DOI:
10.1039/D4RA06510G
(Paper)
RSC Adv., 2025,
15, 5703-5719
Design, synthesis and biological activity of peptidyl β-nitrostyrenes as cysteine protease inhibitors against Leishmania donovani†
Received
9th September 2024
, Accepted 15th December 2024
First published on 19th February 2025
Abstract
Cysteine proteases are essential for the survival of Leishmania parasites that cause several clinical forms of leishmaniases. Inhibiting cysteine protease can be a promising strategy against parasitic diseases because of their essential functions in the life cycles of these pathogens. The aim of the present study was to synthesize and evaluate peptidyl -nitrostyrenes as antipromastigote inhibitors against Leishmania donovani promastigotes. A library of 12 peptidyl β-nitrostyrenes was synthesized and evaluated for anti-promastigote activity. Most of the compounds exhibited comparable activity to the standard, with IC50 values ranging from 1.468 to 16.81 μM. Notably, compounds 14a, 14e, 14f, and 14g showed significant activity against both L. donovani promastigotes and intracellular amastigotes. Compounds 14e and 14f displayed superior anti-promastigote activity with IC50 values of 1.468 μM and 1.551 μM, respectively, compared to the standard (IC50 = 3.073 μM). Moreover, compounds 14e and 14f demonstrated better inhibitory potential against intracellular amastigotes, with IC50 values of 1.28 μM and 0.64 μM, respectively, outperforming AmphoB (IC50 = 3.07 μM). Additionally, compounds 14a and 14g showed negligible cytotoxicity to mammalian macrophages even at a concentration of 28 μM. Given their high activity, favorable safety profiles, and cost-effective synthesis, this class of compounds holds promise for the development of anti-leishmanial drugs.
1. Introduction
Leishmaniasis is a neglected tropical disease caused by parasites from the genus Leishmania. The WHO has identified it as one of 17 such diseases that pose a significant threat to mankind in tropical regions where over a billion people are directly exposed to tropical parasites.1 Major pharmaceutical companies have largely overlooked such diseases in their drug discovery programs due to the lack of financial return.1 The burden of Leishmaniasis is concentrated in a few countries, and environmental factors like massive migrations, urbanization, deforestation, and new irrigation schemes, as well as individual risk factors like HIV, malnutrition, and genetics, contribute to its continued prevalence.2,3 Leishmaniasis can manifest in various ways, with the most common forms being cutaneous and visceral, ranging from asymptomatic to chronic disease. The type of Leishmaniasis a person gets depends on various factors relating to the patient and the parasite causing the disease.4
Cysteine proteases are important enzymes in parasitic organisms and are critical to their replication, virulence, and survival. They are particularly essential to Trypanosoma and Leishmania species, making them attractive targets for developing therapeutic agents against parasitic infections.5 Inhibiting cysteine proteases can be a promising strategy against parasitic diseases because of their essential functions in the life cycles of these pathogens. Researchers have been investigating this approach for years and have made significant strides in developing cysteine protease inhibitors.6,7
Animal trials have shown the potential of these inhibitors in treating diseases caused by parasites like malaria, Leishmania, and Trypanosoma cruzi. Leishmania secretes several cysteine proteases, including cathepsin L-like proteases, cathepsin B-like proteases, and papain-like proteases, which are involved in nutrient acquisition, host cell invasion, and immune evasion.8–10 The mechanism of action of cysteine proteases involves the deprotonation of a nucleophilic cysteine thiol in the enzyme's active site. Michael acceptors, which interact with these thiol groups through a Michael-type addition (Fig. 1), can inhibit the protease's activity, making them potent covalent inhibitors. This property has been exploited in designing inhibitors for leishmaniasis.11,12
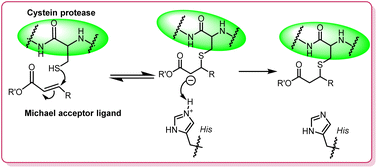 |
| | Fig. 1 Cysteine Protease Inhibitors (CPIs) as chemotherapy for parasitic infections. | |
The versatility and therapeutic potential of β-nitrostyrenes have been demonstrated across a range of applications, from antifungal agents to inhibitors of vital proteases like the SARS-CoV-2 3CL protease.7,13–15 Their effectiveness in disrupting the cellular processes of pathogens positions β-nitrostyrenes as valuable leads in the development of new antimicrobial agents.16,17 Syed Shafi and colleagues created a library of thirty β-nitrostyrenes and evaluated their efficacy against L. donovani, uncovering potent anti-leishmanial activity in both promastigote and amastigote forms. Notably, compound 1a, featuring a methoxy group at the para-position and a methyl substituent at the β-position, demonstrated remarkable anti-leishmanial effects, with IC50 values of 40.50 ± 1.47 nM (promastigotes) and 26.43 ± 2.71 nM (amastigotes). Similarly, compound 1b, distinguished by a hydroxy group at the para-position and an ethyl group at the β-position (Fig. 2), displayed significant activity, with IC50 values of 55.66 ± 2.84 nM (promastigotes) and 61.63 ± 8.02 nM (amastigotes).18 Ze-jun Jia et al. have reported β-nitrostyrenes as inhibitors of SARS-CoV-2 3CL protease, which is a key enzyme in the replication of the COVID-19 virus. The study identified seven β-nitrostyrene derivatives and found that 4-nitro-β-nitrostyrene, 2 showed the lowest IC50 values of 0.7297 μM.19 These findings position these β-nitrostyrenes as promising candidates for further development into anti-leishmanial therapeutics.
 |
| | Fig. 2 β-Nitrostyrenes as therapeutics. | |
Recent studies have further established the significance of peptidyl hybrids as promising cysteine protease inhibitors (Fig. 3). In the realm of anti-leishmanial research, Ahmed H. E. Hassan and his team have made significant contributions with the synthesis of chromone-peptidyl hybrids as potential antileishmanial hits against visceral leishmaniasis. Three hybrids demonstrated notable IC50 values against L. donovani, with values of 9.8, 10, and 12 μM, respectively, rivalling that of erufosine (IC50 = 9.8 μM). Importantly, preliminary cytotoxicity assessments revealed that hybrids 3a and 3b were non-cytotoxic up to 100 μM, unlike erufosine, which exhibited a CC50 value of 19.4 μM. Furthermore, Hassan's group also identified rosmarinic acid-β-amino-α-ketoamide hybrids, with two notable compounds, 3c and 3d, showing promising antileishmanial activity and negligible cytotoxicity, marking them as potential candidates for further development. Additionally, Mayara Castro de Morais and colleagues evaluated the antileishmanial potential of cinnamic acid derivatives against L. infantum. Compounds 3e and 3f stood out, showing strong antileishmanial activity with IC50 values of 33.71 μM and 42.80 μM, respectively, and high selectivity indices, indicating their potential as safer, more targeted treatments compared to traditional drugs like amphotericin B. Compound S9 (3g) displayed excellent leishmanicidal activity20 against L. major promastigotes (IC50 = 37.4 μM) and amastigotes (IC50 = 2.3 μM). Compounds CPB2.8DCTE (3h) exhibited potential anti-leishmanial activity against Leishmania Mexicana cysteine protease (IC50 = 3.7 μM).21 The exploration of peptidyl β-nitrostyrenes and their biological evaluation as cysteine protease inhibitors represents a cutting-edge approach in the ongoing quest for effective treatments against parasitic diseases.
 |
| | Fig. 3 Peptidyl hybrids as anti-leishmanial agents. | |
The rational approach for the designing the new molecules is shown in Fig. 4. The design of the final molecules was inspired by previous studies demonstrating the anti-leishmanial potential of β-nitrostyrenes. Syed Shafi and colleagues synthesized a library of β-nitrostyrenes and identified 4-hydroxy β-nitrostyrene with significant activity against Leishmania donovani.7,18 Building on these findings, we aimed to enhance their therapeutic potential by incorporating the peptidyl motif, which is known to increase selectivity for cysteine proteases. Previous reports have shown that Michael acceptor-type inhibitors, such as peptidyl nitroalkenes, effectively inhibit cysteine proteases like cruzipain and rhodesain with nanomolar potency.22 Additionally, compounds like K11777 and other α,β-unsaturated esters have been successfully used to target viral and parasitic proteases, further validating the use of Michael acceptors for enzyme inhibition.23–27
 |
| | Fig. 4 Rationale design for target molecules. | |
In the present study, we applied the hybrid conjugation of β-nitro styrene with different amino acids with diverse chemical structures allowing detailed characterization of the residue preference of the binding pocket within the substrate-binding site of the protease (Fig. 4).
2. Results and discussion
2.1 Chemistry
The designed peptidyl β-nitrostyrenes were prepared by employing a multistep synthetic strategy. The retrosynthetic analysis of the target molecule results in two important building blocks viz. fragment A, non-peptidyl β-nitrostyrene attached to a carboxylic acid linker and fragment B, a dipeptidyl moiety (Scheme 1).
 |
| | Scheme 1 Retrosynthetic analysis of target molecule. | |
Fragment A (10) was prepared by reacting (E)-4-(2-nitrobut-1-en-1-yl)phenol (9) with bromoacetic acid in the presence of K2CO3. (E)-4-(2-Nitrobut-1-en-1-yl)phenol (9) was obtained by Henry reaction by reacting p-hydroxybenzaldehyde (8) with nitropropane using ammonium acetate as a base (Scheme 2).
 |
| | Scheme 2 Synthesic protocol for acid (fragment A). | |
Boc-protected fragment B was synthesised by coupling various Boc-protected amino acids (12a–k) with morpholine/thiomorpholine/amino acid methyl esters under EDC coupling conditions (Scheme 3). Finally, deprotection of the Boc group resulted in fragment B (13a–k) with the free –NH2 group which was directly used for further coupling reaction, without any purification.
 |
| | Scheme 3 Synthesic protocol for peptides (fragment B). | |
Both the fragments were finally combined by employing EDC coupling to obtain the peptidyl β-nitrostyrenes (14a–k) as shown below in Scheme 4.
 |
| | Scheme 4 Synthesic protocol for peptides (fragment B). | |
By employing the above multistep synthetic strategy, a library of eleven peptidyl β-nitrostyrenes were prepared, as depicted in Fig. 5. The formation of all the synthesized compounds was confirmed by various analytical techniques, including 1H NMR, 13C NMR, Mass and IR.
 |
| | Fig. 5 Synthesized Peptidyl β-nitrostyrenes. | |
The formation of compound 9 was confirmed by the appearance of a characteristic vinyl proton signal at δ 8.0 ppm, ethyl signals at δ 2.88 and 1.28 ppm along with the aromatic signals in 1H NMR. The 1H NMR values for compound 10 are in correlation with the literature values.18 The appearance of two singlets, one corresponding to –COOH proton and the other for -CH2 at δ 13.2 and 4.7 ppm, respectively along with aromatic/aliphatic protons in the 1H NMR confirmed the formation of compound, 10. The formation of peptide fragments 12a–g was confirmed by the appearance of two multiplets between δ 3.73–3.56 and 2.87–2.70 ppm. Further, the formation of dipeptide fragments 12h–k was confirmed by the peaks in the aliphatic region and the –NH peak in 1H NMR spectrum. Finally, the formation of peptidyl β-nitrostyrenes (14a–k) through EDC coupling of 10 and 13(a–k) was confirmed by the missing peak for –COOH proton and the appearance of a new amide peak in the range of δ 3.00–4.00 ppm along with signals belonging to both fragments. The presence of signals at δ 169.51–115.45 ppm and 67.27–12.52 ppm in the 13C NMR further confirms the formation of a new amide bond between both fragments. Finally, the formation of all the peptidyl β-nitrostyrenes, 14a–k was confirmed by mass spectroscopy.
2.2 Pharmacology
2.2.1 Primary screening against L. donovani promastigotes. The in vitro activities were evaluated for all the peptidyl and dipeptidyl β-nitrostyrene derivatives against both L. donovani promastigotes and amastigotes. All the derivatives were first screened against promastigote forms to determine their percentage inhibition at 100 μM concentration. It was observed that nine of the Peptidyl β-nitrostyrenes (14a–i) exhibited a high inhibitory activity at 100 μM concentrations which was better than Amphotericin B (Table 1). The range of % inhibition was (80–85), respectively. Compounds showing 80% and above growth inhibition were screened further to obtain their IC50 values. These IC50 values were obtained using a dose–response curve (Fig. 6 and 9).
Table 1 Primary screening against L. Donovani promastigotes
| S no. |
Test samples |
% inhibition (48 h) |
| 1 |
14a |
84.8 |
| 2 |
14b |
83.7 |
| 3 |
14c |
82.4 |
| 4 |
14d |
80.8 |
| 5 |
14e |
80.4 |
| 6 |
14f |
77.9 |
| 7 |
14g |
78.1 |
| 8 |
14h |
82.2 |
| 9 |
14i |
84.5 |
| 10 |
14j |
65.5 |
| 11 |
14k |
54.8 |
| 12 |
AmphoB |
78.5 |
 |
| | Fig. 6 Primary screening graph. | |
2.2.2 IC50 calculation against L. donovani promastigotes. The IC50 values of the derivatives were obtained using a serial dilution method. Most of the compounds showed significant anti-promastigote activity. All compounds demonstrated comparable anti-promastigote activity as compared to standard with IC50 values ranging between 1.468–16.81 μM as shown below in Table 2 and Fig. 7.
Table 2 IC50 values against L. donovani promastigotes
| Molecules |
IC50 (μM) |
| ● 14a |
11.87 |
| ■ 14b |
16.81 |
| ▲ 14c |
7.236 |
| ▼ 14d |
7.563 |
| ♦ 14e |
1.468 |
| ● 14f |
1.551 |
| ■ 14g |
14.42 |
| ▲ 14h |
10.01 |
| ▼ 14i |
5.298 |
| ♦ AmphoB |
3.073 |
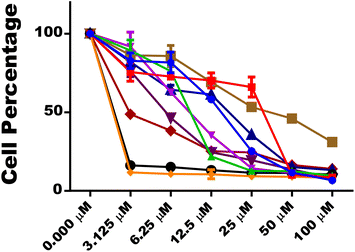 |
| | Fig. 7 IC50 values against L. donovani promastigotes. | |
Compounds 14e and 14f demonstrated better anti-promastigote activity with IC50 values of 1.468 μM and 1.551 μM, respectively, compared to standard (IC50 = 3.073 μM).
2.2.3 Cytotoxicity assay. In vitro cytotoxicity assay was performed with THP-1 human monocytic cells to investigate the cytotoxicity of nine derivatives with greater than 80% inhibition at 100 μM concentrations along with amphotericin B, at varying concentrations. The toxicity assay illustrated that compounds 14a, 14g, and 14h with CC50 are safer than amphotericin B, as shown in Table 3 below.
Table 3 Cytotoxicity values for THP-1 human monocytic cells
| |
Log![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) CC50 CC50 |
CC50 |
|
LogCC50 |
CC50 |
| 14a |
1.458 |
28.68 |
14f |
0.4741 |
2.979 |
| 14b |
0.7734 |
5.934 |
14g |
1.670 |
46.79 |
| 14c |
0.5684 |
3.701 |
14h |
1.109 |
12.86 |
| 14d |
0.5025 |
3.180 |
14i |
0.7120 |
5.153 |
| 14e |
0.3743 |
2.367 |
STD |
1.029 |
10.69 |
Based on determined IC50 values against L. donovani promastigotes and cytotoxicity studies (Fig. 8), it might be stated that derivatives 14a, 14e, 14f and, 14g could act as non-toxic and potent antileishmanial hit compounds for further research.
 |
| | Fig. 8 Cytotoxicity graph against THP-1 human monocytic cells. | |
2.2.4 In vitro antileishmanial activity against intracellular amastigotes. Compounds showing good anti-promastigote activity and lower cytotoxicity, 14a, 14e, 14f, and 14 g were further evaluated for antileishmanial potential in intracellular L. donovani amastigotes in infected macrophages. The percentage reduction in intracellular amastigotes was calculated by counting the macrophages (see Fig. 9) and are tabulated in Table 4. Additionally, the number of amastigotes for every 100 macrophages and the IC50 values of each drug were calculated.
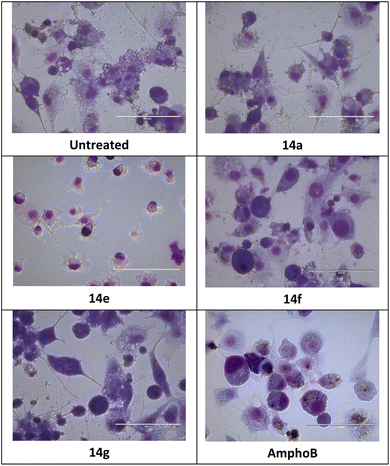 |
| | Fig. 9 Activity against intracellular amastigote. | |
Table 4 Percentage reduction in intracellular amastigotes
| SAMPLE |
Percentage Reductions (%) |
| 3.125 μM |
6.25 μM |
12.5 μM |
IC50 (μM) |
| 14a |
37.2 |
44.1 |
57.5 |
8.07 |
| 14e |
65.4 |
75.1 |
88.5 |
1.28 |
| 14f |
63.2 |
73.4 |
90.3 |
0.64 |
| 14g |
36.7 |
46.8 |
63.2 |
6.37 |
| AmphoB |
52.3 |
63.2 |
85.5 |
3.07 |
The intracellular amastigotes of L. donovani were inhibited by all four compounds, 14a, 14e, 14f and 14g. The inhibitory potential of compounds 14e and 14f with IC50 values of 1.28 μM and 0.64 μM was better than AmphoB with IC50 values of 3.07 μM.
The selectivity data in Table 5 demonstrates that 14a (SI 3.55), 14f (SI 4.65), and 14g (SI 7.34), were equally or more selective for L. donovani amastigotes than the host macrophages as compared to amphoterecin B (SI 3.48).
Table 5 Calculation of selectivity index of 14a, 14e, 14f, 14g and amphoterecin B
| Molecules |
IC50 (μM) |
CC50 (μM) |
SI |
| 14a |
8.07 |
28.68 |
3.55 |
| 14e |
1.28 |
2.367 |
1.85 |
| 14f |
0.64 |
2.979 |
4.65 |
| 14g |
6.37 |
46.79 |
7.34 |
| AmphoB |
3.07 |
10.69 |
3.48 |
2.2.5 Cellular morphological alteration in L.donovani. The cellular morphological changes in Leishmania parasites were revealed through microscopic evaluation of promastigotes treated with 14a, 14e, 14f and 14g. Promastigotes were exposed to the compounds and the standard for 48 hours. Changes were examined using a contrast microscope with 40× objective lens. The promastigotes displayed notable morphological alterations in the parasites, such as cell shrinkage, flagella loss, and cytoplasmic condensation, leading to the complete circularization of promastigotes along with a substantially decreased size as compared to the untreated control. These changes in morphology suggest that the cells experienced apoptosis (Fig. 10), and were consistently observed in the treated promastigotes at both zero and 48 hours. Microscopic assessment of promastigotes treated with compound 14g discloses the cellular morphological changes in Leishmania parasites. Briefly, promastigotes (2 × 106 cells per mL) were incubated in the absence or presence of compound 14g and miltefosine for 48 h at the concentration of 1 μM and observed under a 40× objective lens of contrast microscope. Microscopic observations revealed significant morphological changes in parasites, including cell shrinkage, loss of flagella, and cytoplasmic condensation, resulting in complete circularization of the promastigotes and substantial reduction in size compared to the untreated control. These changes are typical of programmed cell death and were also observed in treated parasites at zero and after 48 hours (Fig. 10).
 |
| | Fig. 10 Analysis of cellular morphology effects of treated promastigotes with compounds. Exponential-phase promastigotes (2 × 106 cells per mL) were incubated with compounds analyzed by light microscopy (200 magnification), as described in Methods. | |
2.2.6 Cysteine protease inhibition assay. Inhibition of cysteine protease (papain) was recorded with an increasing concentration of inhibitor (0–32 μM) using Bz-Arg-AMC hydrochloride as fluorogenic peptide substrate. The activity of papain was recorded for 120 min. As seen in Fig. 11, a decrease in the activity of papain with increasing concentration of inhibitor was observed. Insignificant activity of papain was recorded in the presence of the inhibitor at 32 μM concentration. From the data obtained above, % residual activity was calculated and plotted against the log[inhibitor] to calculate the IC50 value of the inhibitor. Using the equation of the straight line, IC50 for the inhibitor was found and presented in Table 6.
 |
| | Fig. 11 The activity of papain was studied in the absence and presence of an increasing inhibitor concentration. Different concentration of inhibitor is shown by different colors, with dark blue color depicting the activity in the absence of an inhibitor. The excitation wavelength was set at 348 nm and emission was recorded at 440 nm. | |
Table 6 IC50 values against cysteine protease
| Molecules |
14a |
14e |
14f |
14g |
| IC50 (μM) |
9.63 ± 0.28 |
4.44 ± 0.59 |
7.30 ± 0.39 |
11.74 ± 0.48 |
2.2.7 In silico docking studies. In silico docking studies of the most active compounds 14a, 14e, 14f and 14g were conducted against Leishmania mexicana CPB in complex with an aza-nitrile inhibitor using Schrödinger software with PDB ID 6P4E. Docking scores for compounds 14a–i are presented in Table 7.
Table 7 Docking scores of all synthesised compounds (3a–s) in the catalytic binding pocket of cysteine proteases (PDB ID: 6P4E)
| Code |
Docking score [kcal mol−1] |
Code |
Docking score [kcal mol−1] |
| 14a |
−4.195 |
14g |
−4.620 |
| 14b |
−4.463 |
14h |
−6.140 |
| 14c |
−5.300 |
14i |
−5.108 |
| 14d |
−4.452 |
14j |
−5.979 |
| 14e |
−5.480 |
14k |
−6.105 |
| 14f |
−5.515 |
Standard |
−6.007 |
Among all the docked compounds 14e, 14f, 14h, 14j and 14k exhibited good docking scores against the targets (cysteine protease). All docked compounds possess in similar catalytic domain of cysteine protease (Fig. 12 and 13). In the Leishmania mexicana CPB structure (PDB ID 6P4E), both non-covalent hydrophilic and hydrophobic interactions were observed between these compounds and the protein. The compounds demonstrate pi-interactions with TRP 186 and form hydrogen bonds with LEU 162 and GLY 67. Furthermore, they exhibit hydrophobic interactions with LEU 68, MET 69, TRP 27, ALA 140, LEU 162, and TYR 210 amino acid residues. Additionally, they display some polar interactions with SER 65 in chain A, as shown in the 2D & 3D poses in Fig. 12.
 |
| | Fig. 12 (A) and (B) The docking pose of molecule 14e is shown, in PDB ID 6P4E in 2D, and 3D, where it binds with LEU 162 and GLY 67 amino acid residues via hydrogen-bonding as represented by pink line; protein is displayed in ribbon representation (C) and (D) docking pose of 14f is shown, in PDB ID 6P4E in a 2D and 3D structure, where it binds with GLY 67 amino acid residue via hydrogen bonding as represented by the pink line. (E) and (F) docking pose of 14h is shown, in PDB ID 6P4E in a 2D and 3D structure, where it binds with GLY 67 amino acid residue via hydrogen bonding as represented by a pink line (G) The superimposable images of ligand 14h and standard with the catalytic site (H) distance measurement from CYS26 catalytic site (I) superimposition of 14e with E64. | |
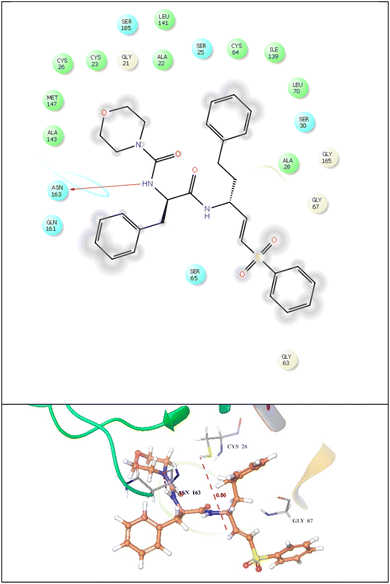 |
| | Fig. 13 2d and 3D Binding interaction of standard ligand (dipeptidyl aza-nitrile) against cysteine protease (PDBID: 6P4E), where it binds with ASN 163 amino acid residue via hydrogen bonding as represented by a pink line. Protein is displayed in ribbon representation. | |
Similarly, the standard exhibited hydrogen bonding with ASN163 amino acid residues in catalytic domain of cysteine protease and all our potent compounds possess similar binding pattern as per standard ligand. It also demonstrated hydrophobic interaction with TRP 27, MET 60, LEU 68, MET 69, ALA 140, LEU 162, TRP 186, and TYR 210 amino acids. Additionally, it displayed some polar interactions with GLN 20, SER 25, SER 65, GLN 71, ASN 163, and HIE 164 in chain A, and the 2D &3D poses are given in Fig. 13. Further, we have superimposed all our potent compounds with standard ligand and found similar binding pattern against catalytic domain of cysteine protease. We have done the distance measurement from CYS26 catalytic site (Fig. 12H) and found that all residues were present within range. After that also compare the known inhibitor E64 with compound 14e (Fig. 12I) and found similar catalytic domain.
2.2.8 Structure activity relationship (SAR). Based on the anti-leishmanial properties of the synthesized peptidyl β-nitrostyrenes; a clear SAR can be drawn as shown in Fig. 14. As illustrated in Fig. 14, there is a clear correlation between the biological activity and the structural modifications at the peptidyl side chain, which appears to be largely favourable across nearly all compounds. The peptide fragment connected to the β-nitrostyrene core is vital for determining both binding affinity and efficacy. Notably, the peptidyl conjugates with heterocyclic moiety (14a–14g) demonstrated better efficacy when compared to the dipeptidyl ester conjugates (14h–14k). Among the peptidyl conjugates (14a–14g), compounds derived from phenyl alanine (14a), tryptophan (14e), and phenylglycine (14g) demonstrated promising antileishmanial activity when compared to the compounds derived from proline (14c and 14d). Further, heterocyclic moiety was also found have greater influence on biological activity. Peptidyl conjugates derived from morpholine (14a, 14e and 14g) were found to demonstrate better activity when compared to the peptidyl conjugates derived from thiomorpholine.
 |
| | Fig. 14 SAR for the synthesized compounds. | |
These varying peptide linkers significantly affect biological activity based on factors like steric hindrance and chain flexibility. For instance, the presence of bulky or flexible linkers, as observed in 14e and 14h, alters the binding geometry, indicating that a more rigid structure could enhance interactions with the target protease's active site.
These modifications collectively emphasize the importance of specific peptide groups in optimizing the biological activity of these peptidyl β-nitrostyrenes. The MD simulations demonstrated that 14e, featuring a combination of tryptophan and morpholine, exhibited remarkable stability, as evidenced by low root mean square fluctuations and consistent hydrogen bond interactions with the protein. This confirms its structural integrity and compatibility within the binding pocket under simulated physiological conditions.
2.2.10 Molecular dynamic simulation. Based on the docking results, molecular dynamic simulation studies were performed for compatibility, stability, conformational strength, and dynamic behaviour of protein–ligand complexes at the scale of atom. The top Glide XP docked compound, having significant non-bonded interactions and binding affinity between the protein and ligand complex, was subjected to MD simulation for a duration of 100 ns using GROMACS 2024.4. The results of the MD simulation are represented as root means square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration, hydrogen bonds, and solvent-accessible surface area (SASA) of protein and ligand complexes.
2.2.11 Root mean square deviation. In this current study, we performed 100 ns of molecular dynamics simulations on the target protein Leishmania mexicana cysteine protease B (LmCPB) system (PDB ID: 6P4E) in a complex with the top-scoring hit compound. The RMSD value for the protein–ligand complexes denotes the high of the protein–ligand complexes system that provides critical insights into the structural dynamics and stability of the system. In the case of MD simulation against LmCPB, the 14e–LmCPB (red trajectory) complex slightly fluctuates, ranging from 0.1–0.3 nm, on the other hand, standard–LmCPB (black trajectory complexes were static and dynamic (0.1–0.2 nm) over the 100 ns simulation period, which provides further insight into the ligand's binding dynamics, as depicted in Fig. 15. Over the 100 ns simulation, compound 14e slightly deviated in initial time from the binding pocket of the LmCPB protein. At the same time, the standard compound showed no deviation from the binding pocket of LmCPB protein systems as represented in Fig. 15. These deviations indicate the ligand's interaction stability within the enzyme's binding pocket throughout the simulation.
 |
| | Fig. 15 MD Simulation LmCPB–ligand complexes root mean square deviation (RMSD) for protein–ligand. | |
2.2.12 Root means square fluctuation. RMSF is a numerical measurement of individual residue flexibility or how much a particular amino acid residue moves (fluctuates) during an MD simulation. RMSF analysis shows a higher fluctuation for standard–LmCPB (black lines) than 14e–LmCPB (red lines) shows the lowest fluctuations over the whole time which ranges from 0–0.55 nm, presented in Fig. 16. The residues indicating the highest fluctuation in the RMSF may be responsible for causing the deviation in the RMSD. This analysis analyzed RMSF for the protein structure complexed with ligands that provided insights into each atom's mean displacement throughout the 100 ns simulation period (Fig. 16).
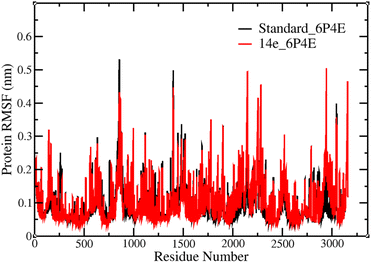 |
| | Fig. 16 MD Simulation LmCPB–ligand complexes root mean square fluctuation (RMSF) for protein. | |
2.2.13 Radius of gyration. The radius of gyration analysis is one of the important calculations for assessing the structural dynamic behaviour of protein, as it reflects changes in the compactness of the protein–ligand complexes throughout the 100 ns simulation. An initial approach to evaluating the stability of the protein is to compute the radius of gyration, a measure of the compactness of the protein structure. When analysing the gyration radius for the whole protein it is evident that the total gyration radius (Rg) does not change significantly over time, implying that the protein is not undergoing major structural changes (Fig. 17).
 |
| | Fig. 17 MD Simulation LmCPB–ligand complexes radius of gyration (Rg) for protein. | |
The radius of gyration for the protein–ligand complexes with 14e and standard were analyzed. The average radius of gyration values was found to be 1.66 ± 0.03 nm for all protein–ligand complexes and can be represented as the arrangement of atoms around the axis and found to be quite stable throughout the 100 ns MD simulation run, depicted in Fig. 17.
2.2.14 Hydrogen bonds. Hydrogen Bonds calculation that the ligand molecules (14e and standard) formed with the target protein as depicted in Fig. 18. The stability and dynamic of the intermolecular hydrogen bonds network between the ligand–protein complexes were analyzed and calculated throughout the 100 ns MD simulation run. The ligand–protein interactions were analyzed to compare the five ligands (Fig. 18) that represent the number of H-bond contacts between the ligands and the protein. It was evident from the plot that we were able to make for compound 14e and standard 1–5 h-bond contacts with target protein LmCPB (Fig. 18).
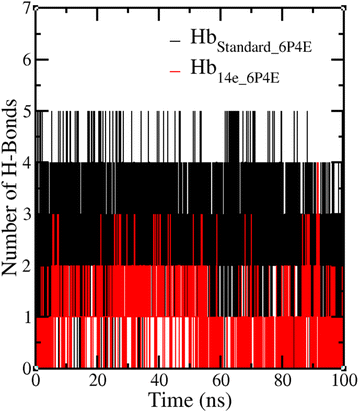 |
| | Fig. 18 MD Simulation LmCPB–ligand complexes: total number of hydrogen bonds for ligand. | |
2.2.15 Solvent accessible surface area. To analyze the structural compactness and solvent accessibility of all complexes (14e–LmCPB and standard–LmCPB) we observe the solvent-accessible surface area (SASA). SASA analysis of protein–ligand complexes during MD simulations showed stable solvent exposure, indicating consistent folding and binding of ligand molecules into the active site of the target protein. It is defined as the surface characterized around a protein by a hypothetical centre of a solvent sphere with the van der Waals contact surface of the molecule. 41e-LmCPB complex depicts the highest value (around 118 nm2) of SASA as compared to standard-LmCPB complex (below 110 nm2) as depicted in Fig. 19. The complexes maintained structural integrity, confirming their stability under simulated physiological conditions (Fig. 19).
 |
| | Fig. 19 MD Simulation LmCPB–ligand complexes: SASA for protein–ligand. | |
3 Conclusion
A library of 12 peptidyl β-nitrostyrenes was synthesized and tested for their efficacy against L. donovani promastigotes and amastigotes. Most of the compounds demonstrated comparable anti-promastigote activity to the standard with IC50 values ranging between 1.468–16.81 μM. Compounds 14a, 14e, 14f, and 14g exhibited significant activity against both L. donovani promastigotes and intracellular amastigotes. Compounds 14e and 14f demonstrated better anti-promastigote activity with IC50 values of 1.468 μM and 1.551 μM respectively as compared to standard (IC50 = 3.073 μM). The inhibitory potential of 14e and 14f against intracellular amastigotes with IC50 values of 1.28 μM and 0.64 μM was better than AmphoB with IC50 value of 3.07 μM. Additionally, insignificant cytotoxicity was seen for compounds 14a and 14g against mammalian macrophages even at a concentration of 28 μM. Given their high activity, safety profiles, and the fact that they can be synthesized cost-effectively, this class of compounds could be potentially valuable for the development of anti-leishmanial drugs.
The use of peptidyl β-nitrostyrenes as inhibitors of cysteine proteases to treat Leishmania donovani represents a new approach compared to current anti-leishmanial therapies like miltefosine and amphotericin B, which target different biological processes. Miltefosine, the first oral drug for leishmaniasis, disrupts cell membrane integrity by affecting lipid metabolism, leading to a type of cell death in the parasite like apoptosis.28,29 While effective, its use is becoming limited due to emerging resistance.29 On the other hand, amphotericin B, which binds to ergosterol in the parasite's membrane, causes cell lysis but has significant nephrotoxicity, making it less practical for widespread use, especially in resource-limited settings.29,30 In contrast, peptidyl β-nitrostyrenes act selectively by inhibiting cysteine proteases that are crucial for the parasite's survival, particularly in intracellular amastigotes. Compounds such as 14e and 14f in this study showed promising efficacy, with IC50 values of 1.28 μM and 0.64 μM against intracellular amastigotes. Additionally, these compounds displayed low toxicity to mammalian macrophages, suggesting a favourable safety profile compared to miltefosine and amphotericin B. Although further optimization is necessary, the selective mechanism and initial efficacy of peptidyl β-nitrostyrenes make them promising candidates for future anti-leishmanial drug development.
4 Materials and methods
4.1 General
All reagents and chemicals used in this research were obtained from GLR, Sigma Aldrich, and Merck (India), and were of AR (analytical reagent) grade quality. The 0.25 mm silica gel 60–120 TLC plates were used to monitor the reactions. UV cabinet was used to visualize spots on the TLC to monitor the reactions. Bruker NMR spectrometer was used for 1H and 13C NMR spectral analysis at 400 MHz and 100 MHz. The spectra were analyzed using Mestrenova software. The HRMS and ESI-MS were obtained on ESI-MS using an Agilent mass spectrometer. The IR spectra were obtained on a Bruker ALPHA FT-IR spectrometer. The Buchi Labortechnik AG 9230 automated melting point apparatus was used for determining the melting points of the derivatives. The identities of the compounds prepared in this study were confirmed through spectral analysis. The compounds were purified through column chromatography using 60–120 and 230–400 mesh sizes of silica gel and recrystallized in ethanol. Additional chemicals used in the in vitro assay including 3-(4,5-dimethylthiazol-2-yl)-2,5-diphenyltetrazolium bromide (MTT) were from BPS Biosciences.
4.2 Chemistry
4.2.1 Typical procedure synthesis of (E)-4-(2-nitrobut-1-en-1-yl)phenol (10). A solution of 4-hydroxybenzaldehyde (1.0 g, 8.20 mmol), 30 eq. of 1-nitropropane (22.0 mL, 250 mmol), and a catalytic amount of sodium acetate was taken in a round-bottom flask and refluxed for 12 hours. After completion of the reaction as confirmed by TLC, the reaction mixture was gradually cooled to ambient temperature, and the volume was reduced using a rotavapor. The reduced reaction mixture was quenched with excess water (30 mL) and extracted with ethyl acetate (3 × 20 mL). The combined organic layers were dried using anhydrous sodium sulfate, filtered, and evaporated to yield the crude product. This crude product was subsequently recrystallised to afford the pure product (1.50 g, 98% yield); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 8.00 (s, 1H), 7.37 (t, J = 8.0 Hz, 2H), 6.91 (t, J = 8.0 Hz, 2H), 5.37 (s, 1H), 2.88 (q, J = 8.0 Hz, 2H), 1.28 (t, J = 8.0 Hz, 3H); HRMS (Q-TOF, ESI) calcd for C9H9N3O [M + H]+ 194.0, found 194.0.
4.2.2 Typical procedure for synthesis of (E)-2-(4-(2-nitrobut-1-en-1-yl)phenoxy)acetic acid (11). To the dry acetone (10 mL) solution of (E)-4-(2-Nitrobut-1-en-1-yl)phenol, 10 (1.0 g, 5.20 mmol), 1.2 eq. of bromoacetic acid (0.86 g, 6.24 mmol) was added. The reaction was then charged with activated K2CO3 (3.60 g, 5 equiv.) in portions. The reaction was refluxed for 11–12 hours. After completion of the reaction as confirmed by TLC, the reaction mixture was gradually cooled to ambient temperature, resulting in the formation of a precipitate. The precipitate obtained was filtered, dissolved in water, and neutralized with 2 M HCl until reaching a pH of 7. The precipitate obtained in the neutralized mixture was filtered off, washed with an excess of water, and subsequently dried under vacuum to obtain the pure off-white solid product (0.12 g, 90% yield); 1H NMR (400 MHz, DMSO-d6) δ (ppm): 13.2 (s, 1H), 8.04 (s, 1H), 7.56 (d, J = 8.0 Hz, 2H), 7.05 (d, J = 12.0 Hz, 2H), 4.77 (s, 2H), 2.83 (q, J = 8.0 Hz, 2H), 1.21 (t, J = 8.0 Hz, 3H); HRMS (Q-TOF, ESI) calcd for C12H13NO5 [M + H]+ 252.0827, found 252.0896.
4.2.3 Typical procedure for the synthesis of (tert-butyl (2-morpholino-2-oxo-1-phenyl ethyl)carbamate) (12a). To the dry DMF solution of 1eq of 2-((tert-butoxycarbonyl)amino)-3-phenylpropanoic acid (0.100 g, 0.40 mmol), 2.2eq of EDC (0.17 g, 0.88 mmol) and a catalytic amount of HOBT (0.10 g) were added. The reaction was stirred for 20 minutes under an inert atmosphere, after which the reaction was charged with 3 eq of morpholine (0.104 g, 1.20 mmol). The reaction was then stirred continuously at ambient temperature for 4–5 h under an inert atmosphere. After completion of the reaction as confirmed by TLC, the reaction mixture was poured into crushed ice, filtered, and then dried under vacuum resulting in a solid product (0.11g, 90% yield); 1H NMR (500 MHz, DMSO-d6) δ (ppm): 7.25–7.16 (m, 6H), 4.50 (q, J = 10.0 Hz, 1H), 3.73–3.56 (m, 4H), 2.87–2.70 (m, 2H), 2.36 (t, J = 10.0 Hz, 1H), 2.14 (d, J = 15.0 Hz, 1H), 1.27 (s, 9H) HRMS (Q-TOF, ESI) calcd for C18H26N2O4 [M + H]+ 335.1926, found 335.1893.
4.2.4 Typical procedure for synthesis of 2-amino-1-morpholino-2-phenylethan-1-one (13a). A solution of tert-butyl (2-morpholino-2-oxo-1-phenylethyl) carbamate (0.10 g, 0.30 mmol) in 20% TFA in chloroform, was stirred at ambient temperature for 4–5 hours. After completion of the reaction as confirmed by TLC, excess TFA was removed by evaporating with hexane as an azeotropic mixture, yielding 2-amino-1-morpholino-2-phenylethan-1-one (14a) in 90% yield. Compound 14a was used in the subsequent step without any further purification.
4.2.5 Typical procedure for synthesis of (E)-N-(2-morpholino-2-oxo-1-phenylethyl)-2-(4-(2-nitrobut-1-en-1-yl)phenoxy) acetamide (14a). To the dry DMF solution of 1eq. of (E)-2-(4-(2-nitrobut-1-en-1-yl)phenoxy)acetic acid, 11 (0.10 g, 0.40 mmol), 2.2 eq. of EDC (0.17 g, 0.88 mmol) and a catalytic amount of HOBT (0.10 g) were added. The reaction was stirred for 20 minutes under an inert atmosphere, after which the reaction was charged with 3 eq of 2-amino-1-morpholino-2-phenylethan-1-one (0.28 g, 1.20 mmol). The reaction was then stirred continuously at ambient temperature for 4–5 h under an inert atmosphere. After completion of the reaction as confirmed by TLC, the reaction mixture was poured into crushed ice, filtered, and then dried under vacuum resulting in a solid product (1.70 g, 90% yield).
5 Characterization data
5.1 (E)-N-(1-Morpholino-1-oxo-3-phenylpropan-2-yl)-2-(4-(2-nitrobut-1-en-1-yl) phenoxy)acetamide (14a)
Light yellowish white solid 91% yield; melting point 119.7–120.8 °C; 1H NMR (400 MHz, CDCl3) δ 8.00 (s, 1H), 7.42 (d, J = 8.0 Hz, 3H), 7.31 (d, J = 8.0 Hz, 3H), 7.20 (d, J = 8.0 Hz, 2H), 7.01 (d, J = 8.0 Hz, 2H), 5.22 (q, J = 8.0 Hz, 1H), 4.54 (s, 2H), 3.63–3.56 (m, 2H), 3.52–3.40 (m, 3H), 3.32–3.27 (m, 1H), 3.10 (d, J = 8.0 Hz, 1H), 3.06 (d, J = 8.0 Hz, 1H), 3.06–2.97 (m, 1H), 2.96–2.85 (m, 3H), 1.28 (t, J = 8.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 169.51, 166.99, 158.55, 152.17, 135.79, 132.74, 131.96, 129.67, 128.87, 127.57, 12.28, 115.45, 67.27, 66.52, 66.11, 49.22, 46.14, 42.43, 40.19, 20.91, 12.52; IR (KBr) ν 3392, 3158, 2933, 2744, 1647, 1609, 1563, 1530, 1511, 1462, 1346, 1285, 1247, 1238, 1131, 1054, 976, 875, 769, 623 cm−1; HRMS (Q-TOF, ESI) calcd for C25H29N3O6 [M + H]+ 468.2056, found 468.2062. Anal. calcd for C25H29N3O6: C, 64.23; H, 6.25; N, 8.99. Found: C, 64.29; H, 6.18; N, 9.04.
5.2 (E)-1-(2-(Morpholine-4-carbonyl)pyrrolidin-1-yl)-2-(4-(2-nitrobut-1-en-1-yl)phenoxy) ethan-1-one (14b)
Light yellowish white solid 81% yield; melting point 108.4–109.1 °C; 1H NMR (400 MHz, CDCl3) δ 7.99 (s, 1H), 7.40 (d, J = 8.0 Hz, 2H), 7.01 (d, J = 8.0 Hz, 2H), 4.91 (dd, J = 4.0, 4.0 Hz, 1H), 4.74 (d, J = 4.0 Hz, 2H), 3.82–3.76 (m, 2H), 3.74–3.60 (m, 6H), 3.57–3.49 (m, 3H), 2.87 (q, J = 8.0 Hz, 2H), 1.27 (t, J = 8.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 170.15, 166.34, 159.66, 151.66, 133.16, 131.98, 129.85, 125.55, 115.41, 115.26, 67.05, 66.92, 56.67, 46.52, 46.28, 42.68, 28.70. 25.70, 20.86, 20.73, 12.47; IR (KBr) ν 3372, 3218, 2927, 2741, 1651, 1609, 1563, 1530, 1511, 1462, 1346, 1285, 1247, 1238, 1131, 1054, 946, 874, 779, 621 cm−1; HRMS (Q-TOF, ESI) calcd for C21H27N3O6 [M + H]+ 484.1861, found 484.1905. Anal. calcd for C21H27N3O6: C, 60.42; H, 6.52; N, 10.07. Found: C, 60.38; H, 6.57; N, 10.11.
5.3 (E)-2-(4-(2-Nitrobut-1-en-1-yl)phenoxy)-N-(1-oxo-3-phenyl-1-thiomorpholinopropan-2-yl)acetamide (14c)
Light yellowish white solid 74% yield; Melting point 86.8–87.9 °C; 1H NMR (400 MHz, CDCl3) δ 8.00 (s, 1H), 7.42 (d, J = 8.0 Hz, 3H), 7.31 (d, J = 8.0 Hz, 3H), 7.18 (d, J = 8.0 Hz, 2H), 7.00 (d, J = 8.0 Hz, 2H), 5.24 (q, J = 8.0 Hz, 1H), 4.52 (s, 2H), 3.83–3.75 (m, 2H), 3.56–3.49 (m, 1H), 3.42–3.37 (m, 1H), 3.04 (d, J = 8.0 Hz, 2H), 2.88 (q, J = 8.0 Hz, 2H), 2.58–2.54 (m, 1H), 2.47–2.38 (m, 2H), 1.90–1.86 (m, 1H), 1.29 (t, J = 8.0 Hz, 3H); 13C NMR (100 MHz, CDCl3) δ 169.56, 166.99, 158.53, 152.18, 135.75, 132.73, 131.96, 129.65, 128.56, 127.55, 126.28, 115.45, 67.24, 49.36, 48.46, 44.97, 40.06, 27.47, 27.34, 20.90, 12.52; IR (KBr) ν 3282, 3148, 2911, 2744, 1647, 1637, 1563, 1530, 1515, 1447, 1346, 1285, 1247, 1238, 1131, 1052, 913, 874, 769, 540 cm−1; HRMS (Q-TOF, ESI) calcd for C25H29N3O5S [M + H]+ 484.1828, found 484.1837. Anal. calcd for C25H29N3O5S: C, 62.09; H, 6.04; N, 8.69; S, 6.63. Found: C, 62.12; H, 5.99; N, 8.73; S, 6.68.
5.4 (E)-2-(4-(2-Nitrobut-1-en-1-yl)phenoxy)-1-(2-(thiomorpholine-4-carbonyl)pyrrolidin-1-yl)ethan-1-one (14d)
Light yellow solid 81% yield; melting point 119.1–120.3 °C; 1H NMR (500 MHz, DMSO) δ 8.06 (s, 1H), 7.56 (d, J = 5.0 Hz, 2H), 7.06 (d, J = 5.0 Hz, 2H), 5.77 (s, 1H), 4.94 (d, J = 15 Hz, 1H), 4.84 (m, 2H), 3.88–3.72 (m, 3H), 3.64–3.57 (m, 3H), 3.47 (d, J = 5.0 Hz, 1H), 2.85 (q, J = 10.0 Hz, 2H), 2.77–2.66 (m, 1H), 2.17–2.13 (m, 1H), 1.98–1.91 (m, 2H), 1.75–1.71 (m, 1H), 1.21 (t, J = 10 Hz, 3H); 13C NMR (100 MHz, DMSO): δ 170.19, 165.52, 160.43, 160.37, 151.12, 133.74, 132.51, 124.66, 115.88, 56.87, 55.40, 45.99, 44.88, 31.52, 28.83, 24.88, 22.04, 20.88, 12.49; IR (KBr) ν 3417, 3391, 3157, 3062, 2931, 2742, 2541, 2211. 1929, 1647, 1609, 1563, 1514, 1462, 1346, 1285, 1238, 1131, 1052, 976, 875, 769 cm−1; HRMS (Q-TOF, ESI) calcd for C21H27N3O5S [M + H]+ 434.1671, found 434.1677. Anal. Calcd for C21H27N3O5S: C, 58.18; H, 6.28; N, 9.69; S, 7.40. Found: C, 58.22; H, 6.26; N, 9.73; S, 7.37.
5.5 (E)-N-(1-(1H-indol-3-yl)-2-morpholino-2-oxoethyl)-2-(4-(2-nitrobut-1-en-1-yl) phenoxy)acetamide (14e)
Light yellow solid 85% yield; melting point 83.1–84.9 °C; 1H NMR (400 MHz, CDCl3) δ 8.14 (s, 1H), 7.99 (s, 1H), 7.66 (d, J = 8.0 Hz, 1H), 7.50 (d, J = 8.0 Hz, 1H), 7.41–7.36 (m, 3H), 7.21 (t, J = 8.0 Hz, 1H), 7.13 (t, J = 8.0 Hz, 1H), 7.07 (s, 1H), 7.00 (d, J = 8.0 Hz, 2H), 5.35 (q, J = 8.0 Hz, 1H), 4.54 (s, 2H), 3.83–3.75 (m, 2H), 3.56–3.49 (m, 1H), 3.42–3.37 (m, 1H), 3.04 (d, J = 8.0 Hz, 2H), 2.88 (q, J = 8.0 Hz, 2H), 2.58–2.54 (m, 1H), 2.47–2.38 (m, 2H), 1.90–1.86 (m, 1H), 1.29 (t, J = 8.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 170.27, 167.05, 158.56, 152.11, 136.18, 132.79, 131.94, 127.54, 126.20, 122.98, 122.67, 120.12, 118.75, 115.46, 111.50, 110.32, 67.29, 48.86, 48.44, 44.86, 30.20, 27.28, 27.21, 20.90, 12.51; IR (KBr) ν 3362, 3178, 2943, 2754, 1637, 1609, 1563, 1516, 1462, 1346, 1284, 1237, 1132, 1054, 975, 871, 769, 591 cm−1; HRMS (Q-TOF, ESI) calcd for C26H28N4O6 [M + H]+ 493.2009, found 493.2012. Anal. calcd for C26H28N4O6: C, 63.40; H, 5.73; N, 11.38. Found: C, 63.45; H, 5.72; N, 11.34.
5.6 (E)-2-(4-(2-nitrobut-1-en-1-yl)phenoxy)-N-(2-oxo-1-phenyl-2-thiomorpholinoethyl) acetamide (14f)
Light yellow solid 82% yield; melting point 107.0–108.2 °C; 1H NMR (400 MHz, CDCl3) δ 8.10 (d, J = 8.0 Hz, 1H), 7.98 (s, 1H), 7.41–7.29 (m, 7H), 6.98 (d, J = 8.0 Hz, 2H), 5.89 (d, J = 8.0 Hz, 1H), 4.51 (q, J = 8.0 Hz, 2H), 4.19–4.15 (m, 1H), 3.73–3.3.60 (m, 3H), 2.91–2.84 (m, 2H), 2.58 (t, J = 8.0 Hz, 2H), 2.32 (dd, J = 4.0, J = 12.0, Hz, 1H), 1.90–1.86 (m, 1H), 1.42–1.35 (m, 1H). 13C NMR (100 MHz, CDCl3) δ 167.69, 166.67, 158.58, 152.02, 136.92, 132.79, 131.88, 129.45, 128.87, 128.01, 126.09, 115.45, 67.29, 53.86, 48.17, 45.26, 29.79, 27.23, 26.92, 20.86, 12.47; IR (KBr) ν 3312, 3154, 2923, 2735, 1800, 1644, 1603, 1583, 1530, 1511, 1440, 1346, 1285, 1247, 1238, 1179, 1054, 976, 873, 779, 613 cm−1; HRMS (Q-TOF, ESI) calcd for C24H27N3O5S [M + H]+ 470.1671, found 470.1678. Anal. Calcd for C24H27N3O5S: C, 61.39; H, 5.80; N, 8.95; S, 6.83. found: C, 61.37; H, 5.83; N, 9.01; S, 6.78.
5.7 (E)-N-(2-morpholino-2-oxo-1-phenylethyl)-2-(4-(2-nitrobut-1-en-1-yl)phenoxy) acetamide (14g)
Light yellow solid 79% yield; melting point 128.5–129.4 °C; 1H NMR (400 MHz, CDCl3) δ 8.08 (d, J = 8.0 Hz, 1H), 7.98 (s, 1H), 7.41–7.31 (m, 7H), 6.98 (d, J = 8.0 Hz, 2H), 5.90 (d, J = 8.0 Hz, 1H), 4.51 (q, J = 8.0 Hz, 2H), 3.78–3.62 (m, 2H), 3.60–3.51 (m, 3H), 3.50–3.42 (m, 1H), 3.29–3.26 (m, 1H), 3.14–3.10 (m, 1H), 2.86 (q, J = 8.0 Hz, 2H), 1.27 (t, J = 4.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 167.81, 166.70, 158.61, 152.06, 136.94, 132.80, 131.90, 129.40, 128.86, 127.95, 126.14, 115.48, 115.16, 67.34, 66.64, 66.07, 53.64, 45.95, 42.86, 20.88, 12.43; IR (KBr) ν 3484.27, 3460.18, 3424.85, 3385.04, 3249.24, 3322.91, 3209.25, 3173.12, 3124.24, 3039.54, 3007.66, 2965.80, 2932.01, 2867.52, 1777.73, 1750.78, 1677.18, 1652.31, 1602.42, 1501.41, 1471.97, 1437.08, 1365.68, 1321.19, 1248.71, 1180.87, 1108.57, 1063.88, 1032.59, 990.68, 945.59, 912.54, 836.85, 811.03, 764.45, 708.25, 662.07, 586.35, 529.62, 484.69, 464.20, 422.40 cm-1; HRMS (Q-TOF, ESI) calcd for C24H27N3O6 [M + H]+ 454.1900, found 454.1906. Anal. Calcd for C24H27N3O6: C, 63.56; H, 6.00; N, 9.27. found: C, 63.53; H, 5.98; N, 9.32.
5.8 Methyl (E)-(2-(1H-indol-3-yl)-2-(2-(4-(2-nitrobut-1-en-1-yl)phenoxy)acetamido)acetyl)valinate (14h)
Off white solid 89% yield; melting point 128.5–129.4 °C; 1H NMR (400 MHz, CDCl3) δ 8.11 (s, 1H), 7.98 (s, 1H), 7.76 (d, J = 8.0 Hz, 1H), 7.37 (d, J = 8.0 Hz, 4H), 7.21 (t, J = 8.0 Hz, 1H), 7.13 (t, J = 8.0 Hz, 2H), 6.90 (d, J = 8.0 Hz, 2H), 6.11 (d, J = 8.0 Hz, 1H), 4.82 (q, J = 8.0 Hz, 1H), 4.56 (s, 2H), 4.39–4.36 (m, 1H), 3.68 (s, 3H), 3.41–3.36 (m, 1H), 3.22–3.16 (m, 1H), 2.87 (q, J = 8.0 Hz, 2H), 2.05–2.00 (m, 1H), 1.29 (t, J = 4.0 Hz, 3H), 0.79 (d, J = 8.0 Hz, 3H), 0.74 (d, J = 8.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 167.81, 166.70, 158.61, 152.06, 136.94, 132.80, 131.90, 129.40, 128.86, 127.95, 126.14, 115.48, 115.16, 67.34, 66.64, 66.07, 53.64, 45.95, 42.86, 20.88, 12.43; IR (KBr) ν 3322, 3148, 2922, 2742, 1645, 1603, 1563, 1516, 1440, 1346, 1285, 1247, 1238, 1180, 1154, 1034, 946, 809, 701, 530 cm−1; HRMS (Q-TOF, ESI) calcd for C29H34N4O7 [M + H]+ 551.2427, found 551.2434. Anal. Calcd for C29H34N4O7: C, 63.26; H, 6.22; N, 10.18. Found: C, 63.23; H, 6.26; N, 10.15.
5.9 Methyl (E)-(2-(4-(2-nitrobut-1-en-1-yl)phenoxy)acetyl)phenylalanylglycinate (14i)
Light yellow white solid 85% yield; melting point 122.5–124.7 °C; 1H NMR (400 MHz, CDCl3) δ 7.99 (s, 1H), 7.40 (d, J = 8.0 Hz, 2H), 7.34 (d, J = 8.0 Hz, 2H), 7.31–7.23 (m, 3H), 7.19 (t, J = 8.0 Hz, 2H), 7.13 (d, J = 8.0 Hz, 1H), 6.94 (d, J = 8.0 Hz, 2H), 4.79 (d, J = 8.0 Hz, 1H), 4.58–4.37 (m, 2H), 4.04–3.89 (m, 2H), 3.73 (s, 2H), 3.16–3.05 (m, 2H), 2.87 (q, J = 8.0 Hz, 2H), 1.28 (t, J = 8 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 170.95, 169.95, 168.04, 158.43, 152.04, 135.99, 132.63, 131.82, 129.26, 128.63, 127.10, 115.29, 115.25, 67.08, 53.75, 52.40, 41.16, 38.21, 20.74, 12.32. IR (KBr) ν 3475, 3440, 3330, 3170, 3134, 3081, 2924, 2853, 1777, 1750, 1646, 1460, 1402, 1337, 1285, 944, 838, 811, 476 cm−1; HRMS (Q-TOF, ESI) calcd for C24H27N3O7 [M + H]+ 470.1849, found 470.1889. Anal. Calcd for C24H27N3O7: C, 61.40; H, 5.80; N, 8.95. Found: C, 61.41; H, 5.77; N, 8.98.
5.10 Methyl (E)-(2-(4-(2-nitrobut-1-en-1-yl)phenoxy)acetyl)tryptophylalaninate (14j)
Light yellow white solid 85% yield; melting point 108.5–109.4 °C; 1H NMR (400 MHz, CDCl3) δ 8.01 (s, 2H), 7.47 (q, J = 4.0 Hz, 2H), 7.42 (d, J = 8.0 Hz, 2H), 7.37 (d, J = 8.0 Hz, 1H), 7.28 (d, J = 4.0 Hz, 2H), 7.23 (d, J = 8.0 Hz, 2H), 6.97 (d, J = 8.0 Hz, 2H), 4.98 (s, 1H), 4.79 (q, J = 8.0 Hz, 1H), 4.54 (s, 1H), 4.45 (q, J = 4.0 Hz, 1H), 3.73 (s, 3H), 3.20–3.08 (m, 2H), 2.12 (q, J = 8.0 Hz, 2H), 1.30 (m, 3H), 0.87 (d, J = 8.0 Hz, 3H). 13C NMR (100 MHz, CDCl3) δ 171.64, 170.28, 167.72, 158.36, 152.13, 135.95, 132.53, 131.84, 131.01, 129.29, 128.78, 127.21, 126.23, 121.94, 116.22, 115.27, 67.17, 57.51, 54.17, 38.18, 31.11, 20.77, 12.39. IR (KBr) ν 3397, 3293, 3220, 3199, 3174, 2975, 2924, 2853, 1779, 1742, 1649, 1602, 1401, 1364, 1177, 1059, 1025, 944, 837, 811, 742, 699, 475, 423 cm−1; HRMS (Q-TOF, ESI) calcd for C27H30N4O7 [M + H]+ 523.2114, found 523.2148. Anal. Calcd for C27H30N4O7: C, 62.06; H, 5.79; N, 10.72. found: C, 62.10; H, 5.82; N, 10.68.
5.11 Methyl (E)-(2-(4-(2-nitrobut-1-en-1-yl)phenoxy)acetyl)phenylalanylalaninate (14k)
Light yellow white solid 85% yield; melting point 118.2–120.4 °C; 1H NMR (400 MHz, CDCl3) δ 8.35 (s, 1H), 7.99 (s, 2H), 7.69 (d, J = 8 Hz, 1H), 7.55 (d, J = 4 Hz, 1H), 7.20 (t, J = 8 Hz, 1H), 7.12 (d, J = 12 Hz, 2H), 7.00 (d, J = 8.0 Hz, 2H), 6.89 (d, J = 8.0 Hz, 2H), 4.85 (q, J = 8.0 Hz, 1H), 4.70 (s, 2H), 4.46 (t, J = 8 Hz, 1H), 3.69 (s, 2H), 3.38 (m, 1H), 3.24 (m, 1H), 2.88 (q, J = 8 Hz, 2H), 1.34–1.24 (m, 6H). 13C NMR (100 MHz, CDCl3) δ 172.77, 170.69, 168.15, 159.12, 151.82, 136.28, 133.49, 132.80, 131.79, 123.59, 122.32, 119.88, 118.63, 116.25, 115.18, 111.37, 77.39, 77.07, 76.75, 67.04, 52.55, 48.42, 28.34, 20.77, 18.05, 12.31. IR (KBr) ν 3328, 3277, 3249, 3198, 2979, 2441, 1776, 1744, 1651, 1602, 1510, 1457, 1402, 1365, 1181, 1063, 1026, 945, 837, 810, 739, 505, 470, 423 cm−1; HRMS (Q-TOF, ESI) calcd for C25H29N3O7 [M + H]+ 483.2006, found 483.2039. Anal. Calcd for C25H29N3O7: C, 62.10; H, 6.05; N, 8.69. found: C, 62.08; H, 6.01; N, 8.63.
5.12 Pharmacology
5.12.1 Leishmania parasite culture and maintenance. The parasite used was Leishmania donovani, strain (MHOM/SD/62/1S (Ahuja et al. 2015). The promastigote stage of the parasite was regularly grown at 26 °C in medium M199 with 10% heat-inactivated fetal bovine serum (FBS), streptomycin (100 μg mL−1), and penicillin 100 (IU ml−1). The promastigotes in log phase were sub-cultured after every 72 hours using a 2 × 106 cells per mL inoculum (Ahuja et al. 2015).
5.13 Cell line culture
THP-1 human macrophages were cultured in RPMI based medium at 37 °C in presence of 5% CO2 for 48 to 72 hours in RPMI-1640 medium (pH 7.4) with 10% heat-inactivated FBS in a 5% CO2 humidified atmosphere (Avishek et al. 2024). Afterwards, an average density of 2 × 105 cells per mL of the cells were transferred to new RPMI-1640 medium.
5.14 MTT assay for anti-promastigote activity
The anti-leishmanial activity of peptidyl β-nitrostyrenes (14a–k) was performed using MTT assay (Sharma et al. 2023). This assay is based on the mitochondrial enzymes NAD and NADH-dehydrogenases forming formazan crystals from the tetrazolium soluble salt. 10000 THP-1 human monocytic cells were seeded at per well in a 96-well plate. The cells were exposed to the synthesized molecules at varying concentrations starting from 100, 50, 25, 12.5, and 6.25 μM, for 24 hours after they reached normal morphology. The 96-well plate was then kept in an incubator with humidified CO2 at 37 °C. Following this, the supernatant was discarded, followed by addition of 0.5 mg mL−1 of MTT solution. It was then incubated for 4 hours at 37 °C in a humidified environment. The media was taken out, followed by the addition of DMSO to dissolve the formazan crystals, and an i3x spectramax molecular device was used to record the absorbance at 570 nm. Compounds showing greater than 50% inhibition were selected for cytotoxicity assay (IC50).31
5.15 Anti-leishmanial activity against L. donovani promastigotes
The synthesized molecules were tested for their in vitro activity against L. donovani promastigotes. A concentration of 2 × 106 cells per mL of L. donovani promastigotes were cultured in RPMI medium for 72 hours at 26 °C with 100 μM concentration of each compound. Amphoterecin B (AmphoB) was used as a reference, and DMSO (0.2%) was the solvent. Parasites in media alone were the control experiment. The viability of parasites was measured using the MTT assay after 72 hours.32
5.16 Determination of IC50 from dose-dependent anti-promastigote activity
The promastigotes of L. donovani were incubated using 2 × 106 cells per mL in the presence and absence of the most active compounds (14a–i) of the series at serial dilutions starting at 25 μM for 72 hours at 26 °C, using AmphoB was as a standard anti-leishmanial drug control. Cell viability was measured using by MTT assay, and the following formula was used to calculate the mean percentage viability: (Mean cell number of treated promastigotes/Mean cell number of untreated promastigotes) × 100. The IC50 values were determined by extrapolation of graph in the graph of % viability vs. concentration of the drug.33
5.17 Cytotoxicity assay
THP-1 human monocytic cells were cultured with 10% FBS in RPMI media, and 1 × 106 cells per mL concentration of cells were seeded. The plate was kept in a humidified CO2 incubator for 24 h at 37 °C. After 24 h, media was removed, and varying concentrations of standard and compounds were added like 100, 50, 25, 12.5, and 6.25 μM for 72 h, before performing the MTT assay.34
5.18 Intracellular amastigotes assay
Macrophages of L.donovani were cultivated on eight-chamber tissue culture slides with 5 × 104 cells in each well and were allowed to adhere in a CO2 incubator containing 5% CO2 for 2 h at 37 °C. RPMI 1640 medium without serum was used to wash the wells twice to eliminate the nonadherent macrophages. The adherent macrophages were subsequently exposed to the metacyclic stage of Leishmania donovani, with a Leishmania to macrophage ratio of 10:1, which was sustained overnight in a 200 μL final solution of complete RPMI 1640 medium. Following a 24 hours incubation period, RPMI 1640 medium with no serum was used to wash free promastigotes. Infected macrophages were then incubated with compounds in duplicate for 72 hours at 37 °C with 5% CO2, except the control well. The number of amastigotes in 100 macrophages were counted at each concentration to evaluate IC50 values of test compounds. Infected macrophage percentages were determined by counting the infected macrophages per 100 macrophages.35
5.19 Calculation of percentage of inhibition and ED50
Using a Neubauer chamber, the extracellular parasites present in treated wells as well as control wells were counted after 24 hours of treatment. For every duplicate well, a microscopic enumeration was conducted at various concentrations. Dead parasites with deteriorated body surfaces were not counted. All counts at varying concentrations were converted to percentages by using the control well mean, which was taken to represent 100% survival. Using Zeiss, AXIO, and Imager A1 light microscope, the slides of the infected macrophage cell line which were stained with Giemsa, were examined, and the infected macrophages/amastigotes per 100 cells were counted.36
5.20 Statistical analysis
IC50 values against L. donovani promastigotes and amastigotes were determined from the triplicate's experiments for the mean and standard error of the mean (SEM). To analyze the differences among groups GraphPad Prism 8.0 software was used. Using the two-way ANOVA, the correlation between drug and their effect was determined. P < 0.05 was regarded as being statistically important for difference and correlation.37
5.21 In Vitro cysteine protease inhibition assay
Inhibition of papain (Roche cat #10108014001) was studied using an increasing concentration of the derivatives. Nα-Benzoyl-L-arginine-7-amido-4-methyl coumarin hydrochloride (Bz-Arg-AMC) from Sigma (cat #B7260), a fluorogenic peptide substrate was used for the papain activity assay. The inhibitors were dissolved in DMSO and an activation buffer containing sodium acetate buffer (pH 5.5), 8 mM DTT, and 4 mM EDTA was used. The reaction mixture (100 μl) also included papain (4 mM), increasing concentration of inhibitor (0–32 μM) and 10 μM substrate. The enzymatic hydrolysis of substrate by papain was monitored using SpectraMax Multi-Mode Microplate Readers with an emission wavelength of 440 nm and an excitation wavelength of 345 nm. The IC50 value for the inhibitor was calculated from the plot of the % residual activity against the log[inhibitor], as described previously.38
5.22 Molecular docking
5.22.1 Protein preparation. The protein structure of cysteine proteases (PDB ID: 6P4E) with a resolution of 1.16 Å was obtained from the Protein Data Bank (PDB) (RCSB). The best resolution values, R-free and R-work, are 0.130 and 0.110, respectively, and the number of resolved residues was considered. The ligand was initially analyzed by Schrödinger's protein preparation wizard panel (Schrödinger). Without undergoing any additional modification to the protein in monomeric form, the ligand was retained and crystallized water molecules were removed. When preparing proteins, the OPLS3 2015 force field was utilised to minimise the amount of protein energy. Following protein preparation, the receptor grid generation tool was employed and a grid surrounding the ligand was created while utilising all of the Glide tool's standard parameters.39,40
5.22.2 Ligand preparation. All the synthesized compounds were drawn in the workspace in Maestro 2D along with the standard. Ligand preparation was initiated after entering all entities. The prepared ligands and receptor grid were docked using the standard procedure of Glide XP docking. The docking scores and poses are presented after the completion of the process.
5.22.3 Ligand docking. For molecular docking or Glide XP docking, the protein was prepared, the grid was generated, and the ligands were prepared. The standard protocol of Glide (Schrödinger's) was employed with a partial charge cut-off of 0.15 and a scaling factor of 0.8 for nonpolar atom's van der Waal radii were employed. Each ligand produced ten poses, and residues within 5 Å of the grid center had per-residue interaction scores calculated.
5.22.4 ADMET analysis. All final compounds were subjected to ADMET analysis using QikProp module of maestro. It provides ranges for comparing the properties of a particular molecule with those of 95% of known drugs. The descriptors calculated were partition coefficient, human oral absorption, Lipinski rule of five etc.
5.22.5 MD simulations. Molecular dynamics simulation is a computational technique employed to investigate the dynamic behavior of molecular systems complex of protein and ligand over time. It provides an analysis of the physical movements of atoms and molecules. The atoms and molecules are allowed to interact for a fixed period, giving a view of the dynamic evolution of the system (protein–ligand complex) at the atomic or molecular level. The top-scoring protein–ligand complexes (Glide docked), with high binding free energies (ΔG Bind), were submitted to a 100 ns MD simulation using the GROMACS suite 2024.4, a widely used MD simulation package to obtain root mean square deviation (RMSD), root mean square fluctuation (RMSF), radius of gyration (Rg), hydrogen bonds, and SASA for the evaluation of dynamic behavior and stability of the protein–ligand complexes. The topology of the protein was generated by GROMACS 2024.4 using the CHARMM27 force field, and the topology and parameters of the ligands were generated from the website SwissParam (https://swissparam.ch/). The systems were solvated using the TIP3P water model and the ions were added to neutralize the system and to achieve the desired ionic strength, using Na+, Cl− at the concentration 0.10 M NaCl. The prepared system was further subjected to energy minimization to remove any steric clashes and ensure a stable starting configuration. Additionally, an index file was generated, and the.mdp file was modified to define the thermal coupling group and adjust position restraints. Minimized systems subjected to equilibration phases (NVT and NPT), including heating to the desired temperature (300 K) and pressure (1.01325 bar), were used to stabilize the system for the final production MD simulations run. Long-range MD simulations were conducted under periodic boundary conditions. The post-MD simulation trajectories were analysed and visualized using the different tools of GROMACS, and VMD, and data plots were generated using Grace.
Data availability
The data supporting this article have been included as part of the ESI.†
Conflicts of interest
The authors declare no conflict of interest.
Acknowledgements
We would like to show our gratitude to SERB (ECR/2017/001067/CS) for financial support and Jamia Hamdard for providing the laboratory facilities.
References
- E. Torres-Guerrero, M. Romano Quintanilla-Cedillo, J. Ruiz-Esmenjaud, R. Arenas, F. Bravo-Puccio, P. Cayetano Heredia and R. J. Hay, F1000Research, 2017, 6, 750 Search PubMed.
- P. Desjeux, Comp. Immunol., Microbiol. Infect. Dis., 2004, 27, 305–318 CrossRef CAS PubMed.
- A. Selvapandiyan, S. L. Croft, S. Rijal, H. L. Nakhasi and N. K. Ganguly, PLoS Neglected Trop. Dis., 2019, 13, e0007616 CrossRef PubMed.
- P. Bhattacharya, R. Dey, P. K. Dagur, A. B. Joshi, N. Ismail, S. Gannavaram, A. Debrabant, A. D. Akue, M. A. KuKuruga, A. Selvapandiyan, J. P. McCoy and H. L. Nakhasi, PLoS Neglected Trop. Dis., 2016, 10, e0004963 CrossRef PubMed.
- M. M. S. Andrade, L. C. Martins, G. V. L. Marques, C. A. Silva, G. Faria, S. Caldas, J. S. C. Dos Santos, V. G. Maltarollo, R. S. Ferreira and R. B. Oliveira, Future Med. Chem., 2020, 12, 571–581 CrossRef CAS PubMed.
- A. Latorre, T. Schirmeister, J. Kesselring, S. Jung, P. Johé, U. A. Hellmich, A. Heilos, B. Engels, R. L. Krauth-Siegel, N. Dirdjaja, L. Bou-Iserte, S. Rodríguez and F. V. González, ACS Med. Chem. Lett., 2016, 7, 1073–1076 CrossRef CAS PubMed.
- S. Sharma, P. A. Yakkala, J. Aboti, I. Latief, M. A. Ansari, W. H. Khan and S. Shafi, ChemistrySelect, 2023, 8, e202300912 CrossRef CAS.
- P. J. Rosenthal, G. K. Lee and R. E. Smith, J. Clin. Invest., 1993, 91, 1052–1056 CrossRef CAS PubMed.
- J. H. McKerrow, Int. J. Parasitol., 1999, 29, 833–837 CrossRef CAS PubMed.
- H. Mahmoudzadeh-Niknam and J. H. McKerrow, Exp. Parasitol., 2004, 106, 158–163 CrossRef CAS PubMed.
- S. T. Liang, C. Chen, R. X. Chen, R. Li, W. L. Chen, G. H. Jiang and L. L. Du, Front. Pharmacol, 2022, 13, 1033003 CrossRef CAS PubMed.
- S. Sharma, P. Anjaneyulu Yakkala, M. A. Beg, S. Tanwar, I. Latief, A. Khan, K. Sharma, S. ur Rehman, A. Selvapandiyan and S. Shafi, ChemistrySelect, 2023, 8, e202302415 CrossRef CAS.
- Evaluation of nitrostyrenes as antifungal agents. I. In vitro studies - PubMed, https://pubmed.ncbi.nlm.nih.gov/24544016/, (accessed 24 March 2024).
- Antifungal activity of beta-nitrostyrenes and some cyclohexane derivatives - PubMed, https://pubmed.ncbi.nlm.nih.gov/24543964/, (accessed 24 March 2024).
- A. Ramzan, S. A. Padder, K. Z. Masoodi, S. Shafi, I. Tahir, R. U. Rehman, R. Prasad and A. H. Shah, Eur. J. Med. Chem., 2022, 240, 114609 CrossRef CAS PubMed.
- Y. Mikami, K. Yazawa, A. Maeda, J. Uno, A. Kubo, N. Saito and N. Kawakami, J. Antibiot., 1991, 44, 1454–1456 CrossRef CAS PubMed.
- N. Milhazes, R. Calheiros, M. P. M. Marques, J. Garrido, M. N. D. S. Cordeiro, C. Rodrigues, S. Quinteira, C. Novais, L. Peixe and F. Borges, Bioorg. Med. Chem., 2006, 14, 4078–4088 CrossRef CAS PubMed.
- S. Shafi, F. Afrin, M. Islamuddin, G. Chouhan, I. Ali, F. Naaz, K. Sharma and M. S. Zaman, Front. Microbiol., 2016, 7, 195169 Search PubMed.
- Z. jun Jia, X. wei Lan, K. Lu, X. Meng, W. jie Jing, S. ru Jia, K. Zhao and Y. jie Dai, J. Mol. Struct., 2023, 1284, 135409 CrossRef PubMed.
- C. Schad, U. Baum, B. Frank, U. Dietzel, F. Mattern, C. Gomes, A. Ponte-Sucre, H. Moll, U. Schurigt and T. Schirmeister, Antimicrob. Agents Chemother., 2015, 60, 797–805 CrossRef PubMed.
- A. Scala, N. Micale, A. Piperno, A. Rescifina, T. Schirmeister, J. Kesselring and G. Grassi, RSC Adv., 2016, 6, 30628–30635 RSC.
- A. Latorre, T. Schirmeister, J. Kesselring, S. Jung, P. Johé, U. A. Hellmich, A. Heilos, B. Engels, R. L. Krauth-Siegel, N. Dirdjaja, L. Bou-Iserte, S. Rodríguez and F. V. González, ACS Med. Chem. Lett., 2016, 7, 1073–1076 CrossRef CAS PubMed.
- Y. Zhou, P. Vedantham, K. Lu, J. Agudelo, R. Carrion, J. W. Nunneley, D. Barnard, S. Pöhlmann, J. H. McKerrow, A. R. Renslo and G. Simmons, Antiviral Res., 2015, 116, 76–84 CrossRef CAS PubMed.
- J. J. Shie, J. M. Fang, T. H. Kuo, C. J. Kuo, P. H. Liang, H. J. Huang, Y. T. Wu, J. T. Jan, Y. S. E. Cheng and C. H. Wong, Bioorg. Med. Chem., 2005, 13, 5240–5252 CrossRef CAS PubMed.
- A. Citarella, A. Scala, A. Piperno and N. Micale, Biomolecules, 2021, 11, 607 CrossRef CAS PubMed.
- K. Akaji, H. Konno, H. Mitsui, K. Teruya, Y. Shimamoto, Y. Hattori, T. Ozaki, M. Kusunoki and A. Sanjoh, J. Med. Chem., 2011, 54, 7962–7973 CrossRef CAS PubMed.
- S. Previti, R. Ettari, E. Calcaterra, S. Di Maro, S. J. Hammerschmidt, C. Müller, J. Ziebuhr, T. Schirmeister, S. Cosconati and M. Zappalà, Eur. J. Med. Chem., 2023, 247, 115021 CrossRef CAS PubMed.
- T. P. C. Dorlo, M. Balasegaram, J. H. Beijnen and P. J. de vries, J. Antimicrob. Chemother., 2012, 67, 2576–2597 CrossRef CAS PubMed.
- The treatment of visceral leishmaniasis: safety and efficacy - PubMed, https://pubmed.ncbi.nlm.nih.gov/25327244/, (accessed 17 October 2024).
- J. D. Herman, Rev. Infect. Dis., 1988, 10, 560–586 CrossRef PubMed.
- P. A. Yakkala, S. R. Panda, S. Shafi, V. G. M. Naidu, M. S. Yar, P. N. Ubanako, S. A. Adeyemi, P. Kumar, Y. E. Choonara, E. v. Radchenko, V. A. Palyulin and A. Kamal, Molecules, 2022, 27, 7642 CrossRef CAS PubMed.
- O. Gupta, T. Pradhan, R. Bhatia and V. Monga, Eur. J. Med. Chem., 2021, 223, 113606 CrossRef CAS PubMed.
- G. Chouhan, M. Islamuddin, M. Y. Want, M. Z. Abdin, H. A. Ozbak, H. A. Hemeg, D. Sahal and F. Afrin, Parasites Vectors, 2015, 8, 183 CrossRef PubMed.
- R. N. Duffin, V. L. Blair, L. Kedzierski and P. C. Andrews, J. Inorg. Biochem., 2020, 203, 110932 CrossRef CAS PubMed.
- K. Vats, R. Tandon, Roshanara, M. A. Beg, R. M. Corrales, A. Yagoubat, E. Reyaz, T. H. Wani, M. S. Baig, A. Chaudhury, A. Krishnan, N. Puri, P. Salotra, Y. Sterkers and A. Selvapandiyan, Biochim. Biophys. Acta, Mol. Cell Res., 2023, 1870, 119416 CrossRef CAS PubMed.
- K. Das Manandhar, T. P. Yadav, V. K. Prajapati, S. Kumar, M. Rai, A. Dube, O. N. Srivastava and S. Sundar, J. Antimicrob. Chemother., 2008, 62, 376–380 CrossRef CAS PubMed.
- V. V. Andrade-Neto, K. M. Rebello, T. M. Pereira and E. C. Torres-Santos, Antimicrob. Agents Chemother., 2021, 65, 1–11 CrossRef PubMed.
- E. Pitsillou, J. Liang, K. Ververis, A. Hung and T. C. Karagiannis, J. Mol. Graphics Modell., 2021, 104, 107851 CrossRef CAS PubMed.
- H. Krishna, P. Anchi, K. Lakshmi, J. Prakash, C. Godugu, N. Shankaraiah and A. Kamal, Bioorg. Chem., 2021, 117, 105461 CrossRef PubMed.
- P. A. Yakkala, S. R. Panda, S. Shafi, V. G. M. Naidu, M. S. Yar, P. N. Ubanako, S. A. Adeyemi, P. Kumar, Y. E. Choonara, E. V. Radchenko, V. A. Palyulin and A. Kamal, Molecules, 2022, 27, 7642 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental procedure and characterization of new compounds (1H and 13C NMR spectra).See DOI: https://doi.org/10.1039/d4ra06510g |
|
| This journal is © The Royal Society of Chemistry 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence e,
Sayeed ur Rehman*d,
Angamuthu Selvapandiyan*b and
Syed Shafi
e,
Sayeed ur Rehman*d,
Angamuthu Selvapandiyan*b and
Syed Shafi





















