DOI:
10.1039/D4RA07467J
(Paper)
RSC Adv., 2025,
15, 3139-3146
Identification of sulfonylated indolo[1,2-a]quinolines as EGFR tyrosine kinase inhibitors†
Received
18th October 2024
, Accepted 15th January 2025
First published on 30th January 2025
Abstract
Two series of indolo[1,2-a]quinolines (IQs), comprising six 6-trifluoromethylthio indolo[1,2-a]quinolines and nine 6-arenesulfonyl indolo[1,2-a]quinolines, were screened for their inhibitory activity against EGFR tyrosine kinase (EGFR-TK) using the ADP-Glo™ kinase assay. Among the 15 IQs screened, four compounds exhibited cytotoxic activity against a lung cancer cell line (A549) that was as potent as the known drug afatinib with lower cytotoxicity in Vero cells. In addition, while they displayed cytotoxic activity against a head and neck squamous cell carcinoma cell line (SCC cells), they were inactive against a colorectal cancer cell line (LS174T cells). Molecular dynamics (MD) simulations revealed that IQSO2R-I (IC50: 0.28 ± 0.05 μM) formed a stable complex with wild-type EGFR through hydrophohic interactions and hydrogen bonding with the K745 residue. Additionally, the compound complied with the extended rule of five. This class of compounds represents a novel class of EGFR-TK inhibitors, which may serve as a novel scaffold for the development of anticancer therapeutics targeting EGFR-TK.
Introduction
Cancer is a major disease that causes high mortality worldwide. Among the various types of cancer, lung cancer is the second most common cancer and has the highest mortality rates.1 Treatment options for patients with lung cancer include surgery, radiotherapy (radiation), chemotherapy, and immunotherapy.2 Nowadays, targeted chemotherapy for cancer treatment has received much attention globally owing to its specificity for cancer cells.3 A number of targeted anticancer drugs are now available for many common cancers, including breast cancer, colorectal, pancreatic cancer, lung cancer, leukemia, lymphoma, and multiple myeloma.4 However, even though targeted therapies are typically better tolerated than traditional chemotherapy, they are still associated with several adverse effects.
Epidermal growth factor receptor (EGFR) is a transmembrane protein and a member of the ErbB family, a subfamily of four closely related receptor tyrosine kinases (TKs).5 Mutations leading to EGFR overexpression are closely associated with the occurrence of several types of cancer, including non-small cell lung cancer (NSCLC), head cancer, breast cancer, ovarian cancer, and bladder carcinoma.6 As a result, targeting the EGFR protein has been recognized as a promising strategy for the targeted therapy of cancer.
Afatinib, a second-generation EGFR-TK inhibitor, is an FDA-approved anticancer drug used for the treatment of EGFR mutation-driven NSCLC.7 Afatinib is used in cancer therapy owing to its inhibitory activity against exon 19 deletion and exon 21 (L858R) substitution mutations. Although afatinib is recommended as a first-line therapy for EGFR-mutated metastatic lung cancer, acquired drug resistance, caused by the T790M mutation of the EGFR-TK domain, unavoidably develops after a median duration of treatment. Additionally, common side effects of tyrosine kinase inhibitors include rash, diarrhea, and liver toxicity. Due to these reasons, new therapeutics that can reduce risks and exhibit fewer adverse side effects are needed.
Nitrogen heterocycles are an important class of compounds found in a number of natural products, drugs and functional materials.8 Among them, indolo[1,2-a]quinolines (IQs) have a unique nitrogen-containing tetracyclic scaffold.9 Afatinib and other approved drugs targeting EGFR, including erlotinib, osimertinib and gefitinib, contain a nitrogen heterocyclic scaffold (Fig. 1). On the basis of our previously reported work10 and the fact that 6-arenesulfonyl indolo[1,2-a]quinolines (IQSO2Rs) and 6-trifluoromethylthio indolo[1,2-a]quinolines (IQSCF3s) bear both indole and quinoline rings in their structures, we hypothesized that IQSO2Rs and IQSCF3s (Fig. 2) might exhibit potential EGFR-TK inhibitory activity. Therefore, our objectives in the present study were to investigate the EGFR-TK inhibitory activity of some selected IQs, focusing on those bearing either a arenesulfonyl group (SO2R) or trifluoromethylthio group (SCF3) in their structures, and to evaluate their effect on the cell viability of EGFR wild-type non-small-cell lung cancer (A549), EGFR wild-type head and neck squamous cell carcinoma (SCC), and EGFR-KRASG12D-mutated colorectal cancer cells (LS174T) and normal cells (Vero cells).
 |
| | Fig. 1 Structures of afatinib, erlotinib, gefitinib, and osimertinib. | |
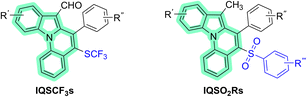 |
| | Fig. 2 Indolo[1,2-a]quinoline (IQ) core (highlighted in green) of 6-trifluoromethylthio indolo[1,2-a]quinolines (IQSCF3s) and 6-arenesulfonyl indolo[1,2-a]quinolines (IQSO2Rs). | |
Results and discussion
Synthesis of IQSCF3s and IQSO2Rs
The syntheses of six 6-trifluoromethylthio indolo[1,2-a]quinolines (IQSCF3-I–IQSCF3-VI) and nine 6-arenesulfonyl indolo[1,2-a]quinolines (IQSO2R-I–IQSO2R-IX) were efficiently achieved by following previously reported protocols (Fig. 3) (see the ESI†).9d,f All the synthesized compounds in this study were purified and characterized (see the ESI†).
 |
| | Fig. 3 Chemical structures of the six IQSCF3s (IQSCF3-I–IQSCF3-VI) and nine IQSO2Rs (IQSO2R-I–IQSO2R-IX) screened toward EGFR-TK inhibitory activity. | |
Inhibition of EGFR-TK by IQs (IQSCF3s and IQSO2Rs)
Primarily, the EGFR-TK inhibitory activity of the six synthesized IQSCF3 series (IQSCF3-I–IQSCF3-VI) and nine synthesized IQSO2R series (IQSO2R-I–IQSO2R-IX) was evaluated. Afatinib, a medication used to treat non-small cell lung carcinoma (NSCLC), was employed as a benchmark. Thus, indolo[1,2-a]quinoline derivatives (IQs) and afatinib were screened at 1 μM using EGFR kinase inhibition assay kits. The results are shown in Fig. 4, and it revealed that all the compounds showed inhibitory activity toward EGFR-TK. Among the 15 compounds screened, six compounds, namely IQSCF3-V, IQSO2R-I, IQSO2R-II, IQSO2R-V, IQSO2R-VI, and IQSO2R-VII showed EGFR-TK inhibitory activity greater than 50% inhibition at 1 μM (96.96% inhibition for afatinib). The preliminary screening results suggested that the IQs could serve as EGFR-TK inhibitors. These six compounds that exhibited greater than 50% inhibition at 1 μM were subjected to further investigation.
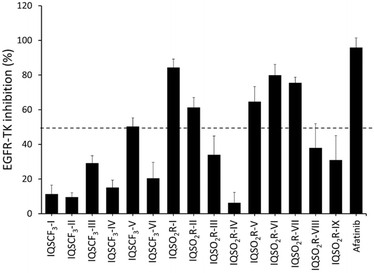 |
| | Fig. 4 Kinase inhibitory activity screening of six synthesized IQSCF3 series (IQSCF3-I–IQSCF3-VI) and nine synthesized IQSO2R series (IQSO2R-I–IQSO2R-IX) toward EGFR-TK at 1 μM as determined by EGFR kinase activity assays. The data are presented as the mean ± standard error of mean (S.E.M.) from three independent experiments (n = 3). | |
Cell viability assay of EGFR-overexpressing cancer cell lines in a lung adenocarcinoma cell line (A549), head and neck squamous cell carcinoma (SCC), colorectal cancer cell (LS174T), and normal kidney epithelial cell (Vero) lines
The six selected compounds, i.e., IQSCF3-V, IQSO2R-I, IQSO2R-II, IQSO2R-V, IQSO2R-VI, and IQSO2R-VII, were subjected to an in vitro cytotoxicity assay against EGFR wild-type non-small-cell lung cancer (A549), EGFR wild-type head and neck squamous cell carcinoma (SCC), and EGFR-KRASG12D-mutated colorectal cancer (LS174T) cells, in comparison with Vero kidney epithelium cells using MTT assays. The cell viability of A549, SCC, LS174T, and Vero cells treated with various concentrations of the six selected compounds or afatinib was evaluated to obtain the half-maximal inhibitory concentration (IC50) values (Table 1). The results revealed that IQSO2R-I, IQSO2R-V, IQSO2R-VI, IQSO2R-VII, and afatinib exhibited cytotoxicity activity against EGFR wild-type A549 cells, with IC50 values of 14.55 ± 3.14, 16.39 ± 2.40, 14.59 ± 0.87, 13.71 ± 1.20, and 13.09 ± 0.80 μM, respectively (Fig. 5A). IQSO2R-I, IQSO2R-V, IQSO2R-VI, and IQSO2R-VII exhibited cytotoxicity effects with IC50 values of 19.80 ± 4.07, 36.71 ± 3.50, 22.58 ± 1.29, and 21.80 ± 2.42 μM in EGFR wild-type SCC cells, respectively (Fig. 5B). Notably, IQSO2R-I, IQSO2R-V, IQSO2R-VI, and IQSO2R-VII displayed slightly lower toxicity in Vero cells with IC50 values in a range of 18–23 μM, whereas the IC50 of afatinib was 6 μM. The six selected compounds did not exhibit cytotoxicity effects against EGFR-KRASG12D LS174T cells.
Table 1 IC50 values of IQSCF3-V, IQSO2R-I, IQSO2R-II, IQSO2R-V, IQSO2R-VI, IQSO2R-VII, and afatinib against A549, SCC, LS174T, and Vero cell linesa
| Compounds |
IC50 (μM) |
| A549 |
SCC |
LS174T |
Vero |
| The data are presented as the mean ± standard error of mean (S.E.M.) from triplicate independent experiments (n = 3). |
| IQSCF3-V |
50.07 ± 1.83 |
46.35 ± 1.31 |
>100 |
17.80 ± 0.31 |
| IQSO2R-I |
14.55 ± 3.14 |
19.80 ± 4.07 |
>100 |
22.64 ± 3.59 |
| IQSO2R-II |
41.13 ± 5.10 |
46.81 ± 5.48 |
>100 |
18.44 ± 0.14 |
| IQSO2R-V |
16.39 ± 2.40 |
36.71 ± 3.50 |
>100 |
18.84 ± 4.31 |
| IQSO2R-VI |
14.59 ± 0.87 |
22.58 ± 1.29 |
>100 |
21.67 ± 1.40 |
| IQSO2R-VII |
13.71 ± 1.20 |
21.80 ± 2.42 |
>100 |
19.43 ± 2.75 |
| Afatinib |
13.09 ± 0.80 |
0.98 ± 0.10 |
4.97 ± 1.09 |
5.77 ± 1.59 |
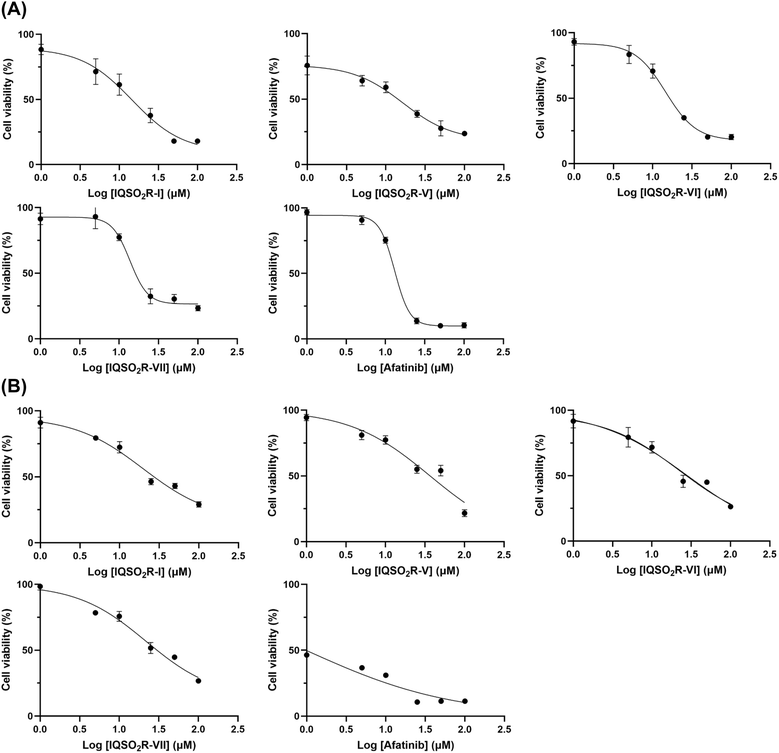 |
| | Fig. 5 Dose–response studies of IQSO2R-I, IQSO2R-V, IQSO2R-VI, and IQSO2R-VII and afatinib in EGFR wild-type non-small-cell lung cancer (A549) (A) and EGFR wild-type head and neck squamous cell carcinoma (SCC) (B). The data are presented as the mean ± standard error of mean (S.E.M.) from triplicate independent experiments (n = 3). | |
Structure–activity relationship (SAR)
To better understand the structure–activity relationship, the inhibitory activity of EGFR-TK and the chemical structure of the IQs were analyzed and revealed that three different locations of substitution on the IQ scaffold, namely (1) the sulfur moiety and the substituent on the arene sulfonyl group (blue color), (2) the substituent on the aryl ring (red color), and (3) the substituents on the indole core (green color), affected the inhibitory potency (Fig. 6). For the sulfur moiety, the IQSCF3 series (IQSCF3-I–IQSCF3-VI) were relatively less active than the IQSO2R series. Among the IQSCF3 series, IQSCF3-V exhibited the highest inhibitory activity (50% inhibition). Among the IQSO2R series, IQSO2R-I, IQSO2R-II, IQSO2R-V, IQSO2R-VI, and IQSO2R-VII, displayed greater than 50% inhibition, with IQSO2R-I exhibiting the highest inhibitory activity (84% inhibition). It should be noted that the IQSO2R series that exhibited inhibitory activity greater than 50% inhibition at 1 μM possessed a para-Me substituent at the arenesulfonyl moiety, except for IQSO2R-II. Thus, the presence of a para-Me moiety at the arenesulfonyl group may play an important role in the EGFR-TK inhibitory activity. For the substituent on the aryl ring, the presence of a para-Me substituent (R2 = Me) at aryl ring had a detrimental effect on the inhibitory activity, with IQSO2R-IV being the least active compound in the IQSO2R series. The electronically different groups (R1 and R3) on the indole scaffold of compounds IQSO2R-V, IQSO2R-VI, and IQSO2R-VII did not show any significant effect.
 |
| | Fig. 6 Analysis of the structure–activity relationship. | |
Kinase inhibitory activity
The results for the EGFR-TK activity obtained from both the EGFR-TK inhibition assay and cell viability analyses against A549 cell lines by MTT assay demonstrated that IQSO2R-I, IQSO2R-V, IQSO2R-VI, and IQSO2R-VII exhibited cytotoxic effects similar to those of afatinib. Therefore, the IC50 values of IQSO2R-I, IQSO2R-V, IQSO2R-VI, and IQSO2R-VII on the kinase inhibitory activity were determined using EGFR-TK inhibition assays to investigate the ligand–protein binding between these compounds and EGFR-TK (Table 2). The IC50 values of IQSO2R-I, IQSO2R-V, IQSO2R-VI, and IQSO2R-VII were 0.28 ± 0.05, 0.38 ± 0.12, 0.44 ± 0.07, and 0.48 ± 0.19 μM, respectively.
Table 2 Kinase inhibitory activity of IQSO2R-I, IQSO2R-V, IQSO2R-VI, and IQSO2R-VIIa
| Compounds |
IC50 (μM) |
| The data are presented as the mean ± standard error of mean (S.E.M.) from triplicate independent experiments (n = 3). |
| IQSO2R-I |
0.28 ± 0.05 |
| IQSO2R-V |
0.38 ± 0.12 |
| IQSO2R-VI |
0.44 ± 0.07 |
| IQSO2R-VII |
0.48 ± 0.19 |
Prediction of the physicochemical properties
The drug-likeness of the four potent IQSO2R derivatives (IQSO2R-I, IQSO2R-V, IQSO2R-VI, and IQSO2R-VII) obtained from the EGFR kinase assay was predicted using the SwissADME web server, based on the extended rule of five and their physicochemical properties.11 This analysis involved assessing various characteristics, such as the molecular weight (MW), number of hydrogen bond acceptors (HBA), number of hydrogen bond donors (HBD), log of octanol-to-water partition coefficient or lipophilicity (LogP), polar surface area (PSA), and the number of rotatable bonds (RB). The results are shown in Table 3. Four IQ derivatives met the acceptable criteria. For validation of the drug-likeness, investigation of the pharmacokinetics properties of these compounds warrants further investigation.
Table 3 Predicted molecular and drug-likeness properties for IQSO2R-I, IQSO2R-V, IQSO2R-VI, and IQSO2R-VIIa
| Compounds |
MW |
HBA |
HBD |
log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) P P |
PSA |
RB |
Drug-likeness |
| Extended rule of five:12 molecular weight (MW) ≤ 700 Da; hydrogen bond acceptors (HBAs) ≤ 10; hydrogen bond donors (HBDs) ≤ 5; logP (octanol–water partition coefficient) ≤ 7.5; polar surface area (PSA) ≤ 200 Å2; and rotatable bonds (RBs) ≤ 20. |
| IQSO2R-I |
461.57 |
2 |
0 |
6.40 |
46.93 |
3 |
Yes |
| IQSO2R-V |
465.54 |
3 |
0 |
6.38 |
46.93 |
3 |
Yes |
| IQSO2R-VI |
481.99 |
2 |
0 |
6.59 |
46.93 |
3 |
Yes |
| IQSO2R-VII |
526.44 |
2 |
0 |
6.68 |
46.93 |
3 |
Yes |
Molecular dynamics study of the IQSO2R-I/EGFR-TK complex
Considering the experimental data and predicted physicochemical properties of IQSO2R derivatives, IQSO2R-I binding susceptibility to the EGFR-TK domain was examined using 500 ns MD simulations with three independent replicates (Fig. 7). The stability of the IQSO2R-I/EGFR complex was assessed by evaluating the all-atom RMSD, the radius of gyration (Rg) of the complex, the number of intermolecular hydrogen bonds (#Hbonds), and the number of atomic contacts (#Atom contacts), as illustrated in Fig. 7A. The RMSD values of the complex only fluctuated during the initial simulations (till ∼100 ns for Run1, ∼250 ns for Run2, and ∼200 ns for Run3), while the Rg (19.9–20.3 Å), #Hbonds (1–2 bonds), and #Atom contacts (10–30 contacts) values remained stable throughout the simulations. The results suggested a high level of system stability and ligand/protein binding.
 |
| | Fig. 7 (A) Structural and dynamic analysis of IQSO2R-I binding to wild-type EGFR-TK over 500 ns MD simulations in terms of the all-atom RMSD, radius of gyration (Rg) of complex, and the number of hydrogen bonds and atomic contacts. (B) Binding pattern and hydrogen bonding of the IQSO2R-I/EGFR-TK complex. | |
The MM/GBSA per-residue binding free energy (ΔGresidue) was obtained based on 100 snapshots taken from the last 100 ns of all the simulations. It can be seen from Fig. 7B that there were 9 amino acids L718, F723, V726, K745, A743, T790, M793, G796, and L844 associated with the IQSO2R-I binding. Notably, the contribution of M793 (hinge region) has also been found in the other reported EGFR-TK inhibitors.13 Additionally, the sulfonyl moiety of IQSO2R-I formed a strong hydrogen bond with K745 (∼72% occupation). As shown in Fig. 8, the presence of the methyl (CH3) substituent at the SO2Ar position in IQSO2R-I facilitated strong hydrophobic interactions with EGFR-TK residues I789 (74%), I744 (41%), and I788 (99%). These interactions likely contribute to the enhanced anticancer efficacy of IQSO2R-I, as evidenced by its IC50 value of 14.55 ± 3.14 μM. In contrast, the absence of the CH3 group at the SO2Ar position in compounds IQSCF3-V and IQSO2R-II may have resulted in the loss of these hydrophobic interactions, potentially leading to reduced anticancer activity. Furthermore, the sulfur moiety in the IQSCF3 series demonstrated lower activity in both the EGFR kinase assay and anticancer evaluation compared to in the IQSO2R series. The replacement of the SO2Ar group with SCF3 may eliminate the critical hydrogen bond with K745 and essential hydrophobic interactions, resulting in the reduced activity for the IQSCF3 series. For example, IQSCF3-V exhibited less than 50% EGFR-TK inhibition and an IC50 of 50.07 ± 1.83 μM against A549. These findings highlight the importance of both hydrophobic interactions and hydrogen bonding in playing a crucial role in enhancing the EGFR-TK and anticancer activity.14
 |
| | Fig. 8 Key interactions of IQSO2R-I with wild-type EGFR-TK depicted in 2D and 3D pharmacophore models derived from the final 100 ns of MD simulations. The red arrow denotes the hydrogen bond acceptor (HBA), while the yellow circles indicate the hydrophobic interactions. | |
Experimental section
Preparation of 6-trifluoromethylthio indolo[1,2-a] quinolines (IQSCF3s) and 6-arenesulfonyl indolo[1,2-a]quinolines (IQSO2Rs)
The investigated 6-trifluoromethylthio indolo[1,2-a]quinolines (IQSCF3-I–IQSCF3-VI) and 6-arenesulfonyl indolo[1,2-a]quinolines (IQSO2R-I–IQSO2R-IX) were prepared by following a previously reported protocol (see ESI†).9d,f
EGFR-TK inhibition assay
The ADP-Glo™ kinase assay kit was obtained from Promega (Madison, WI, USA). The series of IQSCF3s and IQSO2Rs were screened to determine their EGFR tyrosine kinase activity using the ADP-Glo™ kinase assay, as previously described.8 After kinase buffer was added to a well plate, EGFR enzyme at a concentration of 1.25 ng μL−1 and inhibitors were added, followed by a mixture of 25 μM ATP and 12.5 μM poly (Glu–Tyr) and then 1 h incubation. Then, the ADP-Glo reagent and the kinase detection reagent were added and incubated for 40 and 30 min, respectively. The ATP was measured by measuring the luminescence using a microplate reader (BioTek Instruments, VT, USA). The relative inhibition (%) of the inhibitors was calculated compared with the control (no inhibitor), as shown in eqn (1).| |
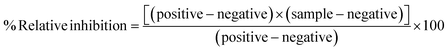 | (1) |
Evaluation of cell cytotoxicity in cancer cell lines
A549 Lung adenocarcinoma, SCC head and neck squamous cell, LS17AT carcinoma colorectal cancer cell, and Vero cell lines were obtained from American Type Culture Collection (Manassas, VA, USA). The A549 cells were maintained in a Kaighn's modification of Ham's F-12 medium (F-12k Medium) together with 10% FBS and 100 U mL−1 penicillin (Life Technologies, Carlsbad, CA, USA). The SCC cells were maintained in Dulbecco's modified Eagle's medium/Ham's F-12 (DMEMF-12) with 10% FBS and 100 U mL−1 penicillin. The LS174T cells were maintained in RPMI-1640 contained with 10% FBS and 100 U mL−1 penicillin. The Vero cells were maintained in eagle's minimum essential medium (EMEM) together with 10% FBS and 100 U mL−1 penicillin. All cells were kept at 37 °C under humidified 95% O2 with 5% CO2 atmosphere.
The cell viability of the A549, SCC, LS174T, and Vero cells was assessed using MTT assays. Briefly, A549 cells (5000 cells per well), SCC cells (10000 cells per well), LS174T cells (7000 cells per well), and Vero cells (4000 cells per well) were seeded in to 96-well plates and incubated at 37 °C for 24 h. After 24 h incubation, the cells were then treated with different concentrations of the second-generation EGFR-TKIs drug (afatinib), IQSCF3s, and IQSO2Rs at 37 °C for 72 h incubation. MTT solution (5 mg ml−1) was added to the cells and kept for 2 h incubation at 37 °C. The reaction was stopped by adding DMSO (100 μL per well). Colorimetric quantification was measured the absorbance at 570 nm using a microplate reader (BioTek Instruments, VT, USA).
Physicochemical property predictions
The physicochemical features, including the number of hydrogen bond donors, hydrogen bond acceptors, and drug-likeness, play a crucial role in the drug discovery and development process. In this study, we calculated these properties for four compounds (IQSO2R-I, IQSO2R-V, IQSO2R-VI, and IQSO2R-VII) using the web-based application SwissADME (http://www.swissadme.ch/).
Molecular dynamic simulations
The crystal structure of erlotinib in complex with the wild-type EGFR-TK was obtained from the Protein Data Bank (PDB ID: 1M17). The IQSO2R-I/EGFR-TK complex was derived by molecular docking using the GOLD program15 according to our previous study on this system.16 The best docking pose (highest GOLD fitness score) of IQSO2R-I binding to EGFR-TK was considered as the starting structure for the all-atom molecular dynamics simulations with three independent runs using different initial velocities. The system was simulated under periodic boundary conditions using the isothermal–isobaric (NPT) ensemble, with a temperature of 310 K and a pressure of 1 atm as per previous studies.17 The AMBER ff14SB force field and the generalized AMBER force field version 2 (GAFF2) were employed to handle the bonded and non-bonded interaction parameters for the protein and ligand, respectively. The system was solvated in the TIP3P water model. Chloride ions were randomly introduced to neutralize the overall charge of the system. The hydrogen atoms and water molecules were subjected to energy minimization using 500 steps of the steepest descent followed by 1500 steps of conjugated gradient methods, while the remaining molecules were kept fixed. The protein–ligand complex (constrained solvents) and the entire complex system were then subjected to further minimization, following the same procedure. Electrostatic interactions were handled using the particle mesh Ewald summation approach, and hydrogen atoms were constrained using the SHAKE algorithm. The temperature was gradually increased from 10 to 310 K using a Langevin thermostat with a collision frequency of 2 ps−1, while the pressure was controlled using the Berendsen barostat. MD simulations were conducted for 500 ns with a time step increment of 2 fs. The MD outputs were analyzed using the cpptraj module, and the per-residue decomposition energy (ΔGbind,residue) was computed using MM/PBSA.py in AMBER20. The interactions of the potent EGFR inhibitor complexed with EGFR-TK were visualized using LigandScout 4.4.9 software.18
Conclusions
In the present work, for the first time, experimental and computational methods were employed to identify potential candidates for EGFR inhibitors based on compounds bearing indolo[1,2-a]quinoline as a core structure. Six 6-trifluoromethylthio indolo[1,2-a]quinolines and nine 6-arenesulfonyl indolo[1,2-a]quinolines, were experimentally screened for their inhibitory activity against EGFR-TK; six compounds (IQSCF3-V, IQSO2R-I, IQSO2R-II, IQSO2R-V, IQSO2R-VI, and IQSO2R-VII) were found to inhibit EGFR-TK activity greater than 50% inhibition at 1 μM (∼90% inhibition for afatinib). From a cell-based assay in human cancer cell lines with EGFR wild-type overexpression (A549 and SCC cell lines), four compounds (IQSO2R-I, IQSO2R-V, IQSO2R-VI, and IQSO2R-VII) exhibited cytotoxic activity against A549 and SCC cells as potent as afatinib, with a slightly lower toxicity in Vero cells. The MD simulation study on the most potent compound, i.e., IQSO2R-I with an IC50 of 0.28 ± 0.05 μM, revealed nine amino acids (L718, F723, V726, K745, A743, T790, M793, G796, and L844) contributed to the formation of the IQSO2R-I/EGFR complex. Therefore, IQSO2R-I represents a novel type of EGFR-TK inhibitor, which may be useful as a novel scaffold for the development of anticancer therapeutics targeting EGFR-TK.
Data availability
The data supporting this article have been included as part of the ESI.†
Author contributions
CK, CM, and TR conceived and designed the experiments. PS, DT and TR conducted theoretical and experimental studies. JP, TU, and NS synthesized all compounds. CK, PS, CM, DT and TR analyzed the data. CK, PS, CM, DT and TR wrote the original manuscript. All authors reviewed and edited the manuscript. All authors have read and agreed to the published version of the manuscript.
Conflicts of interest
Authors declare no conflicts of interest.
Acknowledgements
This research was financially supported by Mahidol University (Basic Research Fund: fiscal year 2023) and the NSRF via the Program Management Unit for Human Resources & Institutional Development, Research and Innovation (grant number B05F650041). C. K. thanks National Research Council of Thailand (NRCT) and Mahidol University (grant number No. N42A650344) and the Center of Excellence for Innovation in Chemistry (PERCH-CIC), Ministry of Higher Education, Science, Research and Innovation for providing scientific instrumental support. D. T. thanks the Second Century Fund (C2F), Chulalongkorn University, for PhD scholarship.
References
- WHO factsheet, https://www.who.int/news-room/fact-sheets/detail/lung-cancer, 2023.
- H. Lemjabbar-Alaoui, O. U. Hassan, Y.-W. Yang and P. Buchanan, Biochim. Biophys. Acta, 2015, 1856, 189 CAS
 .
. - X. Ke and L. Shen, Front. Lab. Med., 2017, 1, 69 CrossRef .
-
(a) S. Di Martino, A. Rainone, A. Troise, M. Di Paolo, S. Pugliese, S. Zappavigna, A. Grimaldi and D. Valente, Overview of FDA-approved anticancer drugs used for targeted therapy, World Cancer Res. J., 2015, 2, e553 Search PubMed ;
(b) F. R. Hirsch, G. V. Scagliotti, J. L. Mulshine, R. Kwon, W. J. Curran, Y.-L. Wu and L. Paz-Ares, Lancet, 2017, 389, 299 CrossRef CAS PubMed .
- R. Roskoski Jr, Pharmacol. Res., 2014, 79, 34 CrossRef PubMed .
-
(a) H. Ogiso, R. Ishitani, O. Nureki, S. Fukai, M. Yamanaka, J. H. Kim, K. Saito, A. Sakamoto, M. Inoue, M. Shirouzu and S. Yokoyama, Cell, 2002, 110, 775 CrossRef CAS PubMed ;
(b) C. H. Yang, H. C. Chou, Y. N. Fu, C. L. Yeh, H. W. Cheng, I. C. Chang, K. J. Liu, G. C. Chang, T. F. Tsai, S. F. Tsai, H. P. Liu, Y. C. Wu, Y. T. Chen, S. F. Huang and Y. R. Chen, Biochim. Biophys. Acta, 2015, 1852, 1540 CrossRef CAS PubMed ;
(c) F. R. Hirsch, M. Varella-Garcia and F. Cappuzzo, F. Oncogene, 2009, 28, S32 CrossRef CAS PubMed .
- V. P. Reddy, Organofluorine Compounds as Anticancer Agents, in Organofluorine Compounds in Biology and Medicine, Elsevier, 2015 Search PubMed .
-
(a) J. P. Michael, Nat. Prod. Rep., 2008, 25, 139 RSC ;
(b) A. F. Pozharskii, A. T. Soldatenkov and A. R. Katritzky, Heterocycles in Life and Society: an Introduction to Heterocyclic Chemistry, Biochemistry and Applications, John Wiley & Sons, 2011 CrossRef ;
(c) T. Eicher, S. Hauptmann and A. Speicher, The Chemistry of Heterocycles: Structure, Reactions, Syntheses, and Applications, Wiley-VCH, 2013 Search PubMed ;
(d) C. T. Walsh, Tetrahedron Lett., 2015, 56, 3075 CrossRef CAS ;
(e) C. Cabrele and O. Reiser, J. Org. Chem., 2016, 81, 10109 CrossRef CAS PubMed ;
(f) A. P. Taylor, R. P. Robinson, Y. M. Fobian, D. C. Blakemore, L. H. Jones and O. Fadeyi, Org. Biomol. Chem., 2016, 14, 6611 RSC ;
(g) M. M. Heravi and V. Zadsirjan, RSC Adv., 2020, 10, 44247 RSC .
-
(a) D. G. Hulcoop and M. Lautens, Org. Lett., 2007, 9, 1761 CrossRef CAS PubMed ;
(b) A. K. Verma, S. P. Shukla, J. Singh and V. Rustagi, J. Org. Chem., 2011, 76, 5670 CrossRef CAS PubMed ;
(c) S. P. Shukla, R. K. Tiwari and A. K. Verma, J. Org. Chem., 2012, 77, 10382 CrossRef CAS PubMed ;
(d) K. Sun, X. L. Chen, Y. L. Zhang, K. Li, X. Q. Huang, Y. Y. Peng, L. B. Qu and B. Yu, Chem. Commun., 2019, 55, 12615 RSC ;
(e) R. Heckershoff, G. May, J. Däumer, L. Eberle, P. Krämer, F. Rominger, M. Rudolph, F. F. Mulks and A. S. K. Hashmi, Chem.–Eur.
J., 2022, 28, e202201816 CrossRef CAS PubMed ;
(f) T. Uppalabat, N. Hassa, N. Sawektreeratana, P. Leowanawat, P. Janthakit, P. Nalaoh, V. Promarak, D. Soorukram, V. Reutrakul and C. Kuhakarn, J. Org. Chem., 2023, 88, 5403 CrossRef CAS PubMed ;
(g) S. Zhang, J. Yuan, G. Huang, C. Ma, J. Yang, L. Yang, Y. Xiao and L. Qu, J. Org. Chem., 2023, 88, 11712 CrossRef CAS PubMed .
- K. Hengphasatporn, T. Aiebchun, P. Mahalapbutr, A. Auepattanapong, O. Khaikate, K. Choowongkomon, C. Kuhakarn, J. Meesin, Y. Shigeta and T. Rungrotmongkol, ACS Omega, 2023, 8, 19645 CrossRef CAS PubMed .
- A. Daina, O. Michielin and V. Zoete, Sci. Rep., 2017, 7, 42717 CrossRef PubMed .
- C. A. Lipinski, Drug Discovery Today:Technol., 2004, 1, 337 CrossRef CAS PubMed .
-
(a) G. P. Doss, B. Rajith, C. Chakraborty, N. NagaSundaram, K. Ali and H. Zhu, Sci. Rep., 2014, 4, 5868 CrossRef PubMed ;
(b) F. Martínez-Jiménez, J. P. Overington, B. Al-Lazikani and M. A. Marti-Renom, Sci. Rep., 2017, 7, 46632 CrossRef PubMed ;
(c) J. Stamos, M. X. Sliwkowski and C. Eigenbrot, J. Biol. Chem., 2002, 277, 46265 CrossRef CAS PubMed .
- W. Nie, L. Tang, H. Zhang, J. Shao, Y. Wang, L. Chen, D. Li and X. Guan, Int. J. Oncol., 2012, 40, 1763 CAS .
- M. L. Verdonk, J. C. Cole, M. J. Hartshorn, C. W. Murray and R. D. Taylor, Proteins, 2003, 52, 609 CrossRef CAS PubMed .
-
(a) D. Todsaporn, A. Zubenko, V. G. Kartsev, P. Mahalapbutr, A. Geronikaki, S. N. Sirakanyan, L. N. Divaeva, V. Chekrisheva, I. Yildiz, K. Choowongkomon and T. Rungrotmongkol, J. Phys. Chem. B, 2024, 128, 12389 CrossRef CAS PubMed ;
(b) T. Aiebchun, P. Mahalapbutr, A. Auepattanapong, O. Khaikate, S. Seetaha, L. Tabtimmai, C. Kuhakarn, K. Choowongkomon and T. Rungrotmongkol, Molecules, 2021, 26, 2211 CrossRef CAS PubMed .
-
(a) D. Todsaporn, A. Zubenko, V. Kartsev, T. Aiebchun, P. Mahalapbutr, A. Petrou, A. Geronikaki, L. Divaeva, V. Chekrisheva, I. Yildiz, K. Choowongkomon and T. Rungrotmongkol, Molecules, 2023, 28, 3014 CrossRef CAS PubMed ;
(b) D. Todsaporn, P. Mahalapbutr, R. P. Poo-arporn, K. Choowongkomon and T. Rungrotmongkol, Comput. Biol. Med., 2022, 147, 105787 CrossRef CAS PubMed .
- G. Wolber and T. Langer, J. Chem. Inf. Model., 2005, 45, 160 CrossRef CAS PubMed .
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence ce,
Chatchai Muanprasat*b and
Chutima Kuhakarn
ce,
Chatchai Muanprasat*b and
Chutima Kuhakarn







