
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicencePalladium-catalyzed chemoselective decarboxylative coupling of alkynyl carboxylic acids with halogenated aryl triflates†
Miao
Wang‡
 ab,
On Ying
Yuen‡
a,
Shan Shan
Ng
a and
Chau Ming
So
*a
ab,
On Ying
Yuen‡
a,
Shan Shan
Ng
a and
Chau Ming
So
*a
aDepartment of Applied Biology and Chemical Technology, The Hong Kong Polytechnic University, Hung Hom, Kowloon, Hong Kong. E-mail: chau.ming.so@polyu.edu.hk
bHenan Key Laboratory of Function-Oriented Porous Materials, College of Chemistry and Chemical Engineering, Luoyang Normal University, Luoyang 471934, People's Republic of China
First published on 16th April 2025
Abstract
A palladium-catalyzed chemoselective decarboxylative coupling of alkynyl carboxylic acids with halogenated aryl triflates has been developed for the efficient synthesis of OTf-arylalkyne scaffolds. Notably, this transformation exhibits an inversion of the conventional chemoselectivity order, favoring C–Br > C–Cl > C–OTf. In addition, a one-pot sequential strategy was established by integrating the decarboxylative coupling with a Suzuki–Miyaura reaction, offering a versatile platform for the synthesis of difunctionalized aromatic compounds. Density functional theory (DFT) calculations indicate that the oxidative addition step governs the chemoselectivity, while the decarboxylation step is proposed to be rate-determining.
Introduction
Decarboxylative coupling is a powerful tool-type strategy for the formation of C–C and C–heteroatom bonds, allowing for the construction of useful heterocyclic skeletons and pharmaceutically active molecules.1 Moreover, this strategy features the utilization of facilely prepared and stable carboxylic acids and low toxicity carbon dioxide as a by-product which has therefore received extensive attention.2 Since Lee and co-workers reported the first example of decarboxylative coupling reaction3 of alkynyl carboxylic acids with aryl halides to synthesize unsymmetric diarylalkynes in 2008, this efficient strategy has drawn wide attention, and has been explored for the construction of C–C,4 C–N,5 C–Br,6 C–P,7 C–S,8 C–Se,9 C–B,10 and C–Si11 bonds. The employment of alkynyl carboxylic acids as surrogates for terminal alkynes offers an advantageous strategy for managing low-boiling-point alkynes, enhancing both the practicality and safety of their manipulation. Although the decarboxylative coupling of alkynyl carboxylic acids has advanced significantly, most studies have focused on substrates with a single electrophilic site (Scheme 1A).3,4 In contrast, chemoselective reactions involving dual electrophilic sites remain underexplored.12 Aryl halides and triflates are among the most commonly used electrophiles, with a generally accepted reactivity order of I > Br ≈ OTf > Cl (Scheme 1B).13 However, cross-coupling reactions involving more than one electrophilic sites continue to face several key challenges, including: (1) a limited substrate scope, (2) difficulty in achieving clean chemoselectivity when two electrophilic sites are present, and (3) the challenge of reversing the conventional reactivity order—such as favoring C–Cl over C–OTf bonds.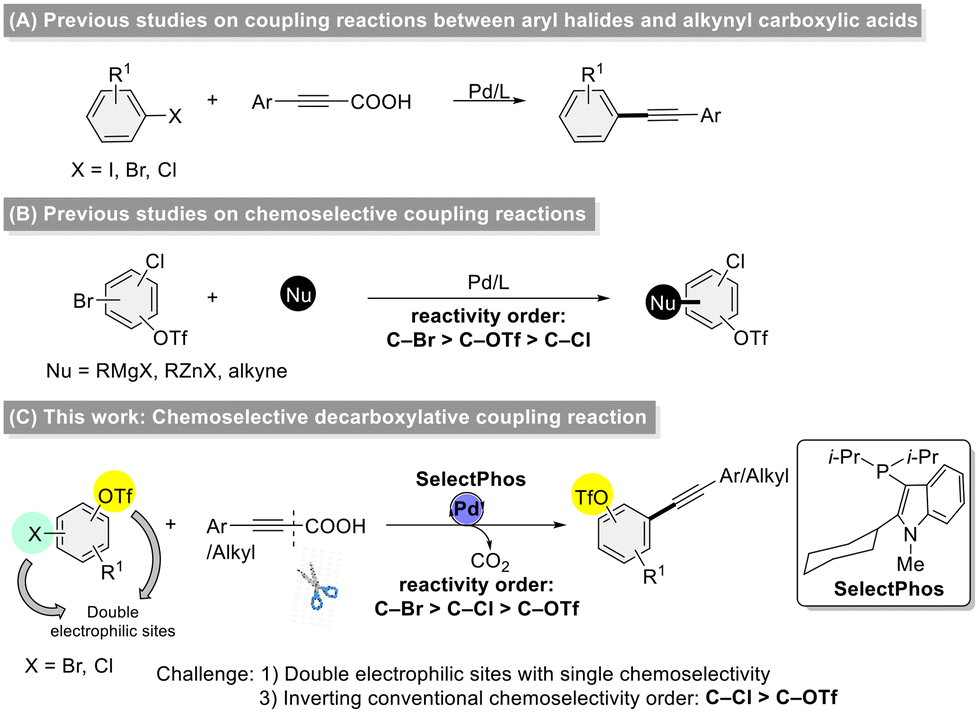 | ||
| Scheme 1 From conventional to chemoselective: coupling strategies using alkynyl carboxylic acids. | ||
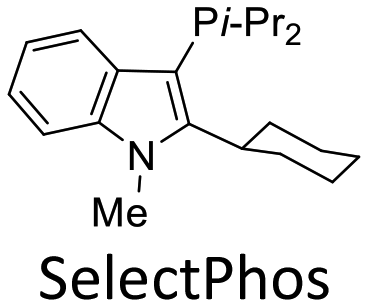
In recent years, we developed a new type of alkyl–heteroaryl-based phosphorus ligand (SelectPhos) and successfully applied it to chemoselective coupling reactions, achieving an inversion of the conventional chemoselectivity order of C–Br > C–Cl > C–OTf.14 In conjunction with our ongoing work on the development of new ligands and chemoselective reactions, we now present our latest research on the application of SelectPhos in the chemoselective decarboxylative coupling of alkynyl carboxylic acids with halogenated aryl triflates, demonstrating a chemoselectivity order of C–Br > C–Cl > C–OTf (Scheme 1C). To the best of our knowledge, this is the first decarboxylative coupling of alkynyl carboxylic acids with chloroaryl triflates at the C–Cl bond over the C–OTf bond.
Results and discussion
We set out to investigate the chemoselective decarboxylative coupling reaction using 3-chloro-5-methylphenyl triflate 1a and 3-phenylpropiolic acid 2a as model substrates, with the systematic screenings of a range of ligands (Table 1). Initially, a series of classical phosphine ligands were screened (L1–L3). PPh3 showed the general reactivity order: OTf > Cl with excellent yield; PCy3 resulted in poor activity, and Pt-Bu3·HBF4 show a certain extent at C–Cl bond over C–OTf bond (C–Cl![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :C–OTf = 10:1), but with low yield. Moreover, Buchwald-type biaryl ligand was also tested (L4), and it showed C–OTf chemoselectivity. Bidentate ligands (L5–L7) were evaluated, and the results indicated that most bidentate ligands provide C–OTf selectivity with excellent activity. Since NHC ligand was reported to show good C–Cl chemoselectivity over C–OTf in the Suzuki–Miyaura coupling,15 we tested the corresponding NHC ligand in this chemoselective decarboxylative coupling reaction, but the result showed poor chemoselectivity and reactivity (L8). To our delight, SelectPhos (L9) gave the result of excellent C–Cl chemoselectivity (C–Cl:C–OTf > 70:1) in 71% yield. Other SelectPhos derivatives (L11–L13) were also tested, both substituents attached to the C2 position on the indole ring and phosphorus atom could directly affect the selectivity and activity, and replacing an aryl group at either position will show the opposite selectivity. The steric hindrance size of substituents attached to the C2 position on the indole ring could affect the selectivity, an adamantly substituent showed roughly C–Cl selectivity but the methyl group showed C–OTf selectivity (L14–L15).
:C–OTf = 10:1), but with low yield. Moreover, Buchwald-type biaryl ligand was also tested (L4), and it showed C–OTf chemoselectivity. Bidentate ligands (L5–L7) were evaluated, and the results indicated that most bidentate ligands provide C–OTf selectivity with excellent activity. Since NHC ligand was reported to show good C–Cl chemoselectivity over C–OTf in the Suzuki–Miyaura coupling,15 we tested the corresponding NHC ligand in this chemoselective decarboxylative coupling reaction, but the result showed poor chemoselectivity and reactivity (L8). To our delight, SelectPhos (L9) gave the result of excellent C–Cl chemoselectivity (C–Cl:C–OTf > 70:1) in 71% yield. Other SelectPhos derivatives (L11–L13) were also tested, both substituents attached to the C2 position on the indole ring and phosphorus atom could directly affect the selectivity and activity, and replacing an aryl group at either position will show the opposite selectivity. The steric hindrance size of substituents attached to the C2 position on the indole ring could affect the selectivity, an adamantly substituent showed roughly C–Cl selectivity but the methyl group showed C–OTf selectivity (L14–L15).
|
|
|||||
|---|---|---|---|---|---|
| Entry | Ligand code | Ligand | Yieldsb (%) | ||
| 3a | 4a | 5a | |||
| a Reaction condition: 3-chloro-5-methylphenyl triflate 1a (0.20 mmol), 3-phenylpropiolic acid 2a (0.36 mmol), Pd(OAc)2 (4 mol%), L (16 mol%), K2CO3 (0.40 mmol) and THF (1.0 mL) were stirred at 100 °C or 3 h. b Calibrated GC yields were reported by using dodecane as an internal standard. | |||||
| 1 | L1 | PPh3 | 0 | 84 | 0 |
| 2 | L2 | PCy3 | 0 | 5 | 0 |
| 3 | L3 | Pt-Bu3·HBF4 | 20 | 2 | 0 |
| 4 | L4 |

|
0 | 35 | 50 |
| 5 | L5 |

|
0 | 73 | 0 |
| 6 | L6 |
|
0 | 99 | 0 |
| 7 | L7 |

|
0 | 98 | 0 |
| 8 | L8 |

|
19 | 3 | 0 |
| 9 | L9 |
|
71 | 1 | 2 |
| 10 | L10 |

|
0 | 63 | Trace |
| 11 | L11 |

|
16 | 1 | 0 |
| 12 | L12 |

|
50 | 2 | 0 |
| 13 | L13 |

|
0 | 77 | 8 |
| 14 | L14 |
|
20 | 7 | Trace |
| 15 | L15 |

|
0 | 27 | 0 |

Next, we were focused on the investigations of the optimal conditions for this chemoselective decarboxylative coupling reaction with SelectPhos (L9). Initially, we evaluated several palladium source (Table S1,† entries 1–3), and dichloro-(1-methylallyl)-dipalladium showed the optimal reactivity, affording the corresponding product in excellent yield (95%). The molar ratio of Pd and L9 were also evaluated (Table S1,† entries 4–6), and 1:4 of Pd and L9 was the best choice. Subsequently, we investigated several bases, including K3PO4, KOAc, KF, and Cs2CO3 (Table S1,† entries 7–10), but these did not lead to a higher yield. Regarding solvent evaluation (Table S1,† entries 11–13), we tested 1,4-dioxane, PhMe, and CPMe, but THF provided the best activity. We tried to reduce the dosage of 3-phenylpropiolic acid, but the yield dropped significantly (Table S1,† entry 14). Finally, the temperature for this reaction were also evaluated, and 100 °C was the best choice (Table S1,† entries 15 and 16).
After obtaining the optimal reaction conditions, we proceeded to investigate the substrate scope of this chemoselective decarboxylative coupling reaction (Scheme 2). Initially, we examined a range of chloroaryl triflates, and the results showed the reaction has good substrate adaptability, no matter whether the C–Cl bond was at positions ortho (3b–3c), meta (3a, 3d–3g, 3i–3j), and para (3h and 3k–p) to the C–OTf bond, affording the corresponding products in moderate to excellent yields (63–95%) with excellent C–Cl chemoselectivity. Broad functional group substituents were tolerated in this reaction, such as methyl (3a, 3e–3h, 3l, 3p), methoxy (3c, and 3i), fluoro (3j and 3m), trifluoromethyl (3n), and ketone (3o).
 | ||
| Scheme 2 Substrate scope for the chemoselective C–Cl (over C–OTf) decarboxylative coupling reaction. (Reaction condition: chloroaryl triflates 1 (0.20 mmol), alkynyl carboxylic acids 2 (0.36 mmol), dichloro-(1-methylallyl)-dipalladium (2 mol%), L9 (16 mol%), K2CO3 (0.40 mmol) and THF (1.0 mL) were stirred at 100 °C for 3 h. Isolated yields based on chloroaryl triflates 1 are reported. aThe reaction was performed on a 1.0 mmol scale.) | ||
Furthermore, this reaction accommodated a wide range of alkynyl carboxylic acids, regardless of the electronically neutral (2,4-di-Me, 3-Me and 4-Me), electron-donating (2-i-Pr), and electron-withdrawing (4-Ph, 3-F, 4-F and 4-CF3) substituents attached to the alkynyl carboxylic acids all reacted smoothly, providing the corresponding OTf-arylalkynes in good to excellent yields with good C–Cl chemoselectivity (3q–3x). Moreover, the naphthyl propargylic acid also adapted to this reaction, affording the corresponding product 3y in 77% yield. Chloroheteroaryl triflates were also applicable as substrates (3z and 3aa).
Encouraged by the positive results mentioned above, we then studied this chemoselective decarboxylative coupling reaction of bromoaryl triflate and bromochloroaryl triflate (Scheme 3). To our delight, when we used 2-bromophenyl triflate (6) as the substrate, the chemoselective decarboxylative coupling reaction proceeded successfully, yielding the corresponding product in 73% with excellent C–Br chemoselectivity over C–OTf (C–Br:C–OTf ≥ 99:1). When using 4-bromo-2-chlorophenyl triflate (7) as the substrate, the reaction also progressed well, providing the corresponding product in 77% yield with excellent C–Br chemoselectivity over C–Cl and C–OTf (C–Br:C–Cl:C–OTf ≥ 99:1:0).
 | ||
| Scheme 3 Pd-Catalyzed chemoselective C–Br (over C–Cl and C–OTf) decarboxylative coupling reaction. (Reaction condition: halogenated aryl triflate 6 or 7 (0.20 mmol), 3-phenylpropiolic acid 2a (0.36 mmol), dichloro-(1-methylallyl)-dipalladium (2 mol%), L9 (16 mol%), K2CO3 (0.40 mmol) and THF (1.0 mL) were stirred at 100 °C for 3 h. Isolated yields based on polyhalogenated aryl triflate are reported.). | ||
To further expand the substrate scope, we next investigated alkyl–alkynyl carboxylic acids as cross-coupling partners (Scheme 4). 2-Pentynoic acid, 2-hexynoic acid, and 2-octynoic acid were found to be effective substrates, affording the desired products in good yields and with excellent C–Cl chemoselectivity.
 | ||
| Scheme 4 Substrate scope for the chemoselective C–Cl (over C–OTf) decarboxylative coupling reaction. (Reaction condition: chloroaryl triflates 1 (0.20 mmol), alkyl–alkynyl carboxylic acids 9 (0.36 mmol), dichloro-(1-methylallyl)-dipalladium (2 mol%), L9 (16 mol%), K2CO3 (0.40 mmol) and THF (1.0 mL) were stirred at 100 °C for 3 h. Isolated yields based on chloroaryl triflates 1 are reported. aToluene was used.). | ||
To further explore the utility of this chemoselective decarboxylative reaction, we investigated its applicability in a sequential synthetic protocol (Scheme 5). As shown, the chloroaryl triflates initially underwent chemoselective decarboxylation to afford the corresponding intermediates. Without isolating these intermediates, aryl boronic acids were subsequently added to the same reaction mixture, enabling a Suzuki–Miyaura coupling at the C–OTf bonds. This one-pot, two-step sequence furnished the corresponding difunctionalized products 11a and 11b in good yields.
 | ||
| Scheme 5 One-pot sequential utilization of chemoselective decarboxylative coupling. (1st step reaction conditions: chloroaryl triflates 1 (0.20 mmol), alkyl or aryl–alkynyl carboxylic acids 2 or 9 (0.36 mmol), dichloro(1-methylallyl)dipalladium (2 mol%), L9 (16 mol%), K2CO3 (0.40 mmol), and THF (1.0 mL) were stirred at 100 °C for 3 h. 2nd step reaction conditions: 4-methoxyphenylboronic acid (0.40 mmol), Pd2(dba)3 (1.5 mol%), SPhos (4.5 mol%), and K3PO4 (0.3 mmol) were added, and the reaction mixture was stirred at 110 °C for 2 h. Isolated yields are reported based on chloroaryl triflates 1.). | ||
To gain deeper mechanistic insight into this chemoselective decarboxylative reaction, a series of competition experiments were conducted (Table 2). The results revealed that the electron-deficient 3-chloro-5-(trifluoromethyl)phenyl triflate afforded the corresponding product in higher yield compared to the electron-rich 3-chloro-5-methylphenyl triflate and the electron-neutral 3-chlorophenyl triflate. These observations suggest that reductive elimination is unlikely to be the rate-limiting step. To further elucidate the reaction mechanism, a series of density functional theory (DFT) calculations were subsequently performed (see the ESI for details, Fig. S1†). The calculations involved monoligated Pd–L9 reacting with 4-chlorophenyl triflate and 3-phenylpropiolic acid. For the reaction proceeding through the C–Cl pathway, consistent with the experimental kinetic studies, the computational results indicated that the reductive elimination step (12K-TS, 7.9 kcal mol−1) was not rate-determining, given its notably lower activation barrier compared to the oxidative addition and decarboxylation steps. Critically, the oxidative addition step determines the chemoselectivity in this reaction. The higher activation barrier for oxidative addition of the C–OTf bond (12N-TS, 22.4 kcal mol−1), compared to that of the C–Cl bond (12D-TS, 11.2 kcal mol−1), strongly disfavors the C–OTf pathway. Indeed, the oxidative addition step for the C–OTf bond exhibits the highest overall energy barrier among all reaction pathways examined, explaining the observed chemoselectivity towards activation of the C–Cl bond. Within the preferred C–Cl reaction pathway, the decarboxylation step (12H-TS, 21.3 kcal mol−1) is proposed as the rate-determining step.
|
|
||||
|---|---|---|---|---|
| Entry | Substrate 1 | % yieldb | Substrate 2 | % yieldb |
| a Reaction condition: substrate 1 (0.1 mmol), substrate 2 (0.1 mmol), 3-phenylpropiolic acid 2a (0.20 mmol), dichloro-(1-methylallyl)-dipalladium (2 mol%), L9 (16 mol%), K2CO3 (0.40 mmol) and THF (1.0 mL) were stirred at 100 °C for 30 min. b Calibrated GC yields were reported using dodecane as the internal standard. Maximum yield for each substrate in the reaction mixture is 50%. | ||||
| 1 | R1 = CF3 | 32 | R2 = Me | 18 |
| 2 | R1 = CF3 | 31 | R2 = H | 20 |

Based on the results discussed herein and supporting literature ref. 4c, a plausible mechanism for the chemoselective decarboxylative coupling reaction is proposed in Scheme 6. This mechanism initiates with the oxidative addition of chloroaryl triflate (1) to the Pd(0)L (A), forming intermediate B. This intermediate then undergoes a ligand-exchange reaction with the alkynyl carboxylic acid anion, produced by the reaction of alkynyl carboxylic acid (2) with K2CO3, yielding intermediate C. Subsequent decarboxylation of intermediate C releases CO2, resulting in the formation of intermediate D. Finally, reductive elimination of intermediate D leads to the formation of the desired products (3), simultaneously regenerating the catalytically active species Pd(0)L, thus completing the catalytic cycle.
 | ||
| Scheme 6 Proposed mechanism. | ||
Conclusions
In summary, we have developed a palladium-catalyzed, chemoselective decarboxylative coupling of alkynyl carboxylic acids with halogenated aryl triflates, enabling the synthesis of OTf-arylalkyne scaffolds for the first time. Notably, this reaction inverts the conventional chemoselectivity order, favoring C–OTf over C–Cl. It also demonstrates remarkable C–Br selectivity over both C–OTf and C–Cl bonds when employing bromoaryl and bromochloroaryl triflates as substrates. Moreover, we introduce a one-pot sequential strategy that integrates decarboxylation with Suzuki–Miyaura coupling, offering a versatile platform for the efficient synthesis of difunctionalized compounds. DFT calculations suggest that chemoselectivity is governed by the oxidative addition step, while the decarboxylation step is rate-determining. We anticipate that this methodology will provide a new approach for the synthesis of alkynyl compounds bearing diverse functional groups, enabling selective modification of alkynyl scaffolds.Data availability
All graphs and chemical structures in this article were created using ChemDraw 22. The data supporting this study have been included in the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
We thank the Research Grants Council of the Hong Kong Special Administrative Region, China (PolyU 15305522) for financial support.References
- For recent reviews on decarboxylation coupling reaction, see: (a) Y. Wei, P. Hu, M. Zhang and W. Su, Metal-Catalyzed Decarboxylative C–H Functionalization, Chem. Rev., 2017, 117, 8864–8907 CrossRef CAS PubMed; (b) F. Penteado, E. F. Lopes, D. Alves, G. Perin, R. G. Jacob and E. J. Lenardão, a-Keto Acids: Acylating Agents in Organic Synthesis, Chem. Rev., 2019, 119, 7113–7278 CrossRef CAS PubMed; (c) A. Varenikov, E. Shapiro and M. Gandelman, Decarboxylative Halogenation of Organic Compounds, Chem. Rev., 2020, 121, 412–484 CrossRef PubMed; (d) N. Rodríguez and L. J. Goossen, Decarboxylative Coupling Reactions: a Modern Strategy for C–C-bond Formation, Chem. Soc. Rev., 2011, 40, 5030–5048 RSC; (e) C. Shen, P. Zhang, Q. Sun, S. Bai, T. S. A. Hor and X. Liu, Recent Advances in C–S Bond Formation via C–H Bond Functionalization and Decarboxylation, Chem. Soc. Rev., 2015, 44, 291–314 RSC; (f) K. Park and S. Lee, Transition Metal-catalyzed Decarboxylative Coupling Reactions of Alkynyl Carboxylic Acids, RSC Adv., 2013, 3, 14165–14182 RSC; (g) T. Patra and D. Maiti, Decarboxylation as the Key Step in C–C Bond-Forming Reactions, Chem. – Eur. J., 2017, 23, 7382–7401 CrossRef CAS PubMed.
- (a) H. Fu, J. Q. Shang, T. Yang, Y. Shen, C. Z. Gao and Y. M. Li, Copper-Catalyzed Decarboxylative Disulfonylation of Alkynyl Carboxylic Acids with Sulfinic Acids, Org. Lett., 2018, 20, 489–492 CrossRef CAS PubMed; (b) Y. Zhang, S. Patel and N. Mainolfi, Copper-catalyzed Decarboxylative C–N Coupling for N-arylation, Chem. Sci., 2012, 3, 3196–3199 RSC; (c) H. Hikawa, F. Kotaki, S. Kikkawa and I. Azumaya, Gold(III)-Catalyzed Decarboxylative C3-Benzylation of Indole-3-carboxylic Acids with Benzylic Alcohols in Water, J. Org. Chem., 2019, 84, 1972–1979 CrossRef CAS PubMed; (d) X. Tan, Z. Liu, H. Shen, P. Zhang, Z. Zhang and C. Li, Silver-Catalyzed Decarboxylative Trifluoromethylation of Aliphatic Carboxylic Acids, J. Am. Chem. Soc., 2017, 139, 12430–12433 CrossRef CAS PubMed.
- J. Moon, M. Jeong, H. Nam, J. Ju, J. H. Moon, H. M. Jung and S. Lee, One-Pot Synthesis of Diarylalkynes Using Palladium-Catalyzed Sonogashira Reaction and Decarboxylative Coupling of Sp Carbon and Sp2 Carbon, Org. Lett., 2008, 10, 945–948 CrossRef CAS PubMed.
- For references utilizing aryl halides as electrophiles, see: (a) J. Moon, M. Jang and S. Lee, Palladium-Catalyzed Decarboxylative Coupling of Alkynyl Carboxylic Acids and Aryl Halides, J. Org. Chem., 2009, 74, 1403–1406 CrossRef CAS PubMed; (b) W.-W. Zhang, X.-G. Zhang and J.-H. Li, Palladium-Catalyzed Decarboxylative Coupling of Alkynyl Carboxylic Acids with Benzyl Halides or Aryl Halides, J. Org. Chem., 2010, 75, 5259–5264 CrossRef CAS PubMed; (c) X. Li, F. Yang and Y. Wu, Palladacycle-Catalyzed Decarboxylative Coupling of Alkynyl Carboxylic Acids with Aryl Chlorides Under Air, J. Org. Chem., 2013, 78, 4543–4550 CrossRef CAS PubMed; (d) F. Sun and Z. Gu, Decarboxylative Alkynyl Termination of Palladium-Catalyzed Catellani Reaction: A Facile Synthesis of α-Alkynyl Anilines via Ortho C–H Amination and Alkynylation, Org. Lett., 2015, 17, 2222–2225 CrossRef CAS PubMed; (e) S. Tartaggia, O. De Lucchi and L. J. Gooßen, Practical synthesis of unsymmetrical diarylacetylenes from propiolic acid and two different aryl bromides, Eur. J. Org. Chem., 2012, 1431–1438 CrossRef CAS.
- (a) W. Jia and N. Jiao, Cu-Catalyzed Oxidative Amidation of Propiolic Acids Under Air via Decarboxylative Coupling, Org. Lett., 2010, 12, 2000–2003 CrossRef CAS PubMed; (b) D. L. Priebbenow, P. Becker and C. Bolm, Copper-Catalyzed Oxidative Decarboxylative Couplings of Sulfoximines and Aryl Propiolic Acids, Org. Lett., 2013, 15, 6155–6157 CrossRef CAS PubMed.
- D. Hazarika and P. Phukan, TsNBr2 Promoted Decarboxylative Bromination of α,β-unsaturated Carboxylic Acids, Tetrahedron Lett., 2018, 59, 4593–4596 CrossRef CAS.
- (a) J. Hu, N. Zhao, B. Yang, G. Wang, L. N. Guo, Y. M. Liang and S. D. Yang, Copper-Catalyzed C-P Coupling Through Decarboxylation, Chem. – Eur. J., 2011, 17, 5516–5521 CrossRef CAS PubMed; (b) X. Li, F. Yang, Y. Wu and Y. Wu, Copper-Mediated Oxidative Decarboxylative Coupling of Arylpropiolic Acids with Dialkyl H-Phosphonates in Water, Org. Lett., 2014, 16, 992–995 CrossRef CAS PubMed.
- (a) G. Rong, J. Mao, H. Yan, Y. Zheng and G. Zhang, Phosphoric Acid-Mediated Synthesis of Vinyl Sulfones Through Decarboxylative Coupling Reactions of Sodium Sulfinates with Phenylpropiolic Acids, J. Org. Chem., 2015, 80, 7652–7657 CrossRef CAS PubMed; (b) H. Fu, J. Q. Shang, T. Yang, Y. Shen, C. Z. Gao and Y. M. Li, Copper-Catalyzed Decarboxylative Disulfonylation of Alkynyl Carboxylic Acids with Sulfinic Acids, Org. Lett., 2018, 20, 489–492 CrossRef CAS PubMed; (c) F. Liu and W. Yi, A Thiol-free Synthesis of Alkynyl Chalcogenides by the Copper-catalyzed C–X (X = S, Se) Cross-coupling of Alkynyl Carboxylic Acids with Bunte Salts, Org. Chem. Front., 2018, 5, 428–433 RSC.
- J. Chen, S. Su, D. Hu, F. Cui, Y. Xu, Y. Chen, X. Ma, Y. Pan and Y. Liang, Copper–Catalyzed Bis– or Trifunctionalization of Alkynyl Carboxylic Acids: An Efficient Route to Bis– and Tris–selenide Alkenes, Asian J. Org. Chem., 2018, 7, 892–896 CrossRef CAS.
- Q. Feng, K. Yang and Q. Song, Highly Selective Copper-catalyzed Trifunctionalization of Alkynyl Carboxylic Acids: an Efficient Route to Bis-deuterated β-borylated α,β-styrene, Chem. Commun., 2015, 51, 15394–15397 RSC.
- L. Zhang, Z. Hang and Z. Q. Liu, A Free-Radical-Promoted Stereospecific Decarboxylative Silylation of α,β-Unsaturated Acids with Silanes, Angew. Chem., Int. Ed., 2015, 55, 236–239 CrossRef PubMed.
- T. Li, X. Qu, Y. Zhu, H. Wang and Z. Zhang, Synthesis of Diarylalkynes by Iron/Copper Co-Catalyzed Decarboxylative sp-sp2 Coupling of Alkynyl Carboxylic Acids and Aryl Halides, Adv. Synth. Catal., 2011, 353, 2731–2738 CrossRef CAS.
- (a) I. Kalvet, G. Magnin and F. Schoenebeck, Rapid Room–Temperature, Chemoselective C–C Coupling of Poly(pseudo)halogenated Arenes Enabled by Palladium(I) Catalysis in Air, Angew. Chem., Int. Ed., 2017, 56, 1581–1585 CrossRef CAS PubMed; (b) G. Kundu, T. Sperger, K. Rissanen and F. Schoenebeck, A Next–Generation Air–Stable Palladium(I) Dimer Enables Olefin Migration and Selective C–C Coupling in Air, Angew. Chem., Int. Ed., 2020, 59, 21930–21934 CrossRef CAS PubMed; (c) Y. Morisaki, R. Sawada, M. Gon, Y. Chujo and T. Inoshita, New Types of Planar Chiral [2.2]Paracyclophanes and Construction of One-Handed Double Helices, Chem. – Asian J., 2016, 11, 2524–2527 CrossRef CAS PubMed; (d) A. D. Finke, E. C. Elleby, M. J. Boyd and C. K. Haley, Zinc Chloride-Promoted Aryl Bromide-Alkyne Cross-Coupling Reactions at Room Temperature, J. Org. Chem., 2009, 74, 8897–8900 CrossRef CAS PubMed.
- (a) C. M. So, O. Y. Yuen, S. S. Ng and Z. Chen, General Chemoselective Suzuki–Miyaura Coupling of Polyhalogenated Aryl Triflates Enabled by an Alkyl-Heteroaryl-Based Phosphine Ligand, ACS Catal., 2021, 11, 7820–7827 CrossRef CAS; (b) S. S. Ng, Z. Chen, O. Y. Yuen and C. M. So, Palladium–Catalyzed Chemoselective Borylation of (Poly)halogenated Aryl Triflates and Their Application in Consecutive Reactions, Adv. Synth. Catal., 2022, 364, 1596–1601 CrossRef CAS; (c) M. Wang and C. M. So, Inverting Conventional Chemoselectivity in the Sonogashira Coupling Reaction of Polyhalogenated Aryl Triflates with TMS-Arylalkynes, Org. Lett., 2022, 24, 681–685 CrossRef CAS PubMed; (d) Z. Chen, C. Gu, O. Y. Yuen and C. M. So, Palladium-catalyzed Chemoselective Direct a-arylation of Carbonyl Compounds with Chloroaryl Triflates at the C–Cl Site, Chem. Sci., 2022, 13, 4762–4769 RSC; (e) M. Wang, O. Y. Yuen and C. M. So, Palladium-Catalyzed Desulfinative Cross-Coupling of Polyhalogenated Aryl Triflates with Aryl Sulfinate Salts: Inversion of Traditional Chemoselectivity, Chin. J. Chem., 2023, 41, 909–914 CrossRef CAS; (f) S. S. Ng, W. H. Pang, O. Y. Yuen and C. M. So, Recent advances in the application of ligands in palladium-catalyzed chemoselective coupling reactions at C–Br, C–OTf, and C–Cl sites, Org. Chem. Front., 2023, 10, 4408–4436 RSC.
- E. K. Reeves, J. N. Humke and S. R. Neufeldt, N-Heterocyclic Carbene Ligand-Controlled Chemodivergent Suzuki–Miyaura Cross Coupling, J. Org. Chem., 2019, 84, 11799–11812 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: Experimental details, and characterization data. See DOI: https://doi.org/10.1039/d5qo00339c |
| ‡ These authors contributed equally to this work. |
| This journal is © the Partner Organisations 2025 |