
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Kinetic resolution of trifluoromethylated heterobenzhydrols via hydrogen-acceptor-free Ir-catalyzed heteroaryl-selective C–H silylation†
Ryu
Tadano
,
Takeshi
Yasui
 and
Yoshihiko
Yamamoto
*
and
Yoshihiko
Yamamoto
*
Department of Basic Medicinal Sciences, Graduate School of Pharmaceutical Sciences, Nagoya University, Chikusa, Nagoya 464-8601, Japan. E-mail: yamamoto.yoshihiko.y9@f.mail.nagoya-u.ac.jp
First published on 11th June 2025
Abstract
Kinetic resolution of benzhydrols via intramolecular C–H silylation is an efficient method for the preparation of chiral benzhydrols. However, the previously reported methods required sterically demanding phenyl rings to achieve group-selective C–H silylation. Herein, we report the kinetic resolution of trifluoromethylated heterobenzhydrols, bearing both phenyl and thiophene rings, via heteroaryl-selective C–H silylation. We conducted computational studies on the factors influencing the enantioselectivity and heteroaryl selectivity.
Introduction
Fluorine-containing compounds have significant applications in pharmaceuticals1 and agrochemicals.2 In particular, the CF3 group is the most common polyfluoroalkyl substituent in newly approved drugs.3 However, most trifluoromethylated pharmaceuticals are benzotrifluoride derivatives. Although numerous chiral pharmaceuticals with the CF3 group on an sp2 carbon atom are commercially available, only a few drugs, namely efavirenz, telotristat ethyl, and suzetrigine, have the CF3 group at the stereogenic carbon center (Fig. 1A).1 Given the numerous reports on racemic bioactive compounds with the CF3 group on a sp3 carbon center chiral trifluoromethylated compounds have high potential value as pharmaceuticals. Notably, trifluoromethylated aryl heteroaryl carbinols (heterobenzhydrols) are significant because they are found in bioactive compounds such as selective androgen receptor modulators (SARMs) and liver X receptor (LXR) agonists (Fig. 1A),4 leading to increasing demand for their enantioselective synthesis. | ||
| Fig. 1 Background of this study. (A) Bioactive molecules bearing the CF3 group on an sp3 carbon. (B) Enantioselective desymmetrization of trifluoromethylated benzhydrols. (C) Kinetic resolution of trifluoromethylated benzhydrols. (D) Kinetic resolution of trifluoromethylated heterobenzhydrols. | ||
Chiral trifluoromethylated benzhydrols are usually synthesized via enantioselective nucleophilic addition reactions.5 However, this method has several limitations. First, organometallic nucleophiles are unstable and have low functional group tolerance. Second, enantioselective nucleophilic addition often requires stoichiometric amounts of chiral ligands. Therefore, we focused on enantioselective desymmetrization via C–H silylation as a novel synthetic approach. Hartwig, Shi, and their coworkers developed enantioselective desymmetrization of benzhydrols using Rh- or Ir-catalyzed intramolecular C–H silylation: however, they did not investigate tertiary alcohol substrates.6 Inspired by their research, we developed enantioselective desymmetrization of trifluoromethylated tertiary benzhydrols, which is the first example of the synthesis of trifluoromethylated benzhydrols via enantioselective desymmetrization (Fig. 1B).7 Notably, no hydrogen acceptor was required for our method, in contrast to the use of norbornene as the hydrogen acceptor that was essential in previous methods. A general limitation of enantioselective desymmetrization is that unsymmetrical benzhydrol substrates with different aryl groups cannot be used. In addition, heterobenzhydrols have not yet been used as substrates for enantioselective desymmetrizations. Another approach for the synthesis of chiral trifluoromethylated benzhydrols is the kinetic resolution of unsymmetrical benzhydrols bearing different aryl groups. Kinetic resolution requires a group-selective reaction involving one of the two aryl groups. Hartwig, Shi, and their coworkers achieved kinetic resolution of unsymmetrical secondary benzhydrols via C–H silylation by introducing a substituent at the ortho position of one phenyl ring to reduce its reactivity by steric replusion.6b Similarly, we succeeded in the kinetic resolution of unsymmetrical tertiary trifluoromethylated benzhydrols bearing a sterically demanding phenyl rings substituted at the 2- or 3,5-positions (Fig. 1C).7 However, group selectivity driven by steric repulsion imposes significant constraints on substrate design owing to the requirement for sterically demanding phenyl groups.
Therefore, we focused on the kinetic resolution of heterobenzhydrols. Previous attempts to achieve enantioselective desymmetrization of bis(1-methyl-3-indolyl)carbinol were unsuccessful. The inferior reactivity of the indole ring in our method suggests that unsymmetrical benzhydrols bearing both phenyl and inactive heteroaryl groups enable group-selective reactions without the introduction of the sterically demanding phenyl group. In this study, we investigated the kinetic resolution of heterobenzhydrols bearing phenyl groups and five-membered heteroaromatic rings (Fig. 1D). Contrary to our expectations, the heteroaromatic rings underwent C–H silylation preferentially over the phenyl groups.
Results and discussion
Reaction development
Among the representative five-membered heteroaromatic compounds, thiophene was selected because of its “chemical proximity” to benzene.8 Trifluoromethylated phenyl thienyl carbinol 1a was prepared via the reaction of trifluoroacetophenone derived from ethyl trifluoroacetate with thienyllithium and subsequent silylation of the hydroxy group with chlorodimethylsilane afforded hydrosilane 2a (see ESI†). The prepared 2a was subjected to C–H silylation under conditions optimized in our previous study; however, to our surprise, thienooxasilole 3a was exclusively obtained instead of the expected benzoxasilole 3a′ (Fig. 2A).9,10 The reaction of the previously employed benzhydrol silyl ethers took 5 h to complete,7 whereas 2a reacted completely within 2 h. This fact suggests that thiophene has a higher reactivity than benzene toward C–H silylation. When the dimethylsilyl group was replaced by a diethylsilyl group, we observed a significantly lower reaction rate even at a higher temperature (80 °C), and partial desilylation was also detected (Fig. S1, ESI†). Based on these results, we shifted our focus toward developing a heteroaryl-selective C–H silylation. We investigated the kinetic resolution of 2a by stopping the reaction at approximately half conversion (Fig. 2B). However, partial desilylation of 2a to 1a occurred during purification by silica gel column chromatography, and the separation of 1a from 3a was difficult. We envisaged that modifying the dimethylsilyl group of unreacted 2a to a trialkylsilyl group would enhance kinetic stability. Therefore, we confirmed that the Ir-catalyzed hydrosilylation of 2a with 4-vinylanisole afforded 4a, completely suppressing its desilylation (Table S1, ESI†).11 Next, we investigated the effect of chiral ligands on the enantioselectivity. Because 3a and 4a were unsuitable for chiral HPLC analysis, we evaluated their enantiopurity after desilylation. Previous studies showed that chiral pyridine-oxazoline (PyOX) ligands that contain indane-fused oxazoline and 6-substituted pyridine rings achieved high enantioselectivity in C–H silylation.6b,7 Therefore, we used the chiral ligands L1–L4 for the kinetic resolution of 2a. PyOX ligands L1 and L2, bearing the ethyl and methyl groups, respectively, on the pyridine ring, afforded nearly identical results, with L2 showing slightly higher enantioselectivity. The use of the previously reported ligand (L4)6b led to slight decrease in enantioselectivity. Generally, 3a was obtained with higher enantiopurity than 4a, independent of the conversion of 2a. In contrast, L3, which bears a bulkier isopropyl group, showed a marked decrease in enantioselectivity. Therefore, L2 was selected as the optimal ligand. We also applied the kinetic resolution conditions developed by Hartwig and Shi to 2a (Fig. 2C).6b In the presence of norbornene as the H2 acceptor, 2a was treated with 2 mol% [Ir(cod)OMe]2 and 4.5 mol% PyOX ligand L4 at 35 °C for 12 h, affording 3a in 49% yield (19F NMR), albeit with several unidentified byproducts being observed; however, substantial amounts of 1a was formed by desilylation of 2a, hampering the separation of 3a. Therefore, we used our acceptor-less conditions for further investigations. | ||
| Fig. 2 Reaction development. (A) Ir-catalyzed intramolecular dehydrogenative silylation of 2a at full conversion. (B) Kinetic resolution of 2a. Enantiomeric ratio (er) was determined by chiral HPLC after desilylation of the products. Selectivity values (s) were determined according to s = ln[1 − c(1 + eep)]/ln[1 − c(1 − eep)] (c: conversion based on product yields, eep: product enantiomeric excess). (C) Reaction of 2a under conditions previously reported (ref. 6b). | ||
Next, we investigated the substrate scope under optimal conditions (Fig. 3). Hydrosilanes 2b–i with substituents on the phenyl ring underwent thiophene-selective C–H silylation, affording 3b–3i with 19![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :81–10:90 er. However, o-tolyl-substituted 3d decomposed during isolation. We assumed that the decomposition of 3d was due to the strain caused by the 2-methyl substituent. Both electron-donating (OMe and SMe) and -withdrawing (Cl and CF3) groups were compatible. The reaction of 2j with a 2-naphthyl group afforded 3j in 32% yield with 13:87 er. Similarly, 5-methylthienyl-substituted 2k and 5-bromothienyl-substituted 2l afforded 3k and 3l, albeit with a slight erosion of er for the latter. Substrates 2m and 2n, bearing CF2H and CF2CO2Et groups at the benzylic position, respectively, underwent thiophene-selective silylation to afford 3m and 3n; however, the er of 3m was lower, indicating that the smaller CF2H group decreases enantioselectivity. Notably, both 3n and unreacted 2n, bearing bulky CF2CO2Et groups, were isolated using column chromatography with diol silica gel, eliminating the need for the hydrosilylation of 2n. The C–H silylation of 2o, which bears a furan ring, proceeded smoothly. However, furooxasilole 3o was unstable and decomposed rapidly upon exposure to air (for details, see ESI†). Therefore, the enantioselectivity was analyzed for 4o, which exhibited a high er of 93:7. The reaction of 2p bearing a N-tosylated 2-pyrrolyl ring also proceeded with a similar heterocycle selectivity. Although 3p was very unstable to afford a complex product mixture after purification, unreacted 2p was recovered without its hydrosilylation. The enantioselectivity of 2p was analyzed after desilylation, which exhibited a much lower er of 64:36. In contrast, the reaction of benzothienyl-substituted 2q was significantly slow at 40 °C, and the reaction at elevated temperatures yielded complex product mixtures.
:81–10:90 er. However, o-tolyl-substituted 3d decomposed during isolation. We assumed that the decomposition of 3d was due to the strain caused by the 2-methyl substituent. Both electron-donating (OMe and SMe) and -withdrawing (Cl and CF3) groups were compatible. The reaction of 2j with a 2-naphthyl group afforded 3j in 32% yield with 13:87 er. Similarly, 5-methylthienyl-substituted 2k and 5-bromothienyl-substituted 2l afforded 3k and 3l, albeit with a slight erosion of er for the latter. Substrates 2m and 2n, bearing CF2H and CF2CO2Et groups at the benzylic position, respectively, underwent thiophene-selective silylation to afford 3m and 3n; however, the er of 3m was lower, indicating that the smaller CF2H group decreases enantioselectivity. Notably, both 3n and unreacted 2n, bearing bulky CF2CO2Et groups, were isolated using column chromatography with diol silica gel, eliminating the need for the hydrosilylation of 2n. The C–H silylation of 2o, which bears a furan ring, proceeded smoothly. However, furooxasilole 3o was unstable and decomposed rapidly upon exposure to air (for details, see ESI†). Therefore, the enantioselectivity was analyzed for 4o, which exhibited a high er of 93:7. The reaction of 2p bearing a N-tosylated 2-pyrrolyl ring also proceeded with a similar heterocycle selectivity. Although 3p was very unstable to afford a complex product mixture after purification, unreacted 2p was recovered without its hydrosilylation. The enantioselectivity of 2p was analyzed after desilylation, which exhibited a much lower er of 64:36. In contrast, the reaction of benzothienyl-substituted 2q was significantly slow at 40 °C, and the reaction at elevated temperatures yielded complex product mixtures.
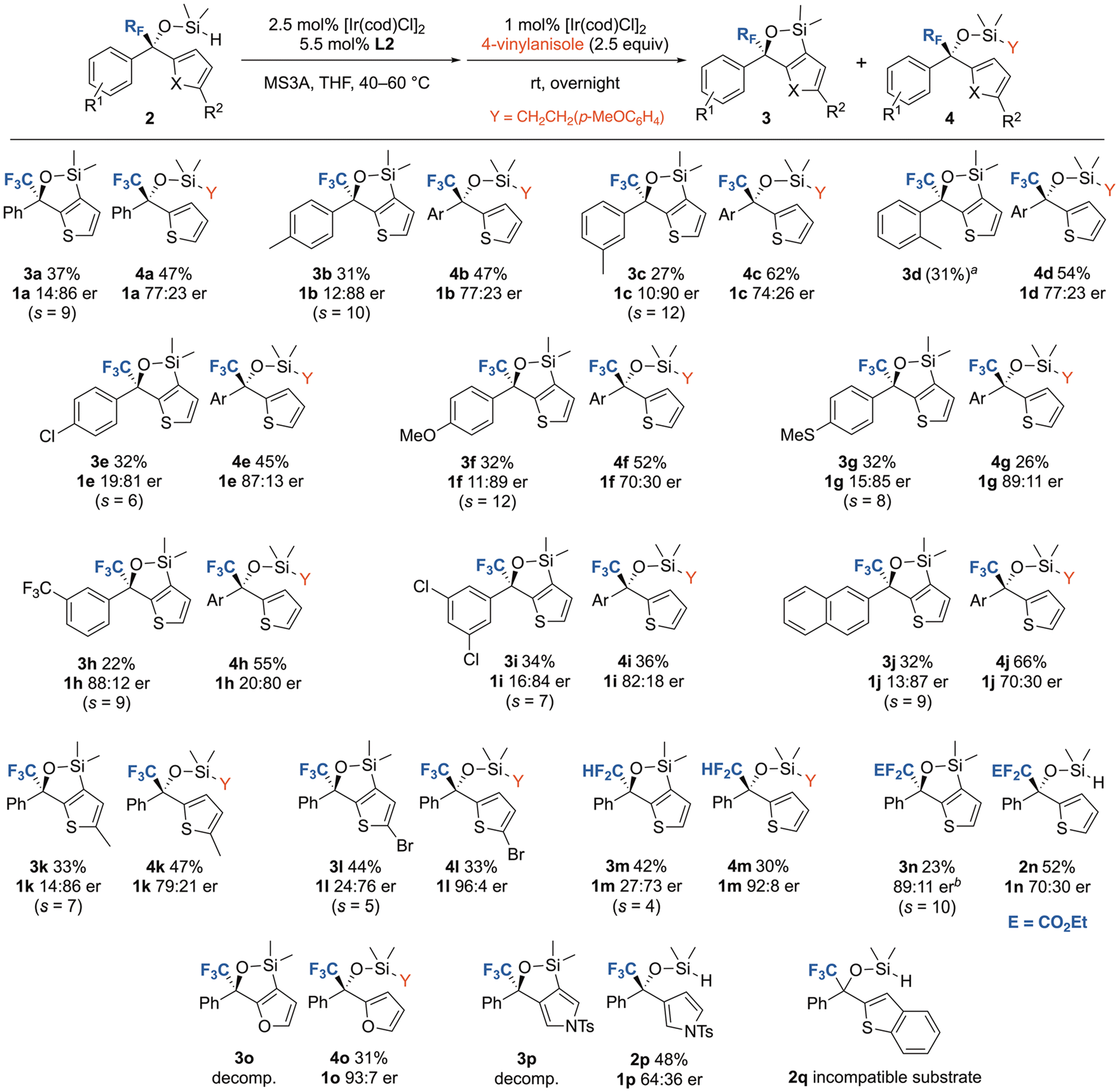 | ||
| Fig. 3 Scope of trifluoromethylated heterobenzhydrol substrates. The er values were determined after desilylation using chiral HPLC. The absolute configurations were assigned by analogy (for details, see ESI†).aYield determined by 19F NMR. bThe er value was determined without desilylation using chiral HPLC. | ||
To explore whether the aryl group, which does not participate in the reaction, can be replaced with alkyl groups, n-butyl-substituted 2r was subjected to C–H silylation (Fig. 4A). However, 3r was obtained with low er (44:56). Because it was hypothesized that the high flexibility of the n-butyl group of 2r led to the decrease in er, heterobenzhydrol derivative 2s, bearing a more constrained c-hexyl group, was examined; however, 3s was obtained with a similarly low er. Moreover, heterobenzhydrol 2t and 2u bearing an ethyl or methyl group instead of the CF3 group was investigated to examine the role of the CF3 group (Fig. 4B). The reaction of 2t with the ethyl group, which has a comparable van der Waals volume with the CF3 group,12 under standard reaction conditions produced the corresponding products 3t and 4t. Although 4t was isolated in 21% yield, thienooxasilole 3t is unstable and decomposed during silica gel chromatography. The enantiomeric ratio of 4t was evaluated after desilylation as 86:14. Similarly, the reaction of 2u with the smaller methyl group afforded 4u in 20% yield, and its enantiomeric ratio evaluated after desilylation was lower (64:36) than that of 4t. Therefore, the CF3 group is beneficial for the stability of thienooxasilole products as well as enantioselectivity; however, it is not essential for the Ir-catalyzed heteroaryl-selective dehydrogenative silylation. This result is in striking contrast to our previous observation that the CF3 group is necessary for efficient reactions in the enantioselective desymmetrization of benzhydrols.7
 | ||
| Fig. 4 Kinetic resolution of other substrates. The yields refer to isolated products, and the er values were determined after desilylation using chiral HPLC. (A) Kinetic resolution of trifluoromethylated thienyl carbinols. (B) Kinetic resolution of heterobenzhydrols without a CF3 group. (C) Kinetic resolution of a bisheteroarylmethanol. | ||
Because the benzothienyl group in 2q did not undergo C–H silylation (Fig. 3), the kinetic resolution of silane 2v, bearing both thienyl and benzothienyl groups, was subjected to the standard Ir-catalyzed C–H silylation conditions. Although the C–H bond on the thienyl group was selectively activated, the reaction was sluggish and partial desilylation of 2v occurred during the prolonged reaction. Therefore, the reaction of 2v was conducted with an increased catalyst loading for 2.5 h (Fig. 4c). Because the separation of thienooxasilole 3v from 1v was impossible, the treatment of an inseparable mixture (3v/1v 91:9) with TBAF afforded 1v with 21:79 er, albeit in a low yield (16%). In contrast, 4v was isolated in 61% yield and its enantiomeric ratio was determined as 60:40, after desilylation.
The kinetic resolution of 2a was performed at a 3 mmol scale to obtain 3a in 60% yield, which was further transformed into deuterated heterobenzhydrol 1a-d in 85% yield with 22:78 er (Fig. 5). Upon treatment of 3a with NBS in the presence of AgF, brominated heterobenzhydrol 5 was also quantitively obtained with 22:78 er.
 | ||
| Fig. 5 Scale-up synthesis of 3a and its transformations. | ||
Computational mechanistic study
In our previous study, we proposed a reaction mechanism for the C–H silylation of trifluoromethylated benzhydrols without a hydrogen acceptor.7 A similar mechanism is expected for heterobenzhydrols; however, C–H activation occurs preferably on the thiophene ring rather than the phenyl ring (Fig. 6). Initially, the in situ-generated Ir(III) dihydride species I13 undergoes the thienyl–H oxidative addition, leading to Ir(V) trihydride species II.14 This is the rate- and enantio-determining step. The subsequent reductive elimination of H2 generates Ir(III) monohydride species III.14,15 The H2 generation in the catalytic reaction was monitored by 1H NMR spectroscopy in our previous study.7 Although the C–H oxidative addition and subsequent reductive elimination of H2 are endergonic and reversible, facile extrusion of H2 gas from the reaction solutions renders the overall reaction irreversible.16 Finally, the Si–H oxidative addition of another substrate is followed by the reductive elimination of thienooxasilole from resulting IV to restore I. | ||
| Fig. 6 Proposed catalytic cycle. | ||
To gain insights into the enantio-determining step, we performed density functional theory (DFT) calculations at the SMD (THF) M06/SDD-6-311+G(d,p)//B3LYP-D3(BJ)/LanL2DZ-6-31G(d) level of theory (for details, see ESI†) on the C–H oxidative addition and subsequent reductive elimination of H2 (Fig. 7A). Ir(III) active species I has a square pyramidal geometry with a silyl ligand at the apical position (Fig. 6).13,17 We assumed that C–H silylation occurs from the same face with the indane moiety of the ligand because the π–π interactions between the indane moiety and thienyl ring stabilize the transition state for the C–H activation steps (vide infra). In fact, C–H silylation from the opposite face with the indane moiety was found to be less efficient (Fig. S2, ESI†). Ir(III) species with the silyl group at the apical position were located as Int-1(S) and Int-1(R), and Int-1(R) is less stable than Int-1(S) by 0.9 kcal mol−1. To provide an empty coordination site for C–H oxidative addition, the geometric isomerization of Int-1(S) and Int-1(R) leads to Int-2(S) and Int-2(R), in which the silyl ligand is located at the basal position. Int-2 is approximately 8 kcal mol−1 less stable than Int-1 owing to the strong trans influence of the silyl group.18 The transition states for the C–H oxidative addition of the thiophene ring were identified as TS-3(S) and TS-3(R), with the activation barriers of 10.5 and 12.7 kcal mol−1 from precursor complex Int-2(S) and Int-2(R), respectively. The subsequent reductive elimination of H2 is almost barrierless (Fig. S3, ESI†).
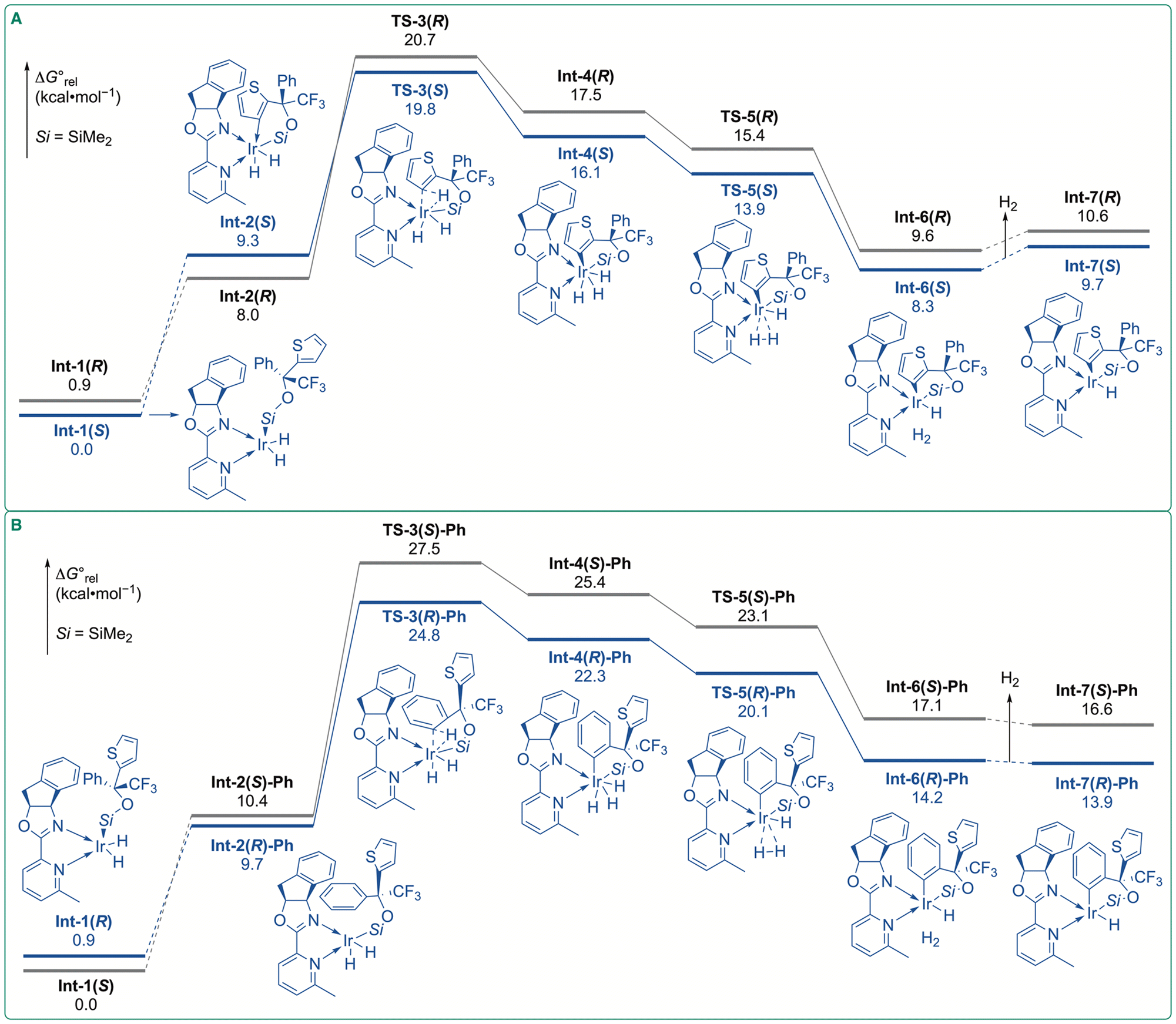 | ||
| Fig. 7 Gibbs energy profiles: [SMD:THF] M06/SDD-6-311+G(d,p)//B3LYP-D3(BJ)/LanL2DZ-6-31G(d) at 298.15 K and 1 atm. (A) Profiles of thienyl–H activation. (B) Profile of Ph–H activation. Structures of stationary points and transition states for the major pathways are shown. | ||
Because the oxidative addition of the silane substrate is facile and reversible,6bInt-1(S) and Int-1(R) are in equilibrium through Ir(V) trihydride species (Fig. S4, ESI†). Because the activation barriers are less than 17 kcal mol−1, this interconversion is more facile than the subsequent thienyl C–H activation. The energetic span between Int-1(S) and TS-3(S) (19.8 kcal mol−1) is lower than that between Int-1(S) and TS-3(R) (20.7 kcal mol−1). Consequently, the (S)-enantiomer was predicted to be formed preferentially, which was in good agreement with the absolute configuration of the experimentally obtained products. The centroid–centroid distance between the indane and thiophene rings in TS-3(S) is shorter than that in TS-3(R) (Fig. 8A). Therefore, the π–π interactions between the indane and thiophene rings are expected to be stronger in TS-3(S) than in TS-3(R). Non-covalent interaction (NCI) analysis19 confirmed that the π–π interactions are more favorable in TS-3(S) (Fig. S5, ESI†).
 | ||
| Fig. 8 Optimized geometries. (A) TS-3(S) and TS-3(R). (B) Thiophene, benzene, Int-4(S), and Int-4(R)-Ph. | ||
Calculations were also performed for the C–H activation of the phenyl ring to compare the reactivity of thiophene and benzene (Fig. 7B). The transition states for Ph–H oxidative addition were identified as TS-3(S)-Ph and TS-3(R)-Ph, with activation barriers of 17.1 and 15.1 kcal mol−1 from precursor complexes Int-2(S)-Ph and Int-2(R)-Ph, respectively. The energetic differences between Int-1(S) and TS-3(S)-Ph (27.5 kcal mol−1) and between Int-1(S) and TS-3(R)-Ph (24.8 kcal mol−1) are higher than those for TS-3(S) and TS-3(R). Thus, C–H activation on the thienyl ring is predicted to be favored compared to that on the phenyl ring, consistent with the experimental results.
Our previous DFT study of the C–H silylation of benzhydrol-derived hydrosilane suggested that the final reductive elimination of the benzoxasilole product occurred via the oxidative addition of the hydrosilane substrate.7 A similar pathway was also found for the reductive elimination of the thienooxasilole product; the oxidative addition of the hydrosilane substrate to Int-7(S) generates Ir(V) metallacyclic intermediate Int-9(S,R) or Int-9(S,S), which undergoes the facile reductive elimination of thienooxasilole with the regeneration of the active catalyst (Fig. S6, ESI†). These results support the proposed Ir(III)/Ir(V) catalytic cycle as outlined in Fig. 6. The reductive elimination of the thienooxasilole product from Int-9(R,R) or Int-9(R,S) also proceeds similarly; however, it is less efficient than that from Int-9(S,R)/Int-9(S,S) as the energetic span between Int-7 and TS-10 is higher for (R)-isomers than for (S)-isomers.
To understand why the activation barrier for the C–H oxidative addition of thiophene is lower than that of benzene, we investigated iridacycle intermediates Int-4 formed from C–H oxidative addition. The relative Gibbs energy 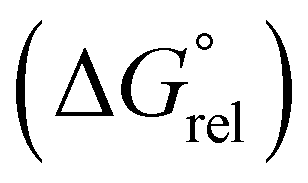 of thienoiridacycle Int-4(S) is 16.1 kcal mol−1, whereas the
of thienoiridacycle Int-4(S) is 16.1 kcal mol−1, whereas the  of benzoiridacycle Int-4(R)-Ph is 22.3 kcal mol−1, indicating that benzoiridacycle Int-4(R)-Ph is less stable by 6 kcal mol−1 than Int-4(S). The C(1)–C(2)–H angle of thiophene is 123.3°, while the corresponding C(1)–C(2)–Ir angle in Int-4(S) is 127.5°, leading to a bond-angle strain of 4.2° (Fig. 8B). In contrast, the C(1)–C(2)–H angle of benzene is 120.0°, while the corresponding C(1)–C(2)–Ir angle in Int-4(R)-Ph is 128.7°, causing a bond-angle strain of 8.7°. Owing to the smaller ring strain of the thienoiridacycle than that of benzoiridacycle, the activation barrier of thienyl–H oxidative addition is reduced.20
of benzoiridacycle Int-4(R)-Ph is 22.3 kcal mol−1, indicating that benzoiridacycle Int-4(R)-Ph is less stable by 6 kcal mol−1 than Int-4(S). The C(1)–C(2)–H angle of thiophene is 123.3°, while the corresponding C(1)–C(2)–Ir angle in Int-4(S) is 127.5°, leading to a bond-angle strain of 4.2° (Fig. 8B). In contrast, the C(1)–C(2)–H angle of benzene is 120.0°, while the corresponding C(1)–C(2)–Ir angle in Int-4(R)-Ph is 128.7°, causing a bond-angle strain of 8.7°. Owing to the smaller ring strain of the thienoiridacycle than that of benzoiridacycle, the activation barrier of thienyl–H oxidative addition is reduced.20
Conclusions
We achieved kinetic resolution of unsymmetrical trifluoromethylated heterobenzhydrols via Ir-catalyzed dehydrogenative C–H silylation. This approach is particularly useful for the synthesis of both enantiomers of heterobenzhydrols bearing the CF3 and thienyl groups at the stereogenic center. DFT calculations revealed that the π–π interactions between the thiophene ring and ligand indenyl moiety play a crucial role in determining the enantioselectivity. Additionally, the smaller ring strain of the six-membered thienoiridacycle intermediate compared to that of the benzoiridacycle intermediate is the cause of the heteroaryl-selective C–H silylation.Author contributions
Y. Y. conceived the project and wrote the manuscript. R. T. carried out the experimental and computational works, analyzed the experimental results, and wrote the manuscript. T. Y. discussed the results.Conflicts of interest
There are no conflicts to declare.Data availability
The data supporting this article have been included as part of the ESI.†Acknowledgements
This research is partially supported by the Platform Project for Supporting Drug Discovery and Life Science Research (Basis for Supporting Innovative Drug Discovery and Life Science Research (BINDS) from AMED under Grant Number JP25ama121044) and JSPS KAKENHI (Grant Number JP22K05110). Computations were performed using the Research Center for Computational Science, Okazaki, Japan (Project: 24-IMS-C119 and 25-IMS-C120). R. T. acknowledges the Inter-disciplinary Frontier Next-Generation Researcher Program of the Tokai Higher Education and Research System.References
- (a) M. Inoue, Y. Sumii and N. Shibata, Contribution of Organofluorine Compounds to Pharmaceuticals, ACS Omega, 2020, 5, 10633 CrossRef CAS PubMed; (b) J. D. Osteen, S. Immani, T. L. Tapley, T. Indersmitten, N. W. Hurst, T. Healey, K. Aergeerts, P. A. Negulescu and S. M. Lechner, Pharmacology and Mechanism of Action of Suzetrigine, a Poetent and Selective NaV1.8 Pain Signal Inhibitor for the Treatment of Moderate to Severe Pain, Pain Ther., 2025, 14, 655 CrossRef PubMed.
- Y. Ogawa, E. Tokunaga, O. Kobayashi, K. Hirai and N. Shibata, Current Contributions of Organofluorine Compounds to the Agrochemical Industry, iScience, 2020, 23, 101467 CrossRef CAS PubMed.
- M. Bassetto, S. Ferla and F. Pertusati, Polyfluorinated groups in medicinal chemistry, Future Med. Chem., 2015, 7, 527 CrossRef CAS PubMed.
- Selected examples: (a) A. L. Handlon, L. T. Schaller, L. M. Leesnitzer, R. V. Merrihew, C. Poole, J. C. Ulrich, J. W. Wilson, R. Cadilla and P. Turnbull, Optimizing Ligand Efficiency of Selective Androgen Receptor Modulators (SARMs), ACS Med. Chem. Lett., 2016, 7, 83 CrossRef CAS PubMed; (b) D. J. Kopecky, X. Y. Jiao, B. Fisher, S. McKendry, M. Labelle, D. E. Piper, P. Coward, A. K. Shiau, P. Escaron, J. Danao, A. Chai, J. Jaen and F. Kayser, Discovery of a New Binding Mode for a Series of Liver X Receptor Agonists, Bioorg. Med. Chem. Lett., 2012, 22, 2407 CrossRef CAS PubMed; (c) X. Jiao, D. J. Kopecky, B. Fisher, D. E. Piper, M. Labelle, S. McKendry, M. Harrison, S. Jones, J. Jaen, A. K. Shiau, P. Escaron, J. Danao, A. Chai, P. Coward and F. Kayser, Discovery and Optimization of a Series of Liver X Receptor Antagonists, Bioorg. Med. Chem. Lett., 2012, 22, 5966 CrossRef CAS PubMed.
- Selected reviews: (a) J.-A. Ma and D. Cahard, Asymmetric Fluorination, Trifluoromethylation, and Perfluoroalkylation Reactions, Chem. Rev., 2004, 104, 6119 CrossRef CAS PubMed; J.-A. Ma and D. Cahard, Asymmetric Fluorination, Trifluoromethylation, and Perfluoroalkylation Reactions, Chem. Rev., 2008, 108, PR1 Search PubMed; (b) Y.-Y. Huang, X. Yang, Z. Chen, F. Verpoort and N. Shibata, Catalytic Asymmetric Synthesis of Enantioenriched Heterocycles Bearing a C–CF3 Stereogenic Center, Chem. – Eur. J., 2015, 21, 8664 CrossRef CAS PubMed; (c) X.-H. He, Y.-L. Ji, C. Peng and B. Han, Organocatalytic Asymmetric Synthesis of Cyclic Compounds Bearing a Trifluoromethylated Stereogenic Center: Recent Developments, Adv. Synth. Catal., 2019, 361, 1923 CrossRef CAS; (d) K. Hirano, Catalytic Asymmetric Construction of CF3-Substituted Chiral sp3 Carbon Centers, Synthesis, 2022, 3708 CrossRef CAS.
- (a) T. Lee, T. W. Wilson, R. Berg, P. Ryberg and J. F. Hartwig, Rhodium-Catalyzed Enantioselective Silylation of Arene C–H Bonds: Desymmetrization of Diarylmethanols, J. Am. Chem. Soc., 2015, 137, 6742 CrossRef CAS PubMed; (b) B. Su, T.-G. Zhou, X.-W. Li, X.-R. Shao, P.-L. Xu, W.-L. Wu, J. F. Hartwig and Z.-J. Shi, A Chiral Nitrogen Ligand for Enantioselective, Iridium-Catalyzed Silylation of Aromatic C–H Bonds, Angew. Chem., Int. Ed., 2017, 56, 1092 CrossRef CAS PubMed; (c) B. Su and J. F. Hartwig, Development of Chiral Ligands for the Transition-Metal-Catalyzed Enantioselective Silylation and Borylation of C–H Bonds, Angew. Chem., Int. Ed., 2022, 61, e202113343 CrossRef CAS PubMed.
- Y. Yamamoto, R. Tadano and T. Yasui, Enantioselective Desymmetrization of Trifluoromethylated Tertiary Benzhydrols via Hydrogen-Acceptor-Free Ir-Catalyzed Fehydrogenative C–H Silylation: Decisive Role of the Trifluoromethyl Group, JACS Au, 2024, 4, 807 CrossRef CAS PubMed.
- K. E. Horner and P. B. Karadakov, Chemical Bonding and Aromacity in Furan, Pyrrole, and Thiophene: A Magnetic Shielding Study, J. Org. Chem., 2013, 78, 8037 CrossRef CAS PubMed.
- (a) T. Ishiyama, K. Sato, Y. Nishio, T. Saiki and N. Miyaura, Regioselective Aromatic C–H Silylation of Five-membered Heteroarenes with Fluorodisilanes Catalyzed by Iridium(I) Complexes, Chem. Commun., 2005, 5065 RSC; (b) H. Sun, Y. Cheng, H. Teng, X. Chen, X. Niu, H. Yang, Y.-M. Cui, L.-W. Xu and L. Yang, 3-Alkyl-2-pyridyl Directing Group-Enabled C2 Selective C–H Silylation of Indoles and Pyrroles via an Iridium Catalyst, J. Org. Chem., 2022, 87, 13346 CrossRef CAS PubMed.
- Single example of intramolecular C–H silylation of thiophene was reported: E. M. Simmons and J. F. Hartwig, Iridium-Catalyzed Arene Ortho-Silylation by Formal Hydroxyl-Directed C–H Activation, J. Am. Chem. Soc., 2010, 132, 17092 CrossRef CAS PubMed.
- S. P. Walsh, A. Lee and R. E. Maleczka, Jr, Iridium-Catalyzed Anti-Markovnikov Hydrosilylation of Vinylbenzenes with a Bis-Silane-Capped Double-Decker Silsesquioxane, Organometallics, 2024, 43, 1085 CrossRef CAS.
- N. A. Meanwell, Fluorine and Fluorinated Motifs in the Design and Application of Bioisosteres for Drug Design, J. Med. Chem., 2018, 61, 5822 CrossRef CAS PubMed.
- (a) J. F. Harrod, D. F. R. Gilson and R. Charles, Oxidative Addition of Silicon Hydrides to Hydridocarbonyltris(triphenylphosphine)iridium(I), Can. J. Chem., 1969, 47, 2205 CrossRef CAS; (b) A. J. A. Chalk, A New Synthesis of Silyliridium Hydrides, J. Chem. Soc. D, 1969, 1207 RSC; (c) M. Zhang, J. Liang and G. Huang, Mechanism and Origins of Enantioselectivity of Iridium-Catalyzed Intramolecular Silylation of Unactivated C(sp3)–H Bonds, J. Org. Chem., 2019, 84, 2372 CrossRef CAS PubMed.
- M. A. Esteruelas, A. Martínez, M. Oliván and E. Oñate, Kinetic Analysis and Sequencing of Si–H and C–H Bond Activation Reactions: Direct Silylation of Arenes Catalyzed by an Iridium-Polyhydrides, J. Am. Chem. Soc., 2020, 142, 19119 CrossRef CAS PubMed.
- (a) P. P. Deutsch and R. Eisenberg, Synthesis and Reactivity of Propionyliridium Complexes. Competitive Reductive Elimination of C–H and H–H Bonds from a Propionyldihydridoiridium Complex, J. Am. Chem. Soc., 1990, 112, 714 CrossRef CAS; (b) P. P. Deutschand and R. Eisenberg, Synthesis and Reactivity of Ethyliridium Complexes. Reductive Elimination of C–H and H–H Bonds from an Ethyldihyridoiridium Complex, Organometallics, 1990, 9, 709 CrossRef.
- T. Ishiyama, T. Saiki, E. Kishida, I. Sasaki, H. Ito and N. Miyaura, Aromatic C–H Silylation of Arenes with 1-Hydrosilatrane Catalyzed by an Iridium(I)/2,9-dimethylphenanthroline (dmphen) Complex, Org. Biomol. Chem., 2013, 11, 8162 RSC.
- C. Karmel and J. F. Hartwig, Mechanism of the Iridium-Catalyzed Silylation of Aromatic C–H Bonds, J. Am. Chem. Soc., 2020, 142, 10494 CrossRef CAS PubMed.
- T. G. Appleton, H. C. Clark and L. E. Manzer, The trans-Influence: Its Measurement and Significance, Coord. Chem. Rev., 1973, 10, 335 CrossRef CAS.
- E. R. Johnson, S. Keinan, P. Mori-Sánchez, J. Contreras-García, A. J. Cohen and W. Yang, Revealing Noncovalent Interactions, J. Am. Chem. Soc., 2010, 132, 6498 CrossRef CAS PubMed.
- W. Guo, T. Zhou and Y. Xia, Mechanistic Understanding of the Aryl-Dependent Ring Formations in Rh(III)-Catalyzed C–H Activation/Cycloaddition of Benzamides and Methylenecyclopropanes by DFT Calculations, Organometallics, 2015, 34, 3012 CrossRef CAS.
Footnote |
| † Electronic supplementary information (ESI) available: Experimental and computational details, compound characterization data, 1H and 13C NMR charts. See DOI: https://doi.org/10.1039/d5qo00725a |
| This journal is © the Partner Organisations 2025 |