglial cells were treated with LPS (500 ng mL−1) with or without AgNPs for a 1 h pulse period. Then, a 24 h chase period was considered. Microglial inflammation was assessed through evaluation of (a) ROS production and (b) TNFα release following AgNP treatment. (c) N9 microglia were treated for 1 h (pulse) with AgNP (50 μg mL−1) and/or LPS (500 ng mL−1). Then, a 24 hours chase period was considered with only LPS present. The medium was then transferred to N27 neurons and incubated for 48 hours. Afterward, the cell viability was assessed through LDH release examination. Adapted from Gonzalez-Carter et al.133 | ||
Consequently, the effects of AgNPs on microglia in the context of neurodegenerative diseases remain controversial. While some studies highlight the proinflammatory and neurotoxic potential of AgNPs, others demonstrate their anti-inflammatory and neuroprotective effects. Therefore, AgNPs act as a double-edged sword in neuroinflammation. Further investigation is required to better perceive their mechanisms and ensure their safe and appropriate use to treat CNS disorders.
6.3. Iron oxide nanoparticles (IONPs)
IONPs are a class of magnetic NPs that have garnered considerable interest for their potential applications in biomedicine and bioengineering. IONPs have recently been explored for drug delivery systems, particularly for their ability to modulate microglia activity and reduce neuroinflammation.135 Along with it, this section reviews several studies to determine the effect of this NP on microglia-mediated neuroinflammation during drug delivery.Iron oxide-based metal NPs are primarily internalized by microglia, mainly through clathrin-mediated endocytosis and macropinocytosis, after which they accumulate in the lysosomal compartment.136 This accumulation in lysosomes plays a crucial role in modulating microglial activity. Interestingly, Wu et al. showed that accumulated IONP attenuated the expression of IL-1β in LPS-stimulated microglia by affecting the secretory lysosomal pathway. In addition, the IL-1β converting enzyme (ICE) was inhibited in microglia following the treatment with IONP by preventing the activity of cathepsin B, an enzyme responsible for IL-1β activation. These findings suggest that IONPs efficiently suppress IL-1β production, highlighting their potential for use in drug-delivery systems that target inflammation.137 Another critical area of investigation is the ability of microglia to alleviate tau pathology in neurodegenerative conditions such as AD. In this regard, Glat et al.138 showed the efficacy of iron oxide (γ-Fe2O3) NPs in delivering fibrin γ377–395 peptides to nervous tissue. Stabilizing the peptide to γ-Fe2O3 NPs significantly decreased the activated microglia in comparison with the same concentration of the free peptides. Therefore, the authors suggested γ-Fe2O3 NPs as suitable carriers for the controlled release of medicine in the CNS. Wang et al.139 also observed that Fe2O3 NPs induced the proliferation of microglia, enhanced phagocytosis, and increased ROS and NO production in microglia. However, the study noted no significant production of inflammatory cytokines, namely, IL-6, IL-1β, and TNF-α, implying that IONPs may boost some microglial functions without causing overt inflammation.
Despite their promising applications, many studies have evidenced the potential detrimental impacts of IONPs on microglia. As reported by Petters et al.,140 IONP exposure triggered ROS production by microglia, causing cellular and tissue damage. Additionally, the IONP accumulation in microglia could lead to changes in microglial morphology and function, potentially disrupting their usual tasks in the brain. Similarly, Luther et al. noted that prolonged exposure to IONPs compromised microglial cell viability, raising concerns about long-term use of these NPs in the brain.136
IONPs offer a noteworthy ability to deliver drugs, particularly in their ability to regulate microglial activity and mitigate neuroinflammation. However, the dual nature of their effects—both as modulators of inflammation and potential toxicity sources—necessitates further investigation. While some studies have shown their efficacy in reducing proinflammatory cytokine production and promoting drug delivery to the CNS, others have indicated possible adverse outcomes, such as the generation of ROS and compromised microglial viability. Consequently, more comprehensive research is required to make sure IONPs are safe and efficient as therapeutic agents for CNS-related conditions.
6.4. Cobalt nanoparticles (CoNPs)
CoNPs have been extensively utilized due to their catalytic, electrical, and magnetic properties. Intriguingly, it has been reported that CoNPs can enter the CNS, possibly due to their resemblance to local air pollutants, which can be inhaled.141 Prolonged exposure to cobalt dust in occupational settings has been associated with cognitive impairments, including reduced memory deficits and attention, showing that cobalt over-intake may contribute to neurodegenerative changes.142 However, the exact effects of CoNPs on triggering neurodegeneration and the underlying mechanisms remain mostly unexplored.Recent studies have paved the way for understanding the potential neurotoxic and pro-inflammatory effects of CoNPs. In this way, Li et al. illustrated that CoNPs induced toxicity and inflammatory responses in microglial BV2 cells by activating NADPH oxidase 2 (NOX2). CoNPs in both BV2 cells and mouse brains (the hippocampus and cortex) further catalyzed ROS production and upregulation of IL-1β and NLRP3, which are inflammation-related proteins. Additionally, CoNP exposure was linked to increased tau phosphorylation, which is a hallmark of neurodegenerative diseases.142 Similarly, a survey by Zheng et al. reported that CoNPs were capable of inducing microglial activation, leading to the expression of oxidative stress-related substances NRF2, heme oxygenase-1 (HO-1), and malondialdehyde (MDA) in the hippocampus and cortex of the rat brain.143 These results indicate that CoNPs can induce significant inflammation and oxidative stress in the brain.
While these studies provide initial insights into the impacts of CoNPs, the limited research on this topic underscores the need for further investigations. The potential proinflammatory effects of CoNPs during drug delivery warrant a deeper understanding of their interactions with microglia and their long-term implications for neurodegenerative processes. Continued research is essential to elucidate how CoNPs may contribute to neuroinflammation and cognitive decline, ultimately identifying their safe application in biomedical contexts.
6.5. Zinc oxide nanoparticles (ZnONPs)
ZnONPs can be easily found in various forms. Some physicochemical properties of ZnONPs, including their surface charge, morphology, concentration, purity, and size impact the interaction of ZnO with microglia. ZnONPs have been found to cause neurotoxicity through microglial activation, which is a significant cause of concern.144,145Several studies have shown that ZnONPs trigger apoptosis in the murine microglial cell line N9 by generating ROS and depleting cellular energy,146,147 leading to neuronal damage148 or self-destructive processes.149 Wei et al. indicated that ZnONPs significantly raised ROS levels and oxidative stress in a time-dependent way in BV-2 cells, which occurred through autophagy and PINK1/parkin-mediated mitophagy.150 Moreover, Sharma et al. found that ZnONPs disrupted matrix metalloproteinases (MMPs) and subsequently activated the apoptotic pathway in microglia via NADPH oxidase-independent ROS generation and ATP depletion. This microglial apoptosis exacerbates the existing neuroinflammation.146 Also, it has been reported that ZnONPs induce a nonapoptotic mode of cell death in microglia, which is probably driven by ROS accumulation, leading to lysosomal destabilization and extensive damage to mitochondria and lysosomes. This nonapoptotic cell death can severely damage the brain by accumulating ROS and releasing lysosomal enzymes and cell debris, resulting in severe neuroinflammation.151 ZnONPs have also triggered several inflammatory responses. For instance, a study claimed that ZnONPs activate NF-κB, Ca2+-dependent extracellular signal-regulated kinase (ERK), and p38 pathways in BV2 microglia following tongue instillation.152 Similarly, Liu et al. revealed that even nontoxic concentrations of ZnONPs led to BV2 proliferation and activation through the Akt (protein kinase B) and ERK signaling pathways.153 Another paper studied the acute outcomes of pulmonary exposure to ZnONPs in a rat model. The results indicated that acute exposure to ZnONPs induces microglial activation, tau protein expression, and oxidative stress in the brain, contributing to neurotoxicity.154
Despite the concerns associated with ZnONPs, some studies have reported potential benefits. In this respect, a survey by Moustafa et al. on diabetic patients revealed that luteolin/ZnONPs could regulate microglial polarization by targeting brain CCAAT/enhancer-binding protein (C/EBPA mRNA). These NPs also alleviated inflammation by modulating redox-sensitive signal transduction pathways. Therefore, it was concluded that luteolin/ZnONPs may offer a novel approach to protecting BBB and preventing neurological complications.155
The adverse effects of ZnONPs on microglial neuroinflammation appear to outweigh their potential benefits in promoting brain health. Urgent and comprehensive studies should be conducted to thoroughly investigate the possible positive and negative effects of ZnONPs on microglia-related neuroinflammation. This is crucial to determine a safer dosage with minimal side effects for patients suffering from neurodegenerative disorders.
6.6. Titanium dioxide nanoparticles (TiO2NPs)
TiO2NPs are widely applied across various sectors, including chemical, electrical, electronic, medical, cosmeceutical, and industrial fields. TiO2NPs can enter the brain directly through the olfactory bulb and accumulate in the hippocampus. Recent studies have raised concerns about the potential harm TiO2NPs pose to biological systems, particularly regarding their toxicity to the CNS.156 Nonetheless, the toxicity of TiO2NPs to the CNS has been poorly investigated so far.Rihane et al. evidenced that TiO2NPs predominantly accumulate in BV-2 cells, promoting mitochondrial dysfunction following oxidative stress. These NPs also cause various side effects, such as damaging the permeability of cell membranes, ROS overproduction, and inhibiting cell adhesion with a loss of mitochondrial transmembrane potential, thereby leading to microglia apoptosis.157 Additionally, Sheng et al. highlighted that TiO2NPs contribute to the apoptosis of primary hippocampal neurons and microglia.158 On top of that, recent reports have indicated that low concentrations of TiO2 stimulate BV2 microglia to undergo immediate and prolonged release of ROS, damaging neurons in brain striatum cultures.148,159 Shin et al.160 reported that ultrafine TiO2NPs stimulate the release of inflammatory mediators, encompassing IL-1β, TNF-α, and mRNA in the brains of LPS-exposed mice. These NPs also enhanced NF-κB binding activity in LPS-stimulated BV2 microglia. Therefore, the study suggests that nanosized TiO2NPs promote exaggerated neuroinflammatory responses by activating microglia. In line with this, another study found that LPS-activated BV-2 cells took more TiO2NPs up compared to non-activated cells, leading to increased IL-6, ROS, MCP-1, and IL-1β.161 Along with it, Xue et al.162 showed that TiO2NPs induced significant iNOS expression and subsequent NO secretion, accompanied by upregulation of chemokines through NF-κB activation in NP-stimulated microglia in vitro. This study also indicated raised levels of pro-inflammatory cytokines.
To conclude, TiO2NPs activate microglia and neuroinflammation using various pathways. Given the small number of papers written on TiO2NPs and the scarcity of positive findings, additional research is essential to comprehensively understand the mechanisms in charge of the harmful effects of TiO2NPs on microglia-related neuroinflammation. These findings will facilitate the evolution of strategies for the secure and efficient employment of TiO2NPs in the treatment of neurodegenerative diseases.
6.7. Silica nanoparticles (SiO2NPs)/Silicon nanoparticles (SiNPs)
Silica nanoparticles (SiO2NPs), one of the most widely employed types of NPs, have been applied across various industries.163 Nanosized SiO2 can cross the BBB, making them valuable for delivering therapeutic and diagnostic agents.164 The exposure to SiO2 does not significantly influence the viability of various neural cells, and it also does not cause neuroinflammation.165 However, long-term exposure to these NPs can cause cognitive impairment, mood dysfunction, and synaptic alterations, potentially by activating mitogen-activated protein kinases (MAPKs).166 Therefore, the potential of SiO2NPs for treating microglia-induced neuroinflammation merits further exploration.SiO2NPs have shown potential in activating microglia. In line with this, in a study, after exposure to fluorescein isothiocyanate-tagged SiO2NPs (FITC-SiO2-NPs),167,168 the number of Iba-1- stained microglia significantly rose in the hippocampus in comparison with the controls.166 Consistently, a study showed that silica-coated magnetic NPs containing rhodamine B isothiocyanate dye (MNPs@SiO2(RITC)) morphologically activated BV2 murine microglia and increased Iba1 expression, an activation marker protein.169 The study also demonstrated that microglia activation elevated serine protein levels in the growth medium. Notably, the secretion of excitotoxic D-serine from primary rat microglia was significantly upregulated, which in turn decreased intracellular ATP and activity of the proteasome in cocultured neuronal cells, particularly in primary cortical neurons. This led to the accumulation of ubiquitinated proteins and the formation of inclusion bodies in cortical and primary dopaminergic neurons cocultured with activated microglia. Thus, the activation of microglia by MNPs@SiO2(RITC) initiates excitotoxicity in neurons through the secretion of D-serine, underscoring the neurotoxic processes triggered by microglial activation.156 SiO2NPs are likely to produce inflammatory agents and cause neuroinflammation. In this way, Xue et al. demonstrated that SiO2NPs enhance the proinflammatory cytokines (TNF-α, IL-1β, and IL-6).162 Additionally, the findings of another study illustrated that deficient SiNP levels could change microglial function. In turn, alteration in proinflammatory genes, cytokine release, and heightened RNS and ROS production adversely affect not only microglial function but also surrounding neurons.170 Correspondingly, MNPs@SiO2(RITC) were shown by another study to enhance ROS production in a dose-dependent manner.171 The findings of another research indicated that the viability of MNPs@SiO2(RITC) was gradually reduced with increasing SiO2NP concentration and exposure duration. The findings indicated that SiNPs could penetrate the cytoplasm, alter the ultrastructure, activate the NLRP3 inflammasome, release a multitude of inflammatory molecules, and start inflammatory reactions. SiNPs were also discovered to induce ferroptosis, increase intracellular ferrous ion levels, and disrupt cellular antioxidant function.170
In summary, the studies reviewed suggest that SiO2NPs possess the potential to induce microglial activation and neuroinflammation, negatively impacting neuronal health. Due to the scarcity of studies on the positive influences of these NPs on microglia-related neuroinflammation, the potential of SiO2NPs for treating neuroinflammation deserves further research to fully elucidate the risks of exploiting this nanoparticle.
Regarding the explained studies, Fig. 4 illustrates the advantages and disadvantages of each metal NP in the inhibition of microglia-mediated neuroinflammation.
 | ||
| Fig. 4 The advantages and disadvantages of drug-loaded metal NPs in microglia-induced neuroinflammation. | ||
7. Conclusion
Microglia are central to neuroinflammation and neurodegeneration, capable of both protecting and harming the CNS. Metal nanoparticles offer exciting potential for targeting microglial activity due to their ability to cross the BBB and move medicines directly to the brain. However, their dual effects, ranging from neuroprotection to exacerbation of inflammation, necessitate careful consideration. While AuNPs and certain IONPs show promise in reducing neuroinflammation, other NPs like AgNPs, ZnONPs, and TiO2NPs have been linked to increased neurotoxicity. As the field progresses, future research is proposed to focus on deciphering the specific mechanisms underlying nanoparticle–microglia interactions, paving the way for targeted and safe interventions for neurodegenerative diseases. A comprehensive understanding of these interactions will enhance the therapeutic potential of nanoparticles and ensure neural homeostasis. In this dynamic landscape, interdisciplinary collaboration and continued exploration are essential to address these challenges and unlock the full therapeutic potential of nanoparticles in neurodegenerative disorders.8. Challenges
Despite the promising potential of metal nanoparticles (NPs) in treating neuroinflammation and modulating microglial activity, several critical challenges remain.8.1. Toxicity and biocompatibility
The biocompatibility of NPs is a key factor in their clinical translation. Although some NPs, such as gold nanoparticles (AuNPs), are considered relatively safe, others, like silver nanoparticles (AgNPs), can induce oxidative stress and pro-inflammatory responses in microglial cells at higher concentrations. The dose-dependent duality of these effects necessitates precise dose optimization and the development of biocompatible coatings to mitigate toxicity.8.2. Blood–brain barrier (BBB) permeability
While the nanoscale size of certain NPs facilitates their ability to cross the BBB, their transport efficiency and distribution in targeted brain regions remain inconsistent. Surface charge, hydrophilicity, and interactions with serum proteins can impact their BBB permeability and bioavailability, limiting therapeutic outcomes.8.3. Stability in biological environments
The physicochemical stability of NPs in the dynamic and complex CNS environment is a significant challenge. Non-functionalized NPs are prone to aggregation and premature clearance, while improperly stabilized particles may lose activity before reaching their target. Effective functionalization strategies are required to enhance stability, circulation time, and targeted delivery.8.4. Long-term safety and accumulation
The long-term effects of NPs, including potential accumulation in brain tissues, remain poorly understood. Chronic exposure could lead to neurotoxicity, inflammation, or immune system interference. Comprehensive in vivo studies are needed to evaluate their safety profiles under prolonged use.8.5. Immune system interactions
NPs may inadvertently activate peripheral or central immune responses, complicating their therapeutic use. Understanding and mitigating these interactions is critical for reducing adverse effects and enhancing therapeutic specificity.9. Future perspective
9.1. Development of advanced nanoparticle designs
The engineering of next-generation NPs with precise targeting capabilities is paramount. Functionalization with ligands specific to inflammatory markers or activated microglia can enhance therapeutic specificity while reducing off-target effects. Stimuli-responsive NPs that release their therapeutic payload in response to pH, temperature, or inflammatory signals could provide controlled and localized treatment.9.2. Exploration of biodegradable nanoparticles
The development of biodegradable NPs that degrade into non-toxic byproducts after delivering their cargo is crucial for minimizing long-term risks. Materials such as polymers, lipid-based carriers, or naturally derived compounds should be further explored to ensure both efficacy and safety.9.3. Combination therapies
Leveraging the co-delivery capabilities of NPs to carry multiple therapeutic agents, such as anti-inflammatory drugs, antioxidants, or gene therapy vectors, can target multiple pathways simultaneously. These approaches could provide synergistic effects, particularly in complex disorders like Alzheimer's disease or Parkinson's disease.9.4. In-depth mechanistic studies
Detailed studies on the molecular mechanisms of NP-microglia interactions are essential. Investigating how NPs influence microglial polarization between pro-inflammatory (M1) and anti-inflammatory (M2) states can guide the design of more effective therapeutic strategies.9.5. Preclinical and clinical translation
Long-term safety, pharmacokinetics, and efficacy studies in animal models are vital for bridging the gap between preclinical research and clinical application. Establishing standardized protocols for NP synthesis, characterization, and biological testing will also facilitate regulatory approval and clinical trials.9.6. Interdisciplinary collaboration
The successful translation of NP-based therapies requires collaboration across disciplines, including materials science, neuroscience, pharmacology, and toxicology. Integrating advanced imaging, computational modeling, and machine learning techniques can accelerate NP design and optimize therapeutic outcomes.By addressing these challenges and exploring these prospective directions, nanoparticle-based therapies could revolutionize the treatment of neuroinflammation and related CNS disorders.
List of abbreviations
| CNS | Central nervous system |
| AuNP | Gold nanoparticle |
| AgNP | Silver nanoparticle |
| IONP | Iron oxide nanoparticle |
| SiO2NP | Silica nanoparticle |
| ZnONP | Zinc oxide nanoparticle |
| CoNP | Cobalt nanoparticle |
| TiO2NP | Titanium oxide nanoparticle |
| TNF-α | Tumour necrosis factor alpha |
| DAMPs | Danger-associated molecular patterns |
| PGE2 | Prostaglandin E2 |
| PAMPs | Pathogen-associated molecular patterns |
| CR and CR | Complement receptors |
| CD | Cluster of differentiation |
| IL | Interleukin |
| ROS | Reactive oxygen species |
| TLR and TLR | Toll-like receptors |
| BACE | Beta-secretase enzyme |
| iNOS | Inducible nitric oxide synthase |
| COX2 | Cyclooxygenase-2 |
| BBB | Blood–brain-barrier |
| ECM | Extracellular matrix |
| NPs | Nanoparticles |
| TGN | Trans-Golgi network |
| Iba-1 | Ionized calcium-binding adapter molecule |
| ALTs | Astrocyte-like |
| N2a | Neuro2a |
| NF-κB | Nuclear factor-κB |
| SPIONPs | Superparamagnetic iron oxide nanoparticles |
| MPI | Magnetic particle imaging |
| DHLA | Dihydrolipoic acid |
| FIB | Fibrinogen |
| BS | Bovine serum |
| PC | Protein corona |
| JAK/STAT | Janus kinase/signal transducers and activators of transcription |
| PEGylated AuNPs | polyethyleneglycol-coupled AuNPs |
| PM-AuNPs | Paeonia moutan to functionalize GNPs |
| CeO2NPs | Cerium oxide nanoparticles |
| Aβ | Amyloid-β |
| GSS | Glutathione synthetase |
| AD | Alzheimer's disease |
| MMP | Matrix metalloproteinases |
| AnPEG-GNPs | Anthocyanins conjugated with PEG-GNPs |
| MAPKs | Mitogen-activated protein kinases |
| MARCO | Macrophage receptor with collagenous structure |
| FITC | Fluorescein isothiocyanate |
| ERK | Extracellular signal-regulated kinase |
Data availability
This article does not include primary research data, software, or code. It is a review article that discusses findings from various studies in the field with all data and figures cited from previously published sources. No new data were generated or analyzed in the preparation of this article.Author contributions
Masood Alaei, Khadijeh Koushki, and Kimia Taebi contributed to the conceptualization and methodology, collected data, and drafted the manuscript. Masood Alaei illustrated the figures and charts. Mahdieh Yousefi Taba and Samaneh Keshavarz Hedayati co-operated in writing and drafting the manuscript. Sanaz Keshavarz Shahbaz participated in supervising, providing critical review, commentary, and revising the final draft of the manuscript.Conflicts of interest
All the authors declare no conflicts of interest related to this manuscript.Acknowledgements
This work was supported by Qazvin University of Medical Sciences.References
- A. Adamu, S. Li, F. Gao and G. Xue, The role of neuroinflammation in neurodegenerative diseases: current understanding and future therapeutic targets, Front. Aging Neurosci., 2024, 16, 1347987 CrossRef CAS.
- S. S. Keshavarz, K. Koushki, S. Keshavarz Hedayati, A. P. McCloskey, P. Kesharwani and Y. Naderi, et al., Polymer nanotherapeutics: A promising approach toward microglial inhibition in neurodegenerative diseases, Med. Res. Rev., 2024, 44(6), 2793–2824 CrossRef PubMed.
- T. Sun, Y. S. Zhang, B. Pang, D. C. Hyun and M. Y. Yang, Xia Engineered nanoparticles for drug delivery in cancer therapy, Angew. Chem., Int. Ed., 2014, 53, 12320–12364 CrossRef CAS.
- E. Hutter, S. Boridy, S. Labrecque, M. Lalancette-Hébert, J. Kriz and F. M. Winnik, et al., Microglial response to gold nanoparticles, ACS Nano, 2010, 4(5), 2595–2606 CrossRef CAS.
- A. Joseph, R. Liao, M. Zhang, H. Helmbrecht, M. McKenna and J. R. Filteau, et al., Nanoparticle-microglial interaction in the ischemic brain is modulated by injury duration and treatment, Bioeng. Transl. Med., 2020, 5(3), e10175 CrossRef CAS.
- M. Prinz, T. Masuda, M. A. Wheeler and F. J. Quintana, Microglia and Central Nervous System-Associated Macrophages-From Origin to Disease Modulation, Annu. Rev. Immunol., 2021, 39, 251–277 CrossRef CAS.
- L. C. Mehl, A. V. Manjally, O. Bouadi, E. M. Gibson and T. L. Tay, Microglia in brain development and regeneration, Development, 2022, 149(8), dev200425 CrossRef CAS.
- V. H. Perry and J. Teeling, Microglia and macrophages of the central nervous system: the contribution of microglia priming and systemic inflammation to chronic neurodegeneration, Semin. Immunopathol., 2013, 35(5), 601–612 CrossRef CAS.
- S. Bilbo and B. Stevens, Microglia: The Brain's First Responders, Cerebrum, 2017, cer-14-17 Search PubMed.
- M. Colonna and O. Butovsky, Microglia Function in the Central Nervous System During Health and Neurodegeneration, Annu. Rev. Immunol., 2017, 35, 441–468 CrossRef CAS PubMed.
- Y.-L. Tan, Y. Yuan and L. Tian, Microglial regional heterogeneity and its role in the brain, Mol. Psychiatry, 2020, 25(2), 351–367 CrossRef PubMed.
- X. Chang, J. Li, S. Niu, Y. Xue and M. Tang, Neurotoxicity of metal-containing nanoparticles and implications in glial cells, J. Appl. Toxicol., 2021, 41(1), 65–81 CrossRef CAS PubMed.
- D. M. Angelova and D. R. Brown, Microglia and the aging brain: are senescent microglia the key to neurodegeneration?, J. Neurochem., 2019, 151(6), 676–688 CrossRef CAS PubMed.
- N. J. Allen and D. A. Lyons, Glia as architects of central nervous system formation and function, Science, 2018, 362(6411), 181–185 CrossRef CAS PubMed.
- S. Jäkel and L. Dimou, Glial Cells and Their Function in the Adult Brain: A Journey through the History of Their Ablation, Front. Cell. Neurosci., 2017, 11, 14 Search PubMed.
- G. C. Brown and J. J. Neher, Microglial phagocytosis of live neurons, Nat. Rev. Neurosci., 2014, 15(4), 209–216 CrossRef CAS PubMed.
- A. N. Hughes and B. Appel, Microglia phagocytose myelin sheaths to modify developmental myelination, Nat. Neurosci., 2020, 23(9), 1055–1066 CrossRef CAS.
- R. Fu, Q. Shen, P. Xu, J. J. Luo and Y. Tang, Phagocytosis of microglia in the central nervous system diseases, Mol. Neurobiol., 2014, 49(3), 1422–1434 CrossRef CAS PubMed.
- C. Herzog, L. Pons Garcia, M. Keatinge, D. Greenald, C. Moritz and F. Peri, et al., Rapid clearance of cellular debris by microglia limits secondary neuronal cell death after brain injury in vivo, Development, 2019, 146(9), dev174698 CrossRef CAS.
- S. Guo, H. Wang and Y. Yin, Microglia Polarization From M1 to M2 in Neurodegenerative Diseases, Front. Aging Neurosci., 2022, 14, 815347 CrossRef CAS PubMed.
- S. Kobashi, T. Terashima, M. Katagi, Y. Nakae, J. Okano and Y. Suzuki, et al., Transplantation of M2-Deviated Microglia Promotes Recovery of Motor Function after Spinal Cord Injury in Mice, Mol. Ther., 2020, 28(1), 254–265 CrossRef CAS PubMed.
- S. R. Subramaniam and H. J. Federoff, Targeting Microglial Activation States as a Therapeutic Avenue in Parkinson’s Disease, Front. Aging Neurosci., 2017, 9, 176 CrossRef.
- N. Zhao, N. L. Francis, H. R. Calvelli and P. V. Moghe, Microglia-targeting nanotherapeutics for neurodegenerative diseases, APL Bioeng., 2020, 4(3), 030902 CrossRef CAS.
- S. Bachiller, I. Jiménez-Ferrer, A. Paulus, Y. Yang, M. Swanberg and T. Deierborg, et al., Microglia in neurological diseases: a road map to brain-disease dependent-inflammatory response, Front. Cell. Neurosci., 2018, 12, 488 CrossRef CAS PubMed.
- G. J. Harry, Microglia in Neurodegenerative Events-An Initiator or a Significant Other?, Int. J. Mol. Sci., 2021, 22(11), 5818 CrossRef CAS PubMed.
- J.-W. Lee, W. Chun, H. J. Lee, S.-M. Kim, J.-H. Min and D.-Y. Kim, et al., The Role of Microglia in the Development of Neurodegenerative Diseases, Biomedicines, 2021, 9(10), 1449 CrossRef CAS PubMed.
- M. E. Lull and M. L. Block, Microglial Activation and Chronic Neurodegeneration, Neurotherapeutics, 2010, 7(4), 354–365 CrossRef CAS.
- D. Doens and P. L. Fernández, Microglia receptors and their implications in the response to amyloid β for Alzheimer's disease pathogenesis, J. Neuroinflammation, 2014, 11(1), 1–14 Search PubMed.
- G. Sita, A. Graziosi, P. Hrelia and F. Morroni, NLRP3 and Infections: β-Amyloid in Inflammasome beyond Neurodegeneration, Int. J. Mol. Sci., 2021, 22(13), 6984 Search PubMed.
- G. Zhang, Z. Wang, H. Hu, M. Zhao and L. Sun, Microglia in Alzheimer's disease: A target for therapeutic intervention, Front. Cell. Neurosci., 2021, 15, 749587 CrossRef CAS PubMed.
- C. Xing and E. H. Lo, Help-me signaling: Non-cell autonomous mechanisms of neuroprotection and neurorecovery, Prog. Neurobiol., 2017, 152, 181–199 Search PubMed.
- R. Von Bernhardi, L. Eugenín-von Bernhardi and J. Eugenín, Microglial cell dysregulation in brain aging and neurodegeneration, Front. Aging Neurosci., 2015, 7, 124 Search PubMed.
- R. A. Feldman, Microglia orchestrate neuroinflammation, Elife, 2022, 11, e81890 Search PubMed.
- G. C. Brown and A. Vilalta, How microglia kill neurons, Brain Res., 2015, 1628, 288–297 CrossRef CAS PubMed.
- U.-K. Hanisch and H. Kettenmann, Microglia: active sensor and versatile effector cells in the normal and pathologic brain, Nat. Neurosci., 2007, 10(11), 1387–1394 CrossRef CAS PubMed.
- H. S. Kwon and S.-H. Koh, Neuroinflammation in neurodegenerative disorders: the roles of microglia and astrocytes, Transl. Neurodegener., 2020, 9, 1–12 CrossRef PubMed.
- D. K. Choi, S. Koppula and K. Suk, Inhibitors of microglial neurotoxicity: focus on natural products, Molecules, 2011, 16(2), 1021–1043 CrossRef CAS PubMed.
- S. K. Maurya, N. Bhattacharya, S. Mishra, A. Bhattacharya, P. Banerjee and S. Senapati, et al., Microglia specific drug targeting using natural products for the regulation of redox imbalance in neurodegeneration, Front. Pharmacol, 2021, 12, 654489 CrossRef CAS PubMed.
- C.-Y. Liu, X. Wang, C. Liu and H.-L. Zhang, Pharmacological targeting of microglial activation: new therapeutic approach, Front. Cell. Neurosci., 2019, 13, 514 CrossRef CAS PubMed.
- Y. S. Gwak, E. D. Crown, G. C. Unabia and C. E. Hulsebosch, Propentofylline attenuates allodynia, glial activation and modulates GABAergic tone after spinal cord injury in the rat, Pain, 2008, 138(2), 410–422 CrossRef CAS PubMed.
- A. V. A. David, R. Arulmoli and S. Parasuraman, Overviews of biological importance of quercetin: A bioactive flavonoid, Pharmacogn. Rev., 2016, 10(20), 84 CrossRef CAS.
- M. Nikodemova, J. J. Watters, S. J. Jackson, S. K. Yang and I. D. Duncan, Minocycline down-regulates MHC II expression in microglia and macrophages through inhibition of IRF-1 and protein kinase C (PKC) α/βII, J. Biol. Chem., 2007, 282(20), 15208–15216 CrossRef CAS PubMed.
- R. Z. Hamza and N. S. El-Shenawy, Anti-inflammatory and antioxidant role of resveratrol on nicotine-induced lung changes in male rats, Toxicol. Rep., 2017, 4, 399–407 CrossRef CAS PubMed.
- T. Farkhondeh, S. L. Folgado, A. M. Pourbagher-Shahri, M. Ashrafizadeh and S. Samarghandian, The therapeutic effect of resveratrol: Focusing on the Nrf2 signaling pathway, Biomed. Pharmacother., 2020, 127, 110234 CrossRef CAS PubMed.
- C. Y. Chang, D.-K. Choi, D. K. Lee, Y. J. Hong and E. J. Park, Resveratrol confers protection against rotenone-induced neurotoxicity by modulating myeloperoxidase levels in glial cells, PLoS One, 2013, 8(4), e60654 CrossRef CAS PubMed.
- Y. Yu, Q. Shen, Y. Lai, S. Y. Park, X. Ou and D. Lin, et al., Anti-inflammatory effects of curcumin in microglial cells, Front. Pharmacol., 2018, 9, 386 CrossRef PubMed.
- T. Akaishi, S. Yamamoto and K. Abe, The Synthetic Curcumin Derivative CNB-001 Attenuates Thrombin-Stimulated Microglial Inflammation by Inhibiting the ERK and p38 MAPK Pathways, Biol. Pharm. Bull., 2020, 43(1), 138–144 CrossRef CAS PubMed.
- Y. Han, R. Chen, Q. Lin, Y. Liu, W. Ge and H. Cao, et al., Curcumin improves memory deficits by inhibiting HMGB1-RAGE/TLR4-NF-κB signalling pathway in APPswe/PS1dE9 transgenic mice hippocampus, J. Cell. Mol. Med., 2021, 25(18), 8947–8956 CrossRef CAS PubMed.
- A. Khan, T. Ali, S. U. Rehman, M. S. Khan, S. I. Alam and M. Ikram, et al., Neuroprotective effect of quercetin against the detrimental effects of LPS in the adult mouse brain, Front. Pharmacol, 2018, 9, 1383 CrossRef CAS PubMed.
- J. Bournival, M. Plouffe, J. Renaud, C. Provencher and M.-G. Martinoli, Quercetin and sesamin protect dopaminergic cells from MPP+-induced neuroinflammation in a microglial (N9)-neuronal (PC12) coculture system, Oxid. Med. Cell. Longevity, 2012, 2012, 921941 Search PubMed.
- Y. Naderi, M. Sabetkasaei, S. Parvardeh and T. Moini Zanjani, Neuroprotective effects of pretreatment with minocycline on memory impairment following cerebral ischemia in rats, Behav. Pharmacol., 2017, 28(2), 214–222 CrossRef CAS PubMed.
- Y. Naderi, M. Sabetkasaei, S. Parvardeh and T. M. Zanjani, Neuroprotective effect of minocycline on cognitive impairments induced by transient cerebral ischemia/reperfusion through its anti-inflammatory and anti-oxidant properties in male rat, Brain Res. Bull., 2017, 131, 207–213 CrossRef CAS PubMed.
- T. Patel, J. Zhou, J. M. Piepmeier and W. M. Saltzman, Polymeric nanoparticles for drug delivery to the central nervous system, Adv. Drug Delivery Rev., 2012, 64(7), 701–705 CrossRef CAS PubMed.
- C. A. Peptu, L. Ochiuz, L. Alupei, C. Peptu and M. Popa, Carbohydrate based nanoparticles for drug delivery across biological barriers, J. Biomed. Nanotechnol., 2014, 10(9), 2107–2148 Search PubMed.
- S. Cramer, R. Rempe and H.-J. Galla, Exploiting the properties of biomolecules for brain targeting of nanoparticulate systems, Curr. Med. Chem., 2012, 19(19), 3163–3187 Search PubMed.
- R. Ansari, S. M. Sadati, N. Mozafari, H. Ashrafi and A. Azadi, Carbohydrate polymer-based nanoparticle application in drug delivery for CNS-related disorders, Eur. Polym. J., 2020, 128, 109607 CrossRef CAS.
- A. Armulik, G. Genové, M. Mäe, M. H. Nisancioglu, E. Wallgard and C. Niaudet, et al., Pericytes regulate the blood–brain barrier, Nature, 2010, 468(7323), 557–561 CrossRef CAS PubMed.
- W. M. Pardridge, Drug targeting to the brain, Pharm. Res., 2007, 24, 1733–1744 CrossRef CAS PubMed.
- W. A. Banks and N. H. Greig, Small molecules as central nervous system therapeutics: old challenges, new directions, and a philosophic divide, Future Sci. OA, 2019, 489–493 CAS.
- W. M. Pardridge, Drug transport across the blood–brain barrier, J. Cereb. Blood Flow Metab., 2012, 32(11), 1959–1972 CrossRef CAS PubMed.
- J. Song, C. Lu, J. Leszek and J. Zhang, Design and development of nanomaterial-based drug carriers to overcome the blood–brain barrier by using different transport mechanisms, Int. J. Mol. Sci., 2021, 22(18), 10118 CrossRef CAS PubMed.
- S. R. Hwang and K. Kim, Nano-enabled delivery systems across the blood–brain barrier, Arch. Pharmacal Res., 2014, 37(1), 24–30 CrossRef CAS PubMed.
- K. Matsuhisa, M. Kondoh, A. Takahashi and K. Yagi, Tight junction modulator and drug delivery, Expert Opin. Drug Delivery, 2009, 6(5), 509–515 CrossRef CAS PubMed.
- E. Nance, S. H. Pun, R. Saigal and D. L. Sellers, Drug delivery to the central nervous system, Nat. Rev. Mater., 2022, 7(4), 314–331 CrossRef CAS PubMed.
- C. Ekhator, M. Q. Qureshi, A. W. Zuberi, M. Hussain, N. Sangroula and S. Yerra, et al., Advances and Opportunities in Nanoparticle Drug Delivery for Central Nervous System Disorders: A Review of Current Advances, Cureus, 2023, 15(8), e44302 Search PubMed.
- J. A. Morris, C. H. Boshoff, N. F. Schor, L. M. Wong, G. Gao and B. L. Davidson, Next-generation strategies for gene-targeted therapies of central nervous system disorders: A workshop summary, Mol. Ther., 2021, 29(12), 3332–3344 CrossRef CAS PubMed.
- A. Sintov, C. Velasco, E. Gallardo-Toledo and E. Araya, Metal Nanoparticles as Targeted Carriers Circumventing the Blood–Brain Barrier, Int. Rev. Neurobiol., 2016, 199–227 CrossRef CAS PubMed.
- A. M. Hersh, S. Alomari and B. M. Tyler, Crossing the Blood–Brain Barrier: Advances in Nanoparticle Technology for Drug Delivery in Neuro-Oncology, Int. J. Mol. Sci., 2022, 23(8), 4153 CrossRef CAS PubMed.
- L. A. Bors and F. Erdő, Overcoming the blood–brain barrier. challenges and tricks for CNS drug delivery, Sci. Pharm., 2019, 87(1), 6 CrossRef.
- T. Siegal, Which drug or drug delivery system can change clinical practice for brain tumor therapy?, Neuro-Oncology, 2013, 15(6), 656–669 CrossRef CAS PubMed.
- J. Kim, S. I. Ahn and Y. Kim, Nanotherapeutics engineered to cross the blood–brain barrier for advanced drug delivery to the central nervous system, J. Ind. Eng. Chem., 2019, 73, 8–18 CrossRef CAS PubMed.
- E. Calzoni, A. Cesaretti, A. Polchi, A. Di Michele, B. Tancini and C. Emiliani, Biocompatible polymer nanoparticles for drug delivery applications in cancer and neurodegenerative disorder therapies, J. Funct. Biomater., 2019, 10(1), 4 CrossRef CAS PubMed.
- C. Medina, M. Santos-Martinez, A. Radomski, O. Corrigan and M. Radomski, Nanoparticles: pharmacological and toxicological significance, Br. J. Pharmacol., 2007, 150(5), 552–558 CrossRef CAS PubMed.
- K. A. Altammar, A review on nanoparticles: characteristics, synthesis, applications, and challenges, Front Microbiol., 2023, 14, 1155622 CrossRef PubMed.
- T. Wu and M. Tang, The inflammatory response to silver and titanium dioxide nanoparticles in the central nervous system, Nanomedicine, 2018, 13(2), 233–249 CrossRef CAS PubMed.
- L. Zhou, Y. Wu, X. Meng, S. Li, J. Zhang and P. Gong, et al., Dye-Anchored MnO Nanoparticles Targeting Tumor and Inducing Enhanced Phototherapy Effect via Mitochondria-Mediated Pathway, Small, 2018, 14(36), 1801008 CrossRef PubMed.
- H. Gao, J. Qian, S. Cao, Z. Yang, Z. Pang and S. Pan, et al., Precise glioma targeting of and penetration by aptamer and peptide dual-functioned nanoparticles, Biomaterials, 2012, 33(20), 5115–5123 CrossRef CAS PubMed.
- W. Zhang, R. Taheri-Ledari, F. Ganjali, S. S. Mirmohammadi, F. S. Qazi and M. Saeidirad, et al., Effects of morphology and size of nanoscale drug carriers on cellular uptake and internalization process: a review, RSC Adv., 2022, 13(1), 80–114 RSC.
- R. Mout, D. F. Moyano, S. Rana and V. M. Rotello, Surface functionalization of nanoparticles for nanomedicine, Chem. Soc. Rev., 2012, 41(7), 2539–2544 RSC.
- N. Zhao, N. L. Francis, H. R. Calvelli and P. V. Moghe, Microglia-targeting nanotherapeutics for neurodegenerative diseases, APL Bioeng., 2020, 4(3), 030902 CrossRef CAS PubMed.
- M. Blank, T. Enzlein and C. Hopf, LPS-induced lipid alterations in microglia revealed by MALDI mass spectrometry-based cell fingerprinting in neuroinflammation studies, Sci. Rep., 2022, 12(1), 2908 CrossRef CAS PubMed.
- C. Qiu, F. Xia, J. Zhang, Q. Shi, Y. Meng and C. Wang, et al. Advanced Strategies for Overcoming Endosomal/Lysosomal Barrier in Nanodrug Delivery. Research, Wash D C. 2023;6:p. 0148 Search PubMed.
- J. Du, L. A. Lane and S. Nie, Stimuli-responsive nanoparticles for targeting the tumor microenvironment, J. Controlled Release, 2015, 219, 205–214 CrossRef CAS PubMed.
- P. Davoodi, L. Y. Lee, Q. Xu, V. Sunil, Y. Sun and S. Soh, et al., Drug delivery systems for programmed and on-demand release, Adv. Drug Delivery Rev., 2018, 132, 104–138 CrossRef CAS PubMed.
- H. Zhu, H. Chen, X. Zeng, Z. Wang, X. Zhang and Y. Wu, et al., Co-delivery of chemotherapeutic drugs with vitamin E TPGS by porous PLGA nanoparticles for enhanced chemotherapy against multi-drug resistance, Biomaterials, 2014, 35(7), 2391–2400 CrossRef CAS PubMed.
- Q. Feng, Y. Shen, Y. Fu, M. E. Muroski, P. Zhang and Q. Wang, et al., Self-assembly of gold nanoparticles shows microenvironment-mediated dynamic switching and enhanced brain tumor targeting, Theranostics, 2017, 7(7), 1875 CrossRef CAS PubMed.
- Y. Zhu, J. Feijen and Z. Zhong, Dual-targeted nanomedicines for enhanced tumor treatment, Nano Today, 2018, 18, 65–85 CrossRef CAS.
- X. Dong, Current strategies for brain drug delivery, Theranostics, 2018, 8(6), 1481 CrossRef CAS PubMed.
- W. H. Chen, G. F. Luo and X. Z. Zhang, Recent advances in subcellular targeted cancer therapy based on functional materials, Adv. Mater., 2019, 31(3), 1802725 CrossRef PubMed.
- Y. Ju-Nam and J. R. Lead, Manufactured nanoparticles: An overview of their chemistry, interactions and potential environmental implications, Sci. Total Environ., 2008, 400(1), 396–414 Search PubMed.
- C. Bai and M. Tang, Toxicological study of metal and metal oxide nanoparticles in zebrafish, J. Appl. Toxicol., 2020, 40(1), 37–63 Search PubMed.
- X. Chang, J. Li, S. Niu, Y. Xue and M. Tang, Neurotoxicity of metal-containing nanoparticles and implications in glial cells, J. Appl. Toxicol., 2021, 41(1), 65–81 Search PubMed.
- V. Kumar, Toll-like receptors in the pathogenesis of neuroinflammation, J. Neuroimmunol., 2019, 332, 16–30 CrossRef CAS PubMed.
- D. S. A. Simpson and P. L. Oliver, ROS Generation in Microglia: Understanding Oxidative Stress and Inflammation in Neurodegenerative Disease, Antioxidants, 2020, 9(8), 743 CrossRef CAS PubMed.
- M. Battaglini, A. Marino, M. Montorsi, A. Carmignani, M. C. Ceccarelli and G. Ciofani, Nanomaterials as Microglia Modulators in the Treatment of Central Nervous System Disorders, Adv. Healthcare Mater., 2024, 13(12), 2304180 CrossRef CAS PubMed.
- X. Feng, A. Chen, Y. Zhang, J. Wang, L. Shao and L. Wei, Central nervous system toxicity of metallic nanoparticles, Int. J. Nanomed., 2015, 4321–4340 CAS.
- M. Tareq, Y. A. Khadrawy, M. M. Rageh and H. S. Mohammed, Dose-dependent biological toxicity of green synthesized silver nanoparticles in rat's brain, Sci. Rep., 2022, 12(1), 22642 Search PubMed.
- A. A. Antsiferova, M. Y. Kopaeva, V. N. Kochkin, A. A. Reshetnikov and P. K. Kashkarov, Neurotoxicity of silver nanoparticles and non-linear development of adaptive homeostasis with age, Micromachines, 2023, 14(5), 984 CrossRef PubMed.
- C. M. Duffy, S. Ahmed, C. Yuan, V. Mavanji, J. P. Nixon and T. Butterick, Microglia as a Surrogate Biosensor to Determine Nanoparticle Neurotoxicity, J. Visualized Exp., 2016,(116), 54662 Search PubMed.
- C.-L. Huang, I.-L. Hsiao, H.-C. Lin, C.-F. Wang, Y.-J. Huang and C.-Y. Chuang, Silver nanoparticles affect on gene expression of inflammatory and neurodegenerative responses in mouse brain neural cells, Environ. Res., 2015, 136, 253–263 Search PubMed.
- L. Xiao, F. Wei, Y. Zhou, G. J. Anderson, D. M. Frazer and Y. C. Lim, et al., Dihydrolipoic acid–gold nanoclusters regulate microglial polarization and have the potential to alter neurogenesis, Nano Lett., 2019, 20(1), 478–495 Search PubMed.
- M. C. Hohnholt, M. Geppert, E. M. Luther, C. Petters, F. Bulcke and R. Dringen, Handling of iron oxide and silver nanoparticles by astrocytes, Neurochem. Res., 2013, 38(2), 227–239 Search PubMed.
- R. Yokel, E. Grulke and R. MacPhail, Metal-based nanoparticle interactions with the nervous system: the challenge of brain entry and the risk of retention in the organism, Wiley Interdiscip. Rev.:Nanomed. Nanobiotechnol., 2013, 5(4), 346–373 Search PubMed.
- J. Milan, K. Niemczyk and M. Kus-Liśkiewicz, Treasure on the Earth—Gold Nanoparticles and Their Biomedical Applications, Materials, 2022, 15(9), 3355 Search PubMed.
- H.-H. Jeong, E. Choi, E. Ellis and T.-C. Lee, Recent advances in gold nanoparticles for biomedical applications: from hybrid structures to multi-functionality, J. Mater. Chem. B, 2019, 7(22), 3480–3496 RSC.
- C. Su, K. Huang, H.-H. Li, Y.-G. Lu and D.-L. Zheng, Antibacterial properties of functionalized gold nanoparticles and their application in oral biology, J. Nanomater., 2020, 2020(1), 5616379 Search PubMed.
- S. Chawla, N. Kerna, N. Carsrud, K. Pruitt, J. Flores, H. Holets and N. J. Obi, et al., Revealing the Health Applica-tions of Gold Nanoparticles for the Cardiovascular System (and in Nerve, Bone, Immune, Skin, and Mental Disorders), EC Cardiology, 2023, 10, 09–23 Search PubMed.
- L. A. Dykman and N. G. Khlebtsov, Gold nanoparticles in biology and medicine: recent advances and prospects, Acta Naturae, 2011, 3(2), 34–55 CrossRef CAS PubMed.
- K. Koushki, S. Keshavarz Shahbaz, M. Keshavarz, E. E. Bezsonov, T. Sathyapalan and A. Sahebkar, Gold nanoparticles: Multifaceted roles in the management of autoimmune disorders, Biomolecules, 2021, 11(9), 1289 CrossRef CAS PubMed.
- Z. D. Ozdal, E. Sahmetlioglu, I. Narin and A. Cumaoglu, Synthesis of gold and silver nanoparticles using flavonoid quercetin and their effects on lipopolysaccharide induced inflammatory response in microglial cells, 3 Biotech, 2019, 9(6), 1–8 Search PubMed.
- I. Kuschnerus, M. Lau, K. Giri, N. Bedford, J. Biazik and J. Ruan, et al., Effect of a protein corona on the fibrinogen induced cellular oxidative stress of gold nanoparticles, Nanoscale, 2020, 12(10), 5898–5905 RSC.
- S. Y. Park, E. H. Yi, Y. Kim and G. Park, Anti-neuroinflammatory effects of Ephedra sinica Stapf extract-capped gold nanoparticles in microglia, Int. J. Nanomed., 2019, 2861–2877 CrossRef CAS PubMed.
- E. Lira-Diaz, M. G. Gonzalez-Pedroza, C. Vasquez, R. A. Morales-Luckie and O. Gonzalez-Perez, Gold nanoparticles produce transient reactive gliosis in the adult brain, Neurosci. Res., 2021, 76–86 CrossRef CAS PubMed.
- E. Hutter, S. Boridy, S. Labrecque, M. Lalancette-Hébert, J. Kriz and F. M. Winnik, et al., Microglial response to gold nanoparticles, ACS Nano, 2010, 4(5), 2595–2606 CrossRef CAS PubMed.
- J. Xue, T. Liu, Y. Liu, Y. Jiang, V. D. D. Seshadri and S. K. Mohan, et al., Neuroprotective effect of biosynthesised gold nanoparticles synthesised from root extract of Paeonia moutan against Parkinson disease–In vitro & In vivo model, J. Photochem. Photobiol., B, 2019, 200, 111635 CrossRef CAS PubMed.
- N. Zhao, X. Yang, H. R. Calvelli, Y. Cao, N. L. Francis and R. A. Chmielowski, et al., Antioxidant nanoparticles for concerted inhibition of α-synuclein fibrillization, and attenuation of microglial intracellular aggregation and activation, Front. Bioeng. Biotechnol., 2020, 8, 112 Search PubMed.
- M.-J. Wang, H.-Y. Huang, W.-F. Chen, H.-F. Chang and J.-S. Kuo, Glycogen synthase kinase-3β inactivation inhibits tumor necrosis factor-α production in microglia by modulating nuclear factor κB and MLK3/JNK signaling cascades, J. Neuroinflammation, 2010, 7(1), 99 Search PubMed.
- M. J. Kim, S. U. Rehman, F. U. Amin and M. O. Kim, Enhanced neuroprotection of anthocyanin-loaded PEG-gold nanoparticles against Aβ1-42-induced neuroinflammation and neurodegeneration via the NF-κB/JNK/GSK3β signaling pathway, Nanomed. Nanotechnol. Biol. Med., 2017, 13(8), 2533–2544 CrossRef CAS PubMed.
- G. Aliev, J. Daza, A. Solís Herrera, M. del Carmen Arias Esparza, L. Morales and V. Echeverria, et al., Nanoparticles as alternative strategies for drug delivery to the Alzheimer brain: electron microscopy ultrastructural analysis, CNS Neurol Disord Drug Targets, 2015, 14(9), 1235–1242 CrossRef CAS PubMed.
- M. R. Pickard and D. M. Chari, Robust uptake of magnetic nanoparticles (MNPs) by central nervous system (CNS) microglia: implications for particle uptake in mixed neural cell populations, Int. J. Mol. Sci., 2010, 11(3), 967–981 CrossRef CAS PubMed.
- I. L. Hsiao, Y. K. Hsieh, C. Y. Chuang, C. F. Wang and Y. J. Huang, Effects of silver nanoparticles on the interactions of neuron-and glia-like cells: Toxicity, uptake mechanisms, and lysosomal tracking, Environ. Toxicol.:Princ. Policies, 2017, 32(6), 1742–1753 CrossRef CAS PubMed.
- C. Sun, N. Yin, R. Wen, W. Liu, Y. Jia and L. Hu, et al., Silver nanoparticles induced neurotoxicity through oxidative stress in rat cerebral astrocytes is distinct from the effects of silver ions, Neurotoxicology, 2016, 52, 210–221 Search PubMed.
- S. Amiri, A. Yousefi-Ahmadipour, M.-J. Hosseini, A. Haj-Mirzaian, M. Momeny and H. Hosseini-Chegeni, et al., Maternal exposure to silver nanoparticles are associated with behavioral abnormalities in adulthood: Role of mitochondria and innate immunity in developmental toxicity, Neurotoxicology, 2018, 66, 66–77 CrossRef CAS PubMed.
- N. Yin, Y. Zhang, Z. Yun, Q. Liu, G. Qu and Q. Zhou, et al., Silver nanoparticle exposure induces rat motor dysfunction through decrease in expression of calcium channel protein in cerebellum, Toxicol. Lett., 2015, 237(2), 112–120 CrossRef CAS PubMed.
- P. Su, J. Zhang, D. Wang, F. Zhao, Z. Cao and M. Aschner, et al., The role of autophagy in modulation of neuroinflammation in microglia, Neuroscience, 2016, 319, 155–167 CrossRef CAS PubMed.
- M. Shang, X. Chang, S. Niu, J. Li, W. Zhang and T. Wu, et al., The Key Role of Autophagy in Silver Nanoparticle-Induced BV2 Cells Inflammation and Polarization, Food Chem. Toxicol., 2021, 112324 CrossRef CAS PubMed.
- M. Shang, S. Niu, X. Chang, J. Li, W. Zhang and M. Guo, et al., Silver nanoparticle-induced impaired autophagic flux and lysosomal dysfunction contribute to the microglia inflammation polarization, Food Chem. Toxicol., 2022, 170, 113469 CrossRef CAS PubMed.
- L. A. Welikovitch, C. S. Do, Z. Maglóczky, J. C. Malcolm, J. Lőke and W. L. Klein, et al., Early intraneuronal amyloid triggers neuron-derived inflammatory signaling in APP transgenic rats and human brain, Proc. Natl. Acad. Sci. U. S. A., 2020, 117(12), 6844–6854 CrossRef CAS PubMed.
- K. Sikorska, I. Grądzka, B. Sochanowicz, A. Presz, S. Męczyńska-Wielgosz and K. Brzóska, et al., Diminished amyloid-β uptake by mouse microglia upon treatment with quantum dots, silver or cerium oxide nanoparticles: Nanoparticles and amyloid-β uptake by microglia, Hum. Exp. Toxicol., 2020, 39(2), 147–158 CrossRef CAS PubMed.
- S. Katarzyna, G. Iwona and W. Iwona, The Effect of Silver Nanoparticles on the Scavenger Receptor-Scara1 on Microglia, Proceedings of the 2nd World Congress on Recent Advances in Nanotechnology (RAN’17), 2017, pp. 2371–5308, DOI:10.11159/icnb17.115.
- K. Sikorska, I. Grądzka, I. Wasyk, K. Brzóska, T. M. Stępkowski and M. Czerwińska, et al., The impact of ag nanoparticles and cdte quantum dots on expression and function of receptors involved in amyloid-β uptake by bv-2 microglial cells, Materials, 2020, 13(14), 3227 CrossRef CAS PubMed.
- Z. Lyu, S. Ghoshdastidar, K. R. Rekha, D. Suresh, J. Mao and N. Bivens, et al., Developmental exposure to silver nanoparticles leads to long term gut dysbiosis and neurobehavioral alterations, Sci. Rep., 2021, 11(1), 6558 CrossRef CAS PubMed.
- D. A. Gonzalez-Carter, B. F. Leo, P. Ruenraroengsak, S. Chen, A. E. Goode and I. G. Theodorou, et al., Silver nanoparticles reduce brain inflammation and related neurotoxicity through induction of H2S-synthesizing enzymes, Sci. Rep., 2017, 7(1), 42871 Search PubMed.
- A. Sharma, V. Jaiswal, M. Park and H.-J. Lee, Biogenic silver NPs alleviate LPS-induced neuroinflammation in a human fetal brain-derived cell line: Molecular switch to the M2 phenotype, modulation of TLR4/MyD88 and Nrf2/HO-1 signaling pathways, and molecular docking analysis, Biomater. Adv., 2023, 148, 213363 CrossRef CAS PubMed.
- I. Khan, K. Saeed and I. Khan, Nanoparticles: Properties, applications and toxicities, Arabian J. Chem., 2019, 12(7), 908–931 CrossRef CAS.
- E. M. Luther, C. Petters, F. Bulcke, A. Kaltz, K. Thiel and U. Bickmeyer, et al., Endocytotic uptake of iron oxide nanoparticles by cultured brain microglial cells, Acta Biomater., 2013, 9(9), 8454–8465 CrossRef CAS PubMed.
- H.-Y. Wu, M.-C. Chung, C.-C. Wang, C.-H. Huang, H.-J. Liang and T.-R. Jan, Iron oxide nanoparticles suppress the production of IL-1beta via the secretory lysosomal pathway in murine microglial cells, Part. Fibre Toxicol., 2013, 10(1), 1–11 CrossRef PubMed.
- M. Glat, H. Skaat, N. Menkes-Caspi, S. Margel and E. A. Stern, Age-dependent effects of microglial inhibition in vivo on Alzheimer's disease neuropathology using bioactive-conjugated iron oxide nanoparticles, J. Nanobiotechnol., 2013, 11(1), 1–12 CrossRef PubMed.
- Y. Wang, B. Wang, M.-T. Zhu, M. Li, H.-J. Wang and M. Wang, et al., Microglial activation, recruitment and phagocytosis as linked phenomena in ferric oxide nanoparticle exposure, Toxicol. Lett., 2011, 205(1), 26–37 CrossRef CAS PubMed.
- C. Petters and R. Dringen, Uptake, metabolism and toxicity of iron oxide nanoparticles in cultured microglia, astrocytes and neurons, Springerplus, 2015, 4(Suppl 1), L32 CrossRef PubMed.
- B. A. Maher, I. A. Ahmed, V. Karloukovski, D. A. MacLaren, P. G. Foulds and D. Allsop, et al., Magnetite pollution nanoparticles in the human brain, Proc. Natl. Acad. Sci. U. S. A., 2016, 113(39), 10797–10801 CrossRef CAS PubMed.
- J. Li, J. Wang, Y.-L. Wang, Z. Luo, C. Zheng and G. Yu, et al., NOX2 activation contributes to cobalt nanoparticles-induced inflammatory responses and Tau phosphorylation in mice and microglia, Ecotoxicol. Environ. Saf., 2021, 225, 112725 CrossRef CAS PubMed.
- F. Zheng, Z. Luo, C. Zheng, J. Li, J. Zeng and H. Yang, et al., Comparison of the neurotoxicity associated with cobalt nanoparticles and cobalt chloride in Wistar rats, Toxicol. Appl. Pharmacol., 2019, 369, 90–99 CrossRef CAS PubMed.
- I.-L. Hsiao and Y.-J. Huang, Effects of various physicochemical characteristics on the toxicities of ZnO and TiO2 nanoparticles toward human lung epithelial cells, Sci. Total Environ., 2011, 409(7), 1219–1228 CrossRef CAS PubMed.
- O. Bondarenko, K. Juganson, A. Ivask, K. Kasemets, M. Mortimer and A. Kahru, Toxicity of Ag, CuO and ZnO nanoparticles to selected environmentally relevant test organisms and mammalian cells in vitro: a critical review, Arch. Toxicol., 2013, 87(7), 1181–1200 CrossRef CAS PubMed.
- A. K. Sharma, V. Singh, R. Gera, M. P. Purohit and D. Ghosh, Zinc oxide nanoparticle induces microglial death by NADPH-oxidase-independent reactive oxygen species as well as energy depletion, Mol. Neurobiol., 2017, 54(8), 6273–6286 CrossRef CAS PubMed.
- R. Dutta, B. P. Nenavathu, M. K. Gangishetty and A. Reddy, Studies on antibacterial activity of ZnO nanoparticles by ROS induced lipid peroxidation, Colloids Surf., B, 2012, 94, 143–150 CrossRef CAS PubMed.
- T. C. Long, J. Tajuba, P. Sama, N. Saleh, C. Swartz and J. Parker, et al., Nanosize titanium dioxide stimulates reactive oxygen species in brain microglia and damages neurons in vitro, Environ. Health Perspect., 2007, 115(11), 1631–1637 CrossRef CAS PubMed.
- J. Choi, Q. Zheng, H. E. Katz and T. R. Guilarte, Silica-based nanoparticle uptake and cellular response by primary microglia, Environ. Health Perspect., 2010, 118(5), 589–595 CrossRef CAS PubMed.
- L. Wei, J. Wang, A. Chen, J. Liu, X. Feng and L. Shao, Involvement of PINK1/parkin-mediated mitophagy in ZnO nanoparticle-induced toxicity in BV-2 cells, Int. J. Nanomed., 2017, 12, 1891 CrossRef CAS PubMed.
- S. Sruthi, T. Nury, N. Millot and G. Lizard, Evidence of a non-apoptotic mode of cell death in microglial BV-2 cells exposed to different concentrations of zinc oxide nanoparticles, Environ. Sci. Pollut. Res., 2021, 28(10), 12500–12520 CrossRef CAS PubMed.
- H. Liang, A. Chen, X. Lai, J. Liu, J. Wu and Y. Kang, et al., Neuroinflammation is induced by tongue-instilled ZnO nanoparticles via the Ca2+-dependent NF-κB and MAPK pathways, Part. Fibre Toxicol., 2018, 15(1), 1–21 CrossRef CAS PubMed.
- J. Liu, Y. Kang, W. Zheng, B. Song, L. Wei and L. Chen, et al., From the cover: ion-shedding zinc oxide nanoparticles induce microglial BV2 cell proliferation via the ERK and Akt signaling pathways, Toxicol. Sci., 2017, 156(1), 167–178 CrossRef CAS PubMed.
- H.-C. Chuang, Y.-T. Yang, H.-C. Chen, Y.-H. Hwang, K.-Y. Wu and T.-F. Chen, et al., Acute effects of pulmonary exposure to zinc oxide nanoparticles on the brain in vivo, Aerosol Air Qual. Res., 2020, 20(7), 1651–1664 CAS.
- E. M. Moustafa, F. S. Moawed and D. F. Elmaghraby, Luteolin/ZnO nanoparticles attenuate neuroinflammation associated with diabetes via regulating MicroRNA-124 by targeting C/EBPA, Environ. Toxicol., 2023, 2691–2704 CrossRef CAS PubMed.
- D. Ziental, B. Czarczynska-Goslinska, D. T. Mlynarczyk, A. Glowacka-Sobotta, B. Stanisz and T. Goslinski, et al., Titanium dioxide nanoparticles: prospects and applications in medicine, Nanomaterials, 2020, 10(2), 387 CrossRef CAS PubMed.
- N. Rihane, T. Nury, I. M’rad, L. El Mir, M. Sakly and S. Amara, et al., Microglial cells (BV-2) internalize titanium dioxide (TiO2) nanoparticles: toxicity and cellular responses, Environ. Sci. Pollut. Res., 2016, 23(10), 9690–9699 CrossRef CAS PubMed.
- L. Sheng, Y. Ze, L. Wang, X. Yu, J. Hong and X. Zhao, et al., Mechanisms of TiO2 nanoparticle-induced neuronal apoptosis in rat primary cultured hippocampal neurons, J. Biomed. Mater. Res., Part A, 2015, 103(3), 1141–1149 CrossRef PubMed.
- T. C. Long, N. Saleh, R. D. Tilton, G. V. Lowry and B. Veronesi, Titanium dioxide (P25) produces reactive oxygen species in immortalized brain microglia (BV2): implications for nanoparticle neurotoxicity, Environ. Sci. Technol., 2006, 40(14), 4346–4352 CrossRef CAS PubMed.
- J. A. Shin, E. J. Lee, S. M. Seo, H. S. Kim, J. L. Kang and E. M. Park, Nanosized titanium dioxide enhanced inflammatory responses in the septic brain of mouse, Neuroscience, 2010, 165(2), 445–454 CrossRef CAS PubMed.
- I.-L. Hsiao, C.-C. Chang, C.-Y. Wu, Y.-K. Hsieh, C.-Y. Chuang and C.-F. Wang, et al., Indirect effects of TiO2 nanoparticle on neuron-glial cell interactions, Chem.-Biol. Interact., 2016, 254, 34–44 CrossRef CAS PubMed.
- Y. Xue, J. Wu and J. Sun, Four types of inorganic nanoparticles stimulate the inflammatory reaction in brain microglia and damage neurons in vitro, Toxicol. Lett., 2012, 214(2), 91–98 CrossRef CAS PubMed.
- M. E. Vance, T. Kuiken, E. P. Vejerano, S. P. McGinnis, Jr M. F. Hochella and D. Rejeski, et al., Nanotechnology in the real world: Redeveloping the nanomaterial consumer products inventory, Beilstein J. Nanotechnol., 2015, 6(1), 1769–1780 CrossRef CAS PubMed.
- D. Liu, B. Lin, W. Shao, Z. Zhu, T. Ji and C. Yang, In vitro and in vivo studies on the transport of PEGylated silica nanoparticles across the blood–brain barrier, ACS Appl. Mater. Interfaces, 2014, 6(3), 2131–2136 CrossRef CAS PubMed.
- A. D. Ducray, A. Stojiljkovic, A. Möller, M. H. Stoffel, H.-R. Widmer and M. Frenz, et al., Uptake of silica nanoparticles in the brain and effects on neuronal differentiation using different in vitro models, Nanomed. Nanotechnol. Biol. Med., 2017, 13(3), 1195–1204 CrossRef CAS PubMed.
- R. You, Y.-S. Ho, C. H.-L. Hung, Y. Liu, C.-X. Huang and H.-N. Chan, et al., Silica nanoparticles induce neurodegeneration-like changes in behavior, neuropathology, and affect synapse through MAPK activation, Part. Fibre Toxicol., 2018, 15(1), 1–18 CrossRef PubMed.
- M. Delaval, S. Boland, B. Solhonne, M.-A. Nicola, S. Mornet and A. Baeza-Squiban, et al., Acute exposure to silica nanoparticles enhances mortality and increases lung permeability in a mouse model of Pseudomonas aeruginosa pneumonia, Part. Fibre Toxicol., 2015, 12(1), 1–13 CrossRef CAS PubMed.
- V. Marzaioli, J. A. Aguilar-Pimentel, I. Weichenmeier, G. Luxenhofer, M. Wiemann and R. Landsiedel, et al., Surface modifications of silica nanoparticles are crucial for their inert versus proinflammatory and immunomodulatory properties, Int. J. Nanomed., 2014, 2815–2832 Search PubMed.
- T. H. Shin, D. Y. Lee, B. Manavalan, S. Basith, Y.-C. Na and C. Yoon, et al., Silica-coated magnetic nanoparticles activate microglia and induce neurotoxic d-serine secretion, Part. Fibre Toxicol., 2021, 18(1), 1–18 CrossRef PubMed.
- S. Hou, C. Li, Y. Wang, J. Sun, Y. Guo and X. Ning, et al., Silica Nanoparticles Cause Activation of NLRP3 Inflammasome in-vitro Model-Using Microglia, Int. J. Nanomed., 2022, 5247–5264 CrossRef CAS PubMed.
- T. H. Shin, B. Manavalan, D. Y. Lee, S. Basith, C. Seo and M. J. Paik, et al., Silica-coated magnetic-nanoparticle-induced cytotoxicity is reduced in microglia by glutathione and citrate identified using integrated omics, Part. Fibre Toxicol., 2021, 18, 1–24 CrossRef PubMed.
- Z. Lyu, S. Ghoshdastidar, K. R. Rekha, D. Suresh, J. Mao and N. Bivens, et al., Developmental exposure to silver nanoparticles leads to long term gut dysbiosis and neurobehavioral alterations, Sci. Rep., 2021, 11(1), 1–14 CrossRef PubMed.
- X. Li, S. Xu, Z. Zhang and H. J. Schluesener, Apoptosis induced by titanium dioxide nanoparticles in cultured murine microglia N9 cells, Chin. Sci. Bull., 2009, 54(20), 3830–3836 CrossRef CAS.
- I. L. Hsiao, C.-C. Chang, C.-Y. Wu, Y.-K. Hsieh, C.-Y. Chuang and C.-F. Wang, et al., Indirect effects of TiO2 nanoparticle on neuron-glial cell interactions, Chem.-Biol. Interact., 2016, 254, 34–44 CrossRef CAS PubMed.
Footnotes |
| † Should be considered joint author. |
| ‡ Current affiliation: The Jackson Laboratory for Genomic Medicine, Farmington, CT, USA Phone (Fax): +982833790620-59 (cell). |
| This journal is © The Royal Society of Chemistry 2025 |