
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
C–H bond halogenation: unlocking regiodivergence and enhancing selectivity through directing group strategies
Palak
Sharma
,
Vijay
Luxami
 and
Kamaldeep
Paul
*
and
Kamaldeep
Paul
*
Department of Chemistry and Biochemistry, Thapar Institute of Engineering and Technology, Patiala-147001, India. E-mail: kpaul@thapar.edu
First published on 3rd June 2025
Abstract
Organohalides represent a crucial class of compounds widely used as key precursors for organometallic reagents, bioactive molecules, and nucleophilic substitution reactions. The increasing prominence of cross-coupling reactions has further elevated the importance of aryl halides, establishing them as key building blocks in organic synthesis. Recent advances in C–H functionalization have underscored the strategic role played by directing groups, in which functional groups act as internal ligands to facilitate C–H activation. This method has emerged as a highly efficient approach for forming C–C and C–X bonds with exceptional regioselectivity directly from otherwise inert C–H bonds. This review highlights recent progress in applying various functional groups, including carboxylic acids, aldehydes, amides, 8-aminoquinoline, N-oxides, PIP, pyridine, and other heterocyclic systems, as directing groups in C–H halogenation reactions over the past five years.
 Palak Sharma | Ms Palak Sharma is pursuing a PhD in Synthetic Organic and Medicinal Chemistry under the supervision of Dr Kamaldeep Paul at the Thapar Institute of Engineering and Technology, Patiala, India. She pursued her Master's at Guru Nanak Dev University, Amritsar, Punjab. Her research focuses on designing and synthesizing hybridized heterocyclic compounds incorporating biologically active moieties, with the aim of evaluating their potential as antibacterial and anticancer agents. |
 Vijay Luxami | Dr Vijay Luxami received her PhD from the Department of Chemistry, Guru Nanak Dev University Amritsar. She joined the School of Chemistry and Biochemistry, Thapar Institute of Engineering and Technology, Patiala, as an Assistant Professor in 2010. She has received various awards including the DST Young Scientist, DST INSPIRE Faculty, and DST-DFG awards. She worked as a Visiting Scientist at the University of Bath, UK. Currently, she is working as a Professor at TIET, Patiala. Her area of research is organic supramolecular materials and their applications in molecular electronics. |
 Kamaldeep Paul | Dr Kamaldeep Paul received his Master's Degree from the Department of Pharmaceutical Sciences in 2000, and PhD in Synthetic Organic and Medicinal Chemistry from the Department of Chemistry, Guru Nanak Dev University, Amritsar, in 2006. Presently, he is working as a Professor in the Department of Chemistry and Biochemistry, Thapar Institute of Engineering & Technology, Patiala, India. He has 125 research publications in peer-reviewed journals. His area of research is synthetic organic and medicinal chemistry, where his research is broadly focused on C–H functionalization, multistep synthesis of heterocyclic molecules, and their in vitro evaluation for anticancer and antimicrobial activities |
1. Introduction
The carbon–halogen (C–X) bond is one of the most crucial functional groups in organic chemistry. It is widely utilized in various applications, including the synthesis of natural products,1–3 pharmaceuticals,4–6 and agrochemicals,7–9 as well as in materials science10–12 and molecular recognition.13,14 Organic halides serve as both synthetic precursors and target compounds in the synthesis of organometallic reagents and their subsequent transformations.15Organohalides have become significantly more important and are now considered essential building blocks in organic synthesis, especially with the emergence of cross-coupling chemistry.16,17 Particularly, aryl and heteroaryl halides are highly valued due to their prevalence in pharmaceutically relevant molecules and high industrial demand.18 Previously, synthetic strategies utilized strong oxidizing agents for preparing organohalides due to the less polarised C–H bond, which has the same electronegativity as carbon and hydrogen.19 This approach poses significant drawbacks and challenges, including harsh reaction conditions, hazardous operations, toxic reagents, poor selectivity, formation of byproducts and over-halogenated substrates. Traditional methods, however, failed to produce highly selective halogenated products, and have been surpassed by emerging new atom-economical direct C–H activation methodologies for obtaining halogenated derivatives.
Over the last decade, C–H activation has emerged as a modern, cost-effective and environmentally friendly tool for organic synthesis, proved to be an effective transformation in many compounds regarding selectivity and milder reaction conditions. Various strategies, including transition-metal catalysis,20 photocatalysis,21,22 enzyme catalysis,23,24 a metal-free approach25,26 and electrochemical methods27,28 have been developed to create C–X bonds. Despite tremendous advancements in C–H functionalization, developing site-selective or regioselective halogenations of organic compounds remains challenging. The most common way to control site selectivity in C–H functionalization is using directing groups such as amides,29 quinolinamides,30 carboxylic acids,31N-oxides32etc. The literature review indicates that nearly all reports on C–H halogenations employ these directing groups to mediate site-selective C–H bond functionalization (Fig. 1).33,34 Very few studies have explored methods that do not rely on a directing group for site selectivity.35,36 Additionally, while significant progress has been made in the C–H halogenation of aromatic compounds, fewer studies focused on the synthesis of vinyl and alkyl halides.37,38
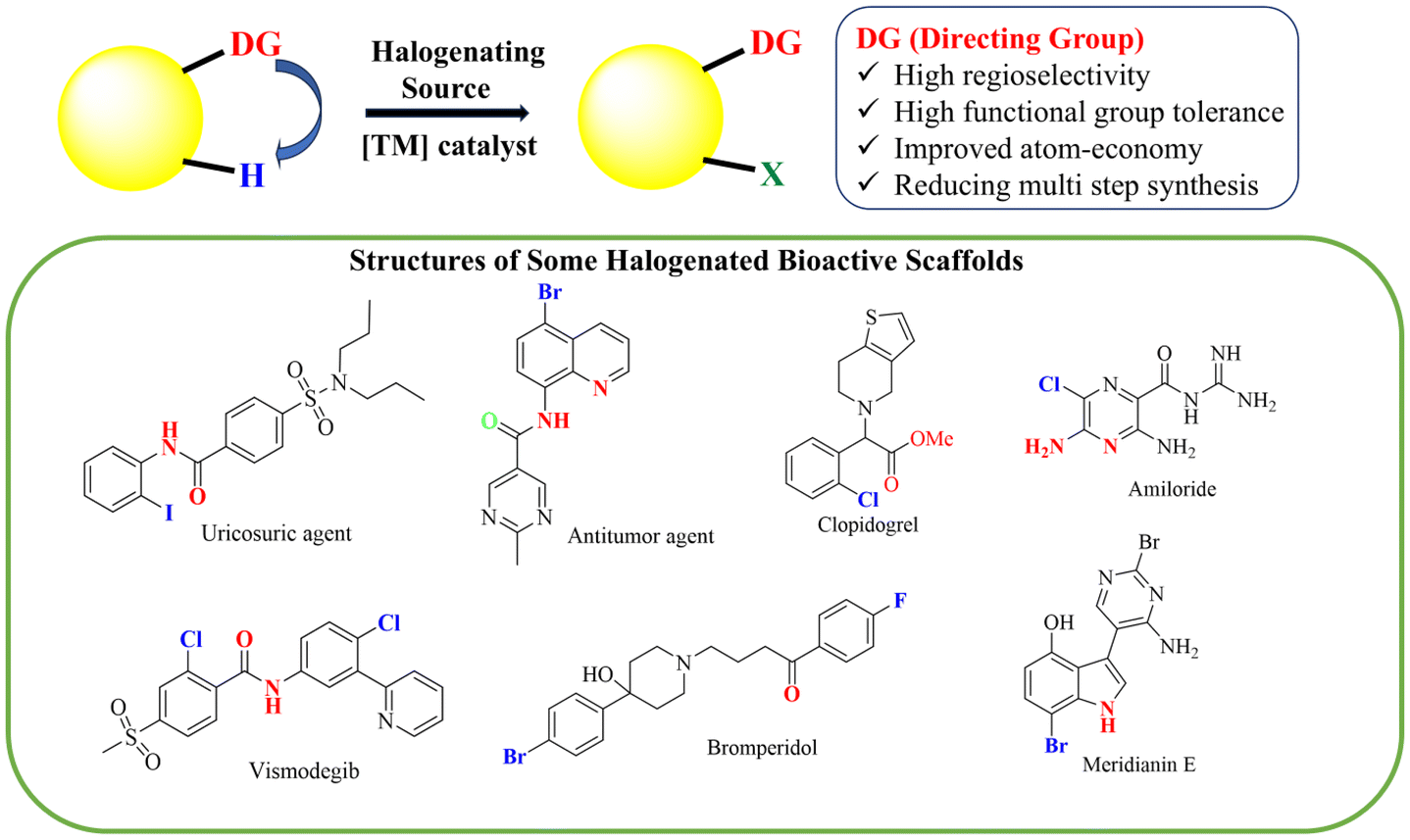 | ||
| Fig. 1 Directing group strategy for C–H halogenation. | ||
The carbon–halogen (C–X) bonds exhibit potency as synthetic intermediates, and facilitate convenient access to various functionalizations achieved through different methodologies such as nucleophilic substitution,39 metal–halogen exchange,40 radical reactions,41 and transition metal-mediated transformations.42 Therefore, valuable information regarding reactivity profiles allows one to plan synthesis where organohalide functionality can be incorporated, which remains intact throughout the entire process until it is either integrated into the target compound or modified. This highlights the importance of carbon–halogen bonds in organic chemistry. Although several specialized reviews and book chapters have focused on C–H functionalization over the past decade, a dedicated literature review on directing group-assisted C–H halogenation (including fluorination, chlorination, bromination, and iodination) has yet to be published. This review aims to fill that gap by providing a comprehensive overview of C–H halogenation reactions facilitated by a variety of directing groups. It categorizes methodologies for the synthesis of aryl, vinyl, alkyl, and heteroaryl halides, drawing from the literature published between 2019 and 2024. Additionally, it highlights recent advancements in this area and offers a comparative analysis with traditional halogenation approaches, emphasizing improvements in efficiency and selectivity.
2. Classification of directing groups
In recent years, the use of Lewis-basic directing groups for regioselective C–H halogenation has emerged as a modern and versatile strategy for constructing C–X bonds. These directing groups are typically classified based on their functional nature and coordination strength—ranging from strongly to weakly coordinating, and including removable, non-removable, traceless, or transient types. This approach has become one of the most powerful tools for achieving site-selectivity in C–H halogenation reactions. These transformations proceed through diverse mechanistic pathways, including transition-metal catalysis, visible-light-induced processes, photocatalysis, and electrochemical methods, offering enhanced efficiency and adaptability across a wide range of synthetic applications.2.1. 8-Aminoquinoline as directing group
The quinoline framework has attracted significant interest from researchers due to its widespread presence in biomedical and organic synthesis. Quinolines serve as essential structural components in numerous pharmaceuticals and biologically active natural products.43 Traditional halogenation methods, such as electrophilic substitution using halogenating reagents, have been surpassed by the direct C–H bond functionalization approach, which provides greater efficiency and selectivity. A pivotal advancement in utilizing the quinoline framework came in 2005 when Daugulis introduced a bidentate directing group, 8-aminoquinoline.44 This innovation has drawn considerable attention in recent years, as it enabled highly selective and efficient C–H bond functionalization in a variety of aromatic, heteroaromatic, and aliphatic compounds.45 By facilitating chelation-assisted coordination with transition metals, 8-aminoquinoline, as a bidentate directing group has proved instrumental in advancing remote and site-selective C–H bond functionalization. The use of bidentate directing groups continues to be a powerful and versatile strategy for activating C–H bonds, making them a cornerstone of modern synthetic protocols.
In 2019, Long et al. developed an efficient method for synthesizing specific halogenated derivatives of 8-amidoquinoline. The optimal results were achieved using an inexpensive iron(III) catalyst (5 mol%), NXS or X2 (X = Br, I) (0.6 mmol) as the halogenating agent, CH3(CH2)5COOH (0.3 mmol) and NaHCO3 (0.3 mmol) as additives, in water as a benign solvent at room temperature for 24 h, using air as the oxidant (Scheme 1). A significant yield improvement of approximately 90% was observed with the addition of CH3(CH2)5COOAg, highlighting the beneficial role played by a long-chain carboxylic acid as potential phase transfer reagent. To extend the utility of the protocol, N-(5-bromoquinolin-8-yl)pivalamide underwent a simple Suzuki coupling reaction with boronic acids, yielding products in moderate to good amounts. Mechanistic investigations revealed that the catalytic cycle proceeded via a radical pathway. Key steps include chelate formation, deprotonation, bromine radical attack via an SET (single-electron transfer) mechanism, oxidation, and metal dissociation from the intermediate via a proton transfer (PT) process, leading to the final product.46
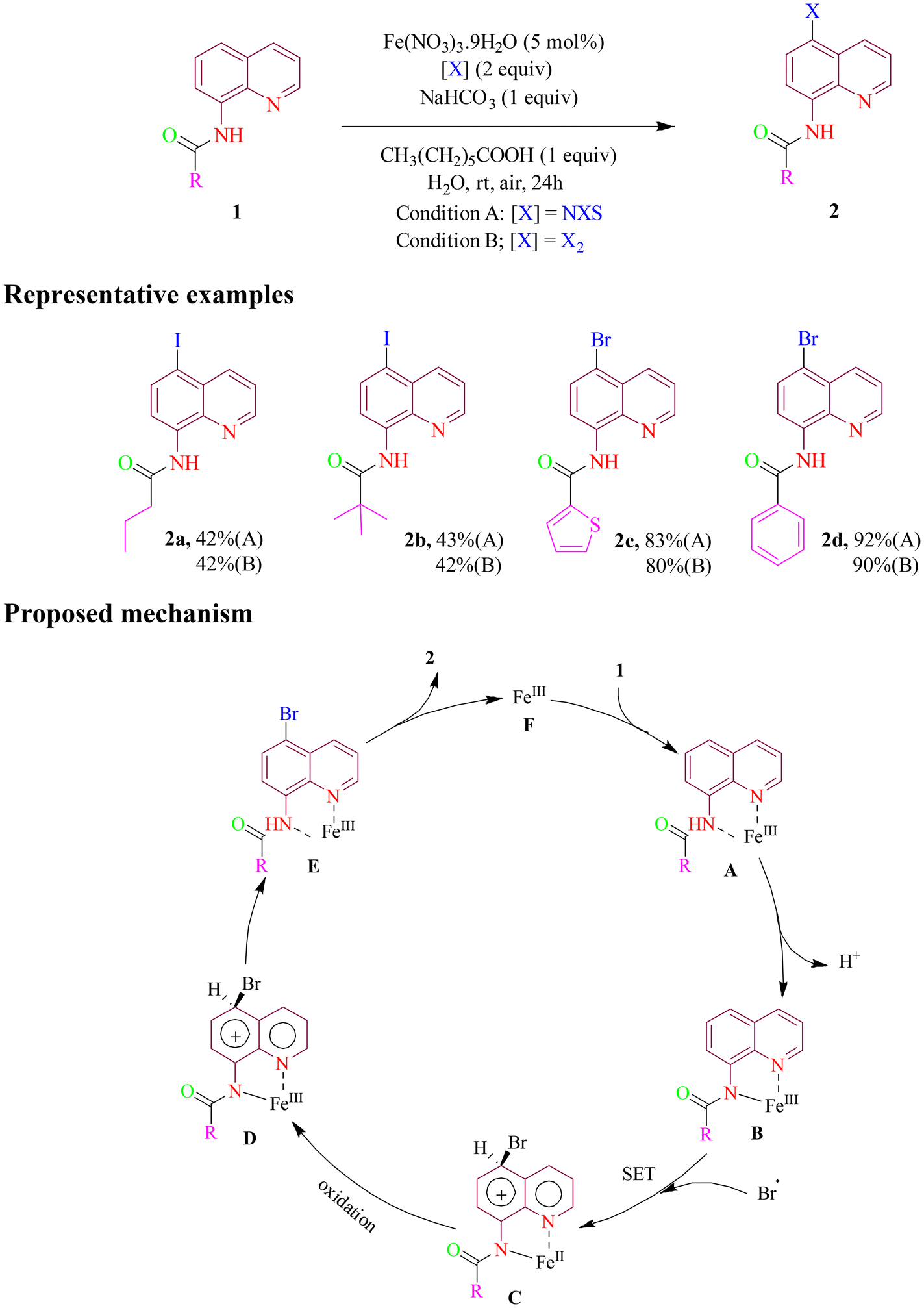 | ||
| Scheme 1 Fe(III)-catalyzed C-5 halogenation of 8-aminoquinoline amide. | ||
Sanford and her team successfully developed a copper-mediated method for regioselective radiofluorination of N-protected 8-aminoquinoline. The approach utilized either K18F or Ag18F as the fluorinating agent, with Ag18F serving as both a nucleophile and base. This dual role enabled fluoride substitution and proton sequestration by activating the C–H bond. The reaction employed N-methylmorpholine N-oxide (NMO) as an oxidant, DBU as a base, and copper catalyst in DMF at 100 °C for 30 min (Scheme 2). Substituting CuI with the more soluble (MeCN)4CuOTf significantly improved the reaction outcomes. The method demonstrated a broad substrate compatibility, effectively fluorinating N-protected 8-aminoquinolines with diverse substituents. It was employed in automated synthesis of high-specific-activity doses of the RARβ2 agonist [18F]AC261066, validating its utility in radiopharmaceutical development.47
 | ||
| Scheme 2 Copper-mediated radiofluorination of N-protected 8-aminoquinoline. | ||
Chen et al., in 2020, developed a new approach for direct chlorination of acrylamides at room temperature using palladium catalyst and N-chlorosuccinamide (NCS) as halogenating reagent. This method resulted in single Z-stereoisomer in excellent yield, formed from a variety of functionalized acrylamides (Scheme 3). The approach proved effective for both α-substituted and α,β-disubstituted acrylamide substrates. Mechanistic studies revealed that the formation of a reversible palladacycle through coordination with a bidentate directing group serves as the rate-determining step. This is followed by oxidative addition and subsequent reductive elimination, ultimately furnishing the desired product.48
 | ||
| Scheme 3 Stereoselective chlorination of acrylamides. | ||
In 2020, the Fang group reported C5 bromination of 8-aminoquinoline amide under green electrochemical conditions. The method utilized inexpensive, non-toxic Cu(OAc)2 as catalyst and NH4Br as both brominating reagent and electrolyte, achieving a high yield of desired products (Scheme 4). Cyclic voltammetry experiments revealed that NH4Br generated Br2 or Br3− species during electrolysis while Cu2+ formed a complex with the 8-aminoquinoline directing group. The proposed mechanistic pathway involves a typical catalytic cycle, beginning with the coordination of 8-aminoquinoline to the metal center. This is followed by the attack of a bromine radical via a single-electron transfer (SET) process, a subsequent proton transfer (PT) step, and finally, metal dissociation to furnish the target product.49
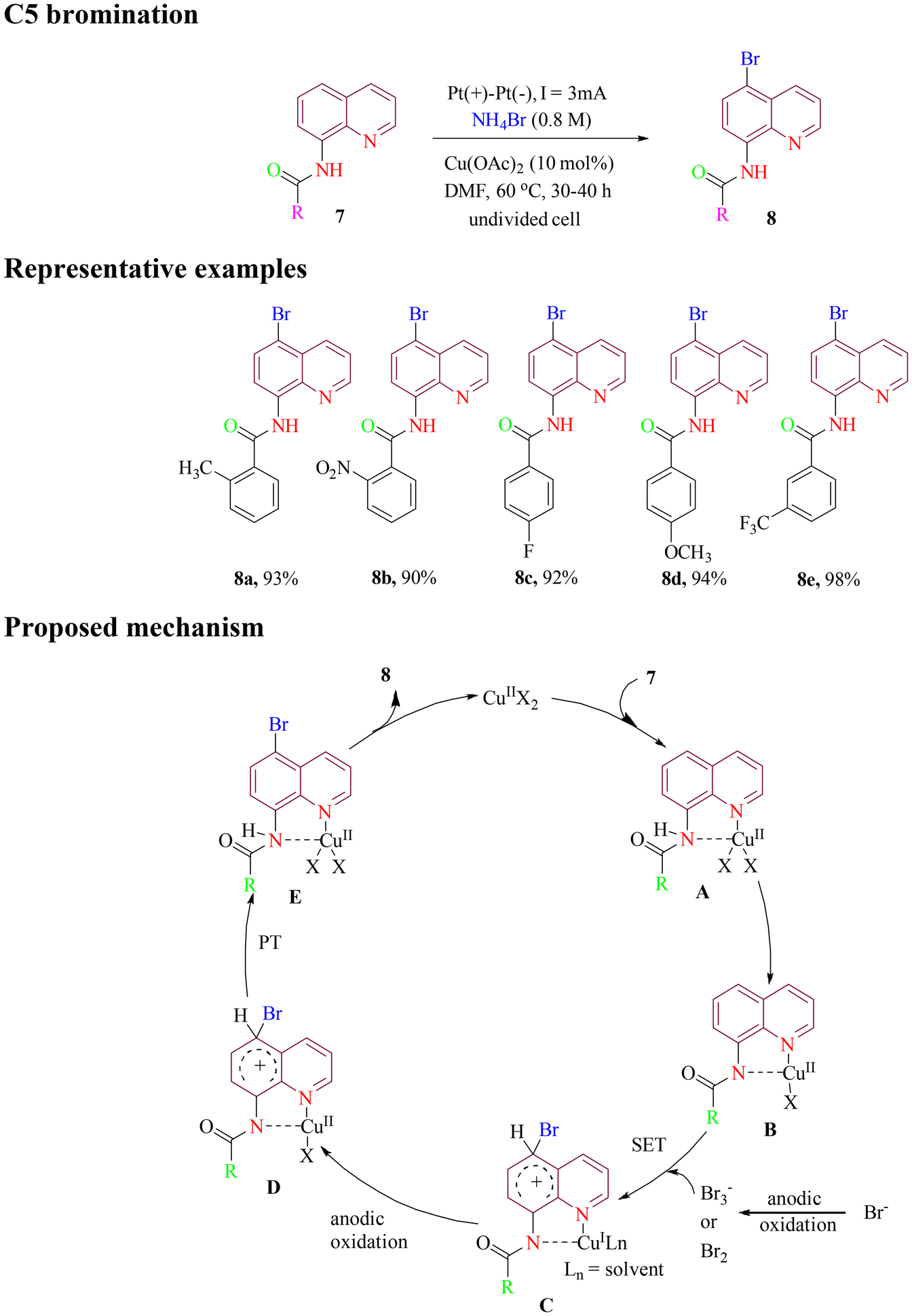 | ||
| Scheme 4 Electrochemical oxidative C5 bromination of 8-aminoquinoline amide. | ||
Subsequently, Guo and colleagues reported that organic halides could serve as source of halogens during electrocatalytic oxidation. They developed an efficient and convenient system for halogenating the C5–H position of 8-aminoquinolines at room temperature by combining electrocatalysis and continuous flow reactions. This system utilized common solvents such as dichloromethane (DCM) and dibromomethane (DBM) as sources of chlorine and bromine, respectively, eliminating the need for additional oxidizing agents (Scheme 5). By applying a constant current of 100 mA to the reaction mixture in continuous flow, the desired products were obtained rapidly with high yields. The method is easily scalable to gram quantities due to the use of a continuous flow device. Notably, the reaction followed the SET mechanistic pathway to achieve the desired product.50
 | ||
| Scheme 5 Copper-catalyzed electrochemical oxidative C-5 halogenation of 8-aminoquinoline amide. | ||
2.2. N-Oxide as directing group
Substrates containing weakly coordinating N-oxides have emerged as valuable synthons for exploring alternative directing groups in C–H functionalization. The use of N-oxide directing groups offers a versatile platform for developing synthetic methods to access diverse heterocyclic compounds. These approaches, however, face challenges related to the effective coordination of the N-oxide with metal catalysts. Regioselectivity in such transformations is typically achieved through the formation of the N-oxide-chelated metallacycle, which plays a critical role in guiding the functionalization to the desired position.51
Dhiman et al. developed a method for bromination at the C8 position of N-oxide quinoline, mediated by [RhCp*Cl2]2 catalyst. The optimized mild reaction conditions yielded the desired products efficiently, with broad substrate scope, tolerating diverse functional groups at other positions of the model substrate (Scheme 6). Mechanistic studies highlighted the active role played by Rh(III) catalyst in the catalytic cycle, with the formation of a key rhodacycle intermediate (A), suggesting that C–H activation is the rate-limiting step. This was confirmed through controlled experiments, deuterium labelling studies, and kinetic isotope effect (KIE) analysis. The desired product 12 was formed from intermediate C, derived from B, through one of two pathways: nucleophilic addition or oxidative addition/reductive elimination.52
 | ||
| Scheme 6 Rh(III)-mediated highly regioselective C–Br formation of quinoline N-oxide through C(8)–H activation. | ||
Next, in 2024, Zhou et al. reported that C–H iodination of 1-aryl pyridine N-oxides, facilitated by Pd(OAc)2, occurred under electrochemical oxidation conditions using I2 as the iodine source (Scheme 7). The reaction involved isoquinoline N-oxides with para or meta-substituted aryl groups at the 1-position and yielded the corresponding iodinated products. Electron-donating groups on the aryl ring enhanced the reaction, producing relatively high product yields. The reaction is considered to proceed through the mechanistic pathway where the palladium catalyst cleaved the ortho-C–H bond at the 1- or 3-position to form a palladacycle, which then reacted with electrochemically generated I+ and produced the final products.53
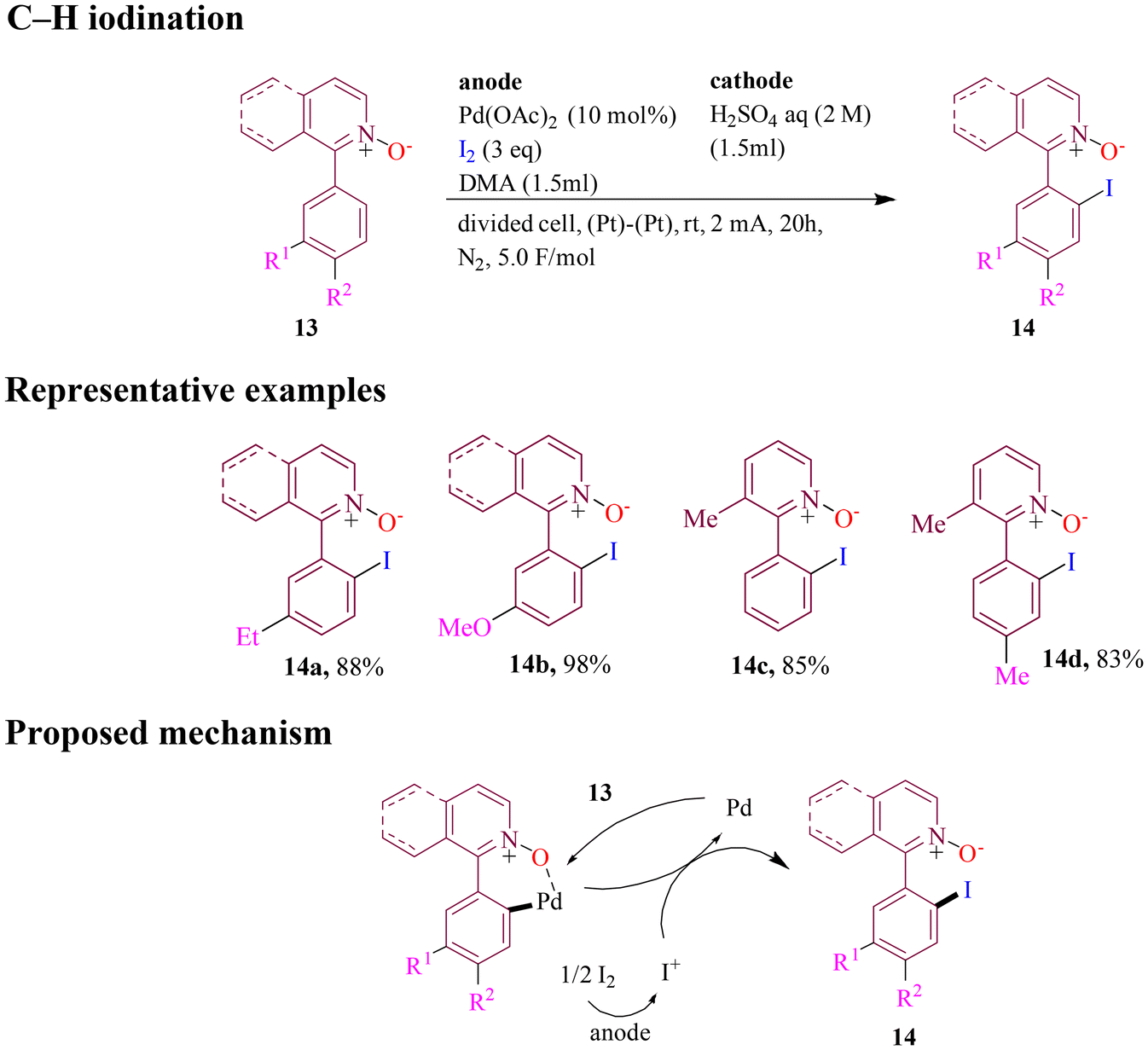 | ||
| Scheme 7 Palladium-assisted electrochemical iodination of 1-arylpyridine N-oxides. | ||
2.3. Amide and Weinreb amide as directing group
Amides are essential functional groups in numerous natural products, polymers, agrochemicals, drug molecules, and synthetic intermediates.54 Due to their planar geometry and the high degree of delocalization of the nitrogen lone pair, amide bonds exhibit notable stability compared with other functional groups. The use of substituted or unsubstituted amide-based directing groups in C–H functionalization was first introduced by Li et al. in 2012.33 This approach gained momentum because the simple directing group operates through a monodentate binding mode and effectively controls reactivity, stereoselectivity, and regioselectivity.55,56 The activity of the amide group is attributed to the carbonyl moiety, which facilitates C–H bond activation by forming an intermediate cyclometallated complex that enables the synthesis of the desired products. Secondary and tertiary amide functionalities have been extensively employed as directing groups for site-selective functionalization of target molecules’ C–H bonds. As a result, amide functional groups are regarded as highly significant and frequently used weak-coordinating directing groups in C–H functionalization chemistry.
In 2019, Li et al. employed this strategy for C–H bond activation to synthesize various regioselective halogenated derivatives of electron-deficient arenes including benzamides, benzoic esters, and sulphonamides. Under optimized reaction conditions, where Ni(OAc)2 was used as a catalyst, NXS (X = Cl, Br, I) served as the halogenating agent and TfOH acted as an acid additive, which afforded satisfactory yields (Scheme 8). Encouraged by these results, the synthesis was extended to other di-halogenated derivatives of electron-deficient arenes, also achieving moderate to good yields. The amide directing group activated the C–H bond by forming a chelate complex with nickel catalyst, which further underwent oxidative addition with N-halosuccinimide followed by reductive elimination to afford the desired products.57
 | ||
| Scheme 8 Ni(II)-catalyzed halogenation of benzamide. | ||
In addition to this, Jaiswal et al. reported regioselective ortho C–H bromination and iodination methods for challenging aliphatic arylacetamide derivatives, utilizing N-halosuccinimides as halogenating agents. This protocol represented the first example of direct bromination and iodination of arenes with primary amide group, achieved using palladium(II) salts without the need for bulky auxiliaries (Scheme 9). Various mechanistic studies indicated that the reaction proceeded through in situ generation of imidic acid from primary amides under Brønsted acidic conditions. This process likely facilitated the formation of six-membered metallacycles, where the nitrogen atom coordinated with palladium(II), aiding in forming C–X bonds. The critical role played by trifluoroacetic acid (TFA) was also identified where it acted as an activator for N-halosuccinimides through protonation and assisted in amide's tautomerization process to imidic acid.58
 | ||
| Scheme 9 Palladium-catalyzed ortho C–H bromination/iodination of aliphatic amides. | ||
Kumar's group developed a method for regioselective halogenation (Br, Cl, and I) of the aromatic ring of benzamide derivatives, using a Brønsted acid-promoted palladium(II) catalyst (Scheme 10). This methodology was effectively carried out with the primary amides, demonstrating compatibility with various benzamides under the established conditions, leading to the formation of halogenated products without the need for an external auxiliary. Based on a series of control experiments, the proposed mechanistic pathway has been followed which involved the coordination of imidic nitrogen atom where base-assisted internal electrophilic substitution (BIES) led to regioselective C–H activation, resulting in formation of a thermodynamically favoured five-membered palladacycle. In this protocol, TFA played three critical roles: (1) facilitating the tautomerization of the amide to imidic acid, (2) activating N-halosuccinimide through protonation, and (3) regenerating the palladium salt.59
 | ||
| Scheme 10 Brønsted acid-promoted palladium-catalyzed ortho C–H halogenation of benzamides. | ||
In 2020, the Ison group explored catalytic C–X bond formation and synthesized ortho-halogenated derivatives of tertiary benzamides, especially, N,N-diisopropylbenzamide. Under optimized reaction conditions involving [Cp*IrCl2]2, AgOTf and AcOH, in 1,2-dichloroethane at 60 °C for 1 h, various iodinated derivatives were developed with good tolerance for both electron-donating and electron-withdrawing groups at the para-position (Scheme 11). Substrates with electron-donating groups yielded better results than those with electron-withdrawing groups. Brominated derivatives of the same substrates were synthesized by increasing the catalyst loading to 6 mol% and extending the reaction time to 4 h. Two possible mechanisms were proposed for the halogenation step, differing in the halogen source. The first mechanism involved direct functionalization with N-halosuccinimide (NXS), while the second suggested functionalization via acetyl hypohalite, generated in situ through an off-cycle equilibrium between acetic acid and halosuccinimide. Coordination of the acetyl hypohalite to the iridium center facilitated product formation via reductive elimination. This different approach of reaction pathway is quite interesting to be considered for further development of halogenated aromatic compounds.60
 | ||
| Scheme 11 Ir-catalyzed iodination and bromination of tert-benzamides. | ||
Tanaka et al. reported a pioneering rhodium-catalyzed method for regioselective bromination of O-phenyl carbamates. The reaction is accelerated by a secondary amide pendant cyclopentadienyl ligand due to hydrogen bonding between the acidic NH group of the ligand and the carbonyl group of NBS (Scheme 12). This strategy demonstrated good functional group tolerance on aromatic rings, producing ortho-brominated product in satisfactory yields. Various mechanistic insights and DFT calculations were elucidated for the possible catalytic cycle. In this cycle, the cleavage of the C–H bond with acetoxy ligand in intermediate A produced aryl rhodium B followed by coordination with N-bromosuccinimide (NBS) to form intermediate C. A concerted bromine transfer occurred through the transition state TS-CD, resulting in the formation of intermediate D. The subsequent formation of the Rh–N bond yielded rhodiumamide E. Coordination of substrate 24 then released product 25 and generated rhodiumamide F. However, an alternative pathway might also exist in the catalytic cycle, where the C–H bond of 24 is cleaved with the succinimidyl ligand in F, creating aryl rhodium Cvia intermediate G.61
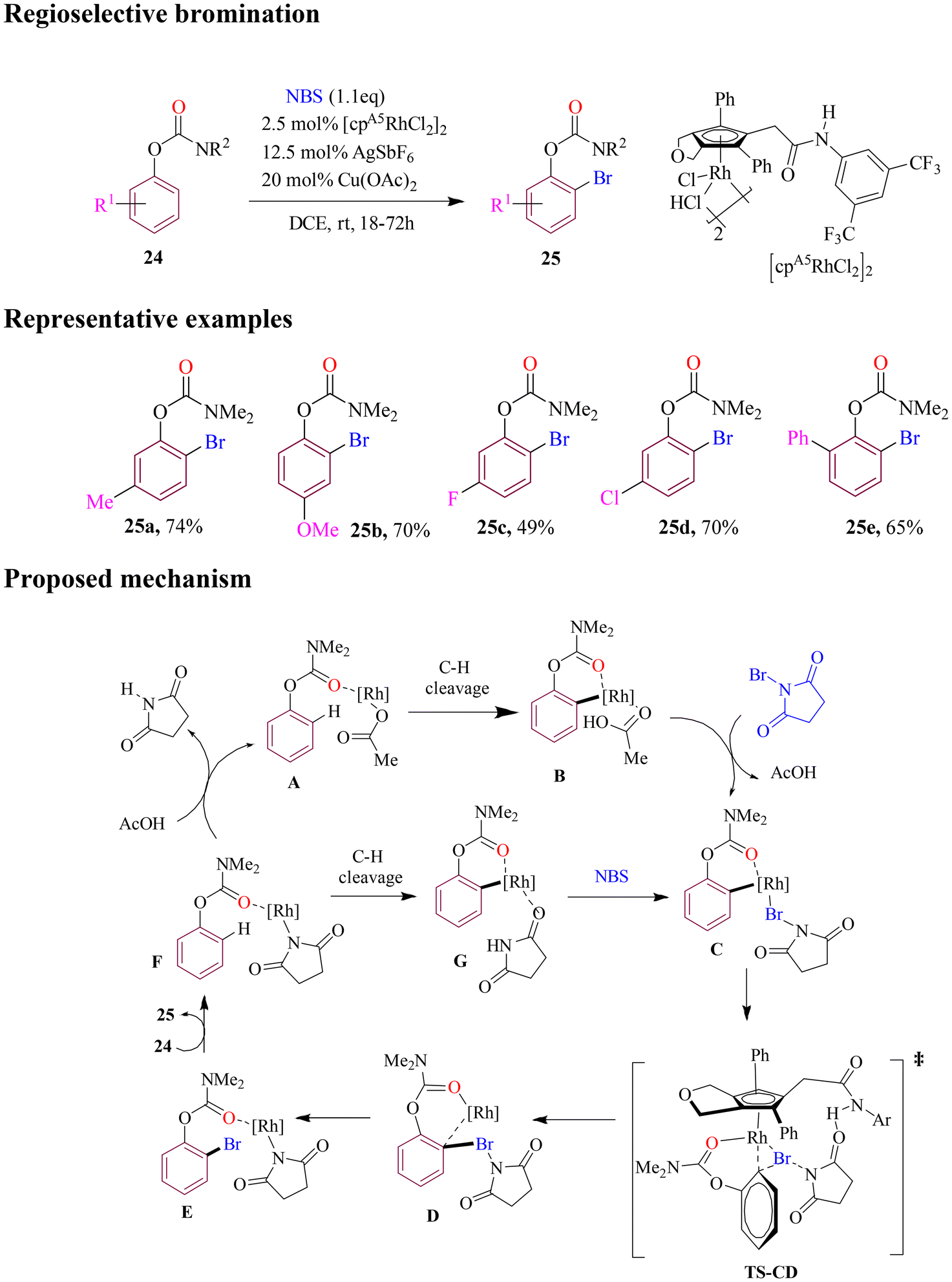 | ||
| Scheme 12 Rhodium-catalyzed carbamate-directed ortho C–H bromination of O-phenyl carbamates. | ||
Recently, Sun et al. reported the regioselective synthesis of brominated derivatives of benzanilide guided by the amide bond as a directing group. The study demonstrated that different brominated regioisomers could be achieved by modulating site-selectivity through promoter choice (Scheme 13). The reaction conditions were optimized and applied to various functionalized substrates, with Pd(II) and HFIP as promoters, and yielded the desired products efficiently. The detailed mechanism for para-bromination is proposed in Scheme 13, whereas ortho-bromination follows the general mechanistic pathway proceeding through C–H bond activation, oxidative addition and reductive elimination.62
 | ||
| Scheme 13 Switchable site-selective bromination of benzanilide. | ||
Weinreb amides (N-methyl-N-methoxyamides) are valuable intermediates in synthetic chemistry due to their distinct reactivity. Typically, the acyl group in the amide directs the metal during C–H activation. However, their weak coordination properties make them challenging substrates for C–H activation reactions.63 These moieties utilized the labile N–O bond as an internal oxidant during C–H activation, eliminating the need for an external oxidant. Weinreb amides are readily synthesized from carboxylic acids, their chlorides, esters, aldehydes, or ketones, making them versatile and accessible building blocks in organic synthesis.
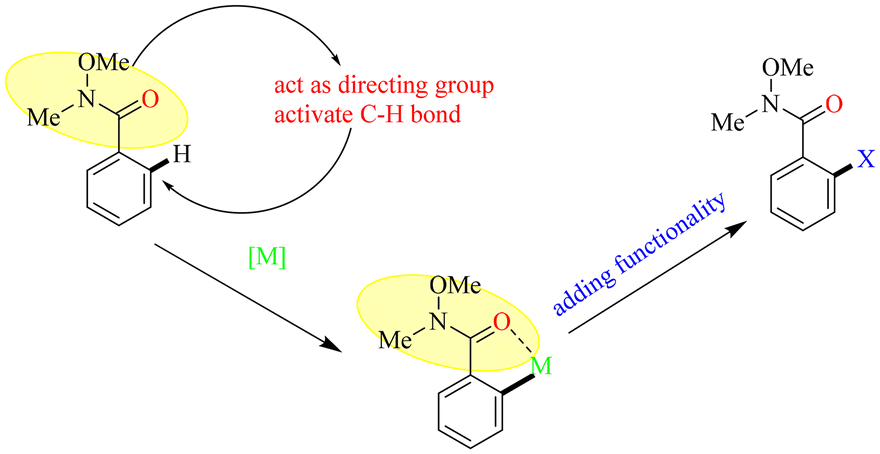
The Matute group recently developed a method for selective ortho C–H iodination of Weinreb amides and benzamides. The approach employed iridium as a catalyst and N-iodosuccinimide (NIS) as the iodinating agent, with acid as a promoter (Scheme 14). The iodination reaction was conducted using mechanochemical methods, which reduced the reliance on HFIP, shortened reaction time, and eliminated the need for consecutive additions of NIS. This alternative approach delivered comparable or even improved yields and selectivity. The introduction of iodine provides a versatile handle for wide range of downstream functionalizations.64
 | ||
| Scheme 14 Iridium-catalyzed regioselective C–H iodination of benzamides and Weinreb amides. | ||
2.4. 2-(Pyridine-2-yl)isopropylamine (PIP) as directing group
Inspired by the structure of aminoquinoline, researchers recognized the importance of an anionic binding site and a strongly coordinating neutral site in activating methylene C–H bonds. Leveraging the Thorpe–Ingold effect, which facilitated cyclometalation through bond angle compression and entropic constraint, the 2-(pyridine-yl)isopropylamine (PIP) was designed as a directing group (DG). This DG features a gem-dimethyl group and a pyridyl group, which upon transformation into the corresponding PIP amide, enables selective β C–H activation. Additionally, modifying the two methyl groups into distinct functional groups or replacing the pyridyl group with a chiral oxazoline offers potential for creating chiral auxiliaries, paving the way for asymmetric C–H functionalization. The versatility of PIP as a bidentate directing group lies in its ability to be readily removed post-reaction via a nitrosylation/hydrolysis sequence or under acidic conditions.65 Since its introduction, PIP has shown immense potential for directing C–H functionalization, demonstrating compatibility with various transition-metal catalysts such as Ni, Cu, Co, and Pd.
In 2019, Mei and co-workers employed a fascinating approach, forming a C–Br bond by activating the C–H bond of a benzamide derivative with the assistance of an optimal PIP directing group, guided by a versatile palladium redox catalyst which could operate under various cycles and oxidation states in a divided cell (Scheme 15). In this transformation, NH4Br served not only as the brominating agent but also acted as the electrolyte, enabling the successful synthesis of arene derivatives substituted with diverse, well-tolerated functional groups at various positions, with yields ranging from good to excellent. The proposed mechanistic pathway involved the coordination of palladium with benzamide, which further proceeded through palladation, delivering Br3− or Br2 generated in situ to high-valent Pd species, followed by reductive elimination, resulting in brominated product after ligand exchange.66
 | ||
| Scheme 15 Bromination of substituted arenes directed by PIP using electrochemical setup. | ||
Similar to this, the Mei group in 2020 performed the same methodology as above for chlorination and bromination of benzamide derivatives possessing PIP as directing group where NH4Br and LiCl acted as electrolytes as well as brominating and chlorinating agents, respectively (Scheme 16). However, the reaction proceeded through a catalytic cycle involving the coordination of benzamide derivative with palladium catalyst, activating ortho C–H bond, in situ generation of Br3− or Br2, followed by reductive elimination to afford the desired brominated product.67
 | ||
| Scheme 16 Chlorination and bromination of substituted benzamide directed by PIP using electrochemical setup. | ||
2.5. Pyridine as directing group
Murai and Chatani pioneered the use of pyridine as a directing group in C–H functionalization, a breakthrough that has since been expanded by various research groups. Pyridine has become a key player in C–H functionalization due to its simplicity and the ease of its incorporation into aliphatic chains or aromatic rings through diverse methods. However, when pyridine is attached via a carbon–carbon (C–C) bond, its removal from the substrate is not feasible; cleavage is only possible when the attachment involves a carbon–heteroatom bond.68 Although significant progress has been made in C–C bond formation chemistry, methods for C–X bond formation remain underdeveloped.
In 2019, the Jiao group developed an efficient method for Pd-catalyzed directed ortho C–H bromination of arenes utilizing a pyridine directing group in which dimethyl sulfoxide (DMSO) was employed as an oxidant with hydrobromic acid (aq) as bromide source (Scheme 17). The optimized method was evaluated for its substrate scope that successfully produced brominated 2-phenylpyridines with good to excellent yields and high selectivity. Various control experiments were performed to gain insight into the mechanistic pathway, revealing the coordination of directing group to the Pd(II) centre, followed by C–H activation, and cleavage of the Pd–C bond through brominating agent [(DMSO)nBr+(DMS)]Br−, which was formed under acidic conditions. Finally, the product dissociated from the Pd(II) centre, regenerating the Pd(II) catalyst and completing the catalytic cycle.69
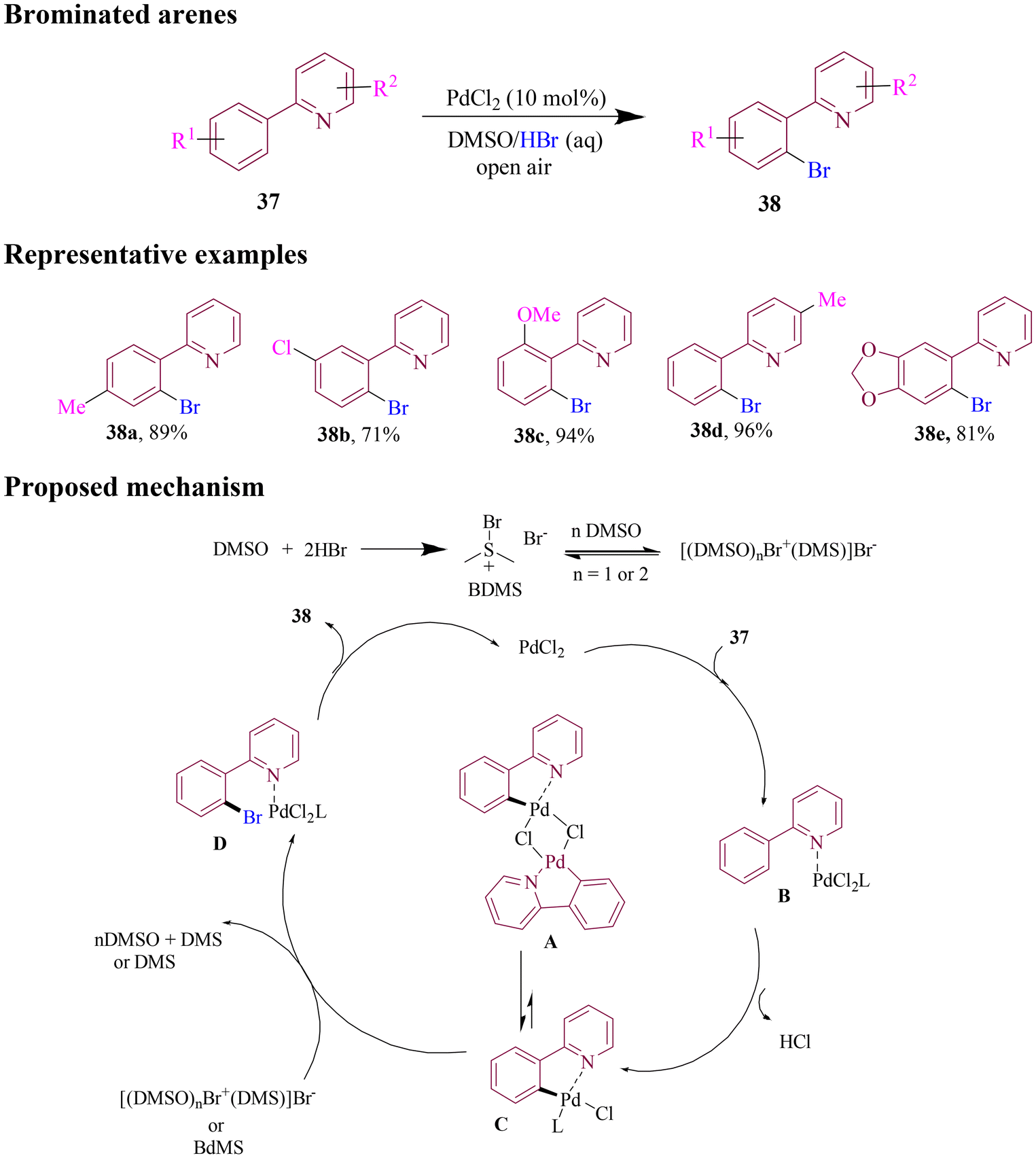 | ||
| Scheme 17 Pd-catalyzed ortho C–H bromination of arenes directed by pyridine. | ||
Guilbaud et al. introduced a method for palladium-catalyzed, pyridyl-directed selective oxidative ortho C–H halogenation of S-unprotected aryl pyridyl sulfides (Scheme 18). This approach avoids both sulfur oxidation and direct electrophilic halogenation of the pyridine moiety, enabling rapid reactions (85% yield in just 40 minutes). This represents a significant improvement over slower C–H halogenation methods that rely on S-oxidized pyridyl sulfoxides and sulfones. The new methodology enabled the direct functionalization of C–H bonds in sulfides and eliminated the need for reduction steps or protection/deprotection of sulfoxides and sulfones, which could be challenging due to their high thermodynamic stability. The reactions were carried out under strictly anhydrous conditions in chlorobenzene, using a controlled amount of halosuccinimide or PIDA as the halogen source. This approach allows for both mono- and ortho-dihalogenation of highly functionalized pyridyl aryl sulfides, offering a versatile tool for halogenation in complex molecular settings.70
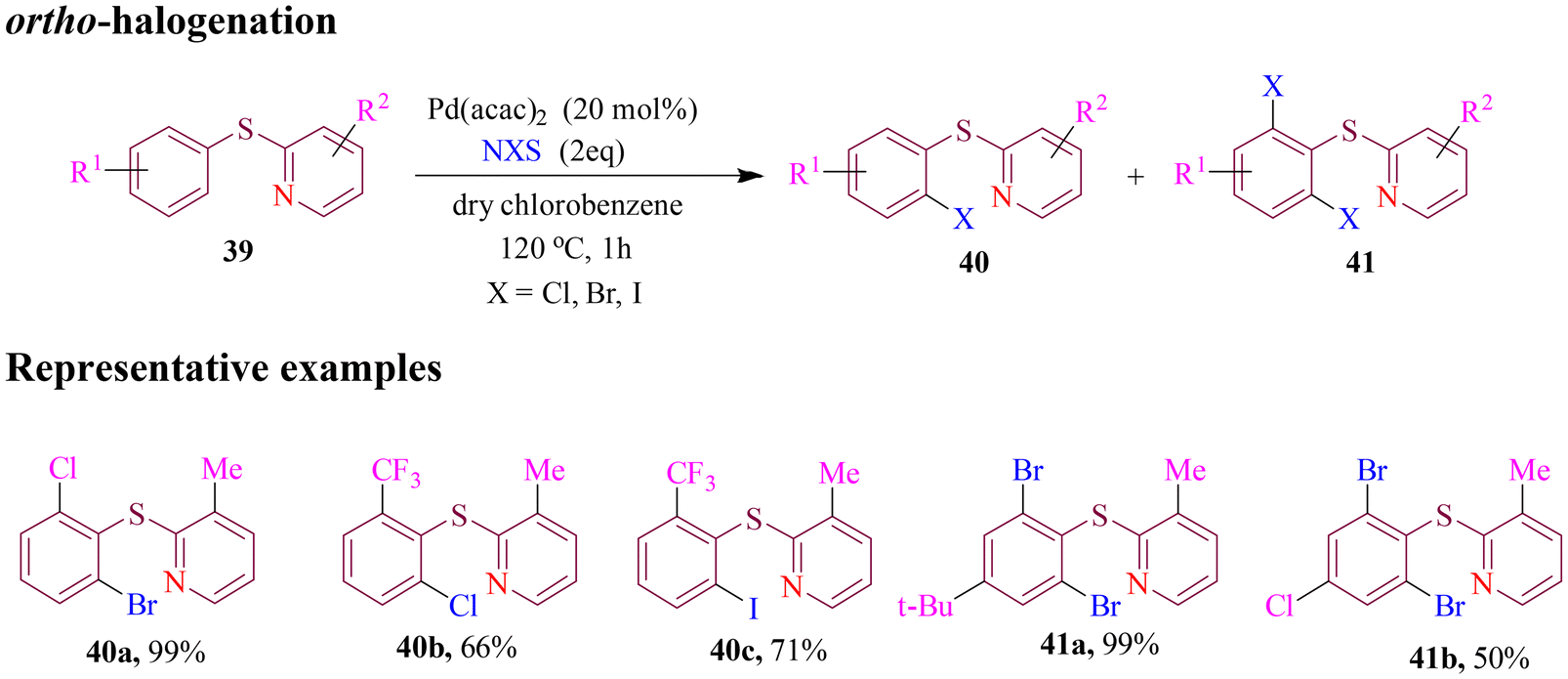 | ||
| Scheme 18 Direct C–H halogenation of pyridyl sulfides. | ||
2.6. Aldehyde and ketone as directing groups
Reactive functional groups such as aldehydes and ketones present significant challenges in C–H functionalization, spurring innovative research into more efficient strategies through the transient directing group strategy. One such approach leverages amines to form an imine in situ, which acts as a directing group (DG) during the reaction.71 This imine can serve as either a monodentate or bidentate ligand, facilitating the crucial C–H activation step.72 Subsequently, metal-catalyzed C–H functionalization occurs on the imine derivative, yielding a newly functionalized product while regenerating the amine. This strategy, referred to as the Transient Directing Group (TDG) approach, simplifies the C–H activation process by eliminating the need for additional DG installation or removal steps, significantly enhancing the overall efficiency and practicality of the method.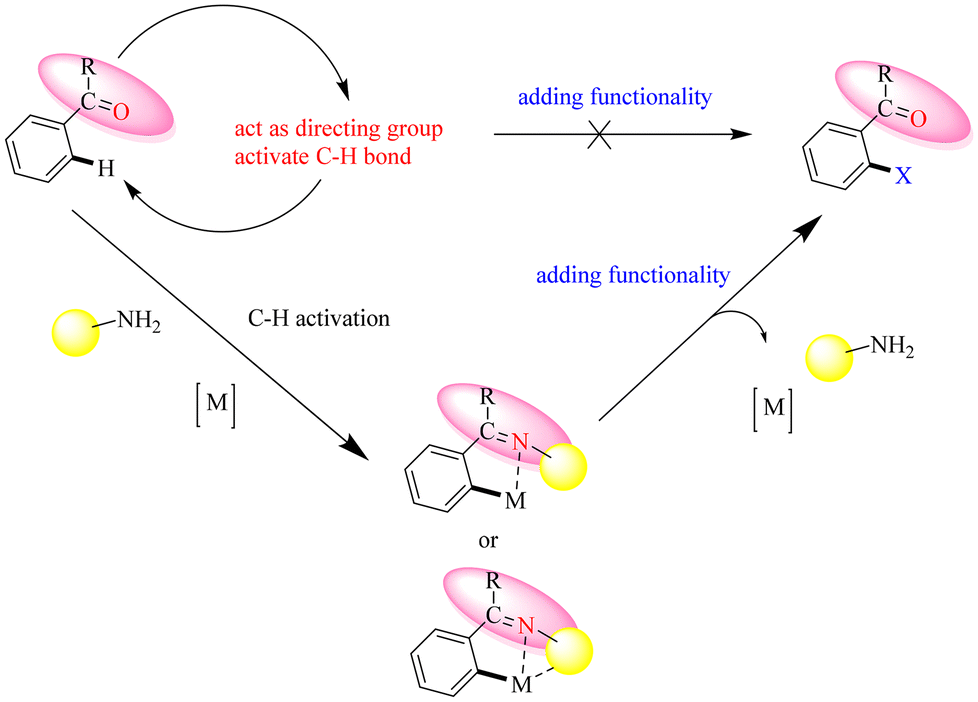
In 2019, Yonga et al. utilized the monodentate transient directing group strategy by employing 2-amino-5-chlorobenzotrifluoride to generate in situ an imine which facilitated the development palladium-catalyzed regioselective C–H bromination of benzaldehydes under mild reaction conditions without the requirement for an external ligand or silver salts (Scheme 19). The protocol was compatible with various electron-donating and electron-withdrawing groups, delivering good to excellent yields of conjugates. Additionally, the strategy was also explored to obtain dibrominated benzaldehyde derivatives, achieving excellent yields when the loading of NBS was increased from 1.5 to 2.5 equivalents.73
 | ||
| Scheme 19 Palladium-catalyzed ortho C–H bromination of benzaldehydes. | ||
The Zhang group developed an efficient method for chlorination utilizing N-chlorosuccinimide (NCS) to functionalize a variety of substituted benzaldehydes, achieving high yields (Scheme 20). By utilizing 4-trifluoromethylaniline as a transient directing group, in combination with a pyridone ligand, the chlorination of benzaldehydes was carried out with excellent yields. The reaction demonstrated broad functional group tolerance, accommodating nitrile, ester, and sulfonamide functionalities on the aldehyde component. Furthermore, a celecoxib derivative could undergo chlorination at a late-stage transformation, where the imine directing group took precedence over any other coordinating functionalities on the substrate. Pyrroles, indoles, and benzothiophene carboxaldehydes were also subjected to chlorination; however, these reactions yielded lower results. Additionally, a dimeric palladacyclic intermediate was isolated, which was amenable to chlorination under the standard conditions used in this study.74
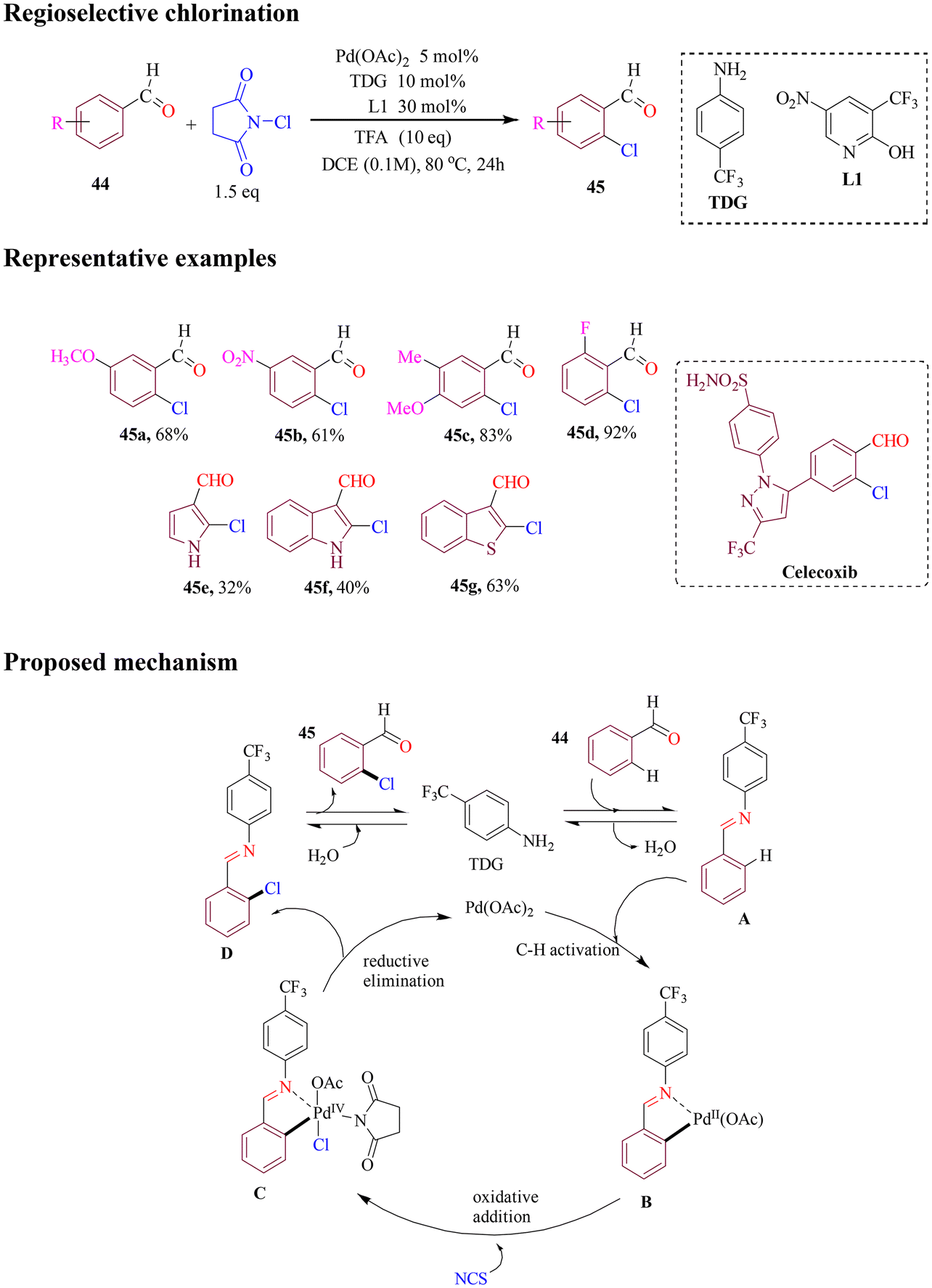 | ||
| Scheme 20 Palladium-catalyzed ortho C–H chlorination of benzaldehydes. | ||
In 2021, La et al. introduced the first palladium-catalyzed direct ortho C–H iodination of benzaldehydes using optimal monodentate transient directing group 2,5-bis(trifluoromethyl)aniline under mild optimized reaction conditions (Scheme 21). The method demonstrated broad substrate scope and practicality, with benzaldehydes and heteroaromatic aldehydes reacting well with readily available N-iodosuccinimide (NIS) to deliver moderate to good yields. The synthetic utility of this methodology was further showcased in two-step total synthesis of the natural product hernandial, involving a subsequent copper-catalyzed cross-coupling reaction. The mechanism involved in situ condensation of benzaldehyde with MonoTDG to form a transient imine, which then coordinated with palladium acetate, leading to ortho C–H activation; oxidative addition, followed by reductive elimination, formed the iodinated product, regenerating both the transient directing group and the palladium catalyst.75
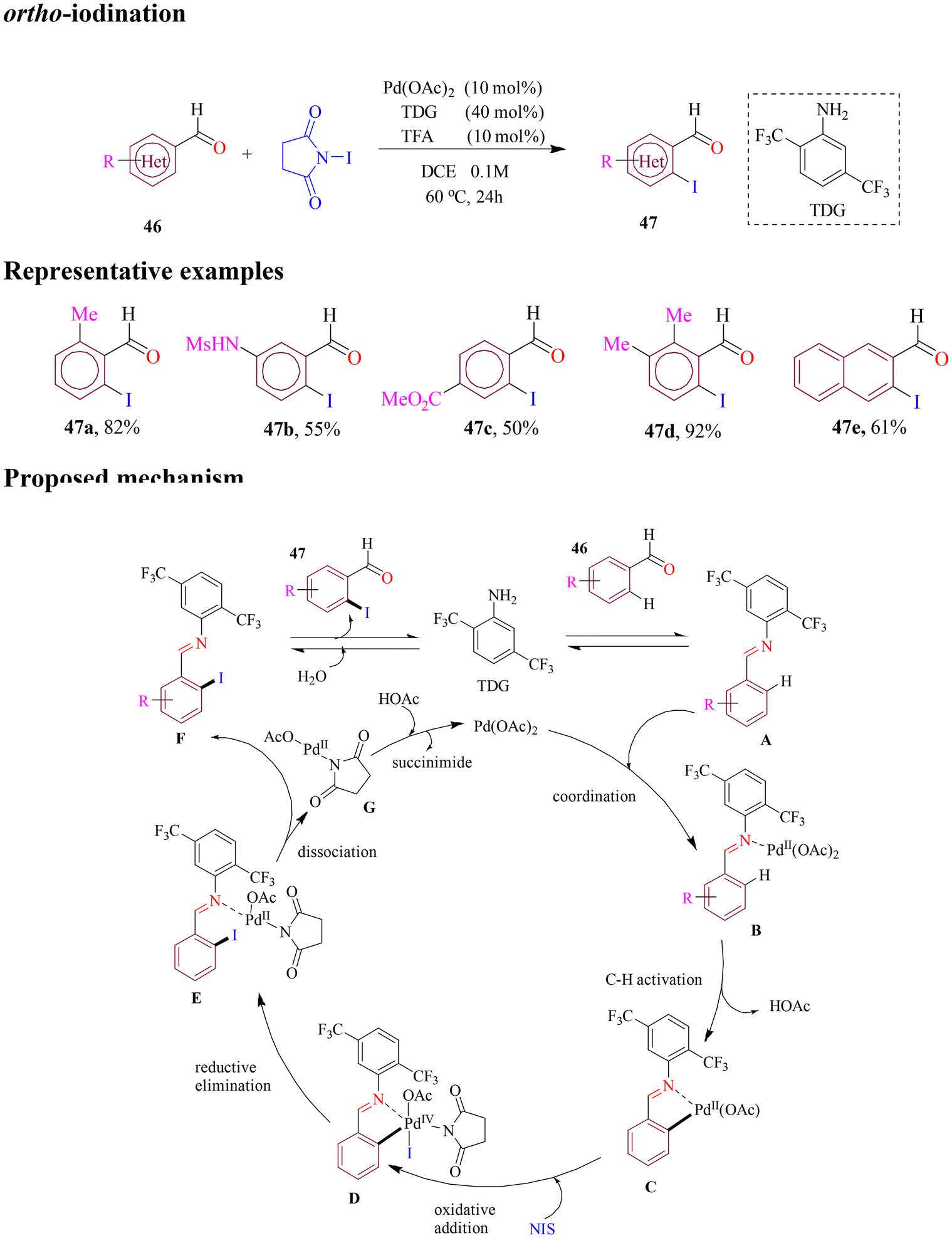 | ||
| Scheme 21 Palladium catalyzed ortho C–H iodination of benzaldehyde and aromatic aldehyde. | ||
In the same year, Zhou and co-workers developed a protocol to direct palladium-catalyzed site-selective C–H chlorination, bromination and iodination of indole-3-carbaldehydes with the assistance of anthranilic acid as a suitable bidentate transient directing group (Scheme 22). The optimized reaction conditions probed for its substrate scope, delivering desired targets in good to excellent yields, possessing various electron-donating and electron-withdrawing groups without the influence of a protecting group on the reaction. It has been found that adding silver salts improved the yields for bromination but had negligible effect on chlorination and iodination. The protocol's applicability was demonstrated in the total synthesis of lysergic acid, Suzuki, Sonogashira, Stille and Miyaura couplings. The experimental results led to the catalytic cycle where imine is generated in situ through condensation, acting as a bidentate coordinating group and coordinating with the palladium centre.76
 | ||
| Scheme 22 Site-selective C–H halogenation of indole-3-carbaldehydes. | ||
In 2021, Wu et al. successfully achieved the ortho-fluorination of aromatic ketones utilizing palladium acetate and the additives carbamate and nitrate (Scheme 23). This developed protocol showed good tolerance for a variety of electron-donating and electron-withdrawing groups, resulting in good to excellent yields. Additionally, experiments were also conducted without carbamate additive, highlighting its significance in promoting fluorination, as it facilitates the in situ generation of imine during the reaction.77
 | ||
| Scheme 23 Palladium-catalyzed ortho C–H fluorination of aromatic ketones. | ||
Wang et al. developed a palladium-catalyzed method for regioselective ortho C–H chlorination of α-ketoesters using commercially available aniline as an efficient monodentate transient directing group (MonoTDG) under mild conditions (Scheme 24). The strategy was tested on a variety of substituted α-ketoesters, yielding good to excellent results. Remarkably, even in cases where the ortho-reactive site was sterically hindered, the C–H chlorination proceeded efficiently with excellent site selectivity, highlighting the robust directing capability of this MonoTDG approach. Based on experimental findings, the proposed catalytic cycle begins with in situ condensation of the aryl α-ketoester with TDG to form a transient imine. This is followed by ortho C–H activation, oxidative addition, and reductive elimination, ultimately delivering the desired chlorinated products.78
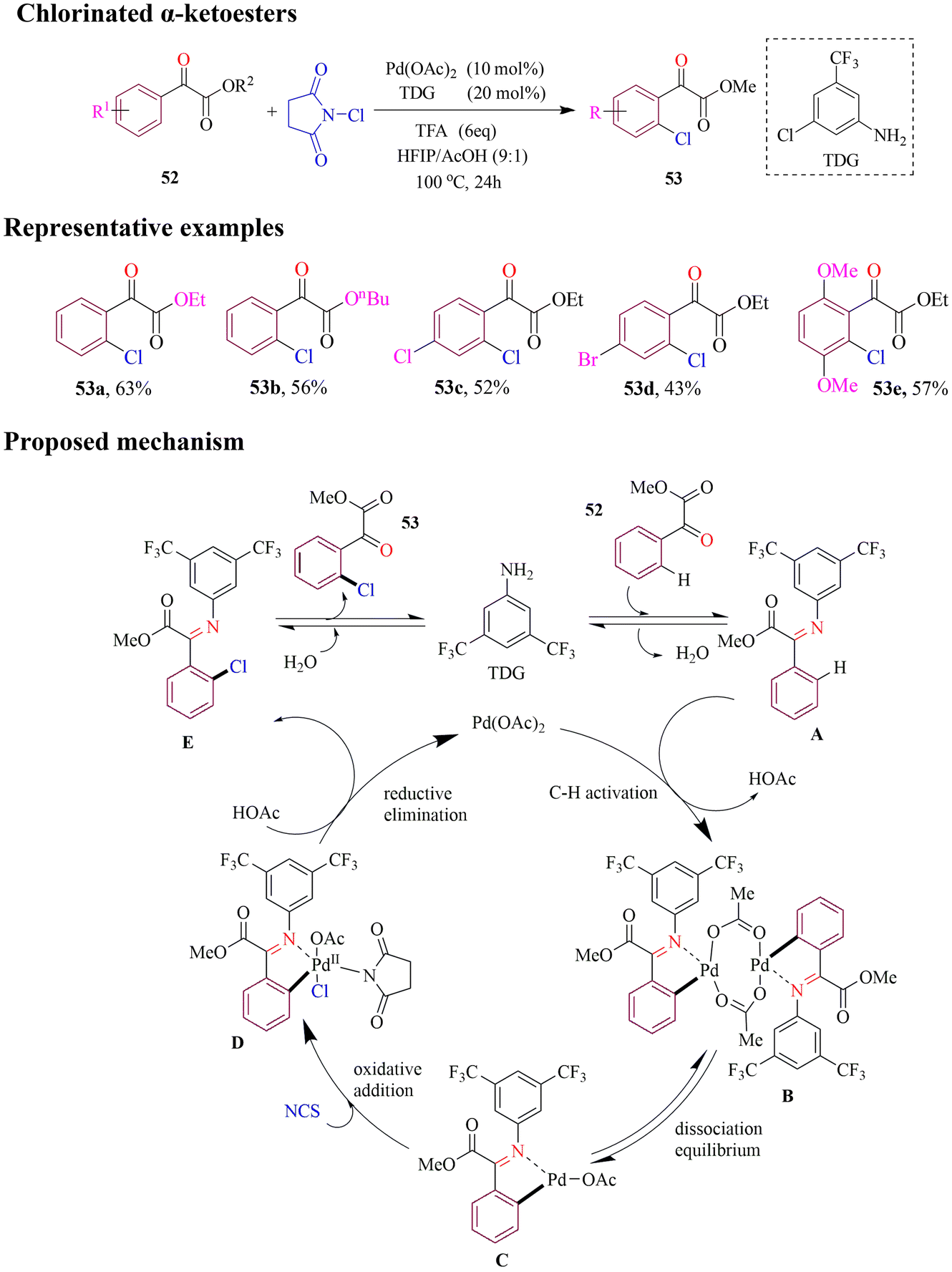 | ||
| Scheme 24 Palladium-catalyzed ortho C–H chlorination of α-ketoesters. | ||
2.7. Carboxylic acid as directing group
Various directing groups are available to achieve C–H activation and further functionalization. The ideal directing group can be considered as omnipresent, small and capable of being easily transformed or removed after the C–H functionalization step. The carboxylate group is considered to have attractive functionality for fulfilling these specifications by regioselective C–H bond activation, and can also act as a traceless directing group. This group shows its versatile activity with its abundance in organic molecules, its ability to be removed via CO2 tracelessly by catalytic extrusion, and its conversion to other moieties. Additionally, the carboxylate group can act as a leaving group in decarboxylative couplings, facilitating the formation of C–C or C–X bonds at the ipso position.79 Although the carboxylate group is inherently meta-directing, transformations mediated by transition metal carboxylates can direct functionalization to the ortho position. This unique behavior further underscores the carboxylate group's utility in directed C–H functionalization.
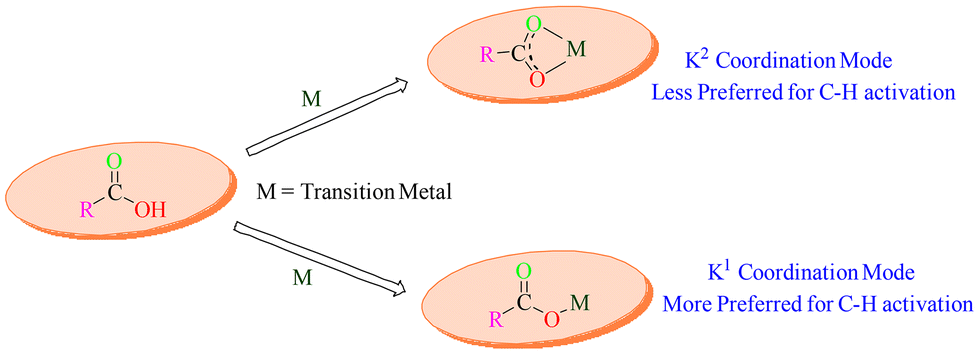
Moreover, there are two coordination modes utilized by the carboxylate group mediated by metal to carry out the transformation, namely K1 and K2 coordination modes. In K1 mode, the metal coordinated with one oxygen and activates the target C–H bond by remaining intact in a better conformation. In contrast, the K2 mode involved coordination of both oxygen atoms to the metal, which spatially isolated the metal centre from the C–H bond, thereby hindering its activation. Different metals prefer different coordination modes, but K1 is considered to be the more effective strategy. From this perspective, to carry out C–H activation, the carboxylate group has proved to be a better alternative.
In 2019, Zhang et al. developed a metal-free method for synthesizing 2-halo pyridine derivatives. This methodology involved decarboxylative bromination and chlorination of 2-picolinic acids using a dihalomethane reagent in the presence of t-BuOCl and NaHCO3 under an air or oxygen atmosphere (Scheme 25). The same reaction conditions could also be extended to decarboxylative halogenation of carboxylic acids derived from quinoline and isoquinoline, yielding halogenated products. However, when performing decarboxylative bromination of heteroaryl carboxylic acids with CH2Br2, the reaction produced the desired heteroaryl bromides along with a small amount of chlorinated biproducts. This is due to in situ generation of a mixed dihalomethane (CH2BrCl) during the reaction. The proposed reaction pathway includes the generation of Cl+ and t-BuO− in the presence of oxygen followed by a decarboxylative process to form a ylide, which transforms into a carbene intermediate, ultimately leading to the desired product.80
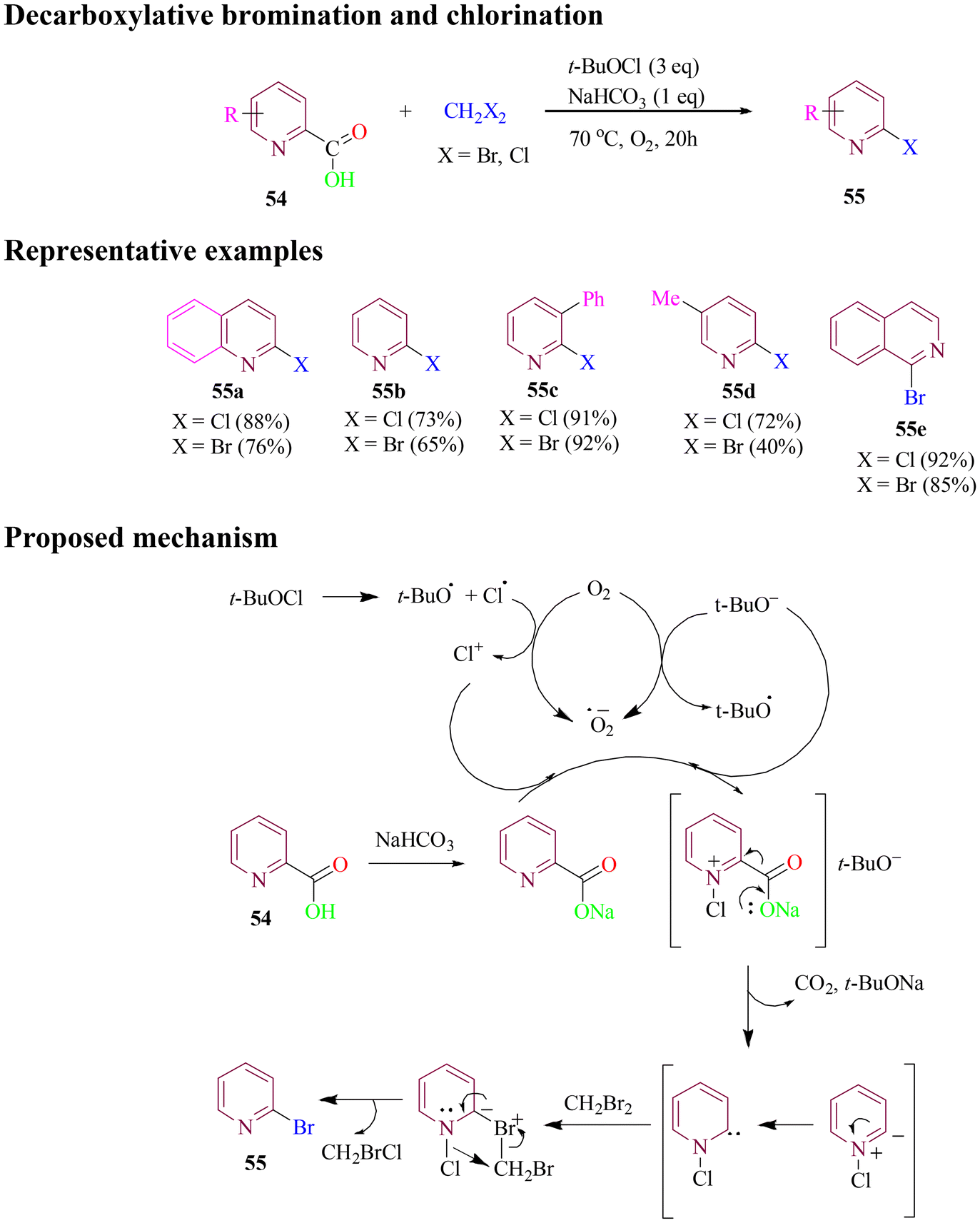 | ||
| Scheme 25 Decarboxylative bromination of 2-picolinic acid. | ||
Barak et al. reported a method for microwave-assisted decarboxylative bromination and iodination of 4-isoxazole carboxylic acids. This approach utilized readily available NXS (where X = I or Br) as the halogen source, with K3PO4 as the base in 1,4-dioxane at 150 °C. This method efficiently yielded bromo- or iodo-isoxazoles in good yields (Scheme 26). Interestingly, proto-decarboxylation products were formed when the reaction was conducted at a higher temperature of 210 °C, in the presence of K3PO4 regardless of whether NCS or NBS was included. The methoxy (OMe) group at the 3-position of isoxazole exhibited significantly higher reactivity compared with fluorine (F), nitro (NO2), and amino (NH2) groups. Moreover, only 3-nitro derivatives could yield iodinated products, while hydrazine and amino groups did not produce any halogenated products. The mechanistic pathway was proposed to proceed via a concerted decarboxylation that led to formation of halogenated products.81
 | ||
| Scheme 26 Decarboxylative bromination and iodination of 4-isoxazole. | ||
In 2020, Matute and co-workers performed a mild and simple method for Ir-catalyzed regioselective monoiodination of benzoic acids. This approach utilized iridium catalyst and iodine as an iodinating agent in protic solvent of HFIP at room temperature for 18 h (Scheme 27). The method was compatible with nearly all functional groups attached to the benzoic acid even in the presence of competing directing groups as substituents in benzoic acids. However, the silver salt was crucial for the selective iodination of the benzoic acid substrates. These reactions outlined a catalytic cycle that proceeded through C–H activation, oxidative addition, and reductive elimination. These findings indicated that the rate-limiting step in C–H activation occurred via a concerted metalation–deprotonation pathway, ultimately leading to the desired products.82
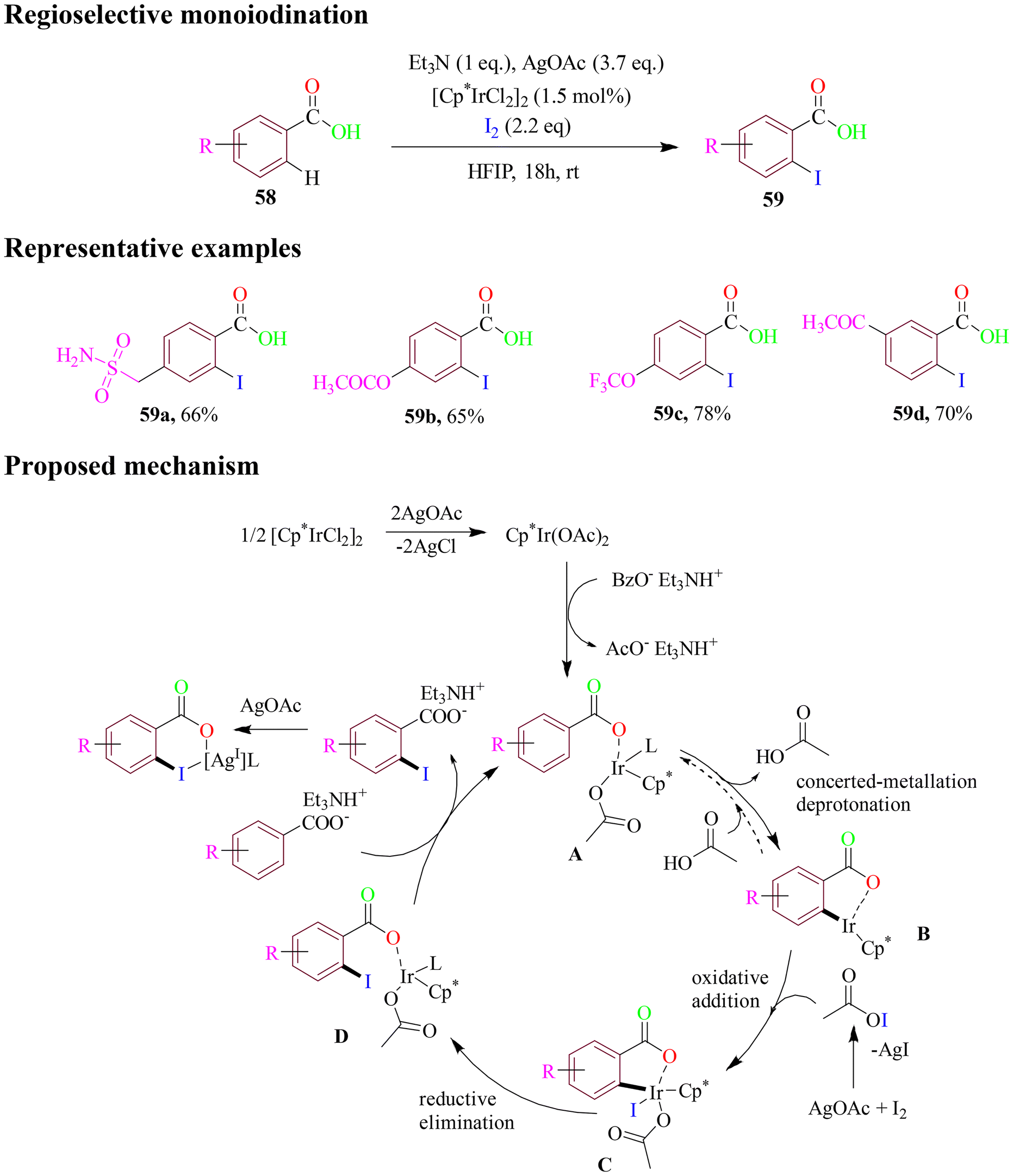 | ||
| Scheme 27 Regioselective iodinated derivatives of various substituted benzoic acid scaffolds. | ||
Ritter and group utilized a mild and versatile approach for aromatic fluorination (Scheme 28) through a low-barrier photoinduced ligand-to-metal charge transfer (LMCT)-enabled radical decarboxylative carbometallation that formed high-valent aryl copper(III) intermediates. The optimized protocol overcame the harsh reaction conditions, limited substrate scope, and the poor functional group tolerance typically associated with aromatic decarboxylation. The selective LMCT excitation–homolysis sequence of copper(II) carboxylates allowed for late-stage fluorination of several complex small molecules.83
 | ||
| Scheme 28 Decarboxylative fluorination of aromatic benzoic acids. | ||
In another approach, the Li group performed Pd(II)-catalyzed selective ortho- and meta-C–H iodination of arenes assisted by aliphatic carboxylic acid directing group via formal metathesis. The protocol utilized 2-nitrophenyl iodide as the iodinating agent where a range of iodinated hydrocinnamic acids and arenes were obtained in good yields (Scheme 29). To gain insights into the mechanistic pathway, a series of control experiments was conducted. These experiments suggested a catalytic cycle that involved the initial coordination of the active palladium catalyst with substrate, followed by a metalation–deprotonation process. Subsequently, oxidative addition occurred, culminating in reductive elimination to yield the desired product.84
 | ||
| Scheme 29 Pd-catalyzed C–H iodination of arenes using aryl iodide. | ||
The Li group reported similar research on the Pd(II)-catalyzed carboxylic group-assisted regioselective and chemoselective iodination of phenethylamines, benzylamines, and 2-aryl anilines (Scheme 30). This method employed 1-iodo-4-methoxy-2-nitrobenzene as a mild iodinating reagent. As a result, various valuable meta-iodinated and even multi-halogenated amines were smoothly synthesized.85
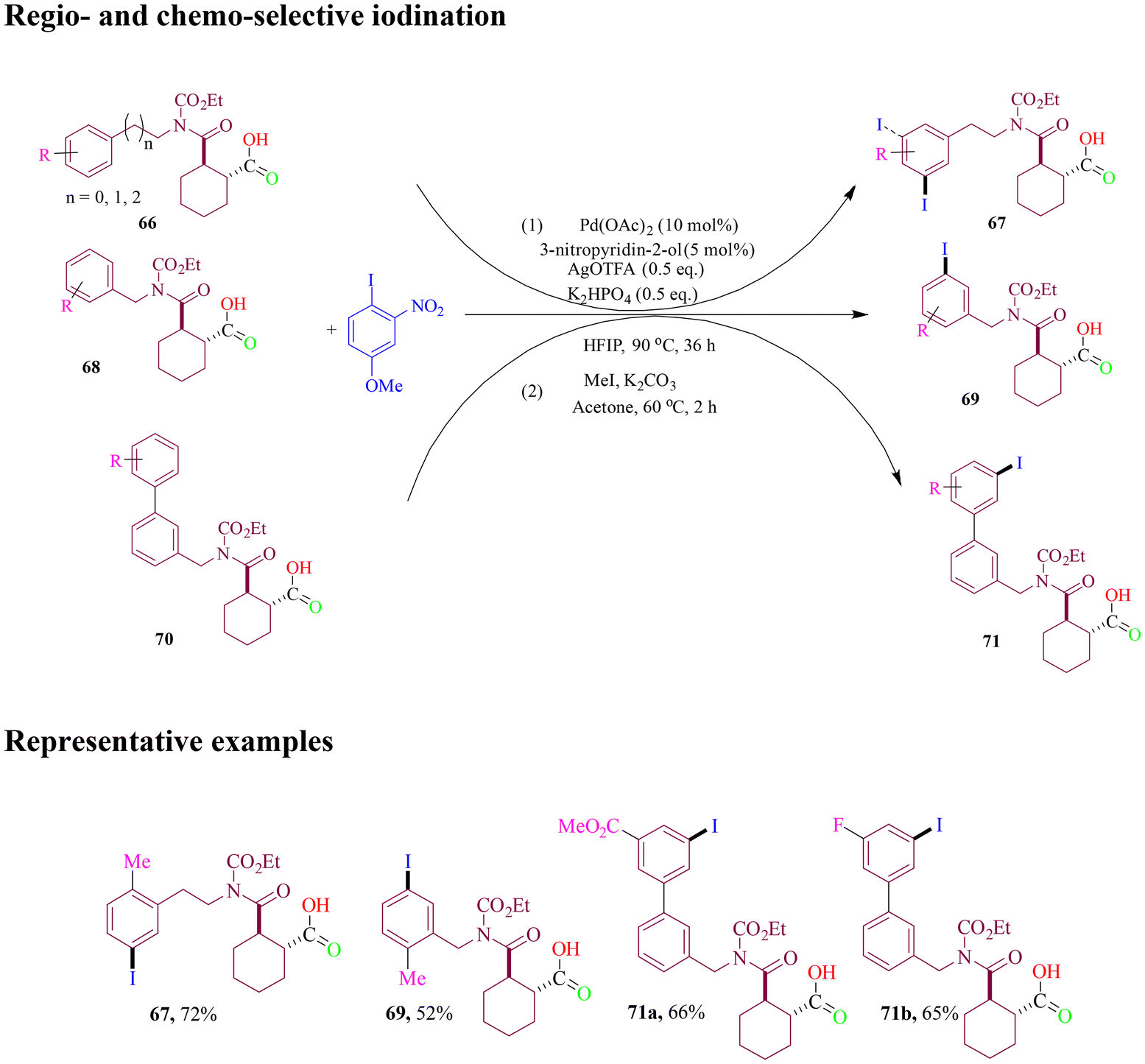 | ||
| Scheme 30 Carboxylic group-assisted meta-iodination of phenethylamines, benzylamines, and 2-arylanilines. | ||
2.8. Other heterocycles as directing group
Like other directing groups discussed so far, heterocycles serve as a common structural motif used to direct C–H functionalization. The C(sp2)-hybridized nitrogen atoms in heterocyclic directing groups act as coordinating sites, facilitating the formation of metallacyclic intermediates crucial for C–H functionalization. This property has garnered significant interest due to the prevalence of heterocycles in bioactive molecules. Heterocyclic directing groups offer advantages such as the absence of a need for installation or removal, making them suitable for late-stage modifications. When installation is required, it can be achieved through various straightforward methods. Additionally, substitutions on heterocyclic scaffolds can modulate their coordinating properties, enabling customization for specific transformations.In 2019, Kommagalla et al. reported cobalt-catalyzed chelation-assisted C–H iodination of aromatic amides using molecular iodine as an iodinating reagent. The reaction was carried out under an atmosphere of air using Co(OAc)2·4H2O as an efficient catalyst with 4-chloro-2-(4,5-dihydrooxazol-2-yl)aniline serving as the directing group (Scheme 31). The reaction showed a wide range of functional group tolerance. Studies revealed that Ag2CO3 was an essential component of the reaction for promoting the iodination and eliminating the formation of byproducts. Kinetic studies and Hammett study revealed that the present C–H iodination approach was likely to proceed through the coordination of 72 with Co(II) species, formation of an arenium ion intermediate, attack of I2 on C, and further the protonation of intermediate E gives the iodination product 73 with the regeneration of the Co(III) species.86
 | ||
| Scheme 31 Cobalt-catalyzed direct C–H iodination of aromatic amides. | ||
Hong et al. designed a method for C–H chlorination, bromination and iodination of 2-arylbenzo[d]oxazoles in different catalytic systems. In the presence of a ruthenium catalyst, halogenation occurred at the C-7 position, whereas when a rhodium catalyst was used, an ortho-selective product was formed with C7-halogenation on 5-methyl-2-(p-substituted)arylbenzo[d]oxazoles as a side reaction (Scheme 32). The established systems delivered products with good to excellent yields, suggesting that the reaction was tolerable to various substrates. Mechanistic experiments and DFT calculations revealed that C7-halogenation catalyzed by Ru proceeded via a single-electron-transfer (SET) radical process, while the Rh-catalyzed ortho-selective halogenation occurred through a redox-neutral SN2 type mechanism. Moreover, the charge difference between benzo[d]oxazolyl and aryl rings led to the different selectivity of Rh-catalyzed halogenations.87
 | ||
| Scheme 32 Ru & Rh-catalyzed C7 and ortho-selective halogenation of 2-arylbenzo[d]oxazoles. | ||
Furthermore, in 2020, He et al. synthesized palladium-catalyzed direct mono- and poly-halogenated derivatives of benzothiadiazole (BTD) where the BTD ring acted as both important nuclei for materials science and as a directing group, which would allow the rational tuning of the physical and chemical properties of BTD derivatives. Here PIDA was used as oxidant, acetic acid as additive, and NaX as halogenating agent for chlorination and iodination, but for bromination NBS gave the best results (Scheme 33). The designed strategy was well tolerated by various substituted benzothiadiazole candidates, where electron-rich substrates reacted much faster than the electron-deficient ones. These derivatives exhibited good solubility and absorbed strongly in the 230 to 380 nm UV range, and have potential applications in materials science.88
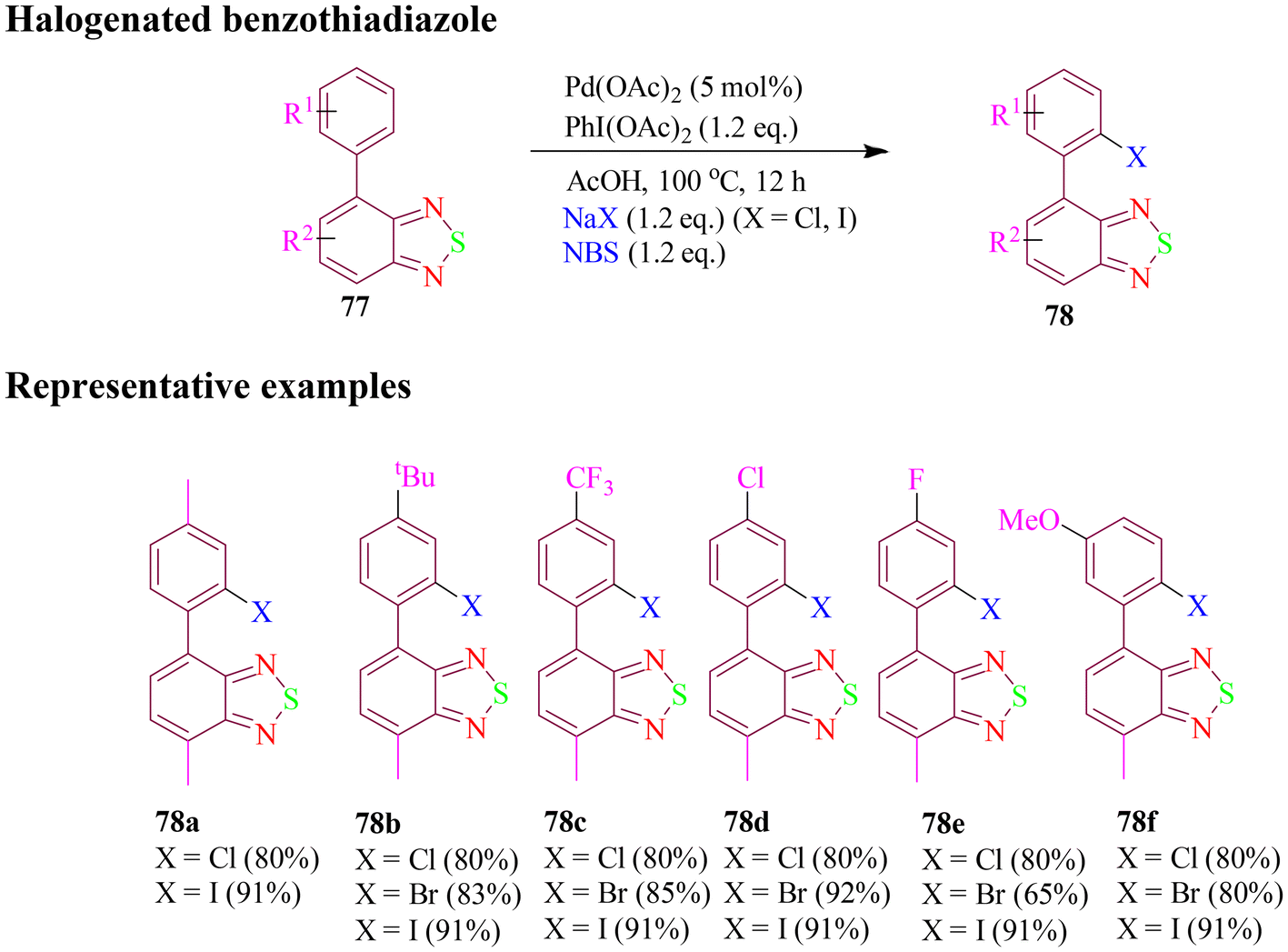 | ||
| Scheme 33 Palladium-catalyzed direct halogenation of benzothiadiazole derivatives. | ||
Koley and co-workers explored palladium-catalyzed regioselective ortho-chlorination and bromination of indolines and tetrahydroquinolines. The method required the addition of CuO to avoid the formation of side products (Scheme 34). The optimized reaction conditions were well tolerated on various substrates bearing electron-donating and electron-withdrawing groups, and obtained good to excellent yields. The applicability of the designed method was demonstrated by synthesizing various valuable synthetic scaffolds, including primaquine and precursors of hippadine and pratosine.89
 | ||
| Scheme 34 Pd-catalyzed chlorination and bromination of indolines and tetrahydroquinolines. | ||
In 2022, the Roger group developed a palladium-catalyzed, N-directed selective ortho-halogenation protocol under atom-economical conditions (Scheme 35). The method is compatible with various functionalized aromatic rings, including pyridine, pyrimidine, pyrazole, oxazoline, naphtho[1,2-d]thiazole, azobenzene, and selectivity-challenging nitrogen-rich s-aryltetrazines. The protocol employed alkali halides as a nucleophilic halogen source and PIDA as the oxidant. The use of microwave irradiation significantly reduced the reaction time and enhanced the efficiency of the synthesis.90
 | ||
| Scheme 35 Selective halogenation of s-tetrazines and other heteroaromatics using alkali halides. | ||
Recently, Zheng et al. disclosed a novel strategy for enantioselective C–H iodination of isoquinolines under mild reaction conditions. NIS was used as the iodinating agent, catalyzed by chiral CpRh(III) complex, affording a series of axially chiral biaryl iodides in excellent yields with enantioselectivity (Scheme 36). The protocol was also compatible with atroposelective C–H chlorination and bromination. The obtained iodide products could be further transformed to other compounds containing C–C, C–N and C–P bonds. Experimental results suggested a plausible catalytic cycle where the desired products comprised two pathways: (a) oxidative addition of NIS to form CpRh(V) complex, followed by reductive elimination assisted by AcOH, or (b) direct nucleophilic substitution of NIS with rhodacycle.91
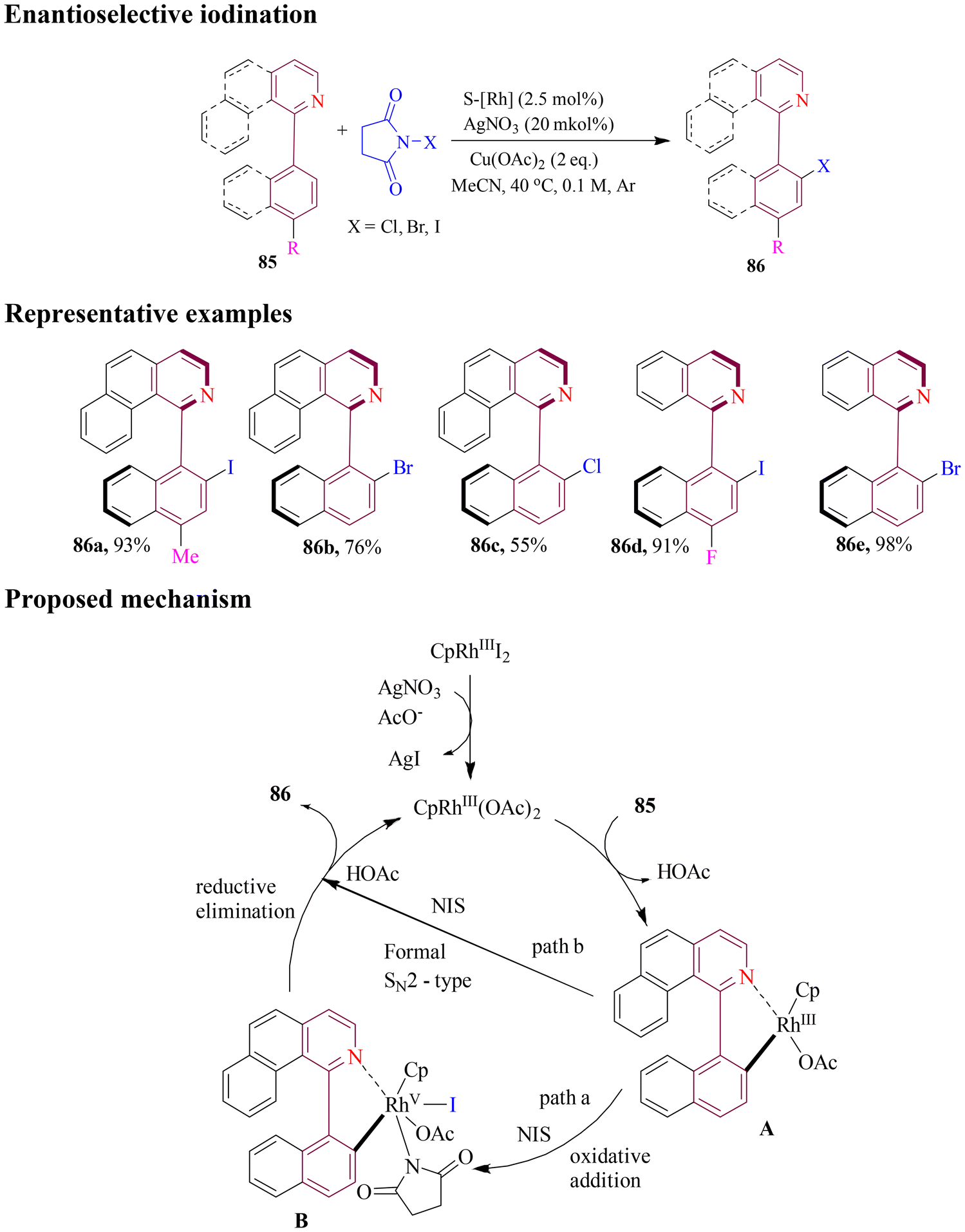 | ||
| Scheme 36 Rh-catalyzed enantioselective C–H iodination of isoquinolines. | ||
2.9. Miscellaneous directing group
Sunoj and Yu reported the fluorination of methylene and methyl groups in aliphatic substrates via F+ oxidants, with distinct methodologies tailored for each substrate class (Scheme 37). Both methods employed 2-hydroxylnicotinaldehyde as a transient directing group and a pyridone ligand. Methylene fluorination required silver salts, while methyl fluorination proceeded without silver, highlighting the contrasting roles played by silver in these reactions. N-Fluorosuccinimide (NFSI) was identified as the most effective oxidant and fluorine source for methylene substrates. It was employed alongside silver trifluoroacetate, pyridone A in superstoichiometric amounts, and a mixed solvent system of HFIP and PhCl. The resulting fluorinated amines were isolated as benzoyl-protected derivatives. Mechanistic studies suggested a Pd(II)/(IV) catalytic cycle involving three key steps: (a) C–H activation, (b) oxidative addition to N–F bond, and (c) reductive elimination, which formed the desired C–F bond. This efficient methodology expanded the toolbox for selective aliphatic fluorination.92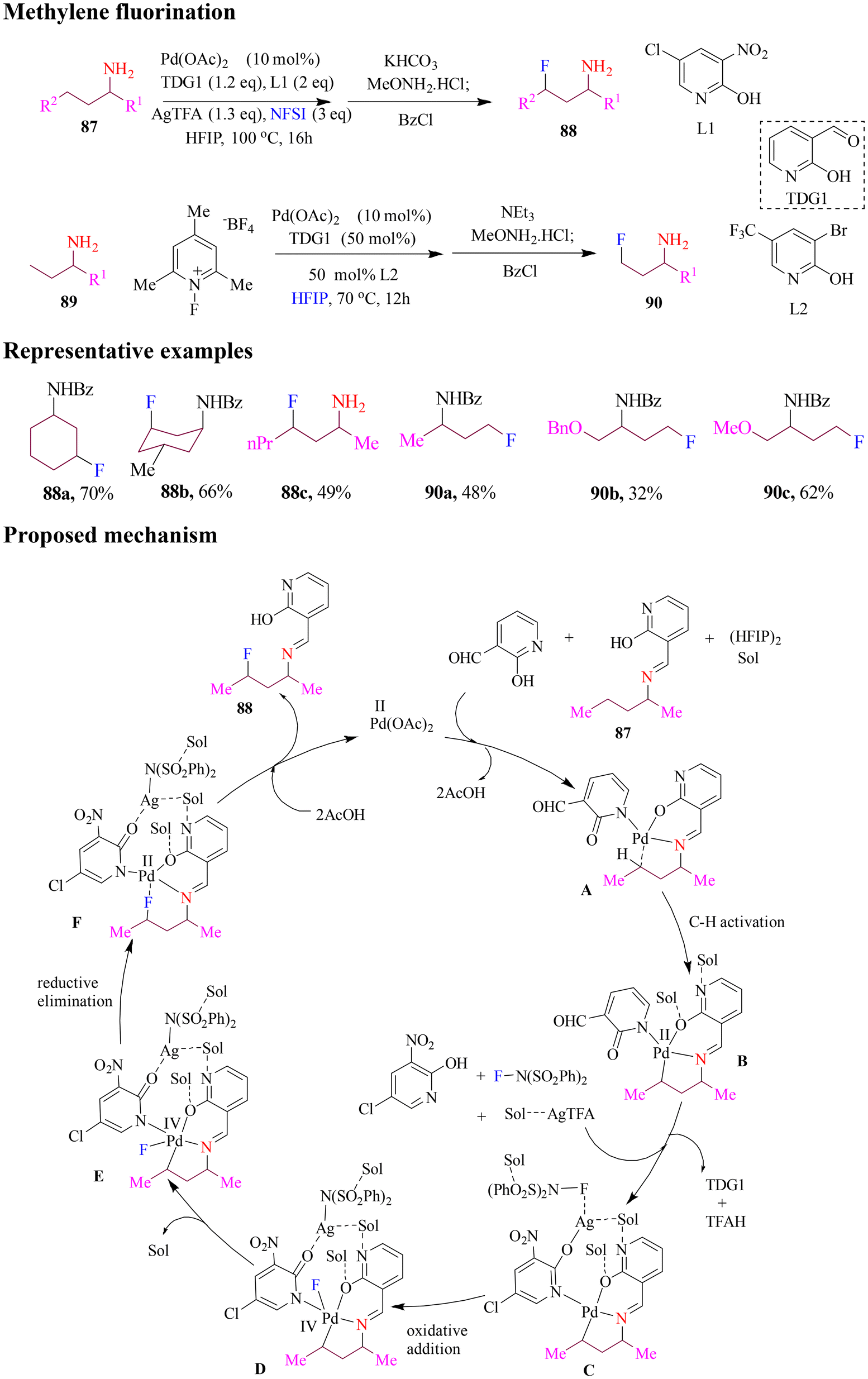 | ||
| Scheme 37 C(sp3)–H fluorination of aliphatic amines catalyzed by palladium. | ||
In 2021, Chan and group designed a fascinating approach for site-selective chlorination of aliphatic γ C–H bonds of ketones, enones and alkylbenzenes without the use of a transient directing group. The cheap and mild protocol utilized copper triflate as a catalyst and dichloramine-T as a chlorinating agent at room temperature (Scheme 38). The optimized reaction conditions’ efficiency was determined by employing a series of secondary or tertiary γ-carbon centres of ketones, (E)-enones and alkylbenzenes. Mechanistic studies supported DFT calculations which proposed that the reaction pathway proceeded through a single-electron mechanism, showing the possibility of two different pathways, i.e., inner-sphere and outer-sphere mechanisms, which competed with the generation of radicals and formation of metal complexes.93
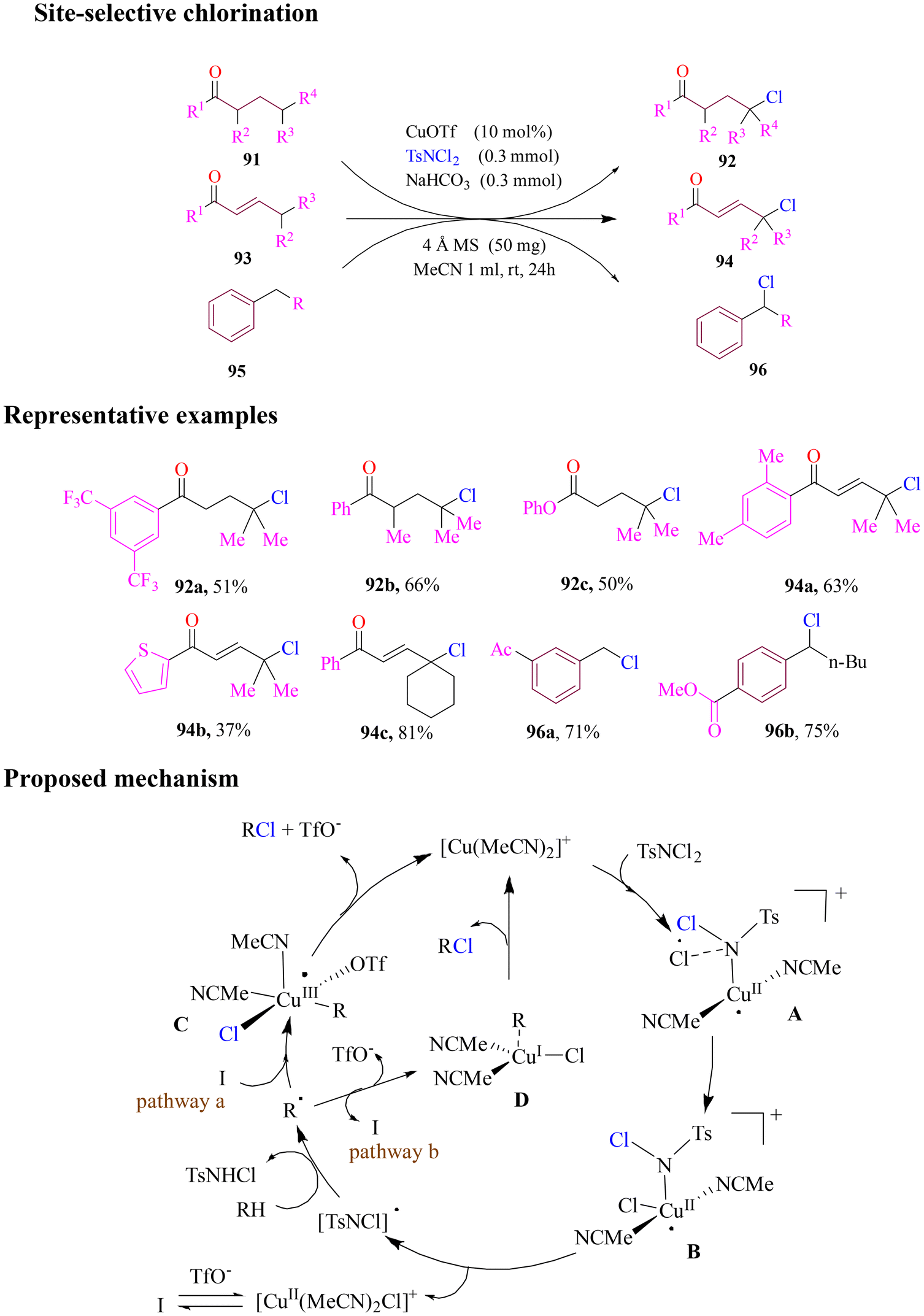 | ||
| Scheme 38 Site-selective chlorination of ketones, (E)-enones and alkylbenzenes by dichloramine-T. | ||
The Bollikolla group established ortho-chlorination, bromination and iodination of arylcyanamide using a Pd(II) catalyst under mild reaction conditions (Scheme 39). The optimized reaction conditions readily delivered the desired products in moderate to good yields with both electron-donating and electron-withdrawing groups. The mechanism proceeds with metal coordinating to the directing group, followed by the formation of a palladacycle through ligand-assisted C–H bond activation. Subsequently, oxidative addition of NXS formed a Pd complex, which underwent reductive elimination to yield the desired product.94
 | ||
| Scheme 39 Pd-catalyzed regioselective halogenation of arylcyanamide. | ||
Recently, Feng et al. reported the first selective ε-C(sp2)–H iodination of 3-arylpropan-1-amines using the unprotected NH2 group as a native directing group, with palladium as the catalyst (Scheme 40). This reaction proceeded via the formation of a seven-membered palladacycle, which was less kinetically favourable than five- or six-membered palladacycles. Under optimized conditions, the reaction displayed a broad substrate scope, with substituted amines delivering the desired iodinated products in excellent yields. Furthermore, the iodination products could undergo copper-catalyzed cyclization in a single step to form 1,2,3,4-tetrahydroquinolone, a key structural motif in drugs, dyes, and natural products.95
 | ||
| Scheme 40 Palladium-assisted ortho-selective C–H iodination of primary amines. | ||
2.10. Regioselective electrophilic halogenation
The direct halogenation of compounds is one of the most fundamental and frequently used reactions in organic chemistry. Regioselective electrophilic halogenation, influenced by directing groups, has gained prominence in recent years. In this approach, the electronics of the substituent guide the substitution process, utilizing various halogenating reagents under mild reaction conditions. This method does not require transition metal chelation and can proceed through different techniques, including photochemical, electrochemical, and radical halogenation.Xiong et al. achieved the simultaneous C5- and C7-dihalogenation and acylation of 8-hydroxyquinolines under catalyst-free conditions using acyl halides as halogenating agent. However, on slightly changing the reaction conditions and directly using O-acylated 8-hydroxyquinoline, regioselective C5-halogenation could be achieved (Scheme 41). Further, the optimized reaction conditions were employed to explore the substrate scope providing the desired products in moderate to good yield. On the basis of various control experiments, it was found that the dihalogenation proceeded through a free radical pathway while the selective halogenation occurred through an electrophilic mechanism.96
 | ||
| Scheme 41 C5- and C7-dihalogenation, and selective C5-halogenation of 8-hydroxyquinoline. | ||
In 2020, Bian et al. developed a transition-metal-free methodology that employed cyclic and acyclic aliphatic ketones as directing groups for site-selective γ-C(sp2)–H iodination of aryl compounds (Scheme 42). This transformation offered a broad substrate scope and operated under mild conditions free from air and moisture. The reaction occurred rapidly and was easy to perform, making it both environmentally friendly and scalable. Based on several control experiments, a possible reaction mechanism was outlined which illustrated that the combination of I2 and PIDA was crucial for the iodination process.97
 | ||
| Scheme 42 Aliphatic ketone directed site-selective mono-iodination of arenes. | ||
In 2021, Rode's group reported a facile protocol for C–H functionalization of imidazo[1,2-a]pyridines with different nucleophilic species including TBAC, TBAB, NaI, KSCN, and NaNO2 (Scheme 43) in acetonitrile. The product showed a dominant functionalization at the 3-position of imidazo[1,2-a]pyridines, giving reaction yields ranging from 62% to 81% under mild conditions. The reaction showed a good group tolerance with 2-aryl substitution. Importantly, the C–H fluorination in the absence of any nucleophile proceeded apparently through an ionic mechanism via electrophilic aromatic substitution. In the presence of water, a hydroxylated-fluorinated product was obtained, being essential for the effective C–H functionalization with a nucleophile. No mechanistic evidence was provided for reactions involving a nucleophile, but it is possible to suppose a radical mechanism. The strategy provided a viable route for the halogenation (e.g., chlorination, bromination, and iodination) of imidazo[1,2-a]pyridines, with potential to be extended to other N-heterocyclic systems.98
 | ||
| Scheme 43 C-3 functionalization of imidazopyridine using Selectfluor. | ||
The Sun group developed an efficient, regioselective and metal-free protocol for para C–H bromination of phenylurea. The method used simple and readily available KBr as bromide source, TFA as an additive and PIDA as an oxidant in acetone at room temperature (Scheme 44). The method tolerated a variety of functional groups and allowed the synthesis of diverse 4-brominated phenylurea derivatives in moderate to excellent yields. A series of control experiments was performed to find out the reaction pathway where, initially, iodonium intermediate A was formed through a nucleophilic attack of urea. Then, cleavage of the N–I bond furnished iodobenzene and nitrenium ion B, which was stabilized by the charge delocalization on the phenyl ring. Finally, the extensively charge-delocalized intermediate C reacted with Br to give the para-brominated products 111.99
 | ||
| Scheme 44 Regioselective C–H bromination of urea. | ||
In 2021, Chen and co-workers developed an economical, user-friendly, and environmentally benign method for selective meta-bromination of pyridines. A sustainable electrochemical approach was developed, combining installation and removal steps using TBAB, NaBr, and LiBr as brominating salts at room temperature. This method operated without the need for catalysts or oxidants (Scheme 45), offering an environmentally friendly alternative. A wide range of brominated pyridine derivatives was synthesized in good to excellent yields. The proposed reaction pathway, supported by various control experiments, showed the electrophilic aromatic substitution of pyridine conjugates.100
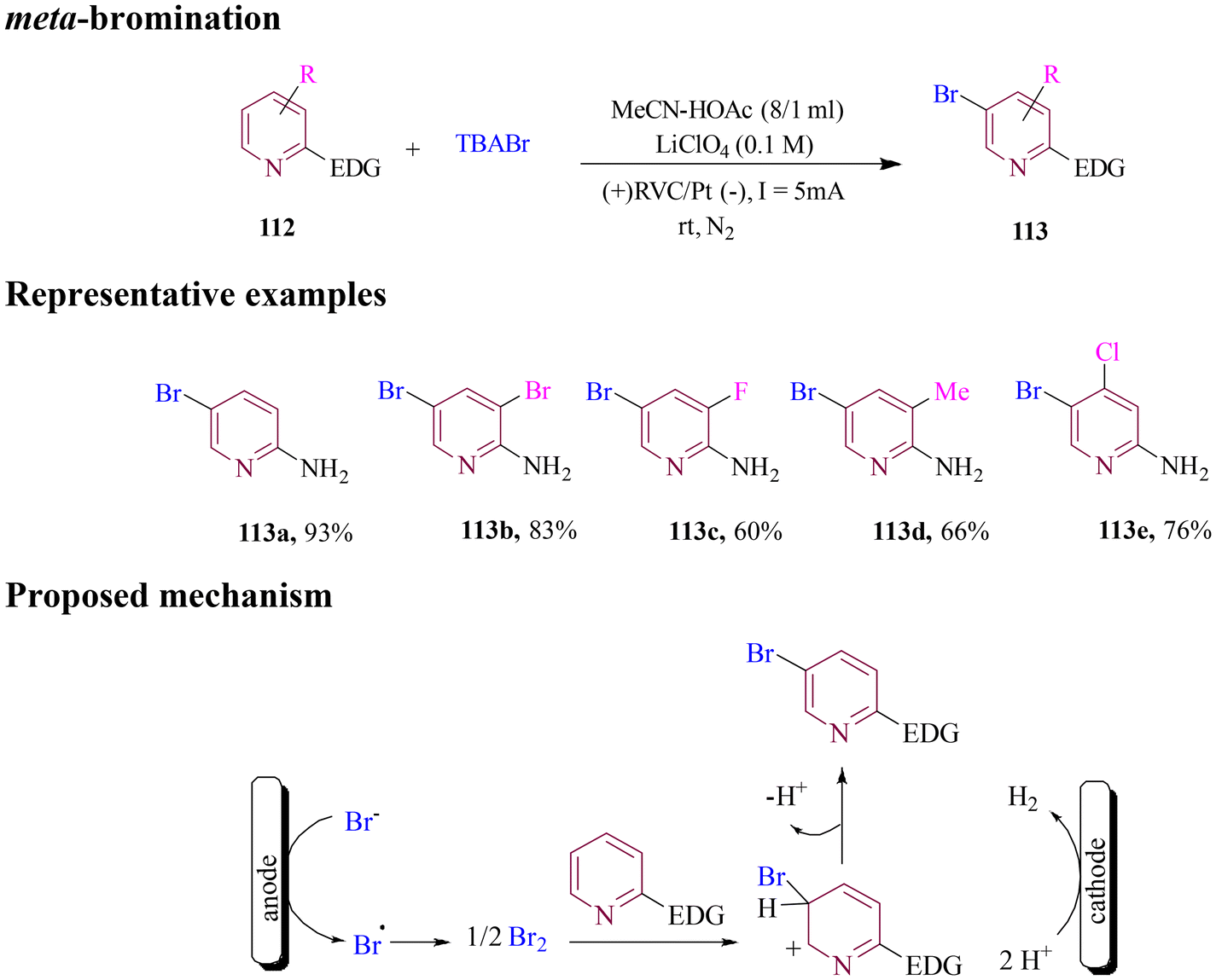 | ||
| Scheme 45 Electrochemical meta-bromination of pyridines. | ||
In 2023, Shinde et al. reported a new protocol for the regioselective ortho-halogenation of N-aryl amides and urea through an oxidative halodeboronation approach. After optimizing the reaction conditions, the scope of the method was explored by testing various para-substituted N-aryl amides containing electron-donating, halogenated, and electron-withdrawing groups. The protocol successfully produced ortho-iodinated and brominated products with moderate to excellent yields (Scheme 46). The method demonstrated versatility by yielding desirable products from electron-rich heteroaromatics and alkyl-based groups. Control experiments and DFT studies revealed a proposed mechanism in which Selectfluor played a dual role as both a fluoride source and an oxidant. The process involved an oxidative ligand exchange-removed boron, followed by ipso-addition and release of the boron species to form the final product. This innovative reactivity of C–B and B–X bonds offers a promising alternative for the functionalization of a broad range of N-heterocycles, opening new avenues in synthetic chemistry.101
 | ||
| Scheme 46 Regioselective ortho-iodination and bromination of N-aryl amides and urea. | ||
Recently, Zhang and group attempted to use activated and un-activated alkyl bromides to synthesize the regioselective brominated derivatives of 8-aminoquinoline amide in the presence of a copper catalyst. The optimal method was employed on various 8-aminoquinoline amides, functionalized with different electron-donating and electron-withdrawing groups (Scheme 47). Activated alkyl bromides gave the desired targets in excellent yield, while un-activated alkyl bromides proceeded with low efficiency following the reactivity order primary > secondary > tertiary. The proposed reaction mechanism began with the dipolar protic solvent DMSO, attacking ethyl bromoacetate to form intermediate A. This intermediate combined with a bromide ion to produce intermediate B, which subsequently interacted with another bromide ion to generate either dimethylsulfonium bromide or a dimethyl thioether/molecular bromine complex (intermediate C). Intermediate C then reacted with 8-aminoquinoline amides via copper-promoted aromatic electrophilic substitution, resulting in the regioselective C5 brominated product.102
 | ||
| Scheme 47 Copper-assisted C5 bromination of 8-aminoquinoline amide using alkyl bromide. | ||
In 2020, the Yu group developed a new photochemical method for the selective chlorination of aliphatic amides. This approach utilized tert-butyl hypochlorite as the chlorinating agent and household CFL as the light source (Scheme 48). The reaction proceeded via a radical mechanism, following a tandem sequence of N-chlorination and photoinduced 1,5-chlorine atom transfer. This process was enabled by the combined action of N-heterocyclic carbene (NHC)·SIPr·HCl and (diacetoxyiodo)benzene (DIB), which promoted N–H chlorination and facilitated the generation of an amide radical through N–Cl cleavage. Under these conditions, a variety of carboxamides and sulfonamides yielded δ-chlorinated products with good efficiency and high selectivity. Notably, the reaction conditions were also compatible with phosphonamides, extending the scope of the methodology. This protocol provides a practical and efficient approach for site-selective chlorination of methyl, methylene, and methine hydrogens, paving the way for advancements in selective aliphatic chlorination.103
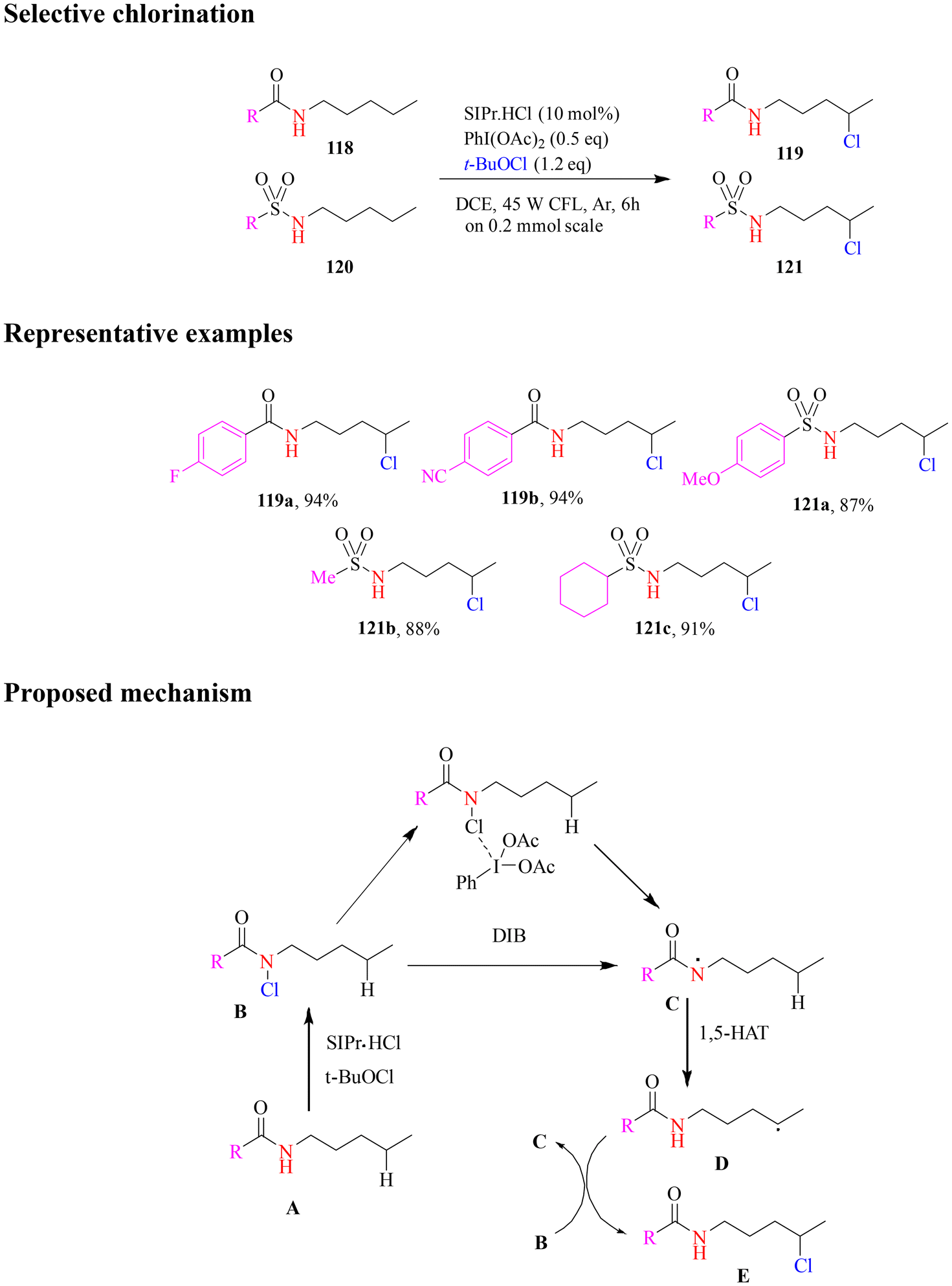 | ||
| Scheme 48 Photoinduced selective chlorination of aliphatic amides. | ||
Under transition-metal-free and mild reaction conditions, Xu and co-workers (2019) achieved one-pot synthesis of highly functionalized N-acetylated and selectively C-5 chlorinated conjugates of 8-aminoquinoline. In this methodology, substituted acetyl chloride acted as both acetylating and halogenating agents (Scheme 49). Additionally, successful results were obtained with brominated derivatives of 8-aminoquinoline by increasing additive loading and extending reaction time. Based on prior literature and experimental findings, a plausible mechanism was proposed. Initially, 8-aminoquinoline (122) reacted with acyl halide to form intermediate A. Subsequently, oxone oxidized the halide anion in the ammonium salt to generate a halogen radical. This radical selectively attacked the C-5 position of the intermediate, forming complex Bvia a single-electron transfer. Finally, the elimination of a proton from complex B yielded the desired product (123).104
 | ||
| Scheme 49 One-pot acetylation and C-5 chlorination of 8-aminoquinoline. | ||
In another notable study, Shu et al. developed a selective method for bromination and chlorination of 8-aminoquinoline using environmentally friendly and cost-efficient visible-light-induced processes. They utilized economical and effective halogenation reagents, specifically 1,3-dibromo-5,5-dimethylhydantoin (DBDMH) and 1,3-dichloro-5,5-dimethylhydantoin (DCDMH), in a continuous flow, which delivered high yields of the desired products within a short reaction time (Scheme 50). The chlorination reaction required an additional oxidizing agent (TBHP), due to the poor reactivity of DCDMH, which extended the reaction time from 0.5 h to 1 h. Moreover, this approach was also successfully applied to sulfonamides, providing moderate to good yields of products. Under visible light irradiation, halogen radicals (Cl˙ or Br˙) and complex A radicals were generated from DXDMH (X = Cl, Br). These radicals reacted with 124 to form radical intermediate B and HBr or complex C. Intermediate B could tautomerize into stable intermediates D and E. Additionally, HBr reacted with complex A to produce X2, which subsequently halogenated intermediates D or E to yield C-5 halogenated product 125. This product could also be obtained through the reactions of halogen radicals with intermediates B or E.105
 | ||
| Scheme 50 Visible light-induced C-5 halogenation of 8-aminoquinoline. | ||
In 2019, Li and co-workers reported bromination and iodination of 8-aminoquinoline or N-acetanilide derivatives. Sodium halides (NaBr, NaI) were employed as halogenating agents at room temperature in the presence of oxidant N-fluorobenzenesulfonimide (NFSI) (Scheme 51). Here, NFSI oxidized NaBr or NaI to obtain Br+ or I+ species which facilitated electrophilic halogenation of arene. This methodology resulted in the formation of dibenzenesulfonimide as a byproduct. However, diphenylsulfonimide is a well-known starting material to synthesize NFSI, making this a potential approach for recycling this stoichiometric oxidant.106
 | ||
| Scheme 51 C–H bromination and iodination of arenes and 8-aminoquinolinamide using sodium halide. | ||
Lei and co-workers developed a selective method for C-5 bromination of 8-aminoquinoline amides using carbon tetrabromide (CBr4) as the bromine source, 10-phenylphenothiazine (PTH) as an organophotoredox catalyst, potassium carbonate (K2CO3) as the base, and a small amount of water. The reaction was performed under blue light irradiation in acetonitrile to produce the expected product (Scheme 52). Both alkyl and aryl-substituted substrates were compatible for this approach. Visible light initially transformed the photocatalyst PTH into its excited form PTH*. A single-electron transfer (SET) process between PTH* and CBr4 generated Br−, ˙CBr3, and PTH˙+. Subsequently, ˙CBr3 oxidized substrate 130 to radical intermediate A and HCBr3. Intermediate A reacted with PTH˙+ to form the carbocation intermediate B, which combined with Br− to generate intermediate C and underwent hydrogen atom transfer (HAT) process to deliver the target product 131.107
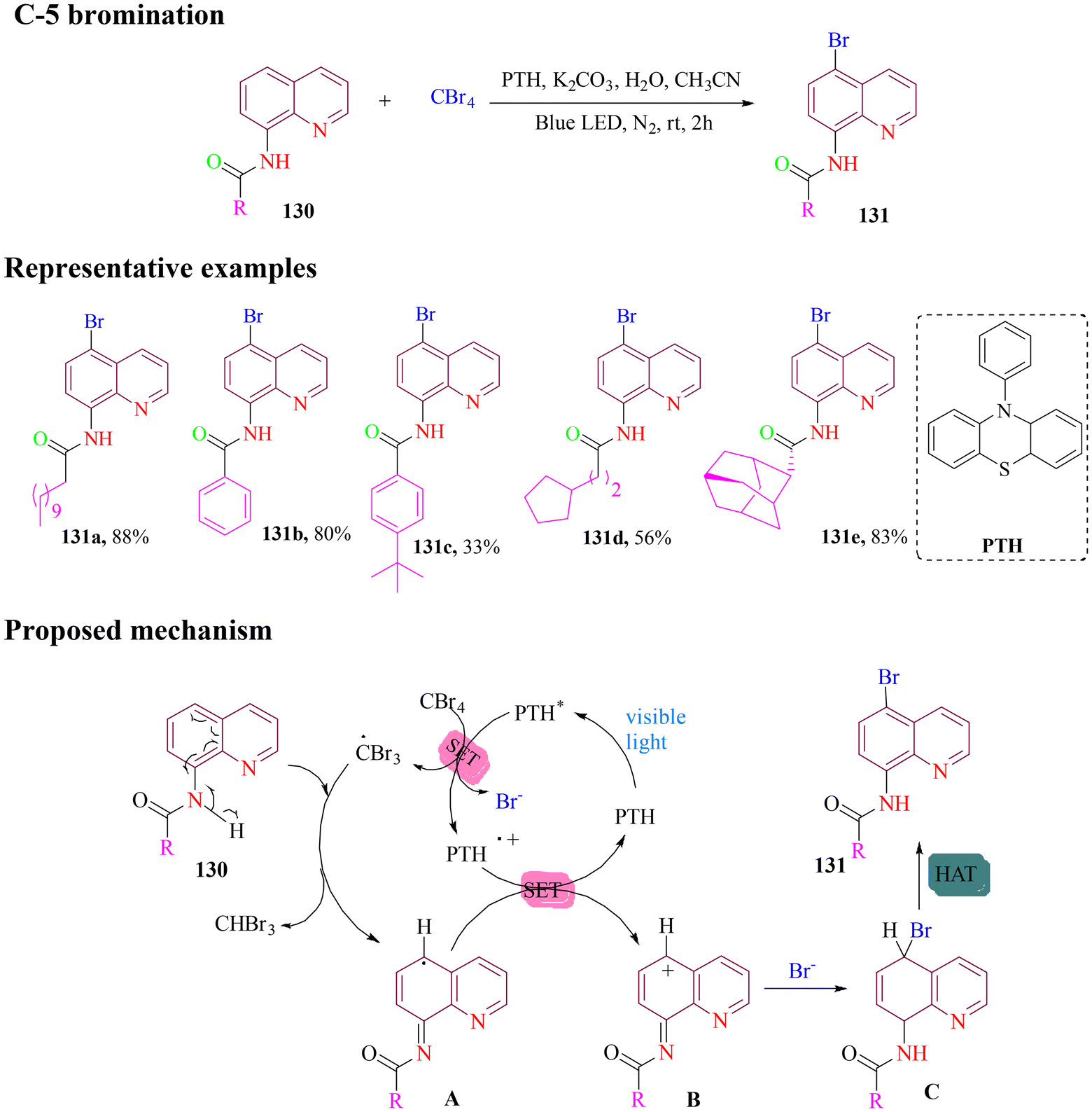 | ||
| Scheme 52 Organophotocatalyst-mediated C5 bromination of 8-aminoquinoline amides. | ||
In 2020, the Xie group successfully achieved the dihalogenation of 8-aminoquinolines at the C5 and C7 positions using NXS (Cl, Br) as the halogenating source through an electrocatalytic process without the use of transition metals and oxidants (Scheme 53). This method circumvents the limitations associated with copper catalysts. Additionally, reducing the reaction time to 3 min under standard conditions achieved a 90% yield of monobrominated product, selectively halogenating the C5 position. Using this monobrominated compound as a reactant in the dihalogenation system led to 97% yield of the target product, demonstrating that the reaction can be controlled stepwise, with the C5 position being more reactive than the C7 position. The proposed mechanism, exemplified with bromination, involves the reduction of NBS at the cathode to generate Br−, which was subsequently oxidized at the anode to produce Br˙. The bromine radical cross-coupled with radical intermediate A, formed via anodic oxidation, to create intermediate B. This intermediate underwent proton transfer to yield the monobrominated product C. The process repeated at the C7 position, forming the dibrominated product 133.108
 | ||
| Scheme 53 Electrochemical oxidative dihalogenation at C5 and C7 positions of 8-aminoquinoline. | ||
Furthermore, Malykhin et al. established a method to access 3-halo-1,2-oxazines from 1,2-oxazine-N-oxides via nucleophilic halogenation utilizing phosphorus oxyhalides or oxalyl halides as halogenating agents. Assisted by TfOH or BF3·Et2O, the halogen substitution in 1,2-oxazines-N-oxide was accomplished under mild conditions (Scheme 54). Moreover, several side processes, including 1,2-oxazine ring contraction and [3,3]-sigmatropic rearrangement, were also observed. Introduction of the azido group at the C-3 position of 1,2-oxazine resulted in cyclization to hitherto unknown tetrazolo[1,5-b][1,2]oxazines. Fragmentation of the 1,2-oxazine ring in 3-halo-1,2-oxazines upon action of strong electrophiles provided a route to unsaturated nitriles and imides.109
 | ||
| Scheme 54 Nucleophilic halogenation of cyclic nitronates. | ||
Wang and co-workers reported novel one-pot synthesis of C3-sulfonate esters and C4-chlorides of quinoline N-oxides that is both chemo- and regioselective. This approach required no metal, oxidant, or additive and employed commercially available sulfonating and chlorinating reagents (Scheme 55). The methodology demonstrated good chemoselectivity and regioselectivity, operated under mild conditions, and exhibited broad functional group tolerance, making it suitable for a wide range of N-oxides. Initially, N-oxide reacted with sulfuryl chloride through an O-acylation step, yielding intermediate A and a chloride ion. Intermediate A then underwent [3,3]-sigmatropic rearrangement to form intermediate B. Next, B was captured by another N-oxide via hydrogen bond interaction, resulting in the formation of intermediate C. Finally, intermediate C was reacted by the chloride ion through nucleophilic substitution, followed by an elimination, resulting in the formation of 137 and 138.110
 | ||
| Scheme 55 Synthesis of C3-sulfonate ester and C4-chloride of quinolines from quinoline N-oxides. | ||
In 2019, Hu et al. successfully generated C–H functionalized chlorinated and brominated products from pyridines and diazines under mild conditions, using lithium chloride (LiCl) and lithium bromide (LiBr) salts, and achieved good to excellent yields with high regioselectivity. The regioselectivity of halogenation in 2-aminopyridines and 2-aminodiazines was significantly influenced by the substituent patterns (Scheme 56). The presence of the nitrogen heterocycle and ortho-amino group adjacent to the heterocycle nitrogen was crucial for converting the starting materials into halogenated products. The authors proposed a radical pathway for the selector-mediated chlorination and bromination of 2-aminopyridines and 2-aminodiazines where a zwitterionic diradical adduct (A–B) was formed through a SET mechanism from N-heteroarene, acting as an electron donor, to Selectfluor which served as the electron acceptor. Meanwhile, the cationic heterocyclic radical (B) underwent deprotonation to form a neutral N-heteroarene radical (C), which could isomerize to yield intermediate D. Intermediate D subsequently reacted with the halogen radical to produce intermediate F, which underwent tautomerization to yield the desired halogenated product.111
 | ||
| Scheme 56 Regioselective halogenation of 2-aminopyridine and 2-aminodiazines in the presence of selectofluor. | ||
Levy et al. developed a strategy for selective chlorination of substituted 2-phenylpyridines and 3-phenylpyridines using a specially designed set of phosphine reagents (Scheme 57). The heterocyclic phosphines were introduced at the 4-position of pyridines as phosphonium salts and were subsequently displaced by halide nucleophiles. In this process, the phosphine was added to triflate, activated 2- and 3,5-disubstituted pyridine rings, forming dearomatized adducts. Computational studies revealed the formation of the C–X bond through a stepwise SNAr pathway, which necessitates the N-activation of the pyridyl group. Steric interactions between the departing phosphine and the pyridyl substituents are particularly significant during the cleavage of C–P bond, which explained the differences in reactivity that were observed between 2- and 3-substituted pyridines. This method allows for halogenation of a wide range of un-activated pyridines and is applicable for late-stage halogenation of complex pharmaceuticals.112
 | ||
| Scheme 57 Selective chlorination of pyridines using phosphine reagents. | ||
The Sekar group developed an atom-economical and facile protocol for electron-catalyzed C–H iodination of various heteroarenes, leveraging halogen-bond interactions at room temperature. The iodination was achieved using only 0.55 equivalents of I2 and 0.50 equivalents of TBHP. Notably, the reaction efficiency improved with the addition of 10 mol% H2O (Scheme 58). The success of the reaction relies on the halogen bond interaction between the heteroaryl substrates (electron donors) and iodine (electron acceptor). This interaction lowered the activation energy for electron transfer, prevented the unwanted Bray–Liebhafsky reaction, and reduced the amount of terminal oxidant required. A combination of control experiments, quantum chemical calculations, and spectroscopic analyses confirmed the formation of a halogen bond, the role it played in lowering the activation barrier, the regioselectivity of the reaction, and the presence of radical intermediates in the reaction mixture. Mechanistic investigations revealed that the reaction proceeds via a radical pathway, with both kinetically controlled and thermodynamically controlled pathways, converging to produce a single regioisomer.113
 | ||
| Scheme 58 Electron-catalyzed iodination of heteroarenes at room temperature. | ||
In another approach, Du et al. developed a facile transition-metal free method for synthesizing ortho-bromoanilides from arylhydroxylamines. Here, thionyl bromide was used as halogenating agent under mild reaction conditions (Scheme 59). To evaluate the substrate scope, the optimized protocol employed on a wide variety of substituted substrates delivered the desired targets in good to moderate yields. Several control experiments ruled out the free radical mechanism involving the intramolecular substitution.114
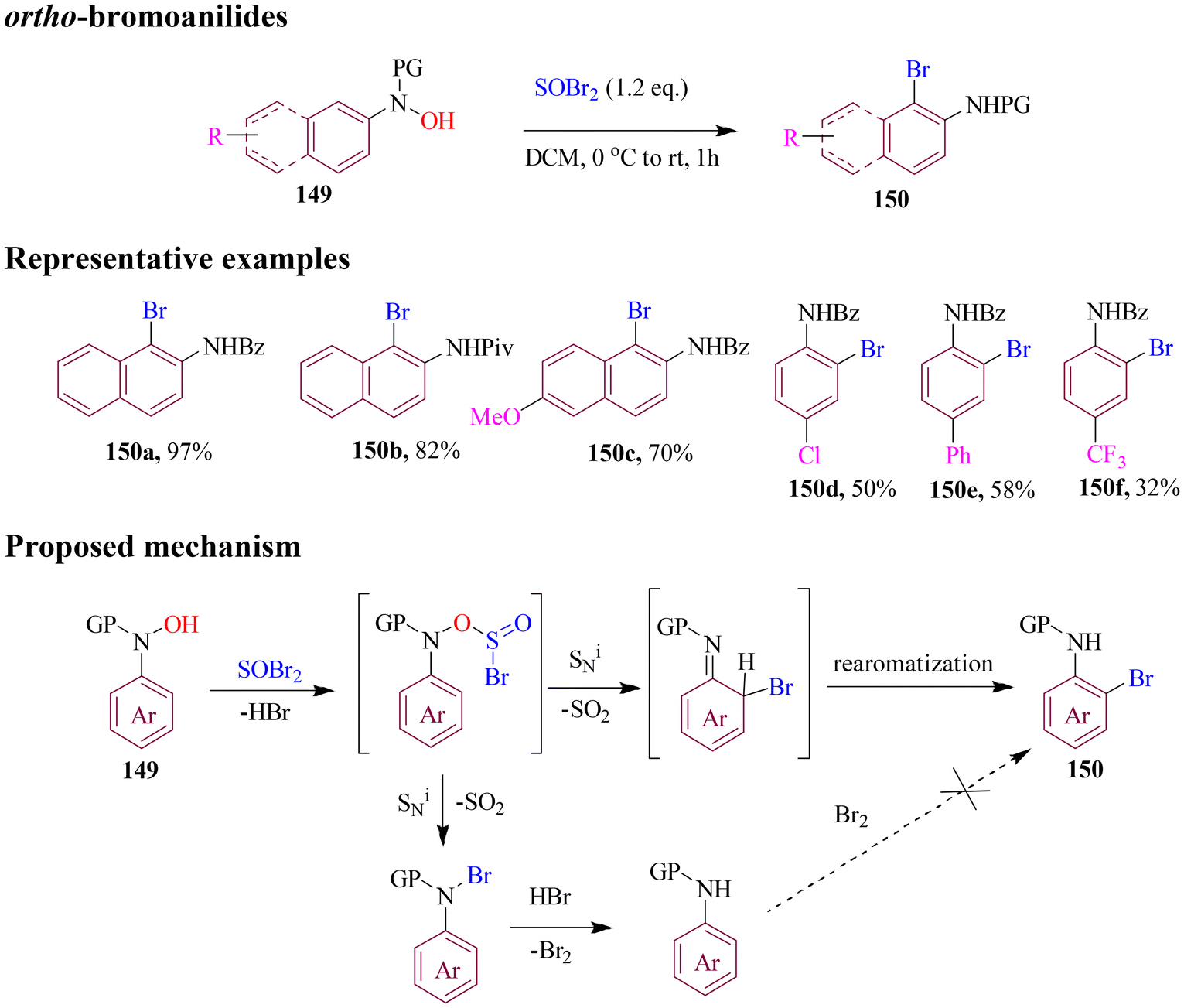 | ||
| Scheme 59 Synthesis of ortho-bromoanilides using thionyl bromide. | ||
Furthermore, Yu et al. performed electrochemical directed chlorination and bromination of electron-deficient C–H bond in different heterocycles such as quinones, coumarins, quinoxalines and 1,3-diketones. The protocol utilized readily available halogen sources such as HCl and KBr under mild reaction conditions, yielding highly site-selective derivatives (Scheme 60). Moreover, a set of control experiments was performed to investigate the mechanistic pathway, suggesting the electrophilic process. Here, first cathodic reduction of benzoquinone could generate the corresponding hydroquinone, which underwent electrophilic chlorination with chlorine (from anodic oxidation of the chloride anion) to afford the chlorinated hydroquinone. Subsequently, anodic oxidation could give the desired product.115
 | ||
| Scheme 60 Electrochemical chlorination and bromination of different heterocycles. | ||
In 2021, the Loh group reported an efficient method for visible-light-induced bromination of electron-rich arenes and heteroarenes. In this approach, the substrate was treated with bromination reagent BrCCl3 under 23 W white light irradiation, resulting in the synthesis of the C4-brominated compound (Scheme 61). Interestingly, unprotected 8-hydroxyquinoline demonstrated greater reactivity than methyl-protected 8-hydroxyquinoline, and both forms were significantly more reactive than 8-aminoquinoline. The photocatalyst Ru(II) was converted into excited Ru(II)* under visible light exposure. Then, Ru(II)* interacted with substrate (159), resulting in the formation of Ru(I) and the amino radical cation (A) via a SET mechanism. Next, Ru(I) was oxidized by oxygen in air, regenerating the initial Ru(II) and completing the photocatalytic cycle. Simultaneously, the intermediate A could undergo tautomerism to form a radical intermediate (B). Subsequently, the radical intermediate (B) reacted with BrCCl3 through SET, forming a cationic intermediate (C). Finally, this cationic intermediate underwent deprotonation to yield the target product 160.116
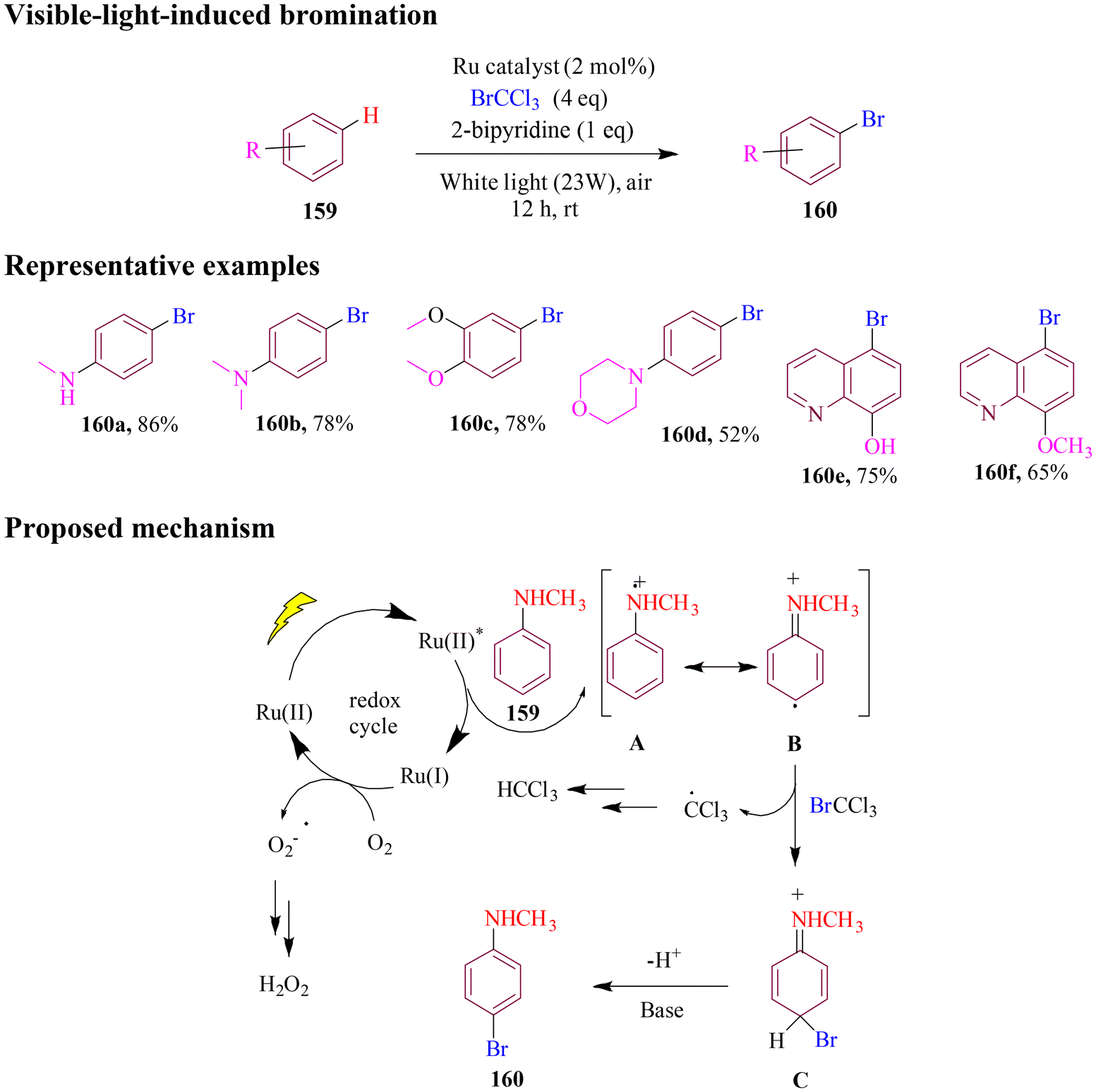 | ||
| Scheme 61 Visible-light induced regioselective bromination of arenes. | ||
Recently, the Fu group incorporated regioselectively bromine and iodine at N-heteroarenes through a FMO modulation strategy using N-benzyl azaarenium salts. In this strategy, dihalogenation was achieved in pyridines and quinolines while monohalogenation was formed in isoquinolines (Scheme 62). The optimized reaction conditions were explored on varied N-benzyl azaarenium salts for meta-bromination of N-heteroarenes, and delivered products in good yields. Furthermore, DFT studies were carried out to elucidate the reaction mechanism, suggesting the electrophilic addition of bromine radical followed by deprotonation and finally the single-electron transfer to provide the final product.117
 | ||
| Scheme 62 meta C–H halogenation of azines. | ||
Recently, the Liu group reported on electrochemical chlorination and bromination of minimally hindered tertiary and secondary benzylic C(sp3)–H bonds (Scheme 63). Their method employed graphite felt (GF) as the anode and a platinum (Pt) sheet as the cathode, using tetraethylammonium tetrafluoroborate (Et4NBF4) as the electrolyte. For chlorination, hydrochloric acid (HCl) was used, while n-butyl tetraalkylammonium bromide (nBu4NBr) served as the brominating agent, with N-hydroxy phthalimide (NHPI) acting as a catalyst. This process was conducted under constant current in an undivided cell. The protocol demonstrated good tolerance for various substitutions, yielding the desired products in moderate to good yields. Mechanistic studies revealed the possible catalytic pathway involved the generation of chlorine radical, single-electron oxidation of NHPI and hydrogen atom transfer from alkane.118
 | ||
| Scheme 63 Electrochemical chlorination and bromination of benzylic C(sp3)–H bonds. | ||
3. Conclusion and future outlook
This review aims to provide a comprehensive overview of recent advancements in directing group-assisted synthesis of organohalides, including aryl, vinyl, alkyl, and heteroaryl halides. Halogen bonds are vital in organic and inorganic compounds, making them essential in materials science, molecular recognition, crystal engineering, catalysis and rational drug design. Additionally, the significance of organohalides as fundamental building blocks for further transformations is emphasized, showcasing how C–H activation strategies with directing groups create a solid mechanistic foundation for future innovations in synthetic chemistry. This review emphasizes the utilization of various types of directing groups to achieve regioselective C–H halogenation (fluorination, chlorination, bromination, and iodination) through various mechanistic pathways, including photocatalysis, visible-light-induced methods, transition-metal catalysis, and the electrochemical approach, which offer efficient alternatives to conventional lengthy synthetic routes and harsh reaction conditions.Besides various advantages, the approach is also associated with various shortcomings, like the requirement for additional steps, additives, and high stoichiometric oxidants, leading to reduced overall efficiency and atom economy. Some directing groups add complexity to the process, generating waste and being highly substrate-specific, limiting their general applicability. Current research shows much potential for this approach, but we have only tapped into a small part. To make these methods more useful, future studies should focus on creating techniques that can handle various functional groups, such as amines, nitro, carbonyl, thioether, hydroxyl, and oxime groups. Moreover, directing groups can be designed to increase their efficiency, such as replacing 8-aminoquinoline with 8-quinolinamide and quinoline N-oxide; converting amide to Weinreb amide, which acts as a weakly coordinating group with transition metal, leading to a more selective C–H activation; and enhancing the ability of aldehyde, ketone through the transient group strategy where imine generated in situ acts as directing group that increases the overall efficiency of the process. Advances in catalyst design, sustainability, and reaction efficiency shape its future outlook. Future research will also likely focus on DGs that can be easily removed or naturally degraded after functionalization to enhance the efficiency of synthetic routes. Using recyclable DGs that can be regenerated and reused will improve cost-effectiveness and sustainability. As medicinal chemistry and late-stage functionalization grow, DGs compatible with biomolecules will become crucial for pharmaceutical applications.
Moreover, significantly less research has focused on fluorination in halogenation despite its increasing demand in drug discovery and agrochemicals, which will catalyze innovation in mild and site-selective fluorination methods. Developing techniques for the selective introduction of chlorine, bromine and iodine will broaden applications in cross-coupling reactions and late-stage functionalization. Adopting metal-free approaches that employ organic catalysts and light-driven halogenation strategies could mitigate toxicity, reduce environmental impact, and offer more sustainable and selective alternatives. Employing computational strategies for better understanding of reaction pathways will facilitate the design of more efficient halogenation processes. Prioritizing the development of catalytic systems that utilize stable, non-toxic and minimal halogen sources which maximize atom economy will make these methods more practical for industrial applications. Thus, by integrating computational tools, greener catalysts, and innovative directing group strategies, the utility of this approach for organic synthesis, pharmaceuticals, and materials sciences can be further expanded. Therefore, ongoing research into C–H halogenation with directing groups presents a valuable opportunity to unlock new reactivity, develop creative synthetic pathways, and achieve transformations with enhanced regio- and stereo-selectivity, sustainability and improved step and atom economy.
Author contributions
P. S.: conceptualization, and drafting. V. L.: visualization, and editing. K. P.: visualization, investigation, supervision and editing.Data availability
No primary research results, software or code have been included and no new data were generated or analysed as part of this review.Conflicts of interest
The authors declare no competing financial interest.Acknowledgements
KP thanks the ANRF, New Delhi (CRG/2023/004080), and CEEMS (project no: TIET/CEEMS/Regular/2021/018), VT-India, for providing funds.References
- D. B. Harpere and D. O'Hagan, The Fluorinated Natural Products, Nat. Prod. Rep., 1994, 11, 123–133 RSC.
- G. W. Gribble, Naturally Occurring Organohalogen Compounds, Acc. Chem. Res., 1998, 31, 141–152 CrossRef CAS.
- J. W. Blunt, B. R. Copp, R. A. Keyzers, M. H. G. Munro and M. R. Prinsep, Marine natural product, Nat. Prod. Rep., 2012, 29, 144–222 RSC.
- D. P. Eldera, A. M. Lipczynskib and A. Teasdale, Control and analysis of alkyl and benzyl halides and other related reactive organohalides as potential genotoxic impurities in active pharmaceutical ingredients (APIs), J. Pharm. Biomed. Anal., 2008, 48, 497–507 CrossRef PubMed.
- R. Wilcken, M. O. Zimmermann, A. Lange, A. C. Joerger and F. M. Boeckler, Principles and Applications of Halogen Bonding in Medicinal Chemistry and Chemical Biology, J. Med. Chem., 2013, 56, 1363–1388 CrossRef CAS PubMed.
- J. Wang, M. S. Rosello, J. L. Acena, C. Pozo, A. E. Sorochinsky, S. Fustero, V. A. Soloshonok and H. Liu, Fluorine in Pharmaceutical Industry: Fluorine-Containing Drugs Introduced to the Market in the Last Decade (2001–2011), Chem. Rev., 2014, 114, 2432–2506 CrossRef CAS PubMed.
- P. Jeschke, The unique role of halogen substituents in the design of modern agrochemicals, Pest Manage. Sci., 2010, 66, 10–27 CrossRef CAS PubMed.
- P. Jeschke, Manufacturing Approaches of New Halogenated Agrochemical, Eur. J. Org. Chem., 2022, e202101513 CrossRef CAS.
- Q. Wang, H. Song and Q. Wang, Fluorine-containing agrochemicals in the last decade and approaches for fluorine incorporation, Chin. Chem. Lett., 2022, 33, 626–642 CrossRef CAS.
- A. Priimagi, G. Cavallo, P. Metrangolo and G. Resnati, The Halogen Bond in the Design of Functional Supramolecular Materials: Recent Advances, Acc. Chem. Res., 2013, 46(11), 2686–2695 CrossRef CAS PubMed.
- B. Dunovica and D. Z. Veljkovic, Halogen bonds as a tool in the design of high energetic materials: evidence from crystal structures and quantum chemical calculations, CrystEngComm, 2021, 23, 6915–6922 RSC.
- F. Yang, C. Li, W. Lai, A. Zhang, H. Huang and W. Li, Halogenated conjugated molecules for ambipolar field-effect transistors and non-fullerene organic solar cells, Mater. Chem. Front., 2017, 1, 1389–1395 RSC.
- G. Berger, P. Frangville and F. Meyer, Halogen bonding for molecular recognition: new developments in materials and biological sciences, Chem. Commun., 2020, 56, 4970–4981 RSC.
- A. Pizzia, C. Pigliacellia, G. Bergamaschib, A. Gorib and P. Metrangolo, Biomimetic engineering of the molecular recognition and self-assembly of peptides and proteins via halogenation, Coord. Chem. Rev., 2020, 411, 213242 CrossRef.
- R. Das and M. Kapur, Transition-Metal-Catalyzed Site-Selective C–H Halogenation Reactions, Asian J. Org. Chem., 2018, 7, 1524–1541 CrossRef CAS.
- C. Bolm, Cross-Coupling Reactions, J. Org. Chem., 2012, 77, 5221–5223 CrossRef CAS PubMed.
- O. Baudoin, Transition metal-catalyzed arylation of unactivated C(sp3)–H bonds, Chem. Soc. Rev., 2011, 40, 4902–4911 RSC.
- N. Kannan, A. R. Patila and A. Sinha, Direct C–H bond halogenation and pseudo halogenation of hydrocarbons mediated by high-valent 3d metal-oxo species, Dalton Trans., 2020, 49(41), 14344–14360 RSC.
- L. C. Allen, Electronegativity is the Average One-Electron Energy of the Valence-Shell Electrons in Ground-State Free Atoms, J. Am. Chem. Soc., 1989, 111, 9003–9014 CrossRef CAS.
- A. Petrone, J. Ye and M. Lautens, Modern Transition-Metal-Catalyzed Carbon–Halogen Bond Formation, Chem. Rev., 2016, 116, 8003–8104 CrossRef PubMed.
- H. Tua, S. Zhub, F. Qinga and L. Chu, Visible-light-induced halogenation of aliphatic C–H bonds, Tetrahedron Lett., 2018, 59, 173–179 CrossRef.
- T. G. Luu, Y. Jung and H. K. Kim, Visible-Light-Induced Catalytic Selective Halogenation with Photocatalyst, Molecules, 2021, 26, 7380 CrossRef CAS PubMed.
- W. Tong, Q. Huang, M. Li and J. Wang, Enzyme-catalyzed C–F bond formation and cleavage, Bioresour. Bioprocess., 2019, 6, 1–8 CrossRef.
- C. Schnepel and N. Sewald, Enzymatic Halogenation: A Timely Strategy for Regioselective C–H Activation, Chem. – Eur. J., 2017, 23, 12064–12086 CrossRef CAS PubMed.
- N. Rong, Y. Yuan, H. Chen, C. Yao, T. Li, Y. Wang and W. Yang, A practical route to 2-iodoanilines via the transition-metal-free and base-free decarboxylative iodination of anthranilic acids under oxygen, Org. Chem. Front., 2021, 8, 4479–4484 RSC.
- D. Guan, H. X. Luan, M. Patiguli, Q. J. Jiao, Q. Q. Yun, Q. S. Chen, C. J. Xu, X. B. Nie, F. P. Hu and G. S. Huang, Metal–free Efficient Method for the Synthesis of N-(2-haloethyl)benzamides through the Ring-opening of 2-oxazolines, ChemistrySelect, 2019, 4, 6668–6671 CrossRef CAS.
- M. D. Karkas, Electrochemical strategies for C–H functionalization and C–N bond formation, Chem. Soc. Rev., 2018, 47, 5786–5865 RSC.
- C. Ma, P. Fang and T. S. Mei, Recent Advances in C–H Functionalization Using Electrochemical Transition Metal Catalysis, ACS Catal., 2018, 8, 7179–7189 CrossRef CAS.
- Y. Jaiswal, Y. Kumar, R. Thakur, J. Pal, R. Subramanian and A. Kumar, Primary Amide Directed Regioselective ortho-C–H-Arylation of (Aryl)Acetamides, J. Org. Chem., 2016, 81, 12499–12505 CrossRef CAS PubMed.
- Y. Dua, Y. Liu and J. P. Wan, Copper-Catalyzed One-Pot N-Acylation and C5–H Halogenation of 8-Aminoquinolines: The Dual Role of Acyl Halides, J. Org. Chem., 2018, 83, 3403–3408 CrossRef PubMed.
- M. Font, J. M. Quibell, G. J. P. Perry and I. Larrosa, The use of carboxylic acids as traceless directing groups for regioselective C–H bond functionalisation, Chem. Commun., 2017, 53, 5584–5597 RSC.
- H. Hwang, J. Kim, J. Jeong and S. Chang, Regioselective Introduction of Heteroatoms at the C-8 Position of Quinoline N-Oxides: Remote C–H Activation Using N-Oxide as a Stepping Stone, J. Am. Chem. Soc., 2014, 136, 10770–10776 CrossRef CAS PubMed.
- C. Sambiagio, D. Schonbauer, R. Blieck, T. Dao-Huy, G. Pototschnig, P. Schaaf, T. Wiesinger, M. F. Zia, J. Wencel-Delord, T. Besset, B. U. W. Maes and M. Schnurch, A comprehensive overview of directing groups applied in metal-catalysed C–H functionalisation Chemistry, Chem. Soc. Rev., 2018, 47, 6603–6743 RSC.
- A. Das and N. Chatani, The Directing Group: A Tool for Efficient and Selective C–F Bond Activation, ACS Catal., 2021, 11, 12915–12930 CrossRef CAS.
- S. Kaltenberger and M. Gemmeren, Controlling Reactivity and Selectivity in the Non directed C–H Activation of Arenes with Palladium, Acc. Chem. Res., 2023, 56, 2459–2472 CrossRef CAS PubMed.
- X. Liu, X. Zhao, F. Liang and B. Ren, t-BuONa-mediated direct C–H halogenation of electron-deficient (hetero)arenes, Org. Biomol. Chem., 2018, 16, 886–890 RSC.
- K. Wang, F. Hu, Y. Zhang and J. Wang, Directing group-assisted transition-metal-catalyzed vinylic C–H bond functionalization, Sci. China: Chem., 2015, 58, 1252–1265 CrossRef CAS.
- J. He, M. Wasa, K. S. L. Chan, Q. Shao and J. Q. Yu, Palladium-Catalyzed Transformations of Alkyl C–H Bonds, Chem. Rev., 2017, 117, 8754–8786 CrossRef CAS PubMed.
- H. Mondal, Halogen and Chalcogen Activation by Nucleophilic Catalysis, Chem. – Eur. J., 2024, 30, e202402261 CrossRef CAS PubMed.
- D. Tilly, F. Chevallier, F. Mongin and P. C. Gros, Bimetallic Combinations for Dehalogenative Metalation Involving Organic Compounds, Chem. Rev., 2014, 114, 1207–1257 CrossRef CAS PubMed.
- R. Lekkala, R. Lekkala, B. Moku, K. P. Rakesh and H. L. Qin, Recent Developments in Radical-Mediated Transformations of Organohalides, Eur. J. Org. Chem., 2019, 2769–2806 CrossRef CAS.
- J. Magano and J. R. Dunetz, Large-Scale Applications of Transition Metal-Catalyzed Couplings for the Synthesis of Pharmaceuticals, Chem. Rev., 2011, 111, 2177–2250 CrossRef CAS PubMed.
- A. Corio, C. G. Pelletier and P. Busca, Regioselective Functionalization of Quinolines through C–H Activation: A Comprehensive Review, Molecules, 2021, 26, 5467 CrossRef CAS PubMed.
- V. G. Zaitsev, D. Shabashov and O. Daugulis, Highly Regioselective arylation of sp3 C–H Bonds Catalyzed by Palladium Acetate, J. Am. Chem. Soc., 2005, 127, 13154–13155 CrossRef CAS PubMed.
- R. Y. Zhu, T. G. S. Denis, Y. Shao, J. He, J. D. Sieber, C. H. Senanayake and J. Q. Yu, Ligand-Enabled Pd(II)-Catalyzed Bromination and Iodination of C(sp3)–H Bonds, J. Am. Chem. Soc., 2017, 139, 5724–5727 CrossRef CAS PubMed.
- Y. Long, L. Pan and X. Zhou, Iron(III)-Catalyzed Highly Regioselective Halogenation of 8-Amidoquinolines in Water, Molecules, 2019, 24, 535 CrossRef PubMed.
- S. J. Lee, K. J. Makaravage, A. F. Brooks, P. J. H. Scott and M. S. Sanford, Copper-Mediated Aminoquinoline-Directed Radiofluorination of Aromatic C–H Bonds with K18F, Angew. Chem., Int. Ed., 2019, 58, 3119–3122 CrossRef CAS PubMed.
- M. Y. Chen, X. Pannecoucke, P. Jubault and T. Besset, Pd-Catalyzed Selective Chlorination of Acrylamides at Room Temperature, Org. Lett., 2020, 22, 7556–7561 CrossRef CAS PubMed.
- X. Yang, Q. L. Yang, X. Y. Wang, H. H. Xu, T. S. Mei, Y. Huang and P. Fang, Copper-Catalyzed Electrochemical Selective Bromination of 8-Aminoquinoline Amide Using NH4Br as the Brominating Reagent, J. Org. Chem., 2020, 85, 3497–3507 CrossRef CAS PubMed.
- X. Lin, C. Zeng, C. Liu, Z. Fang and K. Guo, C-5 selective chlorination of 8-aminoquinoline amides using dichloromethane, Org. Biomol. Chem., 2021, 19, 1352–1357 RSC.
- N. S. Junior, R. S. Gomes, G. A. M. Jardim, Y. F. Liang and L. Ackermann, Weakly-coordinating N-oxide and carbonyl groups for metal-catalyzed C–H activation: the case of A-ring functionalization, Chem. Commun., 2018, 54, 7398–7411 RSC.
- A. K. Dhiman, S. S. Gupta, R. Sharma, R. Kumar and U. Sharma, Rh(III)-Catalyzed C(8)–H Activation of Quinoline N-Oxides: Regioselective C–Br and C–N Bond Formation, J. Org. Chem., 2019, 84, 12871–12880 CrossRef CAS PubMed.
- H. Zhou, M. Miyasaka, Y. H. Wang, T. Kochi and F. Kakiuchi, Palladium-Catalyzed Electrochemical Iodination of 1-Arylpyridine N-Oxides, J. Org. Chem., 2024, 89(22), 16300–16306 CrossRef CAS PubMed.
- R. Thakur, Y. Jaiswal and A. Kumar, Primary amides: Sustainable weakly coordinating groups in transition metal-catalyzed C–H bond functionalization reactions, Tetrahedron, 2021, 93, 132313 CrossRef CAS.
- R. Das, G. S. Kumar and M. Kapur, Amides as Weak Coordinating Groups in Proximal C–H Bond Activation, Eur. J. Org. Chem., 2017, 5439–5459 CrossRef CAS.
- R. Y. Zhu, M. E. Farmer, Y. Q. Chen and J. Q. Yu, A Simple and Versatile Amide Directing Group for C–H Functionalizations, Angew. Chem., Int. Ed., 2016, 55(36), 10578–10599 CrossRef CAS PubMed.
- Z. L. Li, P. Y. Wu and C. Cai, Nickel-catalyzed regioselective C–H halogenation of electron-deficient arenes, New J. Chem., 2019, 43, 3462–3468 RSC.
- Y. Jaiswal, Y. Kumar and A. Kumar, The palladium(II)-catalyzed regioselective ortho, C–H bromination/iodination of arylacetamides with in situ generated imidic acid as the directing group: mechanistic exploration, Org. Biomol. Chem., 2019, 17, 6809–6820 RSC.
- Y. Jaiswal and A. Kumar, Acid-promoted palladium(II)-catalyzed ortho-halogenation of primary benzamides: En route to halo-arenes, Catal. Commun., 2019, 131, 105784 CrossRef.
- A. J. G. Santiago, C. A. Brown, R. D. Sommer and E. A. Ison, Identification of key functionalization species in the Cp*Ir(III)-catalyzed-ortho halogenation of benzamides, Dalton Trans., 2020, 49(45), 16166–16174 RSC.
- J. Tanaka, Y. Shibata, A. Joseph, J. Nogami, J. Terasawa, R. Yoshimura and K. Tanaka, Rhodium-Catalyzed ortho-Bromination of O-Phenyl Carbamates Accelerated by a Secondary Amide-Pendant Cyclopentadienyl Ligand, Chem. – Eur. J., 2020, 26, 5774–5779 CrossRef CAS PubMed.
- Y. Sun, Q. He, X. Lv, N. Zhang, W. Yan, J. Sun and L. Zhuang, Switchable Site-Selective Benzanilide C(sp2)-H Bromination via Promoter Regulation, Molecules, 2024, 29, 2861 CrossRef CAS PubMed.
- J. Kalepu and L. T. Pilarski, Weinreb Amides as Directing Groups for Transition Metal-Catalyzed C–H Functionalizations, Molecules, 2019, 24, 830 CrossRef PubMed.
- A. S. Marco, B. Saavedra, E. Erbing, J. Malmberg, M. J. Johansson and B. M. Matute, Selective C–H Iodination of Weinreb Amides and Benzamides through Iridium Catalysis in Solution and under Mechanochemical Conditions, Org. Lett., 2024, 26, 2800–2805 CrossRef PubMed.
- Q. Zhang and B. F. Shi, From Reactivity and Regioselectivity to Stereoselectivity: An Odyssey of Designing PIP Amine and Related Directing Groups for C–H Activation, Chin. J. Chem., 2019, 37, 647–656 CrossRef CAS.
- Q. L. Yang, X. Y. Wang, T. L. Wang, X. Yang, D. Liu, X. Tong, X. Y. Wu and T. S. Mei, Palladium-Catalyzed Electrochemical C–H Bromination Using NH4Br as the Brominating Reagent, Org. Lett., 2019, 21, 2645–2649 CrossRef CAS PubMed.
- J. Jiao, Y. K. Xing, Q. L. Yang, H. Qiu and T. S. Mei, Site-Selective C–H Functionalization via Synergistic Use of Electrochemistry and Transition Metal Catalysis, Acc. Chem. Res., 2020, 53, 300–310 CrossRef PubMed.
- S. R. Mohanty, N. Prusty, T. Nanda, P. S. Mahulkar and P. C. Ravikumar, Directing group-assisted selective C–H activation of six-membered N-heterocycles and benzo-fused N-heterocycles, Org. Chem. Front., 2024, 11, 540–575 RSC.
- Y. Yuan, Y. Liang, S. Shi, Y. F. Liang and N. Jiao, Efficient Pd-catalyzed C–H Oxidative Bromination of Arenes with DMSO and Hydrobromic Acid, Chin. J. Chem., 2019, 37, 1245–1251 CrossRef.
- J. Guilbaud, A. Selmi, M. Kammoun, S. Contal, C. Montalbetti, N. Pirio, J. Roger and J. C. Hierso, C–H Halogenation of Pyridyl Sulfides Avoiding the Sulfur Oxidation: A Direct Catalytic Access to Sulfanyl Polyhalides and Polyaromatics, ACS Omega, 2019, 4, 20459–20469 CrossRef CAS PubMed.
- X. H. Liu, H. Park, J. H. Hu, Y. Hu, Q. L. Zhang, B. L. Wang, B. Sun, K. S. Yeung, F. L. Zhang and J. Q. Yu, Diverse ortho-C(sp2)–H Functionalization of Benzaldehydes Using Transient Directing Groups, J. Am. Chem. Soc., 2017, 139, 888–896 CrossRef CAS PubMed.
- I. Higham and J. A. Bull, Transient imine directing groups for the C–H functionalisation of aldehydes, ketones and amines: an update 2018–2020, Org. Biomol. Chem., 2020, 18, 7291–7315 RSC.
- Q. Yonga, B. Suna and F. L. Zhang, Palladium-catalyzed ortho-C(sp2)-H bromination of benzaldehydes via a monodentate transient directing group strategy, Tetrahedron Lett., 2019, 60, 151263 CrossRef.
- F. Li, Y. Zhou, H. Yang, Z. Wang, Q. Yu and F. L. Zhang, Monodentate Transient Directing Group Enabled Pd-Catalyzed ortho-C–H Methoxylation and Chlorination of Benzaldehydes, Org. Lett., 2019, 21, 3692–3695 CrossRef CAS PubMed.
- M. La, D. Liu, X. Chen, F. L. Zhang and Y. Zhou, Monodentate Transient Directing Group-Assisted Palladium Catalyzed Direct ortho-C–H Iodination of Benzaldehydes for Total Synthesis of Hernandial, Org. Lett., 2021, 23, 9184–9188 CrossRef CAS PubMed.
- G. Kuang, D. Liu, X. Chen, G. Liu, Y. Fu, Y. Peng, H. Li and Y. Zhou, Transient Directing Group Strategy as a Unified Method for Site Selective Direct C4–H Halogenation of Indoles, Org. Lett., 2021, 23, 8402–8406 CrossRef CAS PubMed.
- Q. Wu, Y. J. Mao, K. Zhou, S. Wang, L. Chen, Z. Y. Xu, S. J. Lou and D. Q. Xu, Pd-Catalysed direct C(sp2)–H fluorination of aromatic ketones: concise access to anacetrapib, Chem. Commun., 2021, 57, 4544–4547 RSC.
- J. Wang, Y. Wu, W. Xu, X. Lu, Y. Wang, G. Liu, B. Sun, Y. Zhou and F. L. Zhang, Monodentate transient directing group promoted Pd-catalyzed direct ortho-C-H arylation and chlorination of α-ketoesters for three-step synthesis of Cloidogrel racemate, Tetrahedron, 2022, 123, 132980 CrossRef CAS.
- R. Sharma and M. R. Yadav, Recent developments in decarboxylative C(aryl)–X bond formation from (hetero)aryl carboxylic acids, Org. Biomol. Chem., 2021, 19, 5476–5500 RSC.
- X. Zhang, X. Feng, H. Zhang, Y. Yamamotoa and M. Bao, Transition-metal-free decarboxylative halogenation of 2-picolinic acids with dihalomethane under oxygen conditions, Green Chem., 2019, 21, 5565–5570 RSC.
- D. S. Barak, D. J. Dahatonde and S. Batra, Microwave-Assisted Metal-Free Decarboxylative Iodination/Bromination of Isoxazole-4-carboxylic Acids, Asian J. Org. Chem., 2019, 8, 2149–2154 CrossRef CAS.
- E. Weis, M. J. Johansson and B. M. Matute, IrIII-Catalyzed Selective ortho-Monoiodination of Benzoic Acids with Unbiased C–H Bonds, Chem. – Eur. J., 2020, 26, 10185–10190 CrossRef CAS PubMed.
- P. Xu, P. L. Rojas and T. Ritter, Radical Decarboxylative Carbometalation of Benzoic Acids: A Solution to Aromatic Decarboxylative Fluorination, J. Am. Chem. Soc., 2021, 143, 5349–5354 CrossRef CAS PubMed.
- S. Li, C. Zhang, L. Fu, H. Wang, L. Cai, X. Chen, X. Wang and G. Li, Arene C–H Iodination Using Aryl Iodides, CCS Chem., 2022, 4, 1889–1900 CrossRef CAS.
- L. Yang, X. Wang, M. Zhang, S. Li, X. Fang and G. Li, Carboxyl group assisted isodesmic meta-C–H iodination of phenethylamines, benzylamines, and 2-aryl anilines, Org. Chem. Front., 2023, 10, 3760–3765 RSC.
- Y. Kommagalla and N. Chatani, Cobalt-Catalyzed C–H Iodination of Aromatic Amides with Molecular Iodine through the Use of a 2-Aminophenyloxazoline Based Bidentate-Chelation System, Org. Lett., 2019, 21, 5971–5976 CrossRef CAS PubMed.
- X. Hong, Q. Zhou, S. Huang, H. Z. Cui, Z. M. Li and X. F. Hou, Transition metal catalysed C7 and ortho-selective halogenation of 2-arylbenzo[d]oxazoles, Org. Chem. Front., 2019, 6, 2226–2233 RSC.
- H. He, J. Guo, W. Sun, B. Yang, F. Zhang and G. Liang, Palladium-Catalyzed Direct Mono- or Polyhalogenation of Benzothiadiazole Derivatives, J. Org. Chem., 2020, 85, 3788–3798 CrossRef CAS PubMed.
- A. Ahmad, H. S. Dutta, M. Kumar, A. A. Khan, Raziullah and D. Koley, Pd-Catalyzed C–H Halogenation of Indolines and Tetrahydroquinolines with Removable Directing Group, Org. Lett., 2020, 22, 5870–5875 CrossRef CAS PubMed.
- A. Daher, O. Abidi, J. C. Hierso and J. Roger, Alkali halides as nucleophilic reagent sources for N-directed palladium-catalysed ortho-C–H halogenation of s-tetrazines and other Heteroaromatics, RSC Adv., 2022, 12, 30691–30695 RSC.
- D. S. Zheng, W. W. Zhang, Q. Gu and S. L. You, Rh(III)-Catalyzed Atroposelective C–H Iodination of 1-Aryl Isoquinolines, ACS Catal., 2023, 13, 5127–5134 CrossRef CAS.
- Y. Q. Chen, S. Singh, Y. Wu, Z. Wang, W. Hao, P. Verma, J. X. Qiao, R. B. Sunoj and J. Q. Yu, Pd-Catalyzed γ-C(sp3)–H Fluorination of Free Amines, J. Am. Chem. Soc., 2020, 142, 9966–9974 CrossRef CAS PubMed.
- J. Jin, Y. Zhao, S. H. Kyne, K. Farshadfar, A. Ariafard and P. W. H. Chan, Copper(I)-catalysed site-selective C(sp3)–H bond chlorination of ketones, (E)-enones and alkylbenzenes by dichloramine-T, Nat. Commun., 2021, 12, 4065 CrossRef CAS PubMed.
- S. N. M. Boddapati, R. Tamminana, M. M. Alam, S. Gugulothu, R. Varalae and H. B. Bollikolla, Efficient Pd(II)-catalyzed regioselective ortho-halogenation of arylcyanamides, New J. Chem., 2021, 45, 17176–17182 RSC.
- Y. Feng, J. Wang, J. Yang, F. Chen, Z. Zhang, C. Ke, J. Lin and H. Lin, Native Amino Group Directed Site-Selective ε-C(sp2)–H of Primary Amines, Org. Lett., 2023, 25, 1348–1352 CrossRef CAS PubMed.
- J. Xiong and Y. Liu, Transition-Metal-free C5, C7-Dihalogenation and the Switchable C5 Halogenation of 8-Hydroxyquinolines, ChemistrySelect, 2019, 4, 693–697 CrossRef CAS.
- L. Bian, S. Z. Tang, M. E. Chen, X. M. Zhang, J. W. Lv, X. W. Chen, F. M. Qi, S. W. Chen and F. M. Zhang, Transition-Metal-Free Site-Selective γ-C(sp2)–H Monoiodination of Arenes Directed by an Aliphatic Keto Group, Org. Lett., 2020, 22, 5314–5319 CrossRef PubMed.
- S. Kalaria, S. Balasubramanianc and H. B. Rode, Difluorinative-hydroxylation and C-3 functionalization (halogenation/SCN/NO) of imidazopyridine using Selectfluor as fluorine source or oxidant respectively, Tetrahedron Lett., 2021, 71, 153028 CrossRef.
- C. M. Wang, J. Y. Du, J. Y. Zhang, K. X. Tang, T. H. Gao, Y. G. Xu and L. P. Sun, Regioselective bromination of aryl ureas with Phenyliodine(III)diacetate and potassium bromide, Tetrahedron, 2019, 75, 130621 CrossRef.
- Y. Wu, S. Xu, H. Wang, D. Shao, Q. Qi, Y. Lu, L. Ma, J. Zhou, W. Hu, W. Gao and J. Chen, Directing Group Enables Electrochemical Selectively Meta-Bromination of Pyridines under Mild Conditions, J. Org. Chem., 2021, 86, 16144–16150 CrossRef CAS PubMed.
- H. Shinde, G. S. Ghotekar, F. M. A. Noa, L. Ohrstrom, P. O. Norrby and H. Sunden, Regioselective ortho halogenation of N-aryl amides and ureas via oxidative halodeboronation: harnessing boron reactivity for efficient C–halogen bond installation, Chem. Sci., 2023, 14, 13429–13436 RSC.
- C. Shao, C. Ma, L. Li, J. Liu, Y. Shen, C. Chen, Q. Yang, T. Xu, Z. Hu, Y. Kan and T. Zhang, Copper-promoted C5-selective bromination of 8-aminoquinoline amides with alkyl bromides, Beilstein J. Org. Chem., 2024, 20, 155–161 CrossRef CAS PubMed.
- Y. Zhu, J. Shi and W. Yu, Photoinduced Site-Selective C(sp3)–H Chlorination of Aliphatic Amides, Org. Lett., 2020, 22, 8899–8903 CrossRef CAS PubMed.
- D. Li, Z. Jia, Y. Jiang, J. Jia, X. Zhao, Z. Li and Z. Xu, One-Pot Functionalization of 8-Aminoquinolines through the Acylation and Regioselective C5-H Halogenation under Transition-Metal-Free Conditions, ChemistrySelect, 2019, 4, 13964–13967 CrossRef CAS.
- Q. Shu, Y. Li, T. Liu, S. Zhang, L. Jiang, K. Jin, R. Zhang and C. Duan, Visible light induced regioselective C5 halogenation of 8-aminoquinolines with 1,3-dihalo-5,5-dimethylhydantoin in continuous flow, Tetrahedron, 2019, 75, 3636–3642 CrossRef CAS.
- C. Shi, Q. Miao, L. Ma, T. Lu, D. Yang, J. Chen and Z. Li, Room-Temperature C–H Bromination and Iodination with Sodium Bromide and Sodium Iodide Using N-Fluorobenzenesulfonimide as an Oxidant, ChemistrySelect, 2019, 4, 6043–6047 CrossRef CAS.
- B. Ma, F. Lu, H. Yang, X. Gu, Z. Li, R. Li, H. Pei, D. Luo, H. Zhang and A. Lei, Visible Light Mediated External Oxidant Free Selective C5 Bromination of 8-Aminoquinoline Amides under Ambient Conditions, Asian J. Org. Chem., 2019, 8, 1136–1140 CrossRef CAS.
- J. Hou, K. Wang, C. Zhang, T. Wei, R. Bai and Y. Xie, Metal-Free Electrochemical Oxidative Dihalogenation of Quinolines on the C5 and C7 Positions Using N-Halosuccinimides, Eur. J. Org. Chem., 2020, 6382–6386 CrossRef CAS.
- R. S. Malykhin, A. O. Kokuev, V. S. Dorokhov, Y. V. Nelyubina, V. A. Tartakovsky, A. A. Tabolin, S. L. Ioffe and A. Y. Sukhorukov, Nucleophilic Halogenation of Cyclic Nitronates: A General Access to 3-Halo-1,2-Oxazines, J. Org. Chem., 2019, 84, 13794–13806 CrossRef CAS PubMed.
- D. Chen, Y. Liu, Z. Lu, H. Wang, M. Li, D. Yue and Z. Wang, Chemo- and regioselective synthesis of C3-sulfonate esters and C4-chlorides of quinolines under metal-free conditions, Org. Chem. Front., 2023, 10, 936–942 RSC.
- J. Hu, G. Zhou, Y. Tian and X. Zhao, Selectfluor-promoted regioselective chlorination/bromination of 2-aminopyridines and 2-aminodiazines using LiCl/LiBr, Org. Biomol. Chem., 2019, 17, 6342–6345 RSC.
- J. N. Levy, J. V. A. Requena, R. Liu, R. S. Paton and A. McNally, Selective Halogenation of Pyridines Using Designed Phosphine Reagents, J. Am. Chem. Soc., 2020, 142, 11295–11305 CrossRef CAS PubMed.
- I. Kazi, S. Guha and G. Sekar, Halogen Bond-Assisted Electron-Catalyzed Atom Economic Iodination of Heteroarenes at Room Temperature, J. Org. Chem., 2019, 84, 6642–6654 CrossRef CAS PubMed.
- Y. Du, Z. Xi, L. Guo, H. Lu, L. Feng and H. Gao, Practical bromination of arylhydroxylamines with SOBr2 towards ortho-bromo-anilides, Tetrahedron Lett., 2021, 72, 153074 CrossRef CAS.
- D. Yu, R. Ji, Z. Sun, W. Li and Z. Q. Liu, Electrochemical chlorination and bromination of electron-deficient C–H bonds in quinones, coumarins, quinoxalines and 1,3-diketones, Tetrahedron Lett., 2021, 86, 153514 CrossRef CAS.
- J. Fan, Q. Wei, E. Zhu, J. Gao, X. Cheng, Y. Lu and T. P. Loh, Visible light-induced mono-bromination of arenes with BrCCl3, Chem. Commun., 2021, 57, 5977–5980 RSC.
- J. Tang, S. Li, Y. Fu, Z. Su, J. Xu, W. Xue, X. Zheng, R. Li, H. Chen and H. Fu, Radical meta-C–H Halogenation of Azines via N-Benzyl Activation Strategy, Org. Lett., 2024, 26, 5899–5904 CrossRef CAS PubMed.
- J. Zhao, J. Zhang, P. Fang, J. Wu, F. Wang and Z. Q. Liu, Electrochemical chlorination of least hindered tertiary and benzylic C(sp3)–H bonds, Green Chem., 2024, 26, 507–512 RSC.
| This journal is © the Partner Organisations 2025 |