
 Open Access Article
Open Access Article This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported Licence
This Open Access Article is licensed under a Creative Commons Attribution-Non Commercial 3.0 Unported LicenceCarbamoylation of azaarenes and olefins with formamides through dual photoredox/HAT catalysis†
Mario
Martos‡
,
Beatriz
Quevedo-Flores‡
,
Loris
Laze
,
Irene
Bosque
 * and
Jose C.
Gonzalez-Gomez
*
* and
Jose C.
Gonzalez-Gomez
*
Instituto de Síntesis Orgánica (ISO) and Departamento de Química Orgánica, Universidad de Alicante, 03080 Alicante, Spain. E-mail: josecarlos.gonzalez@ua.es
First published on 26th November 2024
Abstract
A unique strategy for the photoinduced Minisci and Giese carbamoylation with readily available formamides is described. This approach avoids using less atom-efficient carbamoyl precursors (e.g., oxamic acids) and sacrificial oxidants, relying on dual photoredox/hydrogen atom transfer (HAT) catalysis. The synthetic value of the carboxamide functionality was illustrated with different straightforward transformations that gave access to diverse azaarene derivatives.
Introduction
Known for over a hundred years now,1,2 the formation of amide bonds is still one of the most important transformations in synthetic and bioorganic chemistry. Amide linkages are present in bioactive compounds, including peptides and proteins, and almost a quarter of all commercialized drugs.3 This versatile moiety in synthetic organic chemistry is also present in mass-produced polymers.4 Over the last few decades, research has steered from classic amide formation by coupling acids and amines with significant developments in the direct amidation of compounds via oxidative radical reactions (Scheme 1a).5 This approach mitigates operational issues related to using commonly insoluble carboxylic acids and poorly atom-economic coupling reagents while providing a complementary scope. One seminal contribution to this field is the iron-catalyzed carbamoylation of azaarenes with formamide in the presence of hydrogen peroxide as a sacrificial oxidant, reported by Minisci in 1970 (Scheme 1b).6 Although many different substrates and conditions have been explored since then, the fundamental working principle is mostly based on adding a carbamoyl radical (˙CONR2) to a suitable acceptor.7 These radicals are long-lived and nucleophilic owing to strong conjugation of the non-bonding electrons of the oxygen with the SOMO.8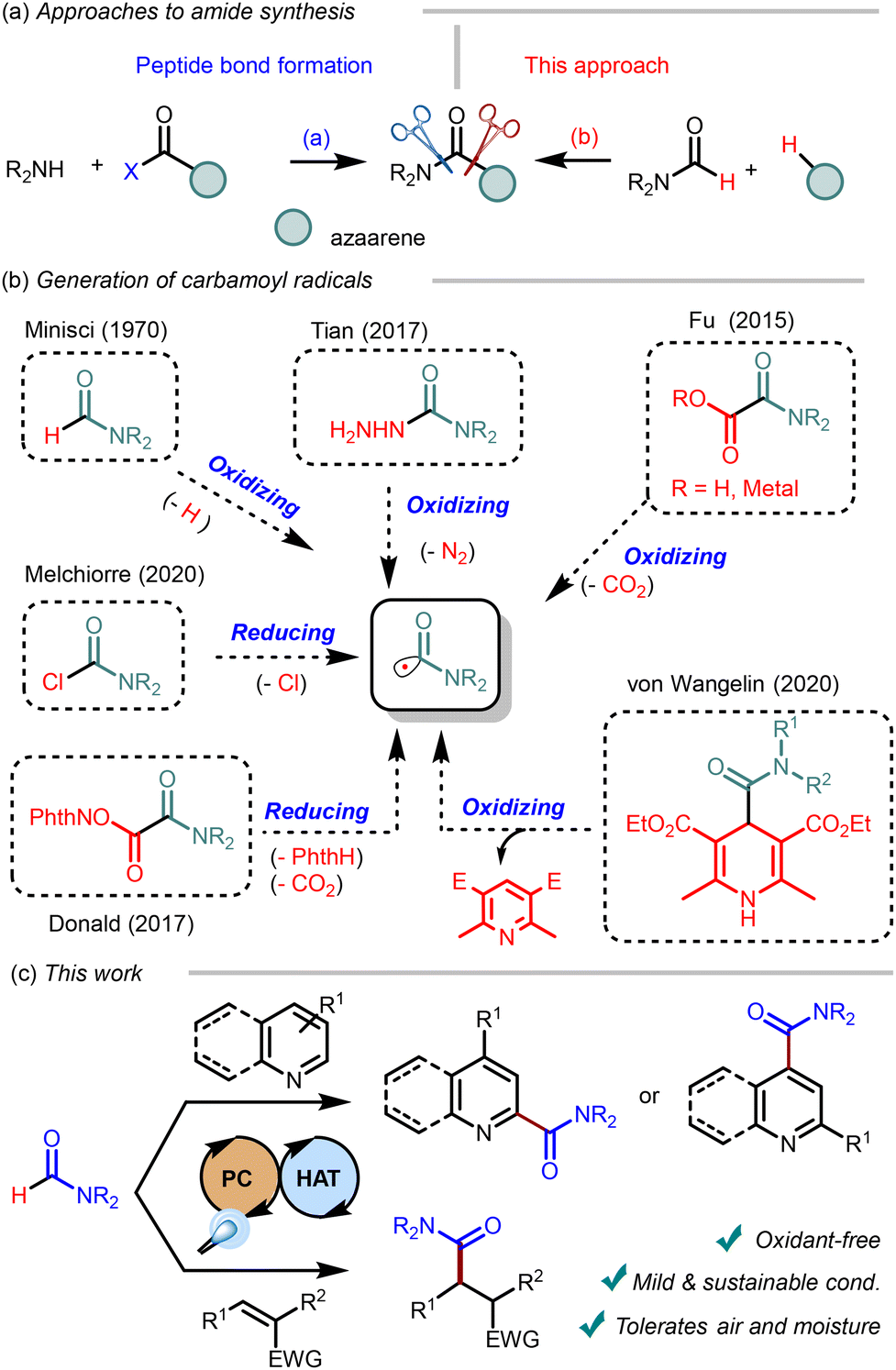 | ||
| Scheme 1 Background of carbamoylation and the present work. | ||
Photocatalytic approaches provide a sustained and controlled radical formation from suitable precursors under milder operating conditions than conventional approaches.9 In this context, a myriad of possibilities has been explored for the photocatalytic generation of carbamoyl radicals (Scheme 1b). For example, the photoinduced oxidation of oxamic acids10–15 with concomitant decarboxylation or semicarbazides with loss of N2![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) 16 has been reported in recent years. On the other hand, photooxidation of 4-carboxamido-1,4-dihydropyridines has proved to be a competent method to obtain carbamoyl radicals but generate the corresponding pyridine as waste.17,18 Moreover, photoinduced reduction of moisture-sensitive carbamoyl chlorides19 or N-hydroxyphtalimido esters are popular approaches.20,21
16 has been reported in recent years. On the other hand, photooxidation of 4-carboxamido-1,4-dihydropyridines has proved to be a competent method to obtain carbamoyl radicals but generate the corresponding pyridine as waste.17,18 Moreover, photoinduced reduction of moisture-sensitive carbamoyl chlorides19 or N-hydroxyphtalimido esters are popular approaches.20,21
Despite the efficiency of many of the above-commented methods, formamides are more readily available and convenient substrates, providing the highest atom economy in generating carbamoyl precursors. The sustainability of the carbamoylation with formamides would be optimal in the absence of sacrificial reagents. In this context, Prieto and Taillefer have recently reported the photoinduced hydrocarbamoylation of styrenes with formamides, relying on decatungstate/disulfide catalysis.22 To our best knowledge, the use of formamide for the carbamoylation of azaarenes without sacrificial oxidants is scarce.23–26 Considering the importance of amide-substituted azaarenes, systematic studies to accomplish this transformation with a reasonably broad substrate scope would be highly desirable.
This work presents a protocol for the direct photoinduced carbamoylation of nitrogenated heterocycles and electron-deficient olefins with formamides.27 Our approach relies on dual photoredox/HAT catalysis, using readily made 9-(2-chlorophenyl)acridine (A) and inexpensive pyridine N-oxide (PyO) as catalysts under blue light. Importantly, this method avoids the need for sacrificial oxidants and tolerates the presence of air and moisture, making this protocol sustainable and user-friendly (Scheme 1c).
Results and discussion
Due to the relevance of nitrogenated heterocyclic scaffolds in medicinal chemistry,28 we selected the Minisci reaction, probably the most relevant pathway towards the radical functionalization of azaarenes,29 to first assay the performance of our system in radical carbamoylation reactions. In an earlier report, we established the capability of A (Ered[A-H+]* = 2.2 V vs. SCE) to efficiently generate a HAT catalyst by oxidation of PyO in the presence of TFA under irradiation at 450 nm.30 The resulting N-oxyl radical (BDE = 99 kcal mol−1)31,32 should then be capable of abstracting a hydrogen atom from the formamide (BDE = 95 kcal mol−1),33 generating the corresponding carbamoyl radical, which would then undergo addition to the protonated heterocycle. Thus, we selected the carbamoylation of lepidine with formamide as the model reaction to test this hypothesis. Using the optimized conditions from our earlier report on the Minisci alkylation of azaarenes,30 we observed the desired product in a modest 26% yield. After some optimization studies (Table 1), we found the optimal conditions to be 10 equivalents of formamide, 5 mol% of A, 30 mol% of PyO and 2 equivalents of TFA at a concentration of 0.1 M in acetonitrile for 48 hours at around 35 C (internal temperature). In contrast to our earlier report, the use of hexafluoroisopropanol (HFIP) was found to be detrimental to the reaction. Other solvents or reaction conditions gave poorer results.|
|
||
|---|---|---|
| Entry | Deviation from above | Yielda (%) |
| a Based on relative GC-MS integral values of the product and starting material. Isolated yield in parentheses. | ||
| 1 | None | 26 |
| 2 | 4-AcPyO instead of PyO | 14 |
| 3 | 127 equiv. of HCONH2 | 17 |
| 4 | MeCN as solvent | 46 |
| 5 | MeCN as solvent, 48 h | 66 (50) |
| 6 | Acetone as solvent, 48 h | 37 |
| 7 | DCE as solvent, 48 h | 60 (52) |
| 8 | EtOH as solvent, 48 h | 0 |
| 9 | EtOAc as solvent, 48 h | 30 |
| 10 | 5 equiv. of HCONH, 60 h | 28 |
| 11 | As entry 5, but 4 equiv. of TFA | 78 (43) |
| 12 | 4 equiv. of HCl, w/o TFA and PyO | Trace |
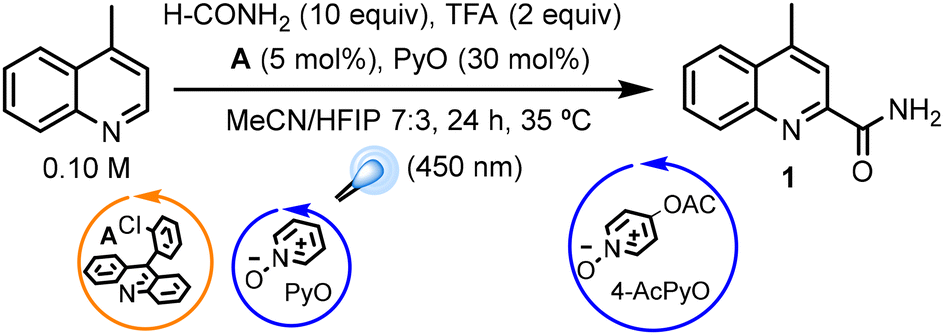
With the optimized conditions set, we explored the scope of the reaction (Scheme 2). Quinolines reacted smoothly, affording the corresponding products (1 to 6) in moderate to good yields with good functional group tolerance. Quinaldine was a particularly convenient substrate, as the low solubility of the product allowed its isolation in pure form after filtration from the reaction mixture and washing with hexane (compound 2). Plain quinoline reacted preferentially at C2, although the C4 derivative and disubstituted products were also obtained (products 5), likely due to the lower steric hindrance at the C2 position. Notably, the DL-menthol functionalized 6 could be obtained in moderate yield. Isoquinolines were challenging substrates in our previous protocol for the alkylation of azaarenes.26 In this case, they reacted cleanly, so we obtained the corresponding 1-carbamoylisoquinolines 7–9 in moderate to good yields. This is further highlighted by obtaining product 8 pure after simple filtration and washing with hexane. Phenanthridines exhibited similar behavior, affording products 10 and 11 in good-to-excellent yields after simple filtration and washing with hexane. Quinoxaline was also well tolerated, affording product 12 in good yield. Pyridines were more challenging substrates, with several failed attempts (Fig. S3†), although the highly activated pyridine ring of roflumilast underwent the desired transformation, affording 13 in moderate yield. In addition, we tested different N-substituted formamides in combination with isoquinoline. Amides bearing alkyl groups, including the heavily sterically hindered diisopropylformamide, were well tolerated (products 14–16). Aryl groups resulted in no reactivity whatsoever (Fig. S3†) towards the desired product. Prieto and Taillefer had previously observed this behavior.22 It could be attributed to the relatively low rate of addition leading to side products, particularly isocyanates obtained by oxidation of the carbamoyl radical, which were detected by GC-MS and are known to be formed under oxidative conditions.34
 | ||
| Scheme 2 Substrate scope of the Minisci carbamoylation. Yields for isolated pure products. a4 equiv. of TFA were used. bAfter 60 h. | ||
After studying the Minisci reaction, we shifted our attention towards the Giese-type addition of carbamoyl radicals to electron-deficient olefins. This redox-neutral process allows for the direct preparation of a variety of masked 1,4-dicarbonyl amides. Thus, we selected the carbamoylation of diethyl ethylidenemalonate as the model reaction. We were delighted to observe the desired product in excellent yield under the standard conditions for the Minisci reaction, but only with 1 equivalent of TFA. Encouraged by this result, we briefly re-examined the reaction conditions (Table 2), and similar good results were obtained with only 5 equivalents of formamide (entry 3). With these conditions in hand, we set on exploring the scope of this transformation (Scheme 3).
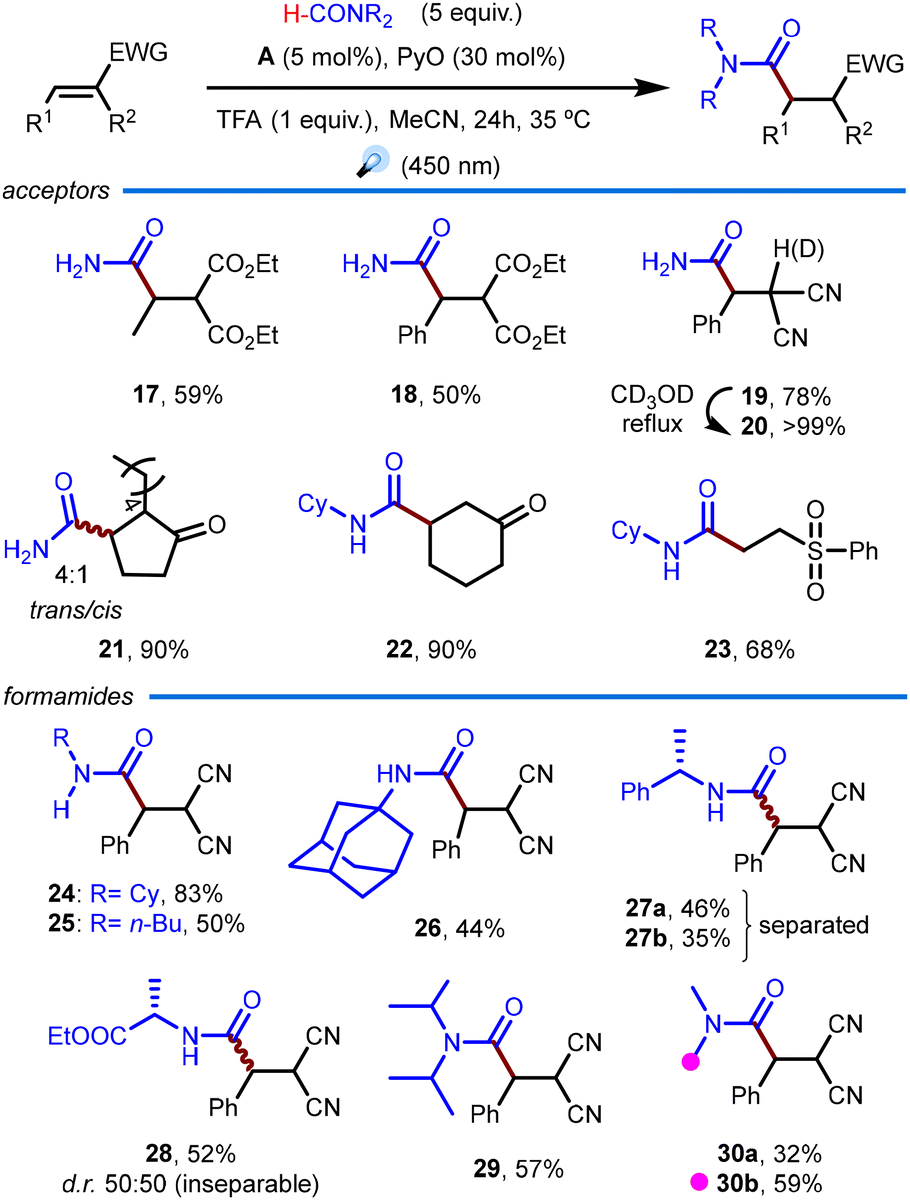 | ||
| Scheme 3 Substrate scope of the Giese-type carbamoylation. Yields for isolated pure products. | ||

In general, good results were obtained across the board. As expected, the more electron-withdrawing benzylidenemalononitrile performed the best among the linear substrates paired with formamide (products 19vs.17 and 18). Interestingly, this product could be easily deuterated between the nitrile groups by refluxing it in deuterated methanol, affording 20 in quantitative yield. Deuteration can be reversed following the same protocol in regular methanol. Cyclic substrates were well tolerated, with 2-pentylcyclopent-2-ene-1-one and cyclohexanone affording 21 and 22 in excellent yield in combination with formamide and N-cyclohexylformamide, respectively. Notably, vinylphenylsulfone was a suitable substrate, affording the synthetically useful product 23 in good yield.35 Products 21 to 23 demonstrate that only one electron-withdrawing group in the substrate is enough for the reaction to proceed smoothly. We then focused on exploring different formamides in combination with benzylidenemalononitrile. This reaction tolerates a broader scope of formamides than the Minisci reaction, although we are still limited to alkyl-substituted formamides (products 24–30). Sterically hindered formamides were successful substrates, affording the desired products in moderate to good yield (compounds 26, 29, and 30). Interestingly, a formamide prepared from (S)-1-phenethylamine was successfully transformed into diastereoisomers 27a and 27b. Although the diastereoselectivity was low, the diastereomers were easily separated by column chromatography, thus offering the opportunity to prepare enantioenriched compounds. Notably, the ethyl ester of N-formyl-L-alanine was fully compatible with this protocol, affording products 28a/28b as an inseparable 1:1 mixture of diastereomers. Finally, N,N-dimethylformamide was also examined as substrate, showing preference towards the methylene substitution (isomer 30b) over the desired product 30a. This selectivity had been previously observed in this type of transformation and is consistent with the number of N–CH bonds (6) overcoming the lower BDE of the OC–H bond.36 It is worth noting that considering the number of C–H bonds (1 vs. 6), the normalized selectivity for the formyl substitution is still high (3 (OCH):1 (NCH)), in line with the selectivity observed for monoalkylformamides and even with diisopropylformamide.
To further demonstrate the applicability and robustness of our dual catalytic system, we decided to perform a series of 1 mmol scale reactions under sunlight, following the same basic protocol for each type of reaction (Scheme 4a). Minisci reactions tolerated well these scaled-up conditions, affording products 7 and 10 in virtually identical yields to those obtained under the general conditions. Product 10 was scaled up further to 5 mmol with only a slight decrease in yield, demonstrating the usefulness of this procedure. Giese-type reactions, on the other hand, were found to be more sensitive as they experienced a significant decrease in yields compared to those observed using the general conditions, obtaining products 19 and 21 in 68% (from 78%) and 57% (from 90%) yield, respectively. They are, however, still satisfactory and useful results from a preparative standpoint.
 | ||
| Scheme 4 (a) Reactions on a 1 mmol scale using solar irradiation. (b) Synthetic versatility of the carboxamide moiety. Isolated yields. | ||
To illustrate the versatility of the amide moiety, we conducted diverse derivatizations of this functionality on substrate 10 (Scheme 4b). The selective methylation of the amidic nitrogen atom was successfully achieved using tetramethylammonium fluoride without affecting the nitrogen atom of the phenanthrene scaffold, likely by a concerted methylation–deprotonation pathway.37 Interestingly, only the monomethyl derivative 31 was isolated under these non-optimized conditions. This methylation prevents the formation of undesired by-products that result from the HAT at the methyl group in a direct coupling with N-methyl amides (e.g., product 30b). On the other hand, dehydration of 10 with POCl3 provided nitrile 32 in 70% yield. Potentially, our carbamoylation methodology, followed by this dehydration, gives access to drug-like nitriles from the corresponding azaarenes.
In addition, the condensation of 10 with acetylacetone afforded enamide 33 with good yield and exclusively Z-selectivity. These Z-enamides have been employed in oxidative cyclization to obtain oxazoles38 and enantioselective copper-catalyzed borylation.39 Finally, we rearranged the carboxamide group to the corresponding carbamate, forming the C(6)–N bond. The Hofmann rearrangement of 10 was conducted according to a modified reported electrochemical protocol, using non-toxic and inexpensive LiBr for the in situ generation of Br2.40 The expected carbamate 34 was obtained in good yield, and its aqueous basic hydrolysis took place smoothly, providing amine 35. This transformation paved the way for other heteroaromatic amines to be obtained from azaarenes with this three-step sequence (carbamoylation → Hofmann rearrangement → hydrolysis). This strategy complements the centenary Chichibabin reaction to introduce amino groups into azaarenes, with better tolerance to basic sensitive moieties.41
To gain insight into the mechanism of the reactions, we performed some control experiments (Scheme 5a). Using phenanthridine as a substrate, removing either A, TFA, or PyO shuts down the reaction completely, as does running it in the dark. As expected, adding two equivalents of TEMPO completely inhibits the reaction. Unfortunately, no carbamoyl adduct could be detected. Deoxygenation experiments ruled out the participation of aerobic O2 as an oxidant since 80% of the product (GC) was formed under these conditions.
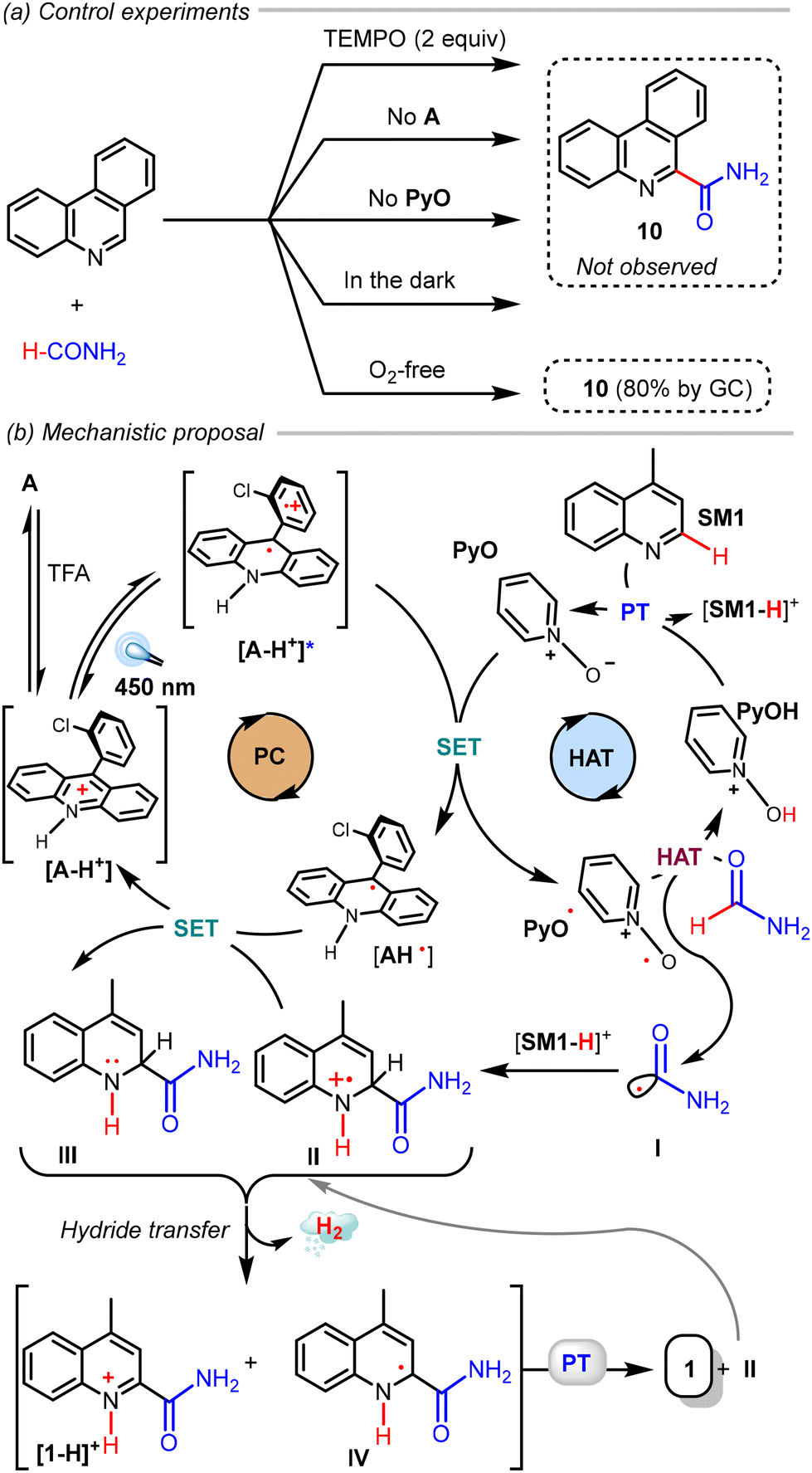 | ||
| Scheme 5 (a) Control experiments. (b) Mechanistic proposal for the Minisci carbamoylation. | ||
Based on these experiments, literature precedents, and our previous investigations,30 we propose the mechanism of the Minisci reaction as depicted in Scheme 5b. The protonated photocatalyst [A-H+] is photoexcited and undergoes single electron oxidation of the PyO, forming the active HAT catalyst. This N-oxyl radical can then abstract a hydrogen atom from formamide, generating the carbamoyl radical I. The radical addition to the protonated heterocycle affords the intermediate radical cation II, which is reduced to intermediate III, cycling back the photocatalyst. Regarding the HAT catalyst, the resulting PyOH is significantly more acidic than TFA and can protonate the azaarene, regenerating the PyO. The dihydroquinoline III can then be oxidized through hydride transfer to II with hydrogen evolution, forming the protonated product and captodative radical IV. A proton transfer between these intermediates should afford the final product while regenerating the intermediate II.
Control experiments for the formation of product 19 revealed that the photocatalyst A, the HAT catalyst PyO, and the light are all indispensable (Scheme 6a). In addition, full conversion to 19 was observed under O2-free conditions, which supports a photoredox catalytic cycle for the turnover of A. Like what was proposed for the Minisci reaction, the interplay of the photocatalyst and the HAT catalyst should facilitate the formation of the carbamoyl radical I (Scheme 6b). The addition of this radical to the electron-deficient olefin should be followed by the reduction of radical intermediate VI to the anionic intermediate VII, regenerating the photocatalyst. Eventually, anion VII can be protonated by the PyOH, regenerating the HAT catalyst and affording the final product. Alternatively, HAT from the acridinyl radical [A-H˙] to VI could directly produce 19 while regenerating the photocatalyst.
 | ||
| Scheme 6 (a) Control experiments. (b) Mechanistic proposal for the Giese carbamoylation. | ||
Conclusions
Our methodology for the oxidant-free carbamoylation of azaarenes and electron-deficient olefins via dual photoredox/HAT catalysis is efficient and user-friendly. It is also compatible with a wide range of substrates, producing valuable carboxamides in good to excellent yields while using readily available catalysts and visible light at room temperature. Compared to other methodologies, our protocol exhibits higher atom economy and minimizes waste generation, making it a sustainable choice. The versatility of the carboxamide functionality as a building block is demonstrated through the straightforward transformation of phenanthridine-6-carboxamide into the corresponding N-methyl amide, nitrile, Z-enamide, and amino derivatives.Author contributions
J. C. G.-G. and I. B. conceived and supervised the project. M. M., B. Q.-F., and L. L. performed the experiments. The manuscript was drafted by M. M. and refined by J. C. G.-G. The ESI was written by M. M. and B. Q.-F. and revised by J. C. G.-G. All authors have approved the final version of the manuscript.Data availability
The data supporting this article have been included as part of the ESI.†Conflicts of interest
There are no conflicts to declare.Acknowledgements
The Generalitat Valenciana financially supported this work (CIAICO/2022/017 and SEJIGENT/2021/005). We also thank MCIN/AEI and the “European Union NextGenerationEU” for the “Consolidación Investigadora” Grant (CNS2022-135161).References
- C. Schotten, Ueber die Oxydation des Piperidins, Ber. Dtsch. Chem. Ges., 1884, 17, 2544–2547 CrossRef.
- E. Baumann, Ueber eine einfache Methode der Darstellung von Benzoësäureäthern, Ber. Dtsch. Chem. Ges., 1886, 19, 3218–3222 CrossRef.
- A. K. Ghose, V. N. Viswanadhan and J. J. Wendoloski, A Knowledge-Based Approach in Designing Combinatorial or Medicinal Chemistry Libraries for Drug Discovery. 1. A Qualitative and Quantitative Characterization of Known Drug Databases, J. Comb. Chem., 1999, 1, 55–68 CrossRef CAS PubMed.
- A. Greenberg, C. M. Breneman and J. F. Liebman, The amide linkage: Structural significance in chemistry, biochemistry, and materials science, John Wiley & Sons, 2000 Search PubMed.
- M. Albert-Soriano and I. M. Pastor, Metal–Organic Framework Based on Copper and Carboxylate-Imidazole as Robust and Effective Catalyst in the Oxidative Amidation of Carboxylic Acids and Formamides, Eur. J. Org. Chem., 2016, 5180–5188 CrossRef CAS.
- F. Minisci, G. P. Gardini, R. Galli and F. Bertini, A new selective type of aromatic substitution: homolytic amidation, Tetrahedron Lett., 1970, 11, 15–16 CrossRef.
- B. A. L. Sacchelli, B. C. Rocha, M. Fantinel and L. H. Andrade, Molecular Construction Using Formamide as a C1 Feedstock, Eur. J. Org. Chem., 2024, e202300930 CrossRef CAS.
- F. Parsaee, M. C. Senarathna, P. B. Kannangara, S. N. Alexander, P. D. E. Arche and E. R. Welin, Radical philicity and its role in selective organic transformations, Nat. Rev. Chem., 2021, 5, 486–499 CrossRef CAS PubMed.
- B. T. Matsuo, P. H. R. Oliveira, E. F. Pissinati, K. B. Vega, I. S. de Jesus, J. T. M. Correia and M. Paixao, Photoinduced carbamoylation reactions: unlocking new reactivities towards amide synthesis, Chem. Commun., 2022, 58, 8322–8339 RSC.
- M. T. Westwood, C. J. C. Lamb, D. R. Sutherland and A.-L. Lee, Metal-, Photocatalyst-, and Light-Free Direct C–H Acylation and Carbamoylation of Heterocycles, Org. Lett., 2019, 21, 7119–7123 CrossRef CAS PubMed.
- D. M. Kitcatt, K. A. Scott, E. Rongione, S. Nicolle and A.-L. Lee, Direct decarboxylative Giese amidations: photocatalytic vs. metal- and light-free, Chem. Sci., 2023, 14, 9806–9813 RSC.
- W.-M. Cheng, R. Shang, H.-Z. Yu and Y. Fu, Room-Temperature Decarboxylative Couplings of α-Oxocarboxylates with Aryl Halides by Merging Photoredox with Palladium Catalysis, Chem. – Eur. J., 2015, 21, 13191–13195 CrossRef CAS PubMed.
- J. D. Williams, S. G. Leach and W. J. Kerr, An Umpolung Approach to Acyclic 1,4-Dicarbonyl Amides via Photoredox-Generated Carbamoyl Radicals, Chem. – Eur. J., 2023, 29, e202300403 CrossRef CAS PubMed.
- I. M. Ogbu, G. Kurtay, F. Robert and Y. Landais, Oxamic acids: useful precursors of carbamoyl radicals, Chem. Commun., 2022, 58, 7593–7607 RSC.
- D. T. Mooney, H. McKee, T. S. Batch, S. Drane, P. R. Moore and A.-L. Lee, Direct C–H amidation of 1,3-azoles: light-mediated, photosensitiser-free vs. thermal, Chem. Commun., 2024, 60, 10752–10755 RSC.
- Z.-Y. He, C.-F. Huang and S.-K. Tian, Highly Regioselective Carbamoylation of Electron-Deficient Nitrogen Heteroarenes with Hydrazinecarboxamides, Org. Lett., 2017, 19, 4850–4853 CrossRef CAS PubMed.
- L. Cardinale, M. O. Konev and A. Jacobi von Wangelin, Photoredox-Catalyzed Addition of Carbamoyl Radicals to Olefins: A 1,4-Dihydropyridine Approach, Chem. – Eur. J., 2020, 26, 8239–8243 CrossRef CAS PubMed.
- Z.-x. Yu, S.-W. Ma, G.-x. Li and L.-j. Ma, Photocatalytic Carbamoyl Radical Transfer to Alkenyl Azaarenes, Synlett, 2024, 35, 1883–1888 CrossRef CAS.
- E. de Pedro Beato, D. Mazzarella, M. Balletti and P. Melchiorre, Photochemical generation of acyl and carbamoyl radicals using a nucleophilic organic catalyst: applications and mechanism thereof, Chem. Sci., 2020, 11, 6312–6324 RSC.
- W. F. Petersen, R. J. K. Taylor and J. R. Donald, Photoredox-catalyzed procedure for carbamoyl radical generation: 3,4-dihydroquinolin-2-one and quinolin-2-one synthesis, Org. Biomol. Chem., 2017, 15, 5831–5845 RSC.
- S. Maiti, S. Roy, P. Ghosh, A. Kasera and D. Maiti, Photo-Excited Nickel-Catalyzed Silyl-Radical-Mediated Direct Activation of Carbamoyl Chlorides To Access (Hetero)aryl Carbamides, Angew. Chem., Int. Ed., 2022, 61, e202207472 CrossRef CAS PubMed.
- A. Prieto and M. Taillefer, Visible-Light Decatungstate/Disulfide Dual Catalysis for the Hydro-Functionalization of Styrenes, Org. Lett., 2021, 23, 1484–1488 CrossRef CAS PubMed.
- P. Xu, P. Chen and H. Xu, Scalable Photoelectrochemical Dehydrogenative Cross–Coupling of Heteroarenes with Aliphatic C–H Bonds, Angew. Chem., Int. Ed., 2020, 59, 14275–14280 CrossRef CAS PubMed.
- C.-Y. Huang, J. Li and C.-J. Li, A cross-dehydrogenative C(sp3)–H heteroarylation via photo-induced catalytic chlorine radical generation, Nat. Commun., 2021, 12, 4010 CrossRef CAS PubMed.
- D.-S. Li, T. Liu, Y. Hong, C.-L. Cao, J. Wu and H.-P. Deng, Stop-Flow Microtubing Reactor-Assisted Visible Light-Induced Hydrogen-Evolution Cross Coupling of Heteroarenes with C(sp3)–H Bonds, ACS Catal., 2022, 12, 4473–4480 CrossRef CAS.
- H. He, Q. Wan, Z.-W. Hou, Q. Zhou and L. Wang, Organoelectrophotocatalytic Generation of Acyl Radicals from Formamides and Aldehydes: Access to Acylated 3-CF3-2-Oxindoles, Org. Lett., 2023, 25, 7014–7019 CrossRef CAS PubMed.
- M. Martos, B. Quevedo-Flores, L. Laze, I. Bosque and J. C. Gonzalez-Gomez, Carbamoylation of azaarenes and olefins with formamides through dual photoredox/HAT catalysis, ChemRxiv, 2024. DOI:10.26434/chemrxiv-2024-0rt1s.
- X. Zhang, S. Li, F. Qiu, H. T. Ang, J. Wu and P. Jia, Photocatalyzed Minisci-type reactions for late-stage functionalization of pharmaceutically relevant compounds, Green Chem., 2024, 26, 3595–3626 RSC.
- M. Martos, I. Bosque and J. C. Gonzalez-Gomez, Synthesis, 2024, 56 DOI:10.1055/s-0043-1775387.
- L. Laze, B. Quevedo-Flores, I. Bosque and J. C. Gonzalez-Gomez, Alkanes in Minisci-Type Reaction under Photocatalytic Conditions with Hydrogen Evolution, Org. Lett., 2023, 25, 8541–8546 CrossRef CAS PubMed.
- M. Schlegel, S. Qian and D. A. Nicewicz, Aliphatic C–H Functionalization Using Pyridine N-Oxides as H-Atom Abstraction Agents, ACS Catal., 2022, 12, 10499–10505 CrossRef CAS PubMed.
- B. Wang, C. Ascenzi Pettenuzzo, J. Singh, G. E. Mccabe, L. Clark, R. Young, J. Pu and Y. Deng, Photoinduced Site-Selective Functionalization of Aliphatic C–H Bonds by Pyridine N-oxide Based HAT Catalysts, ACS Catal., 2022, 12, 10441–10448 CrossRef CAS.
- Y. Zhang, K. B. Teuscher and H. Ji, Direct α-heteroarylation of amides (α to nitrogen) and ethers through a benzaldehyde-mediated photoredox reaction, Chem. Sci., 2016, 7, 2111–2118 RSC.
- F. Minisci, F. Fontana, F. Coppa and Y. M. Yan, Reactivity of Carbamoyl Radicals. A New, General, Convenient Free-Radical Synthesis of Isocyanates from Monoamides of Oxalic Acid, J. Org. Chem., 1995, 60, 5430–5433 CrossRef CAS.
- S.-J. Burlingham, D. Guijarro, I. Bosque, R. Chinchilla and J. C. Gonzalez-Gomez, Visible-light-mediated decarboxylative (E)-alkenylation of aliphatic carboxylic acids with aryl styryl sulfones under metal-free conditions, Org. Biomol. Chem., 2022, 20, 7923–7928 RSC.
- L. Capaldo, D. Merli, M. Fagnoni and D. Ravelli, Visible Light Uranyl Photocatalysis: Direct C–H to C–C Bond Conversion, ACS Catal., 2019, 9, 3054–3058 CrossRef CAS.
- H.-G. Cheng, M. Pu, G. Kundu and F. Schoenebeck, Selective Methylation of Amides, N-Heterocycles, Thiols, and Alcohols with Tetramethylammonium Fluoride, Org. Lett., 2020, 22, 331–334 CrossRef CAS PubMed.
- M. Kamiya, M. Sonoda and S. Tanimori, A rapid access to substituted oxazoles via PIFA-mediated oxidative cyclization of enamides, Tetrahedron, 2017, 73, 1247–1254 CrossRef CAS.
- A. López, T. B. Clark, A. Parra and M. Tortosa, Copper-catalyzed Enantioselective synthesis of β-boron β-amino esters, Org. Lett., 2017, 19, 6272–6275 CrossRef PubMed.
- L. Li, M. Xue, X. Yan, W. Liu, K. Xu and S. Zhang, Electrochemical Hofmann rearrangement mediated by NaBr: practical access to bioactive carbamates, Org. Biomol. Chem., 2018, 16, 4615–4618 RSC.
- D. E. Lewis, Aleksei Yevgen'evich Chichibabin (1871–1945): A Century of Pyridine Chemistry, Angew. Chem., Int. Ed., 2017, 56, 9660–9668 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qo02040e |
| ‡ These authors contributed equally. |
| This journal is © the Partner Organisations 2025 |