
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Mechanism of formation of chiral allyl SCF3 compounds via selenium-catalyzed sulfenofunctionalization of allylboronic acids†
Wen-Jie
Wei
 ,
Kalman J.
Szabo
* and
Fahmi
Himo
*
,
Kalman J.
Szabo
* and
Fahmi
Himo
*
Department of Chemistry, Arrhenius Laboratory, Stockholm University, SE-106 91 Stockholm, Sweden. E-mail: kalman.j.szabo@su.se; fahmi.himo@su.se
First published on 27th February 2025
Abstract
The detailed reaction mechanism of diphenyl selenide-catalyzed sulfenofunctionalization of chiral α-CF3 allylboronic acids is investigated by means of density functional theory calculations. It is demonstrated that the reaction starts with the transfer of the SCF3 group from the (PhSO2)2NSCF3 reagent to the Ph2Se catalyst, a process that is shown to be assisted by the presence of Tf2NH acid. After a proton transfer step, the SCF3 group is transferred to the C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C double bond of the substrate to generate a thiiranium ion. Concerted deborylative opening of the thiiranium ion yields then the final product. Several representative substrates are considered by the calculations, and the origins of the stereoselectivity of the reactions are analyzed by comparing the energies and geometries of the transition states leading to the different products.
C double bond of the substrate to generate a thiiranium ion. Concerted deborylative opening of the thiiranium ion yields then the final product. Several representative substrates are considered by the calculations, and the origins of the stereoselectivity of the reactions are analyzed by comparing the energies and geometries of the transition states leading to the different products.
1. Introduction
Fluorine-containing pharmacophores are often applied in modern drug substances.1 One of five commercial drugs contains at least one C–F bond.2 The main reason for the widespread application of fluorine-containing groups in bioactive small molecules is their beneficial metabolic and pharmacokinetical properties.3 A particularly important pharmacophore is the trifluoromethylthio (SCF3) group, which substantially increases the lipophilicity of the drug substances and also has a pronounced electron-withdrawing character.4–6 As a consequence, several excellent methods appeared for the introduction of the trifluoromethylthio group to organic small molecules.7–21 In particular, the synthesis of chirally enriched SCF3 compounds became an important but challenging area in modern organic chemistry.22–35Very recently, Szabó and co-workers presented an efficient method to introduce the SCF3 group by using (PhSO2)2NSCF3 reagent362 with α-CF3 allylboronic acid derivatives 1 to form a chiral alkenyl SCF3 compound 5 (Scheme 1).37 The reaction relies on the application of selenium-based Lewis base Ph2Se 3 as a catalyst in the presence of Tf2NH 4 as the activator. These reactions have a high degree of functional group tolerance and proceed with excellent stereo-, diastereo- and site-selectivity.
 | ||
| Scheme 1 Se-catalyzed sulfenofunctionalization of allylboronic acid to form chiral allyl SCF3 compound investigated in the present study. | ||
The application of electrophilic sulfenofunctionalization in the presence of selenium catalysis has been documented by the pioneering studies of the groups of Denmark,38 Zhao,26 and others.39 However, the application of allylboronate substrates for trifluoromethylthiolation revealed a couple of new, interesting mechanistic aspects.37 An important feature is the excellent stereochemistry of the reaction, including chirality transfer and high E-selectivity for the formation of the new double bond. The studies also pointed out the benefits of using allylboronic acid type of substrates. Replacement of the B(OH)2 group of 1 with Bpin leads to a significant decrease in the yield of the product, indicating that allyl-Bpin species have a substantially lower reactivity than allylboronic acids.
In the present work, we have performed density functional theory (DFT) calculations to elucidate the mechanism for the formation of allyl SCF3 compounds via the selenium-catalyzed sulfenofunctionalization of allylboronic acids. We consider the reactions of several representative substrates (1a–1d in Scheme 1) and discuss the origins of selectivity for each of them.
2. Computational details
The B3LYP-D3(BJ) functional,40,41 which includes the D3 dispersion correction with the Becke–Johnson damping function,42,43 was used for all calculations presented in this work, and the Gaussian 16 program44 was employed. Geometry optimizations were carried out with the 6-31G(d,p) basis set. To obtain more accurate energies, single-point calculations were performed on the optimized structures using the larger basis set 6-311+G(2d,2p). Analytical frequency calculations were performed at the level of theory of the geometry optimization to calculate the Gibbs free energy corrections. Solvation effects were considered by performing single-point calculations on the optimized structures using the SMD method45 with the parameters of dichloromethane.To evaluate the effect of performing the geometry optimizations with the smaller basis set and in the gas phase, the geometries of the first step of the reaction (2 + 3 + 4 → Int1, see below) were re-optimized twice, in implicit DCM solvent and with the larger basis set. The calculations showed that the geometries and energies were not affected significantly (see ESI†).
Intrinsic reaction coordinate (IRC) calculations were performed on all transition states to confirm the nature of the connecting intermediates. A thorough manual conformation search was carried out on all stationary points to ensure that the structures with the lowest energy were located.
3. Results and discussion
We start the mechanistic investigation by considering the reaction of model substrate 1a, in which the R group is a simple methyl substituent. Although this compound was not included in the experimental studies,37 it can serve as a good reference for γ-alkyl allylboronates. As representatives for different classes of substrates, we subsequently investigate the mechanisms of three other substrates that have been examined explicitly by the experiments,37 namely those in which the R is a relatively bulky benzyl (1b) and tert-butyl (1c) groups, as well as a nitrogen-containing phthalimide derivative 1d (Scheme 1).3.1. Reaction of model substrate 1a
The first step of the reaction mechanism is the transfer of the SCF3 group from the (PhSO2)2NSCF3 transfer reagent 2 to the Ph2Se catalyst 3, generating the SePh2SCF3 cation 7 (Scheme 2a). We found that this step is assisted by the participation of Tf2NH acid 4 that forms a hydrogen bond to the nitrogen of the transfer reagent (TS1, Fig. 1). The calculated barrier is 24.2 kcal mol−1, and the energy of the resulting intermediate Int1, in which acid 4, the (PhSO2)2N− anion 6 and the SePh2SCF3 cation 7 are in complex with each other, lies at 21.3 kcal mol−1 (Fig. 2). The hydrogen bond provided by acid 4 stabilizes the negative charge developing on the nitrogen atom, as seen from the H⋯N distance, which is 1.98 Å at TS1 and 1.86 Å at Int1. Transfer of the SCF3 group without the participation of the acid was also considered and was found to have a much higher energy barrier of 50.4 kcal mol−1 (see ESI†). | ||
| Scheme 2 Reaction steps investigated in the present study. | ||
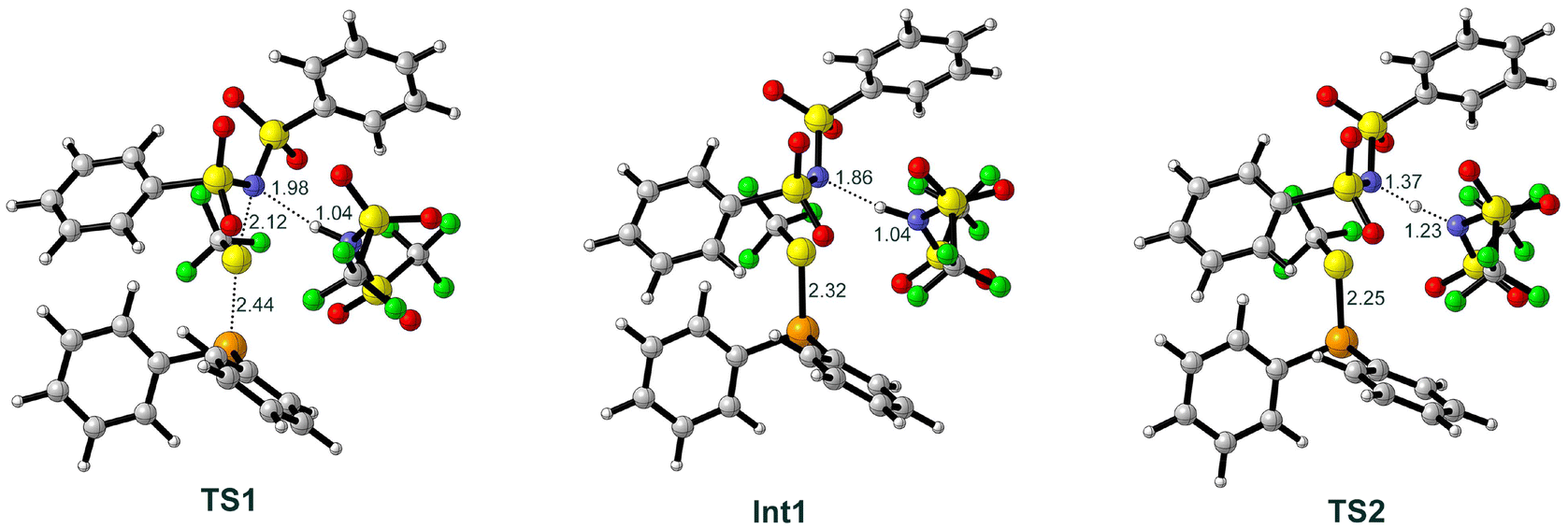 | ||
| Fig. 1 Optimized structures of the transition states and intermediates of TS1, Int1, and TS2. Selected bond distances are indicated in Å. | ||
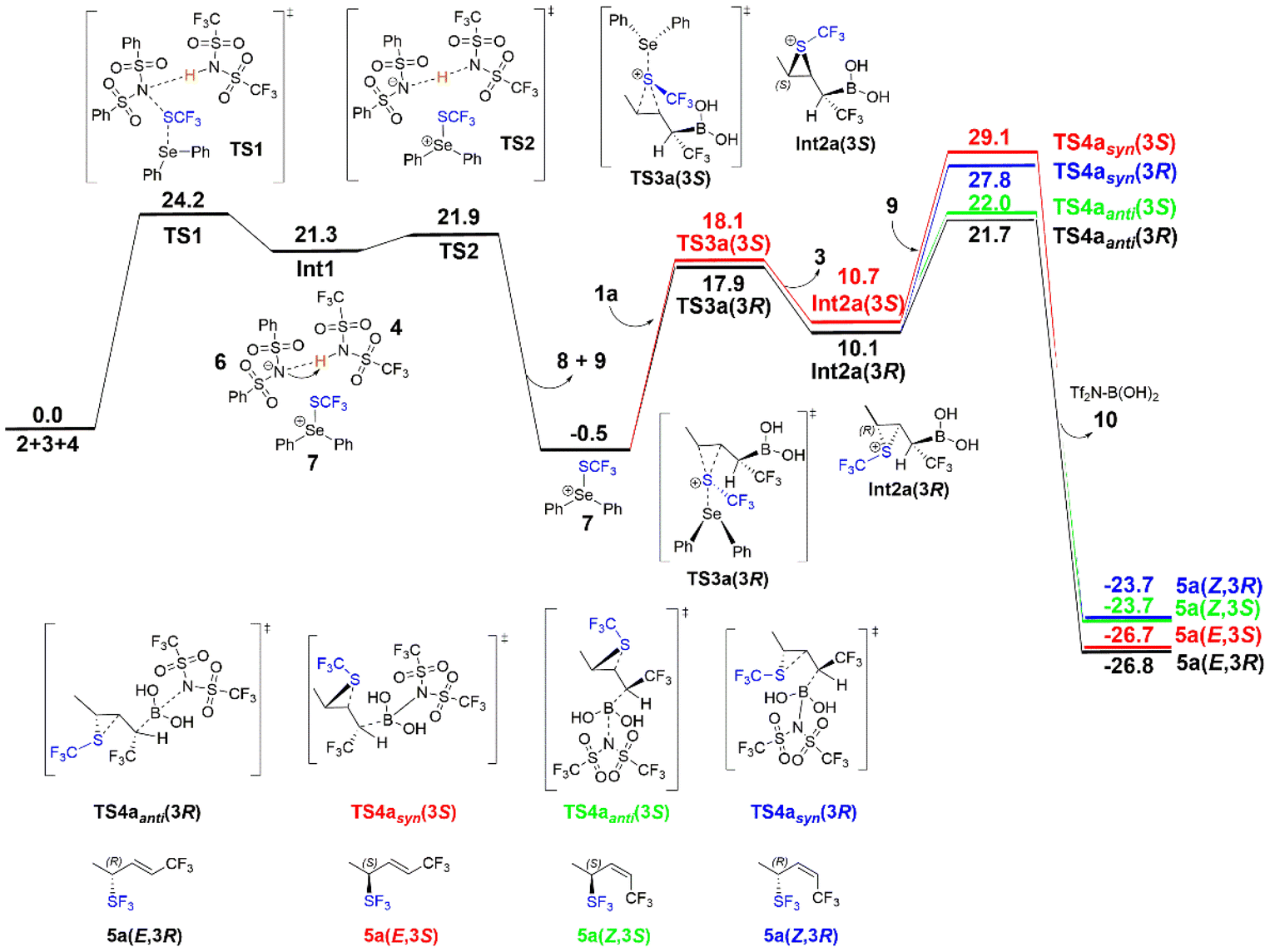 | ||
| Fig. 2 Calculated free energy profile (kcal mol−1) of model substrate 1a. | ||
Next step is a proton transfer from 4 to (PhSO2)2N− anion 6viaTS2, generating (PhSO2)2NH 8 and Tf2N− anion 9 (Scheme 2b). The energy barrier of this step is only 0.6 kcal mol−1 relative to Int1.
We also calculated the alternative pathway with the reversed step order, i.e. in which a proton is first transferred from acid 4 to (PhSO2)2NSCF3, followed by the transfer of the SCF3 group from the generated (PhSO2)2NHSCF3 cation to the Ph2Se catalyst 3. However, the energy barrier of this scenario was found to be very high, 52.9 kcal mol−1 (see ESI†).
In the following step of the mechanism, the SCF3 group of the SePh2SCF3 cation 7 is transferred to the CC double bond of the substrate to generate a thiiranium ion (Scheme 2c). Two competing stereoselective pathways are possible and were investigated, in which the SCF3 group is transferred either to the Re-face of the substrate viaTS3a(3R) to form the (R)-configuration Int2a(3R), or to the Si-face viaTS3a(3S) to generate the (S)-configuration Int2a(3S). The optimized structures of the transition states and intermediates are shown in Fig. 3. The energies of TS3a(3R) and TS3a(3S) are calculated to be 18.4 and 18.6 kcal mol−1, respectively, relative to the previous intermediate, and the resulting Int2a(3R) and Int2a(3S) are 10.6 and 11.2 kcal mol−1 higher, respectively.
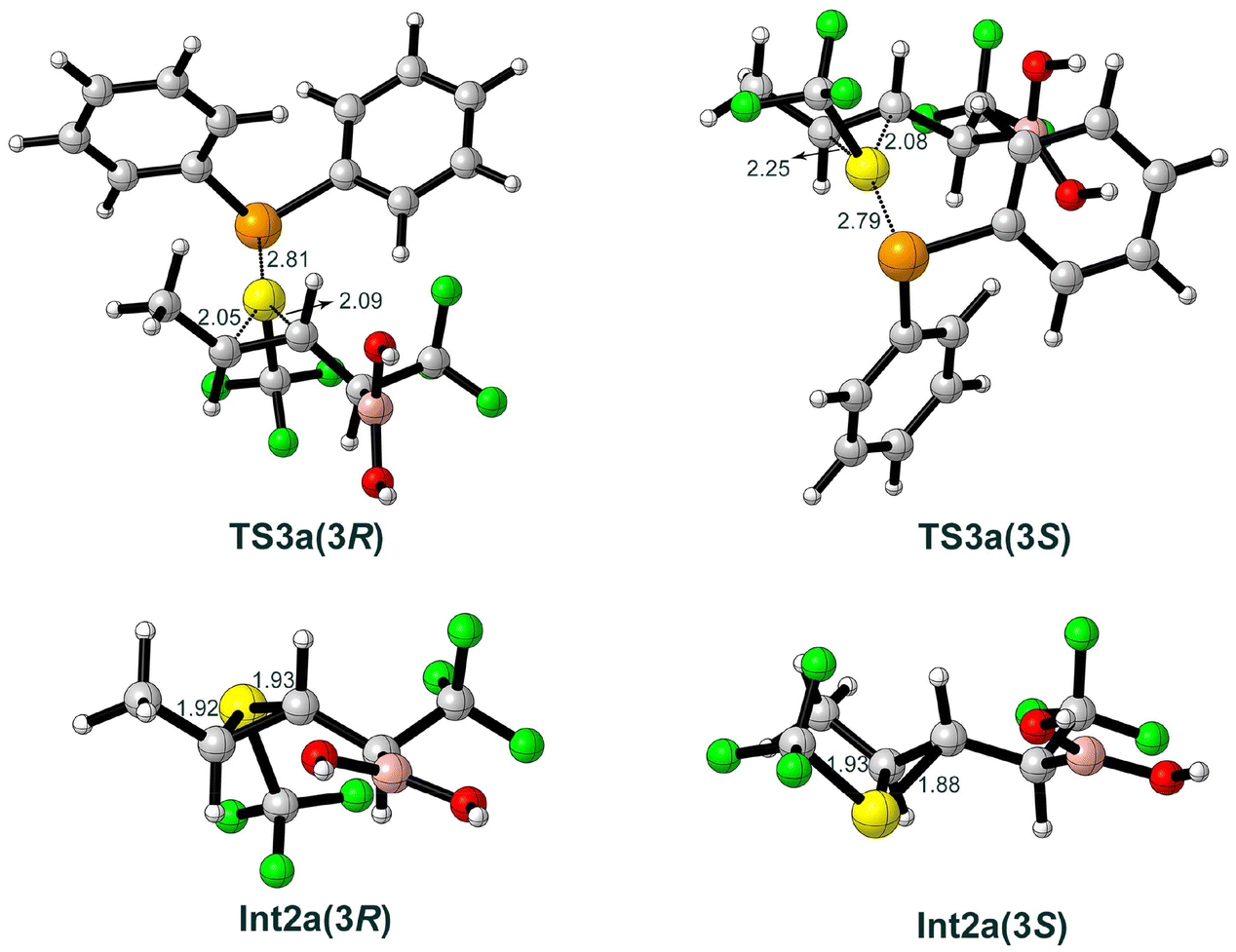 | ||
| Fig. 3 Optimized structures of the transition states and intermediates for the generation of the thiiranium ion Int2a. Selected bond distances are indicated in Å. | ||
For comparison, we also investigated the uncatalyzed reaction, in which the SCF3 group is transferred directly from reagent 2 to substrate 1a, generating Int2a without the participation of catalyst 3 (see ESI†). The energy barrier was calculated to be 29.9 kcal mol−1 for both the 3R- and 3S-pathways, which is significantly higher than the case with catalyst 3. Accordingly, using PhSePh catalyst (3) increases the reactivity of 2. In the positively charged 7 the electrophilicity of SCF3 is substantially increased compared to 2. In addition, the cleavage of the weak Se–S bond in 7 is also easier than the cleavage of the N–S bond of 2. The stability of 7 is poor under ambient conditions, and therefore 2 is converted to 7in situ (in the presence of 4) under the applied reaction conditions.
The final step of the mechanism (Scheme 2d) involves the Tf2N− anion 9 performing a nucleophilic attack on the boronate group of Int2a(3R) or Int2a(3S), triggering the concerted deborylative opening of the thiiranium ion viaTS4 to yield the four possible forms of the final product. The (3R)-configured products are formed from Int2a(3R) and can result in either the E-configuration through the anti-elimination pathway viaTS4anti(3R), or alternatively, rotation of the Cα–Cβ bond leads to the syn-elimination pathway viaTS4syn(3R), resulting in the Z-configuration. Similarly, the E-(3S)- or Z-(3S)-configured products can be achieved viaTS4syn(3S) or TS4anti(3S), respectively. The optimized structures of these transition states are shown in Fig. 4, while the optimized structures of the products are given in the ESI.†
 | ||
| Fig. 4 Optimized structures of the transition states for the step of deborylative elimination in model substrate 1a. Selected bond distances are indicated in Å. | ||
Although the chiral center is formed at TS3, the calculations show that the energy barriers for the eliminations of the boronate group viaTS4 are higher and irreversible, which means that the latter step is the stereoselectivity-determining step of the reaction.
By comparing the energy profiles of the above four pathways (Fig. 2), the calculations of the model substrate 1a show that the energy barriers of anti-eliminations are considerably lower than the syn-eliminations, 21.7 kcal mol−1 in TS4anti(3R)vs. 27.8 kcal mol−1 in TS4syn(3R), and 22.0 kcal mol−1 in TS4anti(3S)vs. 29.1 kcal mol−1 in TS4syn(3S). Inspection of the optimized structures in Fig. 4 shows that the reason for this energy difference is mainly the steric repulsion between the SCF3 group and the leaving boronate group, as these two moieties point toward each other in the syn-elimination, TS4syn(3R) and TS4syn(3S), while in the anti-elimination, TS4anti(3R) and TS4anti(3S), they point away from each other.
The difference in energy between TS4anti(3R) and TS4anti(3S), which lead to the E-(3R)-5a and Z-(3S)-5a products, respectively, is calculated to be only 0.3 kcal mol−1 in favor of the former. The calculations show thus that already the model substrate 1a, with the small methyl substituent, qualitatively reproduces the experimental selectivity trend, in that the formation of the E-(3R)-configured product is associated with the lowest-energy pathway, albeit with a small energy. However, as will be shown below, the calculations using the experimentally employed substrates, with bulkier substituents, yield a more quantitative agreement with the experiments.
To summarize, the mechanism proposed on the basis of the current calculations is given in Scheme 3. The obtained overall energy profile (Fig. 2) indicates that the first step, i.e. the acid-assisted transfer of the SCF3 group viaTS1, is the rate-determining step (RDS) for the model substrate, with a barrier of 24.2 kcal mol−1. However, the final step, i.e. deborylative opening of the thiiranium ring viaTS4, has an overall barrier of 22.2 kcal mol−1, which is quite close in energy, and it is therefore not possible to determine confidently the nature of the RDS based only on the calculations. In particular, various substituents on the substrate may lead to significant changes in the energy of the final step (see below).
 | ||
| Scheme 3 Reaction mechanism suggested on the basis of the current calculations. | ||
Here, it is interesting to mention two previous mechanistic studies on sulfenofunctionalizations of alkenes catalyzed by selenides, where DFT calculations were employed to investigate various aspects of the reactions. However, none of these studies involved a deborylation step, which is a novel aspect of the present study. Denmark and co-workers analyzed the geometries and energies of the transition states for the thiiranium ion formation step, which was assumed to be the enantio-determining step of the reaction,46 while Zhao and co-workers investigated the mechanism of selenide-catalyzed trifluoromethylthiolation of gem-diaryl tethered alkenes to synthesize trifluoromethylthiolated tetrahydronaphthalenes.27
In addition to the results discussed above, we have also considered some other mechanistic alternatives that turned out to have higher energy barriers. As seen from Fig. 2 and 4, catalyst 3 does not participate in the deborylative elimination step in TS4. We have considered whether it can assist this step, but the energy barriers for this scenario were found to be higher (see ESI†). We also considered whether the (PhSO2)2NH species 8 could act as the nucleophile to attack the boronate group of Int2a, but the calculated energy barriers for this pathway were calculated to be very high as compared to when the anionic Tf2N−9 is the nucleophile (see ESI†). Finally, the experiments reveal that replacing the B(OH)2 group of the substrate with Bpin significantly decreases the yield of the product.37 Consistently with this result, the calculations show that the barrier for the case of Bpin is 3.7 kcal mol−1 higher than the case of the B(OH)2 group (see ESI†).
3.2. Reactions with other substrates
Next, we calculated the mechanisms when the R group of the substrate is Bn (1b), tBu (1c), and the phthalimide substituent (1d), all of which have been employed in the experimental study.37 As seen from Fig. 2, the reaction mechanism up to the formation of SePh2SCF3 cation 7 is independent of the substrate, and therefore we investigated the reactions of the other substrates starting from this point.For substrate 1b, with the benzyl substituent, the mechanism was calculated to be very similar to that of the model substrate 1a shown in Scheme 3 (see calculated energy profile in Fig. 5). One small difference is that the formation of the Z-(3R)-configured product through the syn-elimination was found to occur in a stepwise manner (see ESI† for detailed results). The calculations show that the overall barrier for substrate 1b is ca. 2 kcal mol−1 lower than for 1a, and very importantly, the extent of the stereo-differentiation is well-reproduced.
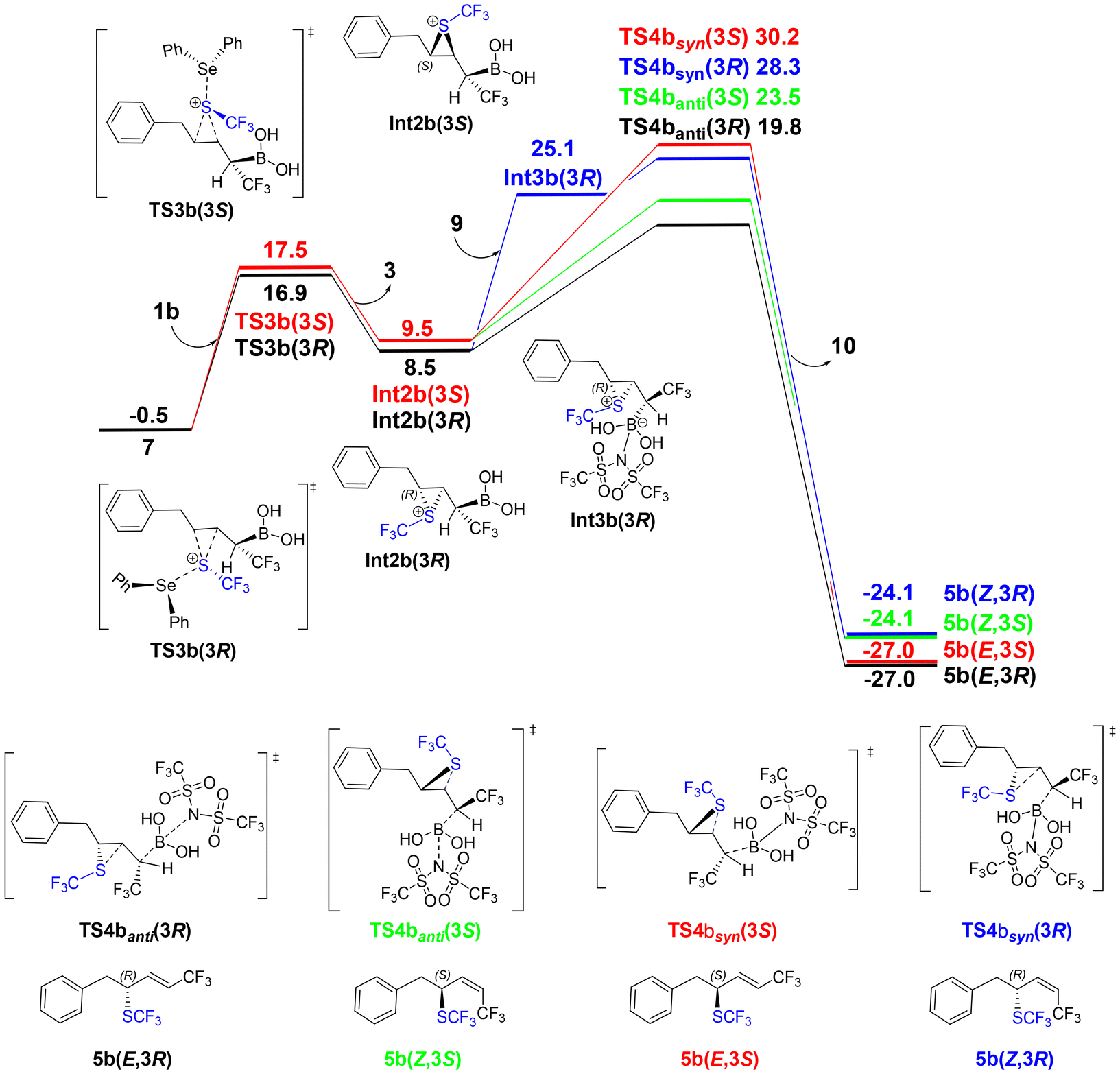 | ||
| Fig. 5 Calculated free energy profile (kcal mol−1) of substrate 1b. | ||
Similarly to substrate 1a, the barriers of anti-eliminations for 1b are considerably lower than the syn-eliminations due to steric repulsion between the SCF3 group and the leaving boronate group. In addition, the pathway leading to the E-(3R)-5b product is now 3.7 kcal mol−1 lower than that leading to the Z-(3S)-5b product (Fig. 5), due to a steric repulsion between the benzyl substituent of the substrate and the SCF3 group (see ESI†). The calculated energy difference is in good agreement with the experimentally observed ee of 93% in favor of the E-(3R) product.
For substrate 1c with the tBu substituent (see ESI†), the situation is very similar to substrate 1b, with both syn-eliminations found to take place in a stepwise manner. The overall barrier was calculated to be ca. 3 kcal mol−1 higher than for 1b, and the selectivity is determined by the same factors, with an energy difference of 4.2 kcal mol−1, in good agreement with the experimental outcome of 98% ee.
Substrate 1d, with the phthalimide substituent, represents an interesting case, because the carbonyl group of 1d may perform a nucleophilic attack at the Cβ atom of the thiiranium ion through an intramolecular mechanism,25,47,48 leading to the opening of the thiiranium ion and yielding a six-membered ring intermediate Int3d′ (see Fig. 6). The calculations show that the energy barrier for this competing intramolecular nucleophilic attack viaTS4d′ is much lower than for the intermolecular reaction of the thiiranium ion with the Tf2N− anion 9viaTS4d, which was found for the other substrates. However, from Int3d′, the barriers for the following steps, which would be the nucleophilic attack of Tf2N− and the deborylative opening of the six-membered ring, were found to be higher in energy compared to the intermolecular reaction (see ESI†), indicating that the intramolecular nucleophilic attack is a reversible process. Thus, formation of Int3d′ can be regarded as an unproductive dead-end for the deborylative trifluoromethylthiolation process.
 | ||
| Fig. 6 Calculated free energy profile (kcal mol−1) of substrate 1d. | ||
The calculations show thus that the reaction of substrate 1d also follows the mechanism of the model substrate 1a. However, as seen from Fig. 6, the energy of Int3d′ is the lowest point on the energy profile before TS4d, which means that the overall barrier should be calculated relative to Int3d′, resulting in a slightly higher barrier as compared to the other substrates (24.9 kcal mol−1 compared to 22.2, 20.3, and 23.2 kcal mol−1, for 1a, 1b and 1c, respectively). Importantly, the stereoselectivity is reproduced also for substrate 1d, with a selectivity-determining energy difference of 2.5 kcal mol−1, in good agreement with the 97% ee observed experimentally. The origins of the selectivity are found to be the same as for the other substrates.
4. Conclusions
In the present work, the reaction mechanism for the formation of chiral allyl SCF3 compounds via diphenyl selenide-catalyzed sulfenofunctionalization of allylboronic acids has been investigated using DFT calculations. Several allylboronic acid substrates were considered, and the mechanism suggested on the basis of the calculations is shown in Scheme 2.The reaction starts with the generation of the catalytically active SePh2SCF3 cation, a process that takes place is in a stepwise manner, with a transfer of the SCF3 group from the (PhSO2)2NSCF3 reagent 2 to the Ph2Se catalyst 3, followed by a proton transfer from acid Tf2NH 4 to the formed (PhSO2)2N− anion 6. Interestingly, the Tf2NH acid stabilizes the negative charge that develops on the nitrogen anion of the (PhSO2)2N− species 6 through a hydrogen bond interaction, lowering thus the barrier for the SCF3 transfer.
Next, the SCF3 group of the formed SePh2SCF3 cation is transferred to the CC bond of the α-CF3 allylboronic acid, generating the thiiranium ion species Int2. Two different pathways are possible, depending on whether the SCF3 group is transferred to the Si or Re face of the CC bond, which eventually lead to the S- or R-configurations of the product, respectively.
Finally, the Tf2N− anion 9 performs a nucleophilic attack at the boronate group of Int2, triggering the opening of the thiiranium ion and the leaving of the boronate group. Rotation around the Cα–Cβ bond of Int2 leads to either the syn- or anti-elimination, generating the E- or Z- configurations of the product. This step constitutes the selectivity-determining step of the reaction, and the calculations show a clear preference for formation of the E-form, in excellent agreement with the high E-selectivity reported in the experimental studies.37 The enantioselectivity is also very well reproduced by the calculations, and analysis of the transition state structures shows that the selectivity is mainly controlled by steric repulsions between the SCF3 group and both the leaving boronate group and the substituent of the α-CF3 allylboronic acid.
The mechanistic insights provided by the current study will be valuable for the development of new regio-, stereo- and diastero-selective selenide-catalyzed, deborylative electrophilic sulfenofunctionalization reactions.
Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The authors thank the Knut and Alice Wallenberg Foundation (Dnr: 2018.0066) and Swedish Research Council (Dnr: 2021-04282 and 2019-04010) for financial support.References
- Y. Zhu, J. Han, J. Wang, N. Shibata, M. Sodeoka, V. A. Soloshonok, J. A. S. Coelho and F. D. Toste, Modern Approaches for Asymmetric Construction of Carbon-Fluorine Quaternary Stereogenic Centers: Synthetic Challenges and Pharmaceutical Needs, Chem. Rev., 2018, 118(7), 3887–3964 CrossRef CAS PubMed.
- H. Mei, J. Han, S. Fustero, M. Medio-Simon, D. M. Sedgwick, C. Santi, R. Ruzziconi and V. A. Soloshonok, Fluorine-Containing Drugs Approved by the FDA in 2018, Chem. – Eur. J., 2019, 25(51), 11797–11819 CrossRef CAS PubMed.
- Y. Zhou, J. Wang, Z. Gu, S. Wang, W. Zhu, J. L. Aceñ, V. A. Soloshonok, K. Izawa and H. Liu, Next Generation of Fluorine-Containing Pharmaceuticals, Compounds Currently in Phase II-III Clinical Trials of MajorPharmaceutical Companies: New Structural Trends and TherapeuticAreas, Chem. Rev., 2016, 116(2), 422–518 CrossRef CAS PubMed.
- C. Hansch, A. Leo and R. W. Taft, A survey of Hammett substituent constants and resonance and field parameters, Chem. Rev., 1991, 91(2), 165–195 CrossRef CAS.
- E. A. Ilardi, E. Vitaku and J. T. Njardarson, Data-Mining for Sulfur and Fluorine: An Evaluation of Pharmaceuticals to Reveal Opportunities for Drug Design and Discovery, J. Med. Chem., 2014, 57(7), 2832–2842 CrossRef CAS PubMed.
- D. O'Hagan, Understanding organofluorine chemistry. An introduction to the C–F bond, Chem. Soc. Rev., 2008, 37(2), 308–319 RSC.
- Q. Shen, A Toolbox of Reagents for Trifluoromethylthiolation: From Serendipitous Findings to Rational Design, J. Org. Chem., 2023, 88(6), 3359–3371 CrossRef CAS PubMed.
- Y.-D. Yang, A. Azuma, E. Tokunaga, M. Yamasaki, M. Shiro and N. Shibata, Trifluoromethanesulfonyl Hypervalent Iodonium Ylide for Copper-Catalyzed Trifluoromethylthiolation of Enamines, Indoles, and β-Keto Esters, J. Am. Chem. Soc., 2013, 135(24), 8782–8785 CrossRef CAS PubMed.
- S. Arimori, M. Takada and N. Shibata, Trifluoromethylthiolation of Allylsilanes and Silyl Enol Ethers with Trifluoromethanesulfonyl Hypervalent Iodonium Ylide under Copper Catalysis, Org. Lett., 2015, 17(5), 1063–1065 CrossRef CAS PubMed.
- Z. Huang, Y.-D. Yang, E. Tokunaga and N. Shibata, Copper-Catalyzed Regioselective Trifluoromethylthiolation of Pyrroles by Trifluoromethanesulfonyl Hypervalent Iodonium Ylide, Org. Lett., 2015, 17(5), 1094–1097 CrossRef CAS PubMed.
- F. Baert, J. Colomb and T. Billard, Electrophilic trifluoromethanesulfanylation of organometallic species with trifluoromethanesulfanamides, Angew. Chem., Int. Ed., 2012, 51(41), 10382–10385 CrossRef CAS PubMed.
- S. Rossi, A. Puglisi, L. Raimondi and M. Benaglia, Synthesis of Alpha-trifluoromethylthio Carbonyl Compounds: A Survey of the Methods for the Direct Introduction of the SCF3 Group on to Organic Molecules, ChemCatChem, 2018, 10(13), 2717–2733 CrossRef CAS.
- S. Alazet, L. Zimmer and T. Billard, Electrophilic Trifluoromethylthiolation of Carbonyl Compounds, Chem. – Eur. J., 2014, 20(28), 8589–8593 CrossRef CAS PubMed.
- W. Tyrra, D. Naumann, B. Hoge and Y. L. Yagupolskii, A New Synthesis of Trifluoromethanethiolates-Characterization and Properties of Tetramethylammonium, Cesium and Di(benzo-15-crown-5)cesium Trifluoromethanethiolates, J. Fluor. Chem., 2003, 119(1), 101–107 CrossRef CAS.
- Z. Weng, W. He, C. Chen, R. Lee, D. Tan, Z. Lai, D. Kong, Y. Yuan and K.-W. Huang, An Air-Stable Copper Reagent for Nucleophilic Trifluoromethylthiolation of Aryl Halides, Angew. Chem., Int. Ed., 2013, 52(5), 1548–1552 CrossRef CAS PubMed.
- D. Kong, Z. Jiang, S. Xin, Z. Bai, Y. Yuan and Z. Weng, Room Temperature Nucleophilic Trifluoromethylthiolation of Benzyl Bromides with (bpy)Cu(SCF3), Tetrahedron, 2013, 69(30), 6046–6050 CrossRef CAS.
- J. Tan, G. Zhang, Y. Ou, Y. Yuan and Z. Weng, Nucleophilic Trifluoromethylthiolation of Allylic Bromides: A Facile Preparation of Allylic Trifluoromethyl Thioethers, Chin. J. Chem., 2013, 31(7), 921–926 CrossRef CAS.
- Q. Lin, L. Chen, Y. Huang, M. Rong, Y. Yuan and Z. Weng, Efficient C(sp3alkyl)-SCF3 Bond Formations via Copper-mediated Trifluoromethylthiolation of Alkyl Halides, Org. Biomol. Chem., 2014, 12(29), 5500–5508 RSC.
- S. Dix, M. Jakob and M. N. Hopkinson, Deoxytrifluoromethylthiolation and Selenylation of Alcohols by Using Benzothiazolium Reagents, Chem. – Eur. J., 2019, 25(32), 7635–7639 CrossRef CAS PubMed.
- X.-H. Xu, K. Matsuzaki and N. Shibata, Synthetic Methods for Compounds Having CF3-S Units on Carbon by Trifluoromethylation, Trifluoromethylthiolation, Triflylation, and Related Reactions, Chem. Rev., 2015, 115(2), 731–764 CrossRef CAS PubMed.
- C. Ghiazza, T. Billard and A. Tlili, Merging Visible-Light Catalysis for the Direct Late-Stage Group-16−Trifluoromethyl Bond Formation, Chem. – Eur. J., 2019, 25(26), 6482–6495 CrossRef CAS PubMed.
- T. Bootwicha, X. Liu, R. Pluta, I. Atodiresei and M. Rueping, N-Trifluoromethylthiophthalimide: A Stable Electrophilic SCF3-Reagent and its Application in the Catalytic Asymmetric Trifluoromethylsulfenylation, Angew. Chem., Int. Ed., 2013, 52(49), 12856–12859 CrossRef CAS PubMed.
- X. Wang, T. Yang, X. Cheng and Q. Shen, Enantioselective Electrophilic Trifluoromethylthiolation of β-Ketoesters: A Case of Reactivity and Selectivity Bias for Organocatalysis, Angew. Chem., Int. Ed., 2013, 52(49), 12860–12864 CrossRef CAS PubMed.
- X.-L. Zhu, J.-H. Xu, D.-J. Cheng, L.-J. Zhao, X.-Y. Liu and B. Tan, In Situ Generation of Electrophilic Trifluoromethylthio Reagents for Enantioselective Trifluoromethylthiolation of Oxindoles, Org. Lett., 2014, 16(8), 2192–2195 CrossRef CAS PubMed.
- X. Liu, R. An, X. Zhang, J. Luo and X. Zhao, Enantioselective Trifluoromethylthiolating Lactonization Catalyzed by an Indane-Based Chiral Sulfide, Angew. Chem., Int. Ed., 2016, 55(19), 5846–5850 CrossRef CAS PubMed.
- X. Liu, Y. Liang, J. Ji, J. Luo and X. Zhao, Chiral Selenide-Catalyzed Enantioselective Allylic Reaction and Intermolecular Difunctionalization of Alkenes: Efficient Construction of C-SCF3 Stereogenic Molecules, J. Am. Chem. Soc., 2018, 140(14), 4782–4786 CrossRef CAS PubMed.
- J. Luo, Q. Cao, X. Cao and X. Zhao, Selenide-catalyzed enantioselective synthesis of trifluoromethylthiolated tetrahydronaphthalenes by merging desymmetrization and trifluoromethylthiolation, Nat. Commun., 2018, 9(1), 527 CrossRef PubMed.
- T. Qin, Q. Jiang, J. Ji, J. Luo and X. Zhao, Chiral Selenide-Catalyzed Enantioselective Synthesis of Trifluoromethylthiolated 2,5-Disubstituted Oxazolines, Org. Biomol. Chem., 2019, 17(7), 1763–1766 RSC.
- M. Y. Jin, J. Li, R. Huang, Y. Zhou, L. W. Chung and J. Wang, Catalytic Asymmetric Trifluoromethylthiolation of Carbonyl Compounds via a Diastereo and Enantioselective Cu-Catalyzed Tandem Reaction, Chem. Commun., 2018, 54(36), 4581–4584 RSC.
- L. Xu, L. Yu, J. Liu, H. Wang, C. Zheng and G. Zhao, Enantioselective Vinylogous Mannich-Type Reactions to Construct CF3S-Containing Stereocenters Catalysed by Chiral Quaternary Phosphonium Salts, Adv. Synth. Catal., 2020, 362(9), 1851–1857 CrossRef CAS.
- H. Hong, C. Zheng, G. Zhao and Y. Shang, Enantioselective Michael Addition Reactions to Construct SCF3-containing Stereocenter Catalyzed by Chiral Quaternary Phosphonium Salts, Adv. Synth. Catal., 2020, 362(24), 5765–5771 CrossRef CAS.
- M. Deliaval, R. Jayarajan, L. Eriksson and K. J. Szabó, Three-Component Approach to Densely Functionalized Trifluoromethyl Allenols by Asymmetric Organocatalysis, J. Am. Chem. Soc., 2023, 145(18), 10001–10006 CrossRef CAS PubMed.
- R. Jayarajan, T. Kireilis, L. Eriksson and K. J. Szabó, Asymmetric Organocatalytic Homologation: Access to Diverse Chiral Trifluoromethyl Organoboron Species, Chem. – Eur. J., 2022, 28(58), e202202059 CrossRef CAS PubMed.
- K. J. Szabó, Fluorination, Trifluoromethylation, and Trifluoromethylthiolation of Alkenes, Cyclopropanes, and Diazo Compounds, in Organofluorine Chemistry, 2021, pp. 201–223 Search PubMed.
- S. J. T. Jonker, R. Jayarajan, T. Kireilis, M. Deliaval, L. Eriksson and K. J. Szabó, Organocatalytic Synthesis of α-Trifluoromethyl Allylboronic Acids by Enantioselective 1,2-Borotropic Migration, J. Am. Chem. Soc., 2020, 142(51), 21254–21259 CrossRef CAS PubMed.
- F. Shen, P. P. Zhang, L. Lu and Q. Shen, [[(Ethoxycarbonyl)difluoromethyl]thio]phthalimide: A Shelf-Stable, Electrophilic Reagent with a Convertible Group for the Synthesis of Diversified Fluoroalkylthiolated Compounds, Org. Lett., 2017, 19(5), 1032–1035 CrossRef CAS PubMed.
- Q. Wang, T. Nilsson, L. Eriksson and K. J. Szabó, Sulfenofunctionalization of Chiral α-Trifluoromethyl Allylboronic Acids: Asymmetric Synthesis of SCF3, SCF2R, SCN and SAr Compounds, Angew. Chem., Int. Ed., 2022, 61(46), e202210509 CrossRef CAS PubMed.
- A. Matviitsuk, J. L. Panger and S. E. Denmark, Catalytic, Enantioselective Sulfenofunctionalization of Alkenes: Development and Recent Advances, Angew. Chem., Int. Ed., 2020, 59(45), 19796–19819 CrossRef CAS PubMed.
- H.-Y. Luo, Z.-H. Li, D. Zhu, Q. Yang, R.-F. Cao, T.-M. Ding and Z.-M. Chen, Chiral Selenide/Achiral Sulfonic Acid Cocatalyzed Atroposelective Sulfenylation of Biaryl Phenols via a Desymmetrization/Kinetic Resolution Sequence, J. Am. Chem. Soc., 2022, 144(7), 2943–2952 CrossRef CAS PubMed.
- A. D. Becke, Density-unctional Exchange-energy Approximation with Correct Asymptotic Behavior, Phys. Rev. A, 1988, 38(6), 3098–3100 CrossRef CAS PubMed.
- A. D. Becke, Density–functional Thermochemistry. III. The Role of Exact Exchange, J. Chem. Phys., 1993, 98(7), 5648–5652 CrossRef CAS.
- S. Grimme, J. Antony, S. Ehrlich and H. Krieg, A Consistent and Accurate ab initio Parametrization of Density Functional Dispersion Correction (DFT-D) for the 94 Elements H-Pu, J. Chem. Phys., 2010, 132(15), 154104 CrossRef PubMed.
- S. Grimme, S. Ehrlich and L. Goerigk, Effect of the Damping Function in Dispersion Corrected Density Functional Theory, J. Comput. Chem., 2011, 32(7), 1456–1465 CrossRef CAS PubMed.
- M. J. Frisch, G. W. Trucks, H. B. Schlegel, G. E. Scuseria, M. A. Robb, J. R. Cheeseman, G. Scalmani, V. Barone, G. A. Petersson, H. Nakatsuji, X. Li, M. Caricato, A. V. Marenich, J. Bloino, B. G. Janesko, R. Gomperts, B. Mennucci, H. P. Hratchian, J. V. Ortiz, A. F. Izmaylov, J. L. Sonnenberg, D. Williams-Young, F. Ding, F. Lipparini, F. Egidi, J. Goings, B. Peng, A. Petrone, T. Henderson, D. Ranasinghe, V. G. Zakrzewski, J. Gao, N. Rega, G. Zheng, W. Liang, M. Hada, M. Ehara, K. Toyota, R. Fukuda, J. Hasegawa, M. Ishida, T. Nakajima, Y. Honda, O. Kitao, H. Nakai, T. Vreven, K. Throssell, J. A. Montgomery Jr., J. E. Peralta, F. Ogliaro, M. J. Bearpark, J. J. Heyd, E. N. Brothers, K. N. Kudin, V. N. Staroverov, T. A. Keith, R. Kobayashi, J. Normand, K. Raghavachari, A. P. Rendell, J. C. Burant, S. S. Iyengar, J. Tomasi, M. Cossi, J. M. Millam, M. Klene, C. Adamo, R. Cammi, J. W. Ochterski, R. L. Martin, K. Morokuma, O. Farkas, J. B. Foresman and D. J. Fox, Gaussian 16 Rev. C.01, Wallingford, CT, 2016 Search PubMed.
- A. V. Marenich, C. J. Cramer and D. G. Truhlar, Universal Solvation Model Based on Solute Electron Density and on a Continuum Model of the Solvent Defined by the Bulk Dielectric Constant and Atomic Surface Tensions, J. Phys. Chem. B, 2009, 113(18), 6378–6396 CrossRef CAS PubMed.
- S. E. Denmark, E. Hartmann, D. J. P. Kornfilt and H. Wang, Mechanistic, Crystallographic, and Computational Studies on the Catalytic, Enantioselective Sulfenofunctionalization of Alkenes, Nat. Chem., 2014, 6(12), 1056–1064 CrossRef CAS PubMed.
- X.-F. Song, T.-M. Ding, D. Zhu, J. Huang and Z.-M. Chen, Lewis-Acid-Mediated Intramolecular Trifluoromethylthiolation of Alkenes with Phenols: Access to SCF3-Containing Chromane and Dihydrobenzofuran Compounds, Org. Lett., 2020, 22(17), 7052–7056 CrossRef CAS PubMed.
- Q. Jiang, H. Li and X. Zhao, Catalytic Electrophilic Thiocarbocyclization of Allenes, Org. Lett., 2021, 23(22), 8777–8782 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available: Additional computational results discussed in the text, absolute energies and energy corrections, and Cartesian coordinates. See DOI: https://doi.org/10.1039/d4qo02170c |
| This journal is © the Partner Organisations 2025 |