
 Open Access Article
Open Access Article This Open Access Article is licensed under a
This Open Access Article is licensed under a Creative Commons Attribution 3.0 Unported Licence
Hydrosilylation and hydrogermylation of white phosphorus†
Jose
Cammarata
a,
Maximilian
Schimpf
a,
Daniel J.
Scott
 *b and
Robert
Wolf
*a
*b and
Robert
Wolf
*a
aInstitute of Inorganic Chemistry, University of Regensburg, 93040 Regensburg, Germany. E-mail: robert.wolf@ur.de
bDepartment of Chemistry, University of Bath, Claverton Down, Bath, BA2 7AY, UK. E-mail: ds2630@bath.ac.uk
First published on 17th April 2025
Abstract
The development of efficient, direct strategies for the transformation of white phosphorus (P4) into useful monophosphorus compounds, as alternatives to the current wasteful and hazardous indirect processes, remains a significant challenge. Encouragingly, recent reports have shown that the reduction of P4 with organotin hydrides and subsequent functionalisation with electrophiles allows for the efficient synthesis of an array of industrially relevant monophosphines in a ‘one-pot’ manner. However, despite the practical and conceptual simplicity, the appeal of this method is limited by the inherent toxicity of most organotin derivatives. Here, we address this problem through experimental and computational studies of the reactivity of lighter and less toxic hydrogermane and hydrosilane homologues of organotin hydrides (R3EH, E = Ge or Si) towards P4. These hydroelementation reactions can be employed to directly transform P4 into useful monophosphorus compounds, in a simple ‘one-pot’ fashion similar to the original organotin-based systems.
Introduction
White phosphorus, P4, is by far the most reactive and industrially relevant allotrope of the element. It serves as the synthetic precursor for all commercially valuable and academically important organophosphorus compounds (OPCs).1–4 Current industrial routes for converting P4 into these useful OPCs involve indirect multi-step processes such as (oxy)chlorination reactions with chlorine gas (Cl2) to produce phosphorus chloride intermediates (PCl3/PCl5/POCl3), or disproportionation reactions under acidic or basic conditions to generate phosphine gas (PH3). The desired OPCs are then obtained by functionalisation of these intermediates with nucleophiles or by hydrophosphination reactions of unsaturated organic compounds, respectively (Scheme 1a).1–6 As a result, significant efforts have been made to develop alternative strategies for the functionalisation of P4 that avoid the use of such hazardous reactants and intermediates.7–11 | ||
| Scheme 1 (a) Current state-of-the-art routes for the transformation of P4 into P1 products. (b) Previously reported hydrostannylation of P4 and subsequent treatment with electrophiles to afford P1 compounds directly from P4. (c) Hydroelemention of P4 with lighter main group hydrides, reported herein. E+ represents a generic electrophile. | ||
Notably, in recent years various strategies have been reported for converting P4directly into one or more P1 products.12–14 These remain at early stages of development, but include the degradation of P4 by divalent silicon species,15 photocatalytic reactions,16,17 electrochemical degradation,18 and oxidative ‘onioation’ of P4.19 Among these new approaches, our group recently reported a simple method that can convert both white and red phosphorus (Pred) into stannyl phosphines (Bu3Sn)xPH3−x (x = 0–3) using the ‘classical’ radical reagent Bu3SnH and initiation by light or a chemical radical initiator such as azobis(isobutyronitrile) (AIBN).20,21 The resulting stannylphosphine mixture serves as a P3− source, affording industrially relevant monophosphorus compounds upon treatment with various electrophiles in a ‘one-pot’ protocol (Scheme 1b). Moreover, simple procedures have been developed for the closed-loop recycling of the tin hydride reagent, thus minimising organotin waste.20,21
Unfortunately, the use of most common organotin compounds raises fundamental concerns around toxicity that cannot be fully mitigated, even by these recycling strategies (although some much less toxic organotin derivatives are known, albeit typically at the cost of significantly higher molecular weight).22 The replacement of R3SnH with more benign options has long been an important goal for synthetic free-radical chemistry, and many alternative reagents have been proposed for a variety of other chemical transformations.23–26 Particularly appealing in the context of P4 reduction are lighter group 14 hydrides, which in other contexts are known to undergo reactions analogous to those of R3SnH, although they display an intrinsically lower propensity for homolysis of the element-hydrogen bonds.27
Here, we present the first examples of hydrogermylation and hydrosilylation of P4. Utilizing LED light irradiation, these reactions produce mixtures of germanyl- and silylphosphanes that can be transformed into valuable P1 products via simple one-pot procedures (see Scheme 1c). Our findings significantly expand the application of hydroelementation as a general method for synthesizing useful organophosphorus compounds in a single reaction step from white phosphorus.
Results and discussion
Inspired by the practical and conceptual simplicity of the hydrostannylation of P4, we anticipated that this radical-based activation of P4 could be extended to the use of lighter R3EH (E = Ge or Si). To evaluate the viability of such a process, we performed a computational investigation at the outset of this project. DFT studies at the PBE-D3(BJ)/def2-TZVP level of theory were focused on the first P–P bond cleavage step of the reaction of P4 with truncated model radicals Me3E˙ (E = Ge, Si, Sn) and the subsequent hydrogen atom abstraction (HAT) step (Fig. 1a). The HAT step was found to be rate limiting for Me3SnH.28 Notably, while the addition of Me3Sn˙ to P4 is energetically uphill (2.2 kcal mol−1), the same process is barrierless and downhill for both other Me3E˙ (E = Ge, Si), forming the ‘butterfly’ P4 radical intermediates (Me3E)P4˙. The subsequent HAT step to (Me3E)P4˙ from another equivalent of Me3EH then proceeds over activation barriers that are significantly higher for Si and Ge than for the heavier analogue Sn (23.4, 18.7 and 10.8 kcal mol−1, respectively) consistent with expected differences in the E–H bond strengths (Fig. 1b, for calculated bond dissociation energies see ESI, Table S1†).24,27,29,30 Nevertheless, all of these barriers should be easily accessible even at room temperature, suggesting that the desired hydro-elementation should be feasible. An analysis of the influence of the substituents in hydrides R3EH (E = Si, R = SiMe3, Ph, Me; E = Ge, R = Ph, Me) revealed the expected trend. The lowest barrier for the hydroelementation of the first P–P bond was observed for (Me3Si)3SiH. The barriers for Me3EH are generally higher than for Ph3EH (see Fig. S2, ESI†). | ||
| Fig. 1 (a) Model hydroelementation of the first P–P bond in P4. (b) Calculated mechanism, via attack of [E]˙ and subsequent HAT (relative free energies in kcal mol−1). [E] = Me3Si, Me3Ge or Me3Sn. For simplicity, stereochemistry is not shown. | ||
Hydrogermylation of P4
Encouraged by the computational results, we began experimentally with investigation of the hydrogermylation of P4. Notably, despite the stronger Ge–H bond, the commercially available organogermanes Bu3GeH and Ph3GeH have both been used as alternatives to replace organotin compounds in various radical reactions.23,25,31 We first tested the reactivity of Bu3GeH towards P4, under similar conditions to those used for Bu3SnH.20 Unfortunately, in sharp contrast to the efficient reaction of P4 with R3SnH, the 31P{1H} NMR spectra of the reaction mixtures mainly showed unconsumed P4 at −521 ppm (for full details see ESI, Section 3.1†). However, when Ph3GeH, which has a weaker Ge–H bond (see ESI, Table S1†),29 was used instead, additional minor resonances were detected at −240.5 and −248.0 ppm. Markedly, significant amounts of orange precipitate had formed at the end of the reactions with both Bu3GeH and Ph3GeH, suggesting the formation of red phosphorus or other insoluble polyphosphorus compounds (presumably (R3Ge)xPyHz; more detailed characterisation is hampered by the insolubility of these species).Further investigations revealed that longer reaction times and irradiation with near UV LED light (365 nm) favoured the conversion of P4 into the new species (Scheme 2a).‡ These can be assigned as (Ph3Ge)2PH (1; −240.5 ppm) and Ph3GePH2 (2; −248.0 ppm), both by analogy to the major products observed in the previous hydrostannylation reactions and by comparison with the chemical shifts reported for related Ge-substituted phosphines.20,32 This assignment is further supported by 1H-coupled 31P NMR spectra, where these resonances appear as a doublet (1J(31P–1H) = 185 Hz) and a triplet (1J(31P–1H) = 181 Hz), respectively (Scheme 2b). Corresponding doublets arising from coupling between 1H and 31P nuclei could also be observed in the 1H NMR spectra (see ESI, Fig. S7†). A single-crystal X-ray diffraction study confirmed the identity of 1, whose structure is analogous to the recently reported (TerMe2Sn)2PH, which is similarly a product of P4 hydroelementation (Ter = 2,6-Mes2C6H3).28 An additional, minor resonance was always also observed during these investigations, appearing as a small singlet at −125.8 ppm in the 31P{1H} NMR spectra. This signal splits into a doublet in the 1H-coupled 31P NMR spectrum (1J(31P–1H) = 199 Hz) and is attributed to Ph3GeP(H)Ph (3) forming as a minor side product (Scheme 2b).§33 Note that unlike with Bu3SnH, no significant tertiary phosphine (i.e. (Ph3Ge)3P) or PH3 products were observed.
 | ||
| Scheme 2 (a) Hydrogermylation of P4 with Ph3GeH promoted by near-UV LED irradiation (365 nm). (b) Insets of the 31P NMR spectrum for the reaction of P4 (0.01 mmol) with Ph3GeH (3 equiv. per P atom) driven by 365 nm LED irradiation. (c) Single-crystal XRD structure of [Ge]2PH (1). [Ge] = Ph3Ge. Thermal ellipsoids are shown at 50%. H atoms, except for the one bound directly to P, are omitted for clarity. C atoms are shown in grey, H is white, P in orange, and Ge in dark green. | ||
Under these reaction conditions, the conversion to the main products (1 and 2) was still relatively limited (<54%), likely due to the continuing formation of the insoluble (Ph3Ge)xPyHz compounds noted above. Gratifyingly, however, using an excess of Ph3GeH and more concentrated reaction mixtures resulted in significantly improved reaction outcomes with conversions up to 87% (for 1 and 2; 95% including 3), reaction times reduced to 24 hours (see Scheme 3a and ESI, Table S2†), and clear yellowish solutions with no observable precipitates at the end of the reaction. Orange precipitates still form at the beginning of these reactions but later disappear,¶ which may suggest that the initially formed (Ph3Ge)xPyHz compounds are in fact still available during the overall reaction course, serving as a source of P atoms in the formation of the final products. A similar degradation process has been described for the reaction of Pred with Bu3SnH.21 Consistent with this suggestion, reactions of Pred with Ph3GeH gave the same hydrogermanylphosphines 1 and 2, albeit with reduced conversion (39%, see ESI, Section 3.4†). Nevertheless, this provides a proof-of-principle confirmation that bench-stable Pred can also be functionalised using germanium hydrides.
 | ||
| Scheme 3 (a) Optimised conditions for the hydrogermylation of P4 promoted by near-UV irradiation (365 nm). (b) Proposed radical chain mechanism for P4 hydrogermylation, where [P]–[P] represents a generic P–P bond. | ||
We propose that the hydrogermylation of P4 follows a radical chain mechanism analogous to the hydrostannylation of P4 and Pred (Scheme 3b).|| This is also consistent with the calculations noted above. Nevertheless, the observation of significant formation of insoluble (R3E)xPyHz is a clear point of contrast between R3GeH and R3SnH.
To understand why this difference might occur, we considered the aggregation of P4 to form larger (R3E)xPyHz moieties computationally. For simplicity, calculations were limited to a single representative step: addition of further P4 to the initial radical intermediate (Me3E)P4˙, which was calculated for all three model systems, E = Sn/Ge/Si. This step would result initially in a P8 “double butterfly” radical intermediate ((Me3E)P8˙), en route to further aggregation and rearrangement steps (Fig. 2). Notably, for all three systems, this step was found to be less energetically demanding than the calculated HAT step that would lead to hydroelementation (cf.Fig. 1b), confirming that aggregation to larger (R3E)xPyHz fragments is likely to be mechanistically relevant. However, for Me3Sn˙ the difference in activation barriers is relatively modest (2.2 kcal mol−1; cf.Fig. 1b and 2 or see ESI, Fig. S1†), whereas for Me3Ge˙ and especially Me3Si˙ it is much greater (10.6 kcal mol−1 and 13.0 kcal mol−1, respectively). This is mostly due to differences in the HAT step, which is sensitive to changes in the E–H bond strength. In comparison, the identity of the Me3E substituent has relatively little impact on the barrier to further P4 addition since it is distant from the active radical site. The results are consistent with the idea that for R3SnH the desired hydrostannylation is sufficiently kinetically competitive with aggregation to prevent significant formation of large, insoluble (R3E)xPyHz fragments. In contrast, for R3GeH and R3SiH there is a much stronger kinetic preference for aggregation, suggesting that large, insoluble (R3E)xPyHz aggregates should be more prevalent and that their precipitation should limit the overall reaction rate, consistent with experimental observations. This problem should become more severe as E–H bond strength increases, consistent with the relative performance of Bu3GeH and Ph3GeH.
 | ||
| Fig. 2 Calculated aggregation between P4 and the initial intermediate [E]P4˙ (relative free energies in kcal mol−1). [E] = Me3Si, Me3Ge or Me3Sn. For simplicity, stereochemistry is not shown. | ||
Functionalization of Ph3GePH2 and (Ph3Ge)2PH
After optimising the hydrogermylation of P4, we next focused on the synthetic utilization of the newly formed Ge-substituted phosphines 1 and 2 in a ‘one-pot’ fashion. Notably, simple acidification of the crude mixture with HCl (in 1,4-dioxane) prompted the cleavage of the P–Ge bonds, resulting in the formation of PH3 (4, Scheme 4, top) with similar efficiency to that achieved using Bu3SnH (≥50% conversion, cf. ≥56% via Bu3SnH). Furthermore, treatment of the crude mixture with benzyl bromide (BnBr) or bromoethane (EtBr) in the presence of base yielded the phosphonium salts [Bn4P]Br (5) and [Et4P]Br (6), with the former being isolated at preparative scale in 77% yield (Scheme 4, cf. 80% via Bu3SnH). These results confirm that the P–Ge bonds in the new P1 intermediate mixture are sufficiently reactive to allow functionalisation into desirable P1 compounds although they require longer reaction times and higher temperatures than with (Bu3Sn)xPH3−x.20 Reactions of the crude mixture of 1 and 2 with other representative electrophiles that reacted successfully with (Bu3Sn)xPH3−x, such as paraformaldehyde or acyl chlorides, have so far only resulted in low conversions and/or incomplete reactions (see ESI, Section 3.7†), indicating that this equivalence is not universal.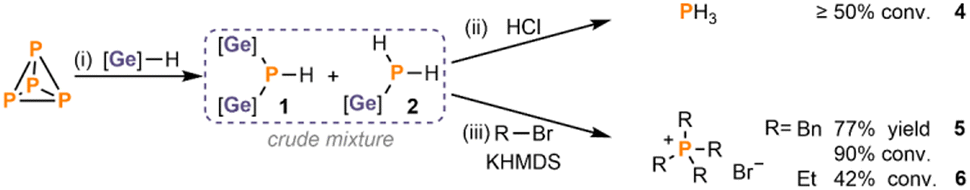 | ||
| Scheme 4 Functionalisation of the crude mixture of 1 and 2 derived from P4 directly into useful P1 products. Equivalents are defined per P atom. (i) P4 (0.25 equiv.), Ph3GeH (3 equiv.), PhMe, 365 nm LEDs, RT, 24 h. (ii) Transformation of P4 into PH3 from crude mixture (1 and 2): 10 equiv. HCl (4.0 M in 1,4-dioxane) RT, 1 (h). (iii) Preparation of phosphonium salts [R4P]Br from crude mixture (1 and 2): 10 equiv. RBr (R = Bn or Et), 2.5 equiv. KHMDS, 100 °C, 3d. [Ge] = Ph3Ge. | ||
Hydrosilylation of P4
Having established successful hydrogermylation of P4, focus was then shifted to the generally cheaper and more readily available hydrosilanes, R3SiH (cf. the more specialised R3GeH). Hydrosilanes, however, are known to be poor radical chain reagents due to their relatively strong Si–H bonds.30,34 Moreover, the calculations discussed above imply that formation of insoluble (R3Si)xPyHz is likely to be an even greater problem for R3SiH than for R3GeH. Indeed, initial reactivity studies confirmed that the combination of R3SiH (R = Ph, Et) with P4 under the same conditions previously established for either R3SnH or R3GeH leads to either no reaction (see ESI, Section 4.1†) or formation of insoluble (R3Si)xPyHz compounds only. Even when the hypersilane (Me3Si)3SiH, known for its higher reactivity as a radical reagent due to the hyperconjugation effect of the Me3Si groups,26,30,35 was used, only trace new resonances (and unreacted P4) were observed in the 31P{1H} NMR spectra when it was reacted with P4 (Scheme 5a). | ||
| Scheme 5 (a) Reactivity studies of silanes R3SiH towards P4 driven by LED irradiation (456 nm or 365 nm). (b) Proposed thiol-assisted hydrosilylation of P4, where [P]–[P] represents a generic P–P bond. | ||
To overcome similar problems in the radical hydrosilylation of unsaturated C![[double bond, length as m-dash]](https://www.rsc.org/images/entities/char_e001.gif) C bonds, organic chemists have developed the use of simple thiols as hydrosilylation catalysts.36,37 The origin of this catalysis lies in the poor electronic compatibility between the hydrosilane and C-centred radical (both nucleophilic), and much better electronic match between these species and the sulfanyl radical and thiol (both electrophilic), respectively.38 Given the well-known, broad chemical similarity between P and C, and the fact that P is even slightly more electropositive than C, we speculated that a similar thiol-assisted reaction could facilitate the key HAT process and enable the direct hydrosilylation of P4 to (R3Si)nPH3−n, as outlined in Scheme 5b.
C bonds, organic chemists have developed the use of simple thiols as hydrosilylation catalysts.36,37 The origin of this catalysis lies in the poor electronic compatibility between the hydrosilane and C-centred radical (both nucleophilic), and much better electronic match between these species and the sulfanyl radical and thiol (both electrophilic), respectively.38 Given the well-known, broad chemical similarity between P and C, and the fact that P is even slightly more electropositive than C, we speculated that a similar thiol-assisted reaction could facilitate the key HAT process and enable the direct hydrosilylation of P4 to (R3Si)nPH3−n, as outlined in Scheme 5b.
Gratifyingly, the reaction of (Me3Si)3SiH and P4 in the presence of an excess of a thiol such as iPr3SiSH led to the full consumption of P4 and the formation of a mixture of Si/H-substituted monophosphorus products (Scheme 6), which appeared as singlets between ca. −210 and −280 ppm in the 31P{1H} NMR spectrum. The main species formed were identified based on the 31P{1H} and 31P NMR spectra (Scheme 6b and c, also see ESI, Fig. S30 and S31†).39,40 The major product was the primary phosphine [(Me3Si)3Si]PH2 (7, −266.5 ppm), followed by [(Me3Si)3Si]P(H)SiMe3 (8, −257.2 ppm), Me3SiPH2 (9, −236.5 ppm) and (Me3Si)2PH (10, −236.3 ppm), which contain Me3Si groups directly bound to the phosphorus atom, indicating Si–Si bond cleavage within the hypersilane motif. Whether this cleavage occurs primarily before or after the hydrosilylation is presently unclear.41 Satisfyingly, upon optimisation, hydrosilylation via this procedure yielded the major reduced P1 products in almost 80% combined yield (Scheme 6d, also see ESI, Table S6†). The hydrosilylation of P4 was also achieved with other thiol derivatives, RSH (R = Cy, Ad, Ph, 4-MePh), and even with 1,4-cyclohexadiene (1,4-CHD; see ESI, Table S7 and Fig. S32–37†).42,43 Optimisation studies revealed that the use of substoichiometric amounts of the hydrogen atom donor was detrimental to overall conversions. Additionally, other, less activated silanes R3SiH (R = Ph, Et, Me3SiO) were found to be ineffective, even when an excess of additive was used (see ESI, Table S8†).
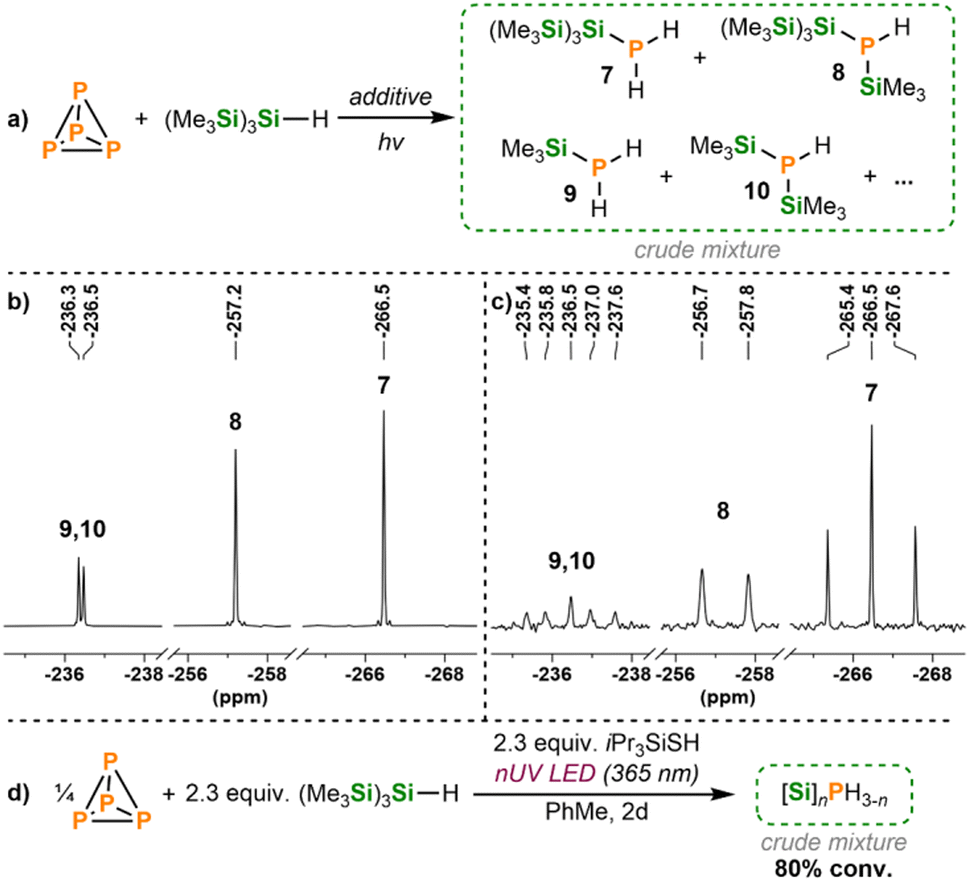 | ||
| Scheme 6 (a) Hydrosilylation of P4 with (Me3Si)3SiH in the presence of a hydrogen atom transfer donor, promoted by LED irradiation. Insets of the 31P{1H} (b) and 31P (c) NMR spectra for the reaction of P4 with (Me3Si)3SiH (3.5 equiv. per P atom) and iPr3SiSH (3.5 equiv. per P atom) in hexane and driven by 365 nm LED irradiation, for the full version of the spectra see ESI, Fig. S30 and S31.† (d) Optimised conditions for the hydrosilylation of P4 in the presence of iPr3SiSH, promoted by near-UV irradiation. | ||
Functionalization of crude [Si]nPH3−n mixture
Finally, we examined the reactivity of the crude phosphine mixture obtained from the hydrosilylation of P4 in the presence of a HAT donor to access monophosphosphorus compounds in ‘one-pot’ transformations. Simple acidification with HCl (in 1,4-dioxane) resulted in the efficient cleavage of the P–Si bonds and the selective formation of PH3 (4) with good conversion (≥64%; Scheme 7, top). Additionally, various phosphonium salts including [Bn4P]Br (5, isolated in 70% yield), [Et4P]Br (6, 54% conversion), and [Bu4P]Br (11, 40% conversion, marketed commercially as CYPHOS 442W) were also accessible directly from P4 after reaction of the [Si]nPH3−n mixture with the corresponding alkyl bromides and base (Scheme 7, middle). Furthermore, treatment of the hydrosilylphosphine mixture with paraformaldehyde in EtOH resulted in exclusive formation of the industrially relevant phosphonium salt THPC (tetrakis(hydroxymethyl)phosphonium chloride, 12, 48% conversion) after quenching with HCl (Scheme 7, bottom). Although these new reactions generally require more forcing conditions than when starting from (Bu3Sn)xPH3−x, they demonstrate that the [Si]nPH3−n mixture has a similar potential to act as an efficient P1 precursor and P3− synthon. | ||
| Scheme 7 Functionalisation of crude [Si]nPH3−n mixture into useful P1 products directly from P4. Equivalents are defined per P atom. (i) P4 (0.25 equiv.), (Me3Si)3SiH (2.25 equiv.), iPr3SiSH or 1,4-CHD (2.25 equiv.), PhMe, 365 nm LEDs, RT, 2d. (ii) One-pot, selective transformation of P4 into PH3 from crude [Si]nPH3−n: 10 equiv. HCl (4.0 M in 1,4-dioxane) RT, 1 (h). (iii) Preparation of phosphonium salts [R4P]Br from crude [Si]nPH3−n: 10 equiv. RBr (R = Bn, Et or Bu), 2.5 equiv. KHMDS, 100 °C, 3d. (iv) preparation of THPC from crude [Si]nPH3−n: EtOH, 12.5 equiv. paraformaldehyde, 50 °C, 2d, then 10 equiv. HCl (4.0 M in 1,4-dioxane), RT, 2h. [Si] = (Me3Si)3Si or Me3Si. | ||
Conclusions
We have described herein our studies into the fundamental reactivity of hydrogermanes and hydrosilanes towards P4, as part of our mission to overcome safety concerns associated with the use of tributyltin compounds. Satisfyingly, the hydroelementation of P4 is successful with both families of lighter homologues R3EH. While our computational and experimental studies both reveal relevant differences in reactivity (which are related to the differing E–H bond strengths), these can be overcome through rational optimisation of the reaction conditions. Furthermore, these hydroelementation reactions yield similarly functionalisable [E]nPH3−n mixtures which can serve as P1 precursors for useful organophosphorus compounds in a ‘one-pot’ reaction. These results clearly show that the desired P4 functionalisation is not limited to organotin derivatives and is in fact generalisable. Extrapolation from these results implies that extension to other known R3SnH alternatives should be possible, many of which are known to compete well with R3SnH in other contexts. Efforts in this direction are currently ongoing in our laboratories. Of particular interest to us is the extension of these results to more robust R3SnH surrogates, to facilitate their recycling and reuse and/or catalytic application (cf. Bu3SnH).20 Besides the obvious efficiency benefits, such (pseudo)catalytic approaches would also remove the need for stoichiometric, ‘upstream’ Cl2 use.**Author contributions
JC and DJS developed the hydrosilylation and hydrogermylation procedures. MS performed preliminary experiments with different R3GeH. JC performed the mechanistic calculations, developed the functionalisation of the crude [R3E]nPH3−n mixtures (E = Si, Ge) and the procedures for product isolation and characterisation. DJS and RW conceived, oversaw, and directed the project. JC and DJS prepared the manuscript, with input from all authors.Data availability
The data supporting the findings of this study are available within the article and its ESI.† Crystallographic information for 1 has been deposited at the CCDC under 2408662.Conflicts of interest
A patent covering all of the results described herein has been filed (as of 13 February 2020) by the University of Regensburg (EP 20,157,197.3; inventors, D. J. S. and R. W.). The authors declare no other competing interests.Acknowledgements
We thank Maria K. Uttendorfer for the acquisition and processing of the XRD data. DJS would like to thank the Alexander von Humboldt foundation for the award of a postdoctoral fellowship and the EPSRC for an Early Career Fellowship (EP/V056069/1). RW would like to thank the DFG (WO 1496/12-1, project number 548830090) for financial support.References
- D. E. C. Corbridge, Phosphorus. Chemistry, Biochemistry and Technology, Elsevier, 2000 Search PubMed.
- J. Svara, N. Weferling and T. Hofmann, Phosphorus Compounds, Organic, Ullmann's Encycl. Ind. Chem., 2012, 27, 20–50 Search PubMed.
- H. Diskowski and T. Hofmann, Phosphorus, Ullmann's Encycl. Ind. Chem., 2012, 26, 725–746 Search PubMed.
- N. Weferling, S. M. Zhang and C. H. Chiang, Commercial Organophosphorus Chemicals: Status and New Developments, Procedia Eng., 2016, 138, 291–301 CrossRef CAS.
- G. Bettermann, W. Krause, G. Riess and T. Hofmann, Phosphorus Compounds, Inorganic, Ullmann's Encycl. Ind. Chem., 2012, 27, 1–18 Search PubMed.
- A. R. Jupp, S. Beijer, G. C. Narain, W. Schipper and J. C. Slootweg, Phosphorus recovery and recycling-closing the loop, Chem. Soc. Rev., 2021, 50, 87–101 RSC.
- J. E. Borger, A. W. Ehlers, J. C. Slootweg and K. Lammertsma, Functionalization of P4 through Direct P–C Bond Formation, Chem. – Eur. J., 2017, 23, 11738–11746 CrossRef CAS PubMed.
- N. K. Gusarova and B. A. Trofimov, Organophosphorus chemistry based on elemental phosphorus: advances and horizons, Russ. Chem. Rev., 2020, 89, 225–249 CrossRef CAS.
- B. M. Cossairt, N. A. Piro and C. C. Cummins, Early-Transition-Metal-Mediated Activation and Transformation of White Phosphorus, Chem. Rev., 2010, 110, 4164–4177 CrossRef CAS PubMed.
- M. Caporali, L. Gonsalvi, A. Rossin and M. Peruzzini, P4 Activation by Late-Transition Metal Complexes, Chem. Rev., 2010, 110, 4178–4235 CrossRef CAS PubMed.
- M. Scheer, G. Balázs and A. Seitz, P4 Activation by Main Group Elements and Compounds, Chem. Rev., 2010, 110, 4236–4256 CrossRef CAS PubMed.
- D. J. Scott, Recent Breakthroughs in P4 Chemistry: Towards Practical, Direct Transformations into P1 Compounds, Angew. Chem., Int. Ed., 2022, 61, e2022050 CrossRef PubMed.
- J. Hu, W. Liu and W. X. Zhang, Direct functionalization of white phosphorus by organolithium reagents to organophosphorus compounds, Phosphorus, Sulfur Silicon Relat. Elem., 2022, 197, 398–407 CrossRef CAS.
- Y. Liu, X. Chen and B. Yu, Sustainable Photo- and Electrochemical Transformation of White Phosphorous (P4) into P1 Organo-Compounds, Chem. – Eur. J., 2023, 29, e202302142 CrossRef CAS PubMed.
- Y. Wang, T. Szilvási, S. Yao and M. Driess, A bis(silylene)-stabilized diphosphorus compound and its reactivity as a monophosphorus anion transfer reagent, Nat. Chem., 2020, 12, 801–807 CrossRef CAS PubMed.
- U. Lennert, P. B. Arockiam, V. Streitferdt, D. J. Scott, C. Rödl, R. M. Gschwind and R. Wolf, Direct catalytic transformation of white phosphorus into arylphosphines and phosphonium salts, Nat. Catal., 2019, 2, 1101–1106 CrossRef CAS PubMed.
- P. B. Arockiam, U. Lennert, C. Graf, R. Rothfelder, D. J. Scott, T. G. Fischer, K. Zeitler and R. Wolf, Versatile Visible-Light-Driven Synthesis of Asymmetrical Phosphines and Phosphonium Salts, Chem. – Eur. J., 2020, 26, 16374–16382 CrossRef CAS PubMed.
- Y. Mei, Z. Yan and L. L. Liu, Facile Synthesis of the Dicyanophosphide Anion via Electrochemical Activation of White Phosphorus: An Avenue to Organophosphorus Compounds, J. Am. Chem. Soc., 2022, 144, 1517–1522 CrossRef CAS PubMed.
- M. Donath, K. Schwedtmann, T. Schneider, F. Hennersdorf, A. Bauzá, A. Frontera and J. J. Weigand, Direct conversion of white phosphorus to versatile phosphorus transfer reagents via oxidative onioation, Nat. Chem., 2022, 14, 384–391 CrossRef CAS PubMed.
- D. J. Scott, J. Cammarata, M. Schimpf and R. Wolf, Synthesis of monophosphines directly from white phosphorus, Nat. Chem., 2021, 13, 458–464 CrossRef CAS PubMed.
- J. Cammarata, D. J. Scott and R. Wolf, Hydrostannylation of Red Phosphorus: A Convenient Route to Monophosphines, Chem. – Eur. J., 2022, 28, e2022024 CrossRef PubMed.
- N. J. Snoeij, A. A. J. van Iersel, A. H. Penninks and W. Seinen, Toxicity of triorganotin compounds: Comparative in vivo studies with a series of trialkyltin compounds and triphenyltin chloride in male rats, Toxicol. Appl. Pharmacol., 1985, 81, 274–286 CrossRef CAS PubMed.
- P. A. Baguley and J. C. Walton, Flight from the Tyranny of Tin: The Quest for Practical Radical Sources Free from Metal Encumbrances, Angew. Chem., Int. Ed., 1998, 37, 3072–3082 CrossRef CAS PubMed.
- B. C. Gilbert and A. F. Parsons, The use of free radical initiators bearing metal-metal, metal-hydrogen and non-metal-hydrogen bonds in synthesis, J. Chem. Soc., Perkin Trans. 1, 2002, 2, 367–387 RSC.
- W. R. Bowman, S. L. Krintel and M. B. Schilling, Tributylgermanium hydride as a replacement for tributyltin hydride in radical reactions, Org. Biomol. Chem., 2004, 2, 585–592 RSC.
- C. Chatgilialoglu, (Me3Si)3SiH: Twenty Years After Its Discovery as a Radical-Based Reducing Agent, Chem. – Eur. J., 2008, 14, 2310–2320 CrossRef CAS PubMed.
- C. Chatgilialoglu and M. Newcomb, Hydrogen Donor Abilities of the Group 14 Hydrides, Adv. Organomet. Chem., 1999, 44, 67–112 CrossRef CAS For comparison of the E–H bond strength and the rate constants for H-abstraction with carbon-centred radicals (e.g. alkyl radicals) from R3EH (E = Si, Ge, Sn), also see refs: 23, 28 and 29.
- J. Cammarata, F. F. Westermair, P. Coburger, D. Duvinage, M. Janssen, M. K. Uttendorfer, J. Beckmann, R. M. Gschwind, R. Wolf and D. J. Scott, Unravelling White Phosphorus: Experimental and Computational Studies Reveal the Mechanisms of P4 Hydrostannylation, Angew. Chem., Int. Ed., 2024, 63, e202408423 CAS.
- C. Chatgilialoglu and M. Ballestri, Hydrogen Donor Abilities of Germanium Hydrides, Organometallics, 1999, 18, 2395–2397 CrossRef CAS.
- C. Chatgilialoglu, Structural and Chemical Properties of Silyl Radicals, Chem. Rev., 1995, 95, 1229–1251 CrossRef CAS.
- H. Yorimitsu and K. Oshima, Recent advances in the use of tri(2-furyl)germane, triphenylgermane and their derivatives in organic synthesis, Inorg. Chem. Commun., 2005, 8, 131–142 CrossRef CAS.
- D. M. Schubert and A. D. Norman, Synthesis of (Trimethylsilyl)- and (Trimethylgermyl)allylphosphine, Inorg. Chem., 1985, 24, 1107–1109 CrossRef CAS.
- J. Escudié, C. Couret and J. Satgé, Reactions d'insertion du phénylphosphinidène dans diverses liaisons germanium–hétéroélément, Recl. Trav. Chim. Pays-Bas, 1979, 98, 461–466 CrossRef.
- C. Chatgilialoglu, Organosilanes in Radical Chemistry, John Wiley & Sons Ltd, 2004 Search PubMed.
- C. Chatgilialoglu and J. Lalevée, Recent applications of the (TMS)3SiH radical-based reagent, Molecules, 2012, 17, 527–555 CrossRef CAS PubMed.
- H. S. Dang and B. P. Roberts, Polarity-reversal catalysis by thiols of radical-chain hydrosilylation of alkenes, Tetrahedron Lett., 1995, 36, 2875–2878 CrossRef CAS.
- B. P. Roberts, Polarity-reversal catalysis of hydrogen-atom abstraction reactions: Concepts and applications in organic chemistry, Chem. Soc. Rev., 1999, 28, 25–35 RSC.
- J. J. A. Garwood, A. D. Chen and D. A. Nagib, Radical Polarity, J. Am. Chem. Soc., 2024, 146, 28034–28059 CAS.
- V. Cappello, J. Baumgartner, A. Dransfeld and K. Hassler, Monophosphanes and diphosphanes with the hypersilyl substituent, Eur. J. Inorg. Chem., 2006, 4589–4599 CrossRef CAS.
- K. X. Bhattacharyya, S. Dreyfuss, N. Saffon-Merceron and N. Mézailles, P4 functionalization by hydrides: Direct synthesis of P–H bonds, Chem. Commun., 2016, 52, 5179–5182 RSC.
- M. Mohamed and M. A. Brook, Can. J. Chem., 2000, 78, 1357–1362 CrossRef.
- I. Chatterjee and M. Oestreich, B(C6F5)3-catalyzed transfer hydrogenation of imines and related heteroarenes using cyclohexa-1,4-dienes as a dihydrogen source, Angew. Chem., Int. Ed., 2015, 54, 1965–1968 CrossRef CAS PubMed.
- H. Bauer, K. Thum, M. Alonso, C. Fischer and S. Harder, Alkene Transfer Hydrogenation with Alkaline-Earth Metal Catalysts, Angew. Chem., Int. Ed., 2019, 58, 4248–4253 CrossRef CAS PubMed.
Footnotes |
| † Electronic supplementary information (ESI) available: General experimental procedures, full reaction optimisation, spectroscopic characterisation, computational details, and procedures for isolation of products. CCDC 2408662. For ESI and crystallographic data in CIF or other electronic format see DOI: https://doi.org/10.1039/d5qi00869g |
| ‡ Analogous resonances were also observed in the reaction with Bu3GeH under these conditions, although with lower conversions. For full details see ESI, Section S3.1.† |
| § Similar side/decomposition products have been observed in our previous study of the P4 hydrostannylation system, in the form of R3SnP(H)R, though in much smaller amounts. The fate of the remaining “Ph2Ge” fragment whose formation is implied by the presence of the P-bound Ph group is currently unclear. |
| ¶ During the optimisation of the reaction, no signals corresponding to soluble polyphosphorus intermediates were observed in the 31P{1H} NMR spectra. |
| || Irradiation with LED light is proposed to induce the formation of an initial R3Ge˙ or R3Si˙ radical which initiates the chain reaction. However, the precise details of this initiation are currently unclear and remain under investigation. Proof-of-concept results have also been achieved using chemical radical initators such as AIBN instead of LED irradiation. However, for these systems we have thus far only been able to achieve low conversions (<28% 1 and 2). For full details see ESI, Section S3.5 and S4.7†. |
| ** Synthesis of R3EH derivatives (E = Si, Ge, Sn) often relies on the use of Cl2 as a stoichiometric reagent (e.g. to prepare early intermediates such as ECl4). Thus, stoichiometric use of these reductants to activate P4 effectively shifts the use of Cl2 ‘upstream’, rather than eliminating it per se. By contrast, recycling of “R3EX” byproducts typically does not require additional Cl2. |
| This journal is © the Partner Organisations 2025 |