
Flexible keratin hydrogels obtained by a reductive method†
María Luz
Peralta Ramos
ab,
Patricia
Rivas-Rojas
 cd,
Hugo
Ascolani
e,
Margherita
Cavallo
f,
Francesca
Bonino
f,
Roberto
Fernandez de Luis
g,
María Ximena
Guerbi
h,
Flabia
Michelini
h,
Celina
Bernal
i,
Juan Manuel
Lázaro-Martínez
ab and
Guillermo
Copello
*ab
cd,
Hugo
Ascolani
e,
Margherita
Cavallo
f,
Francesca
Bonino
f,
Roberto
Fernandez de Luis
g,
María Ximena
Guerbi
h,
Flabia
Michelini
h,
Celina
Bernal
i,
Juan Manuel
Lázaro-Martínez
ab and
Guillermo
Copello
*ab
aDepartamento de Ciencias Químicas, Facultad de Farmacia y Bioquímica, Universidad de Buenos Aires, Buenos Aires, Argentina. E-mail: gcopello@ffyb.uba.ar
bInstituto de Química y Metabolismo del Fármaco (IQUIMEFA), CONICET – Universidad de Buenos Aires, Buenos Aires, Argentina
cLaboratorio de Cristalografía Aplicada, Instituto de Tecnologías Emergentes y Ciencias Aplicadas (ITECA), UNSAM-CONICET, Escuela de Ciencia y Tecnología, San Martín, Buenos Aires, Argentina
dInstituto de Materiales y Procesos Termomecánicos, Universidad Austral de Chile, Valdivia, Chile
eCentro Atómico Bariloche, CNEA y CONICET, R8402AGP Bariloche, Argentina
fDepartment of Chemistry, NIS and INSTM Reference Centers, Università di Torino, Via P. Giuria 7, I-10125 Torino and Via G. Quarello 15/A, Torino, I-10135, Italy
gBCMaterials, Basque Center for Materials, Applications and Nanostructures, UPV/EHU Science Park, 48940 Leioa, Spain
hCentro de Medicina Traslacional, Hospital de Alta Complejidad El Cruce Néstor Kirchner, Buenos Aires, Argentina
iGrupo de Ingeniería en Polímeros y Materiales Compuestos, Instituto de Tecnología en Polímeros y Nanotecnología (ITPN-UBA-CONICET), Facultad de Ingeniería, Universidad de Buenos Aires, Las Heras 2214 (CP 1127AAR), Buenos Aires, Argentina
First published on 1st November 2024
Abstract
Keratin derived materials are still underexploited due to the little understanding of their chemical versatility. Whereas many protein based materials achieve flexibility by crosslinking or interpenetrating with synthetic polymers, we assessed the effect of reductive treatments on aqueous media. Hydrazine sulphate (HZN) and ascorbic acid reduction were compared. The reduced material is bendable and stretchable, whereas the original keratin hydrogel is brittle. This would imply a technological leap in protein materials. Both reductive treatments would achieve reduced keratins by the reduction of oxidised cysteines which leads to a change in the polypeptide chain interaction by a decrease in electrostatic repulsion and swelling. Moreover, in contrast with the ascorbic acid treatment, when higher levels of HZN are employed, the effect of residual sulphates lead to the interchain closeness of the more mobile domains acting as physical crosslinkers, leading to compressed structures with narrower pores. This suggests that the flexible properties of the hydrogel could be related not only to the reduction of the hydrogel but also to the interaction of the sulphate ions with the keratin structure. As a result, the reduction of sulfinic and sulfenic groups to thiol, along with the incorporation of sulphate ions into the structure, impart the material with an elongation at break ranging between 10–25%, nano-scale pores approximately 2 nm in size, swelling capacity of around 50%, all while preserving the biocompatibility observed in the original material tested across two cell lines comprising fibroblasts and keratinocytes.
Introduction
In the present era, the utilization of petroleum-derived materials has gained widespread prominence owing to their remarkable attributes such as versatility, resilience, malleability, ease of use, and cost-effectiveness. However, due to their non-biodegradability and environmental persistence, they have emerged as major global pollutants.1–3 Consequently, one of the solutions to address this issue lies in the adoption of biodegradable materials and waste-derived substances, thereby promoting ecological sustainability and the circular economy. Within this category, biopolymers such as chitin, chitosan, starch, cellulose, collagen, among others, have begun to be harnessed for the production of biomaterials spanning diverse fields. For instance, chitin and chitosan have found applications in food packaging, controlled release systems, tissue engineering, heavy metal adsorption in water, and more. Meanwhile, protein hydrogels have been employed in tissue regeneration, wound dressings, drug delivery, and similar applications.4–8While the merits of biomaterials have generated high expectations, their properties have yet to surpass and outperform those of petroleum derivatives. This is partially attributed to the limited development and investment in biobased polymers compared to synthetic polymers.9–11
In the development of these biomaterials, several obstacles need to be overcome, including the physical–chemical modifications required to enhance their intrinsic properties or achieve new desired characteristics (hydrophobicity, adsorptive capacity, flexibility, rigidity). Within this context, attempts have been made to employ chemical modification treatments to generate functional groups that confer hydrophobic or hydrophilic properties to the material, or to endow it with interaction sites for various compounds.12,13 Additionally, the incorporation of various fillers or the crosslinking of these biomaterials can enhance their mechanical properties such as hardness, rigidity, and flexibility. Various transformations have been investigated, including the oxidation of keratin to keratose, the deacetylation of chitin to chitosan, the esterification of cellulose, and the crosslinking of collagen.14–17 Moreover, composite materials consisting of more than one biopolymer have been produced. These strategies have, for instance, led to Shan Zhang et al. to the synthesis of a composite material composed of silica, calcium alginate, and xanthan gum for lead adsorption in aqueous solutions. Other reported studies by Kuang Li et al. include the synthesis of a film based on soy protein and polyvinylpyrrolidone-functionalized lignosulfonate, exhibiting notable mechanical resistance and UV radiation blocking capabilities, as well as the production of modified cellulose paper reinforced with reduced graphene oxide, possessing improved electrical and mechanical properties, reported by Nguyen Dang Luong et al. Other critical properties such as biocompatibility and biodegradability are essential for the application of these biopolymers in the fields of biomedicine, tissue engineering, prosthetic production, and more. Collagen protein family has been extensively employed in wound dressing to facilitate wound and burn healing18 and as support for bone and cartilage regeneration,19,20 among other applications.
Another challenge in the development of these materials pertains to their processing. While various manipulation techniques such as casting, electrospinning, and the recent incorporation of 3D printing exist, further refinement is still needed in using these techniques for biomaterial production.21–23 Therefore, considering the extensive range of natural polymers available and the numerous fields in which they can be applied, it is of paramount importance to thoroughly investigate the physicochemical and structural characteristics of these materials and delve into the development of techniques and protocols for biomaterial production.
In particular, keratin is a structural protein, the second most abundant after collagen, which can be found as a constituent of skin, hair, feathers, horns, nails, and claws in various animals. It is a protein with a high cysteine residue content and can be classified as α or β keratin based on the prevalence of α helix or β sheets in its secondary structure. It is a semi-crystalline protein composed of crystalline structures called intermediate filaments, which are formed by dimers of α helix chains crosslinked by disulfide and hydrogen bridges and polymerized together. These intermediate filaments are embedded in an amorphous matrix composed of proteins with high and low sulphur content.24,25 Nowadays, keratinic waste is produced in large quantities as a byproduct of the poultry and livestock industry. Keratin extraction for the production of biomaterials would not only add value to these waste products but also promote ecological sustainability.26,27 Due to its biocompatibility, non-toxicity and biodegradability properties, it has been widely used in the development of biomedical materials, tissue engineering and drug delivery. For example, Hanna Lee et al. reported a keratin biofilm derived from human hair, loaded with antibiotics, showing potential for use in periodontal tissue regeneration through implantation within a periodontal pocket.28 Similarly, Aluigi et al. documented a green-friendly synthesis of keratin nanoparticles loaded with Doxorubicin, which were used in vitro assays to inhibit the growth of model tumour cells. Additionally, Miaomiao Zhang et al. reported a PLCL nanofibrous/keratin hydrogel bilayer wound dressing, loaded with FGF-2, promoting skin wound healing. Furthermore, due to its physicochemical properties, keratin possesses numerous functional groups that can serve as binding sites for various molecules, making it suitable for adsorbing water pollutants. For example, a biocomposite based on keratin and graphene oxide with heavy metal adsorption capacity,29 and a keratin-based biomaterial with a high adsorption efficiency of the dye rhodamine B in aqueous acid media have been reported.30 In this line, our research group has already reported the synthesis of a pH-responsive keratin hydrogel, the enhancement of its mechanical properties through the incorporation of GO nanoparticles, and its use in water remediation, as well as oxidative treatments to produce pH and ion-responsive keratose hydrogels.31–33
Considering all of the above, the objective of this work is to study different reductive treatments and their effects on keratin hydrogels. To achieve this objective, keratin gels were treated with hydrazine sulphate and ascorbic acid in aqueous media and then the effectiveness of the reductive treatments was studied. The macroscopic properties of the obtained materials were studied by tensile tests while their physico-chemical properties were studied by spectroscopic techniques such as FTIR-ATR, Raman, 1H HRMAS NMR, 13C CP-MAS NMR, XPS, WAXS and SAXS. The changes in the crystallinity of the crystalline motifs of the keratin hydrogels before and after reductive treatments were also studied by XRD and their thermal properties were studied by TGA and DSC. Finally, the topography of the materials was studied by SEM and swelling tests were performed to check if the reductive treatment affected the previously reported pH-responsive swelling capacity of the hydrogels.
Experimental
Materials
Sodium hydroxide, hydrazine sulphate (HZN), ascorbic acid, tris, sodium acetate, K2HPO4, KH2PO4 were purchased from Anedra (Argentina). Ethanol 96% was acquired from Soria (Avellaneda, Argentina). Ethyl acetate was purchased from biopack (Argentina). Deuterated chloroform was acquired from Sigma-Aldrich (St. Louis, USA). Phosphoric acid 85% from Mallinckrodt (USA). All other reagents were of analytical grade. Whole cow's horns (Bos taurus, Hereford) were kindly donated by Frigorina (La Plata, Buenos Aires, Argentina) and used as keratin source.Preparation of keratin hydrogel (Ker)
Horns were milled and sieved through a 250 μm sieve. Then, the horn powder was washed three times with distilled water and three times with ethyl acetate to remove fat. After that, the powder was dried at 37 °C overnight. Keratin (1.2 g) powder was mixed with 4 ml dilution of NaOH 0.8 N in ethanol (7 ml) and it was left at 37.5 °C for 4 h. After that, the whole mixture was casted into silicon moulds of 6 × 12 cm and left until complete dryness at 37.5 °C. All samples were prepared likewise in order to ensure the obtaining of gels with similar thicknesses. This material was thoroughly washed with distilled water to remove all NaOH residue. After hydration, the hydrogel form of the material was obtained (Ker hydrogel). For assays that required a hydrated sample, immersion in the proper media was carried for 2 h to ensure uniform swelling.Reduction treatment of the keratin hydrogel
Reduced keratin hydrogels (Ker-HZN x%) were obtained by immersing the Ker hydrogel, previously hydrated, in different solutions of HZN (0.1%, 0.25%, 0.5% and 1%) and ascorbic acid 1% with a ratio of 1 g![[thin space (1/6-em)]](https://www.rsc.org/images/entities/char_2009.gif) :138 ml and left it overnight. The concentration range of the reducing agent was selected based on exploratory assays, which revealed a distinct behaviour within this range. Therefore, it was considered optimal for investigating the impact of reduction on keratin hydrogels. The different gels obtained were washed with several volumes of distilled water.
:138 ml and left it overnight. The concentration range of the reducing agent was selected based on exploratory assays, which revealed a distinct behaviour within this range. Therefore, it was considered optimal for investigating the impact of reduction on keratin hydrogels. The different gels obtained were washed with several volumes of distilled water.
Swelling assay
In order to analyse the swelling behaviour of the material after the reduction treatment, 0.05 g of a keratin hydrogel and Ker-HZN x% were submerged in distilled water at pH 7. After equilibrium was reached, the hydrogels were removed from the solution and accurately weighted. The swelling percentage was calculated as follows:| Sw% = (Swelled weight − Dry weight) × 100/Dry weight |
Spectroscopic characterization
ATR-FTIR (diamond attenuated total reflectance) and FT-Raman spectra of keratin and Ker-HZN x% hydrogels were recorded using a Nicolet iS50 advanced spectrometer (Thermo Scientific). ATR-FTIR spectra were acquired with 32 scans and a resolution of 4 cm−1. FT-Raman spectra were acquired with an excitation laser beam of 1064 nm, 0.5 W laser power, resolution of 4 cm−1, 200 scans. To avoid water related bands interference, all samples were previously dried for 24 h at 37.5 °C. For the analysis of the α-helix (1655 cm−1) and β-sheets (1679 cm−1) components, Amide I band (1690–1640 cm−1) was deconvoluted. Spectral baselines were set between 1750 and 1530 cm−1. Spectra decomposition was performed using Fityk software34 assuming a Voight shape for all the peaks.High-resolution magic angle spinning (HRMAS) NMR and solid-state NMR (ss-NMR) spectra were acquired with a Bruker Avance-III HD spectrometer equipped with a 14.1 T magnet operating at Larmor frequencies of 600.09 and 150.91 MHz for 1H and 13C, respectively. Hydrogel materials were studied by 1H HRMAS by packing the sample swelled with D2O into a 4 mm ZrO2 HRMAS rotor with a 50 μL spherical insert. The sample was spun at a magic angle spinning (MAS) rate of 4 kHz. A pre-saturation pulse was used for water-suppression in the 1H experiments. Chemical shifts for 1H (expressed in ppm) are relative to (CH3)4Si. The powdered samples (dry samples) were packed into 3.2 mm ZrO2 rotors and rotated at room temperature at a MAS rate of 15 kHz. 13C CP-MAS (cross-polarization and magic angle spinning) experiments were done in a 3.2 mm MAS probe. Glycine was used as an external reference compound for the recording of the 13C spectra and to set the Hartmann−Hahn matching condition in the CP-MAS experiments in 13C spectra. The contact time during CP was 2 ms. The SPINAL64 sequence (small phase incremental alternation with 64 steps)35 was used for heteronuclear decoupling during acquisition.
The X-ray photoemission spectroscopy data were acquired with a Phoibos 150 (SPECS GmbH) electron energy analyser using monochromatic Al-Kα photons (1486.6 eV). Electrons were collected along the normal to the surface; all the spectra were measured with a pass energy of 30 eV, which results in an estimated energy resolution (analyser plus X-rays) of 0.5 eV. The zero of the binding energy (BE) scale was set by placing the C 1s peak due to adventitious hydrocarbon present on the surface at 285 eV. Wide-scan spectra and detailed S 2p, C 1s, N 1s, and O 1s spectra were measured for each analysed sample. The measured core-level spectra were fitted with Voigt profiles, with a Lorentzian width of 0.2 eV. In the particular case of the S 2p photoemission spectra, each component was described by one S 2p(3/2) and S 2p(1/2) doublet (spin–orbit splitting of 1.18 eV, fixed area ratio 0.5, Voigt peaks of the same shape). The entire set of spectra was adjusted recursively to find the best fit.
The powder X-ray diffractograms of the materials were collected on a PANalytical X′ Pert diffractometer using Cu Kα radiation and a power of 45 kV 40 mA in Bragg configuration (2θ range = 5–50°). Small angle X-ray scattering/wide angle X-ray scattering (SAXS/WAXS) measurements were performed using a XEUSS 2.0 (from XENOCS, France) equipment at the Laboratorio de Cristalografía Aplicada, Instituto de Tecnologias Emergentes y Ciencias Aplicadas (ITECA), EcyT-UNSAM-CONICET, Argentina. Patterns were registered with two synchronous 2D photon counting pixel X-ray detectors for SAXS Pilatus 200k (DECTRIS, Switzerland) and a Pilatus 100k (DECTRIS, Switzerland) placed near the sample with a tilted angle of 36° for WAXS. The scattering intensity, I(q), was recorded in the range of the momentum transfer q, where q = 4π/λ![[thin space (1/6-em)]](https://www.rsc.org/images/entities/i_char_2009.gif) sin(θ), 2θ is the scattering angle and λ = 0.15419 nm is the weighted average of X-ray wavelength of the Cu-Kα12 emission lines. Due to the small beam size at the sample, smearing effects were not considered, with pilatus detector reading background is negligible. All SAXS patterns analysis models are presented in ESI.† (ref. 36 and 37)
sin(θ), 2θ is the scattering angle and λ = 0.15419 nm is the weighted average of X-ray wavelength of the Cu-Kα12 emission lines. Due to the small beam size at the sample, smearing effects were not considered, with pilatus detector reading background is negligible. All SAXS patterns analysis models are presented in ESI.† (ref. 36 and 37)
Microscopic characterization
Electron microscopy images were obtained on a Philips 505 scanning electron microscope (SEM). The hydrogels swelled at neutral pH were freeze-dried and coated with a thin layer of gold before observation.Thermal analysis
Thermogravimetric (TGA) analysis was carried out using a TGA Q600 TA device. Approximately 0.010 g of a dried sample, weighed in an alumina container, was heated at 5 °C min−1 in the temperature range 25–700 °C under nitrogen atmosphere. Differential scanning calorimetry (DSC) was performed using a DSC Q200 instrument. Approximately 0.010 g of a dried sample, weighed in an aluminium container, were tested from −50 °C to 260 °C at a heating rate of 10 °C min−1, under nitrogen flow.Tensile properties
Tensile tests were carried out in an Instron dynamometer model 5985, using a crosshead speed of 5 mm min−1 and a load cell of 0.1 kN. Tensile strength (σm), Young's modulus (E), and elongation at break (εB) were obtained, by following ASTM D1708 – 18 standard recommendations. Hydrated samples as detailed above were removed from the liquid media right before each measurement.Cytotoxicity assay
HT-1080 (ATCC CCL-121) human epithelial cells derived from a Fibrosarcoma connective tissue and human immortalized keratinocyte cell line HaCaT were grown and maintained in DMEM/F12 medium supplemented with 10% inactivated fetal bovine serum.Reduced keratin hydrogels were cut into small pieces and sterilized by alcohol 70% treatment and 1 hour of UV radiation (30 minutes each side). Then, they were submerged in culture medium and incubated during 24 h at 37 °C. Aliquots of the extracts were harvested and tested in the cytotoxicity assay. Cytotoxicity of reduced keratin hydrogels was evaluated on HT1080 and HaCaT cells. Cells seeded in 96 microwell plates were treated with the extracts of keratin hydrogels and incubated during 24 h at 37 °C. Cell viability was determined, as previously reported, by the MTT assay.38 The absorbance of each well was measured on a microplate reader using a test wavelength of 595 nm. Results were plotted as percent absorbance of treated cells with respect to untreated cells. Percentages of viability above 80% were considered non-toxic.
Statistics and graphics
All experiments and their measurements were made in triplicate under identical conditions and statistically analysed by one-way ANOVA. R language environment,39 Minitab 17.1 and Origin 2019B were used for statistical computing and graphics. Molecular representations were drawn using Chemaxon Marvin software (https://chemaxon.com/products/marvin).Results and discussion
Macroscopical and microscopical characterization
The aim was to examine the effect of different reducing reagents on keratin hydrogels. After the reductive treatments with HZN or ascorbic acid, the hydrogel shrinks from its initial volume, but presents a remarkable increase of flexibility with respect to the Ker hydrogel, which is swelled but brittle, as can be seen in Videos S1 and S2 (ESI†) and Fig. 1a. Treatment with HZN at 0.5% and 1% yield hydrogels with similar characteristics (low swelling and noticeable flexibility), whereas treatment with concentrations of 0.1% and 0.25% result in hydrogels with reduced swelling and limited flexibility. On the other hand, treatment with 1% ascorbic acid shows a material with characteristics intermediate between those observed in Ker hydrogel and those treated with HZN, showing increased flexibility with a loss of swelling but still some brittleness. | ||
| Fig. 1 (a) macroscopic properties of Ker-HZN hydrogel (b) SEM images of Ker hydrogel reduced with different concentrations of HZN. | ||
From the microscopic point of view, Fig. 1b shows the SEM images of Ker hydrogel and Ker hydrogel reduced at different concentrations of HZN. It could be observed that the Ker hydrogel has the typical porous structure described in previous works (Peralta Ramos et al., Galaburri et al.).31,32 Furthermore, as the concentration of HZN increases, so does the degree of contraction of the porous structure. These results are in agreement with the loss of swelling of the material and could be probably due to the low pH of the HZN solution and, as below demonstrated, the interaction of the sulphate ions with the polypeptide structure leading to its collapse which hinders the water molecules from entering the pores.
The mechanical properties show to be greatly affected by the HZN treatment. As can be seen in Videos S1 and S2 (ESI†), the Ker hydrogel exhibited high brittleness, whereas treated samples are much more deformable. The Ker hydrogels mechanical response, as well as the water drain due to the compression of the clamps, make these samples unsuitable for tensile tests. Although ascorbic acid treated hydrogels show to gain flexibility, still are brittle enough to make the samples unsuitable for tensile tests. Fig. S1 (ESI†) shows the strain–stress behaviour of the hydrogels treated with HZN, characterised by an initial linear region followed by a non-linear tensile behaviour up to the maximum and the subsequent gradual drop of load to zero. Tensile parameters values are highly variable among samples, and no clear trend or pattern is found regarding the behaviour of the sample as the concentration of HZN is increased. This strain–stress behaviour could be associated with the thickness of the samples (around 0.5 mm) and their high water content which is above 50%.
Spectroscopic characterization of the keratin reduction
With the aim of studying the changes suffered by the keratin hydrogel in the reduction process, FTIR-ATR, Raman, XPS and NMR spectroscopies were employed. In Fig. 2a and Fig. S2 (ESI†) (full spectra) it can be seen the typical bands corresponding to a keratin FT-IR spectrum: stretching vibration of N–H and O–H bonds corresponding to amide A (3300–3200 cm−1, not shown), stretching vibration of C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) O groups corresponding to amide I (1632 cm−1), bending vibration of N–H bond corresponding to amide II (1514 cm−1) and stretching vibration of C–N bond corresponding to amide III (1239 cm−1).40,41 In addition, it can be appreciated the appearance and increase of a broad band around 1085 cm−1 with the increasing of HZN concentration in the treatment, probably corresponding to the presence of remaining SO42− (SO stretching).42 A sulphate related signal can also be detected in the Raman spectra of the treated samples as the appearance and increase of a peak at 975 cm−1 (Fig. 2b and Fig. S3 (full spectra), ESI†) along with the HZN level increase.43 In the IR spectra of the samples (Fig. 2a), it can also be observed the partial or total disappearance of the band around 1390 cm−1 that could be related to keratin Glu and Asp residues (symmetric stretching vibration of COO− groups) promoted by the acid environment (treatment with HZN), and the appearance of a shoulder beside the amide I peak, related to the stretching vibration of protonated COOH groups of Glu and Asp residues due to the COO− protonation.44,45
O groups corresponding to amide I (1632 cm−1), bending vibration of N–H bond corresponding to amide II (1514 cm−1) and stretching vibration of C–N bond corresponding to amide III (1239 cm−1).40,41 In addition, it can be appreciated the appearance and increase of a broad band around 1085 cm−1 with the increasing of HZN concentration in the treatment, probably corresponding to the presence of remaining SO42− (SO stretching).42 A sulphate related signal can also be detected in the Raman spectra of the treated samples as the appearance and increase of a peak at 975 cm−1 (Fig. 2b and Fig. S3 (full spectra), ESI†) along with the HZN level increase.43 In the IR spectra of the samples (Fig. 2a), it can also be observed the partial or total disappearance of the band around 1390 cm−1 that could be related to keratin Glu and Asp residues (symmetric stretching vibration of COO− groups) promoted by the acid environment (treatment with HZN), and the appearance of a shoulder beside the amide I peak, related to the stretching vibration of protonated COOH groups of Glu and Asp residues due to the COO− protonation.44,45
 | ||
| Fig. 2 (a) and (c) FT-IR and (b) and (d) Raman spectra of Ker, Ker-HZN x% hydrogels and ascorbic acid and phosphate buffer treatments. | ||
The ascorbic acid 1% treatment (Fig. 2c) leads to similar changes than the treatment with HZN. It is also observed a decrease of the relative intensity of the band at 1390 cm−1 (Glu and Asp residues) and the appearance of a shoulder beside the amide I peak. The difference between both spectra (treatment with ascorbic acid and HZN) can be related to the absence of sulphate ions and an incomplete protonation of Glu and Asp residues by the ascorbic acid due to similar pKa (4.7, 4.5 and 4.05 respectively)46 while the strong acidity of HZN produces the complete protonation of these residues. In the Raman spectra (Fig. 2d) it could not be seen any significant changes in comparison with the keratin hydrogel spectra. These could imply that both reduction treatments lead to similar chemical modifications (latter supported by NMR and XPS).
For the analysis of the effect of the hydrazine treatment on the keratin secondary structure, the amide I band from the Raman spectra is deconvoluted (Fig. S4, ESI†). In order to simplify the analysis, only the major components of amide I peak (α-helix and β-sheet) are compared.47,48 As the concentration of hydrazine increases, it can be observed a decrease of the α-helix component in relation with the β-sheet component, and this could be related with a conformational change induced by the reduction and the presence of remaining sulphate ions.
In the 13C ss-NMR spectra (Fig. S5a, ESI†), in the range between 35 and 10 ppm overlapped signals are observed for keratin hydrogel and hydrogel treated with 1% HZN, which corresponded to the alkyl components of the side chains, along with the α carbon region around 54 ppm. The signal of β carbons from leucine and cysteine residues forming disulfide bridges can be observed as a peak at 40 ppm. Additionally, the aromatic carbon region between 160 and 130 ppm was distinguishable, as well as a peak at 174 ppm corresponding to the carbonyl region.49–51 Comparing both spectra, it can be observed that the HZN treatment results in a loss of the shoulder in the carbonyl peak and a shift in its maximum from 174 ppm to 172 ppm (Fig. 3a). This can be interpreted as a conformational change in the protein, with a transition from α-helix structures to β-sheet and random coil structures.52,53 This observation is consistent with the results obtained from Raman spectroscopy deconvolution.
 | ||
| Fig. 3 (a) Solid-state 13C CP-MAS NMR spectra of carbonyl zone, XPS S 2p spectra of (a) Ker, Ker-HZN x% hydrogels and (c) ascorbic acid and phosphate buffer treatments. | ||
On the other hand, in the 1H HRMAS NMR spectra (Fig. S5b, ESI†), on the alkyl zone, the only noticeable difference between the keratin hydrogel and those treated with different concentrations of HZN is the loss of signal intensity in the samples starting from 0.25% HZN concentration. This can be attributed to the reduction in swelling of the structure, leading to a restriction in the mobility of the protein chains and consequently resulting in shorter relaxation times and a decrease in signal intensity in the spectrum.
Up to this point, as observed in IR and Raman spectroscopies, aside for the addition of sulphate ions, NMR spectra show that the main chemical structure of keratin remains unaltered, and the main changes produced by the reductive treatment are reflected in conformational reorganization of the polypeptide chain.
In order to study the surface components and the oxidation state of the keratin groups before and after the reduction treatment, an XPS analysis was performed. A typical wide-scan spectrum of keratin hydrogel and the detail of each main component (C 1s, O 1s, N 1s, S 2p) are shown in ESI† (Fig. S6–S8, ESI†). By the deconvolution of S 2p spectra, two oxidation states can be assigned (Table S1, ESI†): 163.7 and 168.2 eV corresponding to the S atoms in S–S or S–H bonds, and S atoms in S–O bonds respectively (Fig. 3b).32,54 After the hydrogel synthesis, the Ker spectrum shows an increase in the relative intensity of the peak corresponding to the SO groups, which could be interpreted as the oxidation of the –SH moieties present in the pristine keratin55 (Table S1, ESI†). After the treatment with the lowest concentration of HZN (0.1%), the relative intensity of the S–O peak decreases (without changing the total sulphur concentration), due to a reduction of these groups this leads to an increase of the 163.7 eV peak. In contrast, when using the higher concentrations of HZN (0.25–1%) the spectra show an increase of the relative intensity of the S–O peak with an increase of the total sulphur concentration. This supports the IR and Raman discussion regarding the presence of remaining SO42− within the keratin structure. In comparison, the treatment with ascorbic acid 1% produces a decrease of the relative intensity of the S–O peak without changing the total sulphur concentration (Table S1, ESI†) which could be attributed entirely to the reduction of the sulfenic and sulfinic groups present in the keratin hydrogel. This confirms the effect of the reduction treatments on the redox state of the sulphur containing moieties.
Thermal analysis
The thermal behaviour of keratin hydrogel and keratin hydrogel treated with different concentrations of HZN was studied by DSC, TGA and DTA (Fig. 4 and Fig. S9, ESI†). The TGA shows two main weight loss processes for all samples. The first one below 150 °C could be attributed to the desorption of water molecules from the structure (Fig. 4a).56 The second weight loss occurs between 200 °C and 500 °C, and it is probably due to a complex process of degradation of the keratin structure (cleavage of disulphide bonds and peptide chain, crosslinking reaction, formation and degradation of aromatic carbons and cyclic amides).57 The only difference between the TGA curves is a decreasing tendency for the remaining residue of the samples as the hydrazine concentration increases. This could be related to the fact that since SO42− ions are completely eliminated as SO2 gas. Thus, the higher the SO42− content, the lower the percentage of remaining residue is. On the DTA (Fig. S9, ESI†) we can observe a change in the degradation pattern of the samples treated with the highest concentrations of HZN, such as an increase in the peak below 300 °C, (similar intensity for ker-HZN 0.25 y 0.5%, and higher intensity for Ker-HZN 1%) related to the formation of inorganic sulphur compounds.58 This could be due to the reduction of sulfonic, sulfenic and sulfinic groups to –SH and the interaction of the sulphate salt with the keratin structure, as it is suggested from the spectroscopic results. | ||
| Fig. 4 (a) TGA, (b) DSC thermograms of Ker, Ker-HZN x% hydrogels. | ||
On the DSC thermogram (Fig. 4b) there are two endothermic events, one between 30 and 150 °C that could be attributed to the evaporation of water molecules (Tw). The second one, between 210 and 250 °C, could be due to the melting transition (Tm) related to the denaturalization of the α-helix structure.31,59 The values of Tw, Tm and enthalpies associated are shown in Table 1. The decrease in Tw values may be due to the contraction of the protein structure caused by the reductive treatment, which reduces the amount of water molecules retained in the structure. This is supported by the SAXS profiles. Furthermore, the decrease in Tm as the concentration of HZN used increases may be attributed to the fact that this treatment reduces the proportion of α-helix structures in keratin.60 This is consistent with the results obtained from NMR and Raman spectroscopy.
| Samples | T w (°C) | ΔH (J g−1) | T m (°C) | ΔH (J g−1) | n | R g (Å) |
|---|---|---|---|---|---|---|
| Ker hydrogel | 80 | 192 | 234 | 6.2 | 2.5 | 29.3 |
| Ker-HZN 0.1% | 86 | 159 | 230 | 9.3 | 2.2 | 53.1 |
| Ker-HZN 0.25% | 72 | 210 | 226 | 3.3 | 3.1 | 17.8 |
| Ker-HZN 0.5% | 70 | 228 | 226 | 3.5 | 3.0 | 18.9 |
| Ker-HZN 1% | 72 | 225 | 225 | 4.7 | 3.2 | 18.0 |
X-ray diffraction, wide angle and small angle X-ray scattering
In order to analyse whether the treatment with HZN alters the crystalline structure of the keratin hydrogel, XRD patterns were collected. In Fig. S10 (ESI†), it can be observed that all samples show the two typical peaks of keratin (∼5–13 2θ for the α-helix domains and ∼16–27 for the β-sheet domains).61,62 It can be seen that the synthesis of the hydrogel reduces the relative intensity of the α-helix peak in comparison with the β-sheet peak, in relation to the ones observed for the keratin powder, possibly due to a loss of crystallinity in the structure caused by basic partial hydrolysis. On the other hand, the treatment with HZN does not alter the relative intensities of those peaks. After comparing this data with the one from Raman and 13C ss-NMR, it can be concluded that the conformational changes endowed by the HZN treatment could be mainly due to an effect in more flexible regions of the polypeptide chain, such as disordered domains and not exclusively to alterations on the α-helix and/or β-sheet domains.WAXS studies (Fig. S11, ESI†) were conducted on the hydrated samples to further analyse the ultrastructure of the reduced hydrogels. As a consequence of hydration, a major halo is observed for all samples centred above 25°, the dominant contribution in the pattern of sample Ker-HZN 0.1%. In the other patterns, the peak related to the β-sheet distance is broadened, and an additional diffraction peak appears that shifts between samples, indicating a larger characteristic distance for samples Ker-HZN 0.25 y 0.5% (≈5.2 Å) respect to Ker-HZN 1% (≈4.4 Å) and Ker (≈4.1 Å).
SAXS patterns of the dry samples are also shown in Fig. 5a as grey dots attached to its corresponding swollen state pattern. In all cases, the low q region exhibits a slope close to −4, associated with collapsed structures as a consequence of the drying process. All SAXS patterns of the swollen samples (Fig. 5a) were fitted employing a combination of a power-law to account for the slope of the low q region, and a constrained Guinier–Porod (GP) model for the “bumps” observed in the mid-to-high q range. This approach is equivalent to the two-stage model recently reported in the literature, focused on the study of the swelling of the nanofiber networks of similar systems.63,64 According to this model, the exponent of the first power-law is related to the complexity of the network, while the radii of gyration are linked to the mean pore size. In this work, the constraints applied to the GP model include fixing the parameter related to the symmetry of the scatterer objects and the Porod slope, assuming as a first approximation spherical pores of rather rough surfaces.
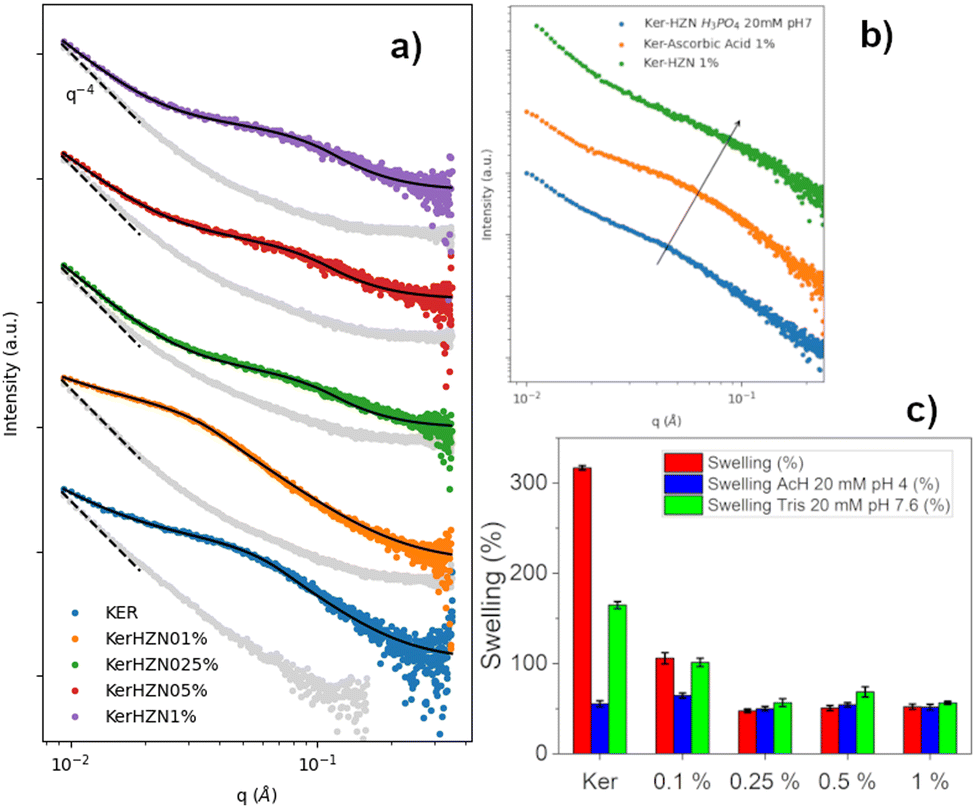 | ||
| Fig. 5 (a) SAXS profiles of Ker, Ker-HZN x% hydrogels, (b) ascorbic acid and phosphate buffer treatments and (c) swelling of Ker, Ker-HZN x% hydrogels. | ||
All in all, the results indicate that treatments using ≥0.25% lead to similar materials, different from the Ker hydrogel. Moreover, the treatment with 0.1% HZN showed a distinctive pattern (Fig. 5a). Therefore, it can be proposed that two effects are produced by HZN. On one side, when 0.1% HZN is used, the reduction of oxidised cysteines (i.e. sulfenic, sulfinic groups) lead to a change in the polypeptide chain interaction from the Ker hydrogel. When higher levels of HZN are employed, along with the effect of the reduced moieties, the effect of residual sulphates lead to the interchain closeness of the more mobile domains acting as physical crosslinkers leading to compressed structures with narrower pores.
As can be seen in Table 1, for the higher HZN concentration (≥0.25%) the n values close to 3 indicate that the network is clustered, leading to smaller mean pore sizes, with no significant differences in the obtained values when varying the HZN percentage. On the other hand, for pure keratin and the lower HZN level (=0.1%), the network exhibits “greater openness”, leading to lower n values and higher pore sizes. This is also in line with 1H-HRMAS NMR and XPS results. The 0.1% HZN treatment achieves a similar extent of reduction as the higher HZN treatments, but the resultant structure is loose enough to allow swelling and a particular conformational organisation. This contrasts with the higher HZN levels (≥0.25%) that would lead to a residual remaining of sulphate ions in the material (seen by XPS) which leads to the contraction of the network and swelling decay (seen by 1H-HRMAS NMR and SAXS).
Influence of reduction and remaining sulphates
In order to understand if the reduction process and the remaining sulphate ions interacting with the keratin structure (detected by FTIR-ATR, Raman and XPS) influence on the hydrogel characteristics, the stability of the reduced hydrogels was assayed by incubating them in phosphate buffer 20 mM pH 7, and the influence of the reduction process was assessed by a treatment with ascorbic acid 1%. Macroscopically, after the incubation with phosphate buffer it was seen that the hydrogel lost its flexibility becoming brittle, similarly to the Ker hydrogel. Also, on the FTIR-ATR and Raman spectra (Fig. 2c and d) it could be observed the disappearance of the band at 1085 cm−1 and the peak at 975 cm−1 respectively (SO stretching). This could be related to the displacement of SO42− ions by the phosphate ions. In addition, this is supported by the XPS spectra (Fig. 3c and Table S1, ESI†). After the incubation of the Ker-HZN 1% sample with phosphate buffer, the relative intensity of S–O peak decreases along with the total concentration of sulphur. Regarding the nanostructure of the hydrogels, in Fig. 5b, it can be seen that the ascorbic acid treatment on Ker hydrogels and the phosphate buffer treatment on Ker-HZN 1% hydrogels lead to similar SAXS profiles highlighting the influence of the sulphate ions on the structuring of the macromolecules. This suggests that the flexible properties of the hydrogel could be related not only to the reduction of the hydrogel but also to the interaction of the sulphate ions with the keratin structure.
Swelling studies
Subsequently, swelling studies were conducted in order to confirm whether the swelling of hydrogels treated with different concentrations of HZN maintained its selective response to pH changes. As can be observed in Fig. 5c, although the hydrogel treated with 0.1% HZN exhibits lower swelling compared to the untreated hydrogel (p < 0.05), it still shows significant swelling variations in its response to pH changes (p < 0.05), resembling the Ker hydrogel. However, hydrogels treated with concentrations of HZN higher than 0.25% lose their ability to change their swelling in response to pH changes presenting no significant differences (p > 0.05). This may be due to the reduction of sulfenic/sulfinic groups in the cysteines of the keratin hydrogel, in addition to the physical crosslinking effect of the present sulphate groups, which decreases the mobility of the protein chains in the keratin structure, preventing water diffusion throughout the pH range studied. Consequently, hydrogels treated with higher concentrations of HZN do not selectively respond to pH. In contrast, hydrogels treated with low concentrations of HZN (0.1%), where only the effect of reducing sulfenic/sulfinic groups is evident, exhibit a loss of swelling compared to the untreated hydrogel, but the selective pH response still prevails. Interestingly, the ascorbic acid reduced hydrogels show swelling of 64 ± 3% similar to the hydrogels treated with concentrations of HZN higher than 0.25%. This also implies that not only contraction of the network but also the presence of remaining sulphates is essential to the overall protein conformation and resulting mechanical behaviour. As a summary of the overall treatment, Fig. 6 shows a representation of the reduction reaction and remaining sulphates.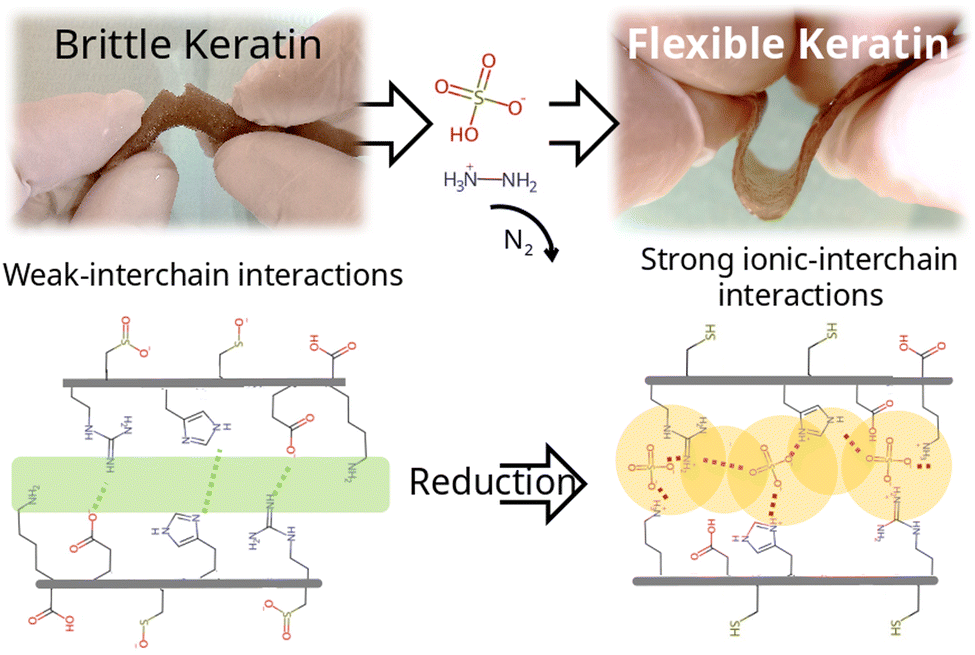 | ||
| Fig. 6 Schematic representation of the reduction reaction and remaining sulphates. | ||
Cytotoxicity assays
In order to verify the biocompatibility of the Ker hydrogels after reductive treatment with hydrazine sulphate, the cytotoxicity of the materials was evaluated by means of the MTT test using 2 types of cell lines, HT1080 (fibroblasts) and HaCat (keratinocytes). In the case of the HT1080 line, there are no significant differences between the control cells and those incubated with Ker hydrogel extracts and those treated with different concentrations of HZN (Fig. S12, ESI†) (Anova + Dunnett test p < 0.05). This behaviour is similar to the HaCaT line, where all treatments show no significant differences to the control cells (Anova + Dunnett test p < 0.05) aside for the ones exposed to Ker-HZN 0.5 and 1% extracts. The cells exposed to 0.5% extracts exhibited lower viability, while those exposed to Ker-HZN 1% demonstrated higher viability than the control, despite being subjected to a higher level of hydrazine during the reduction process. These findings suggest that slight variations among the hydrogel samples may exist, but overall, the HZN-treated hydrogels are not toxic to either cell line (Fig. S13, ESI†).Conclusions
In this study, we successfully synthesised flexible keratin hydrogels through reductive treatments, unveiling a previously unreported behaviour in protein materials. This discovery opens the door to a wider range of potential applications for these hydrogels. The keratin hydrogels were successfully characterised after reductive treatments with hydrazine sulphate and ascorbic acid. The use of both reducing agents lead to the production of hydrogels with a significant increase in flexibility, which can be attributed in part to the reduction of cysteines derived aminoacids (XPS) and a conformational change in the more mobile domains of the protein chains (Raman, ss-NMR, DSC, XRD). On the other hand, the enhanced flexibility is accompanied by a notable reduction in swelling and its response to pH changes compared to the untreated hydrogel, likely due to the crosslinking effect of the remaining sulphate groups in the protein structure (FTIR, Raman, 1H HRMAS NMR, XPS). The presence of sulphate groups resulted in a more compact structure with smaller pores, where the movement of protein chains and water diffusion within the hydrogel are impeded (SEM, swelling, 1H HRMAS NMR, SAXS). Finally, it was observed that the crosslinking effect of sulphate ions is reversible with the treatment of a phosphate buffer at neutral pH, leading to a hydrogel with macro and microscopic characteristics similar to the original keratin hydrogel (SAXS, XPS, FTIR, Raman). The cytotoxicity assays showed that the reduced hydrogels are biocompatible, which extend the applications of these materials to the biomedical field, in example as flexible scaffolds. When comparing treatments, it could be asserted that Ker-HZN 0.5% requires the least amount of reducing agent to achieve similar properties, making it the preferred option for a potential application.Data availability
The authors confirm that the data supporting the findings of this study are available within the article, its ESI,† and raw data is available at https://doi.org/10.5281/zenodo.14019374.Conflicts of interest
There are no conflicts to declare.Acknowledgements
M. L. P. R. is grateful for his doctoral fellowship granted by Universidad de Buenos Aires. The authors would like to acknowledge INTI Mecánica and M. Pianetti for their assistance in SEM analysis. This work was supported by grants from Universidad de Buenos Aires (UBACyT 20020170100125BA) and Agencia Nacional de Promoción Científica y Tecnológica (PICT 2018-01731). The MSCA-RISE-2017 (No 778412) INDESMOF project, which received funding from the European Union's Horizon 2020 research and innovation programme is also acknowledged. F. B. and M. C. acknowledge support from the Project CH4.0 under the MUR program “Dipartimenti di Eccellenza 2023–2027” (CUP: D13C22003520001).References
- A. A. Adeniran and W. Shakantu, The Health and Environmental Impact of Plastic Waste Disposal in South African Townships, Int. J. Environ. Res. Public Health, 2022, 19, 779 CrossRef CAS PubMed.
- N. A. Welden, Plastic Waste and Recycling, Elsevier, 2020, pp. 195–222 Search PubMed.
- L. Liu, M. Xu, Y. Ye and B. Zhang, On the degradation of (micro)plastics, Sci. Total Environ., 2022, 806, 151312 CrossRef CAS PubMed.
- G. Crini, Recent developments in polysaccharide-based materials used as adsorbents in wastewater treatment, Prog. Polym. Sci., 2005, 30, 38–70 CrossRef CAS.
- J. A. M. Ramshaw, J. A. Werkmeister and V. Glattauer, Collagen-based Biomaterials, Biotechnol. Genet. Eng. Rev., 1996, 13, 335–382 CrossRef CAS PubMed.
- M. Kostag and O. A. El Seoud, Sustainable biomaterials based on cellulose, chitin and chitosan composites – A review, Carbohydr. Polym. Technol. Appl., 2021, 2, 100079 CAS.
- B. Aaliya, K. V. Sunooj and M. Lackner, Biopolymer composites, Int. J. Biobased Plast., 2021, 3, 40–84 CrossRef CAS.
- F. Costa, R. Silva and A. R. Boccaccini, Peptides and Proteins as Biomaterials for Tissue Regeneration and Repair, Elsevier, 2018, pp. 175–204 Search PubMed.
- J. Liu, L. Sun, W. Xu, Q. Wang, S. Yu and J. Sun, Current advances and future perspectives of 3D printing natural-derived biopolymers, Carbohydr. Polym., 2019, 207, 297–316 CrossRef CAS PubMed.
- S. Sharma, V. Gupta and D. Mudgal, Current trends, applications, and challenges of coatings on additive manufacturing based biopolymers, Polym. Compos., 2022, 43, 6749–6781 CrossRef CAS.
- A. K. Shrestha and P. J. Halley, Starch Polymers, Elsevier, 2014, pp. 105–143 Search PubMed.
- V. G. Muir and J. A. Burdick, Chemically Modified Biopolymers for the Formation of Biomedical Hydrogels, Chem. Rev., 2021, 121, 10908–10949 CrossRef CAS PubMed.
- M. Haroon, L. Wang, H. Yu, N. M. Abbasi, Z.-A. Zain-ul-Abdin, M. Saleem, R. U. Khan, R. S. Ullah, Q. Chen and J. Wu, Chemical modification of starch and its application as an adsorbent material, RSC Adv., 2016, 6, 78264–78285 RSC.
- Q. Zhang, G. Shan, P. Cao, J. He, Z. Lin, Y. Huang and N. Ao, Mechanical and biological properties of oxidized horn keratin, Mater. Sci. Eng., C, 2015, 47, 123–134 CrossRef CAS.
- A. Kuzuhara, Analysis of structural change in keratin fibers resulting from chemical treatments using Raman spectroscopy, Biopolymers, 2005, 77, 335–344 CrossRef CAS PubMed.
- J. Li, J.-F. Revol and R. H. Marchessault, Effect of degree of deacetylation of chitin on the properties of chitin crystallites, J. Appl. Polym. Sci., 1997, 65, 373–380 CrossRef CAS.
- S. Berlioz, S. Molina-Boisseau, Y. Nishiyama and L. Heux, Gas-Phase Surface Esterification of Cellulose Microfibrils and Whiskers, Biomacromolecules, 2009, 10, 2144–2151 CrossRef CAS PubMed.
- S. Chattopadhyay and R. T. Raines, Collagen-based biomaterials for wound healing, Biopolymers, 2014, 101, 821–833 CrossRef CAS PubMed.
- F. J. O’Brien, Biomaterials & scaffolds for tissue engineering, Mater. Today, 2011, 14, 88–95 CrossRef.
- A. M. Ferreira, P. Gentile, V. Chiono and G. Ciardelli, Collagen for bone tissue regeneration, Acta Biomater., 2012, 8, 3191–3200 CrossRef CAS PubMed.
- N. Li, D. Qiao, S. Zhao, Q. Lin, B. Zhang and F. Xie, 3D printing to innovate biopolymer materials for demanding applications, Mater. Today Chem., 2021, 20, 100459 CrossRef CAS.
- R. M. D. Soares, N. M. Siqueira, M. P. Prabhakaram and S. Ramakrishna, Electrospinning and electrospray of bio-based and natural polymers for biomaterials development, Mater. Sci. Eng., C, 2018, 92, 969–982 CrossRef CAS PubMed.
- M. Meyer, Processing of collagen based biomaterials and the resulting materials properties, Biomed. Eng. OnLine, 2019, 18, 24 CrossRef PubMed.
- Biology of the Integument 2 Vertebrates, ed. J. Bereiter-Hahn, A. G. Matoltsy and K. S. Richards, Springer Berlin, Berlin, Softcover reprint of the original 1986 1st edn, 2014 Search PubMed.
- J. McKittrick, P.-Y. Chen, S. G. Bodde, W. Yang, E. E. Novitskaya and M. A. Meyers, The Structure, Functions, and Mechanical Properties of Keratin, JOM, 2012, 64, 449–468 CrossRef.
- in Keratin as a Protein Biopolymer: Extraction from Waste Biomass and Applications, ed. S. Sharma and A. Kumar, Springer International Publishing, Cham, 2019 Search PubMed.
- Y. Zhang, R. Yang and W. Zhao, Improving Digestibility of Feather Meal by Steam Flash Explosion, J. Agric. Food Chem., 2014, 62, 2745–2751 CrossRef CAS PubMed.
- H. Lee, Y.-S. Hwang, H.-S. Lee, S. Choi, S. Y. Kim, J.-H. Moon, J. H. Kim, K. C. Kim, D.-W. Han, H.-J. Park and H. Bae, Human hair keratin-based biofilm for potent application to periodontal tissue regeneration, Macromol. Res., 2015, 23, 300–308 CrossRef CAS.
- M. Zubair, M. S. Roopesh and A. Ullah, Nano-modified feather keratin derived green and sustainable biosorbents for the remediation of heavy metals from synthetic wastewater, Chemosphere, 2022, 308, 136339 CrossRef CAS PubMed.
- N. D. Tissera, R. N. Wijesena, H. Yasasri, K. M. N. De Silva and R. M. De Silva, Fibrous keratin protein bio micro structure for efficient removal of hazardous dye waste from water, Mater. Chem. Phys., 2020, 246, 122790 CrossRef CAS.
- M. L. Peralta Ramos, J. A. González, L. Fabian, C. J. Pérez, M. E. Villanueva and G. J. Copello, Sustainable and smart keratin hydrogel with pH-sensitive swelling and enhanced mechanical properties, Mater. Sci. Eng., C, 2017, 78, 619–626 CrossRef CAS PubMed.
- G. Galaburri, M. L. Peralta Ramos, J. M. Lázaro-Martínez, R. Fernández de Luis, M. I. Arriortua, M. E. Villanueva and G. J. Copello, pH and ion-selective swelling behaviour of keratin and keratose 3D hydrogels, Eur. Polym. J., 2019, 118, 1–9 CrossRef CAS.
- M. L. Peralta Ramos, G. Galaburri, J. A. González, C. J. Pérez, M. E. Villanueva and G. J. Copello, Influence of GO reinforcement on keratin based smart hydrogel and its application for emerging pollutants removal, J. Environ. Chem. Eng., 2018, 6, 7021–7028 CrossRef CAS.
- M. Wojdyr, Fityk, J. Appl. Crystallogr., 2010, 43, 1126–1128 CrossRef CAS.
- B. M. Fung, A. K. Khitrin and K. Ermolaev, An Improved Broadband Decoupling Sequence for Liquid Crystals and Solids, J. Magn. Reson., 2000, 142, 97–101 CrossRef CAS PubMed.
- guinier_porod—SasView 5.0.5 documentation, https://www.sasview.org/docs/user/models/guinier_porod.html Search PubMed.
- B. Hammouda, Analysis of the Beaucage model, J. Appl. Crystallogr., 2010, 43, 1474–1478 CrossRef CAS.
- F. Denizot and R. Lang, Rapid colorimetric assay for cell growth and survival, J. Immunol. Methods, 1986, 89, 271–277 CrossRef CAS PubMed.
- R: The R Project for Statistical Computing, https://www.r-project.org/ Search PubMed.
- M. Zoccola, A. Aluigi and C. Tonin, Characterisation of keratin biomass from butchery and wool industry wastes, J. Mol. Struct., 2009, 938, 35–40 CrossRef CAS.
- A. Darvishi, H. Bakhshi and A. Heydari, Innovative application of magnetically modified bovine horn as a natural keratin resource in the role of value-added organocatalyst, RSC Adv., 2022, 12, 16535–16543 RSC.
- B. Fernández-d’Arlas, Improved aqueous solubility and stability of wool and feather proteins by reactive-extraction with H2O2 as bisulfide (S S) splitting agent, Eur. Polym. J., 2018, 103, 187–197 CrossRef.
- K. Ben Mabrouk, T. H. Kauffmann, H. Aroui and M. D. Fontana, Raman study of cation effect on sulfate vibration modes in solid state and in aqueous solutions, J. Raman Spectrosc., 2013, 44, 1603–1608 CrossRef CAS.
- A. Barth, The infrared absorption of amino acid side chains, Prog. Biophys. Mol. Biol., 2000, 74, 141–173 CrossRef CAS PubMed.
- A. Barth, Infrared spectroscopy of proteins, Biochim. Biophys. Acta, Bioenerg., 2007, 1767, 1073–1101 CrossRef CAS PubMed.
- B. Bjellqvist, B. Basse, E. Olsen and J. E. Celis, Reference points for comparisons of two-dimensional maps of proteins from different human cell types defined in a pH scale where isoelectric points correlate with polypeptide compositions, Electrophoresis, 1994, 15, 529–539 CrossRef CAS PubMed.
- J. S. Church, G. L. Corino and A. L. Woodhead, The analysis of merino wool cuticle and cortical cells by Fourier transform Raman spectroscopy, Biopolymers, 1997, 42, 7–17 CrossRef CAS.
- R. Paquin and P. Colomban, Nanomechanics of single keratin fibres, J. Raman Spectrosc., 2007, 38, 504–514 CrossRef CAS.
- M. J. Duer, N. McDougal and R. C. Murray, A solid-state NMR study of the structure and molecular mobility of α-keratin, Phys. Chem. Chem. Phys., 2003, 5, 2894–2899 RSC.
- M. Baias, D. E. Demco, D. Istrate, C. Popescu, B. Blümich and M. Möller, Morphology and Molecular Mobility of Fibrous Hard α-Keratins by 1H, 13C, and 129Xe NMR, J. Phys. Chem. B, 2009, 113, 12136–12147 CrossRef CAS PubMed.
- M. Ghosh, B. P. Prajapati, N. Kango and K. K. Dey, A comprehensive and comparative study of the internal structure and dynamics of natural β-keratin and regenerated β-keratin by solid state NMR spectroscopy, Solid State Nucl. Magn. Reson., 2019, 101, 1–11 CrossRef CAS PubMed.
- A. Shavandi, A. Carne, A. A. Bekhit and A. E.-D. A. Bekhit, An improved method for solubilisation of wool keratin using peracetic acid, J. Environ. Chem. Eng., 2017, 5, 1977–1984 CrossRef CAS.
- N. Nishikawa, Y. Tanizawa, S. Tanaka, Y. Horiguchi and T. Asakura, Structural change of keratin protein in human hair by permanent waving treatment, Polymer, 1998, 39, 3835–3840 CrossRef CAS.
- M. J. Richardson and J. H. Johnston, Sorption and binding of nanocrystalline gold by Merino wool fibres—An XPS study, J. Colloid Interface Sci., 2007, 310, 425–430 CrossRef CAS.
- T. M. Florence, Degradation of protein disulphide bonds in dilute alkali, Biochem. J., 1980, 189, 507–520 CrossRef CAS PubMed.
- R. Liu, L. Li, S. Liu, S. Li, X. Zhu, M. Yi and X. Liao, Structure and properties of wool keratin/poly (vinyl alcohol) blended fiber, Adv. Polym. Technol., 2018, 37, 2756–2762 CrossRef CAS.
- E. Senoz, R. P. Wool, C. W. J. McChalicher and C. K. Hong, Physical and chemical changes in feather keratin during pyrolysis, Polym. Degrad. Stab., 2012, 97, 297–307 CrossRef CAS.
- M. Brebu and I. Spiridon, Thermal degradation of keratin waste, J. Anal. Appl. Pyrolysis, 2011, 91, 288–295 CrossRef CAS.
- J. Cao, Melting study of the α-form crystallites in human hair keratin by DSC, Thermochim. Acta, 1999, 335, 5–9 CrossRef CAS.
- X. Wang, Z. Shi, Q. Zhao and Y. Yun, Study on the Structure and Properties of Biofunctional Keratin from Rabbit Hair, Materials, 2021, 14, 379 CrossRef CAS.
- S. Sharma, A. Gupta, A. Kumar, C. G. Kee, H. Kamyab and S. M. Saufi, An efficient conversion of waste feather keratin into ecofriendly bioplastic film, Clean Technol. Environ. Policy, 2018, 20, 2157–2167 CrossRef CAS.
- K. Wang, R. Li, J. H. Ma, Y. K. Jian and J. N. Che, Extracting keratin from wool by using variant, Green Chem., 2016, 18, 476–481 RSC.
- V. Guccini, A. Carlson, S. Yu, G. Lindbergh, R. W. Lindström and G. Salazar-Alvarez, Highly proton conductive membranes based on carboxylated cellulose nanofibres and their performance in proton exchange membrane fuel cells, J. Mater. Chem. A, 2019, 7, 25032–25039 RSC.
- K. Yang, Y. Zhou, Z. Wang, M. Li, D. Shi, X. Wang, T. Jiang, Q. Zhang, B. Ding and J. You, Pseudosolvent Intercalator of Chitin, Adv. Mater., 2021, 33, 2007596 CrossRef CAS PubMed.
Footnote |
| † Electronic supplementary information (ESI) available. See DOI: https://doi.org/10.1039/d4qm00449c |
| This journal is © the Partner Organisations 2025 |