DOI:
10.1039/D5QO00034C
(Research Article)
Org. Chem. Front., 2025,
12, 3230-3238
Pd-catalyzed site-specific heteroaromatic C–H/peri-C–H annulative coupling for synthesis of cyclopenta-fused polycyclic heteroarenes†
Received
7th January 2025
, Accepted 4th March 2025
First published on 5th March 2025
Abstract
Cyclopenta-fused polycyclic heteroarenes (CP-PHAs) composed of acenaphthylene and heteroaromatic units have emerged as intriguing photoelectronic materials in view of the electron-accepting feature of the fused CP rings and the tunable electronic properties of the heteroaromatics. To date, Pd-catalyzed aromatic C–H/peri-C–halogen coupling has been used as one of the most general methods to construct such CP-rings. In contrast, Pd-catalyzed direct aromatic C–H/peri-C–H coupling may provide a shortcut to this CP-ring formation, which is applicable to a wide range of polycyclic arenes bearing peri-C–H bonds, but remains unexploited so far. We herein describe a Pd-catalyzed site-specific C–H/peri-C–H annulative coupling between (benzo)thiophenes or indoles with various polycyclic arenes to access a variety of π-extended CP-PHAs. Benzo[b]thiophenes or thiophenes with a 1-naphthalenyl group at the C3 position gave C2-naphthylated CP-PHAs in the Pd(OPiv)2/AgOPiv system, whereas indoles with a 1-naphthalenyl group at the C2 position gave C3-naphthylated CP-PHAs in the Pd(OPiv)2/AgSbF6 system. Notably, deuteration experiments demonstrated that silver salts play a crucial role in the formation of heteroaryl-Ag species via site-specific C–H metalation of heteroaromatics, which subsequently undergo transmetalation with Pd(II) to form heteroaryl-Pd species and activate the peri-C–H bond of the naphthalene group. Furthermore, this method enables the synthesis of structurally diverse CP-PHAs via activation of peri-C–H bonds in large polycyclic aromatic hydrocarbons such as anthracene, phenanthrene, and pyrene.
Introduction
Cyclopenta-fused polycyclic aromatic hydrocarbons (CP-PAHs) and polycyclic heteroaromatics (CP-PHAs) have emerged as intriguing photoelectronic materials in terms of unique electronic affinity of the fused CP rings and their enhanced stability.1–5 Among them, CP-PHAs composed of acenaphthylene and heteroaromatic units have drawn particular interest due to the electron-accepting nature of acenaphthylene and the tunable electronic properties of the heteroaromatics.4,5 In this context, we are interested in developing new and highly efficient synthetic methods to construct π-extended CP-PHAs fused with electron-rich heteroaromatic and electron-accepting acenaphthylene units to create a new class of donor–acceptor (D–A) type π-electronic molecules. To date, the Pd(0)-catalyzed aromatic peri-C–H/C–Br and heteroaromatic C–H/peri-C–Br annulative couplings are the most efficient methods toward constructing such acenaphthylene-fused CP-PAHs and CP-PHAs (Scheme 1a and b).6–8 However, these methods require brominated arenes, and the extension to other π-extended PAHs with peri-positions is considered problematic, particularly the method using 1,8-dibromonaphthalene (Scheme 1b), due to the difficult in selective prefunctionalization of the peri-positions of such PAHs. Recently, we also developed a novel Pd(II)-catalyzed cascade cyclization via peri-C–H activation for the construction of indole- and acenaphthylene-fused D–A type CP-PHAs (Scheme 1c),9 but this method was limited to the formation of the indole moiety. Considering that this cascade cyclization leads to the formation of an indolyl-Pd intermediate9,10 enabling subsequent peri-C–H bond activation, we envisioned that if heteroaryl-Pd intermediates can be formed site-selectively from readily available naphthyl-substituted benzo[b]thiophene, thiophene, and indole substrates, the desired CP-PHAs fused with naphthalene can be constructed conveniently by direct C–H/peri-C–H annulative coupling (Scheme 1d).6,9,11
 |
| | Scheme 1 Pd-catalyzed synthesis of fused acenaphthylenes via (hetero)aromatic C–H bond activation. | |
Transition-metal-catalyzed oxidative aromatic C–H/C–H coupling reactions, which do not require prefunctionalization of (hetero)aromatic substrates, have been demonstrated to be a promising strategy for the synthesis of biaryl and polycyclic (hetero)aromatic motifs,12 structural fragments frequently used in optoelectronic materials and medicinal chemistry. In particular, depending on the inherent differences in reactivity at the C2 and C3 positions and the catalyst system, a variety of site-selective C–H arylations of (benzo)thiophenes and indoles have been reported to afford biaryls.13–15 However, aromatic C–H/peri-C–H coupling of (benzo)thiophenes or indoles with naphthalenes provides a shortcut to CP ring formation by intramolecular direct coupling, which may be applicable to a wide range of polycyclic arenes bearing peri-C–H bonds, but remains unexplored so far. We herein describe a Pd-catalyzed site-specific heteroaromatic C–H/peri-C–H annulative coupling between (benzo)thiophenes or indoles and various PAHs bearing C–H bonds to access structurally diverse π-extended CP-PHAs (Scheme 1e). Furthermore, we have demonstrated that silver salt additives with different counteranions function not only as oxidants but also as site-specific C–H metalation reagents to form heteroaryl-Ag intermediates followed by Pd(II)-catalyzed transmetalation to generate the desired heteroaryl-Pd intermediates.
Results and discussion
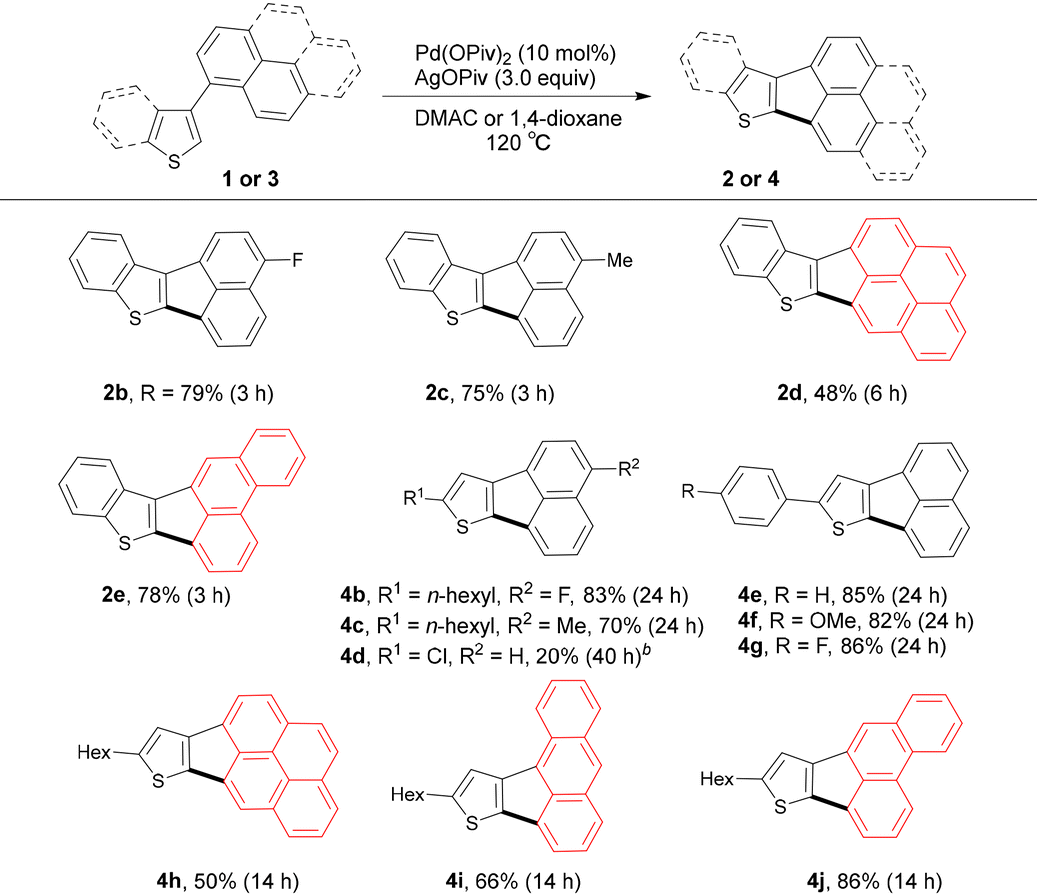
Taking into account the influence in acidity of the C–H bonds at the C2 (pKa = 32.0) and C3 (pKa = 36.8) positions of benzo[b]thiophene on the dehydrometalation reactivity,16 2-(naphthalen-1-yl)benzo[b]thiophene 1a was selected as the substrate to optimize the reaction conditions for the synthesis of benzo[b]thiophene-fused CP-PHA 2a. As shown in Scheme 1c,9 we previously reported that the Pd(OPiv)2 catalyst favors the activation of peri-C–H bonds of PAHs and an additional metal oxidant is required to regenerate active Pd(II) species from the in situ formed Pd(0) species. With these considerations in mind, we evaluated a set of Pd(II) (10 mol%) catalysts and metal oxidants (3 equiv.) by using N,N-dimethyl acetamide (DMAc) as the solvent at 120 °C for 14 h (Table 1). As expected, the use of the Pd(OPiv)2 catalyst in the absence of oxidants, 2a was obtained in a very low yield of 5% (entry 1). The use of cationic silver salts such as AgSbF6 and AgBF4 as oxidants had no obvious effect on improving the yield of 2a (entries 2 and 3). Subsequently, AgOAc oxidant was used to promote the carboxylate-assisted C–H bond metalation, but although a slight increase in efficiency was observed, the yield of 2a was still low (entry 4). To our delight, the reaction efficiency drastically enhanced when AgOPiv oxidant was used, affording 2a in 81% yield in 3 h (entry 5). It was mentioned that the use of a reduced amount of AgOPiv (2 equiv.) resulted in a decrease in the yield of 2a along with the recovered 1a (entry 6) Moreover, the combination of AgOPiv oxidant with PdCl2 or Pd(OAc)2 catalyst instead of Pd(OPiv)2 also showed sufficient efficiency for the formation of 2a (entries 7 and 8). However, the use of Cu(OPiv)2 instead of AgOPiv was ineffective in improving the yield of 2a (entry 9). These results indicate that the existence of high concentrations of pivalate counter anion in AgOPiv is crucial for the implementation of the present C–H/C–H coupling reaction. In addition, the optimized catalyst system in entry 5 showed higher activity in polar solvents such as DMAc, DMF (71%), NMP (65%), i-PrCN (62%), and 1,4-dioxane (70%) than in less polar solvents such as toluene (33%) and chlorobenzene (40%).
Table 1 Optimization of reaction conditions for the coupling of 3-(naphthalen-1-yl)benzo[b]thiophene (1a)a
To verify the difference in reactivity of the C–H bonds at the C2 and C3 positions, the reaction of 2-(naphthalen-1-yl)benzo[b]thiophene 1a′ was performed under the optimized conditions (eqn (1)). As a result, the desired product 2a was obtained in only 3% yield and 1a′ was mainly recovered, indicating that the reactivity of the C–H bond at the C3 position of the benzo[b]thiophene moiety is much lower than that at the C2 position in this catalyst system. Next, the same catalytic system was used to investigate the site-selectivity of C–H bonds at the C2 and C3 positions of thiophene substrates. It has also been reported that the C–H bond acidity is higher at the C2 (pKa = 33.5) position of thiophene than at the C3 (pKa = 39.0) position.16 Similar to the site-selectivity of benzo[b]thiophene substrates, thiophene substrate 3a bearing a 1-naphthyl group at the C3 position produced corresponding product 4a in a high yield of 80% (eqn (2)), whereas thiophene substrate 3a′ bearing a 1-naphthyl group at the C2 position afforded a very poor yield of 4a (eqn (3)). These results indicate that in the present catalytic system, the C–H bond at the C2 position of the thiophene moiety is much more reactive than the C–H bond at the C3 position.
| |  | (1) |
| |  | (2) |
| |  | (3) |
With the site-selective reactivity in hand, the substrate scope of benzo[b]thiophenes 1 and thiophenes 3 was evaluated under optimized conditions (Table 2).17 The electron-withdrawing fluorine group and electron-donating methyl group at the 4-position of the naphthyl moiety in the benzo[b]thiophene substrates, respectively, had negligible electronic effect on the dual C–H bond activation process, yielding corresponding CP-PHAs 2b and 2c in good yields. Remarkably, the peri-C–H bonds in large PAHs such as pyrene and phenanthrene moieties could also be activated to construct π-extended CP-PHAs 2d and 2e in 48% and 78% yields, respectively. Likewise, thiophene substrates bearing fluorine and methyl substituents at the 4-position of the 1-naphthyl moiety, respectively, did not show significant electronic effect on the reactivity difference, affording corresponding CP-PHAs 4b and 4c in good yields. In comparison, the thiophene substrate 3d, 2-chloro-4-(naphthalen-1-yl)thiophene, bearing a chlorine substituent at the 2-position of the thiophene ring, exhibited low reactivity under the standard conditions, affording the annulation product 4d in 20% yield. However, most of the chlorine substituent remained unreacted under the conditions and 3d was recovered in 65% yield albeit with the formation of a trace of the dechlorinated byproduct of 3d. Aryl units such as unsubstituted phenyl, and 4-MeO- and 4-F-substituted phenyls on the thiophene moiety did not affect this C–H/C–H coupling, affording desired products 4e–g in high yields. Gratifyingly, large PAHs such as pyrene, anthracene, and phenanthrene having peri-C–H bonds showed good compatibility for the present C–/C–H coupling, giving the π-extended CP-PHAs 4h–j in good to high yields. Furthermore, large CP-PHA structures fused with two strained CP rings could be constructed by the two-fold C–H/peri-C–H coupling strategy. For example, the reaction of 3k bearing two 1-naphthyl groups at the C3- and C4-positions of the thiophene under the modified conditions led to the cleavage of four C–H bonds to give corresponding product 4k in 58% yield (eqn (4)). Furthermore, with regard to the prospective application of the highly π-extended CP-PHAs as optoelectronic materials,8 the reaction of 3l consisting of a benzo[1,2-b:4,5-b′]dithiophene core and two 1-naphthyl groups was carried out, enabling the two-fold C–H/peri-C–H coupling to afford desired product 4l in 27% yield.
| |  | (4) |
| |  | (5) |
Table 2 Synthesis of various π-extended CP-PHAs fused with benzo[b]thiophenes and thiophenesa
|
Reaction conditions: benzo[b]thiophenes 1 or thiophenes 3 (0.2 mmol), Pd(OPiv)2 (10 mol%), AgOPiv (3 equiv.), DMAc (0.1 M) for substrates 1 and 1,4-dioxane (0.1 M) for substrates 2, 120 °C. Isolated yields are shown.
The starting substrate 3d was recovered in 65% yield.
|

|
To gain insights into the site-selective metalation process, several deuteration reactions were performed. The reaction of 1a with D2O in the presence of 1 equiv. of Pd(OPiv)2 without using AgOPiv afforded the deuterated benzo[b]thiophene 1a-d1 in 3% yield along with the desired coupling product in 25% yield (Scheme 2a). This result implies that C–H metalation with Pd(II) at the C2 position of 1a leads to the formation of a Bt-Pd intermediate, enabling subsequent peri-C–H bond activation, but in much lower yield compared to those using Pd(II) catalyzed conditions in combination with AgOPiv. Sanford and Larrosa demonstrated that silver salts such as AgOPiv and Ag2O promoted the C–H metalation of thiophene and benzo[b]thiophene at the C2 position to form a Bt-Ag intermediate.14b,18 Based on these precedents and our optimized conditions, the reaction of 1a with D2O was carried out using 1 equiv. of AgOPiv in the absence of Pd(II) catalyst. As a result, deuterated 1a-d1 was obtained in 44% yield without forming the coupling product 2a. This H/D exchange indicates the formation of a Bt-Ag intermediate, which should be a key intermediate for the Pd(II)-catalysed transmetalation reaction to produce the Bt-Pd intermediate. Additionally, high resolution mass spectrometry (HRMS) analysis confirmed that no deuterium was incorporated at the 2- or 8-positions of the naphthyl moiety in 1a, 1a-d1, and 2a after these deuteration reactions. In contrast, no obvious H/D exchange was detected when 1a′ was reacted with D2O using AgSbF6 and AgOPiv, respectively (Scheme 2b), which is in line with the result shown in eqn (1), suggesting that the C–H bond at the C3 position of 1a′ is almost inactive in the present catalyst system.
 |
| | Scheme 2 Study for the role of Ag and Pd salts. ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 1H NMR yields are shown using CH2Br2 as an internal standard. 1H NMR yields are shown using CH2Br2 as an internal standard. | |
In addition, parallel kinetic isotope effect (KIE) reactions with protonated 1a and deuterated 1a-d1 were carried out in separated vessels for 30 min under optimized conditions (Scheme 3a). The low KIE value of 1.33 indicates that the C–H bond activation at the C2 position is unlikely to be the rate-determining step. Furthermore, a KIE value of 1.7 was obtained in the intermolecular competition reaction using a 1:1 mixture of 1a and 1a-d7 under optimized conditions in the same vessel for 1 h (Scheme 3b). It should be noted that in the reaction of 1a under standard conditions (shown in Table 1, entry 5), the dimerization product 1a/1a was observed as a byproduct in less than 5% yield. This competition KIE reaction afforded a mixture of dimerization byproducts with a total yield of 12%, including the deuterated homo-coupling dimer 1a-d7/1a-d7 as the major byproduct and the protonated homo-coupling dimer 1a/1a and cross-coupling dimer 1a/1a-d7 as minor byproducts (∼2:1 ratio). The formation of 1a-d7/1a-d7 dimer as the major byproduct and the higher KIE value than the parallel reaction suggest that the peri-C–H bond activation may be the rate-determining step.
 |
| | Scheme 3 Study of deuterium kinetic Isotope effect (KIE) for C–H metalation. 1H NMR yields are shown using CH2Br2 as an internal standard. | |
On the basis of experimental outcomes and the prominent role of the pivalate counteranion in this transformation, a plausible reaction mechanism is outlined in Scheme 4. Both AgOPiv and Pd(OPiv)2 enable the activation of the C–H bond of 1a at the C2 position by a pivalate-assisted concerted metalation deprotonation (CMD) process via transition state TS1.19 In this case, in terms of the C–H metalation efficiency shown in Scheme 2a, the AgOPiv-mediated C–H metalation should predominantly proceed compared to Pd(OPiv)2, affording Bt-Ag intermediate A and Bt-Pd intermediate B, the latter of which also can be formed from the former via transmetalation with Pd(OPiv)2. The peri-C–H bond of intermediate B is then activated by an intramolecular CMD process via transition state TS2, leading to the formation of palladacycle C. Reductive elimination of C produces the corresponding product 2a along with Pd(0) species, the latter of which can be oxidized by AgOPiv oxidant to regenerate the active Pd(OPiv)2 catalyst.
 |
| | Scheme 4 Plausible reaction mechanism for the formation of 2a. | |
The UV-Vis absorption of representative CP-PHAs in chloroform solution showed broad absorption spectra extending up to about 600 nm (Fig. 1). The longest absorption maxima (λmax) and onsets of 2d, 4h, and 4i fused with large π-surfaces of pyrene (2d and 4h) and anthracene (4i), respectively, are redshifted compared to 2a fused with naphthalene due to the large π-extension of the formers (Fig. 1a). Notably, these CP-PHAs with one embedded CP ring exhibit very weak absorbance in the range of 400–600 nm, which can be attributed to the HOMO–LUMO transition with very small oscillator strengths (f = 0.0236–0.0074) as suggested by density functional theory (DFT) calculations at the B3LYP/6-31+G(d) level (Fig. S1, ESI†). In comparison, 4k and 4l fused with two CP rings show increased absorption intensities with λmax of 510 nm and 552 nm, respectively, corresponding to the HOMO–LUMO transitions by DFT calculations (f = 0.1606–0.1141) (Fig. 1b and Fig. S1†). The absorption onset of 590 nm for 4l is redshifted compared to 4k (522 nm) due to the large π-extension system of 4l. The HOMO and LUMO energy levels of CP-PHAs were calculated from the oxidation and reduction potentials by cyclic voltammetry (Table 3 and Fig. S2 in ESI†). Relatively low-lying HOMO and LUMO were observed in the range of −5.59 to −5.37 eV for 2a–4k and −3.61 to −3.48 eV for 2a–4l, respectively, due to the high electron affinity of the fused CP rings, in agreement with the trend of DFT calculations. 4l fused with two CP rings has a high HOMO of −5.16 eV due to the strong electron-donating benzodithiophene core and two alkoxy groups. The electronic structures studied by DFT calculations suggest that the LUMO contours of 2a, 2d, 4h, and 4i are mainly localized at the acenaphthylene core and peripheral polyaromatic units, whereas the HOMOs are delocalized from the (benzo)thiophene moiety to the acenaphthylene moiety. In addition, the HOMO and LUMO of 4l are located at the benzodithiophene core and two acenaphthylene cores, respectively, exhibiting a D–A type electronic structure consistent with the high-lying HOMO and low-lying LUMO energy levels.
 |
| | Fig. 1 UV-Vis absorption spectra in CHCl3, (a) 2a, 2d, 4h, and 4i; (b) 4k and 4l. | |
Table 3 Electrochemical properties of CP-PHAs
| Compd. |
ΔEa [eV] |
HOMOb [eV] |
HOMOc [eV] |
LUMOb [eV] |
LUMOc [eV] |
|
HOMO–LUMO energy gap (ΔE) was calculated from the HOMO and LUMO energy levels.
HOMO and LUMO was calculated from the oxidation and reduction potentials by CV measurement and the Fc/Fc+ (−4.80 eV) was used as an external standard.
DFT calculation at the B3LYP/6-31+G(d) level.
|
|
2a
|
2.07 |
−5.59 |
−5.38 |
−3.52 |
−2.03 |
|
2d
|
2.00 |
−5.48 |
−5.22 |
−3.48 |
−2.23 |
|
4h
|
1.97 |
−5.45 |
−5.22 |
−3.48 |
−2.04 |
|
4i
|
1.97 |
−5.45 |
−5.18 |
−3.48 |
−2.18 |
|
4k
|
1.85 |
−5.37 |
−5.17 |
−3.52 |
−2.08 |
|
4l
|
1.55 |
−5.16 |
−4.81 |
−3.61 |
−2.12 |
Next, we continued to develop the site-specific C–H/peri-C–H coupling of 1-naphthylene-tethered indoles to construct indole-fused CP-PHAs. The indole bearing an N-methyl substituent was investigated to have less acidic C–H bonds at the C2 (pKa = 37.7) and C3 (pKa = 41.4) positions compared to (benzo)thiophenes.16 Therefore, considering the high nucleophilicity of the C3 position of indoles, the 1-(p-tolyl)-1H-indole 5a bearing 1-naphthyl group at the C2 position was selected as the substrate to establish optimal catalyst system. A brief optimization of the conditions showed that the use of the Pd(OPiv)2 catalyst in the absence of an oxidant was almost ineffective in the formation of the corresponding product 6a (Table 4, entry 1). Importantly, cationic AgSbF6 and AgBF4 proved to be highly effective oxidants in improving the yield of 6a, whereas the AgOPiv oxidant, which has been successfully used in the case of (benzo)thiophenes, showed moderate reactivity (entries 2–4). This prominent role of cationic silver salts suggests the involvement of an electrophilic metalation process at the nucleophilic C3 position of 1a. On the other hand, copper salts such as Cu(OPiv)2 and Cu(OTf)2 exhibited lower performance compared to silver salts (entries 5 and 6). In comparison, the combination of PdCl2 with AgSbF6 was found to be ineffective, suggesting that a pivalate counteranion is required to sufficiently implement this C–H/C–H coupling reaction (entry 7).
Table 4 Optimization of reaction conditions for (hetero)aromatic C–H/C–H coupling with indole substratesa
To compare the site-selective reactivity of the C–H bond at the C3 and C2 positions, the indole substrate 5a′ bearing a 1-naphthyl group at the C3 position was subjected to the PdCl2/AgSbF6 system (eqn (6)). As expected, the yield of the corresponding product 6a was very low (14%), indicating that the C3 position of the indole is much more reactive than the C2 position under the present conditions. The N-methyl- and N-hexyl-substituted indoles 5b and 5c were examined to be less effective compared to the N-p-tolyl-substituted indole 5a, affording the corresponding products 6b and 6c in 39% and 30% yields, respectively (eqn (7)). The decreased yields of 6b and 6c could be attributed to the partial decomposition of 5b and 5c, likely due to N-dealkylation of the methyl or hexyl groups on the indole moiety under Pd-catalysed conditions.9 In contrast, the N-p-tolyl group of 5a was highly tolerant to N-dearylation, affording 6a in good yield. It should be noted that we have previously developed an efficient Pd-catalyzed cascade cyclization for the synthesis of N-alkyl-substituted CP-PHAs such as 6c, as shown in Scheme 1c.9 Therefore, the present direct C–H/C–H coupling of N-aryl-substituted indoles can be considered as an alternative method for the synthesis of these indole-fused CP-PHAs. It was noted that the N-unsubstituted indole substrate was almost completely ineffective in yielding the corresponding CP-PHA product. In addition, indoles 5d and 5e bearing an electron-donating methyl group and an electron-withdrawing fluorine group at the 5-position (R) of the indole moiety, respectively, showed no obvious electronic effect in the formation of the corresponding products 6d and 6e in good yields (eqn (8)). Our previous electrochemical studies revealed that the HOMO and LUMO of such indole-fused CP-PHA analogues containing naphthalene, pyrene, and phenanthrene are in the range of −5.25 to –5.12 eV and −3.37 to −2.97 eV, respectively,9 which are higher than those of the (benzo)thiophene analogues as shown in Table 3 due to the stronger electron-donating nature of the indole moiety compared to the (benzo)thiophene moieties.
| |  | (6) |
| |  | (7) |
| |  | (8) |
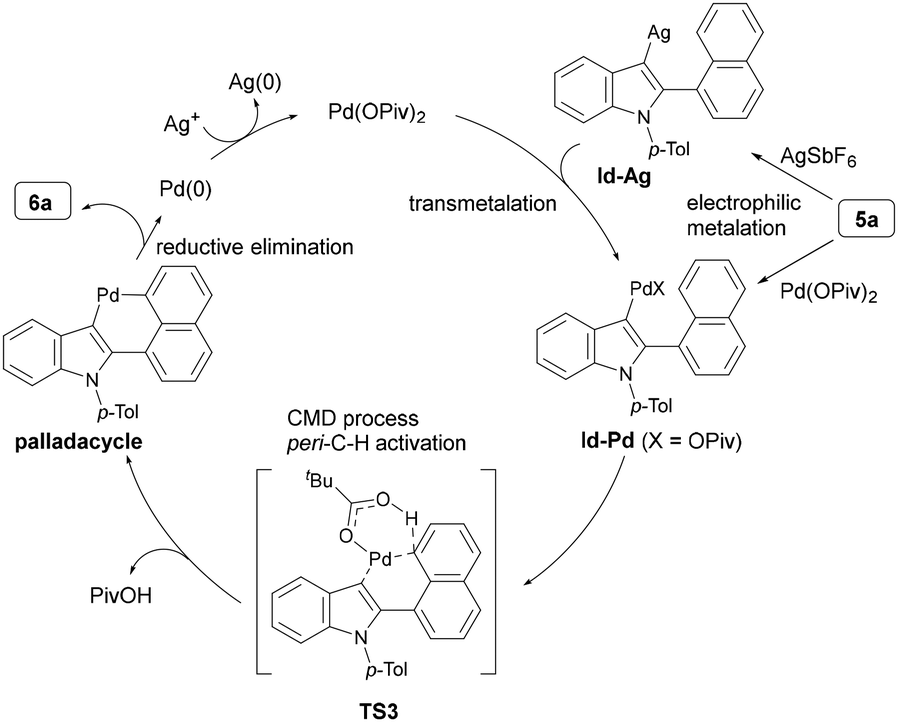
To confirm the involvement of the C–H metalation process, we performed the H/D exchange reaction using 5a and D2O. The reaction with 1 equiv. of Pd(OPiv)2 without using AgSbF6 afforded the deuterated product 5a-d1 in 19% yield together with the corresponding coupling product 6a in 16% yield (Scheme 5a). This result indicates that Pd(OPiv)2 is able to promote C–H metalation at the C3 position of 5avia the formation of an indole-Pd (Id-Pd) intermediate, followed by activation of the peri-C–H bond to give 6a. Meanwhile, the use of 1 equiv. of AgSbF6 afforded 5a in 50% yield without producing 6a, indicating that the C–H metalation reactivity of AgSbF6 at the C3 position of 5a is higher than that of Pd(OPiv)2, leading to the formation of the an indole-Ag (Id-Ag) intermediate. The fact that the combination of Pd(OPiv)2 catalyst with AgSbF6 significantly improved the yield of 6a supports the involvement of the Id-Ag to Id-Pd transmetalation in the present C–H/C–H coupling reaction. Additionally, no deuterium incorporation was observed at the 2- or 8-positions of the naphthyl moiety of 5a, 5a-d1, and 6a after these deuterium reactions. Furthermore, H/D exchange experiments with 5a′ bearing a 1-naphthyl group at the C3 position in the presence of 1 equiv. of AgSbF6 or AgOPiv, respectively, afforded the corresponding deuteration product 5a′-d1 in low yields (Scheme 5b). These results are in line with the hypothesized difference in nucleophilicity between the C2 and C3 positions of indole as well as the difference in C–H bond acidity between benzothiophene and indole.
 |
| | Scheme 5 Study for the role of Ag and Pd salts. 1H NMR yields are shown using CH2Br2 as an internal standard. | |
Based on the experimental outcomes, a plausible reaction mechanism for the direct indole-C–H/peri-C–H coupling was proposed as shown in Scheme 6. Electrophilic metalation at the C3 position of 5a, involving electrophilic substitution of both AgSbF6 and Pd(OPiv)2 and followed by deprotonation, leads to the formation of intermediates Id-Ag and In-Pd, respectively, with the former predominating. The Id-Ag intermediate then undergoes transmetalation with Pd(OPiv)2 to form Id-Pd, which activates the peri-C–H bond via a CMD process with a pivalate counteranion-assisted transition state (TS3) to generate a palladacycle intermediate. Reductive elimination of the palladacycle furnishes the coupling product 6a. Finally, the reduced Pd(0) species is oxidized by a silver oxidant to regenerate the active Pd(II) species.
 |
| | Scheme 6 Plausible reaction mechanism for the formation of 6a. | |
Conclusions
We have disclosed for the first time a new and efficient Pd-catalyzed direct aromatic C–H/peri-C–H coupling reaction via site-specific C–H bond activation of heteroaromatics, such as benzo[b]thiophenes, thiophenes, and indoles, as well as peri-C–H bond activation of various PAHs to construct diverse CP-fused PHAs. The choice of silver salts with respect to the reactive site of the heteroaromatics is found to be crucial for the implementation of this Pd-catalyzed site-specific coupling reaction: AgOPiv with pivalate counteranion enables C–H metalation at the C2 position of benzo[b]thiophenes and thiophenes, whereas the cationic AgSbF6 favors C–H metalation at the C3 position of indoles. The present dual C–H functionalization does not require any directing groups or prehalogenation of the peri-position of polyaromatic unit and the heteroaromatic unit, providing an alternative, convenient, and straightforward approach to construct new CP-PHAs fused with various large PAHs and π-extended CP-PHAs fused with two CP rings, which are difficult to achieve by the reported synthetic methods. Further extension of the current dual C–H annulation strategy to the design and synthesis of novel π-extended PHAs fused with various ring systems is in progress to discover new organic functional materials.
Author contributions
T. J. and M. T. conceived the methodology and wrote the manuscript. M. K. and S. A. performed experiments, measurements, and theoretical calculations. All the authors analysed the data.
Data availability
Experimental procedures, computational data, and characterization of related compounds are provided in the ESI.†
Conflicts of interest
There are no conflicts to declare.
Acknowledgements
This research was supported by a Grant-in-Aid for Scientific Research on Innovative Areas “Hybrid Catalysis for Enabling Molecular Synthesis on Demand” (JP17H06447) from MEXT (Japan) and a Grant-in-Aid for Scientific Research (S) (JP22H04969) from the JSPS.
References
- For review on CP-PAHs, see: Y. T. Wu and J. S. Siegel, Aromatic Molecular-Bowl Hydrocarbons: Synthetic Derivatives, Their Structures, and Physical Properties, Chem. Rev., 2006, 106, 4843–4867 CrossRef CAS PubMed.
- For selected reviews on fullerenes, see:
(a) N. Martín, New challenges in fullerene chemistry, Chem. Commun., 2006, 2093–2104 RSC;
(b) C.-Z. Li, H.-L. Yip and A. K.-Y. Jen, Functional fullerenes for organic photovoltaics, J. Mater. Chem., 2012, 22, 4161–4177 Search PubMed.
- For selected examples on CP-PAHs, see:
(a) F. G. Brunetti, X. Gong, M. Tong, A. J. Heeger and F. Wudl, Strain and Hückel Aromaticity: Driving Forces for a Promising New Generation of Electron Acceptors in Organic Electronics, Angew. Chem., Int. Ed., 2010, 49, 532–536 Search PubMed;
(b) D. T. Chase, A. G. Fix, B. D. Rose, C. D. Weber, S. Nobusue, C. E. Stockwell, L. N. Zakharov, M. C. Lonergan and M. M. Haley, Electron-Accepting 6,12-Diethynylindeno[1,2-b]fluorenes: Synthesis, Crystal Structures, and Photophysical Properties, Angew. Chem., Int. Ed., 2011, 50, 11103–11106 Search PubMed;
(c) C. H. Lee and K. N. Plunkett, Orthogonal Functionalization of Cyclopenta[hi]aceanthrylenes, Org. Lett., 2013, 15, 1202–1205 CrossRef CAS PubMed;
(d) D. T. Chase, A. G. Fix, S. J. Kang, B. D. Rose, C. D. Weber, Y. Zhong, L. N. Zakharov, M. C. Lonergan, C. Nuckolls and M. M. Haley, 6,12-Diarylindeno[1,2-b]fluorenes: Syntheses, Photophysics, and Ambipolar OFETs, J. Am. Chem. Soc., 2012, 134, 10349–10352 CrossRef CAS PubMed;
(e) J. D. Wood, J. L. Jellison, A. D. Finke, L. Wang and K. N. Plunkett, Electron Acceptors Based on Functionalizable Cyclopenta[hi]aceanthrylenes and Dicyclopenta[de,mn]tetracenes, J. Am. Chem. Soc., 2012, 134, 15783–15789 CrossRef CAS PubMed;
(f) H. Xia, D. Liu, X. Xu and Q. Miao, Ambipolar organic semiconductors from electron-accepting cyclopenta-fused anthracene, Chem. Commun., 2013, 49, 4301–4303 RSC;
(g) R. Q. Lu, Y. Q. Zheng, Y. N. Zhou, X. Y. Yan, T. Lei, K. Shi, Y. Zhou, J. Pei, L. Zoppi, K. K. Baldridge, J. S. Siegel and X. Y. Cao, Corannulene derivatives as non-fullerene acceptors in solution-processed bulk heterojunction solar cells, J. Mater. Chem. A, 2014, 2, 20515–20519 RSC;
(h) A. N. Lakshminarayana, J. Chang, J. Luo, B. Zheng, K.-W. Huang and C. Chi, Bisindeno-annulated pentacenes with exceptionally high photo-stability and ordered molecular packing: simple synthesis by a regioselective Scholl reaction, Chem. Commun., 2015, 51, 3604–3607 RSC;
(i) L. J. Purvis, X. Gu, S. Ghosh, Z. Zhang and C. J. Cramer, Douglas, Synthesis and Characterization of Electron-Deficient Asymmetrically Substituted Diarylindenotetracenes, J. Org. Chem., 2018, 83, 1828–1841 Search PubMed;
(j) Q. Xu, C. Wang, Y. Zhao, D. Zheng, C. Shao, W. Guo, X. Deng, Y. Wang, X. Chen, J. Zhu and H. Jiang, Tuning the Properties of Corannulene-Based Polycyclic Aromatic Hydrocarbons by Varying the Fusing Positions of Corannulene, Org. Lett., 2020, 22, 7397–7402 Search PubMed;
(k) Y. Tanaka, N. Fukui and H. Shinokubo, as-Indaceno[3,2,1,8,7,6-ghijklm]terrylene as a near-infrared absorbing C70-fragment, Nat. Commun., 2020, 11, 3873 CrossRef CAS PubMed.
- For reviews on PHAs, see:
(a) M. Stępień, E. Gońka, M. Żyła and N. Sprutta, Heterocyclic Nanographenes and Other Polycyclic Heteroaromatic Compounds: Synthetic Routes, Properties, and Applications, Chem. Rev., 2017, 117, 3479–3716 CrossRef PubMed;
(b) A. Borissov, Y. K. Maurya, L. Moshniaha, W.-S. Wong, M.-K. Żyła-Karwowska and M. Stępień, Recent Advances in Heterocyclic Nanographenes and Other Polycyclic Heteroaromatic Compounds, Chem. Rev., 2022, 122, 565–788 Search PubMed.
- For selected examples on CP-PHAs, see:
(a) Y. Matano, A. Saito, T. Fukushima, Y. Tokudome, F. Suzuki, D. Sakamaki, H. Kaji, A. Ito, K. Tanaka and H. Imahori, Fusion of Phosphole and 1,1′-Biacenaphthene: Phosphorus(V)-Containing Extended π-Systems with High Electron Affinity and Electron Mobility, Angew. Chem., Int. Ed., 2011, 50, 8016–8020 CrossRef CAS PubMed;
(b) A. R. Mohebbi, J. Yuen, J. Fan, C. Munoz, M. F. Wang, R. S. Shirazi, J. Seifter and F. Wudl, Emeraldicene as an Acceptor Moiety: Balanced-Mobility, Ambipolar, Organic Thin-Film Transistors, Adv. Mater., 2011, 23, 4644–4648 Search PubMed;
(c) A. R. Mohebbi and F. Wudl, Electron-Accepting Dithiarubicene (Emeraldicene) and Derivatives Prepared by Unprecedented Nucleophilic Hydrogen Substitution by Alkyllithium Reagents, Chem. – Eur. J., 2011, 17, 2642–2646 Search PubMed;
(d) Y. Nagasaka, C. Kitamura, H. Kurata and T. Kawase, Diacenaphtho[1,2-b;1′,2′-d]silole and -pyrrole, Chem. Lett., 2011, 40, 1437–1439 CrossRef CAS;
(e) M. Mahmut, T. Awut, I. Nurulla and M. Mijit, Synthesis of two novel acenaphthyl-quinoxaline based low-band gap polymers and its electrochromic properties, J. Polym. Res., 2014, 21, 1–9 CAS;
(f) K. Tsukamoto, K. Takagi, S. Nagano, M. Hara, Y. Le, K. Osakada and D. Takeuchi, π-Extension of electron-accepting dithiarubicene with a cyano-substituted electron-withdrawing group and application in air-stable n-channel organic field effect transistors, J. Mater. Chem. C, 2019, 7, 12610–12618 RSC;
(g) W. A. Hussain and K. N. Plunkett, Benzodithiophene-Fused Cyclopentannulated Aromatics via a Palladium-Catalyzed Cyclopentannulation and Scholl Cyclodehydrogenation Strategy, J. Org. Chem., 2021, 86, 12569–12576 CrossRef CAS PubMed.
- For review on peri-C-H annulation, see: T. Jin, Z. Zhao, N. Asao and Y. Yamamoto, Metal-Catalyzed Annulation Reactions for π-Conjugated Polycycles, Chem. – Eur. J., 2014, 20, 3554–3576 Search PubMed.
-
(a) H. A. Wegner, L. T. Scott and A. de Meijere, A New Suzuki-Heck-Type Coupling Cascade: Indeno[1,2,3]-Annelation of Polycyclic Aromatic Hydrocarbons, J. Org. Chem., 2003, 68, 883–887 CrossRef CAS PubMed;
(b) H. A. Wegner, H. Reisch, K. Rauch, A. Demeter, K. A. Zachariasse, A. de Meijere and L. T. Scott, Oligoindenopyrenes: A New Class of Polycyclic Aromatics, J. Org. Chem., 2006, 71, 9080–9087 CrossRef CAS PubMed.
- H. Wu, R. Fang, J. Tao, D. Wang, X. Qiao, X. Yang, F. Hartl and H. Li, Diacenaphthylene-fused benzo[1,2-b:4,5-b’]dithiophenes: polycyclic heteroacenes containing full-carbon five-membered aromatic rings, Chem. Commun., 2017, 53, 751–754 Search PubMed.
- T. Jin, S. Suzuki, H. E. Ho, H. Matsuyama, M. Kawata and M. Terada, Pd-Catalyzed Indolization/peri–C–H Annulation/N–Dealkylation Cascade to Cyclopenta-Fused Acenaphtho[1,2-b]indole Scaffold, Org. Lett., 2021, 23, 9431–9435 Search PubMed.
-
(a) B. Yao, Q. Wang and J. Zhu, Palladium-Catalyzed Coupling of ortho-Alkynylanilines with Terminal Alkynes Under Aerobic Conditions: Efficient Synthesis of 2,3-Disubstituted 3-Alkynylindoles, Angew. Chem., Int. Ed., 2012, 51, 12311–12315 Search PubMed;
(b) B. Yao, Q. Wang and J. Zhu, Palladium(II)-Catalyzed Intramolecular Diamination of Alkynes under Aerobic Oxidative Conditions: Catalytic Turnover of an Iodide Ion, Angew. Chem., Int. Ed., 2012, 51, 5170–5174 Search PubMed;
(c) X.-F. Xia, N. Wang, L.-L. Zhang, X.-R. Song, X.-Y. Liu and Y.-M. Liang, Palladium(II)-Catalyzed Tandem Cyclization/C–H Functionalization of Alkynes for the Synthesis of Functionalized Indoles, J. Org. Chem., 2012, 77, 9163–9170 CrossRef CAS PubMed;
(d) B. Yao, Q. Wang and J. Zhu, Palladium(II)-Catalyzed Cyclizative Cross-Coupling of ortho-Alkynylanilines with ortho-Alkynylbenzamides under Aerobic Conditions, Angew. Chem., Int. Ed., 2013, 52, 12992–12996 CrossRef CAS PubMed;
(e) S. Zhang, H. Ma, H. E. Ho, Y. Yamamoto, M. Bao and T. Jin, Pd-Catalyzed cascade cyclization of o-alkynylanilines via C–H/C–N bond cleavage leading to dibenzo[a,c]carbazoles, Org. Biomol. Chem., 2018, 16, 5236–5240 Search PubMed.
- For selected examples on transition-metal-catalyzed peri-C–H annulation, see:
(a) H. Dang and M. A. Garcia-Garibay, Palladium-Catalyzed Formation of Aceanthrylenes: A Simple Method for Peri-Cyclopentenelation of Aromatic Compounds, J. Am. Chem. Soc., 2001, 123, 355–356 Search PubMed;
(b) S. Mochida, M. Shimizu, K. Hirano, T. Satoh and M. Miura, Synthesis of Naphtho[1,8-bc]pyran Derivatives and Related Compounds through Hydroxy Group Directed C–H Bond Cleavage under Rhodium Catalysis, Chem. – Asian J., 2010, 5, 847–851 Search PubMed;
(c) V. S. Thirunavukkarasu, M. Donati and L. Ackermann, Hydroxyl-Directed Ruthenium-Catalyzed C–H Bond Functionalization: Versatile Access to Fluorescent Pyrans, Org. Lett., 2012, 14, 3416–3419 Search PubMed;
(d) X. Zhang, W. Si, M. Bao, N. Asao, Y. Yamamoto and T. Jin, Rh(III)-Catalyzed Regioselective Functionalization of C–H Bonds of Naphthylcarbamates for Oxidative Annulation with Alkyne, Org. Lett., 2014, 16, 4830–4833 CrossRef CAS PubMed;
(e) J. Yin, M. Tan, D. Wu, R. Jiang, C. Li and J. You, Synthesis of Phenalenyl-Fused Pyrylium Cations: Divergent C–H Activation/Annulation Reaction Sequence of Naphthalene Aldehydes with Alkynes, Angew. Chem., Int. Ed., 2017, 56, 13094–13098 CrossRef CAS PubMed;
(f) M. Shankar, R. K. Rit, S. Sau, K. Mukherjee, V. Gandon and A. K. Sahoo, Double annulation of ortho- and peri-C–H bonds of fused (hetero)arenes to unusual oxepino-pyridines, Chem. Sci., 2020, 11, 10770–10777 RSC.
- For selected reviews on aromatic C–H/C–H coupling, see:
(a) Y. Yang, J. Lan and J. You, Oxidative C–H/C–H Coupling Reactions between Two (Hetero)arenes, Chem. Rev., 2017, 117, 8787–8863 CrossRef CAS PubMed;
(b) I. A. Stepek and K. Itami, Recent Advances in C–H Activation for the Synthesis of π-Extended Materials, ACS Mater. Lett., 2020, 2, 951–974 CrossRef CAS.
- For selected examples on (hetero)aromatic C–H/C–H coupling without using directing groups, see:
(a) D. R. Stuart and K. Fagnou, The Catalytic Cross-Coupling of Unactivated Arenes, Science, 2007, 316, 1172–1175 CrossRef CAS PubMed;
(b) D. R. Stuart, E. Villemure and K. Fagnou, Elements of Regiocontrol in Palladium-Catalyzed Oxidative Arene Cross-Coupling, J. Am. Chem. Soc., 2007, 129, 12072–12073 CrossRef CAS PubMed;
(c) S. J. Tereniak, D. L. Bruns and S. S. Stahl, Pd-Catalyzed Aerobic Oxidative Coupling of Thiophenes: Synergistic Benefits of Phenanthroline Dione and a Cu Cocatalyst, J. Am. Chem. Soc., 2020, 142, 20318–20323 CrossRef CAS PubMed;
(d) T. Doba, L. Ilies, W. Sato, R. Shang and E. Nakamura, Iron catalysed regioselective thienyl C–H/C–H coupling, Nat. Catal., 2021, 4, 631–638 CrossRef CAS.
-
(a) K. Masui, H. Ikegami and A. Mori, Palladium-Catalyzed C–H Homocoupling of Thiophenes: Facile Construction of Bithiophene Structure, J. Am. Chem. Soc., 2004, 126, 5074–5075 CrossRef CAS PubMed;
(b) M. D. Lotz, N. M. Camasso, A. J. Canty and M. S. Sanford, Role of Silver Salts in Palladium-Catalyzed Arene and Heteroarene C–H Functionalization Reactions, Organometallics, 2017, 36, 165–171 CrossRef CAS.
-
(a) B. Liégault, D. Lee, M. P. Huestis, D. R. Stuart and K. Fagnou, Intramolecular Pd(II)-Catalyzed Oxidative Biaryl Synthesis Under Air: Reaction Development and Scope, J. Org. Chem., 2008, 73, 5022–5028 CrossRef PubMed;
(b) K. Saito, P. K. Chikkade, M. Kanai and Y. Kuninobu, Palladium-Catalyzed Construction of Heteroatom-Containing π-Conjugated Systems by Intramolecular Oxidative C–H/C–H Coupling Reaction, Chem. – Eur. J., 2015, 21, 8365–8368 CrossRef CAS PubMed;
(c) D.-D. Guo, B. Li, D.-Y. Wang, Y.-R. Gao, S.-H. Guo, G.-F. Pan and Y.-Q. Wang, Synthesis of 6H-Benzo[c]chromenes via Palladium-Catalyzed Intramolecular Dehydrogenative Coupling of Two Aryl C–H Bonds, Org. Lett., 2017, 19, 798–801 CrossRef CAS PubMed.
-
(a) K. Shen, Y. Fu, J.-N. Li, L. Liu and Q.-X. Gu, What are the pKa values of C–H bonds in aromatic heterocyclic compounds in DMSO?, Tetrahedron, 2007, 63, 1568–1576 CrossRef CAS;
(b) R. R. Fraster, T. S. Mansour and S. Savar, Acidity measurements in THF. V. Heteroaromatic compounds containing 5-membered rings, Can. J. Chem., 1985, 63, 3505–3509 CrossRef.
- It was observed that the benzofuran substrates exhibited very low reactivity in the present C–H/C–H coupling. For example, the coupling of 3-(naphthalen-1-yl)benzofuran afforded the corresponding annulation product, acenaphtho[1,2-b]benzofuran, in only 8–10% yield, with the starting substrate recovered in 84% yield under standard Pd-catalysed conditions using AgOPiv or AgSbF6. Additionally, no reaction was observed when 2-(naphthalen-1-yl)benzofuran was subjected to the standard conditions.
- C. Colletto, A. Panigrahi, J. Fernández-Casado and I. Larrosa, Ag(I)–C–H Activation Enables Near-Room-Temperature Direct α–Arylation of Benzo[b]thiophenes, J. Am. Chem. Soc., 2018, 140, 9638–9643 CrossRef CAS PubMed.
- L. Ackermann, Carboxylate-Assisted Transition-Metal-Catalyzed C-H Bond Functionalizations: Mechanism and Scope, Chem. Rev., 2011, 111, 1315–1345 CrossRef CAS PubMed.
|
| This journal is © the Partner Organisations 2025 |
Click here to see how this site uses Cookies. View our privacy policy here. 
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a  *b,
Masaki
Kawata
a,
Sho
Aida
a and
Masahiro
Terada
*b,
Masaki
Kawata
a,
Sho
Aida
a and
Masahiro
Terada