
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceDiscovery of new benzothiazole-1,2,3-triazole hybrid-based hydrazone/thiosemicarbazone derivatives as potent EGFR inhibitors with cytotoxicity against cancer†
Ateyatallah Aljuhani‡
a,
Mohamed S. Nafie‡ *bc,
Nader R. Albujuq*d,
Mosa Alsehlia,
Sanaa K. Bardaweele,
Khaled M. Darwishfg,
Shaya Y. Alraqaa,
Mohamed Reda Aouad*a and
Nadjet Rezkia
*bc,
Nader R. Albujuq*d,
Mosa Alsehlia,
Sanaa K. Bardaweele,
Khaled M. Darwishfg,
Shaya Y. Alraqaa,
Mohamed Reda Aouad*a and
Nadjet Rezkia
aChemistry Department, College of Sciences, Taibah University, Al-Madinah Al-Munawarah 41477, Saudi Arabia. E-mail: ateyatallah@hotmail.com; mosa_alsehli@hotmail.com; Sqahtani@taibahu.edu.sa; aouadmohamedreda@yahoo.fr; nadjetrezki@yahoo.fr
bDepartment of Chemistry, College of Sciences, University of Sharjah, P.O. 27272, Sharjah, United Arab Emirates. E-mail: mohamed.elsayed@sharjah.ac.ae
cChemistry Department, Faculty of Science, Suez Canal University, P.O. 41522, Ismailia, Egypt. E-mail: mohamed_nafie@science.suez.edu.eg
dDepartment of Chemistry, School of Science, The University of Jordan, Amman 11942, Jordan. E-mail: n.albujuq@ju.edu.jo
eDepartment of Pharmaceutical Sciences, School of Pharmacy, The University of Jordan, Amman 11942, Jordan. E-mail: s.bardaweel@ju.edu.jo
fMedicinal Chemistry Department, Faculty of Pharmacy, Suez Canal University, Ismailia 41522, Egypt. E-mail: khaled_darwish@pharm.suez.edu.eg
gDepartment of Medicinal Chemistry, Faculty of Pharmacy, Galala University, New Galala 43511, Egypt. E-mail: khaled.darwish@gu.edu.eg
First published on 4th February 2025
Abstract
Considering the widespread availability of certain medicines, there is still a critical need for potent anti-cancer agents. It is owing to numerous negative impacts and non-functionality of current drugs, particularly during the late stages of illness. To accomplish this, the new array of 1,2,3-triazole-benzothiazole molecular conjugates tethering hydrazone/thiosemicarbazone linkage 8a–l have been successfully synthesized via the efficient copper-catalyzed 1,3-dipolar cycloaddition of the appropriate un/substituted benzothiazole azides 4a–c with several O-propargylated benzylidene derivatives 7a–d. The newly established 1,2,3-triazole structural hybrids were thoroughly characterized using appropriate spectroscopic techniques (IR, 1H, 13C-NMR & CHN analysis). The cytotoxic features of the investigated triazole hybrids were assessed against three human cancer cell lines, A549, T47-D, and HCT-116 cancer cells, using the MTT assay. Based on the findings, the breast cancer cell line T47D displayed promising results with IC50 values of 13, 17, and 19 μM for the synthesized molecules 8a–c, respectively. Furthermore, the safety assessment of these compounds on normal cell lines revealed a relatively low risk to normal cells, as indicated by their IC50 values exceeding 500 μM, suggesting a reasonable safety margin. Interestingly, the most relevant derivatives 8a, 8b, and 8c, exhibited IC50 values of 0.69, 1.16, and 4.82 μM, respectively, causing inhibition of 98.5%, 96.8%, and 92.3%, compared to Erlotinib (IC50 = 1.3 μM, 98.2% inhibition). Molecular docking results exhibited a good binding affinity of compounds 8a and 8b towards the EGFR active site. Accordingly, these compounds can be further developed as target-oriented EGFR chemotherapeutics against cancer.
1. Introduction
Cancer continues to pose a significant global challenge, contributing to high mortality rates and a growing healthcare burden. Key hurdles in cancer treatment and management stem from the disease's inherent traits, including its aggressiveness, poor prognosis, lack of specificity in anti-cancer drugs, and the persistent development of drug resistance.1 In this regard, applying selective agents or even targeted molecules has been a challenge with low rates of success. Being revolutionary within the medicinal chemistry field, pharmacophoric hybridization arose as an appealing approach to developing novel small molecules with better efficacy, improved binding affinities, and lower toxicity profiles.2,3 The development of hybrid anti-cancer small molecules aims to enhance therapeutic safety profiles while addressing potential drug resistance through multitarget modulation and minimized drug–drug interactions.4,5 Studies on 1,2,3-triazole-containing hybrids have demonstrated their efficacy in overcoming drug resistance by targeting multiple pathways simultaneously, showcasing their versatility in anti-cancer drug design.4,5 Moreover, advancements in structural modification strategies of triazoles have further optimized their pharmacokinetic properties and biological activities, underscoring the utility of pharmacophoric hybridization as a promising approach for creating novel and effective anti-cancer agents.4,5Heterocyclic compounds are well known as most significant organic substances and are commonly found in molecules that are of distinct relevance in medical chemistry. Nitrogen-containing heterocycles are particularly essential to life science research because they are common in nature and serve as building blocks in various vitamins, hormones, and antibiotics as natural products.6 Benzothiazole (BTA) cores are fused heterocyclic molecules. They can be found in an array of therapeutic drugs possessing tremendous relevance in drug development and discovery approaches owing to their vast range of pharmacological properties such as anti-inflammatory,7 fungicidal,8 anti-diabetic,9 analgesics,10 anti-microbial,11 anti-tumor,12 antileishmanial,13 anthelmintic,14 antirheumatic,15 and CNS depressant activity.16 It was observed that the BTA derivatives display unique and extensive biological activity against various kinds of tumors and cancer cell lines with EGFR kinase inhibition17–20
Another interesting nitrogen-based heteroaromatic scaffold has sparked the interest of scientific academia for its distinctive structural features and remarkable bioactivities. The 1,2,3-triazole core harbors the advent of being a stable heterocyclic scaffold capable of maintaining its structural stability under various chemical conditions, including reduction, oxidation, and even hydrolysis events. Additionally, its feasible chemical synthesis through Click chemistry and copper- or ruthenium-catalyzed azide–alkyne cycloaddition methods would permit a high-yield preparation approach.21 In terms of drug development and optimization processes, this nitrogen-containing heterocycle has been considered a valuable bioelectronic isostere for versatile equipartition replacement of different functionalities such as esters, amides, and even diazo scaffolds.22,23 Regarding pharmacodynamics and compound-target affinities, the 1.2.3-triazole exhibits diverse reversible non-covalent binding potentiality towards target key binding sites owing to the ring's polar capacity serving as both hydrogen bond donor and acceptor.24 The dual potentiality of triazole rings made them indispensable for developing a variety of bioactive small molecules at a broad pharmacological spectrum, including antimalarial,25 anti-bacterial,26 antitubercular,27 anti-cancer activity,28 among others. Focusing on the scaffold's potential as anti-cancerous agents, the triazole-based compounds have been heavily reported with diverse mechanistic aspects hampering several signaling pathways, including aromatase, microtubule, bromodomain and extra terminal domain proteins (BETs), as well as tyrosine kinase enzymes.29 Notably, the tyrosine kinase and BET biotargets arose as the primary mechanistic aspects involved with the anti-cancer activities of triazole-based investigational and FDA-approved small molecules.29–31 In particular, several studies illustrated the ability of both 1,2,3-triazole and benzothiazole-containing compounds to exhibit anti-cancer properties by triggering an arrest of the cell cycle and apoptosis of cancer cells via inhibition of epidermal growth factor receptor (EGFR) tyrosine kinase (Fig. 1).32–36
 | ||
| Fig. 1 Literature reported EGFR inhibitors as anti-cancer agents harboring the privilege heteroaromatic scaffold (A) bioactive hybrids containing benzothiazole (compounds I–IV), (B) bioactive hybrids containing 1,2,3-triazole ring moiety (compounds V–VII); (C) molecular design representation between hybrid pharmacophores with benzothiazole and 1,2,3-triazole moities with amide linker. | ||
Reports highlighted the adequacy of these compounds to anchor at the ATP-binding site of EGFR within a competitive inhibition manner. The benzothiazole scaffold represents an https://www.google.com/search?sca_esv=23a6d0305129b91a%26rlz=1C1GCEJ_enIN1138IN1138%26q=efficient%26spell=1%26sa=X%26ved=2ahUKEwiMm7H_l5-LAxWER2wGHToiKWwQkeECKAB6BAgHEAE bioisosteres for the ATP-purine ring, being indispensable for the binding of the benzothiazole-based compound to the central region of the EGFR pocket.31 On the other hand, the triazole-containing EGFR inhibitors offered beneficial binding to EGFR's selectivity pocket via the compounds' triazole scaffold permitting relevant hydrogen bonding with lining residues.32–36
Within the past recent years, triazole-containing small molecule hybrids showed great interest in developing anti-cancerous agents owing to their remarkable ability to hamper multiple cancerous cell lines.37 Incorporating the 1,2,3-triazole nucleus with established anti-cancer pharmacophores not only shows promise for combating drug-resistant cancer but has already demonstrated efficacy in clinical trials against various cancer types.38 Strategically attaching these pharmacophores to the 1,2,3-triazole backbone represents a valuable approach for discovering novel anti-cancer candidates, including hybrids with artemisinin,39 indole,40 and pyridine.41 Such a combination strategy holds the potential of circumventing drug resistance as well as improved pharmacokinetic/dynamics profiles.42 The potential and prospect employment of triazole scaffolds within drug discovery have been profoundly highlighted following the clinical approval of several triazole-based anti-cancerous agents such as tucatinib (EGFR inhibitor), talazoparib (PARP-1 inhibitor) and anastrozole (aromatase inhibitor).43–45 Based on this accumulated evidence, the main aim of the presented study was to develop small anti-cancerous molecules based on the triazole-benzothiazole pharmacophoric hybridization approach (Fig. 1C). Synthesized compounds were evaluated for their anti-cancerous activity through multistage in vitro biological testing including cytotoxicity bioassay against three human cancerous cell lines, antiproliferative assay, anti-migratory wound healing testing, and enzymology inhibition testing against EGFR biotarget. Further, promising compounds were investigated for their molecular affinity towards the cancer-related biotarget through a molecular modeling approach. Combined molecular insights from this study are beneficial for guiding future development and optimization of triazole-benzothiazole hybrids of potential higher activity and less toxic profiles.
2. Results and discussion
2.1. Rationale study design
Hybrid molecules that result from combining several pharmacophores have the ability to overcome the alteration of numerous action mechanisms. The conceivable potential of hybridizing the 1,2,3-triazole core with an alternative anti-cancer pharmacophore holds the promise of generating innovative anti-cancer candidates characterized by outstanding effectiveness and reduced toxicity, particularly among drug-resistant malignancies.46 Throughout the past decade, attractive findings on effective and low-toxicity benzothiazoles and their 2-aminobenzothiazole derivatives were extensively documented. These have gained substantial interest owing to their ubiquitous application as favored scaffolds in medicinal chemistry and drug discovery research. They demonstrate substantial pharmacophoric behavior with a diverse range of biological activities including anti-tumor activity, to develop a fascinating framework of drugs.47 Compound D (Fig. 2) is a very potent tyrosine kinase inhibitor (IC50 = 96 nM) and displayed significant antiproliferative activity against diverse tested cancer cell lines (HT-29, PC-3, A549, and U87MG).48 On the other hand, thiosemicarbazide is now considered a significant linkage in developing biological and pharmacological agents. In recent research, thiosemicarbazide is essential for tyrosine targeting where aryl-thiosemicarbazide significantly inhibited the tyrosinase activity at an IC50 value = 0.05 μM (Fig. 2).49 | ||
| Fig. 2 Rational design of the target compounds; (D) structures of the previously reported compound with benzothiazole; (E) representative example of thiosemicarbazide with biological activity; (F) and (G) target Series I and II are representative examples of hybrid molecules of benzothiazole moiety and thiosemicarbazide derivatives with 1,2,3-triazole linker that exhibits anti-cancer activity; (H) pharmacophoric features of a co-crystalline tyrosine kinase inhibitor exhibiting relevant binding affinity towards the structural/functional motifs of the ATP-binding site (PDB ID: 3W32). | ||
Building upon the aforementioned observations to enhance the anti-tumor efficacy of these reported anti-cancer compounds, we have anticipated designing and synthesizing new hybrid molecules encompassing benzothiazole and triazole moieties as anticipation and continuous pursuit of identifying novel anti-cancer agents. Our adopted ligand-based design strategy focused mainly on the ligation of 2-aminobenzothiazole moiety and a phenoxy fragment linked with thiosemicarbazide derivatives. Furthermore, we investigated the change of various substituents to explore their impact on the anti-cancer activity through the 1,2,3-triazole linker (Fig. 2). Given such findings on the potential bio-features of benzothiazole and 1,2,3-triazole molecular core; endeavors were made to hybrid benzothiazole moiety and the 1,2,3-triazole core through the Click synthesis of the focused alkyne based hydrazone/thiosemicarbazide linkage with the appropriate benzothiazole azides furnishing on the elaboration of the desired hydrazone and thiosemicarbazone derivatives as new Schiff bases tethering benzothiazole-1,2,3-triazole molecular conjugates (Series I and II).
It is worth noting that our designed target compounds were also designed to harbor the key structural features of common tyrosine kinase compounds (Fig. 2). The benzothiazole planner scaffold resembles the ATP-purine core ring being capable of mediating indispensable polar hydrogen bonding with the principal binding residues at the hinge region of the tyrosine kinase catalytic pocket.50 structural diversity on the benzothiazole ring provides prospective binding at the solvent-exposed pocket of the kinase active site. Regarding our designed acetamide triazole central structure, it comprises the heteroatom-aryl spacer being reported as important for occupying the pocket space connecting the ATP-adenine binding site with the tyrosine kinase functional DFG-motif that is typically involved within the enzyme activation machinery.51 Moving towards our thiosemicarbazone or hydrazone moieties in our compounds. These scaffolds resemble the hydrogen bond functionality (acceptors and/or donors) that have been heavily reported within several tyrosine kinase inhibitors capable of mediating polar contacts with kinase's secondary structures such as DFG-motif and αC-helix involved in enzyme's conformational activation.51,52 Finally, the terminal aryl/aroyl groups at Series-I were suggested as beneficial for targeting the buried deep hydrophobic back pocket, which has been correlated to high inhibition profiles and minimal off-target activities of reported tyrosine kinase inhibitors.50 The above-described design approach of our synthesized compounds harbors the advent of combining both ligand and structural-based insights through a holistic approach for increasing the success rate of developing anti-cancerous agents with potential tyrosine kinase inhibition activity. The resulting triazoles-based scaffold 8a–l with bioactive benzothiazole core will proceed for biological activation as cytotoxicity against breast, lung, and colon cancer cells with the EGFR target protein, as EGFR is overexpressed in a variety of human cancers, including lung, head, and neck, colon, pancreas, breast, ovary, bladder and kidney, and gliomas.53
2.2. Chemistry
 | ||
| Scheme 1 Optimized synthetic route for the synthesis of the targeted hydrazone/thiosemicarbazone-based benzothiazole-1,2,3-triazole hybrids 8a–c. | ||
 | ||
| Scheme 2 Synthesis of hydrazone/thiosemicarbazone-based benzothiazole-1,2,3-triazole hybrids 8d–l. | ||
The first route involves the propargylation of the p-hydroxybenzaldehyde (1) with propargyl bromide followed by its cycloaddition with the azidobenzothiazoles 4a–c to afford the corresponding 1,2,3-triazoles bearing benzaldehyde moiety 5a–c, which undergo condensation reaction with benzohydrazide 6a to give the 1,2,3-triazole-benzothiazole molecular conjugates with acetamide linkage 8a–c. The precursor benzothiazole azides 4a–c were prepared via the standard diazotization-azidolysis of the un/substitutedbenzo[d]thiazol-2-amine 3a–c.54 This route was found to produce the targeted adducts 8a–c (Scheme 1: route a) in relatively low yields (30–33%). Conversely, the condensation of the synthesized O-propargylated benzaldehyde 2 with benzohydrazide 6a yielded the alkyne derivative 7a, which was then used as an alkyne in CuAAC reaction with the selected benzothiazole azides 4a–c, yielding the targeted 1,2,3-triazole-benzothiazole molecular conjugates with acetamide linkage 8a–c in 87–91% yields (Scheme 1: route b).
The optimized Click reaction conditions were adopted to synthesize an innovative array of 1,2,3-triazoles carrying benzothiazole moiety and the hydrazone/thiosemicarbazone linkage 8d–l. Thus, the condensation of 4-(prop-2-yn-1-yloxy)benzaldehyde (2) with isoniazide (6b), 3,5-dinitrophenylhydrazine (6c), and/or thiosemicarbazide (6d), in refluxing ethanol with a catalytic quantity of acetic acid as a catalyst, afforded the corresponding hydrazone/thiosemicarbazone-based alkynes 7b–d in 93–95% yields (Table 1).
| Compounds no. | Structure | Mp (°C) | Yield (%) | |
|---|---|---|---|---|
| a Yield from route a in Scheme 1.b Yield from route b in Scheme 1. | ||||
| Alkyne derivatives |  |
96 | ||
| 7a | R |  |
159–160 | 94 |
| 7b | R |  |
180–181 | 94 |
| 7c | R |  |
226–227 | 95 |
| 7d | R | NH2CSNH– | 154–155 | 93 |
| Click adducts route a |  |
|||
| 5a | R1 | –H | 197–198 | 89 |
| 5b | R1 | –CH3 | 210–211 | 88 |
| 5c | R1 | –SO2CH3 | 236–237 | 86 |
| Click adducts route b |  |
|||
| 8a | R1 | –H | 223–224 | 90a |
| R2 |  |
33b | ||
| 8b | R1 | –CH3 | 237–238 | 89a |
| R2 |  |
32b | ||
| 8c | R1 | –SO2CH3 | 270–271 | 88a |
| R2 |  |
30b | ||
| 8d | R1 | –H | 255–256 | 90 |
| R2 |  |
|||
| 8e | R1 | –CH3 | 240–241 | 90 |
| R2 |  |
|||
| 8f | R1 | –SO2CH3 | 289–290 | 88 |
| R2 |  |
|||
| 8g | R1 | –H | 200–201 | 91 |
| R2 |  |
|||
| 8h | R1 | –CH3 | 170–171 | 90 |
| R2 |  |
|||
| 8i | R1 | –SO2CH3 | 190–191 | 89 |
| R2 |  |
|||
| 8j | R1 | –H | 186–187 | 90 |
| R2 | NH2CSNH– | |||
| 8k | R1 | –CH3 | 209–210 | 90 |
| R2 | NH2CSNH– | |||
| 8l | R1 | –SO2CH3 | 173–174 | 88 |
| R2 | NH2CSNH– | |||
Moreover, the ligation of the resulting alkynes 7b–d with benzothiazole azides 4a–c was carried out in the presence of a mixture of copper sulfate (CuSO4) and sodium ascorbate under the 1,3-dipolar cycloaddition conditions and gave selectively 87–91% yields of the Click adduct 8d–l (Table 1).
![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/char_e002.gif) CH) of the starting alkyne 2 supporting its participation in the Click synthesis. In addition, the appearance of a distinct singlet near δH 8.36–8.37 ppm supports the synthesis of the desired 1,2,3-triazole nucleus. The two sets of singlets observed at δH 5.32–5.33 ppm and δH 5.56–5.68 ppm were attributed to the two methylene protons of the –OCH2– and –NCH2– linkages, respectively. While the –CONH– and –CHO protons were recorded as two distinct singlets at δH 12.80–12.86 ppm and δH 9.86–9.88 ppm, respectively. All remaining protons were collected and described in the Experimental part. The 13C NMR spectra showed clearly the absence of the acetylenic carbons and the existence of a downfield signal at δC 191.9–192.5 ppm, assigned to the aldehyde carbonyl carbon.
CH) of the starting alkyne 2 supporting its participation in the Click synthesis. In addition, the appearance of a distinct singlet near δH 8.36–8.37 ppm supports the synthesis of the desired 1,2,3-triazole nucleus. The two sets of singlets observed at δH 5.32–5.33 ppm and δH 5.56–5.68 ppm were attributed to the two methylene protons of the –OCH2– and –NCH2– linkages, respectively. While the –CONH– and –CHO protons were recorded as two distinct singlets at δH 12.80–12.86 ppm and δH 9.86–9.88 ppm, respectively. All remaining protons were collected and described in the Experimental part. The 13C NMR spectra showed clearly the absence of the acetylenic carbons and the existence of a downfield signal at δC 191.9–192.5 ppm, assigned to the aldehyde carbonyl carbon.The structures of the resulting Schiff bases tethering alkyne side chains 7a–d were established using their respective spectroscopic information (IR and NMR spectra). Thus, their IR spectra clearly showed the disappearance of the characteristic aldehyde stretching bands (C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O and C–H Aldehyde) of their starting material 2, confirming the success of the condensation reaction. Their 1H NMR spectra also supported their structures by the absence of the signal attributed to the aldehyde proton and the appearance of new distinct singlets ranging around δH 8.04–8.11 ppm and δH 11.33–11.95 ppm, assigning the azomethine (HCN–) and –CONH– protons, respectively. According to their 1H NMR spectra, the resulting hydrazone/thiosemicarbazone-based alkynes 7a–d comprised of two conformational isomers E/cis and the E/trans as most of the descriptive protons (HCN–, –CONH, OCH2…etc.) were recorded as double signals with different isomeric ratios (see Experimental section). These results were in accordance with our early reported molecules that displayed the existence of cis/trans conformers throughout the amide (CO and N–H) groups and E/Z geometrical isomers within the imine (HCN) linkage.55–58
O and C–H Aldehyde) of their starting material 2, confirming the success of the condensation reaction. Their 1H NMR spectra also supported their structures by the absence of the signal attributed to the aldehyde proton and the appearance of new distinct singlets ranging around δH 8.04–8.11 ppm and δH 11.33–11.95 ppm, assigning the azomethine (HCN–) and –CONH– protons, respectively. According to their 1H NMR spectra, the resulting hydrazone/thiosemicarbazone-based alkynes 7a–d comprised of two conformational isomers E/cis and the E/trans as most of the descriptive protons (HCN–, –CONH, OCH2…etc.) were recorded as double signals with different isomeric ratios (see Experimental section). These results were in accordance with our early reported molecules that displayed the existence of cis/trans conformers throughout the amide (CO and N–H) groups and E/Z geometrical isomers within the imine (HCN) linkage.55–58
The spectra revealed the detection of a new singlet at δH 3.56–3.61 ppm referred to as the terminal alkyne proton. The residual protons were identified in their proper chemical shifts (see Experimental section). In addition, all 13C NMR results confirmed obviously the appearance of new signals caused by to Sp-carbons (C![[triple bond, length as m-dash]](https://www.rsc.org/images/entities/b_char_e002.gif) C) at δC 78.9–79.4 ppm, which proved the production of the desired alkyne precursors 7a–c.
C) at δC 78.9–79.4 ppm, which proved the production of the desired alkyne precursors 7a–c.
The success of the 1,3-dipolar cycloaddition reaction was evidenced by the analysis of the spectral data of the resulting Click products 8a–l. All IR spectra showed the disappearance of the acetylenic stretching bands of absorption (–CC–, C–H), confirming their participation in the formation of triazole ring.
Their 1H NMR spectra also displayed the absence of the acetylenic proton (C–H) and the presence of the diagnostic triazolyl–H-5 proton at δH 8.25–8.42 ppm, evidencing the formation of the targeted 1,2,3-triazole core. Furthermore, the spectra also demonstrated the detection of the distinct singlets recorded at δH 5.27–5.61 ppm and δH 5.26–5.34 ppm attributed to the methylene protons of the –OCH2– and –NCH2– linkages, respectively. The two sets of singlets recorded at δH 8.25–8.63 ppm and 9.88–12.86 ppm were assigned to the –CONH– and –NCH– protons, respectively. The spectral data of the resulting 1,2,3-triazole-based hydrazones/thiosemicarbazones 8a–l revealed the cis/trans conformers on the amide (CO and N–H) and the E/Z geometrical isomers for the imine (HCN) bond, respectively. These findings are congruent with the previously published research that studied this type of isomerism.58,59 The remaining protons resonated with respect to their designated area (see Experimental part). Furthermore, their 13C NMR results fitted their anticipated structures 8a–l and showed the lack of alkyne carbons (–CC–), confirming the accomplished synthesis.
The spectra also showed new signals in the aliphatic area at δC 61.60–61.88 ppm and δC 52.10–53.20 ppm attributed to the –OCH2– and –COCH2– carbons, respectively. The carbonyl carbons (CO) were registered around δC 159.80–166.70 ppm. All carbon signals were recorded with details in the Experimental part.
2.3. Biological assay
| Compound | T47-D | HCT-116 | A549 | Fibroblasts |
|---|---|---|---|---|
| a Positive control for lung cancer A549.b Positive control for breast cancer T47-D.c Positive control for colon cancer HCT-116. | ||||
| 7a | 208 ± 4 | 237 ± 6 | 200 ± 4 | >500 |
| 7b | 347 ± 9 | 395 ± 5 | 298 ± 3 | >500 |
| 7c | 175 ± 6 | 209 ± 5 | 156 ± 4 | >500 |
| 7d | 130 ± 2 | 261 ± 6 | 128 ± 5 | >500 |
| 8a | 17 ± 1 | 84 ± 3 | 18 ± 1 | >500 |
| 8b | 13 ± 1 | 94 ± 1 | 21 ± 1 | >500 |
| 8c | 19 ± 2 | 48 ± 2 | 25 ± 1 | >500 |
| 8d | 30 ± 1 | 100 ± 2 | 38 ± 2 | >500 |
| 8e | 101 ± 2 | 207 ± 5 | 92 ± 2 | >500 |
| 8f | 46 ± 4 | 149 ± 3 | 38 ± 2 | >500 |
| 8g | 95 ± 3 | 109 ± 3 | 103 ± 5 | >500 |
| 8h | 77 ± 2 | 167 ± 4 | 65 ± 3 | >500 |
| 8i | 84 ± 1 | 162 ± 5 | 89 ± 4 | >500 |
| 8j | 54 ± 2 | 190 ± 4 | 63 ± 2 | >500 |
| 8k | 119 ± 4 | 170 ± 7 | 138 ± 4 | >500 |
| 8l | 130 ± 2 | 180 ± 4 | 142 ± 3 | >500 |
| Cisplatina | — | — | 22 ± 1 | |
| Doxorubicinb | 6 ± 0.5 | — | — | |
| 5-FUc | — | 11 ± 0.7 | — | |
Comparing the activity patterns across the cell lines and among the same line itself was highlighted as important to deduce valuable insights regarding the in vitro cytotoxic activity correlation with compounds' structural differences. This structural activity relationship has demonstrated relevant https://www.google.com/search?sca_esv=23a6d0305129b91a%26rlz=1C1GCEJ_enIN1138IN1138%26q=structural%26spell=1%26sa=X%26ved=2ahUKEwjw35K2nZ-LAxVlcGwGHTY2B-gQkeECKAB6BAgHEAE%26cshid=1738301429273491 molecular insights being valuable for guiding future compound optimization and development. As a general observation, the synthesized benzothiazole–triazole adducts (8a–l) exhibited higher cytotoxic activities than their respective penultimate intermediate compounds (7a–d) across all three tested cancerous cell lines. On the other hand, preferential cytotoxic activity towards breast T47-D and lung A549 cell lines has been depicted to all hybrid adducts as compared to colon HCT-116 cancer cells (IC50 range = 13 ± 1 to 130 ± 2 μM @ T47-D; 18 ± 1 to 143 ± 3 μM @ A549; and 48 ± 2 to 207 ± 5 μM @ HCT-116).
The potency values in the two most responsive cell lines, T47-D and A549, showed similar activity patterns across the synthesized benzothiazole–triazole hybrids. Notably, members of the Series-I (8a–i) were assigned with higher potency values (lower IC50 concentrations) as compared to those of the thiosemicarbazone derivatives (Series-II; 8j–l). Compounds with simple unsubstituted aroyl moiety (R2 = benzoyl; 8a–c) demonstrated the highest potency values among all members of Series-I and across both cancerous cell lines. Contrarily, activity values at both T47-D and A549 cell lines were intolerant to terminal substitution with the dinitrophenyl group (8g–i) and, to a lesser extent, towards the pyridinoyl functionality (8d–f).
Moving our attention towards the structural diversity at C6 of the benzothiazole ring scaffold, activity profiles were at quite comparable patterns in both T47-D and A549 cell lines. Series-II compounds were intolerant to any R1 substitutions having better IC50 values for the unsubstituted benzothiazole ring, as seen with compound 8j. Similarly, members of the Series-I pyridinoyl hydrazone terminal groups favored their benzothiazole ring being unsubstituted with 8d for better cytotoxic activity profiles. Contrarily, members of the Series-I dinitrophenyl hydrazone moiety showed preferentiality for small group substitution (methylation) at the benzothiazole C6 position, where 8h showed better IC50s at both cancerous cells. Finally, compounds with benzoyl hydrazone scaffold were quite different across T47-D and A549 cell lines where cytotoxic activity at the earlier cell line was in favor of the R1 methylated analog (8b) while as at lung cancer, activity was intolerant to any R1 substitution. It is worth noting that methylsulphone derivatization at the benzothiazole adduct side was disfavoured for cytotoxic activity for the dinitrophenyl hydrazone and thiosemicarbazone derivatives (8i and 8I) as compared to their unsubstituted benzothiazole parent compounds. Conversely, the introduction of the methylsulphone functionality at the pyridinoylhydrazones (8f versus 8e) was favored for compound 8f cytotoxic activities across the three cancerous cell lines. It was suggested that the latter beneficial boosted cytotoxic activity for 8f could be correlated to the compound's improved physiochemical properties following the sulphone group introduction. Comparative physiochemical properties of 8e and 8f were estimated via the ADMETlab 2.0 web-based platform (https://admetmesh.scbdd.com/; Xiangya School of Pharmaceutical Sciences, Central South University, China). Interestingly, the compounds' hydrophobic characteristics were much favored for compound 8f compared to 8e in terms of log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P (octanol/water partition coefficient) and logD (logP at physiological pH 7.40) indices. Compound 8f showed logP and logD at 2.61 and 2.007, respectively, while higher lipophilicity patterns were seen with 8e (logP = 4.20; logD = 3.17). Generally, a compound's hydrophobicity is the driving force for the compound's permeation across cellular membranes and thus significantly impacts the cellular uptake of compounds.60 Optimal logP and logD values were set at ≤3.0 as per ADMETlab 2.0 thresholds where far below and above these values, the compounds became either potentially incapable of penetrating cellular membranes or being trapped within these membranes disfavouring their partition towards the aqueous https://www.google.com/search?sca_esv=9a5876f960b44ea9%26rlz=1C1GCEJ_enIN1138IN1138%26q=compartments%26spell=1%26sa=X%26ved=2ahUKEwiU5Z2An5-LAxX7SGwGHQuOJe0QkeECKAB6BAgLEAE.61,62 It is worth noting that the predicted aqueous solubility patterns (logS) for both compounds 8e and 8f do not greatly vary (−5.814 versus −5.859, respectively). In these regards, it was suggested that incorporation of methylsulphone group at compound 8f impose balanced hydrophobic/polar characteristics for the compounds being potentially translated into better cancer cell uptake and cytotoxicity activity (lower IC50). Physiochemical properties could further provide a rational for compounds 8i and 8l being intolerant towards methylsulphone structural incorporation. As per in silico calculations, both compounds showed imbalanced polarity/hydrophobicity indices (logS/logD) with significant shift towards higher hydrophilicity characteristics. Generally, both compounds harbor a much polar functionality at their hydrazone terminal side (8i = dinitro; 8l = thiosemicarbazone) as compared to 8f (pyridine ring). Notably, compound 8i depicted a very high polarity index (logS = −6.25) being uncompensated with the estimated low-value hydrophobic characteristic (logD = 2.95). On comparable bases, compound 8l showed a very small hydrophobic parameter with predicted logD at 1.96 under normal physiological conditions (pH 7.40). Both compounds were suggested with compromised cellular uptake the thing that could partially reason for their low IC50 values. All discussed SAR insights are summarized as follows in Fig. 3A.
P (octanol/water partition coefficient) and logD (logP at physiological pH 7.40) indices. Compound 8f showed logP and logD at 2.61 and 2.007, respectively, while higher lipophilicity patterns were seen with 8e (logP = 4.20; logD = 3.17). Generally, a compound's hydrophobicity is the driving force for the compound's permeation across cellular membranes and thus significantly impacts the cellular uptake of compounds.60 Optimal logP and logD values were set at ≤3.0 as per ADMETlab 2.0 thresholds where far below and above these values, the compounds became either potentially incapable of penetrating cellular membranes or being trapped within these membranes disfavouring their partition towards the aqueous https://www.google.com/search?sca_esv=9a5876f960b44ea9%26rlz=1C1GCEJ_enIN1138IN1138%26q=compartments%26spell=1%26sa=X%26ved=2ahUKEwiU5Z2An5-LAxX7SGwGHQuOJe0QkeECKAB6BAgLEAE.61,62 It is worth noting that the predicted aqueous solubility patterns (logS) for both compounds 8e and 8f do not greatly vary (−5.814 versus −5.859, respectively). In these regards, it was suggested that incorporation of methylsulphone group at compound 8f impose balanced hydrophobic/polar characteristics for the compounds being potentially translated into better cancer cell uptake and cytotoxicity activity (lower IC50). Physiochemical properties could further provide a rational for compounds 8i and 8l being intolerant towards methylsulphone structural incorporation. As per in silico calculations, both compounds showed imbalanced polarity/hydrophobicity indices (logS/logD) with significant shift towards higher hydrophilicity characteristics. Generally, both compounds harbor a much polar functionality at their hydrazone terminal side (8i = dinitro; 8l = thiosemicarbazone) as compared to 8f (pyridine ring). Notably, compound 8i depicted a very high polarity index (logS = −6.25) being uncompensated with the estimated low-value hydrophobic characteristic (logD = 2.95). On comparable bases, compound 8l showed a very small hydrophobic parameter with predicted logD at 1.96 under normal physiological conditions (pH 7.40). Both compounds were suggested with compromised cellular uptake the thing that could partially reason for their low IC50 values. All discussed SAR insights are summarized as follows in Fig. 3A.
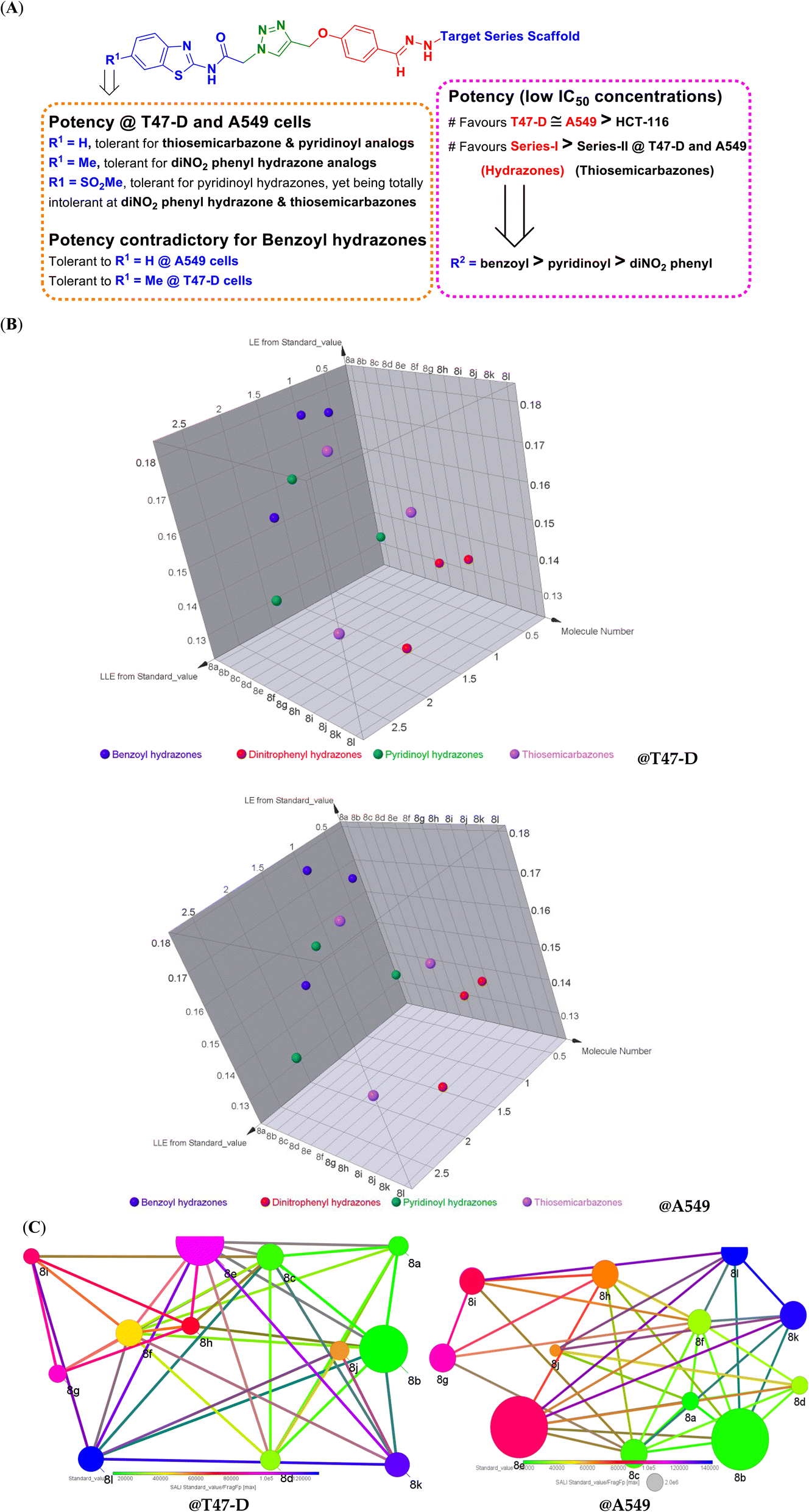 | ||
| Fig. 3 Biological activity correlation to compounds' structural modifications. (A) Summerized SAR insights for the synthesized compounds in terms of their in vitro cytotoxic MTT activity assay; (B) 3D-charts of derived ligand efficiency indices for our synthesized compounds at T47-D and A549 cancer cells. Molecule numbers (compound codes), LEs, and LLEs are shown on x, y, and z cartesians. Compounds belonging to structurally similar scaffolds are color coded; benzoyl hydrazones (blue), dinitrophenyl hydrazones (red), pyridinoyl hydrazones (green), and thiosemicarbazones (purple); (C) Structure–Activity Landscape Index (SALI) for synthesized compounds against H47-D and A549 cell lines. Fragment-based structural similarity is plotted in terms of the compound's IC50. Compounds are represented as labeled nodes, colored in spectrum according to their observed in vitro IC50 standard values (from green to blue color spectrum corresponding to low-value/more potent to high-value/less potent IC50s). Compounds as nodes are connected in pairs if being considered structural-activity cliffs. Significant compound pairs with depicted structural-activity cliffs (high SALI standard_value/FragFp values) are those showing large-sized connected nodes. These large highlighted nodes impose the great impact of minute structural modification on the compound's activity. | ||
The above SAR data could be highly correlated to compounds' physiochemical properties (lipophilicity, heteroatomic compositions, and/or extent of ionization) and, in turn, their pharmacokinetic profiles, the pharmacodynamics of compound-biotarget binding, or even altogether.63 Therefore, we aimed to associate the depicted cytotoxicity IC50 values on top-sensitive cancerous cells (T47-D and A549) with compounds' physiochemical characteristics through estimating more reliable indices such as ligand's efficiencies (LEs) and lipophilic efficiencies (LLEs). These activity-related indices account for compounds' hetero/heavy atom compositions and structural-related lipophilic characteristics (logP), respectively.64 Generally, consensus threshold values of 0.3 for LEs and 3 for LLEs have been correlated to compounds with promising activity profiles.65,66 3D-charts of compounds' LE and LLE values at T47-D and A549 were constructed using DataWarriors V06.02.05 as seen in Fig. 3B. At H47-D cell line, members of the Series-I benzoyl and pyridinoyl hydrazones showed a wide range of LE and LLE indices with 8c and 8f being with the significant activity indices (LE = 0.16 and 0.15; LLE = 1.93 and 2.58, respectively). Regarding the last group members of Series-I, dinitrophenyl hydrazones, compound 8i had moderate activity values: LE = 0.12 and LLE = 1.52. Moving toward the Series-II of thiosemicarbazone analogs, relevant activity values were highlighted for 8j at LE = 0.14 and LLE = 1.81. It is worth noting that activity indices at the A549 cell line were quite comparable to those of H47-D cells, highlighting significant profiles for 8c, 8f, 8i, and 8j compounds (LEs = 0.15, 0.15, 0.12, and 0.13; LLEs = 1.84, 2.66, 1.50, and 1.82, respectively).
To further grasp the impacts of small structural modifications on similar compounds' activity, a simplified quantitative index concerning the structural activity modifications has been employed. Structural-activity cliffs were estimated for the synthesized compounds via DataWarriors V06.02.05 through Structure–Activity Landscape Index (SALI) analysis based on their structural/fragment similarity to their cytotoxicity IC50 values (Fig. 3C). Compounds are represented as nodes and colored in heat map according to their IC50 values. Compounds are to be connected if they are considered structural-activity cliffs, meaning that these connected compounds are most interesting for SAR study being most similar yet with the greatest activity/IC50 changes.67 Within the SALI analysis and at 95% cut-off, certain paired compounds were of the highest SALI values (large-sized nodes) that worth close investigation. First observation was that both Series-I and -II were represented as singular groups owing to great structural/fragment similarity being clustered all together. At H47-D, only members of Series-I (8b and 8e) were represented as highly significant structural-activity cliffs showing the largest sized nodes. The pairing of 8b or 8e was highlighted the most with 8k and 8h at SALI-IC50's highest values (1813500 and 449500, respectively), highlighting the favored impact of the benzoyl hydrazones at the terminal aromatic scaffold (R2). Other significant string pairing of 8e with 8f (SALI-IC50 = 201600) further highlights the beneficial impact of methylsulphone R1 substitution on increasing the IC50 potency only at pyridinoyl hydrazone series. Regarding the A549 cell line, almost comparable SALI findings were highlighted, showing both 8b and 8e as the most significant structural-activity cliffs, being even with larger-size nodes (SALI index) than in case of H47-D cancerous cell line.
 | ||
| Fig. 4 Effect of 8a and 8b treatment on A549 cell migration. A549 cells were treated with 8a and 8b at IC50 and sub-IC50 concentrations for 24 h. *(P ≤ 0.05) significantly different using the unpaired t-test in GraphPad prism. Images of the wound at 0 and 24 h at 10× magnification using EVOS XL Core imaging system is supported in the supplementary (Fig. S25†). | ||
| Compound | % of EGFR inhibition | IC50 ± SDa (μM) |
|---|---|---|
| a Values are expressed as an average of three independent replicates. IC50 values were calculated using sigmoidal non-linear regression curve fit of percentage inhibition against five concentrations of each compound. | ||
| 8a | 98.5 ± 3.1 | 0.69 ± 0.01 |
| 8b | 96.8 ± 2.1 | 1.16 ± 0.02 |
| 8c | 92.3 ± 3.1 | 4.82 ± 0.01 |
| Erlotinib | 98.2 ± 2.4 | 1.3 ± 0.01 |
 | ||
| Fig. 5 Dose–response curve of the % of phosphorylation at each concentration for compounds 8a, 8b and 8c. Compounds were tested using five serial concentrations of 0.01, 0.1, 1, 2.5, and 5 μM. | ||
2.4. In silico studies
Compound 8a was docked inside the protein, and the EGFR active site formed one hydrogen bond as HBA with a bond length/angle of 2.32 Å/179.6°, through its triazole core with the key amino acid with (Met769) like the co-crystallized ligand with binding energy −29.48 kcal mol−1. Besides two hydrogen bond interactions with Lys721 amino acids and the lipophilic interactions by the benzothiazole scaffold (Fig. 6). In addition, compound 8b was docked inside the protein the EGFR active site formed one hydrogen bond as HBA with bond length/angle of 2.15 Å/134.9°, through its triazole moiety with the key amino acid with (Met769) like the co-crystallized ligand with binding energy −27.27 kcal mol−1. Besides the lipophilic interactions by the benzothiazole scaffold (Fig. 7). The depicted few polar interactions between the docked compounds and EGFR protein further highlighted the superior contribution of van der Waal potentials for compound-EGFR stability, making this type of interaction significantly impact the compound's both pharmacokinetic and pharmacodynamic characteristics. Consequently, molecular docking data revealed that compounds 8a and 8b had a high binding affinity as EGFR inhibitors. This might explain the proposed mode of action for anti-cancer efficiency and the proof of concept for the preferential experimental activity of 8a over 8b. In order to account for the compounds' physiochemical properties under experimental investigation, both the physiochemical and ADME parameters of the docked compounds, 8a and 8b, were evaluated in the following section.
 | ||
| Fig. 6 Molecular docking of compound 8a towards EGFR binding site. (A) Surface presentation of binding mode disposition of the co-crystallized ligand (yellow-colored) and compound 8a (cyan-colored) and (B) molecular interactions with the key amino acids. Molecular docking was done using AutoDock Vina, and 3D images were generated using UCSF-Chimera. | ||
 | ||
| Fig. 7 Molecular docking of compound 8b towards EGFR binding site. (A) Surface presentation of binding mode disposition of the co-crystallized ligand (yellow-colored) and compound 8b (cyan-colored) and (B) molecular interactions with the key amino acids. Molecular docking was done using AutoDock Vina, and 3D images were generated using UCSF-Chimera. | ||
| Parameter | 8a | 8b | |
|---|---|---|---|
| a “Mwt = molecular weight; MV = molecular volume; TPSA = total polar surface area; logP = partition coefficient at octanol–water solvent; HBA = hydrogen bond acceptor; HBD = hydrogen bond donor, drug likeness; Lipinski's rule of five (acceptable) = MW ≤ 500, logP ≤ 5, HBA ≤ 10 and HBD ≤ 5”; Pfizer rule (accepted) = logP > 3, TPSA < 75; PAINS = pan assay interference compounds, frequent hitters, alpha-screen artifacts and reactivity compounds; Caco-2 permeability = optimal if > −5.15 log unit; VD = volume of distribution (optimal 0.04–20 l kg−1); BBB penetration = blood–brain barrier penetration; CL = clearance; t1/2 = half-life time (time for the compound blood concentration to decrease to its half). |
|||
| Physiochemical | HBD | 2 | 2 |
| HBA | 10 | 10 | |
| MWt (D) | 511.14 | 525.16 | |
| MV (A3) | 497.78 | 515.08 | |
| PSA (A2) | 123.39 | 123.39 | |
| Solubility (logmol l−1) |
−6.16 | −6.61 | |
| LogP |
4.52 | 4.88 | |
| Medicinal chemistry | Lipinski rule | Accepted | Accepted |
| Pfizer rule | Accepted | Accepted | |
| PAINS | Zero alerts | Zero alerts | |
| Absorption | Caco-2 permeability | −5.15 (optimal) | −4.99 (optimal) |
| MDCK permeability | 1.2 × 10−5 cm s−1 (low-moderate) | 1.2 × 10−5 cm s−1 (low-moderate) | |
| Pgp-inhibitor | Non-inhibitor (99.8% probability) | Non-inhibitor (98.8% probability) | |
| Pgp-substrate | Non-inhibitor (99% probability) | Non-inhibitor (99.99% probability) | |
| Distribution | VD | 1.53 l kg−1 | 1.41 l kg−1 |
| BBB penetration | 13% probability | 5% probability | |
| Metabolism | CYP1A2 | Neither inhibitor/substrate | |
| CYP2C19 | Probable inhibitor | ||
| CYP2C9 | Probable inhibitor/substrate | ||
| CYP2D6 | Neither inhibitor/substrate | ||
| CYP3A4 | Neither inhibitor/substrate | ||
| Excretion | Cl | 4.07 ml min−1 kg−1 | 4.45 ml min−1 kg−1 |
| t1/2 | 10.1% propability of high t1/2 | 9.4% propability of high t1/2 | |
3. Materials and methods
3.1. Chemistry (general methods)
The solvents and reagents used in this study were of the highest quality of analytical reagent grade and were used without any purification. The fine chemicals and solvents were acquired from BDH Chemicals Ltd and Sigma-Aldrich. The melting temperatures were uncorrected and were determined with a Stuart Scientific SMP1. UV fluorescent Silica gel Merck 60 F254 plates were employed for thin layer chromatography (TLC) to monitor the reactions. A UV lamp (254 nm) was used to visualize the resulting spots. Fourier transform infrared spectroscopy (FT-IR) was conducted on the FT-IR spectrometer type PerkinElmer 1430 series. The 1H and 13C nuclear magnetic resonance spectroscopy were gathered on a Bruker spectrometer (400 MHz) using the TMS as an internal reference. A GmbH-Vario EL III Elemental Analyzer was employed to conduct elemental analyses. High-resolution mass spectrometry (HRMS) was measured (in positive ion mode) using electrospray ion trap technique by collision-induced dissociation on Bruker APEX-IV (7 tesla) instrument. The samples were dissolved and infused using a syringe pump with a flow rate of 120 μl min−1. External calibration was conducted using arginine cluster in a mass range m/z 175–871.3.1.1.1. Synthesis of 4-(prop-2-yn-1-yloxy) benzaldehyde (2). Alkyne 2 was prepared via propargylation of 4-hydroxybenzaldehyde (1) in accordance with our previously published work.71
3.1.1.2. General procedure for the synthesis of un/substituted benzothiazole-based azides 4a–c. Azides 4a–c were prepared from the appropriate un/substituted 2-aminobenzothiazole 3a–c in accordance with our previously published work.54
3.1.1.3. General Click procedure for the synthesis of 1,2,3-triazoles 5a–c. To a stirring solution of propargylated benzaldehyde 2 (1 mmol) and azidobenzothiazole 3 (1 mmol) dissolved in DMSO (10 ml), an aqueous solution (10 ml) of CuSO4 (0.10 g) and Na-ascorbate (0.15 g) was added. Stirring was continued for 48 h at 80 °C, until the consumption of the starting material as indicated by thin layer chromatography (TLC). After the completion of the reaction, the mixture was poured onto iced-water. The precipitate thus formed was collected by filtration, washed with saturated solution of ammonium chloride then water and recrystallized from ethanol/DMF to give the targeted 1,2,3-triazoles 5a–c.
3.1.1.4. General procedure for the synthesis of Schiff bases 7a–d. A stirring solution of compound 2 (5 mmol), the appropriate amine 6a–d (5 mmol) and few drops of acetic acid in ethanol (40 ml) was heated under reflux for 4–6 h. After cooling, the resulting precipitate was collected by filtration and recrystallized from ethanol to give the desired Schiff bases 7a–d.
3.1.1.5. General Click procedure for the synthesis of 1,2,3-triazole-benzothiazole molecular conjugates 8a–l. To a stirring solution of propargylated Schiff bases 7a–d (1 mmol) and the appropriate un/substituted benzothiazole-based azides 3a–c (1 mmol) dissolved in DMSO (10 ml), an aqueous solution (10 ml) of CuSO4 (0.10 g) and Na-ascorbate (0.15 g) was added. Stirring was continued for 6–9 h at 80 °C, until the consumption of the starting material as indicated by thin layer chromatography (TLC). After the completion of the reaction, the mixture was poured onto iced-water. The precipitate thus formed was collected by filtration, washed with saturated solution of ammonium chloride then water and recrystallized from ethanol/DMF to give the targeted 1,2,3-triazoles 8a–l.
3.1.2.1. Characterization of 1,2,3-triazoles 5a–c.
3.1.2.1.1 Characterization of N-(benzo[d]thiazol-2-yl)-2-(4-((4-formylphenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetamide (5a). IR (ν, cm−1): 3240 (N–H), 3090 (C–H ar), 1720 (C
O), 1640 (CN), 1560 (CC). 1H NMR (400 MHz, DMSO-d6): δH = 12.81 (1H, s, NH), 9.86 (1H, s, CHO), 8.36 (1H, s, 1,2,3-triazole H-5), 7.99 (2H, d, J = 8 Hz, Ar–H), 7.90 (2H, d, J = 8 Hz, Ar–H), 7.78 (1H, d, J = 4 Hz, Ar–H), 7.46 (1H, d, J = 4 Hz, Ar–H), 7.33–7.28 (2H, m, Ar–H), 5.58 (2H, s, NCH2CO), 5.33 (2H, s, OCH2). 13C NMR (100 MHz, DMSO-d6): δC = 192.5 (HCO); 162.9, 161.5, 152.6, 142.4, 139.5, 136.7, 132.4, 132.2, 131.7, 130.8, 125.9, 122.5, 121.9, 115.8 (Ar–C, 1,2,3-triazole C-5, CN); 62.3 (OCH2); 52.8 (NCH2) ppm. Calculated for C19H15N5O3S: C, 58.01; H, 3.84; N, 17.80. Found: C, 58.19; H, 3.78; N, 17.70.
3.1.2.1.2 Characterization of 2-(4-((4-formylphenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(6-(methylsulfonyl)benzo[d]thiazol-2-yl)acetamide (5b). IR (ν, cm−1): 3290 (N–H), 3060 (C–H ar), 1715 (C
O), 1630 (CN), 1550 (CC). 1H NMR (400 MHz, DMSO-d6): δH = 12.86 (1H, s, NH), 9.88 (s, 1H, CHO), 8.36 (s, 1H, 1,2,3-triazole H-5), 7.90 (d, 2H, J = 8 Hz, Ar–H), 7.77 (s, 1H, Ar–H), 7.65 (d, 1H, Ar–H), 7.27 (d, 3H, J = 4 Hz, Ar–H), 5.56 (s, 2H, NCH2CO), 5.32 (s, 2H, OCH2), 2.40 (s, 3H, CH3). 13C NMR (100 MHz, DMSO-d6): δC = 191.9 (HCO); 163.4, 142.5, 133.8, 132.2, 130.3, 128.1, 127.2, 121.8, 120.8, 115.6 (Ar–C, 1,2,3-triazole C-5, CN); 61.7 (OCH2); 52.1 (NCH2); 21.4 (CH3) ppm. Calculated for C20H17N5O3S: C, 58.96; H, 4.21; N, 17.19. Found: C, 58.74; H, 4.29; N, 17.33.
3.1.2.1.3 Characterization of 2-(4-((4-formylphenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(6-(methylsulfonyl)benzo[d]thiazol-2-yl)acetamide (5c). IR (ν, cm−1): 3220 (N–H), 3050 (C–H ar), 1725 (C
O), 1630 (CN), 1570 (CC). 1H NMR (400 MHz, DMSO-d6): δH = 12.80 (1H, br, NH), 9.88 (s, 1H, CHO), 8.66 (s, 1H, Ar–H), 8.37 (s, 1H, 1,2,3-triazole H-5), 7.97 (s, 2H, Ar–H), 7.90 (d, 2H, J = 8 Hz, Ar–H), 7.28 (d, 2H, J = 8 Hz, Ar–H), 5.62 (s, 2H, NCH2CO), 5.33 (s, 2H, OCH2), 3.25 (s, 3H, CH3). 13C NMR (100 MHz, DMSO-d6): δC = 191.9 (HCO); 166.9, 163.4, 162.1, 152.3, 142.5, 136.0, 132.4, 132.3, 132.2, 130.3, 125.5, 122.8, 121.4, 115.6 (Ar–C, 1,2,3-triazole C-5, CN); 61.7 (OCH2); 52.2 (NCH2); 44.3 (CH3) ppm. Calculated for C20H17N5O5S2: C, 50.95; H, 3.63; N, 14.85. Found: C, 50.77; H, 3.71; N, 14.73.
3.1.2.2. Characterization of Schiff bases 7a–d.
3.1.2.2.1 Characterization of (Z)-N′-(4-(prop-2-yn-1-yloxy)benzylidene)benzohydrazide (7a). IR (ν, cm−1): 3200–3310 (N–H,
CH), 3035 (C–H ar), 2150 (CC), 1700 (CO), 1610 (CN), 1540 (CC). 1H NMR (400 MHz, DMSO-d6): δH = 11.75 (0.90H, s, NH), 11.75 (0.90H, s, NH), 8.41 (0.90H, s, HCN), 8.06 (0.10H, bs, HCN), 7.92 (1.80H, d, J = 8 Hz, Ar–H), 7.81 (0.20H, d, J = 8 Hz, Ar–H), 7.70 (1.80H, d, J = 8 Hz, Ar–H), 7.58–7.50 (3.20H, m, Ar–H), 7.08 (2H, d, J = 8 Hz, Ar–H), 4.86 (2H, s, OCH2), 3.61 (1H, s, CH). 13C NMR (100 MHz, DMSO-d6): δC = 163.4, 159.2, 148.0, 134.0, 132.1, 129.0, 128.9, 128.0, 115.6 (Ar–C, CN, CO); 79.4, 78.9 (CCH); 56.0 (OCH2) ppm. Calculated for C17H14N2O2: C, 73.37; H, 5.07; N, 10.07. Found: C, 73.56; H, 5.13; N, 10.21.
3.1.2.2.2 Characterization of (Z)-N′-(4-(prop-2-yn-1-yloxy)benzylidene)isonicotino-hydrazide (7b). IR (ν, cm−1): 3230–3340 (N–H,
CH), 3055 (C–H ar), 2160 (CC), 1690 (CO), 1620 (CN), 1560 (CC). 1H NMR (400 MHz, DMSO-d6): δH = 11.95 (0.87H, s, NH), 11.91 (0.13H, s, NH), 8.78 (1.74H, d, J = 4 Hz, Ar–H), 8.73 (0.26H, s, Ar–H), 8.41 (0.87H, s, HCN), 8.04 (0.13H, s, HCN), 7.81 (1.74H, d, J = 4 Hz, Ar–H), 7.72 (1.74H, d, J = 8 Hz, Ar–H), 7.66 (0.26H, d, J = 4 Hz, Ar–H), 7.48 (0.25H, d, J = 8 Hz, Ar–H), 7.09 (1.76H, d, J = 8 Hz, Ar–H), 7.01 (0.25H, d, J = 8 Hz, Ar–H), 4.87 (1.75H, s, OCH2), 4.82 (0.25H, s, OCH2), 3.61 and 3.58 (1H, 2 s, CH). 13C NMR (100 MHz, DMSO-d6): δC = 161.9, 159.4, 150.7, 149.2, 141.0, 129.3, 127.7, 121.9, 115.7 (Ar–C, CN, CO); 79.4, 79.0 (CCH); 56.0 (OCH2) ppm. Calculated for C16H13N3O2: C, 68.81; H, 4.69; N, 15.05. Found: C, 68.65; H, 4.61; N, 15.17.
3.1.2.2.3 Characterization of (Z)-1-(2,4-dinitrophenyl)-2-(4-(prop-2-yn-1-yloxy)benzylidene)hydrazine (7c). IR (ν, cm−1): 3240–3320 (N–H,
CH), 3075 (C–H ar), 2140 (CC), 1625 (CN), 1550 (CC). 1H NMR (400 MHz, DMSO-d6): δH = 11.59 (1H, s, NH), 8.85 (1H, d, J = 4 Hz, Ar–H), 8.63 (1H, s, HCN), 8.33 (1H, d, J = 8 Hz, Ar–H), 8.06 (1H, d, J = 8 Hz, Ar–H), 7.76 (2H, d, J = 8 Hz, Ar–H), 7.10 (2H, d, J = 8 Hz, Ar–H), 4.87 (2H, s, OCH2), 3.61 (1H, s, CH). 13C NMR (100 MHz, DMSO-d6): δC = 159.6, 149.7, 144.9, 137.2, 129.6, 127.5, 123.5, 117.1, 115.8 (Ar–C, CN); 79.3, 79.0 (CCH); 56.0 (OCH2) ppm. Calculated for C16H12N4O5: C, 56.47; H, 3.55; N, 16.46. Found: C, 56.59; H, 3.59; N, 16.53.
3.1.2.2.4 Characterization of (Z)-2-(4-(prop-2-yn-1-yloxy)benzylidene)hydrazine-1-carbothioamide (7d). IR (ν, cm−1): 3260–3350 (N–H,
CH), 3065 (C–H ar), 2150 (CC), 1620 (CN), 1570 (CC). 1H NMR (400 MHz, DMSO-d6): δH = 11.33 (1H, s, NH), 8.11 (1H, s, HCN), 7.99 (1H, s, NH), 7.92 (1H, s, NH), 7.75 (2H, d, J = 8 Hz, Ar–H), 7.01 (2H, d, J = 8 Hz, Ar–H), 4.83 (2H, s, OCH2), 3.56 (1H, s, CH). 13C NMR (100 MHz, DMSO-d6): δC = 178.1 (CS); 159.0, 142.5, 129.2, 127.9, 115.4 (Ar–C, CN); 79.4, 78.9 (CCH); 56.0 (OCH2) ppm. Calculated for C11H11N3OS: C, 56.63; H, 4.75; N, 18.01. Found: C, 56.45; H, 4.83; N, 18.14.
3.1.2.3. Characterization of 1,2,3-triazole-benzothiazole molecular conjugates 8a–l.
3.1.2.3.1 Characterization of (E)-N-(benzo[d]thiazol-2-yl)-2-(4-((4-((2-benzoylhydrazono)-methyl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetamide (8a). IR (ν, cm−1): 3290 (N–H), 3050 (C–H ar), 1700 (C
O), 1615 (CN), 1565 (CC). 1H NMR (400 MHz, DMSO-d6): δH = 11.76 (0.80H, s, NH), 9.88 (0.20H, s, NH), 8.42 (1H, s, HCN), 8.35 (1H, s, 1,2,3-triazole H-5), 7.91–7.69 (4H, m, Ar–H), 7.57–7.52 (4H, m, Ar–H), 7.28–7.16 (6H, m, Ar–H), 5.58 (2H, s, NCH2CO), 5.26 (2H, s, OCH2). 13C NMR (100 MHz, DMSO-d6): δC = 168.1, 166.3, 163.4, 160.0 (2 × CO); 148.7, 148.3, 143.0, 129.1, 128.1, 126.7, 123.3, 121.2, 115.6 (Ar–C, 1,2,3-triazole C-5, CN); 61.6 (OCH2); 53.0, 52.2 (NCH2) ppm. Calculated for C26H21N7O3S: C, 61.05; H, 4.14; N, 19.17. Found: C, 61.26; H, 4.19; N, 19.05. HRMS (ESI): m/z [M + Na]+ (calcd for C26H21N7O3SNa: 534.13188; found: 534.13082).
3.1.2.3.2 Characterization of (E)-2-(4-((4-((2-benzoylhydrazono)methyl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(6-methylbenzo[d]thiazol-2-yl)acetamide (8b). IR (ν, cm−1): 3310 (N–H), 3075 (C–H ar), 1710 (C
O), 1620 (CN), 1570 (CC). 1H NMR (400 MHz, DMSO-d6): δH = 11.75 (0.80H, s, NH), 11.61 (0.20H, s, NH), 8.43 (1H, s, HCN), 8.33 (1H, s, 1,2,3-triazole H-5), 7.91 (2H, d, J = 4 Hz, Ar–H), 7.78–7.52 (7H, m, Ar–H), 7.26–7.15 (3H, m, Ar–H), 5.55 (2H, s, NCH2CO), 5.32 (0.20H, s, OCH2), 5.25 (1.80H, s, OCH2), 2.40 (3H, s, CH3). 13C NMR (100 MHz, DMSO-d6): δC = 166.4, 159.8 (2 × CO); 147.8, 145.0, 142.4, 133.6, 131.9, 129.1, 128.9, 115.7 (Ar–C, 1,2,3-triazole C-5, CN); 61.4 (OCH2); 53.3, 52.3 (NCH2); 29.3 (CH3) ppm. Calculated for C27H23N7O3S: C, 61.70; H, 4.41; N, 18.66. Found: C, 61.86; H, 4.47; N, 18.78. HRMS (ESI): m/z [M + 1]+ (calcd for C27H23N7O3S: 526.16558; found: 526.16751).
3.1.2.3.3 Characterization of (E)-2-(4-((4-((2-benzoylhydrazono)methyl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(6-(methylsulfonyl)benzo[d]thiazol-2-yl)acetamide (8c). IR (ν, cm−1): 3330 (N–H), 3040 (C–H ar), 1695 (C
O), 1625 (CN), 1555 (CC). 1H NMR (400 MHz, DMSO-d6): δH = 11.75 (0.80H, s, NH), 9.88 (0.20H, s, NH), 8.66 (1H, s, Ar–H), 8.42 (1H, bs, HCN), 8.35 (1H, bs, 1,2,3-triazole H-5), 7.95–7.89 (3H, m, Ar–H), 7.69 (2H, bs, Ar–H), 7.58–7.52 (3H, m, Ar–H), 7.27–7.16 (3H, m, Ar–H), 5.60 (2H, s, NCH2CO), 5.33 (0.40H, s, OCH2), 5.26 (1.60H, s, OCH2), 3.23 (3H, s, CH3). 13C NMR (100 MHz, DMSO-d6): δC = 160.8, 160.3 (2 × CO); 152.2, 149.2, 142.7, 136.2, 132.2, 129.3, 127.1, 122.8, 121.8, 115.6 (Ar–C, 1,2,3-triazole C-5, CN); 61.6 (OCH2); 53.2 (NCH2); 44.8 (CH3) ppm. Calculated for C27H23N7O5S2: C, 55.00; H, 3.93; N, 16.63. Found: C, 55.26; H, 3.86; N, 16.53. HRMS (ESI): m/z [M + Na]+ (calcd for C27H23N7O5S2Na: 612.10943; found: 612.10743).
3.1.2.3.4 Characterization of (E)-N-(benzo[d]thiazol-2-yl)-2-(4-((4-((2-isonicotinoylhydrazono)methyl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetamide (8d). IR (ν, cm−1): 3300 (N–H), 3035 (C–H ar), 1695 (C
O), 1610 (CN), 1585 (CC). 1H NMR (400 MHz, DMSO-d6): δH = 12.91 (1H, s, CH2CONH), 11.97 (0.90H, s, NH), 11.93 (0.10H, s, NH), 9.88 (0.10H, s, NH), 8.43 (1H, s, HCN), 8.35 (1H, m, 1,2,3-triazole H-5), 7.99–7.88 (3H, m, Ar–H), 7.79–7.72 (4H, m, Ar–H), 7.48–7.33 (2H, m, Ar–H), 7.19–7.10 (3H, m, Ar–H), 5.62 (2H, s, NCH2CO), 5.34 (0.25H, s, OCH2), 5.27 (1.75H, s, OCH2), 3.24 (3H, s, CH3). 13C NMR (100 MHz, DMSO-d6): δC = 162.0, 160.3 (2 × CO); 150.5, 149.3, 142.8, 129.4, 128.9, 127.3, 127.0, 126.7, 124.2, 122.3, 121.0, 115.6 (Ar–C, 1,2,3-triazole C-5, CN); 61.6 (OCH2); 52.3 (NCH2) ppm. Calculated for C25H20N8O3S: C, 58.58; H, 3.93; N, 21.86. Found: C, 58.75; H, 3.99; N, 21.96.
3.1.2.3.5 Characterization of (E)-2-(4-((4-((2-isonicotinoylhydrazono)methyl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(6-methylbenzo[d]thiazol-2-yl)acetamide (8e). IR (ν, cm−1): 3325 (N–H), 3020 (C–H ar), 1700 (C
O), 1610 (CN), 1555 (CC). 1H NMR (400 MHz, DMSO-d6): δH = 12.86 (1H, s, CH2CONH), 12.03 (0.80H, s, NH), 11.95 (0.10H, s, NH), 9.90 (0.10H, s, NH), 8.48 (1H, bs, HCN), 8.35 (1H, m, 1,2,3-triazole H-5), 7.89–7.66 (5H, m, Ar–H), 7.49–7.10 (6H, m, Ar–H), 5.57 (2H, s, NCH2CO), 5.33 (0.25H, s, OCH2), 5.27 (1.75H, s, OCH2), 2.41 (3H, s, CH3). 13C NMR (100 MHz, DMSO-d6): δC = 166.7, 163.0, 159.9 (2 × CO); 147.6, 145.6, 142.6, 133.9, 131.8, 128.6, 125.6, 121.1, 115.7 (Ar–C, 1,2,3-triazole C-5, CN); 61.4 (OCH2); 53.2, 52.7 (NCH2); 29.5 (CH3) ppm. Calculated for C26H22N8O3S: C, 59.31; H, 4.21; N, 21.28. Found: C, 59.55; H, 4.29; N, 21.17.
3.1.2.3.6 Characterization of (E)-2-(4-((4-((2-isonicotinoylhydrazono)methyl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(6-(methylsulfonyl)benzo[d]thiazol-2-yl)acetamide (8f). IR (ν, cm−1): 3350 (N–H), 3055 (C–H ar), 1690 (C
O), 1620 (CN), 1570 (CC). 1H NMR (400 MHz, DMSO-d6): δH = 13.22 (1H, s, CH2CONH), 11.98 (0.80H, s, NH), 11.91 (0.10H, s, NH), 9.88 (0.10H, s, NH), 8.67 (1H, s, Ar–H), 8.44 (0.70H, bs, HCN), 8.32–8.36 (1.30H, m, HCN and 1,2,3-triazole H-5), 7.99–7.88 (5H, m, Ar–H), 7.72 (2H, d, J = 4 Hz, Ar–H), 7.49 (0.30H, d, J = 8 Hz, Ar–H), 7.28–7.16 (2.70H, m, Ar–H), 5.62 (2H, s, NCH2CO), 5.34 (0.25H, s, OCH2), 5.27 (1.75H, s, OCH2), 3.24 (3H, s, CH3). 13C NMR (100 MHz, DMSO-d6): δC = 166.9, 162.0, 160.3 (2 × CO); 152.3, 149.3, 142.9, 136.2, 132.2, 127.3, 125.4, 121.5, 115.6 (Ar–C, 1,2,3-triazole C-5, CN); 61.6 (OCH2); 52.2 (NCH2); 44.8 (CH3) ppm. Calculated for C26H22N8O5S2: C, 52.87; H, 3.75; N, 18.97. Found: C, 52.71; H, 3.80; N, 18.86.
3.1.2.3.7 Characterization of (Z)-N-(benzo[d]thiazol-2-yl)-2-(4-((4-((2-(2,4-dinitrophenyl)hydrazono)methyl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetamide (8g). IR (ν, cm−1): 3320 (N–H), 3055 (C–H ar), 1690 (C
O), 1620 (CN), 1580 (CC). 1H NMR (400 MHz, DMSO-d6): δH = 12.69 (1H, bs, CH2CONH), 11.62 (1H, s, NH), 8.83 (1H, s, Ar–H), 8.63 (1H, s, HCN), 8.31 (2H, s, 1,2,3-triazole H-5 and Ar–H), 8.05 (2H, d, J = 4 Hz, Ar–H), 7.84–7.75 (3H, m, Ar–H), 7.79–7.72 (4H, m, Ar–H), 7.40 (1H, s, Ar–H), 7.19–7.15 (3H, m, Ar–H), 5.51 (2H, bs, NCH2CO), 5.27 (2H, s, OCH2). 13C NMR (100 MHz, DMSO-d6): δC = 165.4 (CO); 149.8, 146.1, 145.2, 142.6, 136.9, 135.0, 130.0, 129.5, 127.1, 126.0, 123.5, 117.1, 115.7 (Ar–C, 1,2,3-triazole C-5, CN); 61.7 (OCH2); 52.9 (NCH2) ppm. Calculated for C25H19N9O6S: C, 52.35; H, 3.34; N, 21.98. Found: C, 52.52; H, 3.39; N, 21.89.
3.1.2.3.8 Characterization of (Z)-2-(4-((4-((2-(2,4-dinitrophenyl)hydrazono)methyl)phenoxy)-methyl)-1H-1,2,3-triazol-1-yl)-N-(6-methylbenzo[d]thiazol-2-yl) acetamide (8h). IR (ν, cm−1): 3340 (N–H), 3055 (C–H ar), 1700 (C
O), 1615 (CN), 1565 (CC). 1H NMR (400 MHz, DMSO-d6): δH = 12.82 (1H, s, CH2CONH), 11.60 (1H, s, NH), 8.85 (1H, s, Ar–H), 8.64 (1H, s, HCN), 8.34 (2H, s, 1,2,3-triazole H-5 and Ar–H), 8.08 (1H, bd, J = 4 Hz, Ar–H), 7.76–7.66 (3H, m, Ar–H), 7.26–7.10 (4H, m, Ar–H), 5.56 (2H, s, NCH2CO), 5.28 (2H, s, OCH2), 2.40 (3H, s, CH3). 13C NMR (100 MHz, DMSO-d6): δC = 160.5 (CO); 149.8, 144.9, 137.2, 133.8, 130.1, 129.5, 128.0, 126.9, 123.5, 121.8, 117.1, 115.7 (Ar–C, 1,2,3-triazole C-5, CN); 61.6 (OCH2); 52.1 (NCH2); 21.4 (CH3) ppm. Calculated for C26H21N9O6S: C, 53.15; H, 3.60; N, 21.45. Found: C, 53.39; H, 3.52; N, 21.57.
3.1.2.3.9 Characterization of (Z)-2-(4-((4-((2-(2,4-dinitrophenyl)hydrazono)methyl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(6-(methylsulfonyl)benzo[d]thiazol-2-yl)acetamide (8i). IR (ν, cm−1): 3315 (N–H), 3080 (C–H ar), 1705 (C
O), 1610 (CN), 1560 (CC). 1H NMR (400 MHz, DMSO-d6): δH = 12.76 (1H, bs, CH2CONH), 11.55 (1H, s, NH), 8.85 (1H, s, Ar–H), 8.64 (2H, s, HCN and Ar–H), 8.35 (2H, s, 1,2,3-triazole H-5 and Ar–H), 8.08–8.76 (4H, m, Ar–H), 7.19–7.10 (3H, m, Ar–H), 5.61 (2H, s, NCH2CO), 5.29 (2H, s, OCH2), 3.62 (3H, s, CH3). 13C NMR (100 MHz, DMSO-d6): δC = 162.7 (CO); 159.6, 149.7, 144.9, 142.8, 137.2, 136.0, 130.2, 129.6, 129.5, 127.5, 123.5, 117.2, 115.8 (Ar–C, 1,2,3-triazole C-5, CN); 61.6 (OCH2); 52.1 (NCH2); 21.4 (CH3) ppm. Calculated for C26H21N9O8S2: C, 47.92; H, 3.25; N, 19.35. Found: C, 47.74; H, 3.17; N, 19.49.
3.1.2.3.10 Characterization of (Z)-N-(benzo[d]thiazol-2-yl)-2-(4-((4-((2-carbamothioylhydrazono)methyl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)acetamide (8j). IR (ν, cm−1): 3280 (N–H), 3050 (C–H ar), 1700 (C
O), 1625 (CN), 1575 (CC). 1H NMR (400 MHz, DMSO-d6): δH = 13.00 (1H, bs, CH2CONH), 11.48 (1H, s, NH), 8.34 (1H, s, 1,2,3-triazole H-5), 8.25 (1H, s, HCN), 8.04 (1H, s, NH), 7.98 (1H, d, J = 8 Hz, NH), 7.76 (3H, t, J = 4 Hz, Ar–H), 7.32–7.25 (3H, m, Ar–H), 7.10 (2H, d, J = 8 Hz, Ar–H), 5.61 (2H, s, NCH2CO), 5.32 (0.20H, s, OCH2), 5.23 (1.80H, s, OCH2). 13C NMR (100 MHz, DMSO-d6): δC = 166.2 (CS); 160.0 (CO); 143.4, 142.8, 131.8, 129.5, 127.3, 126.9, 126.7, 124.3, 122.3, 121.1, 115.3 (Ar–C, 1,2,3-triazole C-5, CN); 61.5 (OCH2); 52.2 (NCH2) ppm. Calculated for C20H18N8O2S2: C, 51.49; H, 3.89; N, 24.02. Found: C, 51.70; H, 3.94; N, 24.17.
3.1.2.3.11 Characterization of (Z)-2-(4-((4-((2-carbamothioylhydrazono)methyl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(6-methylbenzo[d]thiazol-2-yl)acetamide (8k). IR (ν, cm−1): 3260 (N–H), 3040 (C–H ar), 1700 (C
O), 1605 (CN), 1555 (CC). 1H NMR (400 MHz, DMSO-d6): δH = 12.78 (1H, bs, CH2CONH), 11.73 (0.70H, s, NH), 9.87 (0.30H, s, NH), 8.42–8.28 (3H, m, 1,2,3-triazole H-5, HCN and Ar–H), 7.89 (1H, bd, J = 8 Hz, Ar–H), 7.80–7.65 (4H, m, NH and Ar–H), 7.26 (1H, bd, J = 4 Hz, Ar–H), 7.11–7.08 (2H, m, J = 4 Hz, Ar–H), 5.55 (2H, s, NCH2CO), 5.32 (0.60H, s, OCH2), 5.25 (1.40H, s, OCH2), 3.38 (3H, s, CH3). 13C NMR (100 MHz, DMSO-d6): δC = 166.1 (CS); 163.4, 160.3 (CO); 146.7, 133.8, 132.3, 130.3, 129.8, 128.1, 126.9, 121.8, 120.8, 115.6, 115.4 (Ar–C, 1,2,3-triazole C-5, CN); 61.8, 61.6 (OCH2); 52.1 (NCH2), 21.4 (CH3) ppm. Calculated for C21H20N8O2S2: C, 52.49; H, 4.20; N, 23.32. Found: C, 52.76; H, 4.27; N, 23.20.
3.1.2.3.12 Characterization of (Z)-2-(4-((4-((2-carbamothioylhydrazono)methyl)phenoxy)methyl)-1H-1,2,3-triazol-1-yl)-N-(6-(methylsulfonyl)benzo[d]thiazol-2-yl)acetamide (8l). IR (ν, cm−1): 3290 (N–H), 3075 (C–H ar), 1710 (C
O), 1615 (CN), 1570 (CC). 1H NMR (400 MHz, DMSO-d6): δH = 13.31 (1H, bs, CH2CONH), 11.51 (1H, s, NH), 9.89 (1H, s, NH), 8.62 (1H, s, Ar–H), 8.35–8.25 (2H, m, 1,2,3-triazole H-5 and HCN), 8.25 (1H, s, HCN), 8.03 (1H, s, NH), 7.93–7.88 (3H, m, Ar–H), 7.77 (2H, bd, J = 4 Hz, Ar–H), 7.27 (1H, bd, J = 4 Hz, Ar–H), 7.11 (1H, bd, J = 4 Hz, Ar–H), 5.59 (2H, s, NCH2CO), 5.33 (0.60H, s, OCH2), 5.24 (1.40H, s, OCH2), 3.38 (3H, s, CH3). 13C NMR (100 MHz, DMSO-d6): δC = 167.5 (CS); 163.4, 160.0 (CO); 152.6, 143.4, 142.8, 135.7, 132.2, 130.3, 129.5, 127.3, 127.1, 126.9, 125.2, 122.5, 121.2, 115.6, 115.3 (Ar–C, 1,2,3-triazole C-5, CN); 61.8, 61.6 (OCH2); 52.6 (NCH2), 44.5 (CH3) ppm. Calculated for C21H20N8O4S3: C, 46.31; H, 3.70; N, 20.57. Found: C, 46.50; H, 3.65; N, 20.68.
3.2. Biological assay
000 per insert in 70 μl media, A545 cells were incubated for 24 hours in inserts (Ibidi, Germany). Afterward, cells were treated for two hours with 10 μg ml−1 of mitomycin C to inhibit cell proliferation. The media was removed, and cells underwent washing three times before being treated with IC50 and subIC50 concentrations of the investigated compounds. Images were taken at 0 and 24 hours utilizing the EVOS XL Core imaging equipment at 4× magnification. Wound width was assessed with MIPAR software, as previously described (MIPAR Software LLC, OH, USA).723.3. In silico studies
Maestro was used to build and optimize structures of compounds and proteins. Then, the grid-box dimensions around the co-crystallized ligands were used to identify binding locations inside the proteins. Compounds were docked against the protein structures of EGFR (PDB = 1M17) using AutoDock Vina software following routine work.73 Maestro was utilized to optimize protein and ligand structures. Binding activities assessed the molecular docking outcomes according to binding energy and ligand–receptor interactions. Additionally, physicochemical and pharmacokinetic properties were investigated using ADMETlab 2.0 web-based platform (https://admetmesh.scbdd.com/service/evaluation/index; Xiangya School of Pharmaceutical Sciences, Central South University, China).4. Conclusion
An efficient Cu(I)-catalyzed alkyne azide cycloaddition procedure was adopted as a fascinating approach to combine the bioactive benzothiazole and 1,2,3-triazole moieties in a single framework in order to investigate their anti-cancer features. Thus, a new library of 1,2,3-triazole-benzothiazole molecular conjugates 8a–l tethering hydrazone/thiosemicarbaze linkage was successfully designed and generated via the 1,3-dipolar cycloaddition of some un/substituted benzothiazole azides 4a–c with the appropriate O-propargylated benzylidene derivatives 7a–d. Several spectroscopic experiments were well used to characterize the newly generated molecular hybrids. The biological investigation against three human cancer cell lines was carried out and revealed that compounds 8a–c exhibited potent anti-cancer activities against breast cancer cell line T47D while demonstrating reasonable safety on normal cells. In addition, the compounds 8a and 8b significantly inhibited the 2-dimensional cellular migration of the lung cancer cells in a concentration-dependent manner indicating their ability to be further developed into anti-proliferative agents. https://www.google.com/search?sca_esv=9a5876f960b44ea9%26rlz=1C1GCEJ_enIN1138IN1138%26q=Interestingly%26spell=1%26sa=X%26ved=2ahUKEwiQgb3sp5-LAxXaTGwGHZyGM3UQkeECKAB6BAgOEAE, compounds 8a, 8b, and 8c exhibited IC50 values of 0.69, 1.16, and 4.82 μM, respectively. Finally, molecular docking results exhibited a good binding affinity of compounds 8a and 8b towards the EGFR binding site. Accordingly, these compounds can be further developed as target-oriented EGFR chemotherapeutic against cancer.Data availability
The data that support the findings of this study are available in the ESI† of this article.Author contributions
This work was carried out with equal contribution among all authors. All authors contributed to the manuscript writing in their respective parts, read and approved the final manuscript.Conflicts of interest
The authors declare no conflict of interest.References
- J. Zugazagoitia, C. Guedes, S. Ponce, I. Ferrer, S. Molina-Pinelo and L. Paz-Ares, Current Challenges in Cancer Treatment, Clin. Ther., 2016, 38(7), 1551–1566, DOI:10.1016/j.clinthera.2016.03.026.
- G. Bérubé, An overview of molecular hybrids in drug discovery, Expert Opin. Drug Discovery, 2016, 11(3), 281–305, DOI:10.1517/17460441.2016.1135125.
- P. de Sena Murteira Pinheiro, L. S. Franco, T. L. Montagnoli and C. A. M. Fraga, Molecular hybridization: a powerful tool for multitarget drug discovery, Expert Opin. Drug Discovery, 2024, 19(4), 451–470, DOI:10.1080/17460441.2024.2322990.
- Shalini and V. Kumar, Have molecular hybrids delivered effective anti-cancer treatments and what should future drug discovery focus on?, Expert Opin. Drug Discovery, 2021, 16(4), 335–363, DOI:10.1080/17460441.2021.1850686.
- A. K. Singh, A. Kumar, H. Singh, P. Sonawane, H. Paliwal, S. Thareja, P. Pathak, M. Grishina, M. Jaremko and A. H. Emwas, et al., Concept of Hybrid Drugs and Recent Advancements in Anti-cancer Hybrids, Pharmaceuticals, 2022, 15(9), 1071, DOI:10.3390/ph15091071.
- N. Kerru, L. Gummidi, S. Maddila, K. K. Gangu and S. B. Jonnalagadda, A review on recent advances in nitrogen-containing molecules and their biological applications, Molecules, 2020, 25(8), 1909 CrossRef CAS PubMed.
- P. Venkatesh and S. Pandeya, Synthesis, characterisation and anti-inflammatory activity of some 2-amino benzothiazole derivatives, Int. J. ChemTech Res., 2009, 1(4), 1354–1358 Search PubMed.
- Y. Liu, Y. Wang, G. Dong, Y. Zhang, S. Wu, Z. Miao, J. Yao, W. Zhang and C. Sheng, Novel benzothiazole derivatives with a broad antifungal spectrum: Design, synthesis and structure–activity relationships, MedChemComm, 2013, 4(12), 1551–1561 Search PubMed.
- E. Meltzer-Mats, G. Babai-Shani, L. Pasternak, N. Uritsky, T. Getter, O. Viskind, J. r. Eckel, E. Cerasi, H. Senderowitz and S. Sasson, Synthesis and mechanism of hypoglycemic activity of benzothiazole derivatives, J. Med. Chem., 2013, 56(13), 5335–5350 Search PubMed.
- D. Bele and I. Singhvi, Synthesis and analgesic activity of some Mannich bases of 6-substituted-2-aminobenzothiazole, Res. J. Pharm. Technol., 2008, 1(1), 22–24 CAS.
- V. S. Padalkar, V. D. Gupta, K. R. Phatangare, V. S. Patil, P. G. Umape and N. Sekar, Synthesis of novel dipodal-benzimidazole, benzoxazole and benzothiazole from cyanuric chloride: Structural, photophysical and anti-microbial studies, J. Saudi Chem. Soc., 2014, 18(3), 262–268 CrossRef CAS.
- J. Cai, M. Sun, X. Wu, J. Chen, P. Wang, X. Zong and M. Ji, Design and synthesis of novel 4-benzothiazole amino quinazolines Dasatinib derivatives as potential anti-tumor agents, Eur. J. Med. Chem., 2013, 63, 702–712 CrossRef CAS PubMed.
- F. Delmas, A. Avellaneda, C. Di Giorgio, M. Robin, E. De Clercq, P. Timon-David and J.-P. Galy, Synthesis and antileishmanial activity of (1, 3-benzothiazol-2-yl) amino-9-(10H)-acridinone derivatives, Eur. J. Med. Chem., 2004, 39(8), 685–690 CrossRef CAS PubMed.
- D. Munirajasekar, M. Himaja and M. Sunil, Synthesis and anthemintic activity of 2-amino-6-subtituted benzothiazoles, Int. Res. J. Pharm., 2011, 2(1), 114–117 Search PubMed.
- P. S. Yadav, D. Devprakash and G. P. Senthilkumar, Benzothiazole: different methods of synthesis and diverse biological activities, Int. J. Pharm. Sci. Drug Res., 2011, 3(1), 1–7 CrossRef CAS PubMed.
- N. Siddiqui, A. Rana, S. A. Khan, M. A. Bhat and S. E. Haque, Synthesis of benzothiazole semicarbazones as novel anticonvulsants—The role of hydrophobic domain, Bioorg. Med. Chem. Lett., 2007, 17(15), 4178–4182 CrossRef CAS PubMed.
- A. Abd El-Meguid, A. M. Naglah, G. O. Moustafa, H. M. Awad and A. M. El Kerdawy, Novel benzothiazole-based dual VEGFR-2/EGFR inhibitors targeting breast and liver cancers: Synthesis, cytotoxic activity, QSAR and molecular docking studies, Bioorg. Med. Chem. Lett., 2022, 58, 128529, DOI:10.1016/j.bmcl.2022.128529.
- M. Al-Sanea, A. Hamdi, A. A. B. Mohamed, H. W. El-Shafey, M. Moustafa, A. A. Elgazar, W. M. Eldehna, H. Ur Rahman, D. G. T. Parambi, R. M. Elbargisy, S. Selim, S. N. A. Bukhari, O. Magdy Hendawy and S. S. Tawfik, New benzothiazole hybrids as potential VEGFR-2 inhibitors: design, synthesis, anti-cancer evaluation, and in silico study, J. Enzyme Inhib. Med. Chem., 2023, 38, 2166036, DOI:10.1080/14756366.2023.2166036.
- A. El-Meguid, G. O. Moustafa, H. M. Awad, E. R. Zaki and E. S. Nossier, Novel benzothiazole hybrids targeting EGFR: Design, synthesis, biological evaluation and molecular docking studies, J. Mol. Struct., 2021, 1240, 130595, DOI:10.1016/j.molstruc.2021.130595.
- M. Mokhtar, S. M. El-Messery, M. A. Ghaly and G. S. Hassan, Targeting EGFR tyrosine kinase: Synthesis, in vitro anti-tumor evaluation, and molecular modeling studies of benzothiazole-based derivatives, Bioorg. Chem., 2020, 104, 104259, DOI:10.1016/j.bioorg.2020.104259.
- A. Qin, J. W. Lam and B. Z. Tang, Click polymerization, Chem. Soc. Rev., 2010, 39(7), 2522–2544, 10.1039/b909064a.
- M. Akter, K. Rupa and P. Anbarasan, 1,2,3-Triazole and Its Analogues: New Surrogates for Diazo Compounds, Chem. Rev., 2022, 122(15), 13108–13205, DOI:10.1021/acs.chemrev.1c00991.
- E. Bonandi, M. S. Christodoulou, G. Fumagalli, D. Perdicchia, G. Rastelli and D. Passarella, The 1,2,3-triazole ring as a bioisostere in medicinal chemistry, Drug Discov. Today, 2017, 22(10), 1572–1581, DOI:10.1016/j.drudis.2017.05.014.
- H. Y. Guo, Z. A. Chen, Q. K. Shen and Z. S. Quan, Application of triazoles in the structural modification of natural products, J. Enzyme Inhib. Med. Chem., 2021, 36(1), 1115–1144, DOI:10.1080/14756366.2021.1890066.
- X.-M. Chu, C. Wang, W.-L. Wang, L.-L. Liang, W. Liu, K.-K. Gong and K.-L. Sun, Triazole derivatives and their antiplasmodial and antimalarial activities, Eur. J. Med. Chem., 2019, 166, 206–223 CrossRef CAS PubMed.
- B. Zhang, Comprehensive review on the anti-bacterial activity of 1, 2, 3-triazole hybrids, Eur. J. Med. Chem., 2019, 168, 357–372 CrossRef CAS PubMed.
- S. Zhang, Z. Xu, C. Gao, Q.-C. Ren, L. Chang, Z.-S. Lv and L.-S. Feng, Triazole derivatives and their anti-tubercular activity, Eur. J. Med. Chem., 2017, 138, 501–513 CrossRef CAS PubMed.
- K. Lal and P. Yadav, Recent advancements in 1, 4-disubstituted 1H-1, 2, 3-triazoles as potential anti-cancer agents, Anti-Cancer Agents Med. Chem., 2018, 18(1), 21–37 CrossRef CAS PubMed.
- R. K. Mongre, C. B. Mishra, A. K. Shukla, A. Prakash, S. Jung, M. Ashraf-Uz-Zaman and M.-S. Lee, Emerging Importance of Tyrosine Kinase Inhibitors against Cancer: Quo Vadis to Cure?, Int. J. Mol. Sci., 2021, 22(21), 11659 CrossRef CAS PubMed.
- Z.-Q. Wang, Z.-C. Zhang, Y.-Y. Wu, Y.-N. Pi, S.-H. Lou, T.-B. Liu, G. Lou and C. Yang, Bromodomain and extraterminal (BET) proteins: biological functions, diseases and targeted therapy, Signal Transduction Targeted Ther., 2023, 8(1), 420, DOI:10.1038/s41392-023-01647-6.
- O. M. Soltan, M. E. Shoman, S. A. Abdel-Aziz, A. Narumi, H. Konno and M. Abdel-Aziz, Molecular hybrids: A five-year survey on structures of multiple targeted hybrids of protein kinase inhibitors for cancer therapy, Eur. J. Med. Chem., 2021, 225, 113768, DOI:10.1016/j.ejmech.2021.113768.
- D. P. Vala, R. M. Vala and H. M. Patel, Versatile Synthetic Platform for 1, 2, 3-Triazole Chemistry, ACS Omega, 2022, 7(42), 36945–36987 CrossRef CAS PubMed.
- S. R. Bandi, R. Kapavarapu, R. Palabindela, M. Azam, K. Min and S. Narsimha, Synthesis of novel pyrido [2, 3-d] pyrimidine-thiazolidine-1, 2, 3-triazoles: Potent EGFR targeting anti-cancer agents, J. Mol. Struct., 2023, 1294, 136451 CrossRef CAS.
- B. Banerji, K. Chandrasekhar, K. Sreenath, S. Roy, S. Nag and K. D. Saha, Synthesis of triazole-substituted quinazoline hybrids for anti-cancer activity and a lead compound as the EGFR blocker and ROS inducer agent, ACS Omega, 2018, 3(11), 16134–16142 CrossRef CAS PubMed.
- D. I. Othman, A. Hamdi, S. S. Tawfik, A. A. Elgazar and A. S. Mostafa, Identification of new benzimidazole-triazole hybrids as anti-cancer agents: multi-target recognition, in vitro and in silico studies, J. Enzyme Inhib. Med. Chem., 2023, 38(1), 2166037 CrossRef PubMed.
- M. A. Mahmoud, A. F. Mohammed, O. I. Salem, S. M. Rabea and B. G. Youssif, Design, synthesis, and antiproliferative properties of new 1, 2, 3-triazole-carboximidamide derivatives as dual EGFR/VEGFR-2 inhibitors, J. Mol. Struct., 2023, 1282, 135165 CrossRef CAS.
- A. Kamal, S. Prabhakar, M. Janaki Ramaiah, P. Venkat Reddy, C. Ratna Reddy, A. Mallareddy, N. Shankaraiah, T. Lakshmi Narayan Reddy, S. N. Pushpavalli and M. Pal-Bhadra, Synthesis and anti-cancer activity of chalcone-pyrrolobenzodiazepine conjugates linked via 1,2,3-triazole ring side-armed with alkane spacers, Eur. J. Med. Chem., 2011, 46(9), 3820–3831, DOI:10.1016/j.ejmech.2011.05.050.
- T. Liang, X. Sun, W. Li, G. Hou and F. Gao, 1, 2, 3-Triazole-containing compounds as anti–lung cancer agents: Current developments, mechanisms of action, and structure–activity relationship, Front. Pharmacol., 2021, 12, 661173 CrossRef CAS PubMed.
- L. H. Binh, N. T. T. Van, V. T. Kien, N. T. T. My, L. Van Chinh, N. T. Nga, H. X. Tien, D. T. Thao and T. K. Vu, Synthesis and in vitro cytotoxic evaluation of new triazole derivatives based on artemisinin via click chemistry, Med. Chem. Res., 2016, 25, 738–750 CrossRef CAS.
- S. Narsimha, N. S. Kumar, B. K. Swamy, N. V. Reddy, S. A. Hussain and M. S. Rao, Indole-2-carboxylic acid derived mono and bis 1, 4-disubstituted 1, 2, 3-triazoles: Synthesis, characterization and evaluation of anti-cancer, anti-bacterial, and DNA-cleavage activities, Bioorg. Med. Chem. Lett., 2016, 26(6), 1639–1644 CrossRef CAS PubMed.
- B. Prasad, V. L. Nayak, P. Srikanth, M. F. Baig, N. S. Reddy, K. S. Babu and A. Kamal, Synthesis and biological evaluation of 1-benzyl-N-(2-(phenylamino) pyridin-3-yl)-1H-1, 2, 3-triazole-4-carboxamides as antimitotic agents, Bioorg. Chem., 2019, 83, 535–548 CrossRef CAS PubMed.
- S. Wang, Q.-S. Li and M.-G. Su, Solubility of 1 H -1,2,4Triazole in Ethanol, 1Propanol, 2Propanol, 1,2-Propanediol, Ethyl Formate, Methyl Acetate, Ethyl Acetate, and Butyl Acetate at (283 to 363) K, J. Chem. Eng. Data, 2007, 52, 856–858, DOI:10.1021/je060452c.
- K. Litton Jennifer, S. Rugo Hope, J. Ettl, A. Hurvitz Sara, A. Gonçalves, K.-H. Lee, L. Fehrenbacher, R. Yerushalmi, A. Mina Lida and M. Martin, et al., Talazoparib in Patients with Advanced Breast Cancer and a Germline BRCA Mutation, N. Engl. J. Med., 2018, 379(8), 753–763, DOI:10.1056/NEJMoa1802905.
- K. Murthy Rashmi, S. Loi, A. Okines, E. Paplomata, E. Hamilton, A. Hurvitz Sara, U. Lin Nancy, V. Borges, V. Abramson and C. Anders, et al., Tucatinib, Trastuzumab, and Capecitabine for HER2-Positive Metastatic Breast Cancer, N. Engl. J. Med., 2020, 382(7), 597–609, DOI:10.1056/NEJMoa1914609.
- A. J. Knoche, L. B. Michaud and A. U. Buzdar, Efficacy of Anastrozole in a Consecutive Series of Advanced Breast Cancer Patients Treated with Multiple Prior Chemotherapies and Endocrine Agents: M. D. Anderson Cancer Center Experience, Breast J., 1999, 5(3), 176–181, DOI:10.1046/j.1524-4741.1999.98055.x.
- S. Mishra and P. Singh, Hybrid molecules: The privileged scaffolds for various pharmaceuticals, Eur. J. Med. Chem., 2016, 124, 500–536 CrossRef PubMed.
- B. Czako, J. R. Marszalek, J. P. Burke, P. Mandal, P. G. Leonard, J. B. Cross, F. Mseeh, Y. Jiang, E. Q. Chang and E. Suzuki, Discovery of IACS-9439, a potent, exquisitely selective, and orally bioavailable inhibitor of CSF1R, J. Med. Chem., 2020, 63(17), 9888–9911 CrossRef CAS PubMed.
- V. G. Reddy, T. S. Reddy, C. Jadala, M. S. Reddy, F. Sultana, R. Akunuri, S. K. Bhargava, D. Wlodkowic, P. Srihari and A. Kamal, Pyrazolo-benzothiazole hybrids: Synthesis, anti-cancer properties and evaluation of antiangiogenic activity using in vitro VEGFR-2 kinase and in vivo transgenic zebrafish model, Eur. J. Med. Chem., 2019, 182, 111609 CrossRef PubMed.
- H. Hosseinpoor, S. M. Farid, A. Iraji, M. S. Asgari, N. Edraki, S. Hosseini, A. Jamshidzadeh, B. Larijani, M. Attarroshan and S. Pirhadi, Anti-melanogenesis and anti-tyrosinase properties of aryl-substituted acetamides of phenoxy methyl triazole conjugated with thiosemicarbazide: Design, synthesis and biological evaluations, Bioorg. Chem., 2021, 114, 104979 CrossRef CAS PubMed.
- T. Amelia, R. E. Kartasasmita, T. Ohwada and D. H. Tjahjono, Structural Insight and Development of EGFR Tyrosine Kinase Inhibitors, Molecules, 2022, 27(3), 819, DOI:10.3390/molecules27030819.
- E. R. Purba, E. I. Saita and I. N. Maruyama, Activation of the EGF Receptor by Ligand Binding and Oncogenic Mutations: The “Rotation Model”, Cells, 2017, 6(2), 13, DOI:10.3390/cells6020013.
- C. W. Weng, C. H. Wei, J. Y. Tsai, Y. H. Lai, G. C. Chang and J. J. W. Chen, Hybrid Pharmacophore- and Structure-Based Virtual Screening Pipeline to Identify Novel EGFR Inhibitors That Suppress Non-Small Cell Lung Cancer Cell Growth, Int. J. Mol. Sci., 2022, 23(7), 3487, DOI:10.3390/ijms23073487.
- N. Normanno, A. De Luca, C. Bianco, L. Strizzi, M. Mancino, M. R. Maiello, A. Carotenuto, G. De Feo, F. Caponigro and D. S. Salomon, Epidermal growth factor receptor (EGFR) signaling in cancer, Gene, 2006, 366, 2–16, DOI:10.1016/j.gene.2005.10.018.
- N. Rezki, A green ultrasound synthesis, characterization and anti-bacterial evaluation of 1, 4-disubstituted 1, 2, 3-triazoles tethering bioactive benzothiazole nucleus, Molecules, 2016, 21(4), 505 CrossRef PubMed.
- N. Rezki, S. A. Al-Sodies, M. R. Aouad, S. Bardaweel, M. Messali and E. S. H. El Ashry, An eco-friendly ultrasound-assisted synthesis of novel fluorinated pyridinium salts-based hydrazones and anti-microbial and anti-tumor screening, Int. J. Mol. Sci., 2016, 17(5), 766 CrossRef PubMed.
- M. R. Aouad, M. Messali, N. Rezki, A. A.-S. Ali and A. Lesimple, Synthesis and characterization of some novel 1, 2, 4-triazoles, 1, 3, 4-thiadiazoles and Schiff bases incorporating imidazole moiety as potential anti-microbial agents, Acta Pharm., 2015, 65(2), 117–132 CrossRef CAS PubMed.
- V. A. D’yakonov, E. S. Finkelshtein and A. G. Ibragimov, Dzhemilev reaction for the synthesis of spiro [3.3] heptane and spiro [3.4] octanes, Tetrahedron Lett., 2007, 48(49), 8583–8586 CrossRef.
- M. A. Said, D. J. Khan, F. F. Al-Blewi, N. S. Al-Kaff, A. A. Ali, N. Rezki, M. R. Aouad and M. Hagar, New 1, 2, 3-Triazole scaffold schiff bases as potential anti-COVID-19: Design, synthesis, DFT-molecular docking, and cytotoxicity aspects, Vaccines, 2021, 9(9), 1012 CrossRef CAS PubMed.
- M. R. Aouad, D. J. Khan, M. A. Said, N. S. Al-Kaff, N. Rezki, A. A. Ali, N. Bouqellah and M. Hagar, Novel 1, 2, 3-Triazole Derivatives as Potential Inhibitors against Covid-19 Main Protease: Synthesis, Characterization, Molecular Docking and DFT Studies, ChemistrySelect, 2021, 6(14), 3468–3486 CrossRef CAS PubMed.
- M. J. McKeage, S. J. Berners-Price, P. Galettis, R. J. Bowen, W. Brouwer, L. Ding, L. Zhuang and B. C. Baguley, Role of lipophilicity in determining cellular uptake and antitumour activity of gold phosphine complexes, Cancer Chemother. Pharmacol., 2000, 46(5), 343–350, DOI:10.1007/s002800000166.
- J. Dong, N. N. Wang, Z. J. Yao, L. Zhang, Y. Cheng, D. Ouyang, A. P. Lu and D. S. Cao, ADMETlab: a platform for systematic ADMET evaluation based on a comprehensively collected ADMET database, J. Cheminf., 2018, 10(1), 29, DOI:10.1186/s13321-018-0283-x.
- G. Xiong, Z. Wu, J. Yi, L. Fu, Z. Yang, C. Hsieh, M. Yin, X. Zeng, C. Wu and A. Lu, et al., ADMETlab 2.0: an integrated online platform for accurate and comprehensive predictions of ADMET properties, Nucleic Acids Res., 2021, 49(W1), W5–w14, DOI:10.1093/nar/gkab255.
- T. Tuntland, B. Ethell, T. Kosaka, F. Blasco, R. X. Zang, M. Jain, T. Gould and K. Hoffmaster, Implementation of pharmacokinetic and pharmacodynamic strategies in early research phases of drug discovery and development at Novartis Institute of Biomedical Research, Front. Pharmacol., 2014, 5, 174, DOI:10.3389/fphar.2014.00174.
- A. L. Hopkins, G. M. Keserü, P. D. Leeson, D. C. Rees and C. H. Reynolds, The role of ligand efficiency metrics in drug discovery, Nat. Rev. Drug Discovery, 2014, 13(2), 105–121, DOI:10.1038/nrd4163.
- P. D. Leeson and B. Springthorpe, The influence of drug-like concepts on decision-making in medicinal chemistry, Nat. Rev. Drug Discovery, 2007, 6(11), 881–890, DOI:10.1038/nrd2445.
- S. Schultes, C. de Graaf, E. E. J. Haaksma, I. J. P. de Esch, R. Leurs and O. Krämer, Ligand efficiency as a guide in fragment hit selection and optimization, Drug Discov. Today, 2010, 7(3), e157–e162, DOI:10.1016/j.ddtec.2010.11.003.
- R. Guha and J. H. Van Drie, Structure-activity landscape index: identifying and quantifying activity cliffs, J. Chem. Inf. Model., 2008, 48(3), 646–658, DOI:10.1021/ci7004093.
- C. Deng, J. Xiong, X. Gu, X. Chen, S. Wu, Z. Wang, D. Wang, J. Tu and J. Xie, Novel recombinant immunotoxin of EGFR specific nanobody fused with cucurmosin, construction and anti-tumor efficiency in vitro, Oncotarget, 2017, 8(24), 38568–38580, DOI:10.18632/oncotarget.16930.
- J. L. Yang, X. J. Qu, P. J. Russell and D. Goldstein, Regulation of epidermal growth factor receptor in human colon cancer cell lines by interferon alpha, Gut, 2004, 53(1), 123–129, DOI:10.1136/gut.53.1.123.
- J. Stamos, M. X. Sliwkowski and C. Eigenbrot, Structure of the epidermal growth factor receptor kinase domain alone and in complex with a 4-anilinoquinazoline inhibitor, J. Biol. Chem., 2002, 277(48), 46265–46272, DOI:10.1074/jbc.M207135200.
- F. Alam, M. Khan and M. Ateeq, Synthesis of Triazole-Based Nonionic Surfactants for Nanostructured Drug Delivery: Investigation of Their Physicochemical and Biological Aspects, J. Surfactants Deterg., 2019, 22(6), 1419–1427 CrossRef CAS.
- A. Alkhawaldeh and S. Bardaweel, Molecular Investigation of the Antitumor Effects of Monoamine Oxidase Inhibitors in Breast Cancer Cells, BioMed Res. Int., 2023, 2023(1), 2592691 Search PubMed.
- M. S. Nafie, M. A. Tantawy and G. A. Elmgeed, Screening of different drug design tools to predict the mode of action of steroidal derivatives as anti-cancer agents, Steroids, 2019, 152, 108485 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Full characterization analyses (Fig. S1–S30). See DOI: https://doi.org/10.1039/d4ra07540d |
| ‡ Both authors shared equally to this manuscript with shared first authorship. |
| This journal is © The Royal Society of Chemistry 2025 |